

Abstract
Iron-sulfur cluster proteins play key roles in a multitude of physiological processes; including gene expression, nitrogen and oxygen sensing, electron transfer, and DNA repair. Biosynthesis of iron-sulfur clusters occurs in mitochondria on iron-sulfur cluster scaffold proteins in the form of [2Fe-2S] cores that are then transferred to apo targets within metabolic or respiratory pathways. The mechanism by which cytosolic Fe-S cluster proteins mature to their holo forms remains controversial. The mitochondrial inner membrane protein Atm1p can transport glutathione-coordinated iron-sulfur clusters, which may connect the mitochondrial and cytosolic iron-sulfur cluster assembly systems. Herein we describe experiments on the yeast Atm1p/ABCB7 exporter that provide additional support for a glutathione-complexed cluster as the natural physiological substrate and a reflection of the endosymbiotic model of mitochondrial evolution. These studies provide insight on the mechanism of cluster transport and the molecular basis of human disease conditions related to ABCB7. Recruitment of MgATP following cluster binding promotes a structural transition from closed to open conformations that is mediated by coupling helices, with MgATP hydrolysis facilitating the return to the closed state.
Table of Contents Entry
The mechanism of Atm1p mediated mitochondrial Fe-S cluster export is addressed, providing support for a glutathione-complexed [2Fe-2S] cluster as a physiologically relevant substrate. Binding of [2Fe-2S](GS)4 reduces KM for Mg-ATP ~ 100-fold, triggering the closed to open transition. A carboxylate residue in the binding site serves a key role in communicating structural change after substrate binding and is implicated in a disease-causing role in a natural human mutation. A detailed mechanism of cluster export, combining kinetic, thermodynamic and structural data is presented.
Introduction
Iron-sulfur clusters are essential cofactors that function in electron transport, transcriptional and translational regulation, and enzymatic substrate binding and catalysis. However, the mechanisms of in vivo assembly and trafficking are not fully understood. Iron and sulfur would be toxic if allowed to freely exist in the cell,3 and in the case of eukaryotes the majority of these clusters are assembled in mitochondria via a suite of assembly proteins.4 Essential to this model is the requirement for membrane-bound transporters to facilitate cluster delivery for cytosolic proteins. One family of transporters, the ATP “binding cassette” (ABC) transporters, utilize the energy of MgATP to move solutes across the membrane. The ABC transporters are integral membrane proteins1–2 that exhibit a conserved core structure with two transmembrane (TM) domains that confer substrate binding, and two highly conserved nucleotide binding domains (NBD) that execute MgATP hydrolysis to initiate conformational changes to promote substrate transport.5 Coupling helices found in the interconnecting loops (ICL) mediate communication between the TMD and NBD.6 Although the ABC transporters have been extensively studied, many questions remain regarding the transport cycle and molecular mechanism. Functionally, they tend to be mechanistically diverse,6b and prior studies have debated whether one or two nucleotide hydrolysis events are required for substrate transport, with varying results.7
Atm1p is a Saccharomyces cerevisiae ABC transporter that is homodimeric, consisting of an amino-terminal transmembrane domain and a carboxy-terminal nucleotide binding domain.8 Yeast cells deficient in Atm1p have a general deficiency of heme and iron-sulfur-cluster-containing proteins, are sensitive to oxidizing agents, and show increased levels of both glutathione and especially mitochondrial iron levels, indicating a crucial role for Atm1p in iron homeostasis.9 To date, the substrate for this transporter is believed to contain cysteine, with much of the evidence suggesting a species involving glutathione.10 Our laboratory has demonstrated glutathione-bound iron-sulfur cluster, [2Fe-2S](GS4), as a potential substrate, because these clusters have been shown to cause a significant increase in the ATPase activity of Atm1p,11 are biosynthetically accessible via glutathione extraction from the ISU scaffold protein,11 and are actively transported in proteoliposome models.12 An alternative polysulfide adduct of oxidized glutathione dimer has also been proposed,13 but the physiological relevance is unclear. Moreover, prior studies have shown that heavy metal complexes with glutathione are transported by ABC transporters in the context of heavy metal removal, consistent with the endosymbiotic model.14 Crystal structures of the homologous Atm1p from Novosphingobium aromaticivorans (NaAtm1p), an alphaproteobacterium and a key phylum underpinning the endosymbiotic model, have been solved. The crystal structure with GSSG revealed two separate binding locations for the oxidized glutathione molecule, separated by 5 Å, suggesting a potential binding pocket for a [2Fe-2S](GS)4 cluster and further supporting the cluster as a natural substrate.14d The [2Fe-2S](GS)4 complex is hydrolytically stable and can be taken up by certain apo iron-sulfur cluster proteins, suggesting a plausible cellular role in the physiological labile iron pool.15
ABCB7 is the human ortholog of the ABC transporter Atm1p; sharing 43% sequence identity and 72% similar amino acid composition.16 ABCB7 has been linked to X-linked sideroblastic anemia, a disease caused by a single amino acid mutation.17 Four different ABCB7 residues (D208, I400, V411, and E433) are susceptible to a point mutation that is capable of causing the anemia, and all of them are in different locations of the protein structure.18 All residues implicated in disease-causing mutations are conserved, indicating a major functional or structural role for each.
Of these disease-causing mutations, the human E433K mutation is the most phenotypically severe. The corresponding residue in S. cerevisiae (D398) is one of three residues implicated in glutathione binding in a crystal structure of Atm1p in complex with the peptide and appears to serve an important functional role based on its proposed binding interaction with glutathione. It is also a disease-causing mutation in the human homologue of the corresponding residue. A series of S. cerevisiae Atm1p mutants (D398A/E/K/N/R) were created to analyze the impact of these substitutions on structure-function properties of the transporter that might provide mechanistic insight on the functional role of this residue and the molecular basis of disease in the corresponding of E433 position of the human ABCB7 transporter.
Finally, to address the question of whether one or two nucleotide hydrolysis events are required for cluster transport,7 we report transport studies of an E598A derivative, an essential residue for MgATP turnover, and compare observed activity levels for various combinations of wild type and mutant export dimer relative to theoretical expectations.
Experimental
Site-Directed Mutagenesis, Protein Growth and Purification
These standard methods are more fully detailed in supplementary information and primers to make amino acid substitutions are summarized (Table S1). Briefly, Rosetta cells containing the Atm1p gene were grown in LB with 100 mg/L ampicillin until an OD600 of approximately 0.6 was reached, induced with 20 μg/L anhydrotetracycline, pelleted after a 24 h induction period, and the stored at −80°C until use.
Approximately 2.5 g of cell pellet was incubated for 30 min in 20 mM Tris, 75 mM NaCl, 10 mM EDTA, pH 8.0 with 1 mg/mL lysozyme, and then sonicated and centrifuged at 6,000 rpm (3000 g) for 30 min at 4°C. The supernatant was removed and centrifuged at 67,000 rpm (150,000 g) for 30 min at 4°C. The supernatant was again removed, and the pellet resuspended in 10 mL of 20 mM MOPS, 200 mM NaCl, 20% sorbitol, 0.5% n-dodecyl β-D-maltoside (ddm), pH 6.5. The solution was homogenized, incubated on ice for 30 min, and centrifuged at 67,000 rpm for 60 min at 4°C. The supernatant was then removed and diluted 10-fold with 100 mM Tris, 150 mM NaCl, 0.25% ddm, pH 8.0 (buffer W). This was then applied to a StrepTactin superflow column equilibrated with buffer W. The column was then washed with 50 mL buffer W and the protein was eluted with buffer W with 10 mM D-desthiobiotin and evaluated to be > 95% pure by SDS-PAGE. Protein samples were concentrated to 2.5 μM, aliquoted, flash frozen, and stored at −20°C until used.
IAEDANS labeling of Atm1p
An aliquot of Atm1p was incubated at 4°C overnight with 300x TCEP in buffer W with 60x IAEDANS (5-({2-[(iodoacetyl)amino]ethyl}amino)naphthalene-1-sulfonic acid) dissolved in DMSO. The reaction mixture was applied to a PD-10 column equilibrated with buffer W the following morning and eluted with buffer W. One mL fractions were collected and analyzed by electronic absorbance. Fractions that showed a peak at both 280 nm and 349 nm contained labeled Atm1p. Labeled Atm1p fractions were then flash frozen and stored for later use. Labeling efficiency was calculating by use of an extinction coefficient of 5700 M−1 cm−1 at 336 nm, while the extinction coefficient of the label at 280 nm is 1900 M−1 cm−1. The calculated protein extinction coefficient of 67840 M−1 cm−1 was used to calculate the labeling efficiency (X). The labeling efficiency was approximately 30%, as only 1 solvent exposed cysteine (C160) exists, which would be likely the only residue to be labeled.
Liposome Synthesis
DOPG (Dioleoylphosphatidyl-Glycerol), DOPC (Dioleoyl-phosphatidyl-Choline), and DOPE (Dioleoylphosphatidyl-Ethanolamine) were purchased from Avanti Polar Lipids, Inc. An equimolar mixture of DOPG, DOPC and DOPE (133 μL, 131 μL and 124 μL, respectively) were vortexed and dried over Argon gas. The lipid mixture was stored under vacuum for one day to dry them completely. The lipid residue was dissolved in 1 mL of 50 mM HEPES, 100 mM NaCl, pH 7.5 and extruded 21 times through a 400 nm membrane. For acrylamide quenching experiments the lipid residue was dissolved in 500 μL of the same buffer and then 500 μL of buffer containing MgCl2 plus ADP, ATP, or ATPγS was added to the dissolved lipid solution to a final concentration of 3 mM MgCl2 plus nucleotide. For the sample containing MgCl2, ATP, and vanadate, vanadate was added for a final concentration of 100 μM prior to extrusion. Vanadate is used to trap the protein in the MgADP plus inorganic phosphate state.19
Atm1p Incorporation into Proteoliposomes
Incorporation of Atm1p into proteoliposomes was based on prior protocols.12, 20 The previously described liposomes were diluted two-fold with 50 mM HEPES, 100 mM NaCl, pH 7.5 and OD550 measured and baselined. A solution of 10% Triton-X was titrated into the liposome mixture in 4 μL increments until a maximum OD550 was attained. Additional 2 μL increments of 10% Triton-X were added until the “loose” state was reached, at which point the OD550 reached approximately −0.05, where the negative relative absorbance reflects the earlier baselining to a higher starting absorbance. Purified Atm1p (80 μL of 2.5 μM) was added per 1 mL of liposome and incubated for 15 min at 4°C. For a negative control, 50 mM HEPES, 100 mM NaCl, pH 7.5 was added at the same volume as the protein. Aliquots of 75 mg of BioBeads were added to the proteoliposome solution at 4°C at 0.5, 1, and 2 h and a final aliquot was added the next morning. Two hours after the final addition of BioBeads, the BioBeads were removed by centrifugation at 3,000 rpm for 1 min. The reconstituted proteoliposome was removed from the buffer by ultracentrifugation at 80,000 rpm for 20 min, and the pellet was resuspended in 1 mL of 50 mM HEPES, 100 mM NaCl, pH 7.5. The resulting proteoliposome was maintained on ice and typically used immediately in the transport assay. In acrylamide quenching assays, the liposome pellet was resuspended in 100 μL instead of 1 mL.
