

Abstract
Neonatal CD4+ and CD8+ T cells have historically been characterized as immature or defective. However, recent studies prompt a reinterpretation of the functions of neonatal T cells. Rather than a population of cells always falling short of expectations set by their adult counterparts, neonatal T cells are gaining recognition as a distinct population of lymphocytes well suited for the rapidly changing environment in early life. In this review, I will highlight new evidence indicating that neonatal T cells are not inert or less potent versions of adult T cells but instead are a broadly reactive layer of T cells poised to quickly develop into regulatory or effector cells, depending on the needs of the host. In this way, neonatal T cells are well adapted to provide fast-acting immune protection against foreign pathogens, while also sustaining tolerance to self-antigens.
Keywords: neonate, immune development, immunological memory, adaptive immunity, CD4+ helper T cells, CD8+ cytotoxic T cells
INTRODUCTION
Immunologists have long been fascinated by the differences between CD4+ and CD8+ T cell functions in early life compared to adulthood. Seminal studies by Sir Peter Medawar’s group in the 1950s demonstrated that fetal exposure to antigen leads to the inactivation or absence of T cells capable of mounting an immune response (1). Since this phenomenon, referred to as neonatal tolerance (2), was only observed when antigen was introduced before or during the neonatal period of life, neonatal T cells were considered uniquely susceptible to becoming unresponsive or tolerant to specific antigens. In the 1980s and early 1990s, in vitro experiments further confirmed the view that neonatal T cells were immunodeficient, or simply immature versions of their adult counterparts, based on their reduced ability to produce IL-2 and IFN-γ following activation.
Studies performed in the late 1990s proposed that CD4+ T cells in neonates were not, in fact, impaired at responding to stimulation. Instead, they preferentially made T helper type 2 (Th2) cytokines (IL-4, IL-5, IL-13), which were not measured in previous studies (3–5). These findings led to the idea that neonatal T cells were not immunodeficient but rather an immunodeviant version of their adult counterparts. This theory better explained why individuals were more susceptible to both infection and allergies during early stages of development (6). Also, a Th2-biased immune system in early life made teleological sense, since excessive Th1 inflammation can be detrimental to the developing fetus (7). For these reasons, the immunodeviant theory was and continues to be the widely accepted paradigm in the field, with most reviews on neonatal immunity devoting a significant amount of time to discussing the cellular and molecular studies that support it.
However, accumulating evidence suggests we are in the midst of another paradigm shift in our understanding of T cell functions in the neonatal period. A new model, supported by work from numerous labs, proposes that neonatal T cells are not simply immature versions of adult T cells with blunted or deviant responses but rather a distinct population of T cells with unique functional properties well suited to perinatal life (8–12). This significant departure from the current dogma is supported by experiments showing that neonatal and adult T cells are derived from separate progenitors and exhibit distinct gene expression profiles even prior to stimulation (8–10). In fact, neonatal T cells possess a transcriptome/epigenetic landscape that makes them more responsive to instructive cues after activation (9, 10, 13–15). As a result, neonatal T cells appear to be poised to establish a state of tolerance to self and allogenic antigens under nonthreatening conditions. However, in the presence of life-threatening infections, this poising allows neonatal T cells to rapidly mount an effector response, albeit at the expense of forming memory, to keep the host alive.
The central question of this review is whether existing evidence supports an entirely new model whereby neonatal CD4+ and CD8+ T cells are neither defective nor deficient but rather uniquely suited to the purpose of protecting the host in early life. Here, I highlight the growing evidence suggesting that neonatal T cells are a distinct population of lymphocytes programmed differently than adult T cells, attempting to reconcile the differing and sometimes conflicting studies of neonatal T cell function, as well as put the new developments into historical perspective to provide a more complete picture of the biology of neonatal T cells.
NEONATAL T CELLS ARE DERIVED FROM DISTINCT PROGENITORS
To understand the biology of neonatal T cells, it is important to first trace their developmental pathway and consider their position in the broad architecture of immune development (see the sidebar titled When Is a Mouse Neonatal?). Previous work has demonstrated that the ontogeny of the immune system does not progress in a linear manner from fetal life to adulthood. Rather, the immune system is stratified into layers of distinct immune cells that develop sequentially from distinct waves of hematopoietic stem cells (HSCs) (16–19). For many years, this model, referred to as the layered immune system model (20), was only applied to different lineages of murine γδ T cells (18, 19) and B cells (16, 17), which are functionally distinct and arise in succession. CD4+ and CD8+ T cells are also derived from fetal liver and adult bone marrow HSCs (21–24), but they have historically been viewed as single lineages of lymphocytes that mature only after stimulation with foreign antigen. In the last 5–10 years, however, a number of groups have found compelling evidence (in mice and humans) to extend the layered immune system model to CD4+ and CD8+ T cells (8, 9, 25, 26) (Figure 1). These studies have raised the provocative idea that neonatal T cells represent a distinct lineage of cells hiding in plain sight.
