

Abstract
Hepatocellular carcinoma (HCC) is the major form of liver cancer for which there is no effective therapy. Genetic modification with T-cell receptors (TCRs) specific for HCC-associated antigens, such as α-fetoprotein (AFP), can potentially redirect human T cells to specifically recognize and kill HCC tumor cells to achieve antitumor effects. In this study, using lentivector and peptide immunization, we identified a population of cluster of differentiation 8 (CD8) T cells in human leukocyte antigen (HLA)-A2 transgenic AAD mice that recognized AFP158 epitope on human HCC cells. Adoptive transfer of the AFP158-specific mouse CD8 T cells eradicated HepG2 tumor xenografts as large as 2 cm in diameter in immunocompromised nonobese diabetic severe combined immunodeficient gamma knockout (NSG) mice. We then established T-cell hybridoma clones from the AFP158-specific mouse CD8 T cells and identified three sets of paired TCR genes out of five hybridomas. Expression of the murine TCR genes redirected primary human T cells to bind HLA-A2/AFP158 tetramer. TCR gene-engineered human T (TCR-T) cells also specifically recognized HLA-A2+AFP+ HepG2 HCC tumor cells and produced effector cytokines. Importantly, the TCR-T cells could specifically kill HLA-A2+AFP+ HepG2 tumor cells without significant toxicity to normal primary hepatocytes in vitro. Adoptive transfer of the AFP-specific TCR-T cells could eradicate HepG2 tumors in NSG mice.
Conclusion:
We have identified AFP-specific murine TCR genes that can redirect human T cells to specifically recognize and kill HCC tumor cells, and those AFP158-specific TCRs have a great potential to engineer a patient’s autologous T cells to treat HCC tumors.
Approximately 854,000 new cases of liver cancer, 85%−90% of which are hepatocellular carcinoma (HCC), are diagnosed annually, making it the sixth most common cancer worldwide.(1) According to the American Cancer Society, in the United States, the incidence rate of liver cancer increases the fastest among all cancers, and the number of liver cancer cases has doubled in the last decade, likely due to obesity/diabetes.(2) Liver cancer is the second leading cause of cancer death in adult men due to a lack of therapies.(3,4) Thus, new therapeutic approaches for liver cancer are urgently needed. Activation of the host immune system has been shown to generate significant antitumor effects.(5,6) For example, check point blockade unleashes a patient’s immune responses, generating therapeutic effects in a variety of cancers,(7) including HCC.(8) While that is promising, the antitumor efficacy of programmed death 1 (PD-1) blockade depends on the preexistence of tumor-specific infiltrating T cells that are not always available.(9) Additionally, unleashing nonspecific immunity by checkpoint blockade may cause autoimmune destruction. On the other hand, immunotherapy targeting tumor-specific or tumor-associated antigens is less likely to cause autoimmune diseases.
Human α-fetoprotein (AFP) is reexpressed in approximately 70%−80% of HCC tumors,(10,11) serving as a biomarker for diagnosis and a potential target for immunotherapy. Butterfield et al. identified four human leukocyte antigen (HLA)-A2-restricted AFP epitopes and developed peptide-based and vector-based vaccines.(12,13) Though modest AFP-specific cluster of differentiation 8 (CD8) responses were detected, no antitumor effect was observed in HCC patients.(14) A more effective, specific immunotherapy is adoptive transfer of tumor-specific autologous T cells.(15,16) Unfortunately, most tumor-specific T cells are of low affinity or cannot be expanded from patients. One way to obtain high-affinity antigen-specific T cells is to genetically modify a patient’s T cells with tumor-specific T-cell receptors (TCRs).(17–19) Identification of tumor antigen-specific TCRs is the first critical step in T-cell engineering and adoptive cell therapy. Recently, Sun et al. cloned an AFP158 epitope-specific TCR from human T cells.(20) Not surprisingly, this AFP158-specific TCR has little antitumor effect because of its low affinity. In a meeting abstract, Gerry et al.(21) reported that mutations in the complementarity-determining region (CDR) of an AFP158-specific human TCR(22) could increase its affinity and showed that human T cells transduced with the affinity-enhanced TCR (TCR-T cells) could better recognize HCC tumor cells. The antitumor efficacy of this AFP-specific TCR-T cell is being tested in a recent approved clinical trial. However, reports of severe on-target and off-target toxicity of melanoma-associated antigen A3 (MAGE-A3) TCR-T(23,24) suggest that more TCRs are needed in order to select the optimal TCRs that can provide human T cells the tumor-killing activity without toxicity to normal cells. Thus, there is a strong demand for more effective and safe AFP-specific TCRs for HCC immunotherapy.
In this study, we developed an effective strategy to identify TCRs for HLA-A2 presented AFP158 epitope. We first established an immunization approach of lentivector (lv)-prime and peptide-boost to elicit high-level CD8 responses in HLA-A2 transgenic AAD mice. A large number of AFP158-specific CD8T cells generated in mice without repeated in vitro restimulation should keep their TCR diversity and enabled us to study their antitumor effects prior to TCR identification. We found that the AFP158-specifc mouse CD8 T cells recognized and killed human HCC tumor cells in vitro and eradicated large human HepG2 tumors in nonobese diabetic severe combined immunodeficient gamma knockout (NSG) mice. We then identified three pairs of TCRs out of five hybridomas. Transduction of human T cells with the mouse TCR genes redirected them to specifically recognize and kill HepG2 tumor cells without toxicity to normal hepatocytes in vitro. Adoptive transfer of the human TCR-T cells eradicated HepG2 tumor in NSG mice. In conclusion, we have identified AFP158-specific TCRs with a great potential to redirect a patient’s autologous T cells for HCC treatment.
Materials and Methods
MICE
HLA-A2 transgenic AAD mice (female 7–8 weeks) and immunocompromised NSG mice (female 7–10 weeks) were obtained from Jackson Laboratory. The AAD mice express a chimeric major histocompatibility complex I (MHCI) of human HLA-A0201 α1-α2 domain and mouse H-2Dd α3 domain (binding mouse CD8).(25) Animal protocols were approved by the Institutional Animal Care and Use Committee of Augusta University.
CELL LINES AND NORMAL HEPATOCYTES
HEK293T, T2, and HepG2 cells were from the American Type Culture Collection; Huh7 cells were from Dr. Satyanayana Ande of the Georgia Cancer Center, Hep3B cells were from Dr. Mitchell Ho of the National Cancer Institute, and AFP− HepG2 cells were provided by Dr. Jingxiong She of Augusta University. The expression of AFP and HLA was verified by western blot and immunological staining. The cells were cultured for no more than eight passages, to assure their authenticity. Cells were checked for myco-plasma by a PCR test (Fisher Scientific). Normal primary hepatocytes were purchased from Sekisui XenoTech (Kansas City, KS).
TUMORS
Five million HepG2 cells were inoculated into the flank of NSG mice. Tumor growth was monitored by measuring the length, width, and height of the tumors. Tumor volume was calculated as 1/2(length × width × height).
RECOMBINANT lv AND IMMUNIZATION
The AFP and influenza virus PR8 M1 genes were cloned into lv to generate AFP-lv and Flu-M-lv. Lvs were prepared and titrated.(26,27) Mice were subcutaneously immunized with AFP-lv(28) or Flu-M-lv and boosted 12–15 days later with AFP158 (FMNKFIYEI) or M158 (GILGFVFTL) peptide.(29)
TETRAMER STAINING
Splenocytes were stained with indicated surface markers, Tet158 tetramers (NIH Tetramer Core Facility), and/or the anti-Vβ antibody panel (BD Biosciences). Tetramer staining was conducted before anti-Vβ antibody staining.
T-CELL HYBRIDOMAS
T-cell hybridomas were created by fusing the sorted mouse CD8+Tet158+ cells with BW-Lyt2.4 cells that lacked the TCR α and β chains and selected in HAT medium as described.(30) Single-hybridoma clones were obtained by serial dilution.
IDENTIFICATION OF PAIRED TCRα AND β CHAINS
The technique 5′ rapid amplification of complementary DNA ends(31) was conducted to amplify the TCR α and β genes. Briefly, total RNA was isolated from hybridomas, and complementary DNA was made with an oligo dT primer. PolyC was added to the 5′ end of complementary DNA by terminal transferase. PCR was conducted using the 5′ pGI primer (CACCGGGIIGGGIIGGGIIGG) and 3′ primers corresponding to the constant (C) region of the α (GGCATCACAGGGAACG) or β (CCAGAAGGTAGCAGAGACCC) chain. Based on the obtained sequence of α and β variable (V) regions, specific primers corresponding to the V region of the α (ATGAAATCC TTTAGTATTTCCC) or β (ATGGGCTCCAGG CTCTTTCTG) chain were used with an internal primer of the α C region (GCACATTGATTTGGGAGTC) or the β C region (GGGTAGCCTTTT GTTTGTTTG) to amplify the V region. Nucleotide sequences of the TCR α and β chains were obtained.
TCR GENES AND RECOMBINANT lv
TCR α and β genes were designed from the above-identified V-D-J region. The C region of the TCRα chain and the C2 region of the TCRβ chain were used to create the full-length TCRs. A P2A sequence(32) was inserted between the α and β chains. The entire TCR genes were codon-optimized, synthesized, and cloned into lv.
TRANSDUCTION OF HUMAN T CELLS
Human T cells were isolated from the buffy coat of healthy donors by negative selection and activated by the CD3/CD28 tetrameric antibody complex (Stem-cell Technologies) for 2 days before they were transduced with lv. The CD3/CD28 antibody complex was rinsed away 2 days after stimulation. Twenty units of interleukin-2 (IL-2) was in the culture through the process.