Proteoliposomes containing a mixture of native and the E598A derivative contain the same total amount of protein, however protein was mixed in different ratios (100% WT, 0% E598A; 75% WT, 25% E598A; 50% WT, 50%; 25% WT, 75% E598A; 0% WT, 100% E598A) prior to addition into liposomes to allow for equilibration of dimers. The E598A derivative was used as it is unable to hydrolyze MgATP, so only one of the monomer would be active in a native / E598A heterodimer.21
ATPase Assay
Experiments to quantify the stimulation of Atm1p ATPase activity by glutathione and cluster were performed with WT Atm1p and mutants. ATPase activity was monitored by use of the ENZChek Phosphate Assay Kit with 7.2 μL of 2.5 μM protein, 3 μL of 20x buffer, 3 μL of 1 mM MESG, 1.2 μL of MgATP (prepared by mixing 100 mM ATP in 1x buffer and 100 mM MgCl2 in 1x buffer 1:1), 0.6 μL of 500 μM purine nucleoside phosphorylase, varying amounts of GSH/cluster, and water up 60 μL. The concentrations of GSH used were 0, 0.031, 0.061, 0.13, 0.25, 0.50, and 2.5 mM. The concentrations of cluster used were 0, 0.1, 1, 5, 10, 25, and 50 μM. To inhibit cluster breakdown in the absence of additional glutathione, the cluster was dissolved in degassed water and a fresh batch of cluster was used for each set of experiments.
The reactions were monitored over 10 min at 360 nm, and the change in ATPase activity (y), relative to the basal activity in the absence of cluster, was calculated from the slope from 5–10 min. As previously described,11–12 the measured activity difference (y) was then plotted versus concentration (x) of GSH or cluster, and equation (1) was fit to the data (Fig. 1). KD is the apparent dissociation constant for the cluster/glutathione to the binding pocket, d is a linear term to account for magnesium-induced cluster degradation at high cluster concentrations,11 and ΔVmax represents the maximum (or limiting) change in reaction velocity when the transporter is saturated with cluster or glutathione.
Figure 1:
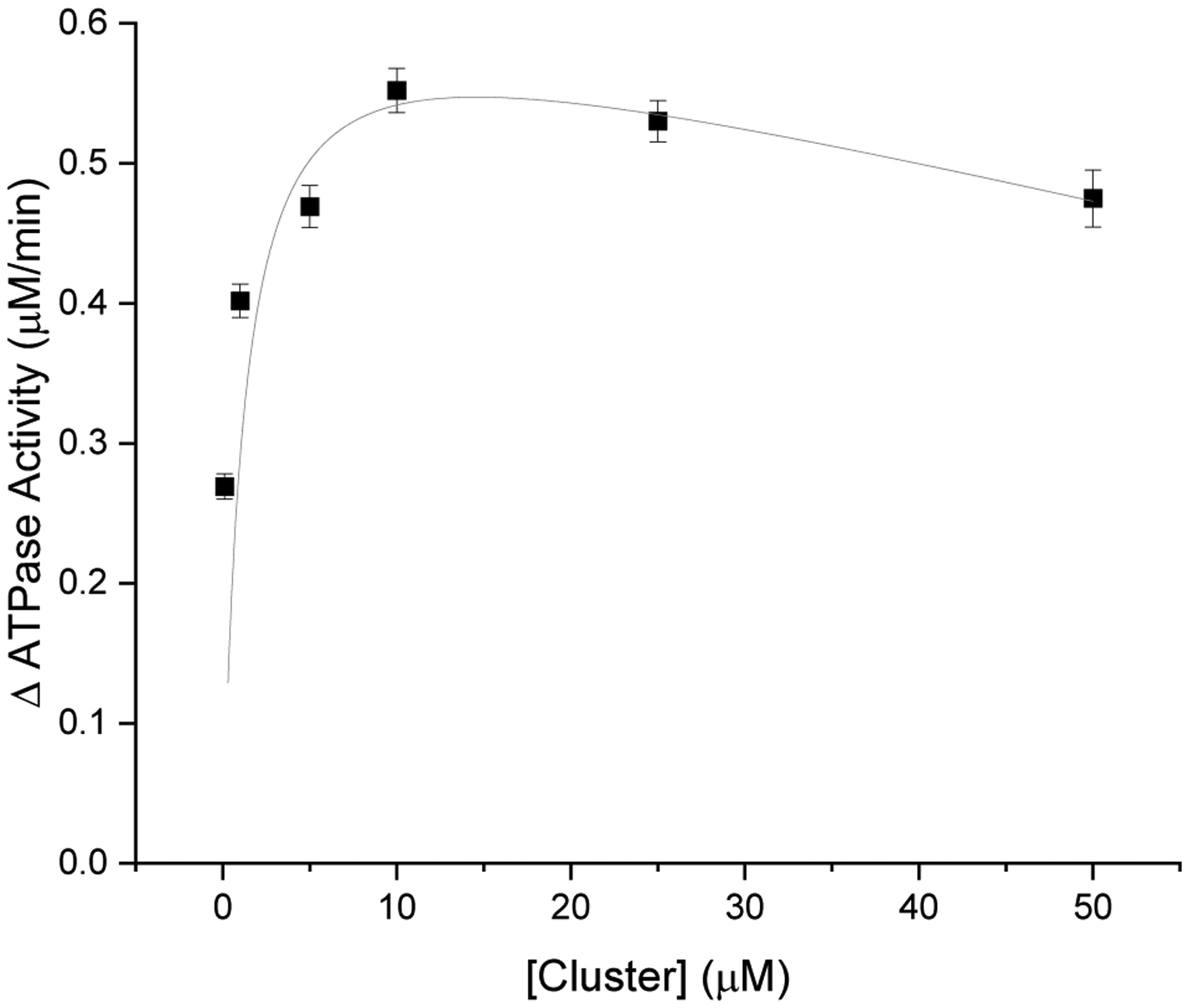
ATPase activity of WT Atm1p measured at different concentrations of cluster, relative to the basal level in the absence of added cluster (Fig. 2). KD for cluster was calculated to be 910 ± 60 nM. The increase in ATPase activity, relative to basal, was determined to be 0.64 ±0.04 μM/min under these reaction conditions.
| (1) |
In separate experiments, Michaelis-Menten plots were also generated by varying the concentration of substrate MgATP (S) in both the presence and the absence of cluster (Fig. 2). The final concentrations of MgATP used were 0, 0.1, 0.5, 1.0, and 5.0 mM, with 1.7 μM Atm1p and either 0.0 or 2.5 μM cluster. Reactions were performed as described above, and again the data from the 0 mM trial was subtracted from all other trials in that specific series of experiments. The standard Michaelis-Menten equation (equation 2) was then fit to the data, where V is the reaction velocity, Vmax is the maximum reaction velocity at saturating MgATP, KM is the Michaelis constant, and S is the [MgATP]. Michaelis-Menten parameters were also determined following addition of 0.1, 0.25, and 0.5 μM cluster concentrations (such as shown in Fig 2).
Figure 2:

A Michaelis-Menten plot for MgATP turnover in the absence (o) and presence (□) of 2.5 μM [2Fe-2S](GS)4 cluster. A large change in KM is observed in the presence of cluster, which underlies the observed stimulation of ATPase activity in the presence of non-saturating MgATP when cluster is added.
| (2) |
Transport Assay
To a solution of freshly made proteoliposome (0.5 mL in 50 mM HEPES and 100 mM NaCl at pH 7.5), MgATP and [2Fe-2S](GS)4 cluster in GSH solution, pH 8.6, were added to obtain a final concentration of 11.5 mM MgATP and 10 mM [2Fe-2S](GS)4 cluster in 1 mM GSH. The solution was mixed and incubated for 1 h at 25 °C, and the reaction mixture was centrifuged for 5 min at 6,000 rpm at 25 °C to remove precipitate formed from cluster hydrolysis. The decantate was centrifuged further at 80,000 rpm for 20 min at 4 °C to isolate the proteoliposome. The resulting pellet was resuspended in 275 μL of 50 mM HEPES, 100 mM NaCl, pH 7.5, and the proteoliposome was denatured by the addition of 82.5 μL of 2 M HCl and boiling at 95 °C for 15 min to release iron. To isolate the iron in solution, the denatured proteoliposome solution was centrifuged at 13,000 rpm for 10 min, and the solution was neutralized by the addition of 800 μL of 10 mM, pH 6.5 MES buffer and 9.2 μL of 5 M NaOH to 100 μL of the decantate. A stock tiron solution (100 mM, 100 μL in MES buffer) was added to chelate the released ferric ions, and the solution was incubated at room temperature in the dark for 10 min before measuring the absorbance at 550 nm by use of a Cary Win UV Spectrophotometer. The negative control was subtracted from each trial and normalized to WT. The transport assay was also conducted with a final concentration of 1.35 mM MgATP.
To investigate if MgATP hydrolysis in both NBD is required for substrate transport, the assay was conducted with the different ratios of the native protein to the E598A derivative as described earlier.
Cluster titration into Atm1p
Titrations were conducted with 2.5 μM Atm1p in 100 mM Tris, 150 mM NaCl, 0.25% ddm, pH 8.0 with a starting volume of 100 μL on a Cary Eclipse. Data was collected continuously for 30 sec and then averaged, using an excitation wavelength of 280 nm, and a 350 nm emission wavelength of nm, and a slit width of 5 nm to monitor the natural Trp fluorescence from Atm1p. The [2Fe-2S](GS)4 cluster was dissolved in argon-purged water at a concentration of 5 mM, and 0.2 μL volumes of cluster were then titrated in until the fluorescence change had leveled off. Data was fit by plotting fluorescence change vs. cluster concentration by application of Origin software.