Figure 1.
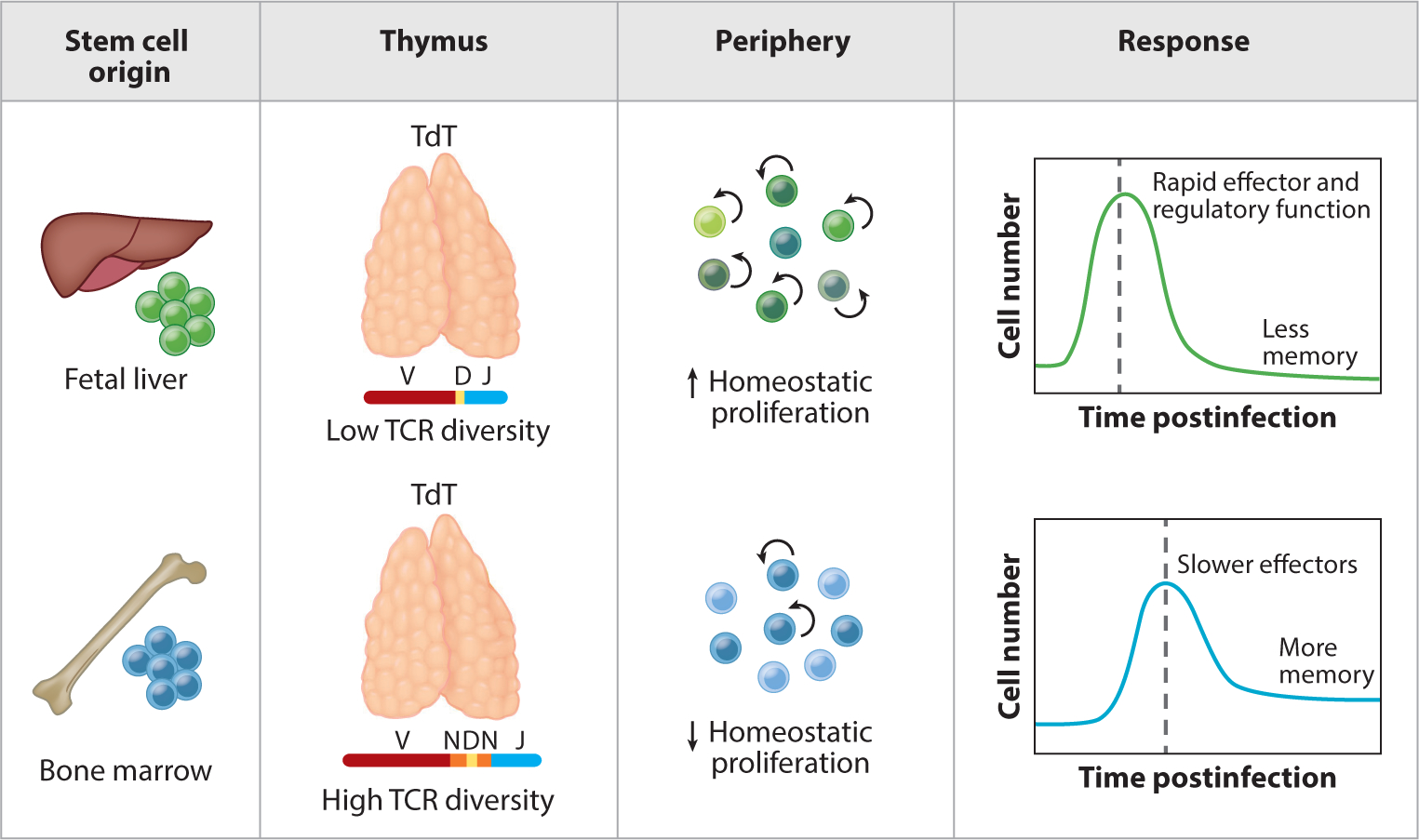
Neonatal and adult T cells have different origins and functions. This figure depicts the layered immune system model for CD4+ and CD8+ T cells. Unlike adult T cells, neonatal T cells are derived from fetal hematopoietic stem cells, exhibit shorter and more restricted T cell receptors in the absence of TdT, and undergo higher rates of homeostatic proliferation in the periphery. Following stimulation, neonatal T cells more quickly differentiate into effector or regulatory T cells than their adult counterparts, albeit at the expense of forming long-lived memory cells. Abbreviation: TCR, T cell receptor.
