

Abstract
Measurements of very low levels of biomolecules, including proteins and nucleic acids, remain a critical challenge in many clinical diagnostic applications due to insufficient sensitivity. While digital measurement methods such as Single Molecule Arrays (Simoa), or digital ELISA, have made significant advances in sensitivity, there are still many potential disease biomarkers that exist in accessible biofluids at levels below the detection limits of these techniques. To overcome this barrier, we have developed a simple strategy for single molecule counting, dropcast single molecule assays (dSimoa), that enables more target molecules to be counted through increased sampling efficiency and with a simpler workflow. In this approach, beads are simply dropcast onto a microscope slide and dried into a monolayer film for digital signal readout. The dSimoa platform achieves attomolar limits of detection, with an up to 25-fold improvement in sensitivity over Simoa, the current state of the art for ultrasensitive protein detection. Furthermore, due to its simple readout process and improved cost-effectiveness compared to existing digital bioassays, dSimoa increases amenability to integration into point-of-care platforms. As an illustration of the potential utility of dSimoa, we demonstrate its ability to measure previously undetectable levels of Brachyury, a tissue biomarker for chordoma, in plasma samples. With its significantly enhanced sensitivity and simplicity, dSimoa can pave the way toward the discovery of new biomarkers for early disease diagnosis and improved health outcomes.
Graphical Abstract
INTRODUCTION
The ability to accurately measure extremely low levels of biomolecules, such as proteins, nucleic acids, and metabolites, is essential for a wide range of clinical and environmental applications, including disease diagnostics, drug discovery, pathogen detection in food, environmental toxin detection, and bioprocess control. Ultrasensitive measurement techniques are especially critical in clinical diagnostics, as many potential biomarkers exist in accessible biofluids at levels well below the detection limits of current laboratory methods.1 Digital measurement methods, such as digital enzyme-linked immunosorbent assay (ELISA), have vastly improved measurement sensitivities by up to 1000-fold over traditionally used analytical techniques such as conventional ELISA.2–5 However, the sensitivities of digital measurement techniques remain insufficient for many diagnostic applications, particularly for measuring disease-related proteins. For instance, while several protein biomarkers for neurological disorders have been shown to be upregulated in cerebrospinal fluid, highly invasive lumbar punctures are required for these measurements, thus making it impractical to screen individuals for early disease detection.6–9 As only a small fraction of brain-derived proteins passes through the blood-brain barrier into circulation, highly sensitive techniques that can detect and identify rare protein biomarkers through a simple blood test are crucial for addressing this unmet diagnostic need.10–12 Improving analytical sensitivity is also a major challenge in other diseases for which rapid point-of-care (POC) diagnosis is essential for effective medical intervention but where easily accessible biofluids, such as saliva or urine, are required. These biofluids contain only a minimal serumnal component, necessitating ultrasensitive techniques for protein biomarker detection.
One main barrier toward increasing sensitivity in digital ELISA is low sampling efficiency. While digital ELISA methods utilize single molecule counting to improve measurement sensitivity, low sampling efficiencies limit the number of target molecules that are counted. At very low target concentrations, the Poisson noise from counting single events, √N, where N is the number of counted molecules, contributes significantly to measurement error. As an example, at a sampling efficiency of 5%, only 30 out of the 600 target molecules in 100 μL of a 10 aM sample will be counted, assuming perfect capture efficiencies. The theoretical Poisson noise-associated coefficient of variation (CV), √N/N, is 18% at this low sampling efficiency and in reality much higher when accounting for capture efficiencies well below 100% and experimental error. This high measurement uncertainty therefore poses a major limitation for detecting rare molecules. Increasing sampling efficiencies to count more target molecules can thus greatly improve measurement precision and sensitivity but remains a challenge in digital ELISA.
Existing digital ELISA approaches utilize microwells or water-in-oil droplets to isolate individual beads carrying single target protein molecules.2,5,13–15 The current state of the art for digital ELISA is Single Molecule Arrays (Simoa), which captures single target molecules on antibody-coated paramagnetic beads and isolates individual beads into femtoliter-sized microwells for single molecule counting.2 A large excess number of beads over the number of target molecules in the sample is used to ensure digital measurements, where each bead has either zero or one captured target molecule and follows the Poisson distribution. Each captured molecule is labeled with a biotinylated detector antibody to form an immunocomplex sandwich, which is then labeled with the enzyme conjugate streptavidin-β-galactosidase (SβG). The beads are subsequently loaded, along with fluorogenic enzyme substrate into the microwells, each of which can fit at most one bead. Upon sealing of the microwells with oil, a high local concentration of fluorescent product is catalytically generated in each well that contains a bead carrying an SβG molecule. Thus, the number of target molecules is measured by counting “on” and “off” wells.
While Simoa can achieve subfemtomolar limits of detection and is the current gold standard for ultrasensitive protein detection, its sensitivity is limited by low sampling efficiencies. Only about 5% of the total number of beads can be loaded by gravity into the microwells and analyzed.16 While an external magnetic force is utilized for bead loading in the most recently developed Simoa instrument, the HD-X Analyzer, the percentage of analyzed beads remains around 5%. Other methods to improve bead loading have also been explored, including electric field-directed bead loading, hydrophilic-in-hydrophobic microwell arrays, and digital microfluidics.14,15,17–19 While these methods have increased bead loading efficiencies, demonstrations of their improvements in digital immunoassay sensitivities remain limited. Furthermore, complex fabrication methods and workflows limit the use of these approaches in POC applications. Another strategy for improving sampling efficiency in digital bioassays is bead encapsulation in water-in-oil droplets. Digital droplet-based immunoassays have been demonstrated with up to 60% bead loading efficiencies and have shown equal or improved sensitivities of up to an order of magnitude higher than that of the current Simoa technology.5,13 While droplet microfluidic systems are well established for diverse applications, the need for highly controlled, high-throughput droplet generation introduces additional fabrication and processing steps that increase complexity when integrating into POC systems. Furthermore, as a significant fraction of droplets do not contain beads but must still be imaged, improving imaging throughput remains another challenge toward POC implementation.
Here, we report a greatly simplified, more sensitive digital ELISA platform, dropcast single molecule assays (dSimoa), that addresses the above-mentioned challenges, using on-bead signal generation combined with bead dropcasting into a monolayer film for single molecule counting. With localization of a nondiffusible fluorescent signal to each bead carrying a target molecule, this platform not only eliminates the need for bead loading into microwells or droplets for signal compartmentalization, but also enables significantly more beads to be analyzed for improved sampling efficiency and thereby enhanced sensitivity. Furthermore, the vastly simplified readout process and improved cost-effectiveness of dSimoa, which requires only a microscope slide for bead loading and a simple optical setup for signal readout, can facilitate potential integration into a POC system. The dSimoa platform achieves attomolar limits of detection, with an up to 25-fold increase in sensitivity over the current Simoa technology. As a proof of concept, we demonstrate the ability of dSimoa to measure previously undetectable levels of Brachyury, a tissue biomarker for chordoma, a rare form of bone cancer, in plasma. The enhanced sensitivity and simplicity of dSimoa thus make it a highly promising platform for biomarker discovery and future POC diagnostic development.
RESULTS
Development of Dropcast Single Molecule Assays.
To enable bead dropcasting for counting of “on” and “off” beads, we first developed a strategy for generating a localized signal on each bead carrying a full immunocomplex sandwich. Rolling circle amplification (RCA), an isothermal DNA amplification method based on the processive action of a polymerase around a circular DNA template, generates long concatemers of DNA repeats to provide rapid and strong signal amplification. As RCA has been successfully used for the detection of individual protein–protein complexes and nucleic acids, we hypothesized that RCA would enable detection of single immunocomplex sandwiches captured on beads.20–23 RCA has also been performed on immunocomplexes on beads isolated in microwell arrays to enable multiplexed protein detection.24 To incorporate RCA into our single molecule detection platform, we labeled each immunocomplex sandwich with an RCA primer annealed with a circular DNA template (Figure 1). After RCA is performed, the generated DNA concatemer attached to each immunocomplex can be hybridized with a large number of complementary fluorescently labeled DNA probes for visualization.
Figure 1.
