

Abstract
Multiple ion fragmentation methods involving collision-induced dissociation (CID), higher-energy collisional dissociation (HCD) with regular and very high energy settings, and electron-transfer dissociation (ETD) with supplementary HCD (EThcD) were implemented to improve the confidence of cross-link identifications. Three different S. cerevisiae proteasome samples cross-linked by diethyl suberthioimidate (DEST) or bis(sulfosuccinimidyl)suberate (BS3) were analyzed. Two approaches are introduced to combine interpretations from the above four methods. Working with cleavable cross-linkers such as DEST, the first approach searches for cross-link diagnostic ions and consistency among the best interpretations derived from all four MS2 spectra associated with each precursor ion. Better agreement leads to a more definitive identification. Compatible with both cleavable and non-cleavable cross-linkers such as BS3, the second approach multiplies scoring metrics from a number of fragmentation experiments to derive an overall best match. This significantly increases the scoring gap between the target and decoy matches. Validity of cross-links fragmented by HCD alone and identified by Kojak, MeroX, pLink, and Xi was evaluated using multiple fragmentation data. Possible ways to improve identification credibility are discussed. Data are available via ProteomeXchange with identifier PXD018310.
Keywords: ion fragmentation methods, cross-linked peptides, proteasome, cross-linking mass spectrometry, proteome
Graphical Abstract
INTRODUCTION
Proteins are key players involved in virtually all activities taking place in cells. Independent of whether they are components of macromolecular complexes, proteins interact with each other, and as a result, protein-protein interactions play crucial roles in their cellular functions. Direct observations of protein structures and their interactions with other molecules are required to understand the detailed processes that occur inside living cells. While X-ray crystallography and NMR spectroscopy provide the highest resolution and most definitive structural information, the shortcomings of these methods in terms of sensitivity and sample state have been well documented.1–4 It is also difficult to crystallize large macromolecular complexes due to their dynamic nature.5 Disordered regions of proteins that do not appear in crystal structures can also play roles in their function.6 Cryo-electron microscopy technology has rapidly improved to the point where its resolution is competitive with that of x-ray crystallography.7 However, the freezing of samples takes them out of their natural state and could perturb protein structure. To study low abundance proteins, particularly in vivo, higher-sensitivity mass spectrometry-based structural probes are proving attractive. Although pull-down assays can identify what proteins interact, they do not identify how they interact. The conjunction of covalent cross-linking with mass spectrometry provides additional information. This evolving and rapidly growing subfield of proteomics not only enables pairs of interacting proteins to be linked together for subsequent analysis; the length of the cross-linker provides distance constraints and the process of cross-link interpretation implicitly determines the residues where linkages occur. This provides topological information about the interacting protein surfaces. Despite its appeal, there are major challenges associated with this type of experiment that have been summarized in several reviews.8–13 Most significantly, the concentration of linked peptides is low relative to that of unlinked proteolytic peptides and “dead-ends” (peptides attached to just one end of a linker). Furthermore, the bioinformatics problem of interpreting cross-linked spectra is one of high combinatorial complexity: instead of just considering all of the peptides that a proteome might engender, cross-linking interpretation programs must consider all combinations of these peptides. In addition, there is a rather subtle problem: one peptide of a cross-linked pair often fragments better than the other. The credibility of a cross-link identification is usually measured by target/decoy database matching. For any precursor ion that is fragmented, all theoretical peptide pairs whose linked mass matches that of the precursor ion to within some error tolerance are considered. Theoretical fragments from each such pair are compared with experimental MS2 fragment ion masses. The tendency for one peptide (that might be referred to as “alpha”) to fragment better than the other (peptide “beta”) leads to a good identification of one but not the other.14 In fact, one of the peptides often exhibits so few cleavages that multiple target sequences and even decoy sequences provide equivalently good assignments for that peptide. Whenever decoy sequences match proteomic data approximately as well as target sequences, the credibility of identifications is low and the false discovery rate climbs. These challenges tend to be exacerbated when in vivo cross-linking is attempted since the number of different proteins in the sample is so large. Iacobucci and Sinz have specifically commented on the proliferation of mis-assigned cross-links in publications.15 One solution to these problems that is growing in popularity is to employ cleavable cross-linkers.16–23 In this approach the low energy fragmentation of a cross-linked pair of peptides leads to individual peptide ions, each containing part of the cross-linker. These are subsequently fragmented in MS3 experiments to ascertain their identities. The great advantage is that the MS3 data can be interpreted by normal proteomics informatics tools and the database needed for this does not grow as the square of the number of peptides. Often overlooked is that due to the lower sensitivity of orbitraps, MS3 mass measurements are usually performed in ion traps at lower mass accuracy and these results are not further checked. This is unfortunate because only a fraction of peptide ion trap spectra yield definitive identifications.24–25 Users have also reported that CID cleavable linkers do not always cleave to produce the expected mass pairs.26 In a community study of a single protein, bovine serum albumin (BSA), it appeared that non-cleavable cross-linkers can lead to as many identifications as a cleavable linker.27 However, this might be less true in cross-linking studies of complex systems. An additional problem is that the solubility of currently available cleavable cross-linkers is poor. Succinimidyl esters are typically dissolved in dimethyl sulfoxide before being added to water. The limited solubility may be a problem for efficient cross-linking of proteins within cells.28 All of this suggests that this is not a solved problem and further cross-linker developments are warranted.
The recently published community study mentioned above aimed at summarizing the wide variety of approaches that have been applied to cross-linking.27 In an attempt to provide some basis for comparison they chose to investigate cross-links in a single protein BSA. Nevertheless, because different cross-linking reagents were utilized, different data interpretation algorithms were employed and data were recorded with different chromatography and mass spectrometry instrumentation, this community study serves more as an overview than a critical analysis of different approaches. While copious results from different groups were summarized, conclusions about best approaches were not drawn. In fact, to discourage comparison of different methods, results were not directly associated with specific research groups or interpretation algorithms. While the identification of the maximum number of credible identifications is normally the goal in cross-linking studies, it was impossible to infer from the data presented which experimental methods and which data interpretation algorithms were most successful. Some provided numerous identifications, some yielded not so many, but whether the identification of a particularly large number of cross-links should be viewed as a great experimental method or an over-zealous data interpretation algorithm was not addressed. More recently, Beveridge et al. compared several popular cross-linking data interpretation algorithms by studying cross-linked synthetic peptides.29 One of the goals of the present work is to use biological samples to shed light on this subject.
The use of more than one ion fragmentation method to increase data interpretation confidence has been demonstrated in conventional proteomics experiments.30–36 Exploitation of multiple methods would be expected to improve cross-link identification confidence, particularly if complementary methods could provide useful information about the less definitively identified (“beta”) peptide. Indeed, some groups have applied ETD and CID and observed some improvement in identifications.20, 26 Several years ago we demonstrated that diethylsuberthioimidate (DEST) is an effective homobifunctional non-cleavable cross-linker.37 Since it is amine-reactive, this molecule is quite analogous to commercial succinimidyl ester-based reagents. The two comparative advantages of DEST are its improved water solubility and the fact that reaction with amines yields amidino rather than amide linkages. Amidino groups are positively charged at neutral pH as are primary amines so replacing one charged moiety with another should help to preserve protein structures.38 In addition, the charged amidino linkages would be expected to facilitate ion exchange chromatography that is often used to separate cross-links from peptides.39 Recently, we reported that electron-transfer dissociation (ETD) cleaves cross-links both along peptide backbones and particularly at the amidino linkages.40 This is interesting since collision-induced dissociation (CID) and higher-energy collisional dissociation (HCD) only cleave DEST cross-links along the peptide backbones. This difference suggests the intriguing possibility of simultaneously having a cleavable and a non-cleavable cross-linker that can produce complementary and quite orthogonal cross-link data sets using the three different fragmentation methods. Finally, we have found some value in using particularly high energy HCD conditions to enhance the production of immonium ions that identify residues found in the cross-link and this is further explored in the present work.
Naturally, the obvious question that arises is how to combine the results from fragmenting cross-link precursors with multiple methods to increase the confidence of cross-link identifications. The present study utilizes two approaches. In the first, the EThcD experiment must produce mass pairs that establish the masses of the two peptides and at least two of the fragmentation methods must lead to the same cross-link interpretation. In the second approach, we consider the top identifications of each of the four MS2 spectra and multiply scoring metrics for the four in order to determine which cross-link hit is the best overall match to the four spectra.
The samples that are investigated in the present work are of modest complexity: there are 14 and 19 different proteins in the yeast proteasome core and regulatory particles and their average masses are 27 and 49 kDa respectively. These samples are much more complex than the single BSA protein but less complex than whole-cell lysates. They therefore provide an excellent basis for a comparative study. HCD is the most popular fragmentation method that is commonly employed in cross-linking studies.27, 41 For this reason, HCD mass spectrometry data are provided to four cross-linking interpretation algorithms, Kojak,42 MeroX,43–44 pLink,45–47 and Xi48–49 that were selected because they all calculate and report false discovery rates (FDRs), are publicly available, are easy to use and have been employed in a number of publications. The same HCD data, along with complementary EThcD, CID and very high energy HCD data obtained with the same cross-link precursor ions are also interpreted by our own data analysis program. This approach offers a fair head-to-head comparison of four popular cross-linking interpretation algorithms using HCD data that they are normally provided with. By reserving additional complementary EThcD, CID and high energy HCD data that these programs were not given, we have the possibility of independently confirming the validity of conclusions that they reached from HCD data alone. We are also able to explore whether the additional EThcD, CID and high energy HCD data can offer alternative insights about data interpretation or help to improve the confidence of cross-link identifications.
EXPERIMENTAL SECTION
Sample Preparation, Cross-linking, Proteolytic Digestion and Fractionation
The 19S regulatory particle and 20S core particle of the proteasome were isolated as previously described.50 Polyubiquitin was synthesized following reported procedures.51 Three samples derived from yeast proteasome were investigated in this work. The first containing the core particle and the second containing the regulatory particle were cross-linked with BS3. The third involving the regulatory particle and polyubiquitin was cross-linked with DEST. Cross-linked protein samples were digested with trypsin. Because DEST cross-links tend to be highly charged, tryptic digests from DEST experiments were fractionated by strong cation exchange chromatography. More details are presented in Supporting Information.