The first evidence for the layered immune system model came from a seminal study done in humans by the McCune group (8). They showed that in the human CD4+ T cell compartment, fetal-derived CD4+ T cells proliferate more rapidly than adult-derived CD4+ T cells and preferentially become regulatory T cells (Tregs). This was demonstrated using an elegant humanized mouse model, where fetal and adult stem and progenitor cells (HSPCs) were injected into SCID-hu mice following stimulation with alloantigen in vitro. The authors observed a distinct transcriptome in fetal Tregs compared to their adult counterparts, suggesting that these cells are made differently in early life. More recently, studies in neonatal mice have demonstrated the propensity for CD4+ T cells to exhibit rapid proliferation and differentiation in Tregs after T cell receptor (TCR) stimulation (11). As for other lineages of CD4+ T cells, Adkins performed fetal thymic transplant experiments and found that fetal-derived CD4+ T cells preferentially made Th2 cytokines when stimulated with low amounts of antigen (26), which recapitulated previous observations made with CD4+ T cells from humans (27, 28). Collectively, these studies have provided a basis for why CD4+ T cells behave differently in early life and suggest that the Treg and Th2 paths are the default tracks of differentiation for neonatal CD4+ T cells.
There is also evidence for the layered immune system model in CD8+ T cells. Recently, Wang et al. (25) suggested that neonatal CD8+ T cells represent a distinct lineage of cells, as they exhibit a distinct gene expression profile from the time they are created in the thymus, and they maintain this difference even after being exported into the periphery (13, 29). Other compelling evidence for the existence of developmental layers in the CD8+ T cell compartment was obtained by comparing the progeny of fetal and adult precursors (25). In these studies, fetal and adult progenitors were injected into an adult thymus to generate populations of CD8+ T cells that developed in the same thymic and peripheral environment. Here, the fetal-derived CD8+ T cells exhibited an enhanced capacity to proliferate and gave rise to more short-lived effectors after infection, whereas the adult-derived CD8+ T cells responded with slower kinetics but gave rise to more memory CD8+ T cells. Consistent with results observed in mice, purified subsets of naive CD8+ T cells from newborn humans have also been shown to divide sooner than those from adults after in vitro stimulation (30).
The enhanced capacity of neonatal CD4+ and CD8+ T cells (both human and mouse) to divide after stimulation is reminiscent of other immune cells derived from fetal HSCs, such as B1 B cells. However, a question remains: Why would the immune system retain fetal HSCs during evolution to provide the host with both CD4+ T cells with a bias toward becoming Tregs/Th2 cells and CD8+ T cells with an inherent propensity for generating effector cytotoxic T lymphocytes? One way to reconcile these seemingly dichotomous findings is to consider the differences in experimental design of the CD4+ and CD8+ T cell studies. In the case of CD4+ T cells, the readout for their behavior has typically been examined under noninflammatory conditions (e.g., anti-CD3/CD28, alloantigens), whereas most of the CD8+ T cell work has been performed in mouse models of infections with pathogenic microbes (e.g., vaccinia virus, Listeria monocytogenes). Thus, it is possible that the different fates displayed by neonatal CD4+ and CD8+ T cells have less to do with cell type and more to do with the different conditions under which they were stimulated. Indeed, addition of IL-12 to in vitro cultures enabled neonatal CD4+ T cells to produce adult-like levels of IFN-γ (31), and vaccination of neonates with the more inflammatory BCG and whole-cell pertussis vaccines resulted in adult-like Th1 responses (32, 33). Moreover, neonatal CD8+ T cells differentiate into Tregs when exposed to self-antigens in the periphery (34). Thus, it is possible that the immune system has retained fetal HSCs to generate a layer of fast-acting CD4+ and CD8+ T cells that can more quickly differentiate into the cells most useful to the host, depending on the circumstances in which they were primed.