3-(4,5-DIMETHYLTHIAZOL-2-YL)-2,5-DIPHENYLTETRAZOLIUM BROMIDE ASSAY
To measure the live HepG2 cells after overnight coculture with mouse splenocytes, a 3-(4,5-dime-thylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was performed as described.(33)
INTRACELLULAR STAINING AND ENZYME-LINKED IMMUNOSORBENT ASSAY
Peripheral blood cells or splenocytes were restimulated with indicated peptides in the presence of Golgi-Stop (Biolegend) for 4 hours and intracellularly stained for interferon-gamma (IFNγ) or IL-2 as described.(34) Enzyme-linked immunosorbent assay (ELISA) of IFNγ and IL-2 was conducted per the instructions (Biolegend).
LACTATE DEHYDROGENASE ASSAY AND PROPIDIUM IODIDE STAINING
TCR-T cells were cocultured with HepG2 tumor cells (5 × 104) at indicated effector to target cell (E/T) ratios overnight. The cytotoxicity of TCR-T cells was determined by measuring the lactate dehydrogenase (LDH) activity in the coculture media as instructed (Promega). The HepG2 cells after coculture with TCR-T were then stained with propidium iodide (PI; BD Biosciences).
ADOPTIVE CELL TRANSFER
The indicated numbers of splenocytes, T-cell populations, or human TCR-T cells were transferred into NSG mice bearing human HepG2 tumors. TCR-T cells in mouse blood were monitored by immunological staining.
STATISTICAL ANALYSIS
Statistical analyses were performed using t test or analysis of variance (GraphPad Inc.).
Results
IMMUNIZATION OF AAD MICE ELICITS A HIGH LEVEL OF AFP158-SPECIFIC CD8 T CELLS THAT RECOGNIZE AND KILL HUMAN HepG2 CELLS
To induce CD8 T cells that can recognize the HLA-A2/AFP158 complex, we immunized AAD mice with AFP-lv or AFP peptide. We found that AFP-lv immunization induced a modest level of AFP158 epitope-specific CD8 responses, whereas peptide did not (Fig. 1A). However, AFP158 peptide significantly boosted the lv-primed CD8 responses (Fig. 1A). Critically, mouse CD8 T cells produced IFNγ after coculture with AFP+, but not AFP−, HepG2 cells (Fig. 1B), suggesting that the vaccine-activated mouse CD8 T cells could specifically recognize AFP+ HepG2 tumor cells. In addition, after coculture with the immunized splenocytes, the AFP+ HepG2 cells were killed in a dose-dependent manner (Fig. 1C,D). Together, the data suggest that immunization of AAD mice with lv-prime and peptide-boost elicits a high level of AFP158-specific CD8 T cells that recognize and kill HepG2 tumor cells.
FIG. 1.
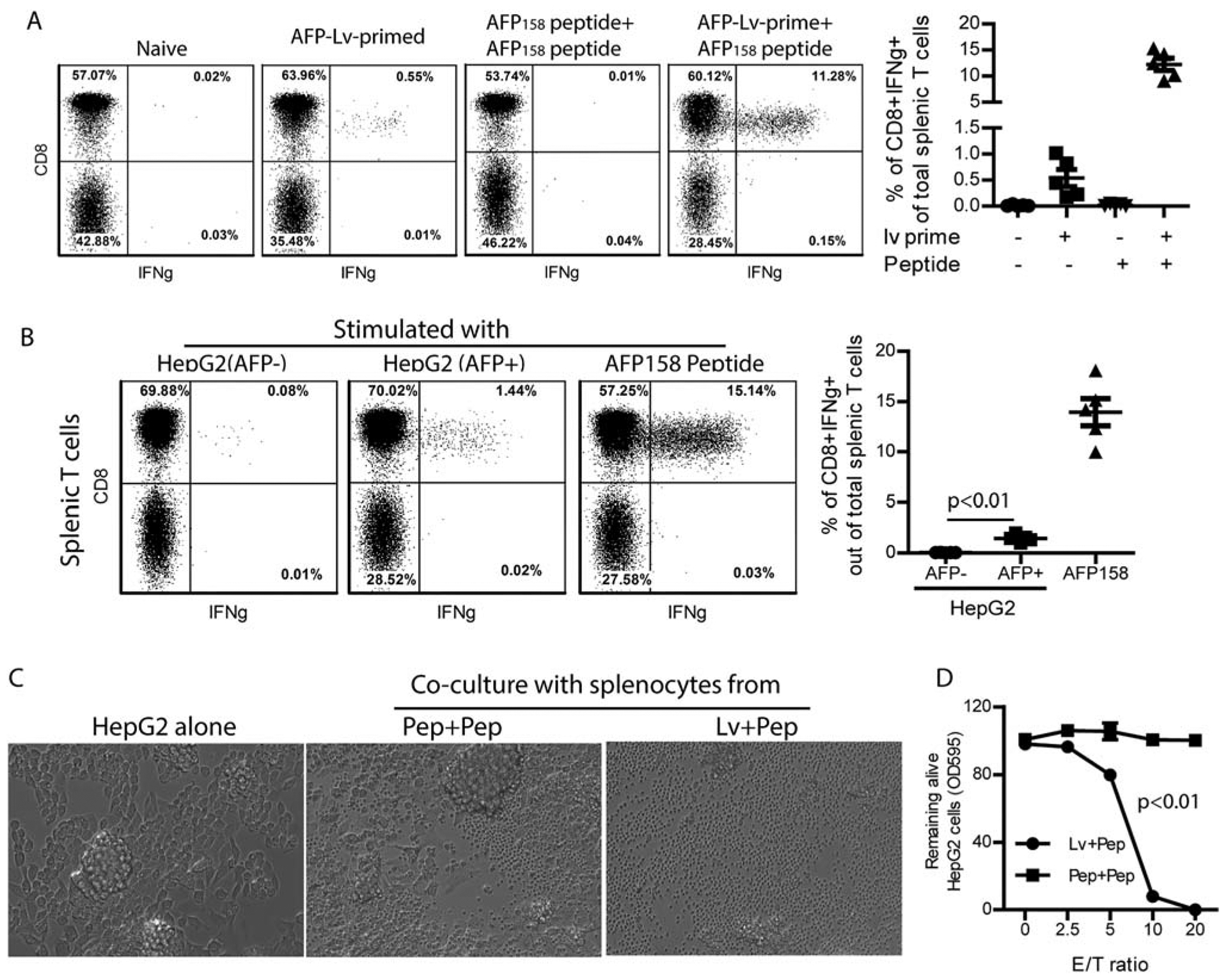
Immunization of AAD mice elicits a high level AFP158-specific CD8 T cells that recognize and kill human HepG2 cells. (A) HLA-A2 transgenic AAD mice were primed with AFP-lv and boosted with AFP158 peptide. Peripheral blood cells from the indicated mice were analyzed for CD8 and IFNγ 12 days after prime and 5 days after boost by gating on the Thy1.2+ T cells after ex vivo stimulation with AFP158 peptide. Representative dot plots and a summary of five mice are shown. (B) Splenocytes of the lv-primed and peptide-boosted mice were cocultured with AFP− or AFP+ HepG2 cells for 4 hours and analyzed for IFNγ. AFP158 peptide stimulation was used as positive control. Representative plots and a summary of five mice are shown. A t test was used for statistical analysis. (C,D) The cytotoxicity of the immunized mouse splenocytes was studied by coculturing them with HepG2 cells. Pictures were taken after overnight coculture (C). An MTT assay was then performed to determine the remaining live HepG2 cells after splenocytes were rinsed away. The MTT data of HepG2 tumor cells cocultured with different ratios of splenocytes was compared to MTT data of HepG2 tumor cells alone to calculate the percentage of live cells. Shown is the dose-dependent killing of HepG2 cells by splenocytes. Analysis of variance was used for statistical analysis. The experiment was done three times with similar data. Abbreviations: Lv+Pep, lv-primed and peptide-boost; Pep+Pep, peptide-prime and peptide-boost; OD, optical density.
ADOPTIVE TRANSFER OF IMMUNIZED AAD MOUSE SPLENOCYTES PREVENTS AND ERADICATES LARGE HepG2 TUMORS
In this adoptive transfer experiment, we found that splenocytes from AFP-immunized AAD mice completely prevented HepG2 tumor challenge in NSG mice (Fig. 2A,B). Strikingly, adoptive transfer of immunized AAD splenocytes was able to eradicate HepG2 xenografts as large as 2 cm in diameter (Fig. 2C).
FIG. 2.