The affinity of [2Fe-2S](GS)4 for Atm1p in the presence MgADP, MgATPγS, MgATP plus vanadate, and in the absence of nucleotide was also investigated. Aliquots (0.2 μL) of cluster were titrated into 60 μL of IAEDANS-labeled Atm1p reconstituted into liposomes until fluorescence change leveled off. Data was collected as stated above except with an excitation wavelength of 334 nm and an emission wavelength of 492 nm. A standard one-site binding equation (3) was then fit to all cluster titration data, where If is the measured fluorescence, B corresponds to the overall change in fluorescence intensity between the cluster free and cluster saturated states, KD is cluster affinity, and x is the concentration of cluster added.
| (3) |
Stern-Volmer acrylamide quenching
Acrylamide quenching assays were used to analyze states of the protein in the presence of different nucleotides.22 Titrations were conducted with Atm1p in liposomes in 50 mM HEPES, 100 mM NaCl, pH 7.5 with a starting volume of 60 μL on a Cary Eclipse. Acrylamide was added in 0.2 μL aliquots from a 3 M stock and emission intensity measured by scanning from 325–335 nm with an excitation wavelength of 280 nm and a slit width of 5 nm. Data was averaged over the 10 nm window and collected in triplicate. The standard linear equation (4) was then used for fitting in Origin, where is the initial fluorescence intensity, If is the fluorescence at a given quencher concentration, kq is the quenching coefficient, τ0 is the excited state lifetime, and Q is the concentration of the quencher.
| (4) |
Cluster binding concentration dependence
To determine the order of cluster binding, cluster binding kinetics were analyzed on a SX20 stopped-flow spectrometer from Applied Photophysics. Reactions were carried out with IAEDANS labeled Atm1p (190 nM) in one syringe and varying amounts of cluster (from 0.5 – 60 μM) in the other syringe. A second set of reactions with GSH (0.00625 – 10 mM) replacing the cluster. The fluorescence emission signal at 492 nm was monitored over a course of 10 – 30 sec, based on when the change in fluorescence leveled off. An excitation wavelength of 334 nm was used. Data was collected in triplicate and averaged.
| (5) |
Data was processed using DynaFit4 and the model defined by equation (5) used for fitting, where A is Atm1p, B is the [2Fe-2S](GS)4 cluster, C is the Atm1p-bound cluster complex, and D is the Atm1p complex after structural changes. Rate constant ks is the observed first-order rate constant for cluster binding to Atm1p, where ks = k2[cluster] and k2 is the second-order rate constant, while ki is the first-order rate constant for intramolecular structural rearrangement. DynaFit4 was used to solve for ks and ki, and the observed rate constants were then plotted against cluster concentration in Origin and equation (6) used for fitting, where y is one or other of the two fitted observed rate constants (ks or ki), x is the cluster concentration, m is the order of the reaction for cluster, and b is a y offset to account for direct quenching of the IAEDANS fluorescence by cluster. When fitting the GSH dependence data no second-order term (ks) was observed because GSH binding did not quench the fluorescence. Only the first-order intramolecular transition (ki) was detected.
| (6) |
Structure Comparison
The shift in residue position for each individual residue was calculated using UCSF Chimera 1.11.2. The structure of N. aromaticivorans Atm1p with no ligand bound in the proposed binding pocket (PDB: 4MRN) was compared to Atm1p with S-mercury glutathione (PDB: 4MRV). Shifts were calculated between each pair of structures for both chain A and chain B by aligning the chain of each pair by structures being compared and then using the match tool.
Results
ATPase Assay
Native Atm1p and substituted derivatives all showed a basal level of ATPase activity of roughly 0.5 μM/min. Stimulation by both glutathione and [2Fe-2S](GS)4 cluster was determined for both WT and substituted derivatives. Under the conditions used, glutathione stimulated the ATPase activity of WT Atm1p by 0.54 ± 0.03 μM/min (Fig. S1), while [2Fe-2S](GS)4 cluster stimulated the activity by 0.64 ± 0.05 μM/min (Fig. 1). The D398N derivative behaved similar to WT, with glutathione causing a 0.32 ± 0.01 μM/min increase in ATPase activity (Fig. S2) and the [2Fe-2S](GS)4 cluster causing a 0.36 ± 0.02 μM/min increase (Fig. S3). All other mutants showed no stimulation in the presence of glutathione or cluster (Table 1).
Table 1:
ATPase stimulation in the presence of [2Fe-2S](GS)4 and GSH. Stimulation of ATPase activity of Atm1p variants was measured in the presence of glutathione and [2Fe-2S](GS)4 cluster. In the cases where stimulation was observed, the apparent KD for cluster or glutathione binding was calculated based on the substrate dependence curve. The extent of stimulation reflects the relative change in Michaelis-Menten profiles for ATPase activity in the presence or absence of possible transporter substrates. nd – could not be determined in the absence of stimulation.
| GSH stimulation | GSH KD | cluster stimulation | cluster KD | |
|---|---|---|---|---|
| Wild-Type | 0.54 ± 0.03 μM/min | 32 ± 9 μM | 0.64 ± 0.04 μM/min | 910 ± 60 nM |
| D398A | no stimulation | nd | no stimulation | nd |
| D398E | no stimulation | nd | no stimulation | nd |
| D398K | no stimulation | nd | no stimulation | nd |
| D398N | 0.32 ± 0.01 μM/min | 74 ± 10 μM | 0.36 ± 0.02 μM/min | 5.6 ± 0.8 μM |
| D398R | no stimulation | nd | no stimulation | nd |
As seen from the Michaelis-Menten profiles for MgATP activity (Fig. 2, Figs S4 – S18, and Table 2), stimulation reflects the increased activity at lower MgATP concentrations in the presence of cluster (or glutathione), reflecting a decrease in KM. For wild-type Atm1p, a range of concentrations of cluster were tested to obtain an understanding of how cluster concentration affected the KM of MgATP. The general observed trend was a decrease in KM as cluster concentration increased, while Vmax remained relatively unchanged between 0.66 and 0.84 μM/min. KM decreased from 2.5 ± 0.3 mM in the absence of cluster to 0.027 ± 0.016 mM in the presence of 2.5 μM [2Fe-2S](GS)4. The Michalis-Menten profile for WT Atm1p is shifted in the presence of cluster due to the increase in ATPase activity (arising predominantly from a decrease in KM) in the presence of cluster at lower substrate concentrations compared to activity in the absence of cluster. Due the increase in activity at lower MgATP concentrations, the Vmax is reached at lower substrate concentrations, reflecting the lower Km for MgATP.
Table 2:
MgATP hydrolysis in the presence and absence of cluster. ATPase activity was measured as a function of MgATP in the presence and absence of [2Fe-2S](GS)4 cluster. Michaelis-Menten parameters were extracted, showing a decrease in KM in the presence of cluster. The increase in kcat/KM in the presence of cluster provides an indication of the effect of cluster on the efficiency of MgATP hydrolysis.
| 0 μM Cluster KM (mM) | 0 μM Cluster Vmax (μM / min) | 2.5 μM Cluster KM (mM) | 2.5 μM Cluster Vmax (μM / min) | fold decrease in KM | |
|---|---|---|---|---|---|
| Wild-Type | 2.5 ± 0.3 | 0.82 ± 0.03 | 0.027 ± 0.016 | 0.70 ± 0.03 | 93 |
| D398A | 1.0 ± 0.1 | 0.52 ± 0.02 | 1.5 ± 0.3 | 0.58 ± 0.04 | 0.67 |
| D398E | 1.2 ± 0.2 | 0.60 ±0.02 | 0.58 ± 0.18 | 0.33 ± 0.03 | 2.1 |
| D398K | 2.8 ± 0.3 | 0.90 ± 0.02 | 0.54 ± 0.13 | 0.46 ± 0.03 | 5.2 |
| D398N | 2.8 ± 0.5 | 0.63 ± 0.04 | 0.74 ± 0.18 | 0.43 ± 0.03 | 3.8 |
| D398R | 0.80 ± 0.10 | 0.62 ± 0.02 | 0.23 ± 0.04 | 0.62 ± 0.02 | 3.5 |
ATPase activity for Atm1p was also measured as a function of MgATP concentration in the presence and absence of cluster for D398 derivative. Unlike the native protein there were minimal changes in the KM for MgATP (Table 2). The greatest changes in KM for MgATP for the tested D398 substitutions were observed for the D398K and D398N derivatives. The KM for MgATP decreased from 2.8 ± 0.3 mM in the absence of cluster to 0.54 ± 0.13 mM in the presence of 2.5 μM cluster for the D398K derivative. Similarly, the KM for MgATP decreased from 2.8 ± 0.5 mM in the absence of cluster to 0.74 ± 0.18 in the presence of 2.5 μM cluster for the D398K derivative. The D398A, E, and R derivatives showed little to no decrease in the KM for MgATP in the presence 2.5 μM cluster.
By use of equation (1), the apparent dissociation constants for substrate KD’s were calculated for glutathione and cluster binding, with cluster displaying an affinity of 910 ± 60 nM for WT and 5.6 ± 0.8 μM for D398N. The binding affinities for GSH were 32 ± 9 μM for WT and 74 ± 0.10 μM for D398N (Table 1).
Transport Assay
The relative ability of Atm1p variants to transport [2Fe-2S](GS)4 was analyzed by proteoliposome transport assays conducted under two sets of conditions. First, with saturating [MgATP], and second, with a more physiologically relevant rate-limiting [MgATP]. All results were normalized to WT, relative to a defined time for transport. After 60 min the D398N mutant transported 60 ± 5% as much cluster as WT Atm1p, while D398K transported 40 ± 3% of the amount of cluster compared to WT. The other three mutants showed little to no transport, all transporting less than 8% compared to WT (Fig. 3, left). When the transport assay was repeated with native Atm1p, and D398N and D398K derivatives with a lower concentration of MgATP, the substituted proteins transferred cluster at a lower rate relative to WT than observed with higher concentrations of MgATP (~ 20% for the D398N derivative and 12 % for the D398K derivative, Fig. 3, right).