NEONATAL T CELLS PERSIST INTO ADULTHOOD AND RETAIN THEIR CELL-INTRINSIC PROPERTIES
An important technological advancement that has changed our perception of neonatal T cells is the development of fate-mapping mouse models. In the past, the study of neonatal T cells was limited to how they respond in early life. However, we now have the ability to permanently label CD4+ and CD8+ T cells produced in neonatal mice and examine their behavior in adulthood. Studies using fate-mapping models demonstrate that the enhanced ability of neonatal T cells to rapidly differentiate into effector and regulatory cells are of significant value to the host later in life. Thus, rather than dispensing of neonatal T cells altogether, the immune system maintains this developmental layer in adulthood to serve as early effectors against foreign pathogens and sustain tolerance to self-antigens.
In the case of CD4+ T cells, Yang et al. (35) used a fate-mapping model involving Foxp3-driven tamoxifen-inducible Cre (Foxp3cre-ERT2) mice to label a wave of Tregs produced at either 0–10 days of age or 35–45 days of age. They found that Tregs produced in early life are stably maintained in adulthood and exhibit a unique transcriptome. Interestingly, the genes upregulated in neonatal-derived Tregs were associated with Treg function and cell division, consistent with their more potent suppressive activity and enhanced ability to proliferate. Moreover, adoptive transfer of neonatal-derived Tregs, but not adult-derived Tregs, into newborn Aire−/− mice inhibited the progression of multi-organ autoimmune disease. Therefore, Tregs produced during a specific ontogenic window in early life have unique functions and are required for maintaining self-tolerance. Although more work is required to translate such findings to humans, Mold et al. (36) found evidence to suggest that human Tregs made against maternal antigens in utero persist into at least the teenage years.
For neonatal CD8+ T cells, Smith et al. developed a strategy using mice with CD4-driven tamoxifen-inducible Cre (CD4cre-ERT2) to induce expression of TdTomato and permanently label or time-stamp a wave of CD8+ T cells made in the thymus at the time of tamoxifen exposure (9). Using this approach, they found that CD8+ T cells made near birth persist into adulthood and express higher levels of effector genes prior to stimulation, whereas CD8+ T cells made later in life were enriched for genes found in naive cells. In vitro studies also demonstrated that neonatal-derived CD8+ T cells retained their enhanced capacity to proliferate after TCR stimulation and underwent bystander activation in response to innate cytokines (IL-12 and IL-18). Following infection, neonatal CD8+ T cells still gave rise to terminally differentiated effectors, but CD8+ T cells made later in life preferentially gave rise to long-lived memory cells. In fact, the first cells made during early stages of development were the first cells to respond to infection and become effectors in adulthood. These studies, along with recent work done by Reynaldi et al. (37), suggest that age-related differences in the CD8+ T cell response to infection may be linked to the developmental composition of cells in the starting population. Collectively, these data demonstrate that neonatal CD4+ and CD8+ T cells are maintained in adulthood as a distinct developmental layer and have important roles in mediating immune homeostasis and protection (see sidebar titled Do Neonatal T Cells Represent a Distinct Lineage of Lymphocytes?).
NEONATAL T CELLS ARE POISED FOR RAPID DIFFERENTIATION
Another major factor that has changed our perception of neonatal T cells is the rapid growth of next-generation sequencing. In the past, immunologists tested hypotheses based upon their experience, instincts, and published data, focusing on single genes (e.g., Nfat) at a time. Now, with the advent of new technology, we have the ability to examine the entire transcriptome in equivalent populations of neonatal and adult T cells in an unbiased manner. Using this new technology, recent studies have shown that neonatal cells are poised for rapid differentiation (9, 10, 13–15, 25). For example, neonatal murine CD8+ T cells have been shown to express more effector-like genes during thymic development (25), suggesting they follow a developmental trajectory similar to innate-like lymphocytes, such as natural killer (NK) cells, mucosa-associated invariant T (MAIT) cells, and γδ T cells, which often do not exist in a classical naive state (46). Naive neonatal CD8+ T cells in the periphery exhibit a more effector-like chromatin landscape, with increased accessibility to genes that favor effector cell differentiation (9). They are essentially hardwired for rapid proliferation and differentiation. In contrast, adult T cells express genes typically associated with naive T cells (9, 25). This naive state in adult T cells is likely important for internalizing contextual information from antigen-presenting cells in the lymph node and may leave open more differentiation pathways for them to travel down. However, increased plasticity comes at the cost of time, which is a luxury that likely cannot be afforded in early life.