HPLC/Nano-ESI MS2 Analysis with Multiple Ion Fragmentation Methods
Tryptic digests were analyzed with an EASY-nLC 1200 liquid chromatograph (ThermoFisher Scientific) coupled with an Orbitrap Fusion Lumos Tribrid mass spectrometer (ThermoFisher Scientific). Each precursor was activated by four ion fragmentation events: 1) EThcD with ETD reaction time of 50, 70 or 100 ms followed by a supplementary activation with HCD at a low collision energy of 15 or 20%, 2) CID with a 35% normalized collision energy, 3) HCD with a collision energy setting of 30 or 35% and recording fragments from 140 m/z, or 4) HCD with a collision energy setting of 50% and recording fragments as small as 70 m/z. In this manuscript, the normal and very high energy HCD fragmentation events are referred to as HCD and hcd respectively. These two events were recorded separately in order to detect both small and large ions with high sensitivity. Mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE52 partner repository with the dataset identifier PXD018310. More details about instrument setups are in Supporting Information.
Data Analysis
Proteome Discoverer 2.1 software (Thermo Fisher Scientific, Waltham, MA) converted each *.raw orbitrap data file to four *.mgf files, one for each ion fragmentation method. HCD is a popular27 and relatively efficient41 fragmentation method that is commonly used to identify cross-linked peptides. Therefore *.mgf files containing HCD spectra only were submitted to four cross-link identification algorithms: Kojak (1.6.1 working together with Percolator53 2.9), MeroX (2.0.1.1), pLink (2.3.8), and Xi (comprised of XiSearch 1.7.0 and XiFDR 1.4.1. Search parameters are specified in Supporting Information. The cross-linked peptides identified from the three samples by each of the algorithms at 1% and 5% FDRs were tabulated in Tables S1–S3 of Supporting Information. Venn diagrams comparing cross-links identified by these algorithms were plotted using Venny 2.1.54
In order to independently validate the interpretations of HCD data derived by the four publicly available programs, spectra obtained from fragmenting the same precursor ions by four methods were interpreted by an in-house program that searched for different ion types for each fragmentation method. These are detailed in Table S4. Our in-house program computes metrics such as Correlation Score, number of peaks matched (Matches), and percentage of fragment ion intensity matched (%Int) to each peptide of each tentative identification. In other words, cross-linked peptides receive Correlation Score1, Matches1 and %Int1 for peptide α, and Correlation Score2, Matches2 and %Int2 for peptide β. Since regular peptides and dead-ends are comprised of only single peptides, Correlation Score2, Matches2, and %Int2 are all zero for these species. The sums of Correlation Score1 and Correlation Score2, Matches1 and Matches2, %Int1 and %Int2 are referred to as Correlation Score, Matches, and %Int, respectively. Our in-house program is further described in Supporting Information.
Two different approaches were used to look for consistency between the tentative cross-link identifications derived from the four ion fragmentation methods. The first approach worked only with the DEST ETD cleavable cross-linker since it required the detection of ETD diagnostic mass pairs.40 Our program checked whether the identifications derived from EThcD spectra were consistent with observed mass pair peaks and then determined how many of the best interpretations of the four fragmentation spectra were the same. DEST cross-links with at least two ETD mass pairs that were interpreted as the best match from at least two fragmentation methods were tabulated in Table S3 and used to validate the DEST cross-links identified by other algorithms using HCD data alone.
Our second approach for combining the interpretations of different types of spectra did not rely on cross-link diagnostic ions, and therefore could be applied to both cleavable (DEST) and non-cleavable (BS3) cross-linkers. Our program compared the top ten best interpretations of each EThcD, CID, HCD and hcd spectrum seeking consistencies. For each case in which the same cross-link identification was found among the top ten interpretations of all four fragmentation spectra, Composite and Overall Scores were calculated as follows:
The only difference between these is that Composite Score includes scoring metrics for the entire cross-link while Overall Score includes metrics associated with each peptide component of each cross-link. Composite Score and Overall Score histograms comparing target-target, target-decoy, and decoy-decoy score distributions are plotted in Figure S1 of Supporting Information. We generally found Overall Score to be more accurate and illuminating for evaluating the quality of a cross-link identification because it yields larger values for matches in which both peptides contribute significantly to the score. Nevertheless, this distinction was not always perfectly clear because virtually every spectrum contains peaks that can be assigned to cleavages in peptide α or peptide β. Therefore, evaluating the contributions of peptides α and peptide β to each spectrum can be somewhat arbitrary. For this reason, we found that in certain cases Composite Score better distinguished target and decoy hits. When the Overall Score matched a decoy hit but the Composite Score matched a target hit, this spectrum interpretation was not considered definitive and neither hit was used in FDR computations.
Regular peptides and dead-ends were identified based on Composite Scores. These species would always receive an Overall Score of zero due to the lack of a second peptide. When Composite Score was larger for a cross-link or dead-link interpretation than for any peptide or dead-end, Overall Score was then used to provide a more refined determination of the best peptide α and β. Cross-links from the three samples were identified at 1% and 5% FDRs based on the familiar equation55
where NTT, NTD, and NDD denote the numbers of target-target, target-decoy and decoy-decoy cross-links. Identified cross-links were tabulated in Tables S1–S3 in Supporting Information along with results from other algorithms.
RESULTS
Identification of Cross-links by Different Interpretation Algorithms Based on HCD Data
HCD is commonly considered to be the most efficient fragmentation method for cross-link matches.27, 41 Therefore, to mimic conventional workflows, three sets of HCD MS2 spectra were submitted to Kojak, MeroX, pLink, and Xi. Database search parameters were outlined in Supporting Information. The first and second sets of data were acquired from BS3 cross-linked 20S core particle and 19S regulatory particle of S. cerevisiae proteasome, respectively. The third sample involved a mixture of the proteasome regulatory particle and polyubiquitin at a 1:1 weight ratio cross-linked by DEST. Due to the large molecular mass of the regulatory particle and the multiple ubiquitin subunits in polyubiquitin, the molar concentration of ubiquitin was roughly 100 times that of the regulatory particle proteins. The numbers of cross-link spectrum matches identified by the four programs from the three datasets at FDRs of 1% and 5% are displayed in Figure 1. (Note that XiFDR excluded cross-links with peptides shorter than six residues and Kojak apparently does not consider cross-links comprised of two identical peptides, which was somewhat common in our third sample that contained polyubiquitin.) Discrepancies among these four programs varied with sample complexity: from the first set of data, about 51% of cross-link spectrum matches were consistently identified by all four programs and 19% of cross-link spectrum matches were only identified by one program; from the second dataset, 52% of the cross-links were commonly identified and about 15% were uniquely identified. In the third dataset, only 20% of cross-link spectrum matches were identified by all programs and 30% of the cross-link spectrum matches were found by just a single program. Because of these discrepancies, it is desirable to employ independent information about the spectra to validate assignments and possibly derive alternatives. This can be accomplished using EThcD, CID and hcd data that were not provided to the above programs. A table and Venn diagrams comparing the performance of these four programs will be discussed in a later section.
Figure 1.
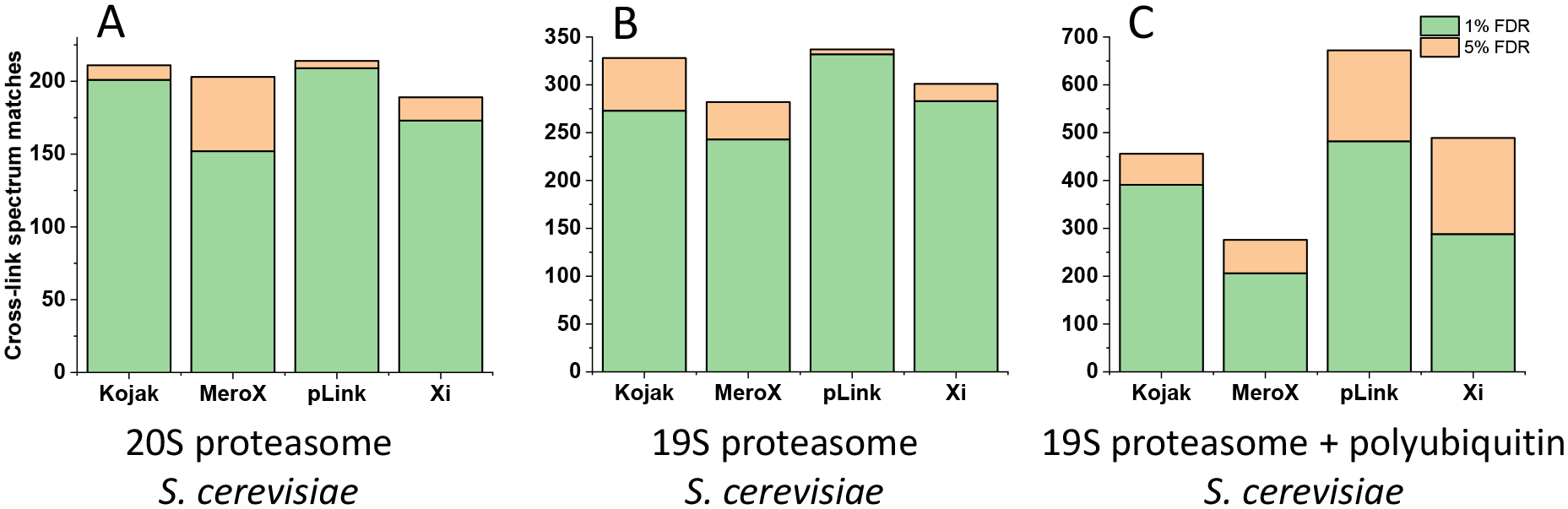
Numbers of cross-link spectrum matches identified by different data interpretation algorithms at 1% FDR (green) plus the additional matches identified when FDR was increased to 5% (orange) from (A) 20S core particle of proteasome, (B) 19S regulatory particle of proteasome, and (C) 19S regulatory particle of proteasome with polyubiquitin.
Dissociation of Precursors by Multiple Ion Fragmentation Methods
To generate novel fragmentation data that were interpreted only by our own analysis program, precursor ions were activated by EThcD, CID, HCD and hcd. Figure 2 displays an example of a DEST proteasome cross-link. The +4 charged precursor ion at 599.316 m/z activated by EThcD yielded the spectrum displayed in Figure 2A that was best matched to a cross-link AQFQELDS[K]K―YDDQL[K]QR. c and z• ions were predominantly observed. c4 to c6, c9, z+15 to z+19, and b2 ions identified peptide α while c6, c+16, c7, c+17, z+15-NH3, z+16, and w2 ions supported the assignment of peptide β. Uniquely, due to the ETD favored cleavages at amidino groups, DEST cross-links yielded mass pairs of P-NH2 and P+L+NH3 ions for both constituent peptides.40 In this EThcD spectrum, diagnostic mass pairs were found as the most intense peaks; their masses further supported this cross-link identification.
Figure 2.