Figure 3:

Proteoliposome transport assays. Proteoliposome transport assays were conducted with Atm1p and [2Fe-2S](GS)4. Iron quantitation was then performed to determine how much cluster was transported. The substitutions D398A, D398E, and D398R knocked out transport, while substitutions D398N and D398K showed decreased transport relative to WT for assays conducted at a MgATP concentration of 11.3 mM (left). Assays conducted at a MgATP concentration of 1.35 mM show a greater decrease in the amount of cluster transported relative to native protein (right).
Cluster titration into Atm1p
Titrations of the Fe-S cluster complex to fluorophore-labeled protein were conducted to provide an alternative method to obtain cluster binding affinities. For WT Atm1p, the KD for cluster binding was determined to be 23.2 ± 3.0 μM (Fig. 4). This differs from the 910 nM affinity determined in stimulation experiments but reflects the differing solution conditions. The latter was obtained from turnover experiments in the presence of MgATP, while the 23 μM affinity was obtained from direct titration of fluorophor-labeled protein in the absence of nucleotide. Each of the D398A, E, K, N, and R substitutions also demonstrated affinities for the cluster that were similar to WT, with KDs ranging from 26.4 μM to 43.7 μM (Figs. S19–S24, Table S3).
Figure 4:
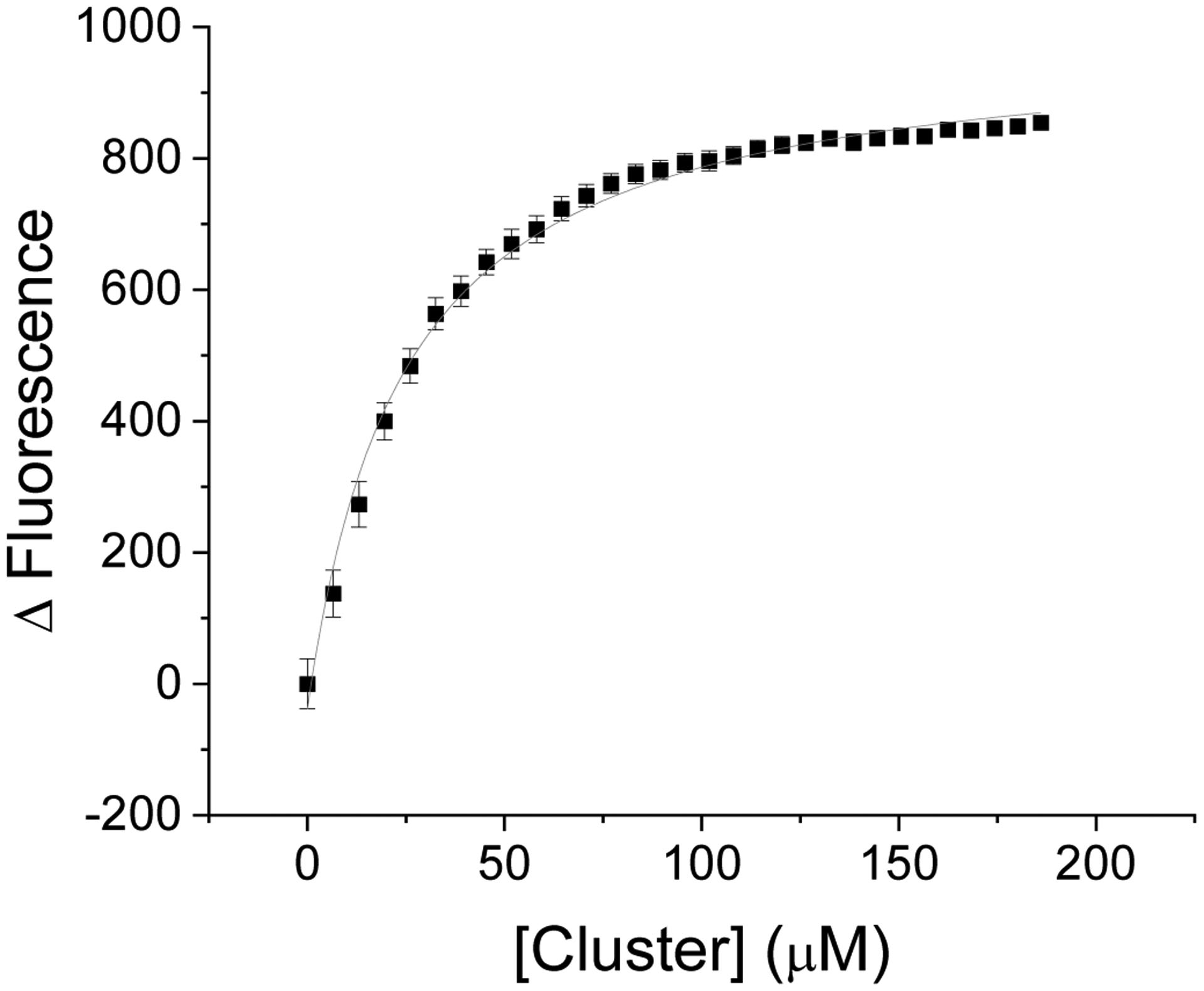
[2Fe-2S](GS)4 binding to native Atm1p. Cluster binding affinity for WT Atm1p was determined by titrating cluster into labeled Atm1p and was determined to be 23 ± 3 μM.
Cluster titrations were also conducted in the presence of different nucleotides to investigate cluster affinity in different states of the protein. Binding affinity was assayed in the presence of MgATPγS, MgADP, and MgATP plus vanadate, as well as in the absence of nucleotide (Figs S25–S28). The affinity for cluster in the absence of nucleotide and in the presence of MgADP were similar, with KD’s of 119 ± 2 μM and 120 ± 1 μM, respectively. The binding affinity for the cluster in the presence of MgATPγS and MgATP plus vanadate were also very similar, with KD’s of 179 ± 7 μM and 179 ± 1 μM, respectively.
Influence of E598A substitution on cluster transport
To determine if MgATP hydrolysis at one or both dimers is necessary for substrate transport, a solution of native protein was mixed with the E598A derivative, which knocks out MgATP hydrolysis at one of the MgATP binding sites in the dimer.21 For experiments involving varying ratios of WT:E598A, the results were normalized to 100% WT. The 75% WT : 25% E598A sample transferred 55 ± 5% of the amount of cluster transferred in the 100 % WT sample. The 50% WT : 50% E598A sample and 25% WT : 75% E598A transferred 29 ± 0% and 14 ± 1% of the amount of cluster transferred by the 100% WT sample, respectively (Fig. 5). The amount of cluster transferred closely matched a model in which only the WT dimer was able to transfer cluster (Fig 5 and Table S2). This model assumes the equilibration of the mixed WT and mutant proteins, and the good fit of the experimental data to the model validates this assumption.
Figure 5:

Cluster transport in of WT/E598A Atm1p. Proteoliposome transport assays were conducted at different ratios of WT to E598A Atm1p to determine the number of hydrolysis events required to transport cluster. Expected amount of cluster transported was calculated by determining the percent of native Atm1p homodimers at different ratios of native to E398A Atm1p if two WT monomers are required for substrate transport (dark gray), or percent of native homodimers and heterodimers if only one active monomer is required for substrate transport (light gray). The amount of cluster transported was normalized to the amount of cluster transported by the all native protein sample (black), which follows closely the model of two active monomers required for substrate transport.
Acrylamide quenching
Acrylamide quenching assays were conducted to determine if Atm1p is in different states in the presence of different nucleotides. Initial fluorescence intensity (), divided by the fluorescence at a given quencher concentration (If)was plotted against concentration of acrylamide in the presence of difference nucleotides (Fig. 6). In the absence of nucleotide, the slope of the curve was 0.0085 ± 0.0008, which is similar to the slope obtained in the presence of MgADP, 0.0074 ± 0.0005 mM−1. These are distinct from the slopes in the presence of MgATPγS and MgATP plus vanadate, each of which displayed similar slopes of 0.0051 ± 0.0009 mM−1 and 0.0055 ± 0.0007 mM−1 (Fig. 6), respectively. This suggests that the protein is in a different state to that obtained with either with no nucleotide added or in the presence of MgADP, as the only thing that changed is the quenching coefficient (equation 4).22 For this experiment, and τ0 are constants, while kq and therefore If are dependent upon the state of the protein and concentration of quencher.
Figure 6:

Acrylamide quenching assays in the presence of different nucleotides. Acrylamide quenching assays were conducted on WT Atm1p in liposomes in the presence of Mg-ADP, Mg-ATPγS, Mg-ATP plus vanadate, and in the absence of nucleotide, to differentiate the state of the protein in the presence of different nucleotides. The slopes in the absence of nucleotide and in the presence of Mg-ADP were similar, as were the slopes in the presence of Mg-ATPγS and Mg-ATP plus vanadate, suggesting that the protein is in a different state from that obtained in the absence of nucleotide or in the presence of Mg-ADP. Equation (4) was used to fit the data, where F corresponds to If and FO corresponds to .
Stopped-flow evaluation of cluster binding kinetics
Rapid single-turnover stopped-flow methods were used to evaluate cluster binding kinetics at different cluster concentrations and were analyzed to determine the order of the reaction with respect to cluster (Figs 7 and S29–S31). IAEDANS-labeled Atm1p from one syringe was mixed with [2Fe-2S](GS)4 dissolved in degassed water in the other syringe. When analyzing the change in rate versus cluster concentration the order of the reaction with respect to cluster was calculated to be 0.98 ± 0.02, suggesting that rate dependence of cluster is first order (ks = k2[cluster]), with a fitted overall second-order rate constant k2 of 1.2 ± 0.1 ×10−4 μM−1s−1. Minimal changes in rate were observed for ki as cluster concentration increased, suggesting an intramolecular conformational change with a rate constant ki of 1.4 ± 0.2 × 10−4 s−1. 23 This intramolecular conformation change was also observed with GSH (Figs. S30 and S31). Because glutathione alone does not quench the fluorescence in a concentration dependent manner, the ks and constant terms were removed from the model in equation 5. The rate constant ki was determined to be 7.9 ± 4.3 × 10−9 s−1, which is slower than the conformation change observed in the presence of cluster, possibly due to cluster occupying multiple GSH binding sites, making it easier for the TMD to communicate to the NBD when cluster is bound.
Figure 7:

[2Fe-2S](GS)4 cluster binding kinetics. Cluster binding kinetics from stopped flow experiments were analyzed using DynaFit4 software to determine the effective rate constants for binding of cluster and subsequent intramolecular structural change. Observed first-order rate constants for each phase of the binding profile (equations 5 and 6, Fig. S29) were plotted as a function of cluster concentration. Cluster binding was determined to be first-order with respect to cluster (black squares) as ks increased linearly with cluster concentration (m = 0.98 ± 0.02). The observed rate constant ki remained relatively unchanged as cluster concentration increased (red circles), consistent with an intramolecular change that can be attributed to changes in the protein conformation following cluster binding.