Mechanistically, recent work suggests that naive CD4+ and CD8+ T cells are transcriptionally shifted to a more effector-like state in early life because of developmental differences in miRNA expression (13, 14) (Figure 2). The miRNAs that are preferentially expressed in adult T cells may allow them to exist in a naive state by actively suppressing genes involved in differentiation. For example, in adult CD8+ T cells (both mice and humans), miR-29 is enriched and represses drivers of effector cell differentiation (e.g., T-bet, Eomes) (13, 14). By contrast, in neonatal T cells, lower expression of miR-29 positions them further along the effector cell differentiation continuum and potentially explains their inherent bias toward the short-lived effector lineage after infection. Another miRNA that is upregulated in adult T cells and helps to maintain their naive phenotype is let-7 (14, 25, 47). The let-7 family of miRNAs is downregulated in fetal-derived T cells because its expression is blocked by a developmentally regulated RNA-binding protein (Lin28b) in fetal HSCs (25, 44, 48), creating a metabolic profile highly conducive to rapid growth and proliferation (47, 49, 50). A number of papers have suggested that Lin28b may be responsible for the developmental switch between neonatal and adult T cell functions. Overexpression of Lin28b in adult murine CD8+ T cells, for example, results in reduced levels of let-7 expression and increased terminal differentiation after infection, similar to neonatal CD8+ T cells (25). Likewise, inhibiting Lin28b in human fetal CD4+ T cells leads to the upregulation of let-7 and a reduced ability to differentiate into FOXP3+CD25+ Tregs, akin to adult CD4+ T cells (51).
Figure 2.
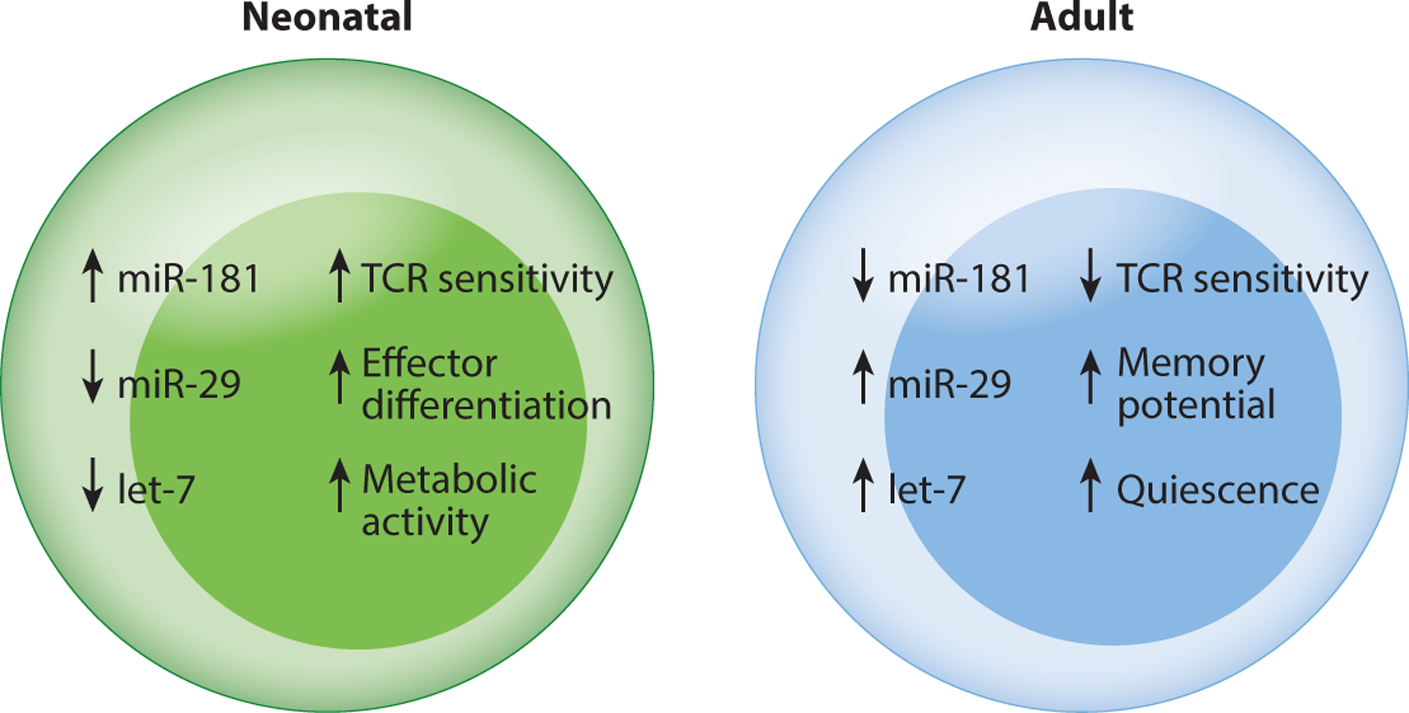
Neonatal and adult T cells are programmed differently. This diagram shows how neonatal T cells are transcriptionally shifted to a more effector-like state in early life by developmentally regulated miRNAs. Importantly, these miRNAs (e.g., miR-29, miR-181, and let-7) do not function as on-off switches, but rather as fine-tuners of gene expression in naive CD4+ and CD8+ T cells at different stages of life. In this way, different miRNAs serve as rheostats for activation (miR-181), effector cell differentiation (miR-29), and metabolism (let-7) in T cells, adjusting various thresholds based upon the need to mount rapid effector or regulatory responses. Abbreviation: TCR, T cell receptor.
There are also examples of miRNAs that are upregulated in neonatal T cells (13, 14), which may keep them in a more activated state. For example, miR-181 expression plays an important role in tuning the threshold for TCR signaling during T cell development by limiting expression of multiple phosphatases (52). Thus, its expression correlates with TCR sensitivity. During T cell development, miR-181 expression is highest in CD4+CD8+ double-positive thymocytes to facilitate interaction with low-affinity ligands but is then downregulated to increase the TCR activation threshold in mature naive T cells. Neonatal CD4+ and CD8+ T cells express elevated amounts of miR-181 (13–15), leaving them more sensitive to TCR activation. Consistent