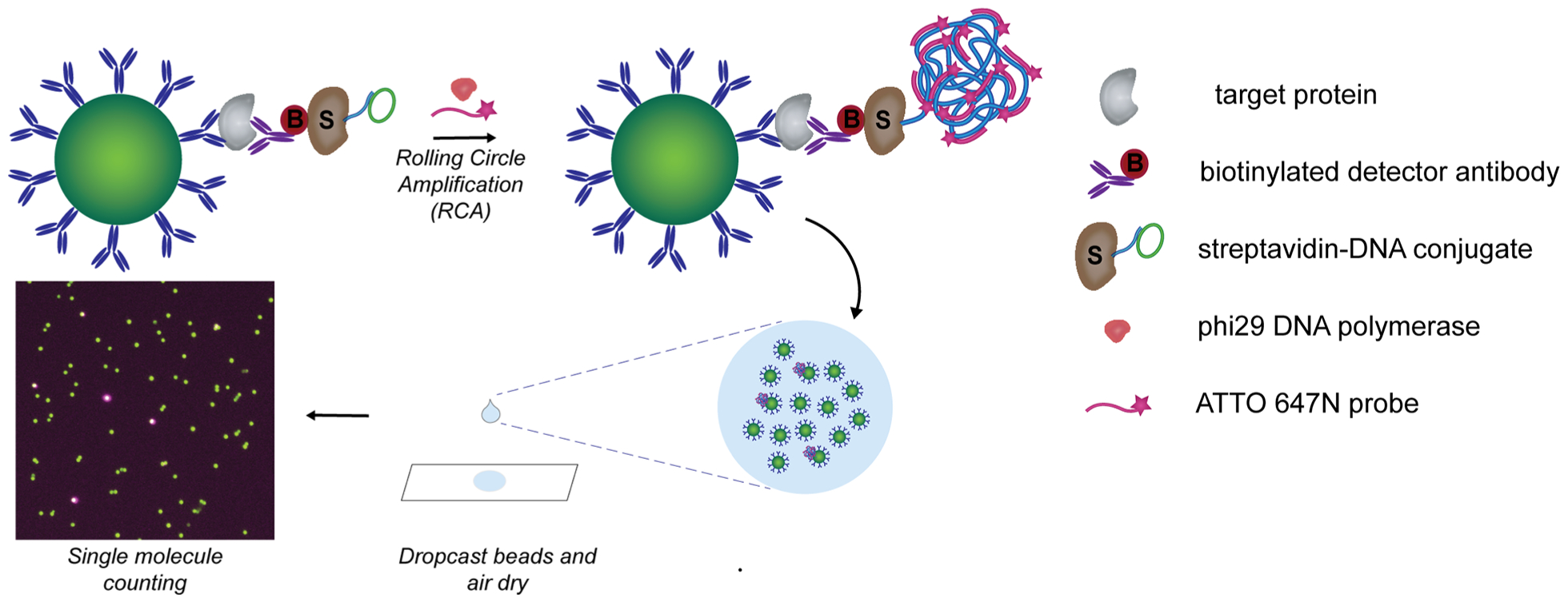
Schematic of dropcast single molecule assays. Upon formation of single immunocomplex sandwiches on antibody-coated paramagnetic beads and labeling with a streptavidin-DNA conjugate, rolling circle amplification (RCA) is performed on the beads to generate a long concatemer attached to each immunocomplex. Fluorescently labeled DNA probes are hybridized to the concatemer during RCA to produce a localized fluorescent signal on beads carrying a full immunocomplex sandwich. After RCA, the beads are concentrated, dropcast onto a microscope slide, and allowed to dry to form a monolayer film. Single target molecules are counted by fluorescence imaging of the dropcast film and counting “on” and “off” beads.
The dSimoa method utilizes the same target capture steps as conventional Simoa in which antibody-coated paramagnetic beads are incubated with the sample and biotinylated detector antibody to form an immunocomplex sandwich. However, instead of labeling the immunocomplex sandwich with streptavidin-β-galactosidase (SβG), streptavidin conjugated to a preannealed primer-template pair is used to label the immunocomplex sandwich. RCA is then carried out on each labeled immunocomplex sandwich at 37 °C for signal amplification. Furthermore, a fluorescently labeled DNA probe is added into the RCA reaction for in situ hybridization. After the RCA reaction, the beads are washed, concentrated, dropcast onto a microscope slide, and allowed to dry to form a monolayer film for imaging. As our preliminary attempts of directly using a detector antibody-DNA conjugate for immunocomplex formation followed by RCA resulted in high background signals (data not shown), we used a streptavidin-DNA conjugate for all dSimoa assays.
To evaluate the signal amplification and bead distribution in the dropcast films, we used dSimoa to detect interleukin-1 beta (IL-1β) as a model analyte, with the same capture and detector antibody pair used in a previously validated Simoa assay. Fluorescent dye-encoded beads (488 nm) were used to facilitate bead identification in the dropcast film for analysis, as salt crystal formation from the dropcast buffer could interfere with bead identification in brightfield images. With 100000 assay beads and a dropcast volume of approximately 15 μL, the dropcast bead films show minimal bead aggregation and high, uniform bead densities across the film (Figure 2A,B). Furthermore, the dropcast process is rapid, with 15 μL dropcast volumes drying into films of 12–15 mm diameter within 15 min. The presence of a captured target analyte on a bead is indicated by a fluorescent signal covering all or part of a bead (Figure 2C–E). As inclusion of aggregated beads in image analysis can affect the accuracy of the calculated fraction of “on” beads, bead aggregates of two or more beads, which constituted about 20–25% of the beads in each film, were separated by watershed segmentation in the image analysis algorithm, and any remaining bead aggregates were excluded from analysis via a size threshold. Representative histograms of the maximum fluorescence intensities on all imaged beads in the dropcast film show a wide range of “on” bead signal intensities, due to the broad size distribution of concatemers generated by RCA (Figure 2F,G). The number of “on” and “off” beads in each dropcast film was calculated by fitting a normal distribution to the maximum fluorescence intensities of each bead and assigning a threshold for “on” beads as five standard deviations above the mean. The average target molecule per bead (AMB), analogous to the average enzyme per bead (AEB) calculated in conventional Simoa, was determined using the Poisson distribution equation.25
Figure 2.
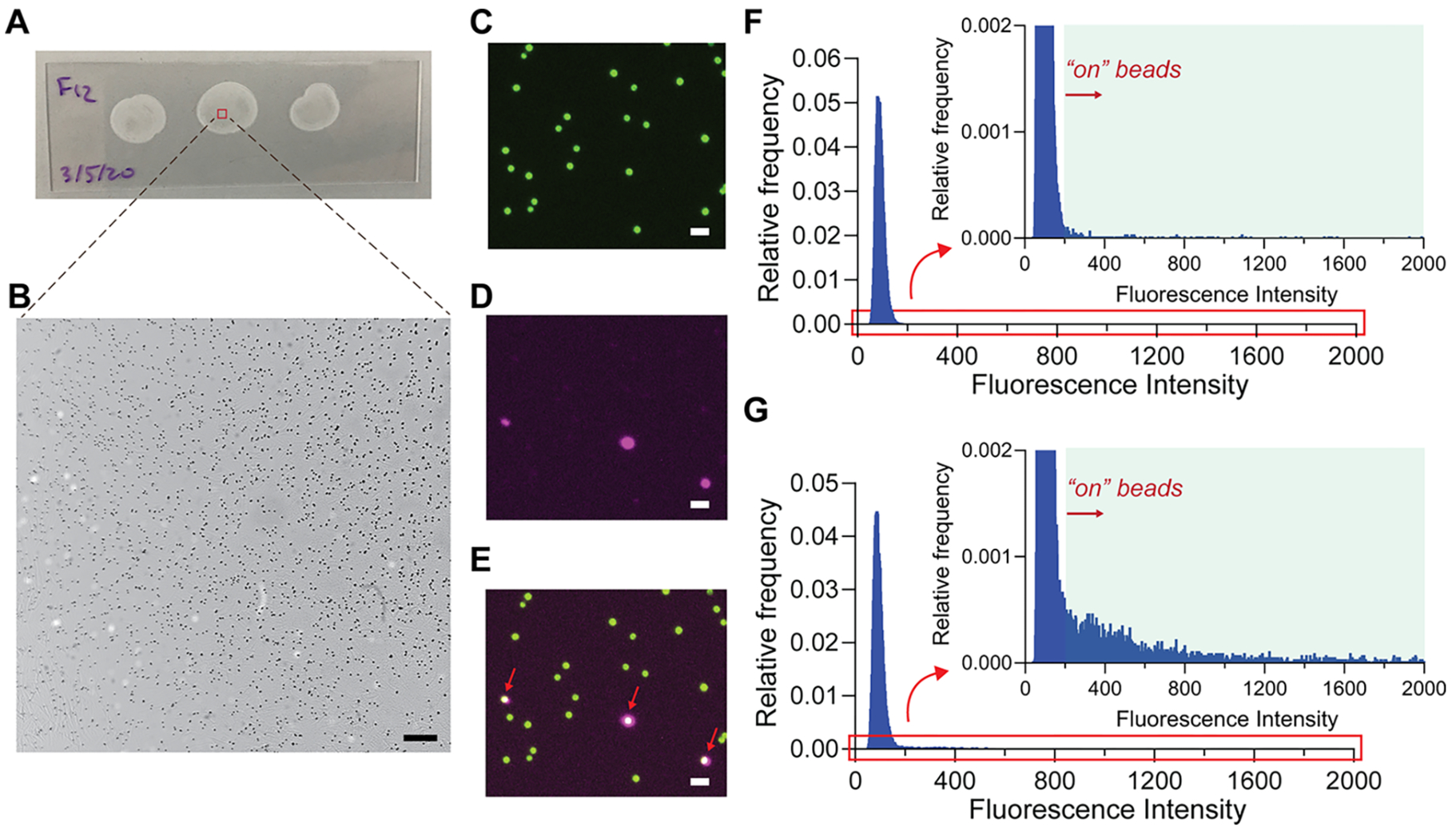
Imaging of dSimoa films. (A) Representative photograph and (B) brightfield image of dropcast bead films on a microscope slide. Approximately 2000–2500 beads are analyzed in each frame. Scale bar = 100 μm. (C–E) Representative images of “on” and “off” dye-encoded beads in dropcast film: bead fluorescence (488 nm; C), ATTO 647N probe (647 nm; D), and merged (E). Red arrows indicate “on” beads. Scale bar = 10 μm. (F, G) Representative histograms of the maximum fluorescence intensity values (subtracted from background fluorescence intensity in the image) on each bead for 0 fM IL-1β (F) and 10 fM IL-1β (G) samples. A normal distribution was fitted to the fluorescence intensity values, and the cutoff for an “on” versus “off” bead was determined as five standard deviations above the mean.