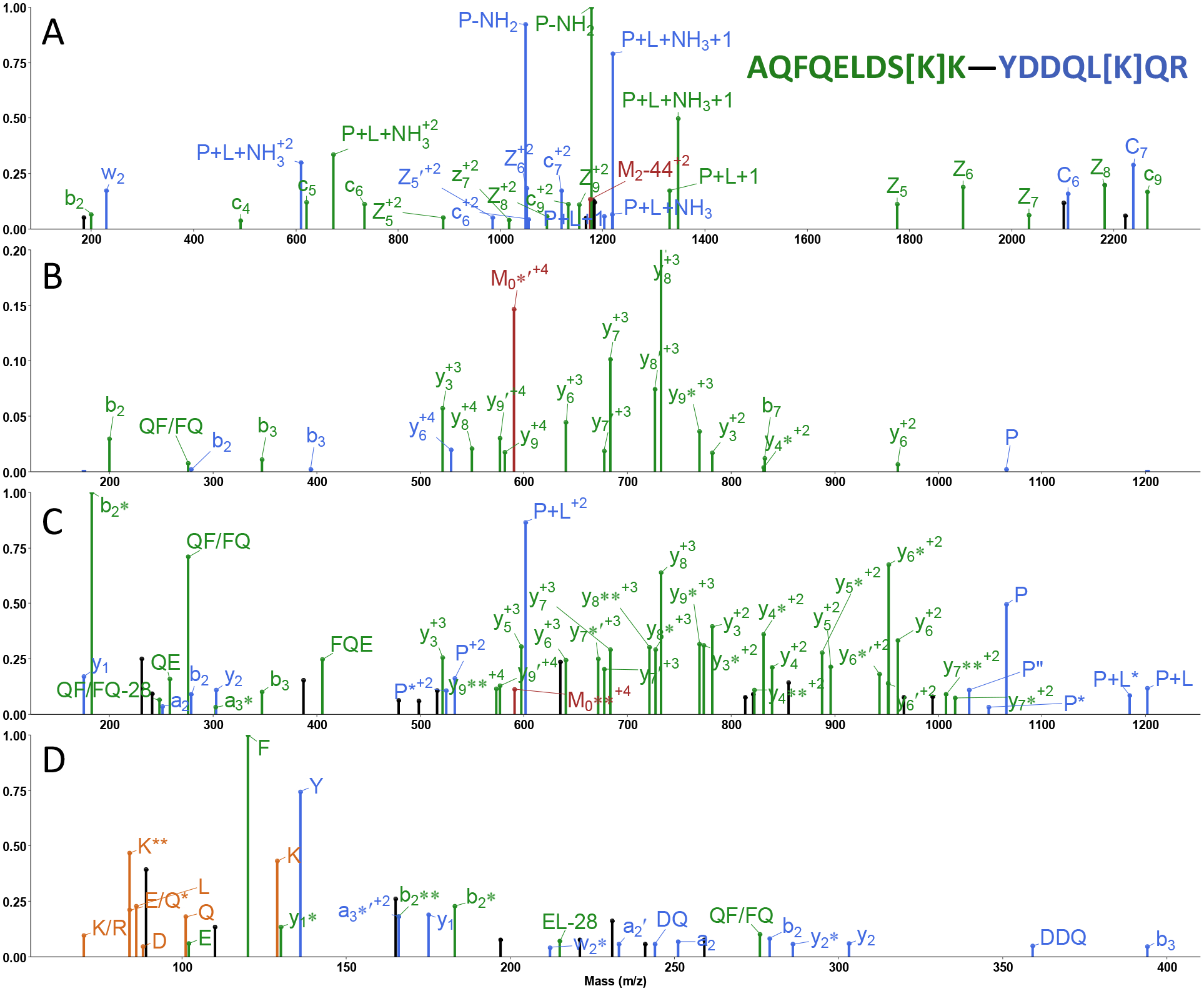
MS2 spectra of the cross-link AQFQELDS[K]K―YDDQL[K]QR between proteasome proteins Rpn9 and Rpt4 fragmented by (A) EThcD, (B) CID, (C) HCD and (D) hcd methods. Green and blue peaks are formed following cleavage of peptides α and β, respectively; peaks associated with the precursor ion are in red; orange peaks are immonium ions attributable to either peptide; unassignable peaks are in black. Neutral losses of ammonia and water are denoted by an asterisk (*) and prime (‘) respectively. c+1 and z+1 ions are represented as C and Z ions. Subscripts following M represent the number of hydrogen atoms lost from the precursor ion after electron capture.
Through low energy pathways, CID is somewhat more selective with preferential cleavages on the N-terminal side of Pro when a mobile proton is available and C-terminal to Asp or Glu when charge is sequestered.56–58 The CID MS2 spectrum of the same precursor ion, as plotted in Figure 2B, was best matched to the same cross-link. Peptide α was identified based on b2, b3, b7, y3, and y6 to y9 ions; peptide β was identified from b2, b3, and y6 ions. Note that the y8 ion of peptide α was plotted off-scale since it was much more intense than any other fragment. The high abundance of this peak results from the favored a2/b2 CID ion cleavage pathway.59–60
HCD is a beam-type CID in which fragment ions are provided with additional activation. As a result, HCD spectra are less likely to be dominated by a single feature but have a more even distribution of peak intensities, making it a popular method to identify cross-links. When the same 599.316 m/z precursor ion was activated by HCD, the fragmentation pattern shown in Figure 2C yielded the same AQFQELDS[K]K―YDDQL[K]QR cross-link interpretation. Peptide α was matched based on y3 to y9, b2 and b3 ions; peptide β was identified from y1, y2, a2 and b2 ions. In this spectrum, neutral losses of ammonia and water from fragment ions and internal backbone cleavages were also prevalent.
The higher energy HCD spectrum that we refer to as hcd is displayed in Figure 2D. In this case, the high collisional energy caused b- and y- ions to be very efficiently fragmented to small internal and immonium ions that only provide amino acid composition information. Matching immonium ions, that are particularly intense for aromatic residues, has been shown to be an alternative means to identify peptides61–63 and the complementarity determining regions of antibodies.64 As depicted in Figure 2D, the same cross-link was identified based on immonium ions of F, Y, K, Q, E, D, I/L, R, internal ions EL, QF or FQ of peptide α, DQ and DDQ of peptide β, as well as several small b and y ions associated with each peptide. From these four orthogonal MS2 spectra, this cross-link identification appears to be definitive. This assignment was initially surprising since the two linked lysine residues are from the lid (Rpn9) and base (Rpt4) of the proteasome with a Cα distance of over 53Å,65 greatly exceeding the 24Å constraint of DEST.37 However, a deep cryo-EM classification of the proteasome exposed an intermediate state with these two lysine residues only 13Å apart. Evidently, a distance constraint from a single PDB structure may not be the best way to validate a cross-link identification.
Overall, Figure 2 illustrates that the four ion activation methods yield complementary fragment ions. The orthogonality of the four spectra should facilitate cross-link identifications when the information that they convey is combined. Two approaches to combine this information are discussed next.
Search for Consistent Best Matches from Different Ion Fragmentation Methods
For each group of four MS2 spectra associated with a precursor ion, spectra generated by each fragmentation method were first interpreted independently. Then, an in-house script looked for agreement among the four interpretations. The supposition is that if a match to a spectrum derived from one fragmentation method is correct, data from other methods should support this identification. Random matches, including false target-target matches, are not expected to be consistent from one fragmentation method to another. In experiments involving DEST, its ETD cleavable characteristic was also exploited in this approach by requiring that a cross-link must have at least two diagnostic peaks to support its identification.
Different degrees of consistency were observed. At the highest level, all four orthogonal spectra yielded the same interpretation as exemplified in Figure 3A. These are very confident cross-links. From the third dataset acquired from DEST cross-linked proteins, 241 cross-links of this type were found, none of which were decoy hits, which leads to 0.0% FDR.
Figure 3.
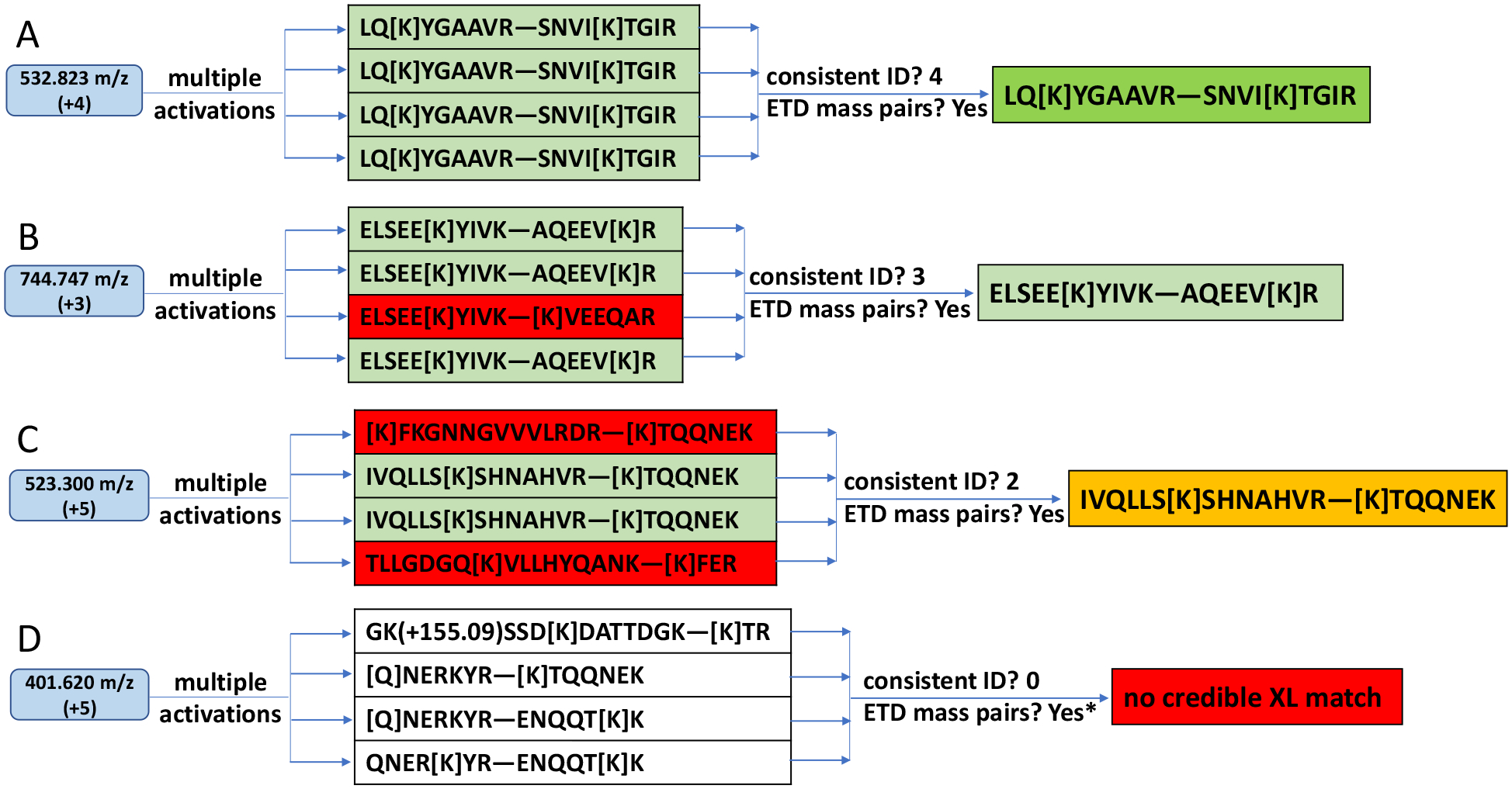
Examples displaying varying degrees of consistency among EThcD, CID, HCD and hcd interpretations for four tentative cross-links. ETD mass pairs are required in our first cross-link identification approach. (A) fragmentation of 532.823 m/z precursor ions yielding four consistent interpretations. (B) fragmentation of 744.747 m/z precursor ions yielding three consistent interpretations. (C) fragmentation of 523.300 m/z precursor ions yielding two consistent interpretations. (D) fragmentation of 401.620 m/z precursor ions yielding no consistent interpretation. In (A)-(C) the ETD mass pairs are concordant with the final interpretations. In (D) ETD mass pairs do not support interpretations derived from other methods, as indicated by *.