with this idea, Palin et al. (15) reported that increased expression of miR-181 in cord blood CD4+ T cells is responsible for their enhanced calcium signaling and higher Erk phosphorylation after TCR activation. In contrast, CD4+ T cells from older individuals exhibit low levels of miR-181 and reduced Erk phosphorylation after TCR stimulation (53). Expression of miR-181 is also increased in tolerized T cells, as shown by Schietinger et al. (54). Thus, higher expression of miR-181 in naive neonatal T cells provides them with higher TCR sensitivity, which may enable them to respond more strongly to both self-antigens and foreign antigens.
Why would it be advantageous for neonatal T cells to exist in a more effector-like state? It seems counterintuitive that neonatal T cells are transcriptionally and epigenetically poised for differentiation during a time when they need to limit autoimmune reactions and be flexible in how they respond to different types of antigens. One possibility is that neonatal T cells are more traveled along the axis of differentiation and have a lower threshold for activation, so that they can more quickly become tolerized or activated depending on the signals encountered during priming. For example, if neonatal T cells are primed via the TCR with antigen alone, they will quickly become tolerized. However, if costimulation and inflammatory cytokines are present, neonatal T cells will rapidly differentiate into short-lived effectors. In this way, neonatal T cells are poised for both activation and tolerance, ensuring that they quickly give rise to the types of T cells most beneficial for the host.
NEONATAL T CELLS USE MORE BROADLY REACTIVE TCRs
It is well known that the recombination program for generating TCRs is different in early life (40, 55–58). In the past, the focus was on how neonatal T cells had a more restricted TCR repertoire (59), which supported the idea that neonatal T cells were immunodeficient. However, recent studies have shown that the neonatal TCR repertoire is also biased toward self-reactive TCRs, which has important bearing on how we interpret their functions. Support for a more self-reactive TCR repertoire in neonatal T cells comes from studies done in mice (60) and humans (61) that show higher amounts of CD5, a marker of TCR avidity for self-pMHC molecules, on neonatal T cells compared to their adult counterparts. These findings are significant because the affinity between TCRs and self-pMHC molecules influences their ability to undergo homeostatic proliferation and react to foreign antigens. For example, CD5hi cells are able to outcompete CD5lo cells for self-pMHC trophic signals and proliferate more rapidly under homeostatic conditions (62, 63). Studies by Fulton et al. (64) have also demonstrated that CD5hi cells have enhanced reactivity to inflammatory cues and are poised to respond to infection, akin to neonatal T cells. These data are consistent with studies looking at B1 B cells, which also express a more self-reactive repertoire and respond rapidly to infection (65). In this way, the usage of different TCRs may help promote, rather than limit, immune functionality in early life.