Structure comparison
The crystal structures of NaAtm1p with and without S-mercury glutathione were overlaid and compared using the match tool in Chimera (Fig. 8). In chain A the greatest change occurs in the nucleotide binding domain between residues 351 and 450, which contains the Walker A motif, and from residues 467 – 482 and 515 – 613. The RMSF (root mean square fluctuation) of residues in these areas are generally 1.5 – 2.0 Å. Another set of residues from 122–142 also showed greater change (1.0 – 1.9 Å) than the surrounding residues. Embedded in these residues is interconnecting loop 1 (ICL1), which contains the coupling helices that link the transmembrane helices to NBD (Fig. 9). In chain B there is a large conformational change that includes ICL 2 (residues 232–251, shifts ranging between 1.2–2.0 Å), which connects the transmembrane domains to the NBD of chain A (Fig. 10). The binding of the S-mercury glutathione to the protein causes these structural changes that, based on RMSF, connects changes in the TM helices to changes to the NBD via the 2 ICLs. One of the key ICL residues that may play an important role in fostering communication between the TMD and NBD of NaAtm1p is R136. In the absence of S-mercury glutathione R136 interacts with E493 of the same chain, but E493 of the opposite chain when S-mercury glutathione is bound in the TMD.
Figure 8:

Structural changes in NaAtm1p with S-mercury glutathione bound. Structures of NaAtm1p with and without S-mercury glutathione (Hg(GS)2) were overlaid and the shifts in each residue were determined by use of Chimera. A,B: RMSF for each residue in chains A and B. The graphs are color-coded to match their region on the NaAtm1p monomer (C). Transmembrane domain (TMD) helices are colored in red, ICLs in yellow, nucleotide binding domains (NBD) in green, and Walker A motif, ABC signature motif, and Walker B motif in blue. The ICLs contain the connecting helices that mediate communication between the TMDs and NBDs. The residues that show the greatest shifts were those in and around the ICLs and NBDs. This supports a transport mechanism where binding of substrate in the TM domain is communicated to the NBD through the ICLs to initiate the transport cycle, thereby avoiding MgATP hydrolysis when no cluster is bound.
Figure 9:

Interactions between R136 with surrounding residues. Interactions of R136 in NaAtm1p with E139 and E493 in the absence (top, PDB ID: 4mrn) and presence (bottom, PDB ID: 4mrv) of Hg(GS)2. Residue R136 is shown in yellow, E139 and E493 in pink, and ABC signature motif in cyan.
Figure 10:
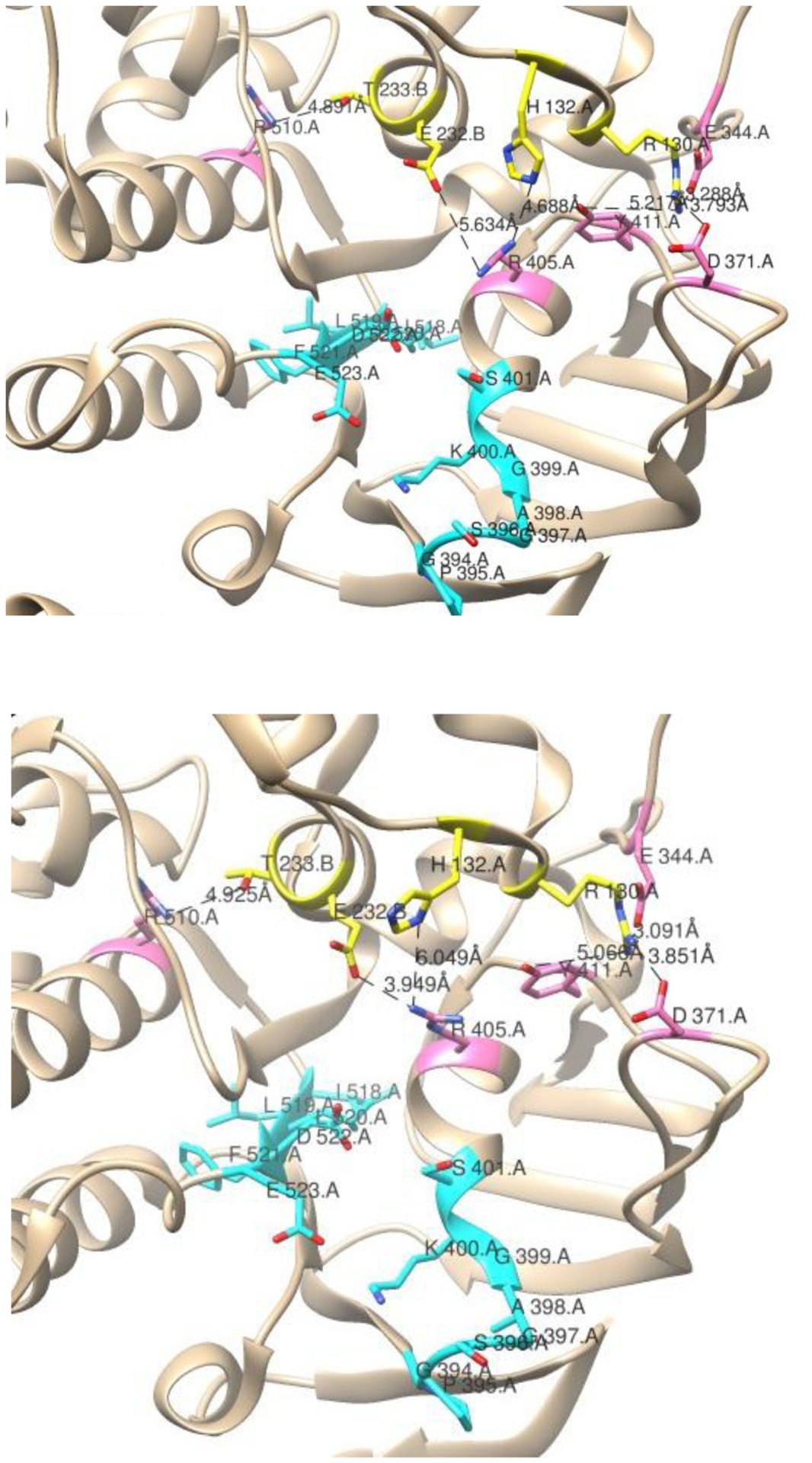
Interactions between ICL1 and ICL2 and the NBD. Interactions of ICL1 and ICL2 in NaAtm1p interacting with residues of the NBD in chain A in the absence (top, PDB ID: 4mrn) and presence (bottom, PDB ID: 4mrv) of Hg(GS)2. ICL1 residues R130 and H132 in chain A and ICL2 residues E232 and T233 of chain B are shown in yellow. Chain A residues that interact with ICL residues E344, D371, R405, Y411, and R510 are shown in pink, and Walker A and Walker B motifs of chain A in cyan.
There are two distinct binding sites for oxidized glutathione on NaAtm1p. The higher affinity site, which includes residues Y156, N269, Q272, D316, M317, G319, and M320, while the residues implicated in binding at the other site are R206, R210, AR323, and T324.14d In chain A, residues G319 and M320 shift 1.76 and 2.79 Å respectively, while in chain B the residues that show the greatest shift are Y156 and M320. The shift in these residues most likely helps communicate the structural changes to the rest of the protein following substrate binding.
Discussion
ATPase activity of Atm1p was stimulated in the presence of both glutathione and [2Fe-2S](GS)4, as previously shown,11 supporting a model where the physiological role of Atm1p/ABCB7 is to export glutathione-coordinated iron-sulfur clusters from the mitochondria. The D398 residue appears to play a key role in the function of Atm1p as it is one of 3 residues implicated in glutathione binding in a crystal structure of Atm1p.18 In the human homologue, ABCB7, the residue corresponding to D398, E433, is one of 5 residues where mutations have been linked to X-linked sideroblastic anemia.17a Thus, substitutional analysis was conducted on the D398 residue to assess the importance of the residue on protein function and provide insight into how cluster binding impacts ATPase activity.
[2Fe-2S](GS)4 Cluster promotes MgATP binding to the NBD
It is known that the ATPase activity of an ABC transporter is stimulated in the presence of substrate.24 Michaelis-Menten profiles for Atm1p ATPase activity were measured in the presence and absence of cluster. WT Atm1p displayed a similar Vmax in the presence and absence of cluster, but the KM was observed to decrease nearly 100-fold in the presence of 2.5 μM cluster. KM did not show such a dramatic change in the case of the substituted derivatives. A comparison of kcat/KM in the presence and absence of cluster provides insight on the molecular mechanism that results in an increase in MgATP hydrolysis in the presence of cluster. The concentration of MgATP in the mitochondria is 520 ± 60 μM,25 therefore the shift in KM of MgATP from 2.5 ± 0.3 mM to 0.027 ± 0.016 mM for WT Atm1p in the presence of cluster regulates transporter function to be “on” only when cluster is bound. Consistent with observations in the transport assays, the D398N and D398K derivatives showed a small change in KM in the presence of cluster. None of the other mutants displayed a significant change in KM nor significant cluster transport.
Studies of other ABC-type transporters have shown either an increase in Vmax for ATPase activity in the presence of substrate, or an increase in Vmax accompanied by a decrease in KM.26 However, Atm1p shows essentially no change in Vmax values at different concentrations of cluster, but a significant change in KM. The most likely explanation is that the binding of cluster causes structural changes in the substrate binding domain that then causes structural changes in the NBD via the ICL’s that leads to a decrease in the KM of MgATP hydrolysis. When the crystal structures of Na Atm1p with and without S-mercury glutathione were aligned and overlaid, it appears that the binding of the substrate is communicating structural changes to the NBD (Figs. 9 and 10). Previous studies have shown that changes in the transmembrane helices can be communicated to the nucleotide-binding domain through interconnecting loops,27 and a similar mechanism is most likely used here.