At the next level of consistency, three out of the four spectra yielded the same interpretation. The final identification, chosen to be the match supported by three spectra, was likely to be correct but less unambiguous. Among 178 cross-links identified this way, four were decoy matches, leading to a 2.3% FDR within this group. After combining with all cross-links identified previously based on four consistent interpretations, a total of 415 target cross-links were identified with FDR of 0.96%. Figure 3B illustrates a typical example of this kind of identification.
As exemplified in Figure 3C, sometimes two fragmentation spectra led to one interpretation while the other two led to alternative matches. Not surprisingly, relying on just two consistent spectral interpretations was less reliable. In our dataset, 95 cross-links were identified in this way, but 7 were decoy hits, yielding an 8.0% FDR within this group and a total of 503 target matches at 2.2% FDR after combining the above results for four and three consistent matches.
As exemplified in Figure 3D, some sets of four spectra generated by fragmenting the same precursor ion were matched to four different structures and these identifications were simply rejected. As mentioned in the Experimental Section, this approach of searching for consistent best matches required the detection of cross-link diagnostic ions as a prerequisite to recognizing cross-links. Therefore, this approach was not applicable to the first two sets of data cross-linked by BS3.
Combining Scores to Find a Best Overall Match
An alternative approach to identifying the best match to multiple sets of data is based on the premise that a false positive may match one spectrum better than the true positive, but it is unlikely to match four orthogonal spectra better than the correct identification. In addition, it is reasonable to expect that a true identification should rank among the top matches for individual fragmentation spectra, even if it is not always the best. In this work, we considered the top ten matches to each set of MS2 spectra involving the same precursor mass. For each cross-link identification that was found among these top ten hits our data interpretation program generated Overall Scores by multiplying scoring metrics associated with the interpretation of each MS2 spectrum as outlined above. Figure 4 illustrates an example of the +4 charged precursor ion at 536.029 m/z from the DEST cross-linked regulatory particle sample that was fragmented by the four methods. Eight potential cross-link identifications (designated A-H) were found among the top ten matches to all four MS2 spectra. The two highest Overall Scores were cross-links B and A that shared the same peptide α. B was the best overall match due to its highest Overall Score. Note that the 9th and 10th best matches to each of these spectra were not commonly found among the interpretations of other spectra and thus were automatically excluded from best overall match consideration. Unlike our first approach, ETD mass pairs were not required, although with DEST most of the best overall matches did have ETD mass pairs. Not demanding the detection of mass pairs make this approach applicable to non-cleavable linkers such as BS3.
Figure 4.
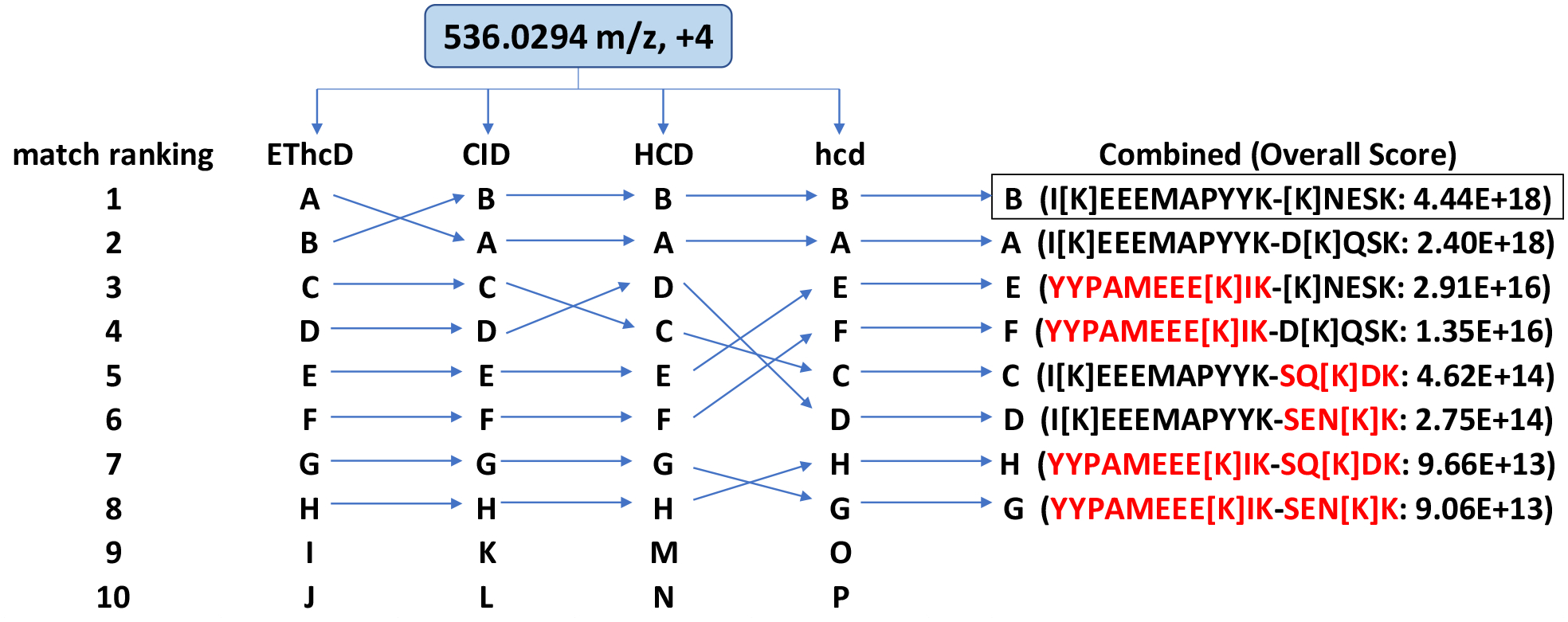
Second approach for deriving the best overall match of a cross-link by combining results from multiple ion fragmentation methods. The best overall match, B, is the second-best match derived from EThcD but the best match of CID, HCD and hcd. It received the highest Overall Score. Red peptides are from decoy database.
Having outlined our approach of combining individual MS2 interpretation scores to derive the best overall matches, the next question is whether this will ultimately improve our ability to distinguish true positives from false positives compared with interpreting data derived from a single fragmentation experiment. Due to combinatorial complexity, for any sample, a cross-link database is much larger than an analogous peptide or dead-end database. Therefore, a majority of individual MS2 spectra are initially assigned as cross-links (even though most of the assignments are wrong). For example, with our BS3 cross-linked proteasome regulatory particle data, roughly 2000 spectra were recorded with each fragmentation method. Approximately 600 to 700 target-target matches and roughly 600 to 1000 target-decoy or decoy-decoy matches were found with each fragmentation method. The rest were identified as regular peptides or dead-ends or they failed to match anything from the proteome. Histograms plotted in Figure 5A–D display the number of cross-link spectrum matches as a function of Overall Score on log scales for the four individual ion fragmentation methods. Target-target matches (green bars), tend to have higher Overall Scores than target-decoy matches (red bars) or decoy-decoy matches (blue bars). Scores for the best target and decoy matches for each individual fragmentation method differ by about an order of magnitude. When the results of all four fragmentation methods are combined to find the best overall match, as shown in Figure 5E, the total number of decoy matches, as reflected in the integrated area under all red and blue bars, decreases significantly to approximately 100. In contrast, the number of highly scored target matches in green bars does not diminish. This is because many decoy matches found as the best interpretation of one fragmentation experiment are lower-scoring matches for another MS2 experiment and may not even be among the top ten matches for another MS2 spectrum. Thus, when Overall Scores are computed, decoy matches often drop out from the list of best matches. The Overall Score for the credible target matches when combining the four methods are in the realm of 1019 to 1025. This is comparable to numbers obtained by multiplying best-scored target matches from individual fragmentation methods, indicating, as we proposed above, that true target matches of cross-linked peptides can be consistently identified as top matches with good scores no matter what ion fragmentation method is implemented. Figure 5E shows that the Overall Scores of the best decoy matches are often inferior to the best target matches by several orders of magnitude, enlarging the target-decoy gap. Since decoy matches are supposed to provide information about false target matches, this result implies that a fortuitous target match identified in one type of MS2 spectrum is unlikely to be validated by other fragmentation methods as originally hypothesized. The similarity of the score distributions of target and decoy matches in Figure 5D suggests that the hcd method often fails to distinguish target and decoy matches for reasons that will be discussed below. Therefore, in addition to calculating Overall Scores from all four methods, we also computed Overall Scores derived from only EThcD, CID and HCD data. A display of the distribution of Overall Scores based on only these three methods was plotted in Figure 5F, which looks quite similar to Figure 5E. The advantages of combining results from multiple fragmentation methods, such as a reduced number of decoy matches and a larger gap between target and decoy matches, are again apparent.
Figure 5.