By simply transferring the entire volume of beads to a microscope slide, we are able to image and analyze 40–50% of the total number of assay beads on average, with most of the remaining beads either lost during wash or transfer steps or excluded from analysis due to aggregate formation. Thus, the sampling efficiency in dSimoa represents a significant improvement over the ~5% of beads analyzed using the current Simoa technology. In addition to eliminating the requirement for microwells, dSimoa also enables far fewer beads to be used for target capture due to the increased percentage of beads that can be analyzed, thus improving sampling of rare target molecules while minimizing Poisson noise. The current Simoa technology uses 500000 beads, while dSimoa uses 100000 beads. Decreasing the number of beads can increase the signal to background, as there will be more “on” beads relative to the total number of beads and thereby a higher AMB. Furthermore, the fluorescent signal remains highly stable in the dropcast film in its dry state, with no decreases in measured AMB values even after one month (Table S1).
Digital Detection of Proteins with dSimoa.
To assess the sensitivity of dSimoa, we generated calibration curves for two human cytokines, IL-1β and interleukin-10 (IL-10), using the same antibody pairs previously used in the corresponding Simoa assays. These dSimoa assays attained low- to midattomolar limits of detection (LODs), showing 25- and 15-fold improvements in sensitivity over the corresponding conventional Simoa assays for IL-10 and IL-1β, respectively (Figure 3; Table 1). The limits of quantification (LOQs), calculated as ten standard deviations above the background (AMB or AEB of the blank), were also improved by an order of magnitude in the dSimoa assays compared to the conventional Simoa assays. By substantially increasing the percentage of beads that can be analyzed, dSimoa enhances sampling efficiencies of low abundance molecules and enables far fewer beads to be used. In addition, the 5-fold reduction in the number of beads increased the signal to background, contributing to the significant enhancements in sensitivity (Figure 3C,F).
Figure 3.
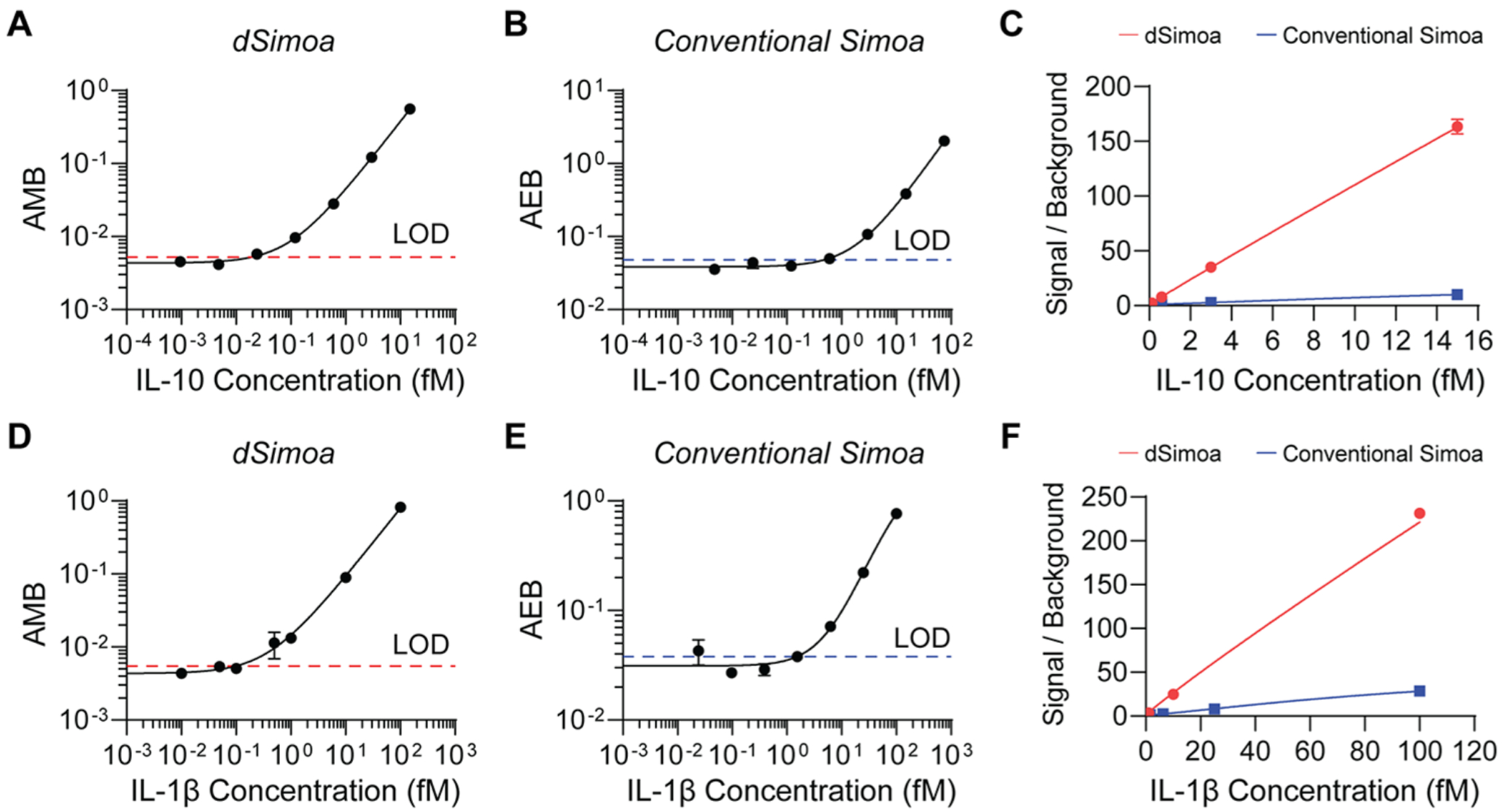
Comparisons of dSimoa and conventional Simoa assay sensitivities. (A) dSimoa and (B) conventional Simoa calibration curves for human IL-10. Dashed lines indicate the calculated limits of detection (LODs). (C) Comparison of signal to background ratios between dSimoa and conventional Simoa across the IL-10 calibration curve range. (D) dSimoa and (E) conventional Simoa calibration curves for human IL-1β. (F) Comparison of signal to background ratios between dSimoa and conventional Simoa across the IL-1β calibration curve range.
Table 1.
Calculated Limits of Detection (LODs) and Limits of Quantification (LOQs) for IL-10 and IL-1β Using dSimoa and Conventional Simoaa
| LOD | LOQ | ||||
|---|---|---|---|---|---|
| Target | dSimoa | Conventional Simoa | Quanterix | dSimoa | Conventional Simoa |
| IL-10 | 19.2 aM | 485.0 aM | 204.3 aM | 64.5 aM | 1.57 fM |
| IL-1β | 99.6 aM | 1.52 fM | 941 fM | 384.4 aM | 5.46 fM |
LOD and LOQ values were calculated as three and ten standard deviations above the background, respectively. The reported LOD values by the corresponding Quanterix Simoa assays were calculated as 2.5 standard deviations above the background.