Structural basis for KM regulation and coupling to the substrate binding site
Comparison of the crystal structure of NaAtm1p obtained with (PDB ID: 4mrv) and without (PDB ID: 4mrn) bound substrate reveals five residues in the coupling helices that show changes in their contacts to surrounding residues following substrate binding (three in ICL1 and two in ICL2) (Fig. 9). Residue R136 in ICL1 of both chains lies at the interface of the two NBD, and the contacts are observed to change significantly in the presence of substrate. For example, in the absence of bound Hg(GSH)2 residue R136 of chain A is located 3.116 Å from residue E493 on chain A, while only lying 2.974 Å from R136 of chain B, which mirrors chain A in its other interactions. Following substrate binding, residue R136 of chain A distances itself from R136 of chain B, as they are located 12.375 Å from each other. R136 of chain A no longer interacts with residue E493 of chain A, as it is closest to E493 of chain B (5.024 Å) and E139 of chain A (2.232 Å) (Table S4, Fig. S39). Changes in contacts to R136 could possibly communicate changes to the ABC signature motif, (residues 498–502) as it is located near residue E493.
The remaining residues that change contact (R130, H132, E232, and T233) interact with residues that border the Walker A and Walker B motifs (Fig. 10). Potential electrostatic interactions near the Walker A and Walker B motifs of chain A occur between R130 of chain A and E344, D371, and Y411 of chain A, H132 of chain A with R405 of chain A, E232 of chain B with R405 of chain A, and T233 of chain B with R510 of chain A (Fig 10). These interactions are mirrored in the NBD of chain B. The distances between each of these sets of residues changes upon substrate binding (Table S4) and, combined with the changes that take place around residue R136, provide insight into how the TMD domain communicates with the NBD.
[2Fe-2S](GS)4 Cluster binding triggers intramolecular structural transitions.
While these structural perturbations occur locally, they can communicate changes to other areas of the protein. It was observed that the greatest change in atomic positions arise in the ICLs and the NBD (Fig. 8). These structural changes are reflected in the fitting of the stopped flow kinetic traces. The data fit poorly to a simple A + B → C model, where A is Atm1p, B is [2Fe-2S](GS)4, and C is the complex of cluster bound to Atm1p. Based on the hypothesis that structural changes were occurring following cluster binding we added the additional first-order term seen in equation 6 to account for such intramolecular structural transitions. Cluster binding was determined to be first order with respect to cluster as ks increased linearly as cluster concentration increased (m = 0.98 ± 0.02). Rate constant ki remained relatively unchanged as cluster concentration increased, consistent with an intramolecular reaction that can be attributed to changes in protein conformation following cluster binding
Correlation of substrate transport with efficiency of coupling to the NBD
Modest stimulation of ATPase activity in the D398N derivative was observed in the presence of substrate. Glutathione binding sites on Atm1p show hydrogen bonding interactions between the carboxyl group of the D398 sidechain and the carboxyl group of the glycine residue of GSH, suggesting that at least one of the oxygens of the D398 sidechain is protonated, acting as a H-bond donor.18 It would therefore be possible for one of the hydrogens of the amine in the D398N mutant to act as an H-bond donor, mimicking the wild-type protein. Although the E, K, and R could also potentially be H-bond donors, the side chains are significantly longer than D and N, potentially putting the carboxyl group of the glycine out of reach. Therefore, the shape of the residue and its ability to act as an H-bond donor appear to be important for the molecular mechanism that stimulates ATPase activity.
Proteoliposome transport assays were conducted to determine the relative efficiencies of [2Fe-2S](GS)4 cluster transport for each derivative. At saturating MgATP levels (11.3 mM), WT protein showed the greatest ability to transport cluster, while the A, E, and R mutants showed little to no cluster transport. The D398N derivative transported clusters 60 ± 6 % as well as WT, while the D398K derivative also showed the ability to transport cluster, transporting 40 ± 4% as much cluster as WT. In humans, the E433K mutation, the residue homologous to the D398 residue, is the most severe of ABCB7 mutants that result in sideroblastic anemia. It was therefore surprising that the D398K mutant was able to transport cluster although at a reduced rate compared to native Atm1p. However, at more physiologically relevant levels of 1.35 mM MgATP the D398K mutant transferred little cluster relative to WT (10 ± 0%) (Fig. 3). This suggests that in the cell MgATP levels are sufficient to transport substrate through the native protein, but when the disease-causing mutation is present the transporter can no longer transport cluster sufficiently at the physiological level of MgATP, which could be the underlying cause of the E433K substitution resulting in disease.
Even though the WT, D398K, and D398N constructs all had similar affinities for the cluster, the stimulation profiles for all three were different, which explains the difference in the amount of cluster transported when the assays were conducted with differing concentrations of MgATP (Fig. 3).
Mechanism of cluster transport
Cluster export appears to follow an accepted MgATP switch model.28,29 The first step involves binding of the [2Fe-2S](GS)4 in the substrate binding pocket found in the transmembrane helices. Binding of the substrate causes structural change in the nucleotide binding of chain A,18 which enhances the affinity for MgATP. This also demonstrates how substrate binding in the TM domain influences MgATP hydrolysis in the NBD.29–30. Following binding of MgATP, the classical ATP switch model is continued as the two NBDs dimerize, resulting in conformational changes in the TM domain that allow the substrate to be transported across the membrane. In the third step MgATP is then hydrolyzed, the inorganic phosphate and MgADP dissociate, and the transporter returns to its initial state with no substrate or nucleotide bound (Fig. 11).29
Figure 11:

[2Fe-2S](GS)4 cluster transport cycle. The proposed transport cycle starts with cluster (yellow) and MgATP (blue) in the mitochondrial matrix. Cluster binds to Atm1p, communicating structural changes to the NBD (1). MgATP then binds, causing the conformational change in the protein that allows for cluster transport (2). MgATP hydrolysis occurs, and the protein in still in the open outward state with both MgADP plus inorganic phosphate bound (3). Phosphate is released, allowing for the protein to return to the original open outward conformation (4).1 MgADP is release to complete the cycle (5).2
This model is supported by the cluster binding affinities and fluorescence quenching experiments performed in the presence of various nucleotides. Cluster binding affinities were found to be similar (119 ± 2 μM and 120 ± 1 μM, respectively), either in the absence or presence of nucleotide. Fluorescence quenching assays yielded similar slopes (0.0085 ± 0.0008, 0.0074 ± 0.0005), suggesting the protein to be in the same structural state for the case with no nucleotide bound, and in the presence of MgADP (Fig 8, top, bottom left). In the presence of either MgATPγS or MgATP plus vanadate, the cluster affinity was again similar (179 ± 7 μM, 179 ± 1 μM), and the fluorescence quenching assays yielded similar results (0.0051 ± 0.0009, 0.0055 ± 0.0007). Taken together these results suggest that the protein is in the open-outward state with either MgATPγS or MgATP bound, where MgATPγS mimics the MgATP-bound state, while the MgATP plus vanadate mimics the MgADP + Pi state after MgATP hydrolysis (Fig 11, bottom middle, bottom right).19, 22
Two MgATP hydrolysis events are required per equivalent of cluster transport
Even in ABC transporters that exist as a homodimer, it is possible that the two NBDs have different affinities for MgATP, and it has been debated whether one or two hydrolysis events are required for substrate transport.7, 31 When comparing the structures of substrate free and S-mercury glutathione bound NaAtm1p, it is of interest to note that the NBD of chain A was shifted more significantly than the NBD of chain B. The underlying reason for this reflects greater movement of the ICL1 of chain A and ICL2 of chain B, relative to ICL2 of chain A and ICL1 of chain B following substrate binding. ICL1 (Fig. 9) of chain A and ICL2 (Fig. 10) of chain B are the two ICL’s that connect the transmembrane helices to the NBD of chain A. In addition to MgATP binding being asymmetric, MgATP hydrolysis can also be asymmetric in a homodimer as seen previously with other ABC transporters such as P-gp, MutS, and bacterial transporters of histidine and maltose, suggesting the number of MgATP hydrolysis events required for transport is dependent on the system.32 The transport assays conducted with the E598A derivative suggest that two hydrolysis events are required for cluster transport, as the data fit to a model where the heterodimer and the E598A homodimer was incapable of substrate transport observed in the transport assays with the E598A mutants as only the dimers consisting of two native monomers were able to transport cluster (Fig. 5).
Substitutional analysis of the residue implies that hydrogen bonds between glutathione and the D398 residue most likely plays an important role in communicating structural changes from the substrate binding site to the NBD via the interconnecting helices. A D398N substitution can partially retain cluster transport and ATPase stimulation because of the similar shape of side chains and their ability to participate in H-bonding as H-bond donors. Although the rate-limiting step of the transport cycle remains controversial and may vary between different proteins,29 recent studies suggest that either the rearrangement of the transport protein or MgATP hydrolysis may determine the rate of the cycle.33 Either way the D398 residue appears to serve a key role in enhancing the rate of the cycle by communicating structural changes to the NBD as the kcat/KM is increased 68-fold in the presence of [2Fe-2S](GS)4 cluster.
Active or passive transport?
While ABC transporters are typically thought of as transporting substrates against the concentration gradient, it has been shown in the case of Coptis japonica ABCB2 and PGP4 mediated transport of berberine,34 and auxin,35 respectively occurs down the concentration gradient. ABCB14 uptake of malate in Arabidopsis thaliana36 how substrate can move passively down a concentration gradient, while utilizing MgATP hydrolysis to effect structural change, as suggested by the data presented herein for [2Fe-2S](GS)4 export from mitochondria to the cytosol. In fact, higher concentrations of [2Fe-2S](GS)4 complex cluster would be anticipated in the mitochondria, the major location for cluster biosynthesis. Accordingly, while the overall transport process requires the energy of MgATP hydrolysis to execute structural change, it is likely that no energy is required to move substrate cluster against a concentration gradient, consistent with the similarity in binding affinities determined for cluster in the open and closed conformational states.
Supplementary Material
Significance to Metallomics.
Herein we investigate the mechanism of mitochondrial export of [2Fe-2S] cofactors. Glutathione-coordinated iron-sulfur clusters are shown to be a viable substrate for ABC7-type mitochondrial transporters, connecting mitochondrial and cytosolic iron-sulfur cluster assembly systems, consistent with an endosymbiotic model of mitochondrial evolution. These studies provide insight on the mechanism of cellular cluster trafficking and the molecular basis of human disease conditions related to ABCB7.
Acknowledgements
This work was supported by a grant from the National Institutes of Health [AI072443].