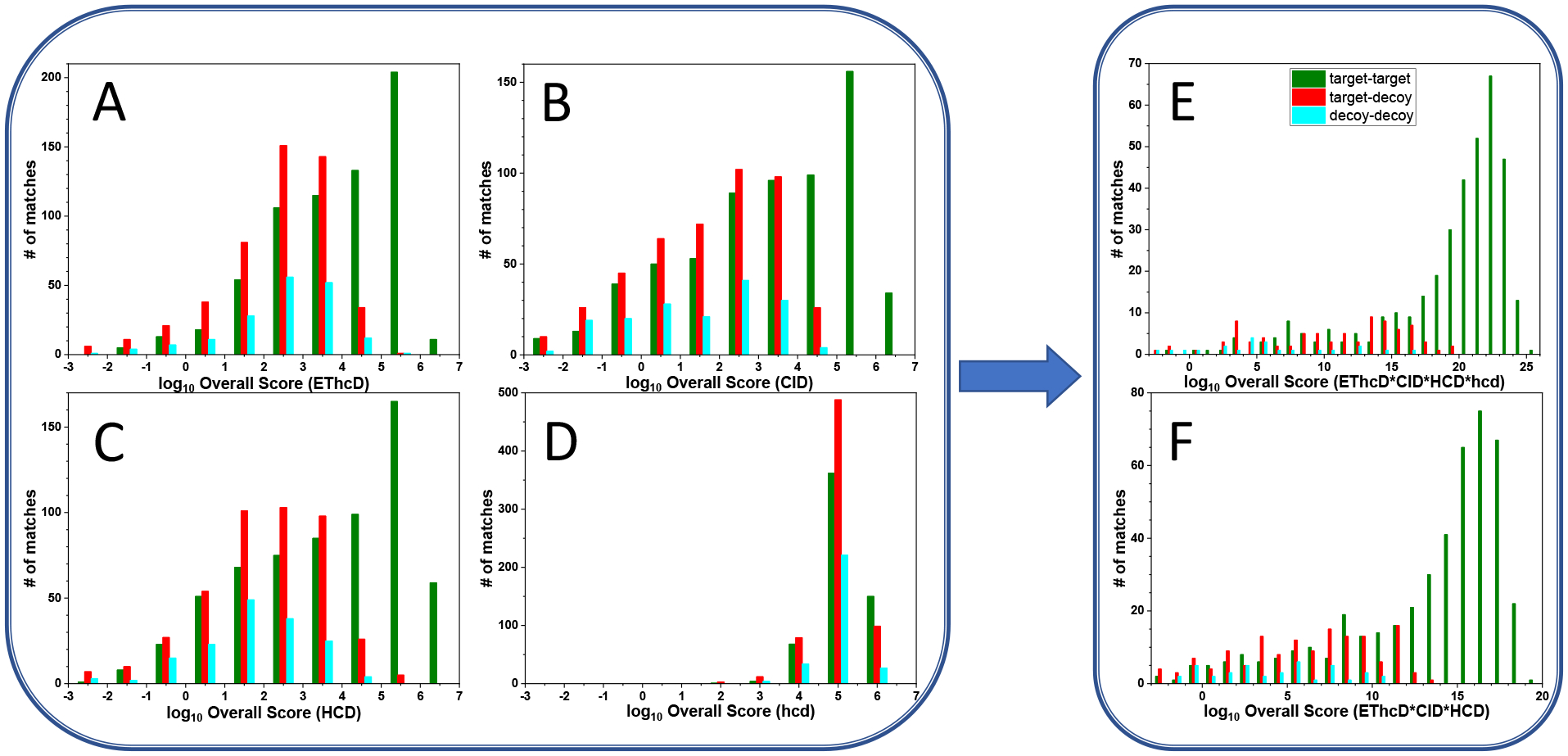
Histograms showing the numbers of target-target (green), target-decoy (red) and decoy-decoy matches (blue) of cross-linked peptides as a function of log Overall Score.
Interpretations are from (A) EThcD, (B) CID, (C) HCD, (D) hcd, or from our second approach combining results of (E) all four methods and (F) the first three methods.
The example shown in Figure 2 yielded an Overall Score of 1.47×1017 from combining four methods or 1.13×1012 from the first three. These Overall Scores are typical of the cross-links identified with 1% FDR from the third sample. Note that for the first two samples, Overall Scores were somewhat higher because spectra derived from these samples typically contained more peaks and this led to more peak matches. Through the second approach, from the samples of core particle, regulatory particle or regulatory particle with polyubiquitin, we identified 165, 258 and 347 cross-links, respectively, at 1% FDR when all four methods were combined. Likewise, we identified 206, 283 and 474 cross-links at 5% FDR. With only three methods combined, we identified 185 and 216, 283 and 347, or 383 and 543 cross-linked peptides at 1% and 5% FDRs, respectively. All cross-linked peptides identified through the second approach are tabulated in Tables S1–S3 of Supporting Information and used to validate those found by other data interpretation algorithms.
Comparison of Data Interpretation Algorithms
The extra fragment information obtained from the EThcD, CID and hcd experiments provided us a means to validate the cross-link identifications found by other algorithms that interpreted only HCD data. A few examples illustrating the process of evaluating the validity of cross-link identifications are discussed in Supporting Information.
Based on the additional EThcD, CID, and hcd spectra recorded for each precursor ion, the validity of all Kojak, MeroX, pLink and Xi cross-link identifications was checked. If our analysis yielded identical peptides but the linkage site differed, a cross-link was still considered validated. Numbers of cross-link spectrum matches identified at 1% and 5% FDRs by the four programs and validated by our approaches are listed in Table 1. The cross-link spectrum matches that agreed with our best interpretations are highlighted in green in Tables S1–S3 of Supporting Information. Venn diagrams showing the overlap of cross-link spectrum matches identified by our methods and each of the four selected public algorithms are displayed in Figure S3 of Supporting Information. In particular, using our first approach, at least two of the four fragmentation methods must have yielded the same result and ETD mass pairs must have been observed. Alternatively, with our second approach, the Overall Scores derived from the four methods (or at least from EThcD, CID and HCD) must have exceeded the 5% FDR cutoff. Some cross-link spectrum matches found by other programs could not be validated based on the above criteria. However, if they were found to involve the same linkages that were validated in other spectra, these identifications were nevertheless considered credible through manual checks and these are highlighted in yellow in Tables S1–S3. Finally, for some precursor ions, EThcD spectra were uniquely poor; for example, they might contain only ten or fewer fragment masses. In these cases, neither of our approaches worked well because the EThcD spectra did not contain diagnostic mass pairs and the cross-link interpretation that we were trying to validate was not among the top ten EThcD hits. Nevertheless, manual checks revealed that two or three of the other methods sometimes yielded a consistent validating interpretation. These cases are also considered credible though less definitive and are highlighted in salmon in Tables S1–S3. In the “# Validated” columns of Table 1, we tabulate the number of green-highlighted validations + the number of yellow and salmon highlighted validations, followed by the sum of all three. By confirming the interpretations of other programs in such a variety of ways, we avoid favoring one cross-link interpretation program over another just because the details of its scoring algorithm might be similar to ours. In the first two datasets, most cross-links were directly validated and are highlighted in green, whereas in the third dataset, a larger fraction was indirectly validated as reflected in yellow and salmon highlighting in Tables S1–S3. The values in “% Validated” columns in Table 1 represent the percent of cross-links identified by each program that were validated by any of the methods outlined above. Venn diagrams in Figure 6 summarize numbers of cross-links identified by different programs at 5% FDR along with, in parentheses, the total numbers of validations listed in Table 1 and detailed in Tables S1–S3.
Table 1.
Comparison of Cross-link Data Interpretation Algorithms
| Kojak | MeroX | pLink | Xi | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| # Found | # Validateda | % Validatedb | # Found | # Validateda | % Validatedb | # Found | # Validateda | % Validatedb | # Found | # Validateda | % Validatedb | |
| 20S Proteasome | ||||||||||||
| 1% FDR | 201 | 178+9=187 | 93.0% | 152 | 148+3=151 | 99.3% | 209 | 198+9=207 | 99.0% | 173 | 164+9=173 | 100.0% |
| 5% FDR | 211 | 181+9=190 | 90.0% | 203 | 195+7=202 | 99.5% | 214 | 200+10=210 | 98.1% | 189 | 167+13=180 | 95.2% |
| 19S Proteasome | ||||||||||||
| 1% FDR | 273 | 254+7=261 | 95.6% | 243 | 234+3=237 | 97.5% | 332 | 310+12=322 | 97.0% | 283 | 270+9=279 | 98.6% |
| 5% FDR | 328 | 296+8=304 | 92.7% | 282 | 268+5=273 | 96.8% | 337 | 315+12=327 | 97.0% | 301 | 278+9=287 | 95.3% |
| 19S Proteasome + polyubiquitin | ||||||||||||
| 1% FDR | 391 | 265+105=370 | 94.6% | 206 | 158+44=202 | 98.1% | 482 | 357+123=480 | 99.6% | 288 | 216+70=286 | 99.3% |
| 5% FDR | 456 | 303+120=423 | 92.8% | 276 | 209+61=270 | 97.8% | 672 | 449+200=649 | 96.6% | 489 | 325+158=483 | 98.8% |
number of green-highlighted validations (see Supporting Information) + the number of yellow and salmon highlighted validations, followed by the sum of all three
calculated as # Validated/# Found
Figure 6.
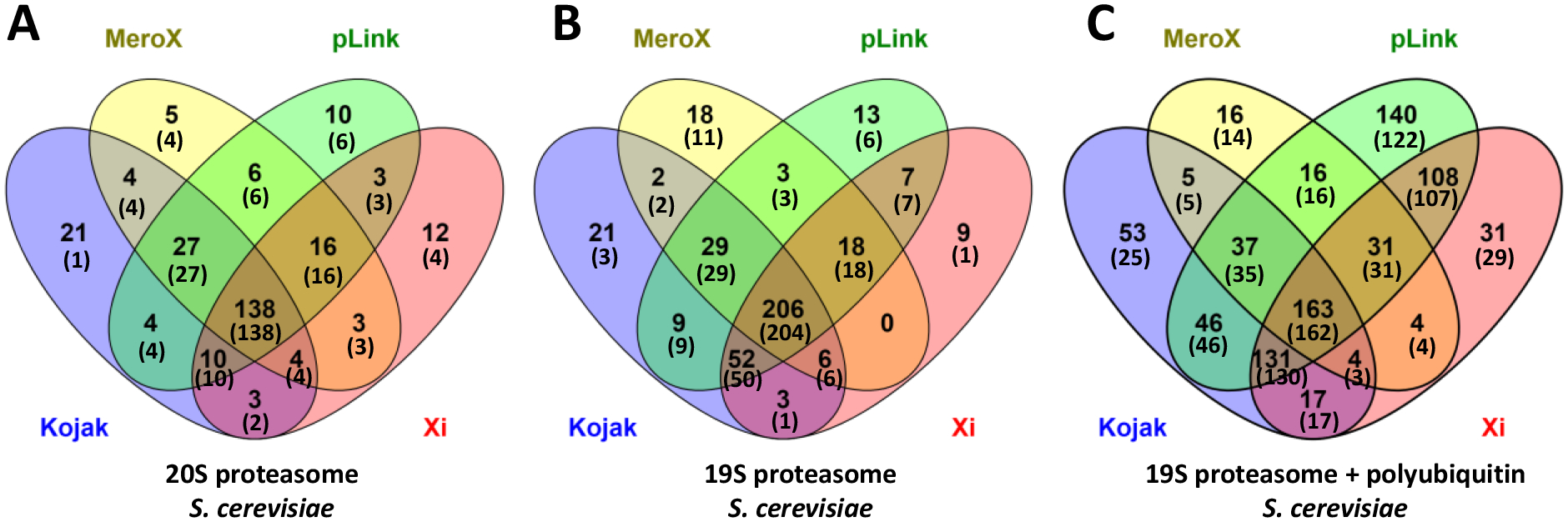
Venn diagrams showing the numbers of cross-link spectrum matches identified by the four publicly available algorithms at 5% FDR and the numbers (in parentheses) of these cross-links that were validated by our multiple ion fragmentation methods from the samples of (A) proteasome core particle, (B) proteasome regulatory particle, and (C) proteasome regulatory particle and polyubiquitin.