In addition to improving the signal to background, we hypothesized that increasing the sampling efficiencies of the captured target molecules also helped to achieve lower LODs with dSimoa, by reducing measurement imprecision due to Poisson noise. To determine whether our experimental results supported this hypothesis, we randomly selected subsets of the beads analyzed in the IL-10 calibration curve and determined the LODs and coefficients of variation (CVs) of the background measurements at varying percentages of total assay beads analyzed (Figure 4). When few beads (below 10% of the total beads) were analyzed, the imprecision of the background measurements was very high, with CVs of greater than 20%, which corresponded to poorer LODs, as expected from Poisson sampling noise. Moreover, there were high variations in the obtained LODs among different random samplings when low percentages of beads were analyzed. These observations thus demonstrate the important role of sampling efficiency in the precision and sensitivity of digital measurements. The calculated LOD values did not increase much further when 20% or more of the beads were analyzed, suggesting that close to maximal sensitivity can be attained upon imaging at least 20% of the assay beads.
Figure 4.
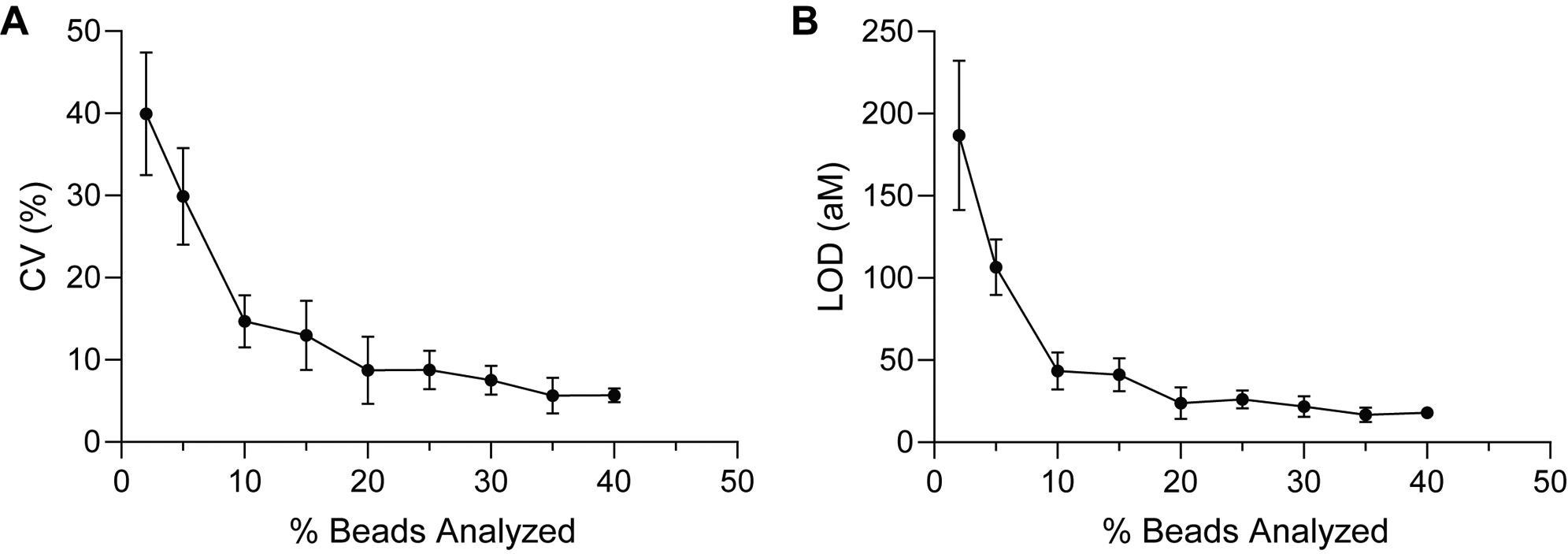
Effect of sampling efficiency on measurement precision and sensitivity. (A) Measurement CVs of the background signal and (B) calculated LODs for randomly selected subsets of beads imaged in the dSimoa calibration curve for IL-10. The percentage of beads analyzed represents the percentage of total assay beads. Each point represents the average of four different randomly selected subsets of beads.
To validate the performance of dSimoa in biological fluids, we performed spike and recovery experiments in human saliva for IL-10. Recovery rates of various concentrations of recombinant human IL-10 protein spiked into pooled human saliva ranged from 76% to 122%, thus demonstrating that dSimoa can reliably detect proteins in saliva (Figure S1A). Furthermore, dSimoa measurements of IL-10 in serial dilutions of human saliva showed linear dilution, indicating minimal interference from the saliva matrix on the dSimoa assay (Figure S1B). The dSimoa assay also showed high measurement precision, with CVs well below 10% across all the saliva samples.
To explore the potential diagnostic utility of the improved sensitivity of dSimoa, we developed a dSimoa assay for Brachyury, a T-box transcription factor that is strongly linked to chordoma, a primary bone cancer in the spine or skull base.26 While elevated levels of Brachyury expression have been found in chordoma tumors, there have been no reports on the measurement of Brachyury in plasma, to the best of our knowledge.27–30 The calibration curves generated by the dSimoa and conventional Simoa assays for Brachyury yielded LODs of 244.6 aM and 841.4 aM, respectively (Figure 5A,B). This dSimoa assay provides only a 3-fold improvement in sensitivity over the conventional Simoa assay. The relatively small improvement in LOD may be attributed to the smaller increase in the signal to background compared to the conventional Simoa assay (Figure 5C). As reducing the number of total assay beads also reduces the capture antibody concentration, the extent of improvement in signal to background from decreasing the number of assay beads may be smaller for antibodies with lower binding affinities, which can lead to decreased capture efficiencies despite the higher ratio of target molecules to beads. To validate the performance of the dSimoa assay in plasma and serum matrices, we performed spike and recovery experiments, obtaining recoveries of at least 65–70% in most of the spiked plasma and serum samples, with the majority of measurement CVs below 10% (Figure S2A). We further validated the accuracy of the dSimoa assay in plasma by confirming acceptable dilution linearity (Figure S2B).
Figure 5.
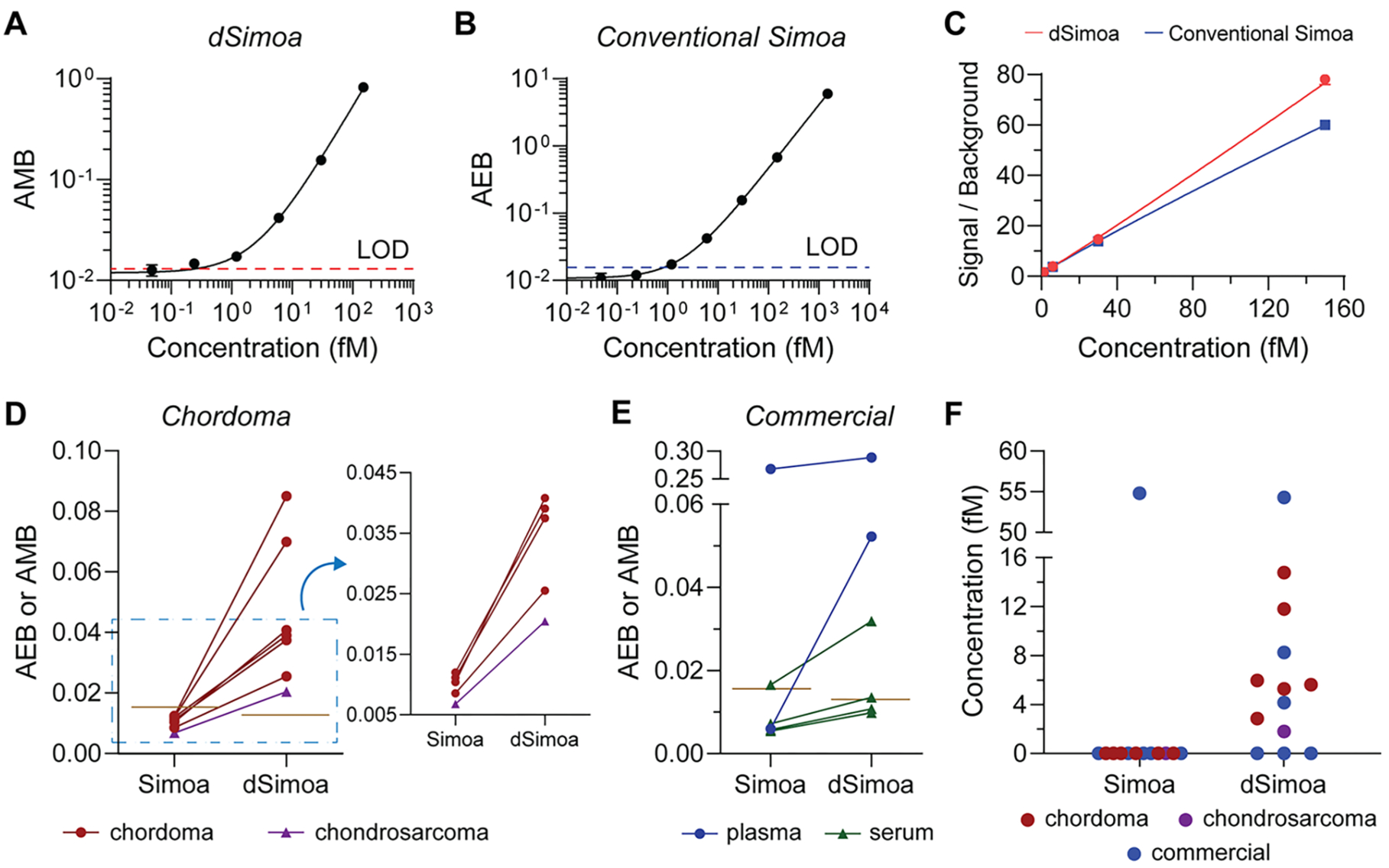
Measurements of Brachyury in plasma. (A) dSimoa and (B) conventional Simoa calibration curves for human Brachyury. Dashed lines indicate the calculated limits of detection (LODs). (C) Comparison of signal to background ratios between dSimoa and conventional Simoa across the calibration curve range. (D, E) Average enzyme per bead (AEB) and average molecule per bead (AMB) values measured by conventional Simoa and dSimoa, respectively, in chordoma and chondrosarcoma patient plasma samples (D) and commercial plasma and serum samples (E). Brown lines indicate LODs of the assays. (F) Measured concentrations in chordoma, chondrosarcoma, and commercial plasma and serum samples using dSimoa and conventional Simoa. Measurements below the LOD were assigned a value of zero.