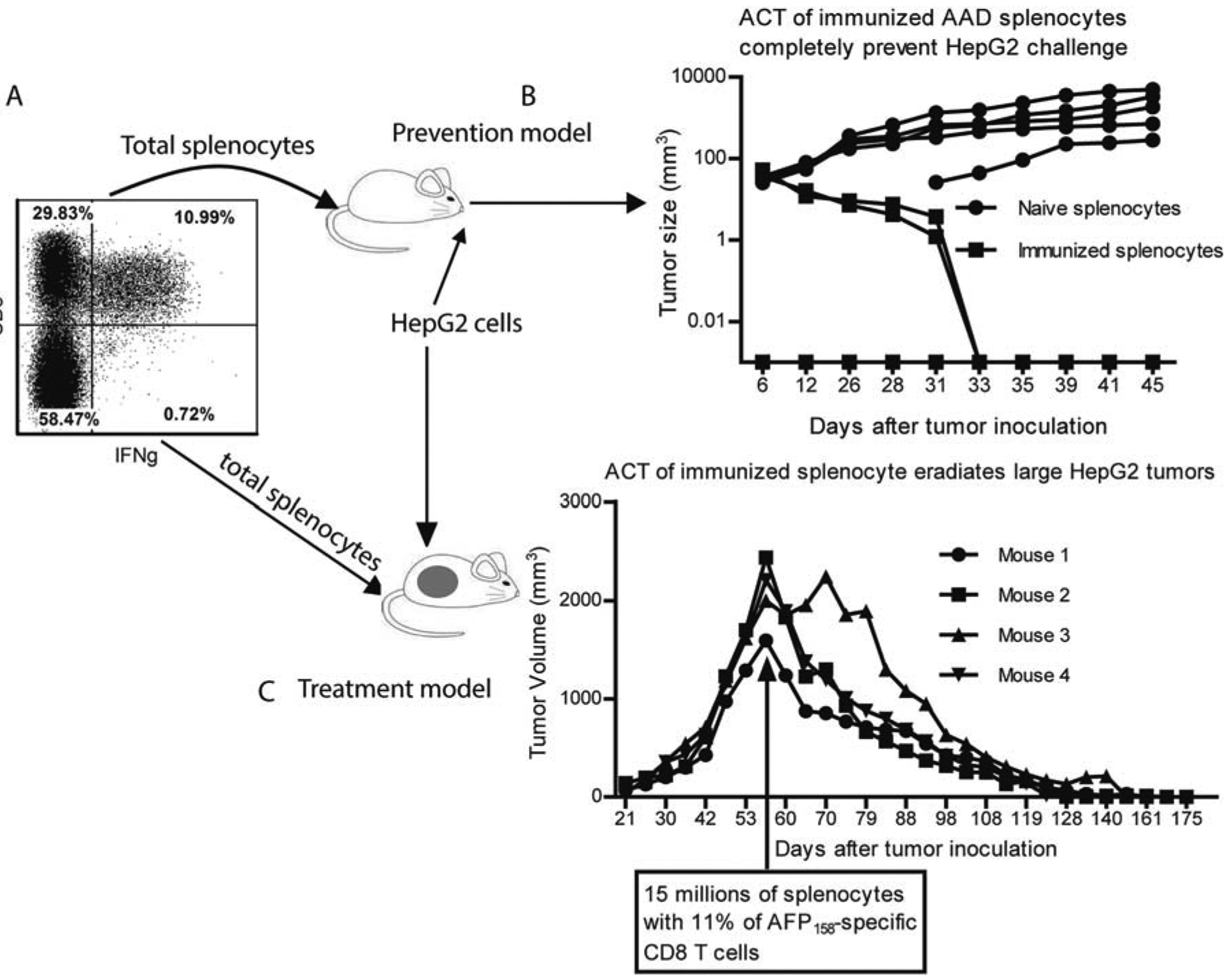
Adoptive transfer of the immunized AAD mouse splenocytes prevents and eradicates large HepG2 tumors in NSG mice. (A) Approximately 11% of the prime-boosted AAD mouse splenocytes produced IFNγ in response to AFP158 peptide. (B) In the preventive model, 15 million total splenocytes of the naive or immunized mice were injected into NSG mice, which was followed by HepG2 tumor cell challenge 2 days later. The tumor growth curve is shown. (C) In the therapeutic model, NSG mice were injected with 15 million total splenocytes of the immunized mice when HepG2 tumor size reached 2 cm in diameter. Shown is the tumor volume curve. The experiment was done twice with similar data. Abbreviation: ACT, Adoptive cell transfer.
ADOPTIVE TRANSFER OF AFP158-SPECIFIC CD8 T CELLS ERADICATES HepG2 TUMORS
To further define the immune cells with an antitumor effect, we isolated CD8 T cells from AFP-immunized AAD mice (Fig. 3A). CD8 T cells from AAD mice immunized with influenza virus M1 antigen were used as the control. We found that adoptive transfer of CD8 T cells from AFP-immunized mice eradicated HepG2 tumors (Fig. 3B). In contrast, the HepG2 tumors continued growing in the NSG mice that received CD8 T cells from M1 antigen-immunized mice. Next, we isolated AFP158-specific CD8 T cells by tetramer sorting (Fig. 3C). After adoptive transfer, only the Tet158+ CD8 T cells could eradicate HepG2 tumors in NSG mice (Fig. 3D). Together, the in vitro and in vivo data verify that AAD mouse CD8 T cells specific for the HLA-A2/ AFP158 complex can recognize and kill human HCC tumor cells.
FIG. 3.
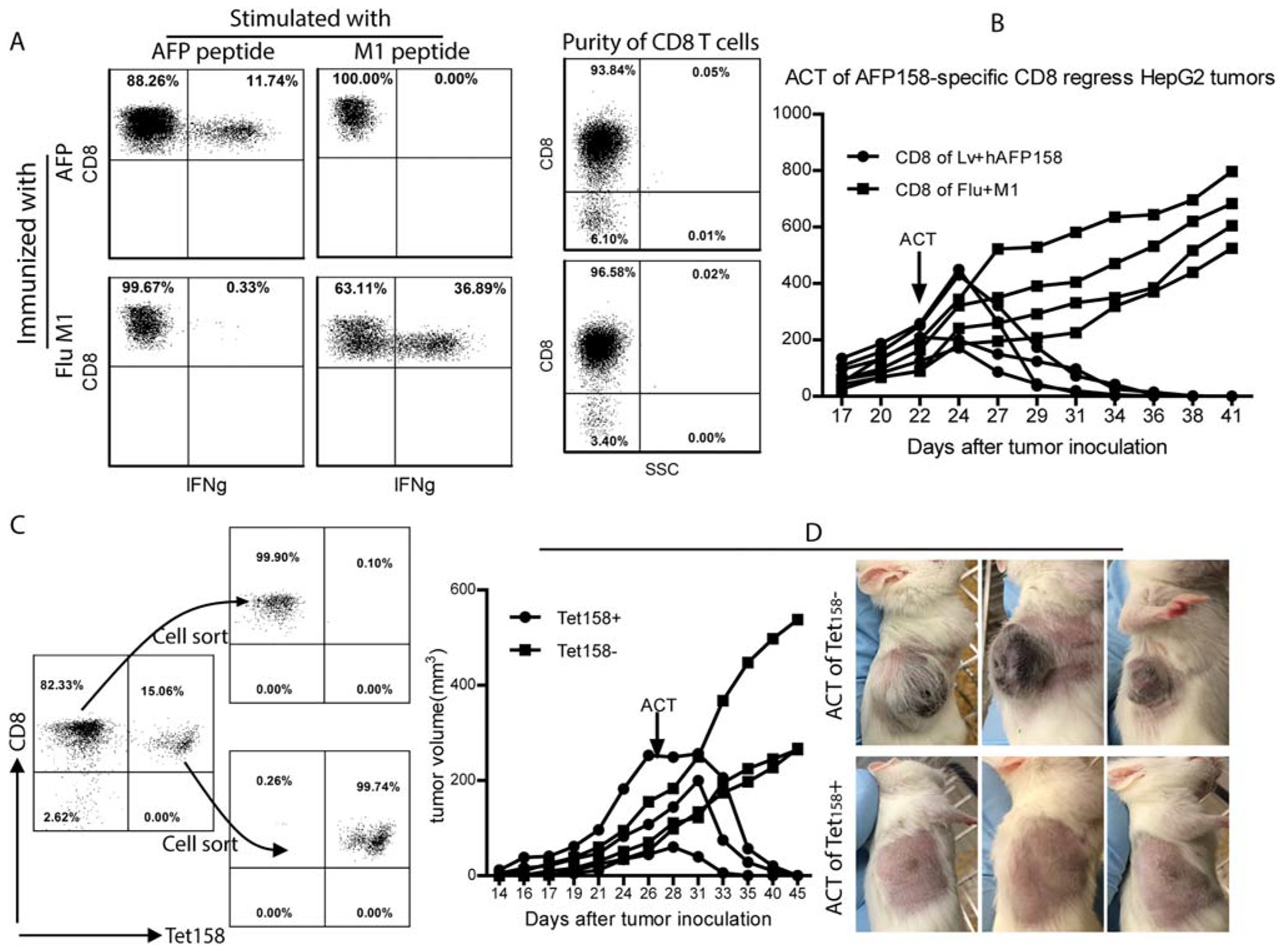
Adoptive transfer of AFP158-specific CD8 T cells eradicates HepG2 tumors in NSG mice. (A) CD8 T cells from AFP-immunized mice were isolated by magnetic beads. CD8 T cells from mice immunized with influenza virus M1 antigen peptide were used as control. The purity of CD8 T cells and the percentage of IFNγ-producing cells are shown. (B) NSG mice bearing HepG2 tumors were injected with 5 million CD8 T cells from AFP-immunized or M1-immunized mice (~0.5 million AFP-specific and 1.5 million M1-specific CD8 T cells, respectively). Tumor growth curve is shown. (C) Magnetic bead-purified CD8 T cells from AFP immunized mice were further separated into Tet158+ and Tet158− cells by cell sorting after Tet158 tetramer staining. The purity of Tet158+ and Tet158− CD8 cells before and after sorting is presented. (D) NSG mice bearing HepG2 tumors were injected with 0.5 million Tet158+ or Tet158− CD8 T cells. Tumor growth curve and pictures at the end of the experiment are presented. Abbreviations: ACT, Adoptive cell transfer; SSC, side scatter.
T-CELL HYBRIDOMAS CREATED FROM Tet158+ CD8 T CELLS BIND TO Tet158 TETRAMER
To identify paired TCR α and β chains, we created T-cell hybridomas. Prior to generating T-cell hybridomas, the TCR Vβ chains of Tet158+ cells were characterized. Approximately 90% of the Tet158+ CD8 T cells could be stained by anti-Vβ8.3 antibody (Supporting Fig. S1); the other 10% of Tet158+ CD8T cells were stained with antibodies against Vβ2, Vβ4, Vβ5.1/5.2, Vβ6, or Vβ11. We obtained 39 T-cell hybridomas that were stained by Tet158 tetramers (Fig. 4A). Twenty-five of them could be stained by anti-Vβ8.3 antibody (data not shown). Further experiments showed that five of the anti-Vβ8.3+ T-cell hybridomas responded to AFP+HepG2 tumor cells and produced IL-2 (Fig. 4B).