Footnotes
Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Mi W, Li YY, Yoon SH, Ernst RK, Walz T, Liao MF, Structural basis of MsbA-mediated lipopolysaccharide transport. Nature 2017, 549 233–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Priess M, Goddeke H, Groenhof G, Schafer LV, Molecular mechanism of ATP hydrolysis in an ABC transporter. ACS Cent. Sci 2018, 4 1334–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson DC, Dean DR, Smith AD, Johnson MK, Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem 2005, 74 247–81. [DOI] [PubMed] [Google Scholar]
- 4.Ye H, Rouault TA, Human iron-sulfur cluster assembly, cellular iron homeostasis, and disease. Biochemistry 2010, 49 4945–4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones PM, George AM, The ABC transporter structure and mechanism: Perspectives on recent research. Cell. Mol. Life. Sci 2004, 61 682–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doshi R, Ali A, Shi W, Freeman EV, Fagg LA, van Veen HW, Molecular disruption of the power stroke in the ATP-binding cassette transport protein Msba. J. Biol. Chem 2013, 288 6801–6813; [DOI] [PMC free article] [PubMed] [Google Scholar]; Szollosi D, Rose-Sperling D, Hellmich UA, Stockner T, Comparison of mechanistic transport cycle models of ABC exporters. Biochim. Biophys. Acta 2018, 1860 818–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barsony O, Szaloki G, Turk D, Tarapcsak S, Gutay-Toth Z, Bacso Z, Holb IJ, Szekvolgyi L, Szabo G, Csanady L, Szakacs G, Goda K, A single active catalytic site is sufficient to promote transport in P-glycoprotein. Sci. Rep 2016, 6; [DOI] [PMC free article] [PubMed] [Google Scholar]; Sauna ZE, Ambudkar SV, About a switch: How P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol. Cancer Ther 2007, 6 13–23; [DOI] [PubMed] [Google Scholar]; Tombline G, Bartholomew LA, Urbatsch IL, Senior AE, Combined mutation of catalytic glutamate residues in the two nucleotide binding domains of P-glycoprotein generates a conformation that binds ATP and ADP tightly. J. Biol. Chem 2004, 279 31212–31220; [DOI] [PubMed] [Google Scholar]; Martin C, Higgins CF, Callaghan R, The vinblastine binding site adopts high and low-affinity conformations during a transport cycle of P-glycoprotein. Biochemistry 2001, 40 15733–15742; [DOI] [PubMed] [Google Scholar]; Urbatsch IL, Tyndall GA, Tombline G, Senior AE, P-glycoprotein catalytic mechanism - Studies of the ADP-vanadate inhibited state. J. Biol. Chem 2003, 278 23171–23179. [DOI] [PubMed] [Google Scholar]
- 8.Chloupkova M, Reaves SK, Lebard LM, Koeller DM, The mitochondrial ABC transporter Atm1p functions as a homodimer. FEBS Lett. 2004, 569 65–69. [DOI] [PubMed] [Google Scholar]
- 9.Kispal G, Csere P, Guiard B, Lill R, The ABC transporter Atm1p is required for mitochondrial iron homeostasis. FEBS Lett. 1997, 418 346–350; [DOI] [PubMed] [Google Scholar]; Miao R, Kim H, Koppolu UMK, Ellis EA, Scott RA, Lindahl PA, Biophysical characterization of the iron in mitochondria from Atm1p-depleted Saccharomyces cerevisiae. Biochemistry 2009, 48 9556–9568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lill R, Dutkiewicz R, Freibert SA, Heidenreich T, Mascarenhas J, Netz DJ, Paul VD, Pierik AJ, Richter N, Stumpfig M, Srinivasan V, Stehling O, Milhlenhoff U, The role of mitochondria and the CIA machinery in the maturation of cytosolic and nuclear iron-sulfur proteins. Eur. J. Cell. Biol 2015, 94 280–291. [DOI] [PubMed] [Google Scholar]
- 11.Qi WB, Li JW, Cowan JA, A structural model for glutathione-complexed iron-sulfur cluster as a substrate for ABCB7-type transporters. ChemComm 2014, 50 3795–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li JW, Cowan JA, Glutathione-coordinated 2Fe-2S cluster: a viable physiological substrate for mitochondrial ABCB7 transport. ChemComm 2015, 51 2253–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaedler TA, Thornton JD, Kruse I, Schwarzländer M, Meyer AJ, H. W. v. Veen, J. Balk, A conserved mitochondrial ATP-binding cassette transporter exports glutathione polysulfide for cytosolic metal cofactor assembly. J. Biol. Chem 2014, 289 23264–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bosnjak I, Uhlinger KR, Heim W, Smital T, Franekic-Colic J, Coale K, Epel D, Hamdoun A, Multidrug efflux transporters limit accumulation of inorganic, but not organic, mercury in sea urchin embryos. Environ. Sci. Technol 2009, 43 8374–8380; [DOI] [PMC free article] [PubMed] [Google Scholar]; Long Y, Li Q, Wang YH, Cui ZB, MRP proteins as potential mediators of heavy metal resistance in zebrafish cells. Comp. Biochem. Physiol. C, Pharmacol. Toxicol. Endocrinol 2011, 153 310–317; [DOI] [PubMed] [Google Scholar]; Leslie EM, Deeley RG, Cole SPC, Multidrug resistance proteins: Role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol. Appl. Pharmacol 2005, 204 216–237; [DOI] [PubMed] [Google Scholar]; Lee JY, Yang JG, Zhitnitsky D, Lewinson O, Rees DC, Structural basis for heavy metal detoxification by an Atm1-type ABC exporter. Science 2014, 343 1133–1136; [DOI] [PMC free article] [PubMed] [Google Scholar]; Parker LJ, Bocedi A, Ascher DB, Aitken JB, Harris HH, Lo Bello M, Ricci G, Morton CJ, Parker MW, Glutathione transferase P1–1 as an arsenic drug-sequestering enzyme. Protein Sci. 2017, 26 317–326; [DOI] [PMC free article] [PubMed] [Google Scholar]; Perez RR, Sousa CA, Vankeersbilck T, Machado MD, Soares EV, Evaluation of the role of glutathione in the lead-induced toxicity in Saccharomyces cerevisiae. Curr. Microbiol 2013, 67 300–305; [DOI] [PubMed] [Google Scholar]; Salerno M, Petroutsa M, Garnier-Suillerot A, The MRP1-mediated effluxes of arsenic and antimony do not require arsenic-glutathione and antimony-glutathione complex formation. J. Bioenerg. Biomembr 2002, 34 135–145; [DOI] [PubMed] [Google Scholar]; Leverrier P, Montigny C, Garrigos M, Champeil P, Metal binding to ligands: Cadmium complexes with glutathione revisited. Anal. Biochem 2007, 371 215–228; [DOI] [PubMed] [Google Scholar]; Dijkstra M, Havinga R, Vonk RJ, Kuipers F, Bile secretion of cadmium, silver, zinc and copper in the rat. Involvement of various transport systems. Life Sci. 1996, 59 1237–1246; [DOI] [PubMed] [Google Scholar]; Fardel O, Kolasa E, Le Vee M, Environmental chemicals as substrates, inhibitors or inducers of drug transporters: implication for toxicokinetics, toxicity and pharmacokinetics. Expert Opin. Drug Metab. Toxicol 2012, 8 29–46; [DOI] [PubMed] [Google Scholar]; Odermatt A, Suter H, Krapf R, Solioz M, Primary structure of 2 p-type ATPases involved in copper homeostasis in Enterococcus-hirae. J. Biol. Chem 1993, 268 12775–12779; [PubMed] [Google Scholar]; Cui ZF, Hirata D, Tsuchiya E, Osada H, Miyakawa T, The multidrug resistance-associated protein (MRP) subfamily (Yrs1/Yor1) of Saccharomyces cerevisiae is important for the tolerance to a broad range of organic anions. J. Biol. Chem 1996, 271 14712–14716; [DOI] [PubMed] [Google Scholar]; Hong YF, Lai YT, Chan GCF, Sun HZ, Glutathione and multidrug resistance protein transporter mediate a self-propelled disposal of bismuth in human cells. Proc. Natl. Acad. Sci. U.S.A 2015, 112 3211–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi WB, Li JW, Chain CY, Pasquevich GA, Pasquevich AF, Cowan JA, Glutathione complexed Fe-S centers. J. Am. Chem. Soc 2012, 134 10745–10748; [DOI] [PMC free article] [PubMed] [Google Scholar]; Qi WB, Li JW, Chain CY, Pasquevich GA, Pasquevich AF, Cowan JA, Glutathione-complexed iron-sulfur clusters. Reaction intermediates and evidence for a template effect promoting assembly and stability. ChemComm 2013, 49 6313–6315; [DOI] [PMC free article] [PubMed] [Google Scholar]; Fidai I, Wachnowsky C, Cowan JA, Mapping cellular Fe-S cluster uptake and exchange reactions - divergent pathways for iron-sulfur cluster delivery to human ferredoxins. Metallomics 2016, 8 1283–1293; [DOI] [PMC free article] [PubMed] [Google Scholar]; Fidai I, Wachnowsky C, Cowan JA, Glutathione-complexed 2Fe-2S clusters function in Fe-S cluster storage and trafficking. J. Biol. Inorg. Chem 2016, 21 887–901. [DOI] [PubMed] [Google Scholar]
- 16.Csere P, Lill R, Kispal G, Identification of a human mitochondrial ABC transporter, the functional orthologue of yeast Atm1p. FEBS Lett. 1998, 441 266–270. [DOI] [PubMed] [Google Scholar]
- 17.Bekri S, Kispal G, Lange H, Fitzsimons E, Tolmie J, Lill R, Bishop DF, Human ABC7 transporter: gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron-sulfur protein maturation. Blood 2000, 96 3256–3264; [PubMed] [Google Scholar]; Shimada Y, Okuno S, Kawai A, Shinomiya H, Saito A, Suzuki M, Omori Y, Nishino N, Kanemoto N, Fujiwara T, Horie M, Takahashi E, Cloning and chromosomal mapping of a novel ABC transporter gene (hABC7), a candidate for X-linked sideroblastic anemia with spinocerebellar ataxia. J. Hum. Genet 1998, 43 115–122. [DOI] [PubMed] [Google Scholar]
- 18.Srinivasan V, Pierik AJ, Lill R, Crystal structures of nucleotide-free and glutathione-bound mitochondrial ABC transporter Atm1. Science 2014, 343 1137–1140. [DOI] [PubMed] [Google Scholar]