Finally, we compared the abilities of dSimoa and conventional Simoa to detect endogenous Brachyury in several chordoma patient plasma samples as well as commercial plasma and serum samples from healthy donors. While the conventional Simoa measurements fell below its LOD for all six chordoma patient samples, dSimoa was able to measure detectable levels of Brachyury in all six samples (Figure 5D), demonstrating that even a relatively small improvement in sensitivity is sometimes sufficient to measure clinically important biomarkers. We also tested one chondrosarcoma patient sample, which was undetectable by conventional Simoa but detectable by dSimoa. Among the six commercial plasma and serum samples, Brachyury was detectable in one sample using conventional Simoa and in three samples using dSimoa (Figure 5E). Notably, although the measured concentrations by dSimoa in many of the samples were in the low femtomolar range, above the calculated LOD of the conventional Simoa assay, measurements of these samples by conventional Simoa still fell below its LOD (Figure 5F). However, among the AEBs that were below the conventional Simoa LOD, higher AEB values generally correlated to higher AMB values and measured concentrations in the dSimoa assay. Furthermore, at higher sample concentrations, dSimoa and conventional Simoa yielded similar measured concentrations. The superior performance of dSimoa in plasma and serum at low concentrations may be attributed to several factors, including the improved LOD of dSimoa and better sampling efficiencies, which can increase measurement precision particularly at low concentrations. Another possibility is that dSimoa performs more accurately in plasma and serum matrices than conventional Simoa, with higher signal to background and recoveries in plasma and serum using 5-fold fewer beads. In addition, conventional Simoa employs a large enzyme label that may exhibit higher nonspecific binding than the much smaller oligonucleotide label used in dSimoa. Finally, the greater amount of washing during the dSimoa assay compared to the conventional Simoa assay may have further reduced interference from plasma and serum components.
DISCUSSION
We have developed an innovative, simple single molecule measurement platform that can detect low- to midattomolar protein concentrations. By addressing the challenge of low efficiencies in sampling rare target molecules in digital ELISA approaches, we enhanced sensitivity by up to 25-fold over the current Simoa technology, which is presently the gold standard for ultrasensitive protein detection. The attomolar LODs achieved by dSimoa correspond to an over 10000-fold increase in sensitivity over conventional immunoassays. Localization of a nondiffusible amplified signal to each bead eliminates the need for signal compartmentalization into microwells or droplets, allowing direct dropcasting of all the beads onto a slide for rapid drying and formation of a monolayer film. This simple approach allows 40–50% on average of the total assay beads to be analyzed–an 8- to 10-fold increase over the ~5% sampling efficiency of the current Simoa technology. At low sample concentrations, particularly with capture efficiencies well below 100% (~1–3% across all capture and labeling steps in the dSimoa assays developed in this work), improved sampling is critical for minimizing Poisson noise-associated measurement CVs. Although some beads were lost during washing steps or excluded from analysis if overlapping or aggregated, we found from our experimental results that analyzing 20% of the total assay beads already improves LODs by about an order of magnitude, with small further improvements in measurement CVs and the LOD as more beads are analyzed. The significantly improved sampling efficiency of dSimoa also allows the use of fewer assay beads compared to conventional Simoa, increasing the fraction of “on” beads” and thereby the signal to background. Further improvements in sensitivity can be attained by using affinity reagents with lower dissociation constants and decreasing nonspecific binding of the affinity reagents and streptavidin-DNA label. With the development of better affinity reagents and methods to reduce nonspecific binding, dSimoa can potentially detect down to zeptomolar protein concentrations.
With its attomolar sensitivity, dSimoa can pave the way toward discovery of new biomarkers and biological mechanisms underlying various diseases. As a proof of principle, we demonstrated that dSimoa can measure low concentrations of the T-box-family transcription factor Brachyury that were previously undetectable by the current Simoa technology in plasma samples from chordoma patients. While Brachyury has been shown to be highly overexpressed in the tumors of chordoma patients, its levels in plasma have not been assessed.26–30 As the diagnosis of chordoma requires an invasive needle or incisional biopsy into the skull base or spine, a blood-based test would provide a significantly lower-risk diagnostic procedure and potentially facilitate early diagnosis of chordoma.31,32 While our measurements were performed in only a small sample cohort, the significantly improved detectability of Brachyury in chordoma patient plasma samples using dSimoa opens new possibilities for a potential blood test and the discovery of new biological mechanisms. Achieving an order of magnitude or more improvement in sensitivity with dSimoa also holds important implications for the discovery of new blood-based biomarkers for many other cancer types and neurological disorders. A diagnostic blood test for neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases would prove especially critical for widespread screening and early diagnosis, which are currently very difficult due to the need for highly invasive lumbar punctures. In many cases, where biomarker levels become detectable only after significant disease progression, the enhanced sensitivity of dSimoa can accelerate disease diagnosis in early stages for improved health outcomes.
Importantly, dSimoa also increases the simplicity of digital bioassay signal readout, which upon further development can potentially be integrated into a POC platform and thus address challenges of low sensitivity in current POC diagnostics. While increasing sampling efficiency for enhanced sensitivity in digital immunoassays has also been demonstrated in bead droplet arrays and droplet digital ELISA methods, these methods introduce additional complexity in fabrication and processing steps.13,14 In contrast, the digital readout process for dSimoa requires only a microscope slide for bead loading and a simple optical setup, with no additional materials or complex instrumentation needed. Furthermore, the dropcasting process is remarkably simple and rapid. While further work is required to integrate the sample processing steps in the dSimoa assay into an automated, streamlined microfluidic system for POC implementation, the dropcast method simplifies the single molecule detection readout process and increases cost-effectiveness compared to current microwell- or droplet-based digital ELISA methods. The enhanced sensitivity of dSimoa can potentially enable the detection of various biomarkers that exist at very low concentrations and have not previously been measured in easily accessible biofluids such as saliva and urine. An additional interesting aspect of dSimoa is the long-term signal stability in the dropcast films, which increases flexibility in the assay process. For instance, in resource-limited settings, where a suitable optical setup may not be readily available, the dropcast films can be easily shipped to facilities for imaging and analysis, with no signal loss for at least one month.
Future work will focus on streamlining and automating the dSimoa workflow to increase amenability to POC application, including integration into a microfluidic device for sample processing and incorporation of portable imaging. Automation of the sample processing workflow in a microfluidic system, combined with the single molecule resolution and high sampling efficiency of dSimoa, can potentially reduce assay times compared to the current 3 h workflow, while still achieving high sensitivities for detecting low abundance biomarkers that are currently undetectable by existing POC platforms. Furthermore, due to the rapid kinetics of RCA and the reduced diffusional distances in small microfluidic reaction volumes, we anticipate that the times for each sample processing step, including RCA, can be further shortened. Although RCA was carried out for 1 h in this work, we began observing detectable signals after 15 min. RCA signal amplification time can be further reduced by increasing the spatial density of fluorescent labels on each concatemer. As several automated and streamlined microfluidic-based methods have been developed for bead-based immunoassays, incorporation of the dSimoa sample processing steps into an automated microfluidic platform is highly feasible.5,33–36 In addition, many portable fluorescence imaging platforms have been developed, including smartphone attachments for imaging single fluorescent nanoparticles and RCA products, and can be readily adapted for dSimoa.5,37–41 While multiple frames were required to completely capture each dropcast film in this work, shorter imaging times should be possible using an automated hand-held reader or a wide field-of-view camera. Moreover, as supported by our sampling analysis of the dSimoa results, only ~20% of the total assay beads may need to be imaged to achieve close to maximal sensitivity. Future integration of dSimoa into an ultrasensitive, portable, and automated platform can potentially facilitate widespread screening, early detection, and monitoring of many diseases, including infectious diseases such as the recent SARS-CoV-2 pandemic and tuberculosis, traumatic brain injuries, and myocardial infarction.