FIG. 4.
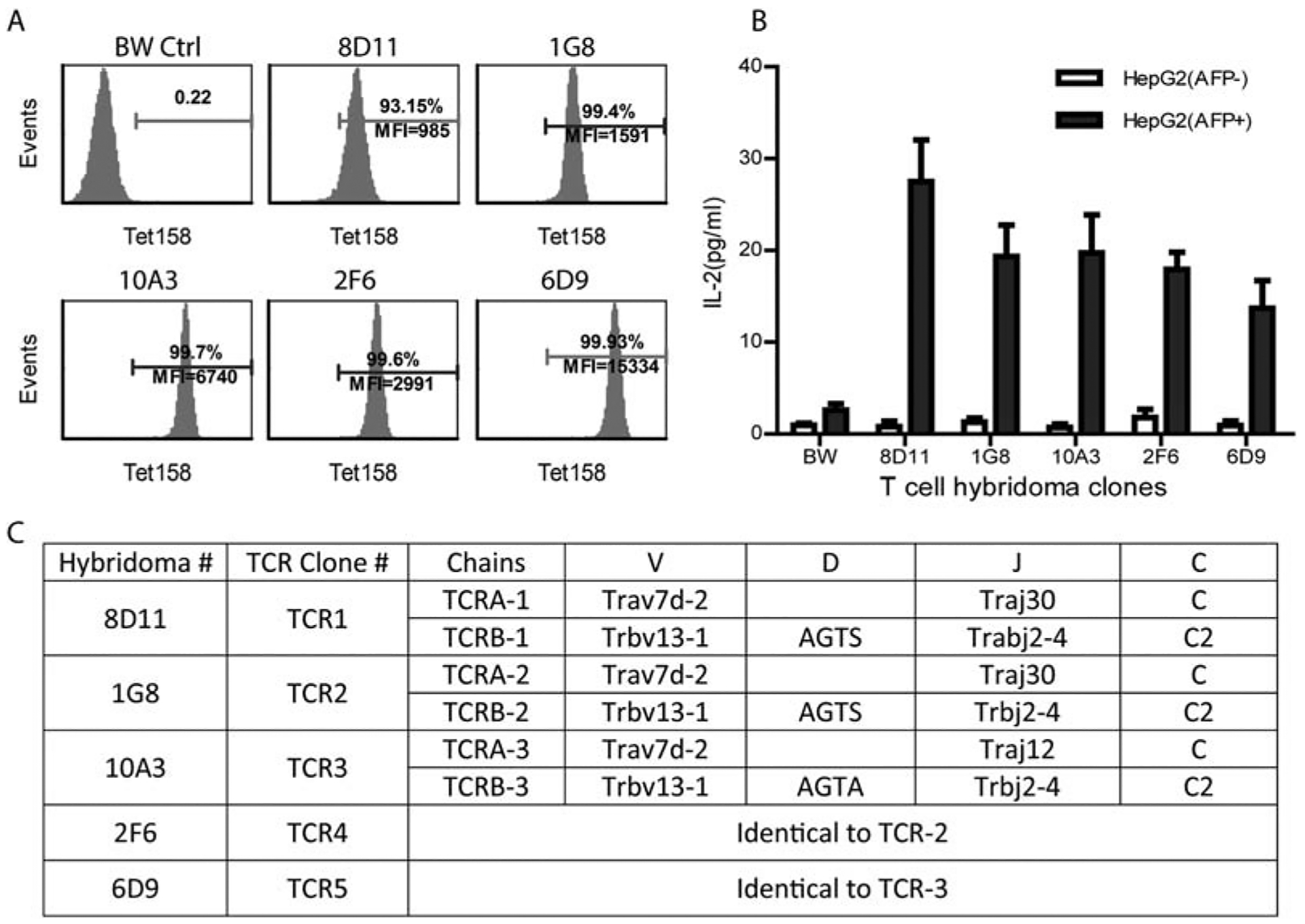
Paired TCRα and β chains are identified from the T-cell hybridomas that bind to Tet158 tetramer. (A) T-cell hybridomas were established from sorted Tet158+ cells. Representative tetramer staining of five hybridomas is shown. BW-Lyt2.4 partner cells were negative control. (B) T-cell hybridomas produced IL-2 in response to AFP+, but not AFP−, HepG2 tumor cells. (C) Paired TCRα and β chains were identified from five hybridomas and their TCR genes are summarized. TCR4 and TCR2 are identical, as are TCR5 and TCR3. The protein sequences of TCR-1 and TCR-2 α chains have a two-AA difference in the V-J junction, whereas the TCR-3 α chain has a 10-AA difference from TCR-1 and TCR-2 in the CDR3 region. The protein sequences of TCRβ chain are identical except that TCR-3 has a one-AA difference from TCR-1 and TCR-2 in the D-J junction. Abbreviation: BW, BW-Lyt2.4 cell.
PAIRED TCR α AND β CHAINS WERE IDENTIFIED FROM HYBRIDOMAS
The paired TCR α and β chains from the five IL2-producing hybridomas after coculture with HepG2 cells were amplified and sequenced. The result is summarized in Fig. 4C. Out of these five hybridomas, three unique sets of TCR genes were identified. The amino acid (AA) sequences of the TCR α and β chains were compared to the National Center for Biotechnology Information and the International Immunogenetics Information System data-banks. The data show that all three TCRα chains have the same variable region (TRAV7D-2) and that the TCRβ chains share the same TRBV13–1 variable region (corresponding to Vβ8.3 antibody staining). Detailed analysis revealed that TCR1 and TCR2 are similar. The β chains of TCR1 and TCR2 were identical; the α chains of TCR1 and TCR2 had two-AA differences in the V-J junction. On the other hand, TCR3 is more divergent from TCR1 and TCR2. Although the β chain of TCR3 differs from that of TCR1 and TCR2 by one AA, the α chain J region of TCR3 is encoded by a different gene, which makes the TCR3 α chain 10 AA different from the TCR1 and TCR2 α chains in the CDR3 region.
EXPRESSION OF TCR α AND β CHAINS IN HUMAN T CELLS FORMS FUNCTIONAL TCRs
To study whether the mouse TCR genes could engineer human T cells to create TCR-T cells that recognize the HLA-A2/AFP158 complex, we synthesized the TCR1, TCR2, and TCR3 genes and cloned them into lv (Fig. 5A). First, transduction of Jurkat cells with TCR-lv showed that expression of the TCR genes enabled them to bind Tet158 tetramer. The mean fluorescence intensity (MFI) of Tet158 staining on the TCR3-transduced cells was higher (Fig. 5B). Then, we studied whether the TCR genes could redirect primary human T cells to recognize the HLA-A2/ AFP158 complex. Primary T cells were isolated from healthy donor buffy coat and transduced by lv after CD3/CD28 activation (Fig. 5C). Using green fluorescent protein (GFP) as a reporter, we found that approximately 60% of human T cells were transduced by GFP-lv at a multiplicity of infection of 10 (Supporting Fig. S2). Transduction of human T cells with TCR genes enabled 20%−30% of them to bind Tet158 tetramer (Fig. 5D). Although different donors’ T cells were transduced with different efficacies, there was no significant variation among the TCRs. Again, the MFI of Tet158 staining was consistently higher on the TCR3-T cells, indicating that TCR3 may have a higher affinity for the HLA-A2/AFP158 complex. Both CD8 and CD4 TCR-T cells could bind the Tet158 tetramer. However, the MFI of Tet158 staining on CD8 TCR-T cells was higher (Fig. 5E), suggesting that CD8 enhanced binding of the TCR to the HLA-A2/AFP158. When the TCR-transduced T cells were stained with anti-Vβ8.3 or the Tet158 tetramer separately, the percent of Tet158+ cells was nearly identical to the percent of anti-Vβ8.3+ cells (Supporting Fig. S3), suggesting that there was no mispairing with endogenous human TCRs.
FIG. 5.