Despite the generally excellent agreement between the HCD interpretations of the four algorithms and our own interpretations of complementary ion fragmentation spectra, we were not able to confirm all of their identifications. In some cases, we found alternative and convincing interpretations that proved that identifications found by one of the programs were incorrect. In other cases, we were not able to derive better and convincing interpretations but the lack of consistency among our multiple ion fragmentation causes us to doubt their conclusions. The values in the “% Validated” column of Table 1 should be a reasonable representation of the percentage of true identifications found by each program at each designated FDR.
We found that Kojak, MeroX, pLink and Xi all work quite well and all of the reported false discovery rates were reasonable. Among the four programs, pLink always identified the most cross-links, especially when FDR was set at 1%. pLink yielded remarkably good results with the third dataset identifying the most cross-links with appropriately estimated FDRs. Kojak also identified large numbers of cross-links, but a smaller fraction of them could be validated. Nevertheless, based on our % Validation numbers, the FDRs may improve somewhat as the sizes of the datasets and proteomes increase. This may be due to the machine learning algorithm used by Kojak/Percolator. MeroX is overall the most accurate of the four programs based on the three datasets we tested. Their 5% FDR appears to be overestimated; it is probably better than this, which limited the total numbers that it identified. The quadratic mode of MeroX we tested is recommended for use with up to ten proteins in the proteome. For our simplest sample, the first dataset, at 5% FDR MeroX yielded comparable numbers of cross-links to pLink with the smallest number of unvalidated matches among all four programs. Xi obtained roughly comparable numbers of cross-links as Kojak with accurate FDR estimates. Our conclusions that pLink identifies the most cross-links and that FDR rates derived by the four algorithms are approximately correct are quite consistent with the results of a cross-linking study based on a synthetic peptide library.29 Mechtler and coworkers also reported that adding contaminants to increase the size of proteome could improve FDR estimates with some of the algorithms.29
Inspection of Figure 6 suggests that among all cross-links identified at 5% FDR, over 98% of those found by at least two of the four programs were validated. The very few that were not validated tend to be top matches from at least one ion fragmentation method as indicated by the rankings of these cross-links from each fragmentation method provided in Tables S1–S3. This suggests that they are still possible true matches. In contrast, only about 60% of the cross-links that were identified by only a single program were validated. Details showing the number of cross-link spectrum matches that were identified by only a single algorithm with 1% and 5% FDR along with the number of these that were validated are tabulated in Table S6 of Supporting Information. Note that a few cross-links assigned to 1% FDR were not validated. Most of the cross-links that could not be validated tended to not be among the top matches identified by our program for any fragmentation method, strongly suggesting that they are false. These results indicate that there would be a cooperative advantage of using Kojak, MeroX, pLink and Xi to analyze all data. Drawing conclusions from consistent interpretations should lead to improved confidence.
DISCUSSION
Our two approaches for combining data from four ion fragmentation methods have been shown to be effective at identifying cross-linked peptides and evaluating the validity of cross-links found by four publicly available algorithms. About 90% of all identifications were validated using multiple ion fragmentation methods, although more than half of the cross-link spectrum matches identified by one or more of the algorithms were not found by all four. For some of the 10% that were not validated, our approaches suggested better interpretations, and these are listed in Tables S1–S3. For the rest, no interpretation appears to be particularly credible.
A few limitations of our approaches have been encountered and improvements are under investigation. For example, with the first approach, some cross-links were not identified because ETD mass pairs did not appear in poor EThcD spectra, even though CID, HCD, and hcd led to credible cross-link identifications. Likewise, with the second approach, a correct hit might not even receive an Overall Score if it is not listed as one of the top ten interpretations of every fragmentation experiment. This happened most often with hcd. The use of reverse decoy databases is particularly problematic for hcd experiments. When protein sequences are simply reversed, target and decoy peptides have identical or nearly identical immonium and internal fragment ion masses. This is most likely the origin of the poor hcd performance displayed in Figure 5D. Use of randomized protein sequences as the decoy database may alleviate this issue. Calculating the Overall Score using only EThcD, CID and HCD results allowed about 10% more cross-link spectrum matches to be identified. The second approach might be improved by considering more than ten tentative interpretations from each method when deriving the best overall match. For more complicated samples, more top hits may need to be considered. Alternatively, instead of using hcd data to derive a fourth independent identification of a precursor ion, it might be better to use the immonium and small internal fragments found in hcd spectra to simply confirm identifications derived from EThcD, CID and HCD spectra. In other words, hcd data may not be informative enough to provide identification selectivity. In the future, additional approaches for combining multiple sets of MS2 spectra and interpretations, including machine learning, will be tested.
Spectrum quality significantly impacts the performance of all data interpretation algorithms. Spectrum quality appears to correlate with the number of peaks and the fraction of them that are in isotope clusters. For example, HCD spectra in our first and second sets of data contained on the order of 1000 peaks; more than 60% of features were in isotope clusters. All programs performed well and identified similar numbers of cross-links. However, in the third dataset, only about 100 peaks on average appeared in spectra and only about 40% were in isotope clusters. This may have been because of the overabundance of polyubiquitin in this sample. The lower information content in these spectra challenged the algorithms leading to less consistent results. pLink identified 100 and 200 more cross-link spectrum matches at 1% and 5% FDR respectively than other algorithms. Nevertheless, as displayed in Figure 6C, even with its high sensitivity, pLink overlooked about 100 cross-link spectrum matches that were found by the other three programs. Likewise, as shown in Table S3, pLink failed to recognize about 50 cross-link spectrum matches that we were able to identify by exploiting complementary EThcD, CID and hcd data. Similarly, our combined method also suffered from poor spectrum quality, as shown by the large second numbers in the “# Validated” columns of Table 1.
Besides mass spectrometer performance and precursor ion abundance, instrument settings associated with each fragmentation method can also affect spectrum quality. For instance, with a 21 to 39 ms ETD reaction time, the EThcD method has been reported to be effective at identifying highly charged and large cross-links.41 However, we used 100 ms ETD reaction time in acquiring our third set of data. In this case, we found that small precursors in +3 and +4 charge states yielded informative spectra leading to good identifications. However, large precursors in +5 to +7 charge states often yielded poor spectra with a very limited number of peaks that were not identified. Apparently, the long reaction time applied to ions that strongly attracted electrons neutralized the ion fragments and eventually yielded non-detectable species. ETD reaction duration has been suggested as a pivotal factor that can impact spectral quality.66 Varying the ETD reaction times based on precursor charge state and mass may be necessary to improve the quality of EThcD spectra. In summary, high-quality spectra are an underlying requirement to obtain satisfactory cross-linking results independent of which data analysis algorithm is employed.
Because of the time that it takes to execute multiple ion fragmentation experiments, fewer precursors can be selected for fragmentation and it is possible that fewer cross-links may be identified. It is natural to ask whether it is worthwhile to do this. In our experience, although some less abundant precursors were not fragmented when fewer precursors were selected, the most abundant precursors that were selected for fragmentation yielded highly informative MS2 spectra, leading to high identification probabilities. For instance, without enrichment and fractionation, we used a 60-min LC gradient to analyze about 1μg of tryptic digest prepared from a BS3 cross-linked proteasome sample. Among 2132 precursor ions selected for fragmentation, about 350 were identified as cross-links, leading to almost 250 unique cross-linked peptide pair identifications. This is a good rate of identification considering that cross-linked species are expected to be much less abundant than peptides.13, 67 Recording more spectra that are of lower quality is probably not advantageous.
Occasionally, even high-quality spectra from multiple ion fragmentation experiments lead to ambiguities in interpretation. In such cases, additional MS3 experiments that generate fragments associated with each cross-linked peptide may help to distinguish the most credible match from a few candidates. Such experiments can easily be executed on the distinctive mass pairs generated by fragmentation of DEST cross-links since each member of a mass pair is associated with one of the two peptides. Note that hcd fragmentation would be particularly attractive to employ in MS3 experiments; its high sensitivity, simple spectra and small ion fragments provide useful information about each peptide’s amino acid composition and its sensitivity to spectral corruption that was discussed above should not be a factor in MS3 experiments. A systematic assessment of how MS3 can be combined with multiple fragmentation MS2 experiments to identify cross-links will be performed in the future.
In the present work, we evaluated the performance of Kojak, MeroX, pLink, and Xi using only MS2 data in an HCD workflow. Some algorithms have additional unique functions that were not utilized in this study. For example, MeroX has RISE, RISEUP and Proteome-wide modes compatible with cleavable cross-links.44 pLink enables cross-linked peptide quantification from stable isotope-labeled datasets.47 The boost mode of XiFDR has demonstrated enhanced identification of credible cross-links at the residue pair level with 5% FDR.68
This study aimed to demonstrate that the complementary data observed with different ion fragmentation methods provide information that can improve cross-link peptide identifications and can validate identifications derived from a single fragmentation method. While our DEST cross-linker affords some unique ETD fragment pathways, the use of multiple ion fragmentation techniques should be applicable to other cross-linkers. Although in this work results from the interpretation of individual mass spectra were combined in two ways, in the future we will investigate alternative approaches in which we will first combine the data from multiple complementary spectra of the same precursor ion and then attempt to derive identifications. It is likely that other data interpretation algorithms would also benefit from using complementary fragment ion data generated by multiple ion fragmentation methods to achieve improved overall results.