CONCLUSION
In summary, we have developed a simple, ultrasensitive single molecule detection platform, dSimoa, that enhances sensitivity by up to 25-fold over the current state-of-the-art digital ELISA technology. By improving sampling of rare target molecules, this approach enables protein detection in the attomolar range, thus opening a window into a wide range of potential disease biomarkers that were previously unmeasurable. Importantly, dSimoa also simplifies the digital assay readout process and is therefore more amenable to future integration into a POC system. The dSimoa platform can also be readily adopted to measure other disease-related biomolecules, including micro-RNAs and small molecules, in simpler and more sensitive assays compared to the previously developed Simoa assays.42,43 By measuring very low concentrations of biomolecules that are undetectable by current methods, dSimoa provides a highly promising and simple platform for ultrasensitive detection that can facilitate early disease diagnosis.
METHODS
Materials.
All antibodies, recombinant proteins, and DNA sequences used in this study are listed in the Supporting Information. DNA primer, template, and probe were obtained from Integrated DNA Technologies or MilliporeSigma. Conjugation and assay buffers, as well as dye-encoded carboxylated 2.7-μm paramagnetic beads (Homebrew Multiplex Beads 488), were purchased from Quanterix Corporation.
Preparation of Antibody-Coated Capture Beads.
For each target, capture antibody was buffer exchanged into Bead Conjugation Buffer (Quanterix), using a 50K Amicon centrifugal filter (0.5 mL, MilliporeSigma). Bead Conjugation Buffer was added to antibody solution in the filter up to 500 μL, followed by centrifugation at 14000×g for 5 min. The effluent was discarded and the process was repeated twice. Buffer-exchanged antibody was recovered by inverting the filter into a new tube and centrifuging at 1000×g for 2 min, followed by a 50 μL Bead Conjugation Buffer rinse and a second centrifugation at 1000×g for 2 min. Antibody concentration was measured with a NanoDrop spectrophotometer, and antibody was diluted to 0.5 mg/mL (IL-10), 0.3 mg/mL (Brachyury), or 0.2 mg/mL (IL-1β) in Bead Conjugation Buffer for subsequent bead coupling. Dye-encoded paramagnetic beads (2.8 × 108) were washed three times with 200 μL of Bead Wash Buffer (Quanterix) and two times with 200 μL of Bead Conjugation Buffer, and resuspended in 190 μL cold Bead Conjugation Buffer. A 1 mg vial of 1-ethyl-3-(3-(dimethylamino)propyl) carbodiimide hydrochloride (EDC) (Thermo Fisher Scientific) was then reconstituted in 100 μL of cold Bead Conjugation Buffer, and 10 μL was immediately added to the beads. The beads were activated for 30 min under shaking. After activation, the beads were washed with 200 μL of cold Bead Conjugation Buffer, resuspended in 200 μL of capture antibody solution, and placed on a shaker for 2 h for antibody coupling. The antibody-coupled beads were subsequently washed two times with 200 μL of Bead Wash Buffer and blocked with 200 μL of Bead Blocking Buffer (Quanterix) for 30 min under shaking. After blocking, the beads were washed with 200 μL of Bead Wash Buffer and then with 200 μL of Bead Diluent (Quanterix), before resuspension in 200 μL Bead Diluent. For IL-1β, the EDC activation and antibody coupling steps were performed at 4 °C, with 4.2 × 108 beads, 9 μL of EDC for bead activation, and 300 μL of 0.2 mg/mL antibody for conjugation. A Beckman Coulter Z1 Particle Counter was used to count the beads, which were stored at 4 °C for subsequent use in assays.
Preparation of Streptavidin-DNA Conjugate.
The RCA template (MilliporeSigma) was first annealed to a 5′ azide-modified primer (Integrated DNA technologies) by heating a solution of 45 μL of 100 μM primer, 54 μL of 100 μM template, and 26.6 μL of 5x NEBNextQuick Ligation reaction buffer (New England Biolabs) at 95 °C for 2 min and allowing the solution to slowly cool to room temperature over 90 min. The template was then ligated by the addition of 7.5 μL of T4 DNA ligase (2000000 units/mL, New England Biolabs) and incubation of the reaction at room temperature for 3 h. The ligation reaction was then buffer exchanged into PBS with 1 mM ethylenediaminetetraacetic acid (EDTA) using a Zeba spin desalting column (7K MWCO, Thermo Fisher Scientific). Streptavidin (Biolegend 280302) was buffer exchanged into phosphate-buffered saline (PBS) with a 10K Amicon centrifugal filter (0.5 mL, MilliporeSigma), following the same buffer exchange procedure as described above for capture antibodies, and then diluted to 1 mg/mL in PBS. Dibenzocyclooctyne-PEG4-N-hydroxysuccinimidyl ester (DBCO-PEG4-NHS, 1 mg, MilliporeSigma) was dissolved in 200 μL of dimethyl sulfoxide, and a 20-fold molar excess was added to the buffer-exchanged streptavidin. The conjugation reaction was allowed to incubate for 30 min at room temperature and was then purified with a 10K Amicon centrifugal filter. The conjugated streptavidin was washed with PBS with 1 mM EDTA in five centrifugations at 14000×g for 5 min followed by one centrifugation at 14000×g for 15 min. The purified DBCO-conjugated streptavidin was then recovered by inverting the filter and centrifuging at 1000×g for 2 min. Annealed primer-template was added to the DBCO-conjugated streptavidin at a 2-fold molar excess, and the conjugation reaction was allowed to proceed overnight at 4 °C. The streptavidin-DNA conjugate was then stored in aliquots at −80 °C with 0.1% bovine serum albumin (BSA), 5 mM EDTA, and 0.02% sodium azide, without further purification.
Dropcast Single Molecule Assays.
All dSimoa assays were performed in 96-well plates (Greiner Bio-One, 655096). Antibody-coated beads, recombinant proteins, and biotinylated detector antibodies were diluted in Sample Diluent (Quanterix) to the desired concentrations. Detector antibody and streptavidin-DNA concentrations for each assay are listed in the Supporting Information. For each assay, 10 μL of antibody-coated beads (100000 beads total) and 10 μL of biotinylated detector antibody were added to 100 μL of protein sample. The plate was then sealed and shaken for 1 h for immunocomplex formation. The beads were washed six times with System Wash Buffer 1 (Quanterix) using a BioTek 405 TS Microplate Washer, followed by resuspension in 100 μL of streptavidin-DNA conjugate diluted in Sample Diluent with 5 mM EDTA. The plate was shaken for 15 min for streptavidin-DNA labeling of the immunocomplexes and then washed eight times with System Wash Buffer 1 using the microplate washer. After washing, the beads were transferred to a new 96-well plate and washed an additional time with 200 μL of System Wash Buffer 1 before resuspension in 60 μL of RCA solution. The RCA solution consisted of 0.5 mM deoxynucleotide mix (New England Biolabs), 0.33 U/uL phi29 DNA polymerase (Lucigen), 0.2 mg/mL BSA, 1 nM ATTO 647N-labeled DNA probe (Integrated DNA Technologies), and 0.1% Tween-20 in a reaction buffer comprising 50 mM Tris-HCl (pH 7.5), 10 mM (NH4)2SO4, and 10 mM MgCl2. Dithiothreitol (DTT) was removed from the phi29 polymerase solution received from the manufacturer using a Zeba spin desalting column (7K MWCO, Thermo Fisher Scientific). RCA was performed at 37 °C for 1 h with shaking of the plate, after which 150 μL of PBS with 5 mM EDTA was added to each sample to stop the RCA reaction. The beads were washed two times with 200 μL of dropcast buffer (50 mM Tris-HCl, 50 mM NaCl, 0.1% Tween-20, 0.5% BSA), concentrated to 10–15 μL, and then resuspended and dropcast onto a microscope slide. The dropcast beads were allowed to dry for 10–15 min to form monolayer films.
For saliva samples, pooled human saliva (BioIVT) was centrifuged at 13150×g for 20 min at 4 °C. In dilution linearity experiments, the desired volume of supernatant was serially diluted 2- to 32-fold in Sample Diluent with protease inhibitor (Halt Protease Inhibitor Cocktail, Thermo Fisher Scientific). For spike and recovery experiments, recombinant human IL-10 protein was spiked into 4-fold diluted saliva samples at 100, 10, and 1 fM.