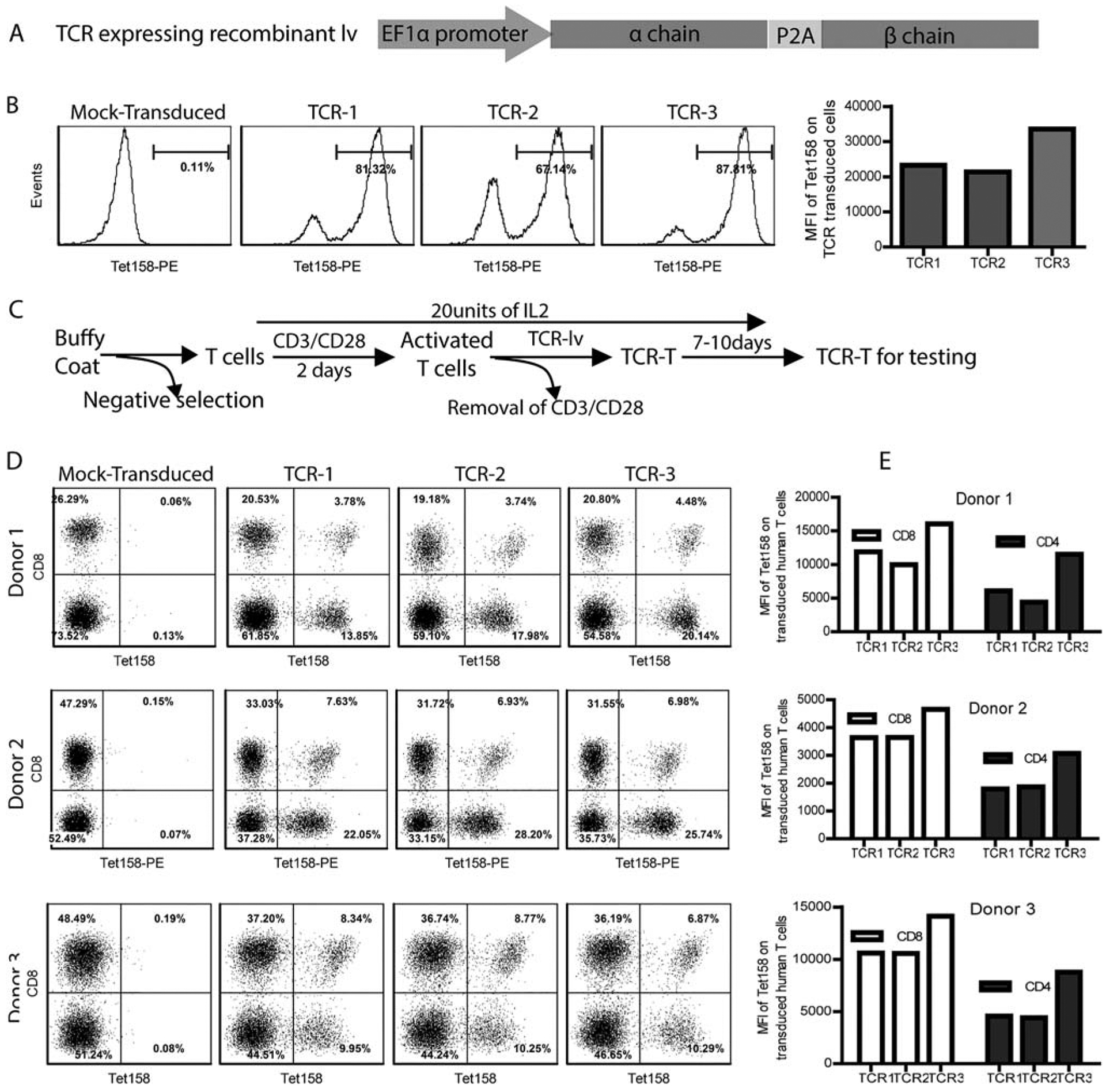
Expression of the paired TCR α and β chains forms TCRs that bind Tet158 tetramer. (A) Sketch of a recombinant lv-expressing TCR gene is shown. Paired TCR α and β chain genes were expressed as a single molecule under the control of the EF1α promoter. A P2A sequence was inserted in between to generate equal numbers of TCR α and β chains. (B) Tet158 tetramer staining of the Jurkat cells after transduction with TCR-lv was shown. Histogram and MFI are presented. (C) The process of generating human TCR-T cells is presented. Mock transduced T cells had undergone same CD3/CD28 treatment without lv transduction. (D,E) Primary human T cells from 3 donors were transduced with TCR-lvs and examined 7–10 days after lv transduction. Both the percent (D) and MFI (E) of Tet158+ CD8 and CD4 T cells are presented.
HUMAN TCR-T CELLS CAN SPECIFICALLY RECOGNIZE HLA-A2-PRESENTED AFP158 PEPTIDE AND AFP+ HepG2 TUMOR CELLS
First, we studied whether the human TCR-T cells could specifically recognize HLA-A2-presented AFP158 peptide. We measured the cytokine production by TCR-T cells after stimulation with T2 cells pulsed with AFP158, glypican 3 (GPC3), or M1 peptide. The data showed that TCR-T cells could be stimulated to produce IFNγ by T2 cells pulsed with AFP158 peptide. In contrast, no IFNγ was detected in the TCR-T culture after stimulation by T2 cells pulsed with M158, GPC3326, or GPC3367 peptide (Supporting Fig. S4). The mock T cells did not respond to AFP158 peptide. Next, we found that TCR-T cells also recognized HLA-A2+AFP+ HepG2 tumor cells and produced IFNγ but did not recognize HLA-A2+AFP−, HLAA11+AFP+ Huh7, or HLA-A28+AFP+ Hep3B cells (Fig. 6A). Intracellular staining showed that the IFNγ was mainly produced in CD8 TCR-T cells (Fig. 6B). However, both CD4 (Fig. 6C) and CD8 (Fig. 6B) TCR-T cells were able to produce IL-2. There was no significant difference among the three TCRs. Furthermore, the TCR-T, especially the CD8 TCR-T, cells underwent significant proliferation after coculture with HepG2 cells (Fig. 6D).
FIG. 6.
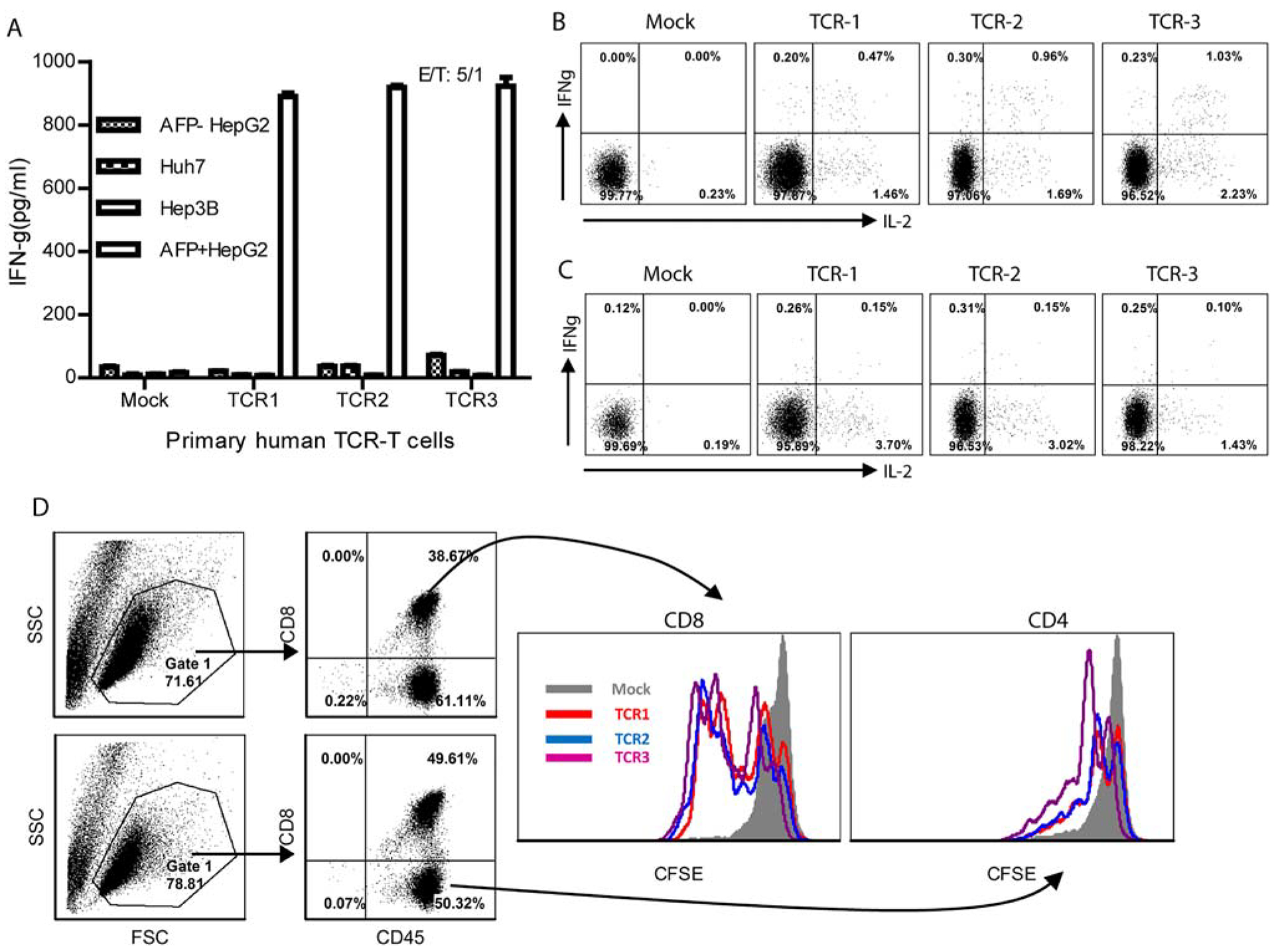
Human TCR-T cells can specifically recognize HLA-A2 presented AFP158 peptide and AFP+ HepG2 tumor cells. (A) One hundred thousand human TCR-T cells were cocultured overnight with HLA-A2+AFP+ HepG2 cells, HLA-A2+AFP− HepG2 cells, or HLA-A2−AFP+ Huh7 and Hep3B tumor cells; and the IFNγ level in the medium was measured by ELISA. (B,C) To measure IFNγ and IL-2 by intracellular staining, TCR-T cells were stimulated with HLA-A2+AFP+ HepG2 cells for 6 hours in the presence of GolgiStop. Only CD8 or CD4 T cells were gated and are shown in the plots. (D) Induction of CD8 and CD4 TCR-T cell proliferation was examined after 2-day coculture with AFP+ HepG2 tumor cells. The experiment was repeated twice with similar data. Abbreviations: CFSE, carboxyfluorescein succinimidyl ester; FSC, forward scatter; SSC, side scatter.