- 19.Sharma S, Davidson AL, Vanadate-induced trapping of nucleotides by purified maltose transport complex requires ATP hydrolysis. J. Bacteriol 2000, 182 6570–6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poolman B, Doeven MK, Geertsma ER, Biemans-Oldehinkel E, Konings WN, Rees DC, Functional analysis of detergent-solubilized and membrane-reconstituted ATP-binding cassette transporters. Methods Enzymol. 2005, 400 429-+; [DOI] [PubMed] [Google Scholar]; Geertsma ER, Mahmood N, Schuurman-Wolters GK, Poolman B, Membrane reconstitution of ABC transporters and assays of translocator function. Nat. Protoc 2008, 3 256–266; [DOI] [PubMed] [Google Scholar]; Kuhnke G, Neumann K, Mühlenhoff U, Lill R, Stimulation of the ATPase activity of the yeast mitochondrial ABC transporter Atm1p by thiol compounds. Mol. Membr. Biol 2006, 23, 173–184 [DOI] [PubMed] [Google Scholar]
- 21.Orelle C, Dalmas O, Gros P, Di Pietro A, Jault JM, The conserved glutamate residue adjacent to the Walker-B motif is the catalytic base for ATP hydrolysis in the ATP-binding cassette transporter BmrA. J. Biol. Chem 2003, 278 47002–47008. [DOI] [PubMed] [Google Scholar]
- 22.Vigano C, Margolles A, van Veen HW, Konings WN, Ruysschaert JM, Secondary and tertiary structure changes of reconstituted LmrA induced by nucleotide binding or hydrolysis - A Fourier transform attenuated total reflection infrared spectroscopy and tryptophan fluorescence quenching analysis. J. Biol. Chem 2000, 275 10962–10967. [DOI] [PubMed] [Google Scholar]
- 23.Cohen BE, Pralle A, Yao XJ, Swaminath G, Gandhi CS, Jan YN, Kobilka BK, Isacoff EY, Jan LY, A fluorescent probe designed for studying protein conformational change. Proc. Natl. Acad. Sci. U.S.A 2005, 102 965–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorbulev S, Abele R, Tampe R, Allosteric crosstalk between peptide-binding, transport, and ATP hydrolysis of the ABC transporter TAP. Proc. Natl. Acad. Sci. U.S.A 2001, 98 3732–3737; [DOI] [PMC free article] [PubMed] [Google Scholar]; Ketchum CJ, Schmidt WK, Rajendrakumar GV, Michaelis S, Maloney PC, The yeast a-factor transporter Ste6p, a member of the ABC superfamily, couples ATP hydrolysis to pheromone export. J. Biol. Chem 2001, 276 29007–29011. [DOI] [PubMed] [Google Scholar]
- 25.Gout E, Rebeille F, Douce R, Bligny R, Interplay of Mg2+, ADP, and ATP in the cytosol and mitochondria: Unravelling the role of Mg2+ in cell respiration. Proc. Natl. Acad. Sci. U.S.A 2014, 111 E4560–E4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharom FJ, Yu XH, Chu JWK, Doige CA, Characterization of the ATPase activity of p-glycoprotein from multidrug-resistant Chinese-hamster ovary cells. Biochem. J 1995, 308 381–390; [DOI] [PMC free article] [PubMed] [Google Scholar]; Cho C, Reck-Peterson SL, Vale RD, Regulatory ATPase sites of cytoplasmic dynein affect processivity and force generation. J. Biol. Chem 2008, 283 25839–25845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dawson RJP, Locher KP, Structure of a bacterial multidrug ABC transporter. Nature 2006, 443 180–185; [DOI] [PubMed] [Google Scholar]; Cotten JF, Ostedgaard LS, Carson MR, Welsh MJ, Effect of cystic fibrosis-associated mutations in the fourth intracellular loop of cystic fibrosis transmembrane conductance regulator. J. Biol. Chem 1996, 271 21279–21284; [DOI] [PubMed] [Google Scholar]; Currier SJ, Kane SE, Willingham MC, Cardarelli CO, Pastan I, Gottesman MM, Identification of residues in the 1st cytoplasmic loop of p-glycoprotein involved in the function of chimeric human MDR1-MDR2 transporters. J. Biol. Chem 1992, 267 25153–25159; [PubMed] [Google Scholar]; Lankat-Buttgereit B, Tampe R, The transporter associated with antigen processing: Function and implications in human diseases. Physiol. Rev 2002, 82 187–204. [DOI] [PubMed] [Google Scholar]
- 28.Deeley RG, Westlake C, Cole SPC, Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol. Rev 2006, 86 849–899; [DOI] [PubMed] [Google Scholar]; Shintre CA, Pike ACW, Li Q, Kim JI, Barr AJ, Goubin S, Shrestha L, Yang J, Berridge G, Ross J, Stansfeld PJ, Sansom MSP, Edwards AM, Bountra C, Marsden BD, von Delft F, Bullock AN, Gileadi O, Burgess-Brown NA, Carpenter EP, Structures of ABCB10, a human ATP-binding cassette transporter in apo- and nucleotide-bound states. Proc. Natl. Acad. Sci. U.S.A 2013, 110 9710–9715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Higgins CF, Linton KJ, The ATP switch model for ABC transporters. Nat. Struct. Mol. Biol 2004, 11 918–926; [DOI] [PubMed] [Google Scholar]; Linton KJ, Higgins CF, Structure and function of ABC transporters: The ATP switch provides flexible control. Pflugers Arch. 2007, 453 555–567. [DOI] [PubMed] [Google Scholar]
- 30.Petronilli V, Ames GF, Binding protein-independent histidine permease mutants - Uncoupling of ATP hydrolysis from transmembrane signaling. J. Biol. Chem 1991, 266 16293–16296; [PubMed] [Google Scholar]; Davidson AL, Shuman HA, Nikaido H, Mechanism of maltose transport in Escherichia-coli - Transmembrane signaling by periplasmic binding-proteins. Proc. Natl. Acad. Sci. U.S.A 1992, 89 2360–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mittal A, Bohm S, Grutter MG, Bordignon E, Seeger MA, Asymmetry in the homodimeric ABC Transporter MsbA Recognized by a DARPin. J. Biol. Chem 2012, 287 20395–20406; [DOI] [PMC free article] [PubMed] [Google Scholar]; Jones PM, George AM, Opening of the ADP-bound active site in the ABC transporter ATPase dimer: Evidence for a constant contact, alternating sites model for the catalytic cycle. Proteins 2009, 75 387–396. [DOI] [PubMed] [Google Scholar]
- 32.Senior AE, AlShawi MK, Urbatsch IL, The catalytic cycle of P-glycoprotein. FEBS Lett. 1995, 377 285–289; [DOI] [PubMed] [Google Scholar]; Liu CE, Liu PQ, Ames GFL, Characterization of the adenosine triphosphatase activity of the periplasmic histidine permease, a traffic ATPase (ABC transporter). J. Biol. Chem 1997, 272 21883–21891; [DOI] [PubMed] [Google Scholar]; Nikaido K, Liu PQ, Ames GFL, Purification and characterization of HisP, the ATP-binding subunit of a traffic ATPase (ABC transporter), the histidine permease of Salmonella typhimurium - Solubility, dimerization, and ATPase activity. J. Biol. Chem 1997, 272 27745–27752; [DOI] [PubMed] [Google Scholar]; Hrycyna CA, Ramachandra M, Ambudkar SV, Ko YH, Pedersen PL, Pastan I, Gottesman MM, Mechanism of action of human P-glycoprotein ATPase activity - Photochemical cleavage during a catalytic transition state using orthovanadate reveals cross-talk between the two ATP sites. J. Biol. Chem 1998, 273 16631–16634; [DOI] [PubMed] [Google Scholar]; Davidson AL, Laghaeian SS, Mannering DE, The maltose transport system of Escherichia coli displays positive cooperativity in ATP hydrolysis. J. Biol. Chem 1996, 271 4858–4863; [PubMed] [Google Scholar]; Urbatsch IL, Sankaran B, Weber J, Senior AE, P-glycoprotein is stably inhibited by vanadate-induced trapping of nucleotide at a single catalytic site. J. Biol. Chem 1995, 270 19383–19390; [DOI] [PubMed] [Google Scholar]; Davidson AL, Sharma S, Mutation of a single MalK subunit severely impairs maltose transport activity in Escherichia coli. J. Bacteriol 1997, 179 5458–5464; [DOI] [PMC free article] [PubMed] [Google Scholar]; Lamers MH, Winterwerp HHK, Sixma TK, The alternating ATPase domains of MutS control DNA mismatch repair. EMBO J. 2003, 22 746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Al-Shawi MK, Polar MK, Omote H, Figler RA, Transition state analysis of the coupling of drug transport to ATP hydrolysis by P-glycoprotein. J. Biol. Chem 2003, 278 52629–52640; [DOI] [PubMed] [Google Scholar]; Syberg F, Suveyzdis Y, Kotting C, Gerwert K, Hofmann E, Time-resolved Fourier transform infrared spectroscopy of the nucleotide-binding domain from the ATP-binding cassette transporter MsbA ATP hydrolysis is the rate-limiting step in the catalytic cycle. J. Biol. Chem 2012, 287 23923–23931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shitan N, Dalmas F, Dan K, Kato N, Ueda K, Sato F, Forestier C, Yazaki K, Characterization of Coptis japonica CjABCB2, an ATP-binding cassette protein involved in alkaloid transport. Phytochemistry 2013, 91 109–116. [DOI] [PubMed] [Google Scholar]
- 35.Santelia D, Vincenzetti V, Azzarello E, Bovet L, Fukao Y, Duchtig P, Mancuso S, Martinoia E, Geisler M, MDR-like ABC transporter AtPGP4 is involved in auxin-mediated lateral root and root hair development. FEBS Lett. 2005, 579 5399–5406; [DOI] [PubMed] [Google Scholar]; Terasaka K, Blakeslee JJ, Titapiwatanakun B, Peer WA, Bandyopadhyay A, Makam SN, Lee OR, Richards EL, Murphy AS, Sato F, Yazaki K, PGP4, an ATP binding cassette P-glycoprotein, catalyzes auxin transport in Arabidopsis thaliana roots. Plant Cell 2005, 17 2922–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee M, Choi Y, Burla B, Kim YY, Jeon B, Maeshima M, Yoo JY, Martinoia E, Lee Y, The ABC transporter AtABCB14 is a malate importer and modulates stomatal response to CO2. Nat. Cell Biol 2008, 10 1217–1223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.