Plasma samples from chordoma patients were obtained from Dr. Sandro Santagata and Dr. Keith Ligon (Brigham and Women’s Hospital) and centrifuged at 2000×g for 10 min at 4 °C, and the supernatant was aliquoted to prevent freeze–thaw cycles. Commercial plasma and serum samples were obtained from BioIVT. All samples were diluted 8-fold in Sample Diluent for measurements.
Imaging and Analysis.
Brightfield and fluorescent images of the dropcast bead films were acquired using an Olympus IX81 inverted microscope, with a scientific CMOS camera (ORCA-Flash4.0 LT+, Hamamatsu) and 10x objective. Fluorescence images obtained with a GFP filter (1 s exposure) were used to locate the dye-encoded beads, while fluorescence images obtained with a Cy5 filter (1 s exposure) were used to identify “on” versus “off” beads. Commercial software (cellSens) was used to control the stage and camera. Brightfield and fluorescence images were acquired for each frame, and multiple frames were acquired to capture the entire dropcast film, excluding the film edges. About 20–25 frames were acquired per dropcast film, with average total imaging times of approximately 15 min.
Image analysis was performed in MATLAB. The beads were first located in the 488 nm fluorescent image using a disk-shaped morphological structuring element, with top-hat filtering to correct for uneven illumination. Overlapping or aggregated beads were separated by watershed segmentation, and any remaining aggregated beads were removed by a size cutoff. The maximum signal intensity on each identified bead was determined in the corresponding Cy5 fluorescent image, which first underwent a top-hat filter to correct for uneven illumination. A Gaussian distribution was fitted to the bead fluorescence intensities, and the cutoff intensity value for an “on” versus “off” bead was determined as five standard deviations above the mean of the distribution. Thus, all beads with intensities above the cutoff value were counted as “on” beads. The fraction of on beads was calculated as the total number of “on” beads over the total number of beads, and the average molecule per bead (AMB) was subsequently calculated from the Poisson distribution.
Calibration curves were fit using a four parameter logistic (4PL) fit in GraphPad Prism and used to determine unknown sample concentrations. The R2 values of the calibration curve fits can be found in the Supporting Information. All measurements were performed in 3–4 replicates, except dilution linearity and spike and recovery assays, which were performed in duplicates. The limit of detection (LOD) of each assay was calculated as the concentration corresponding to three standard deviations above the background AMB.
Simoa Assays.
Conventional Simoa assays were performed on an HD-X Analyzer (Quanterix), using the same antibody-coated capture beads (500000 beads per assay) and biotinylated detector antibodies at the same concentrations as in the corresponding dSimoa assays and 100 μL sample volumes. Streptavidin-β-galactosidase (SβG) Concentrate (Quanterix) was diluted in SβG Diluent (Quanterix) to the desired concentration. The same incubation time of 1 h was used for the antibody capture step in which the beads, sample, and detector antibody are incubated for immunocomplex sandwich formation. For each target, two assay conditions were performed: one assay with the same SβG concentrations and incubation times as in the corresponding dSimoa assay, and one assay with a standard SβG concentration and incubation time used on the HD-X (150 pM SβG for 5 min). Beads, detector antibody, and SβG were placed in plastic bottles (Quanterix), and samples were added to a 96-well plate, all of which were loaded into the HD-X Analyzer. The enzyme substrate (resorufin β-D-galactopyranoside), Wash Buffer 1, Wash Buffer 2, and Simoa Sealing Oil were loaded into the HD-X Analyzer according to the manufacturer’s instructions. All assay steps, image analyses, and calculations of average enzyme per bead (AEB) were automated, as previously described in detail.13
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Dr. Sandro Santagata and Dr. Keith Ligon (Brigham and Women’s Hospital) for providing the chordoma patient samples measured in this study and Mark Zaki for suggestions of Brachyury measurements in plasma. The authors would also like to thank Limor Cohen and Adam Maley for helpful discussions on assay development and analysis. This work was supported by Good Ventures (Open Philanthropy Project). Connie Wu would also like to acknowledge support by a postdoctoral fellowship from the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health (grant no. 1F32EB029777-01).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c04331.
Signal stability, validation of human IL-10 dSimoa assay in pooled human saliva, validation of human Brachyury dSimoa assay in human plasma and serum, conventional Simoa assays, AMB and AEB values for calibration curves, summary of dSimoa assay conditions, antibodies and recombinant protein standards, DNA sequences used in dSimoa assays (PDF)
The authors declare the following competing financial interest(s): David R. Walt is a founder and equity holder of Quanterix Corporation, and also serves on its Board of Directors. Dr. Walt’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict of interest policies. All other authors declare no competing financial interest.
REFERENCES
- (1).Cohen L; Walt DR Highly Sensitive and Multiplexed Protein Measurements. Chem. Rev 2019, 119 (1), 293–321. [DOI] [PubMed] [Google Scholar]
- (2).Rissin DM; Kan CW; Campbell TG; Howes SC; Fournier DR; Song L; Piech T; Patel PP; Chang L; Rivnak AJ; Ferrell EP; Randall JD; Provuncher GK; Walt DR; Duffy DC Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol 2010, 28, 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Rissin DM; Walt DR Digital Readout of Target Binding with Attomole Detection Limits via Enzyme Amplification in Femtoliter Arrays. J. Am. Chem. Soc 2006, 128 (19), 6286–6287. [DOI] [PubMed] [Google Scholar]
- (4).Rissin DM; Walt DR Digital Concentration Readout of Single Enzyme Molecules Using Femtoliter Arrays and Poisson Statistics. Nano Lett. 2006, 6 (3), 520–523. [DOI] [PubMed] [Google Scholar]
- (5).Yelleswarapu V; Buser JR; Haber M; Baron J; Inapuri E; Issadore D Mobile platform for rapid sub-picogram-per-milliliter, multiplexed, digital droplet detection of proteins. Proc. Natl. Acad. Sci. U. S. A 2019, 116 (10), 4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Robey TT; Panegyres PK Cerebrospinal fluid biomarkers in neurodegenerative disorders. Future Neurol. 2019, 14 (1), FNL6. [Google Scholar]
- (7).Olsson B; Lautner R; Andreasson U; Öhrfelt A; Portelius E; Bjerke M; Hölttä M; Rosén C; Olsson C; Strobel G; Wu E; Dakin K; Petzold M; Blennow K; Zetterberg H CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol. 2016, 15 (7), 673–684. [DOI] [PubMed] [Google Scholar]
- (8).Galasko DR; Shaw LM CSF biomarkers for Alzheimer disease — approaching consensus. Nat. Rev. Neurol 2017, 13 (3), 131–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Fortea J; Carmona-Iragui M; Benejam B; Fernández S; Videla L; Barroeta I; Alcolea D; Pegueroles J; Muñoz L; Belbin O; de Leon MJ; Maceski AM; Hirtz C; Clarimón J; Videla S; Delaby C; Lehmann S; Blesa R; Lleó A Plasma and CSF biomarkers for the diagnosis of Alzheimer’s disease in adults with Down syndrome: a cross-sectional study. Lancet Neurol. 2018, 17 (10), 860–869. [DOI] [PubMed] [Google Scholar]
- (10).Hampel H; O’Bryant SE; Molinuevo JL; Zetterberg H; Masters CL; Lista S; Kiddle SJ; Batrla R; Blennow K Blood-based biomarkers for Alzheimer disease: mapping the road to the clinic. Nat. Rev. Neurol 2018, 14 (11), 639–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Simrén J; Ashton NJ; Blennow K; Zetterberg H An update on fluid biomarkers for neurodegenerative diseases: recent success and challenges ahead. Curr. Opin. Neurobiol 2020, 61, 29–39. [DOI] [PubMed] [Google Scholar]