HUMAN TCR-T CELLS HAVE POTENT AND SPECIFIC CYTOTOXICITY AGAINST HepG2 TUMOR CELLS
In this study, we investigated the cytotoxicity of TCR-T cells. Using an LDH assay, we found that TCR-T cells killed ~80% of HepG2 tumor cells in 24 hours at a low E/T ratio, whereas they did not generate a significant cytotoxic effect on the AFP− or HLA-A2− tumor cells (Fig. 7A). The cytotoxicity against HepG2 cells was dose-dependent (Fig. 7B). At an E/T ratio of 0.5 (the real E/T ratio was 0.15 because only ~30% of T cells were Tet158+ cells), ~50% of HepG2 cells were killed, suggesting that TCR-T cells could have multiple killing of HepG2 cells. Using purified T subsets, we showed that the tumor killing activity was mainly from the CD8 TCR-T cells (Fig. 7C–E). However, CD4 TCR-T cells also had a lower level of cytotoxicity, resulting in 15% of specific killing at a ratio of 1.5/1 (Fig. 7E). The CD8 TCR-T cells encircled the HepG2 cells to form clusters (Fig. 7D). On the other hand, the CD4 TCR-T and mock-transduced T cells did not form obvious clusters. The PI staining revealed that the cluster contained mainly dead cells (Fig. 7F). PI staining also showed that, whereas CD8 TCR-T cells may have the dominant cytotoxicity, CD4 TCR-T cells also had a lower activity of killing HepG2 tumor cells, in concordance with the LDH assay. Mock-transduced human T cells did not induce death of HepG2 tumor cells in all assays. The specificity of TCR-T cells was further studied in vitro by examining whether they could recognize and kill normal hepatocytes. The data showed that all three TCR-T cell types did not recognize and kill normal hepatocytes (Supporting Fig. S5).
FIG. 7.
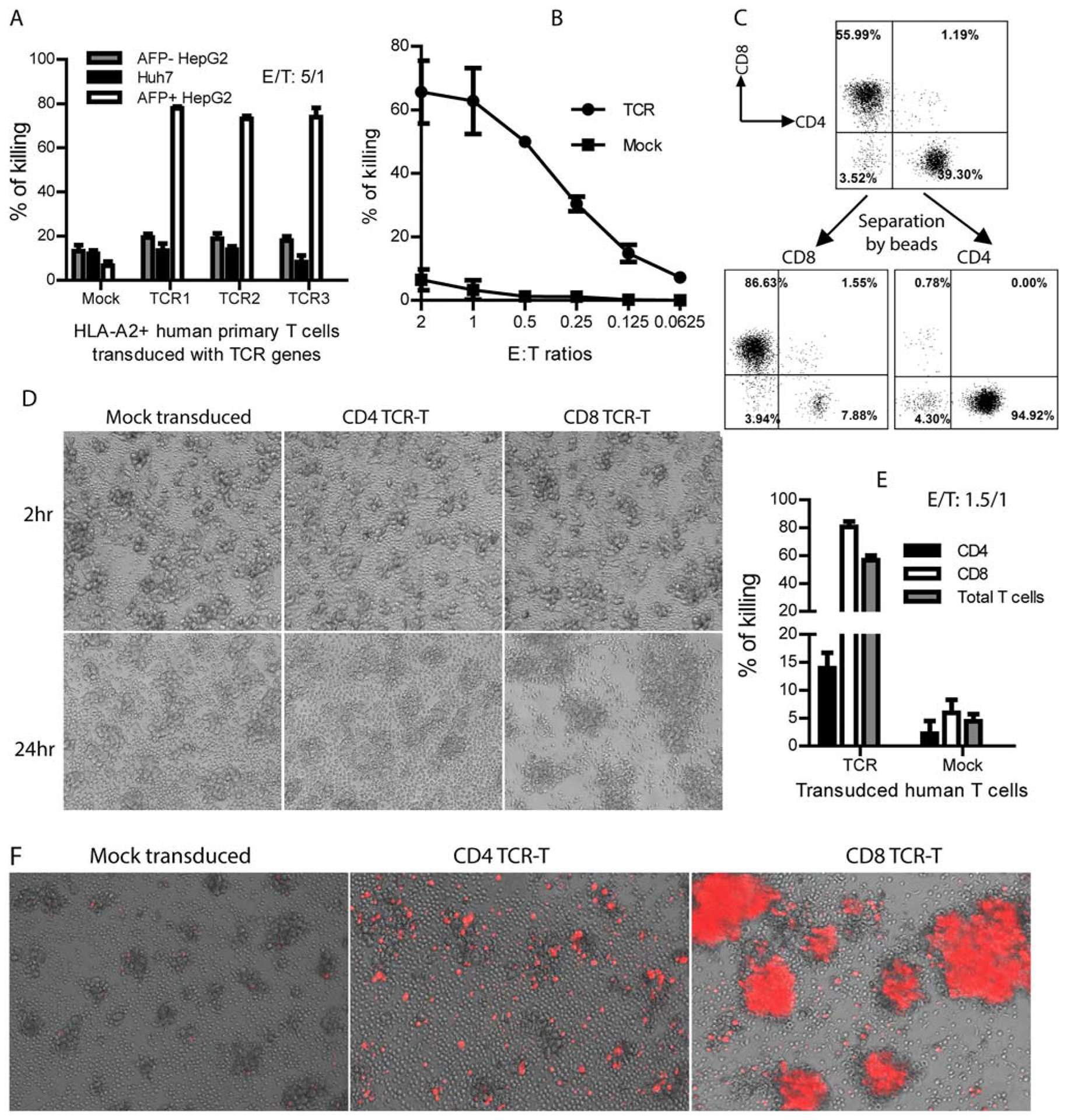
Human TCR-T cells have potent and specific cytotoxicity against HepG2 tumor cells. (A) One hundred thousand TCR-T cells were cocultured overnight in triplicate with the indicated tumor cells at E/T ratios. The killing efficacy of HepG2 tumor cells by human TCR-T (both CD8 and CD4) cells was measured by LDH assay. (B) Dose-dependent killing of AFP+HepG2 tumor cells was shown by TCR-T cells. (C) Donor CD8 and CD4 TCR-T cells were separated by magnetic beads after TCR transduction. (D) Coculture of the mock-transduced, CD4, or CD8 TCR-T cells with HepG2 tumor cells. Pictures were taken at 2 and 24 hours after coculture. (E) LDH assay was conducted at 24 hours after coculture to measure the killing effect. Statistical analysis was done with t test. (F) PI staining of TCR-T and HepG2 tumor cell cocultures after 24 hours to reveal dead cells. The experiment was repeated trice with similar data.
ADOPTIVE TRANSFER OF TCR-T CELLS GENERATES ANTITUMOR EFFECT IN NSG MICE
To evaluate the TCR-T cells’ in vivo antitumor effect, NSG mice bearing HepG2 tumors were adoptively transferred with human TCR-T cells (Fig. 8A). We found that adoptive transfer of TCR-T cells inhibited HepG2 tumor outgrowth in NSG mice (Fig. 8B). One of the TCR-T cell-treated mice developed a tumor, but the tumor was eradicated by 3 weeks posttransfer (Fig. 8C,D). In contrast, HepG2 tumors continued growth after transfer of the mock-transduced T cells. Both the mock-T and TCR-T cells survived approximately 3–4 weeks in the absence of human IL-2 administration (Fig. 8E). There was, however, a significant increase in Tet158+ cells among the TCR-T cells on day 8 posttransfer (Fig. 8F). To examine the in vivo antitumor effect of different TCR-T subsets, we separated CD4 and CD8 TCR-T cells after transduction and transferred them separately into HepG2 tumor-bearing NSG mice. We found that adoptive transfer of either T-cell subset could generate an antitumor effect (Supporting Fig. S6A). Also, the kinetics showed that the percent of Tet+ cells increased after adoptive transfer into HepG2 tumor-bearing mice (Supporting Fig. S6B), consistent with data in Fig. 8F, suggesting that there may be an antigen-induced TCR-T expansion. Phenotype staining showed that nearly half of the total and CD8 TCR-T cells and a third of CD4 TCR-T cells in NSG mice were of naive-like phenotype (CD45RA+CD62L+) (Supporting Fig. S6C).
FIG. 8.