Summary and Conclusions
Two approaches were introduced to improve cross-link identifications by combining the results from orthogonal EThcD, CID, HCD and hcd fragmentation experiments. In one approach, complete consistency among the top interpretations derived from four different MS2 spectra associated with each precursor ion led to our most confident identifications. This approach worked better with a cleavable cross-linker, DEST, since its ETD fragmentation yielded diagnostic ions that identified peptide masses. In a second approach, we derived the best overall match for every precursor ion by combining the results of the top ten tentative identifications found using each ion fragmentation method. In some cases, it appears to be advantageous to use only EThcD, CID and HCD data and not hcd when calculating Overall Scores. hcd spectra may be better used to confirm EThcD, CID and HCD results instead of being interpreted independently. Overall Scores were larger when there was spectral evidence supporting the identification of both peptides α and β. Because cross-link diagnostic ions were not required, this second approach could be applied to both cleavable and non-cleavable cross-linkers.
The complementary fragmentation information acquired from EThcD, CID and hcd enabled us to examine the validity of cross-link identifications that Kojak, MeroX, pLink and Xi arrived at using only HCD data. Nearly 90% of the cross-link spectrum matches identified by one or more programs were consistent with conclusions that we reached from interpreting multiple fragmentation experiments. We therefore consider these spectrum matches to be validated. In general, all programs worked very well. pLink identified the largest number of cross-links in the three datasets we tested. Our somewhat limited evidence suggests that Kojak may work better with large datasets recorded from complicated samples; MeroX (quadratic mode) may work better with smaller datasets acquired from simpler samples; pLink and Xi appear to work generally well with all types of data. More than 98% of cross-link spectrum matches identified by at least two of the four programs with 5% FDR were validated, suggesting that the use of all four of these algorithms to analyze datasets with subsequent harvesting of cross-links identified by at least two of these four programs could improve identification credibility.
Finally, spectrum quality plays an essential role in the identification of cross-links. The number of fragment peaks and percentage of peaks involved in isotope clusters are related to spectrum quality and impact the performance of data interpretation algorithms. High-quality spectra are an underlying requirement to obtain satisfactory cross-linking results independent of which data analysis algorithm is employed.
Supplementary Material
Acknowledgments
We would like to thank Dr. Jon Meek for the spectrum plotting program. This work was supported by the National Science Foundation grant CHE-1904749, the National Institutes of Health grant R01 GM135264-01 and the Indiana Core CTSI Pilot Grant Program.
Footnotes
Supporting Information
- Supporting Experimental Section, Examples Illustrating the Validation Process, Figure S1. Comparison between Composite Score and Overall Score distributions, Figure S2. Ambiguous cross-link identification of an HCD MS2 spectrum, Figure S3. Venn diagrams showing the overlap of cross-link spectrum matches identified by our methods and each of the four selected public algorithms, Table S4. Ion Types Considered by Our In-house Program, Table S5. Metrics Comparing Two Tentative Interpretations of One Cross-link, Table S6. Validity Evaluation of Unique Cross-Link Spectrum Matches Identified by Only One Algorithm (PDF)
- Table S1. BS3 Cross-links Identified from S. cerevisiae Proteasome Core Particle, Table S2. BS3 Cross-links Identified from S. cerevisiae Proteasome Regulatory Particle, Table S3. DEST Cross-links Identified from S. cerevisiae Proteasome Regulatory Particle and Polyubiquitin (XLSX)
REFERENCES
- 1.Acharya KR; Lloyd MD The advantages and limitations of protein crystal structures. Trends Pharmacol. Sci 2005, 26, 10–14. [DOI] [PubMed] [Google Scholar]
- 2.Ardenkjaer-Larsen JH; Boebinger GS; Comment A; Duckett S; Edison AS; Engelke F; Griesinger C; Griffin RG; Hilty C; Maeda H; Parigi G; Prisner T; Ravera E; van Bentum J; Vega S; Webb A; Luchinat C; Schwalbe H; Frydman L Facing and Overcoming Sensitivity Challenges in Biomolecular NMR Spectroscopy. Angew. Chem. Int. Ed 2015, 54, 9162–9185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Elipe MVS Advantages and disadvantages of nuclear magnetic resonance spectroscopy as a hyphenated technique. Anal. Chim. Acta 2003, 497, 1–25. [Google Scholar]
- 4.Davis AM; Teague SJ; Kleywegt GJ Application and limitations of X-ray crystallographic data in structure-based ligand and drug design. Angew. Chem. Int. Ed 2003, 42, 2718–2736. [DOI] [PubMed] [Google Scholar]
- 5.Russell RB; Alber F; Aloy P; Davis FP; Korkin D; Pichaud M; Topf M; Sali A A structural perspective on protein-protein interactions. Curr. Opin. Struct. Biol 2004, 14, 313–324. [DOI] [PubMed] [Google Scholar]
- 6.Oldfield CJ; Dunker AK Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Annu. Rev. Biochem 2014, 83, 553–584. [DOI] [PubMed] [Google Scholar]
- 7.Bai XC; McMullan G; Scheres SHW How cryo-EM is revolutionizing structural biology. Trends Biochem. Sci 2015, 40, 49–57. [DOI] [PubMed] [Google Scholar]
- 8.Sinz A Chemical cross-linking and mass spectrometry to map three-dimensional protein structures and protein-protein interactions. Mass Spectrom. Rev 2006, 25, 663–682. [DOI] [PubMed] [Google Scholar]
- 9.Petrotchenko EV; Borchers CH Crosslinking Combined with Mass Spectrometry for Structural Proteomics. Mass Spectrom. Rev 2010, 29, 862–876. [DOI] [PubMed] [Google Scholar]
- 10.Singh P; Panchaud A; Goodlett DR Chemical cross-linking and mass spectrometry as a low-resolution protein structure determination technique. Anal. Chem 2010, 82, 2636–2642. [DOI] [PubMed] [Google Scholar]
- 11.Rappsilber J The beginning of a beautiful friendship: cross-linking/mass spectrometry and modelling of proteins and multi-protein complexes. J. Struct. Biol 2011, 173, 530–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miteva YV; Budayeva HG; Cristea IM Proteomics-based methods for discovery, quantification, and validation of protein-protein interactions. Anal. Chem 2013, 85, 749–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Merkley ED; Cort JR; Adkins JN Cross-linking and mass spectrometry methodologies to facilitate structural biology: finding a path through the maze. J. Struct. Funct. Genomics 2013, 14, 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trnka MJ; Baker PR; Robinson PJ; Burlingame AL; Chalkley RJ Matching cross-linked peptide spectra: only as good as the worse identification. Mol. Cell. Proteomics 2014, 13, 420–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iacobucci C; Sinz A To Be or Not to Be? Five Guidelines to Avoid Misassignments in Cross-Linking/Mass Spectrometry. Anal. Chem 2017, 89, 7832–7835. [DOI] [PubMed] [Google Scholar]
- 16.Petrotchenko EV; Olkhovik VK; Borchers CH Isotopically coded cleavable cross-linker for studying protein-protein interaction and protein complexes. Mol. Cell. Proteomics 2005, 4, 1167–1179. [DOI] [PubMed] [Google Scholar]
- 17.Muller MQ; Dreiocker F; Ihling CH; Schafer M; Sinz A Cleavable cross-linker for protein structure analysis: reliable identification of cross-linking products by tandem MS. Anal. Chem 2010, 82, 6958–6968. [DOI] [PubMed] [Google Scholar]
- 18.Petrotchenko EV; Serpa JJ; Borchers CH An isotopically coded CID-cleavable biotinylated cross-linker for structural proteomics. Mol. Cell. Proteomics 2011, 10, M110 001420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaake RM; Wang X; Burke A; Yu C; Kandur W; Yang Y; Novtisky EJ; Second T; Duan J; Kao A; Guan S; Vellucci D; Rychnovsky SD; Huang L A new in vivo cross-linking mass spectrometry platform to define protein-protein interactions in living cells. Mol. Cell. Proteomics 2014, 13, 3533–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu F; Rijkers DT; Post H; Heck AJ Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat. Methods 2015, 12, 1179–1184. [DOI] [PubMed] [Google Scholar]
- 21.Chakrabarty JK; Naik AG; Fessler MB; Munske GR; Chowdhury SM Differential Tandem Mass Spectrometry-Based Cross-Linker: A New Approach for High Confidence in Identifying Protein Cross-Linking. Anal. Chem 2016, 88, 10215–10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kamal AM; Aloor JJ; Fessler MB; Chowdhury SM Cross-linking Proteomics Indicates Effects of Simvastatin on the TLR2 Interactome and Reveals ACTR1A as a Novel Regulator of the TLR2 Signal Cascade. Mol. Cell. Proteomics 2019, 18, 1732–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chakrabarty JK; Sadananda SC; Bhat A; Naik AJ; Ostwal DV; Chowdhury SM High Confidence Identification of Cross-Linked Peptides by an Enrichment-Based Dual Cleavable Cross-Linking Technology and Data Analysis tool Cleave-XL. J. Am. Soc. Mass Spectrom 2020, 31, 173–182. [DOI] [PubMed] [Google Scholar]
- 24.Cox J; Hubner NC; Mann M How Much Peptide Sequence Information Is Contained in Ion Trap Tandem Mass Spectra? J. Am. Soc. Mass Spectrom 2008, 19, 1813–1820. [DOI] [PubMed] [Google Scholar]
- 25.Deutsch EW; Lam H; Aebersold R Data analysis and bioinformatics tools for tandem mass spectrometry in proteomics. Physiol. Genomics 2008, 33, 18–25. [DOI] [PubMed] [Google Scholar]