- (12).Parnetti L; Gaetani L; Eusebi P; Paciotti S; Hansson O; El-Agnaf O; Mollenhauer B; Blennow K; Calabresi P CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol. 2019, 18 (6), 573–586. [DOI] [PubMed] [Google Scholar]
- (13).Cohen L; Cui N; Cai Y; Garden PM; Li X; Weitz DA; Walt DR, Single Molecule Protein Detection with Attomolar Sensitivity using Droplet Digital ELISA. Under revision. [DOI] [PubMed] [Google Scholar]
- (14).Kim SH; Iwai S; Araki S; Sakakihara S; Iino R; Noji H Large-scale femtoliter droplet array for digital counting of single biomolecules. Lab Chip 2012, 12 (23), 4986–4991. [DOI] [PubMed] [Google Scholar]
- (15).Witters D; Knez K; Ceyssens F; Puers R; Lammertyn J Digital microfluidics-enabled single-molecule detection by printing and sealing single magnetic beads in femtoliter droplets. Lab Chip 2013, 13 (11), 2047–2054. [DOI] [PubMed] [Google Scholar]
- (16).Wilson DH; Rissin DM; Kan CW; Fournier DR; Piech T; Campbell TG; Meyer RE; Fishburn MW; Cabrera C; Patel PP; Frew E; Chen Y; Chang L; Ferrell EP; von Einem V; McGuigan W; Reinhardt M; Sayer H; Vielsack C; Duffy DC The Simoa HD-1 Analyzer: A Novel Fully Automated Digital Immunoassay Analyzer with Single-Molecule Sensitivity and Multiplexing. Journal of Laboratory Automation 2016, 21 (4), 533–547. [DOI] [PubMed] [Google Scholar]
- (17).Barbee KD; Hsiao AP; Heller MJ; Huang X Electric field directed assembly of high-density microbead arrays. Lab Chip 2009, 9 (22), 3268–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Decrop D; Pardon G; Brancato L; Kil D; Zandi Shafagh R; Kokalj T; Haraldsson T; Puers R; van der Wijngaart W; Lammertyn J Single-Step Imprinting of Femtoliter Microwell Arrays Allows Digital Bioassays with Attomolar Limit of Detection. ACS Appl. Mater. Interfaces 2017, 9 (12), 10418–10426. [DOI] [PubMed] [Google Scholar]
- (19).Decrop D; Brans T; Gijsenbergh P; Lu J; Spasic D; Kokalj T; Beunis F; Goos P; Puers R; Lammertyn J Optical Manipulation of Single Magnetic Beads in a Microwell Array on a Digital Microfluidic Chip. Anal. Chem 2016, 88 (17), 8596–8603. [DOI] [PubMed] [Google Scholar]
- (20).Jarvius J; Melin J; Göransson J; Stenberg J; Fredriksson S; Gonzalez-Rey C; Bertilsson S; Nilsson M Digital quantification using amplified single-molecule detection. Nat. Methods 2006, 3 (9), 725–727. [DOI] [PubMed] [Google Scholar]
- (21).Söderberg O; Gullberg M; Jarvius M; Ridderstråle K; Leuchowius K-J; Jarvius J; Wester K; Hydbring P; Bahram F; Larsson L-G; Landegren U Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 2006, 3 (12), 995–1000. [DOI] [PubMed] [Google Scholar]
- (22).Kuhnemund M; Hernandez-Neuta I; Sharif MI; Cornaglia M; Gijs MAM; Nilsson M Sensitive and inexpensive digital DNA analysis by microfluidic enrichment of rolling circle amplified single-molecules. Nucleic Acids Res. 2017, 45 (8), No. e59–e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Lizardi PM; Huang X; Zhu Z; Bray-Ward P; Thomas DC; Ward DC Mutation detection and single-molecule counting using isothermal rolling-circle amplification. Nat. Genet 1998, 19 (3), 225–232. [DOI] [PubMed] [Google Scholar]
- (24).Konry T; Hayman RB; Walt DR Microsphere-Based Rolling Circle Amplification Microarray for the Detection of DNA and Proteins in a Single Assay. Anal. Chem 2009, 81 (14), 5777–5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Rissin DM; Fournier DR; Piech T; Kan CW; Campbell TG; Song L; Chang L; Rivnak AJ; Patel PP; Provuncher GK; Ferrell EP; Howes SC; Pink BA; Minnehan KA; Wilson DH; Duffy DC Simultaneous Detection of Single Molecules and Singulated Ensembles of Molecules Enables Immunoassays with Broad Dynamic Range. Anal. Chem 2011, 83 (6), 2279–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Vujovic S; Henderson S; Presneau N; Odell E; Jacques TS; Tirabosco R; Boshoff C; Flanagan AM Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J. Pathol 2006, 209 (2), 157–165. [DOI] [PubMed] [Google Scholar]
- (27).Presneau N; Shalaby A; Ye H; Pillay N; Halai D; Idowu B; Tirabosco R; Whitwell D; Jacques TS; Kindblom L-G; Brüderlein S; Möller P; Leithner A; Liegl B; Amary FM; Athanasou NN; Hogendoorn PCW; Mertens F; Szuhai K; Flanagan AM Role of the transcription factor T (brachyury) in the pathogenesis of sporadic chordoma: a genetic and functional-based study. Journal of Pathology 2011, 223 (3), 327–335. [DOI] [PubMed] [Google Scholar]
- (28).Yang XR; Ng D; Alcorta DA; Liebsch NJ; Sheridan E; Li S; Goldstein AM; Parry DM; Kelley MJT (brachyury) gene duplication confers major susceptibility to familial chordoma. Nat. Genet 2009, 41 (11), 1176–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Shen J; Li C-D; Yang H-L; Lu J; Zou T-M; Wang D-L; Deng M Classic chordoma coexisting with benign notochordal cell rest demonstrating different immunohistological expression patterns of brachyury and galectin-3. J. Clin. Neurosci 2011, 18 (1), 96–99. [DOI] [PubMed] [Google Scholar]
- (30).Miettinen M; Wang Z; Lasota J; Heery C; Schlom J; Palena C Nuclear Brachyury Expression Is Consistent in Chordoma, Common in Germ Cell Tumors and Small Cell Carcinomas, and Rare in Other Carcinomas and Sarcomas: An Immunohistochemical Study of 5229 Cases. American Journal of Surgical Pathology 2015, 39 (10), 1305–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Walcott BP; Nahed BV; Mohyeldin A; Coumans J-V; Kahle KT; Ferreira MJ Chordoma: current concepts, management, and future directions. Lancet Oncol. 2012, 13 (2), No. e69–e76. [DOI] [PubMed] [Google Scholar]
- (32).Bergh P; Kindblom L-G; Gunterberg B; Remotti F; Ryd W; Meis-Kindblom JM Prognostic factors in chordoma of the sacrum and mobile spine. Cancer 2000, 88 (9), 2122–2134. [DOI] [PubMed] [Google Scholar]
- (33).Boyd-Moss M; Baratchi S; Di Venere M; Khoshmanesh K Self-contained microfluidic systems: a review. Lab Chip 2016, 16 (17), 3177–3192. [DOI] [PubMed] [Google Scholar]
- (34).Zhang Y; Nguyen N-T Magnetic digital microfluidics - a review. Lab Chip 2017, 17 (6), 994–1008. [DOI] [PubMed] [Google Scholar]
- (35).Nie S; Henley WH; Miller SE; Zhang H; Mayer KM; Dennis PJ; Oblath EA; Alarie JP; Wu Y; Oppenheim FG; Little FF; Uluer AZ; Wang P; Ramsey JM; Walt DR An automated integrated platform for rapid and sensitive multiplexed protein profiling using human saliva samples. Lab Chip 2014, 14 (6), 1087–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sista RS; Eckhardt AE; Srinivasan V; Pollack MG; Palanki S; Pamula VK Heterogeneous immunoassays using magnetic beads on a digital microfluidic platform. Lab Chip 2008, 8 (12), 2188–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Wei Q; Qi H; Luo W; Tseng D; Ki SJ; Wan Z; Göröcs Z; Bentolila LA; Wu T-T; Sun R; Ozcan A Fluorescent Imaging of Single Nanoparticles and Viruses on a Smart Phone. ACS Nano 2013, 7 (10), 9147–9155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kühnemund M; Wei Q; Darai E; Wang Y; Hernández-Neuta I; Yang Z; Tseng D; Ahlford A; Mathot L; Sjöblom T; Ozcan A; Nilsson M Targeted DNA sequencing and in situ mutation analysis using mobile phone microscopy. Nat. Commun 2017, 8, 13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kong JE; Wei Q; Tseng D; Zhang J; Pan E; Lewinski M; Garner OB; Ozcan A; Di Carlo D Highly Stable and Sensitive Nucleic Acid Amplification and Cell-Phone-Based Readout. ACS Nano 2017, 11 (3), 2934–2943. [DOI] [PubMed] [Google Scholar]
- (40).Minagawa Y; Ueno H; Tabata KV; Noji H Mobile imaging platform for digital influenza virus counting. Lab Chip 2019, 19 (16), 2678–2687. [DOI] [PubMed] [Google Scholar]
- (41).Ghosh KK; Burns LD; Cocker ED; Nimmerjahn A; Ziv Y; Gamal AE; Schnitzer MJ Miniaturized integration of a fluorescence microscope. Nat. Methods 2011, 8 (10), 871–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Cohen L; Hartman MR; Amardey-Wellington A; Walt DR Digital direct detection of microRNAs using single molecule arrays. Nucleic Acids Res. 2017, 45 (14), No. e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wang X; Cohen L; Wang J; Walt DR Competitive Immunoassays for the Detection of Small Molecules Using Single Molecule Arrays. J. Am. Chem. Soc 2018, 140 (51), 18132–18139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.