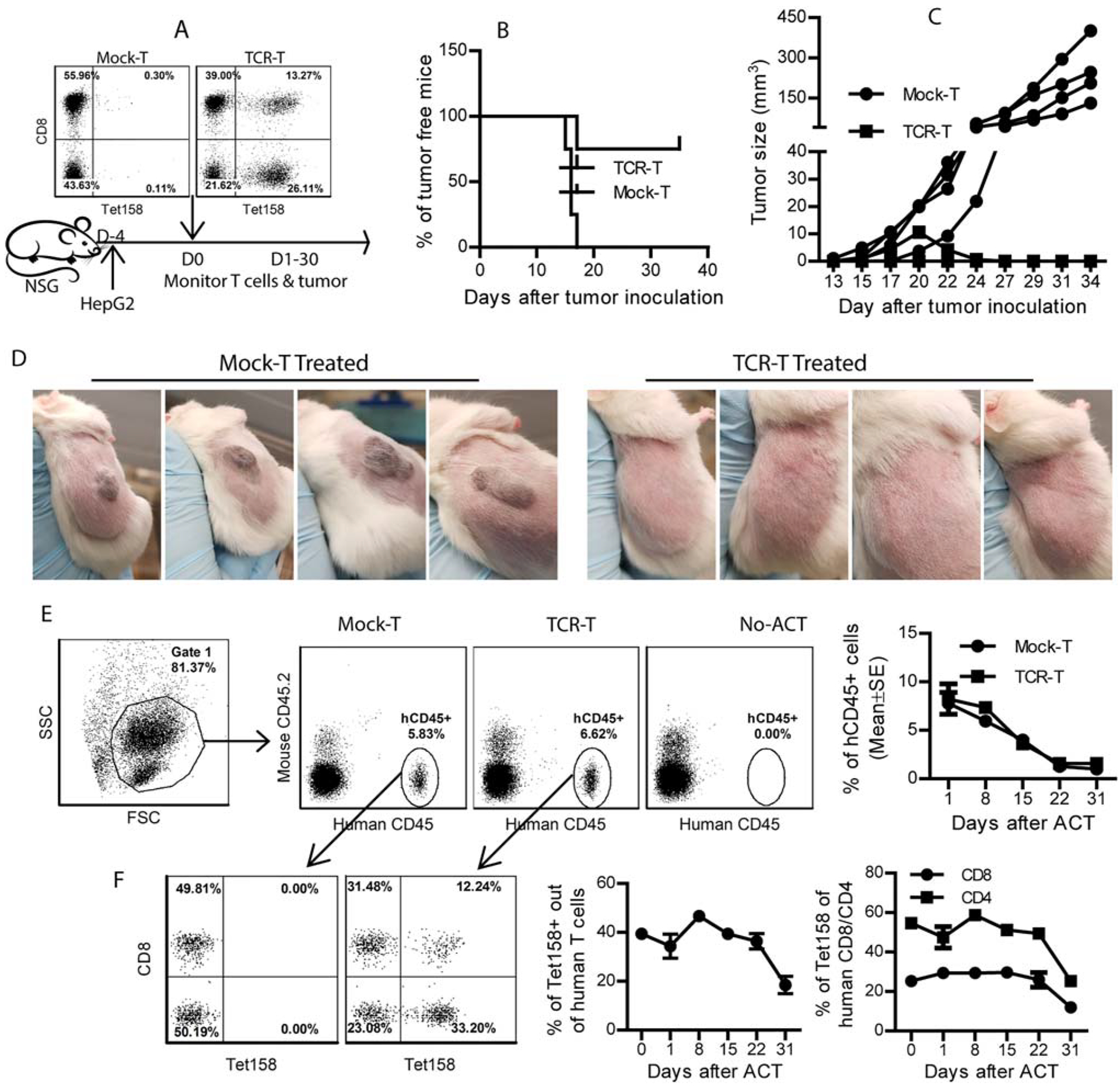
Adoptive transfer of TCR-T prevents and regresses HepG2 tumors in NSG mice. (A) NSG mice bearing 4-day HepG2 tumors were adoptively transferred with 20 million TCR-T cells. Percentage of Tet158+ cells in the TCR-T cells is shown. Tumor outgrowth and TCR-T cells in the mice were monitored. (B,C) The outgrowth and volume of each tumor are presented. (D) Pictures of HepG2 tumors at day 34 after inoculation. (E) Representative dot plots showed the percentage of hCD45+ cells among mouse blood cells. The kinetics of hCD45+ cells in NSG mice is summarized from four mice. (F) Percentage of Tet158+ out of the transferred human T cells in NSG mice is presented. Kinetics of Tet158+ cells among total transferred total human T cells, CD8 cells, and CD4 T cells is summarized from four mice. The experiment was repeated twice with similar data. Abbreviations: ACT, Adoptive cell transfer; FSC, forward scatter; SSC, side scatter.
Discussion
In this study, we established an effective immunization strategy to obtain a large number of AFP158-specific (Tet158) mouse CD8 T cells that specifically recognized and killed human HepG2 tumor cells in vitro and eradicated large HepG2 tumor xenografts in NSG mice. Subsequently, we established T-cell hybridomas from the Tet158 CD8 T cells, which allowed us to identify three pairs of TCR α and β chains that bound the HLA-A2/AFP158 complex. The TCR genes enabled human CD8 T cells to specifically recognize and kill HepG2 tumor cells in vitro. Furthermore, adoptive transfer of the TCR-T cells could eradicate HepG2 tumors in NSG mice.
TCR-T cells have been studied in a number of clinical trials,(35) which include TCRs specific for tumor-associated antigens of p53,(36) carcinoembryonic antigen,(37) MAGE3,(23,24) GP100,(16) MART1,(38) and NY-ESO.(39) Most of the TCR genes, including the recently reported AFP158-specific TCRs,(20,22) were isolated from T-cell clones established by repeated in vitro antigen stimulation, which might significantly reduce TCR diversity. For example, after 4 weeks of in vitro stimulation, only one TCR sequence was found in 19 of the GPC3367-specific T-cell clones.(40) Compared to this conventional approach of repeated in vitro antigen stimulation, we established an effective in vivo immunization strategy in the HLA-A2 mice using one lv-prime and one peptide-boost. This approach generates a large number of AFP158-specific CD8 T cells, which allows us to study their specific killing of tumor cells in vitro and to validate their antitumor function in vivo. Importantly, the large number of T cells enabled us to create T-cell hybridomas without repeated restimulation, which should maintain the diversity of the TCR repertoire. Not surprisingly, we were able to identify three unique sets of TCRs out of five T-cell hybridomas. There are an additional 34 Tet158+ T-cell hybridomas that can also be the resource for finding an optimal TCR of strong antitumor activity without on-target and off-target toxicity.
TCRs specific for human tumor antigens can be identified from human T cells or HLA-A2 transgenic mice. Human TCR is inherently of low affinity because of negative selection and may require affinity enhancement to achieve effective recognition of tumor antigens.(21,41) On the other hand, murine TCRs can recognize human antigenic epitopes with high affinity. However, high-affinity TCRs have a higher chance of cross-recognizing off-target antigens,(42) resulting in toxicity. It has been demonstrated that the TCRs derived from conventional HLA-A2 mice had a higher affinity than the TCRs derived from the HLA-A2/Kb mice with chimeric MHCI.(43) Thus, in our study, we intentionally used AAD mice, in which the MHCI is a chimeric molecule consisting of the α1-α2 domain of HLA-A2 and the α3 domain of mouse H-2Dd. This chimeric MHCI molecule makes the mice more responsive to antigen immunization because mouse CD8 molecules bind the α3 domain of MHC to help T cells engage with antigen-presenting cells. At the same time, the provision of CD8 assistance should increase the likelihood of generating CD8 T cells with optimal TCR affinity (not very high affinity). Additionally, the use of murine TCRs minimizes mispairing with endogenous human TCRs,(44) reducing the chance of generating off-target toxicity. We found that the percent of TCR Vβ chain+ cells is very close to the percent of Tet+ cells, suggesting that, indeed, there is no TCR mispairing in the transduced human T cells. However, murine TCR was found to be immunogenic in a portion of patients, though the immunogenicity did not correlate to a lower antitumor effect(36); thus, humanization may be needed for repeated use of TCR-T cells in patients.
The three sets of AFP158-specific murine TCRs were also able to render CD4 T cells capable of binding the Tet158 tetramer. The production of IL-2 by CD4 TCR-T cells in response to HepG2 tumor cells may provide the cytokine necessary for maintaining T-cell proliferation in vivo. Additionally, CD4 TCR-T cells demonstrated low cytotoxicity against HepG2 tumor cells. In fact, adoptive transfer of sufficient CD4 TCR-T cells resulted in potent antitumor effects in NSG mice. Thus, although CD8 TCR-T cells may be the major player in the killing of AFP+ tumor cells, CD4 TCR-T cells can also generate an antitumor effect.
Another factor that affects the TCR-T antitumor efficacy is the T cells themselves. One recent study found the CD26hi T cells were more persistent after transfer and generated a potent antitumor effect,(45) which may be the better T-cell subset for TCR engineering. On the other hand, we found that stem-like memory T cells with naive-like phenotype generated a better antigen response in vivo.(46) In the current study, we found that a significant portion of TCR-T cells were of naive-like phenotype (CD45RA+CD62L+) in NSG mice. Whether the CD45RA+CD62L+ cells are more responsive to antigen and thus generate a better antitumor effect on TCR-T cells remains to be studied.
Adoptive transfer of TCR-T cells has generated remarkable antitumor effects in multiple myeloma(47) and in synovial cell sarcomas and melanoma,(48) suggesting that TCR-engineered T cells have a great potential to control solid tumors. However, the liver is equipped with a number of immunosuppression mechanisms,(49) posing a significant barrier to the success of HCC immunotherapy. But a recent report showed that some immune suppression in the liver could be overcome by checkpoint blockade,(5′) opening the possibility that the combination of TCR-T cells with PD-1 blockade may enhance the antitumor effect in an immunosuppressive microenvironment. Though our initial study showed no significant toxicity on normal primary hepatocytes by our AFP-specific TCR-T cells, in light of the severe toxicity recently observed after adoptive transfer of MAGE-A3-specific TCR-T cells,(23,24,51) the on-target and off-target toxicity of our TCR-T cells should be extensively examined in preclinical models(52) before conducting clinical trials to test their antitumor effect. The multiple TCRs identified in this study provide an increased arsenal for T-cell engineering and HCC immunotherapy. And the diverse TCR repertoire pool gives us the resources and a higher chance of finding the optimal TCR for a strong antitumor effect without causing severe on-target and off-target toxicity. Considering that the Food and Drug Administration has approved two chimeric antigen receptor gene-modified T cells for immunotherapy of hematological tumors, the AFP-specific TCR genes identified in this study, if proved to be safe, should have a great potential of being used to modify patients’ autologous T cells for adoptive cell therapy of HCC.(50)
Supplementary Material
Acknowledgment:
We acknowledge the advice of Dr. Ingatowicz and Dr. Singh of Augusta University on creating T-cell hybridomas and thank the National Institutes of Health’s Tetramer Core Facility for synthesizing the HLA-A2/AFP158 tetramer. We thank Jessica Ho of the University of Georgia for editing the manuscript.
Supported by a National Institutes of Health/National Cancer Institute grant (R01 CA168912, to Y.H.) and a Georgia Cancer Center grant (to Y.H., T.M., and E.K.).
Abbreviations:
- AA
amino acid
- AFP
α-fetoprotein
- CD
cluster of differentiation
- E/T
effector to target cell ratio
- GPC3
glypican 3
- HCC
hepatocellular carcinoma
- HLA
human leukocyte antigen
- IFNγ
interferon-gamma
- IL
interleukin
- LDH
lactate dehydrogenase
- lv
lentivector
- MFI
mean fluorescence intensity
- MHCI
major histocompatibility complex I
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NSG
nonobese diabetic severe combined immunodeficient gamma knockout
- PI
propidium iodide
- TCR
T-cell receptor
- TCR-T
TCR gene-modified T cell
Footnotes
Supporting Information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.29844/suppinfo.