- 26.Liu F; Lossl P; Scheltema R; Viner R; Heck AJR Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification. Nat. Commun 2017, 8, 15473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iacobucci C; Piotrowski C; Aebersold R; Amaral BC; Andrews P; Bernfur K; Borchers C; Brodie NI; Bruce JE; Cao Y; Chaignepain S; Chavez JD; Claverol S; Cox J; Davis T; Degliesposti G; Dong MQ; Edinger N; Emanuelsson C; Gay M; Gotze M; Gomes-Neto F; Gozzo FC; Gutierrez C; Haupt C; Heck AJR; Herzog F; Huang L; Hoopmann MR; Kalisman N; Klykov O; Kukacka Z; Liu F; MacCoss MJ; Mechtler K; Mesika R; Moritz RL; Nagaraj N; Nesati V; Neves-Ferreira AGC; Ninnis R; Novak P; O’Reilly FJ; Pelzing M; Petrotchenko E; Piersimoni L; Plasencia M; Pukala T; Rand KD; Rappsilber J; Reichmann D; Sailer C; Sarnowski CP; Scheltema RA; Schmidt C; Schriemer DC; Shi Y; Skehel JM; Slavin M; Sobott F; Solis-Mezarino V; Stephanowitz H; Stengel F; Stieger CE; Trabjerg E; Trnka M; Vilaseca M; Viner R; Xiang Y; Yilmaz S; Zelter A; Ziemianowicz D; Leitner A; Sinz A First Community-Wide, Comparative Cross-Linking Mass Spectrometry Study. Anal. Chem 2019, 91, 6953–6961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bruce JE In vivo protein complex topologies: sights through a cross-linking lens. Proteomics 2012, 12, 1565–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beveridge R; Stadlmann J; Penninger JM; Mechtler K A synthetic peptide library for benchmarking crosslinking-mass spectrometry search engines for proteins and protein complexes. Nat. Commun 2020, 11, 742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nielsen ML; Savitski MM; Zubarev RA Improving protein identification using complementary fragmentation techniques in Fourier transform mass spectrometry. Mol. Cell. Proteomics 2005, 4, 835–845. [DOI] [PubMed] [Google Scholar]
- 31.Wu SL; Huhmer AF; Hao Z; Karger BL On-line LC-MS approach combining collision-induced dissociation (CID), electron-transfer dissociation (ETD), and CID of an isolated charge-reduced species for the trace-level characterization of proteins with post-translational modifications. J. Proteome Res 2007, 6, 4230–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zubarev RA; Zubarev AR; Savitski MM Electron capture/transfer versus collisionally activated/induced dissociations: solo or duet? J. Am. Soc. Mass Spectrom 2008, 19, 753–761. [DOI] [PubMed] [Google Scholar]
- 33.Kim S; Mischerikow N; Bandeira N; Navarro JD; Wich L; Mohammed S; Heck AJ; Pevzner PA The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Mol. Cell. Proteomics 2010, 9, 2840–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frese CK; Altelaar AF; van den Toorn H; Nolting D; Griep-Raming J; Heck AJ; Mohammed S Toward full peptide sequence coverage by dual fragmentation combining electron-transfer and higher-energy collision dissociation tandem mass spectrometry. Anal. Chem 2012, 84, 9668–9673. [DOI] [PubMed] [Google Scholar]
- 35.Hernandez-Alba O; Houel S; Hessmann S; Erb S; Rabuka D; Huguet R; Josephs J; Beck A; Drake PM; Cianferani S A Case Study to Identify the Drug Conjugation Site of a Site-Specific Antibody-Drug-Conjugate Using Middle-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom 2019, 30, 2419–2429. [DOI] [PubMed] [Google Scholar]
- 36.Gomes FP; Diedrich JK; Saviola AJ; Memili E; Moura AA; Yates JR 3rd EThcD and 213 nm UVPD for Top-Down Analysis of Bovine Seminal Plasma Proteoforms on Electrophoretic and Chromatographic Time Frames. Anal. Chem 2020, 92, 2979–2987. [DOI] [PubMed] [Google Scholar]
- 37.Lauber MA; Reilly JP Novel amidinating cross-linker for facilitating analyses of protein structures and interactions. Anal. Chem 2010, 82, 7736–7743. [DOI] [PubMed] [Google Scholar]
- 38.Lauber MA; Rappsilber J; Reilly JP Dynamics of ribosomal protein S1 on a bacterial ribosome with cross-linking and mass spectrometry. Mol. Cell. Proteomics 2012, 11, 1965–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lauber MA; Reilly JP Structural analysis of a prokaryotic ribosome using a novel amidinating cross-linker and mass spectrometry. J. Proteome Res 2011, 10, 3604–3616. [DOI] [PubMed] [Google Scholar]
- 40.Zhao B; Reilly CP; Reilly JP ETD-Cleavable Linker for Confident Cross-linked Peptide Identifications. J. Am. Soc. Mass Spectrom 2019, 30, 1631–1642. [DOI] [PubMed] [Google Scholar]
- 41.Kolbowski L; Mendes ML; Rappsilber J Optimizing the Parameters Governing the Fragmentation of Cross-Linked Peptides in a Tribrid Mass Spectrometer. Anal. Chem 2017, 89, 5311–5318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hoopmann MR; Zelter A; Johnson RS; Riffle M; MacCoss MJ; Davis TN; Moritz RL Kojak: Efficient Analysis of Chemically Cross-Linked Protein Complexes. J. Proteome Res 2015, 14, 2190–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gotze M; Pettelkau J; Schaks S; Bosse K; Ihling CH; Krauth F; Fritzsche R; Kuhn U; Sinz A StavroX--a software for analyzing crosslinked products in protein interaction studies. J. Am. Soc. Mass Spectrom 2012, 23, 76–87. [DOI] [PubMed] [Google Scholar]
- 44.Gotze M; Pettelkau J; Fritzsche R; Ihling CH; Schafer M; Sinz A Automated assignment of MS/MS cleavable cross-links in protein 3D-structure analysis. J. Am. Soc. Mass Spectrom 2015, 26, 83–97. [DOI] [PubMed] [Google Scholar]
- 45.Yang B; Wu YJ; Zhu M; Fan SB; Lin JZ; Zhang K; Li S; Chi H; Li YX; Chen HF; Luo SK; Ding YH; Wang LH; Hao ZQ; Xiu LY; Chen S; Ye KQ; He SM; Dong MQ Identification of cross-linked peptides from complex samples. Nat. Methods 2012, 9, 904–906. [DOI] [PubMed] [Google Scholar]
- 46.Lu S; Fan SB; Yang B; Li YX; Meng JM; Wu L; Li P; Zhang K; Zhang MJ; Fu Y; Luo JC; Sun RX; He SM; Dong MQ Mapping native disulfide bonds at a proteome scale. Nat. Methods 2015, 12, 329–311. [DOI] [PubMed] [Google Scholar]
- 47.Chen ZL; Meng JM; Cao Y; Yin JL; Fang RQ; Fan SB; Liu C; Zeng WF; Ding YH; Tan D; Wu L; Zhou WJ; Chi H; Sun RX; Dong MQ; He SM A high-speed search engine pLink 2 with systematic evaluation for proteome-scale identification of cross-linked peptides. Nat. Commun 2019, 10, 3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Giese SH; Fischer L; Rappsilber J A Study into the Collision-induced Dissociation (CID) Behavior of Cross-Linked Peptides. Mol. Cell. Proteomics 2016, 15, 1094–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mendes ML; Fischer L; Chen ZA; Barbon M; O’Reilly FJ; Giese SH; Bohlke-Schneider M; Belsom A; Dau T; Combe CW; Graham M; Eisele MR; Baumeister W; Speck C; Rappsilber J An integrated workflow for crosslinking mass spectrometry. Mol. Syst. Biol 2019, 15, e8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gautam AKS; Martinez-Fonts K; Matouschek A, Scalable In Vitro Proteasome Activity Assay. 2018; Vol. 1844, p 321–341. [DOI] [PubMed] [Google Scholar]
- 51.Martinez-Fonts K; Matouschek A A Rapid and Versatile Method for Generating Proteins with Defined Ubiquitin Chains. Biochemistry 2016, 55, 1898–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vizcaino JA; Cote RG; Csordas A; Dianes JA; Fabregat A; Foster JM; Griss J; Alpi E; Birim M; Contell J; O’Kelly G; Schoenegger A; Ovelleiro D; Perez-Riverol Y; Reisinger F; Rios D; Wang R; Hermjakob H The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2013, 41, D1063–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brosch M; Yu L; Hubbard T; Choudhary J Accurate and sensitive peptide identification with Mascot Percolator. J. Proteome Res 2009, 8, 3176–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oliveros JC Venny. An interactive tool for comparing lists with Venn’s diagrams.
- 55.Walzthoeni T; Claassen M; Leitner A; Herzog F; Bohn S; Forster F; Beck M; Aebersold R False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nat. Methods 2012, 9, 901–903. [DOI] [PubMed] [Google Scholar]
- 56.Breci LA; Tabb DL; Yates JR; Wysocki VH Cleavage N-terminal to proline: analysis of a database of peptide tandem mass spectra. Anal. Chem 2003, 75, 1963–1971. [DOI] [PubMed] [Google Scholar]
- 57.Wysocki VH; Tsaprailis G; Smith LL; Breci LA Mobile and localized protons: a framework for understanding peptide dissociation. J. Mass Spectrom 2000, 35, 1399–1406. [DOI] [PubMed] [Google Scholar]
- 58.Gu C; Tsaprailis G; Breci L; Wysocki VH Selective gas-phase cleavage at the peptide bond C-terminal to aspartic acid in fixed-charge derivatives of Asp-containing peptides. Anal. Chem 2000, 72, 5804–5813. [DOI] [PubMed] [Google Scholar]
- 59.Paizs B; Suhai S Fragmentation pathways of protonated peptides. Mass Spectrom. Rev 2005, 24, 508–548. [DOI] [PubMed] [Google Scholar]
- 60.Medzihradszky KF; Chalkley RJ Lessons in de novo peptide sequencing by tandem mass spectrometry. Mass Spectrom. Rev 2015, 34, 43–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chi H; Sun RX; Yang B; Song CQ; Wang LH; Liu C; Fu Y; Yuan ZF; Wang HP; He SM; Dong MQ pNovo: De novo Peptide Sequencing and Identification Using HCD Spectra. J. Proteome Res 2010, 9, 2713–2724. [DOI] [PubMed] [Google Scholar]
- 62.Hohmann LJ; Eng JK; Gemmill A; Klimek J; Vitek O; Reid GE; Martin DB Quantification of the compositional information provided by immonium ions on a quadrupole-time-of-flight mass spectrometer. Anal. Chem 2008, 80, 5596–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Y; Li SM; He MW Fragmentation Characteristics and Utility of Immonium Ions for Peptide Identification by MALDI-TOF/TOF-Mass Spectrometry. Chinese J. Anal. Chem 2014, 42, 1010–1016. [Google Scholar]
- 64.DeGraan-Weber N; Ashley DC; Keijzer K; Baik MH; Reilly JP Factors Affecting the Production of Aromatic Immonium Ions in MALDI 157 nm Photodissociation Studies. J. Am. Soc. Mass Spectrom 2016, 27, 834–846. [DOI] [PubMed] [Google Scholar]
- 65.Unverdorben P; Beck F; Sledz P; Schweitzer A; Pfeifer G; Plitzko JM; Baumeister W; Forster F Deep classification of a large cryo-EM dataset defines the conformational landscape of the 26S proteasome. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 5544–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rose CM; Rush MJ; Riley NM; Merrill AE; Kwiecien NW; Holden DD; Mullen C; Westphall MS; Coon JJ A calibration routine for efficient ETD in large-scale proteomics. J. Am. Soc. Mass Spectrom 2015, 26, 1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Steigenberger B; Pieters RJ; Heck AJR; Scheltema RA PhoX: An IMAC-Enrichable Cross-Linking Reagent. ACS Cent. Sci 2019, 5, 1514–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fischer L; Rappsilber J Quirks of Error Estimation in Cross-Linking/Mass Spectrometry. Anal. Chem 2017, 89, 3829–3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.