View this article online at wileyonlinelibrary.com.
Potential conflict of interest:
Nothing to report.
REFERENCES
- 1).Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the Global Burden of Disease Study. JAMA Oncol 2017;3:524–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Welzel TM, Graubard BI, Quraishi S, Zeuzem S, Davila JA, El-Serag HB, et al. Population-attributable fractions of risk factors for hepatocellular carcinoma in the United States. Am J Gastroenterol 2013;108:1314–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Kao WY, Su CW, Chau GY, Lui WY, Wu CW, Wu JC. A comparison of prognosis between patients with hepatitis B and C virus-related hepatocellular carcinoma undergoing resection surgery. World J Surg 2011;35:858–867. [DOI] [PubMed] [Google Scholar]
- 4).Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015;65: 87–108. [DOI] [PubMed] [Google Scholar]
- 5).Yang Y Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest 2015;125:3335–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).Pardee AD, Butterfield LH. Immunotherapy of hepatocellular carcinoma: unique challenges and clinical opportunities. Oncoimmunology 2012;1:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Kudo M Immune checkpoint blockade in hepatocellular carcinoma. Liver Cancer 2015;4:201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Pico de Coana Y, Choudhury A, Kiessling R. Checkpoint blockade for cancer therapy: revitalizing a suppressed immune system. Trends Mol Med 2015;21:482–491. [DOI] [PubMed] [Google Scholar]
- 10).Shao YY, Lin ZZ, Hsu C, Shen YC, Hsu CH, Cheng AL. Early alpha-fetoprotein response predicts treatment efficacy of antiangiogenic systemic therapy in patients with advanced hepatocellular carcinoma. Cancer 2010;116:4590–4596. [DOI] [PubMed] [Google Scholar]
- 11).Vora SR, Zheng H, Stadler ZK, Fuchs CS, Zhu AX. Serum alpha-fetoprotein response as a surrogate for clinical outcome in patients receiving systemic therapy for advanced hepatocellular carcinoma. Oncologist 2009;14:717–725. [DOI] [PubMed] [Google Scholar]
- 12).Butterfield LH, Meng WS, Koh A, Vollmer CM, Ribas A, Dissette VB, et al. T cell responses to HLA-A*0201-restricted peptides derived from human alpha fetoprotein. J Immunol 2001;166:5300–5308. [DOI] [PubMed] [Google Scholar]
- 13).Butterfield LH, Ribas A, Meng WS, Dissette VB, Amarnani S, Vu HT, et al. T-cell responses to HLA-A*0201 immunodominant peptides derived from alpha-fetoprotein in patients with hepatocellular cancer. Clin Cancer Res 2003;9:5902–5908. [PubMed] [Google Scholar]
- 14).Butterfield LH, Ribas A, Dissette VB, Lee Y, Yang JQ, De la Rocha P, et al. A phase I/II trial testing immunization of hepatocellular carcinoma patients with dendritic cells pulsed with four alpha-fetoprotein peptides. Clin Cancer Res 2006;12:2817–2825. [DOI] [PubMed] [Google Scholar]
- 15).Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 2012;12:269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood 2009;114:535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Schumacher TN. T-cell-receptor gene therapy. Nat Rev Immunol 2002;2:512–519. [DOI] [PubMed] [Google Scholar]
- 18).Sharpe M, Mount N. Genetically modified T cells in cancer therapy: opportunities and challenges. Dis Model Mech 2015;8: 337–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med 2016;22:26–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Sun L, Guo H, Jiang R, Lu L, Liu T, He X. Engineered cytotoxic T lymphocytes with AFP-specific TCR gene for adoptive immunotherapy in hepatocellular carcinoma. Tumour Biol 2016; 37:799–806. [DOI] [PubMed] [Google Scholar]
- 21).Gerry AB, Pumphrey NJ, Docta RY, Brewer JE, Bennett AD, Jakobsen BK. Improved affinity AFP-specific T cell receptor for hepatocellular carcinoma. J Immunother Cancer 2013;1(Suppl. 1):P10. [Google Scholar]
- 22).Pichard V, Royer PJ, Richou C, Cauchin E, Goebes K, Gaignerie A, et al. Detection, isolation, and characterization of alpha-fetoprotein-specific T cell populations and clones using MHC class I multimer magnetic sorting. J Immunother 2008;31: 246–253. [DOI] [PubMed] [Google Scholar]
- 23).Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother 2013;36:133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 2013;122:863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Newberg MH, Smith DH, Haertel SB, Vining DR, Lacy E, Engelhard VH. Importance of MHC class 1 alpha2 and alpha3 domains in the recognition of self and non-self MHC molecules. J Immunol 1996;156:2473–2480. [PubMed] [Google Scholar]
- 26).He Y, Zhang J, Mi Z, Robbins P, Falo LD Jr. Immunization with lentiviral vector-transduced dendritic cells induces strong and long-lasting T cell responses and therapeutic immunity. J Immunol 2005;174:3808–3817. [DOI] [PubMed] [Google Scholar]
- 27).He Y, Zhang J, Donahue C, Falo LD Jr. Skin-derived dendritic cells induce potent CD8+ T cell immunity in recombinant lentivector-mediated genetic immunization. Immunity 2006;24: 643–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Hong Y, Peng Y, Guo ZS, Guevara-Patino J, Pang J, Butterfield LH, et al. Epitope-optimized alpha-fetoprotein genetic vaccines prevent carcinogen-induced murine autochthonous hepatocellular carcinoma. HEPATOLOGY 2014;59:1448–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Cho HI, Celis E. Optimized peptide vaccines eliciting extensive CD8 T-cell responses with therapeutic antitumor effects. Cancer Res 2009;69:9012–9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).White J, Kappler J, Marrack P. Production and characterization of T cell hybridomas. Methods Mol Biol 2000;134:185–193. [DOI] [PubMed] [Google Scholar]
- 31).Walchli S, Loset GA, Kumari S, Johansen JN, Yang W, Sandlie I, et al. A practical approach to T-cell receptor cloning and expression. PLoS One 2011;6:e27930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Kim JH, Lee SR, Li LH, Park HJ, Park JH, Lee KY, et al. High cleavage efficiency of a 2A peptide derived from porcine teschovirus-1 in human cell lines, zebrafish and mice. PLoS One 2011;6:e18556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).He Y, Huang L. Growth inhibition of human papillomavirus 16 DNA-positive mouse tumor by antisense RNA transcribed from U6 promoter. Cancer Res 1997;57:3993–3999. [PubMed] [Google Scholar]
- 34).Liu Y, Peng Y, Mi M, Guevara-Patino J, Munn DH, Fu N, et al. Lentivector immunization stimulates potent CD8 T cell responses against melanoma self-antigen tyrosinase-related protein 1 and generates antitumor immunity in mice. J Immunol 2009;182:5960–5969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).Kunert A, Straetemans T, Govers C, Lamers C, Mathijssen R, Sleijfer S, et al. TCR-engineered T cells meet new challenges to treat solid tumors: choice of antigen, T cell fitness, and sensitization of tumor milieu. Front Immunol 2013;4:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36).Davis JL, Theoret MR, Zheng Z, Lamers CH, Rosenberg SA, Morgan RA. Development of human anti-murine T-cell receptor antibodies in both responding and nonresponding patients enrolled in TCR gene therapy trials. Clin Cancer Res 2010;16: 5852–5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 2011;19:620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 2006;314:126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39).Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol 2011;29: 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Dargel C, Bassani-Sternberg M, Hasreiter J, Zani F, Bockmann JH, Thiele F, et al. T cells engineered to express a T-cell receptor specific for glypican-3 to recognize and kill hepatoma cells in vitro and in mice. Gastroenterology 2015;149:1042–1052. [DOI] [PubMed] [Google Scholar]
- 41).Robbins PF, Li YF, El-Gamil M, Zhao Y, Wargo JA, Zheng Z, et al. Single and dual amino acid substitutions in TCR CDRs can enhance antigen-specific T cell functions. J Immunol 2008; 180:6116–6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42).Zhao Y, Bennett AD, Zheng Z, Wang QJ, Robbins PF, Yu LY, et al. High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. J Immunol 2007;179:5845–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43).Theobald M, Biggs J, Dittmer D, Levine AJ, Sherman LA. Targeting p53 as a general tumor antigen. Proc Natl Acad Sci USA 1995;92:11993–11997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44).Cohen CJ, Zhao Y, Zheng Z, Rosenberg SA, Morgan RA. Enhanced antitumor activity of murine-human hybrid T-cell receptor (TCR) in human lymphocytes is associated with improved pairing and TCR/CD3 stability. Cancer Res 2006;66: 8878–8886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45).Bailey SR, Nelson MH, Majchrzak K, Bowers JS, Wyatt MM, Smith AS, et al. Human CD26high T cells elicit tumor immunity against multiple malignancies via enhanced migration and persistence. Nat Commun 2017;8:1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46).Wu S, Zhu W, Peng Y, Wang L, Hong Y, Huang L, et al. The antitumor effects of vaccine-activated CD8+ T cells associate with weak TCR signaling and induction of stem-like memory T cells. Cancer Immunol Res 2017;5:908–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47).Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med 2015;21:914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48).Robbins PF, Kassim SH, Tran TL, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res 2015;21:1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49).Robinson MW, Harmon C, O’Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol 2016;13:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50).Shalapour S, Lin XJ, Bastian IN, Brain J, Burt AD, Aksenov AA, et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017;551:340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51).Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med 2013;5:197ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52).Gerry A, Sanderson J, Maroto M, Ferronha T, Ranganathan S, Norry E, et al. Targeting alpha-fetoprotein with TCR engineered T cells in HCC [Abstract]. J Clin Oncol 2016;34(15_Suppl): 3051–3051. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.