Neonatal and adult T cells also express different TCRs due to a delay in expression of TdT, the enzyme responsible for insertion of random N-additions (55, 56). As a result, the TCR repertoire in neonatal mice is less diverse and comprised of more germ line–encoded clonotypes (66–70). It was initially assumed that TCR repertoire diversity was restricted in early life to limit pathogenic T cell responses during critical periods of growth and development. However, when Mathis and colleagues examined immune competence in adult mice lacking TdT expression, they found no major immune defects in TdT knockout mice following immunization or infection with several different viral pathogens (71). Gavin & Bevan (72) then proposed that TdT-deficient mice were not more susceptible to infection because germ line–encoded clonotypes were more cross-reactive. In their study, they showed that CD8+ T cell clonotypes from TdT-deficient mice were more peptide promiscuous and capable of responding to many more different peptides than wild-type clonotypes. It appears, therefore, that restriction of TdT expression in early life benefits the host by providing the fetus/neonate with broader recognition capabilities during a time when fewer T cells are present.
Is there a cost to having a more peptide-promiscuous or self-reactive repertoire in early life? To answer this question, a number of groups have crossed TdT knockout mice to different strains of autoimmune-prone mice (73–75). Instead of an increase in autoimmune pathology, the TdT knockout mice had a lower incidence of autoimmune disease. The authors suggested that the lack of TdT conferred protection against autoimmune disease because TdT knockout mice had a more restricted TCR repertoire. However, in the presence of infection, the more peptide-promiscuous repertoire may be sufficient to elicit a protective response. Another possibility is that TdT knockout mice have a lower incidence of autoimmune disease because the germ line–encoded clonotypes are more prone to tolerance. In fact, previous studies have demonstrated that shorter TCRs are more easily tolerized (76, 77). Although this work was done in adult T cells, one recent study used an autoimmune mouse model to demonstrate that neonatal T cells are indeed more susceptible to tolerance than adult T cells (78). Thus, an additional evolutionary benefit to delaying TdT expression in early life may be that it enables the host to more rapidly fill the peripheral pool (79) with T cells best equipped to counter the many different types of antigens (self, commensal, pathogen) encountered before and after birth.
NEONATAL T CELLS HAVE AN ENHANCED ABILITY TO RESPOND TO INFLAMMATION AND DANGER SIGNALS
Our understanding of neonatal T cells has been shaped by the use of assays and metrics that work best for assessing the functions of adult T cells. Indeed, the initial characterization of neonatal T cell behavior was based largely on in vitro studies comparing the ability of cord blood T cells and adult peripheral blood T cells to produce IFN-γ after stimulation via the TCR. However, recent studies suggest that neonatal T cells may be less dependent on TCR recognition and instead have an enhanced ability to respond to inflammation and danger signals (Figure 3). For example, there is an antigen-inexperienced population of CD8+ T cells (denoted virtual memory cells) that preferentially develop from fetal progenitors and can produce IFN-γ in response to innate cytokines alone (e.g., IL-12 and IL-18) (9, 25). These findings put (some) naive neonatal CD8+ T cells in the same camp as other innate-like lymphocytes, such as MAIT cells, NK T (NKT) cells, γδ T cells, innate lymphoid cells, and NK cells, all of which can be activated by cytokines alone. Second, CD4+ and CD8+ T cells express NK cell receptors in early life (9, 25, 29, 80), some of which (CD161) have been recently shown to exhibit important regulatory functions (80). Third, neonatal CD8+ T cells are capable of responding to pathogen-associated molecular patterns (PAMPs) via Toll-like receptors (TLRs). For example, human CD8+ T cells isolated from cord blood have been shown to express TLR2 and TLR5, and stimulation with their respective ligands (Pam3Cys and flagellin) results in increased proliferation and production of IFN-γ (81, 82). Human neonatal CD4+ T cells also respond to stimulation with TLR1/2 ligands (83, 84), raising the possibility that recognition of PAMPs may be a general feature of neonatal T cells.
Figure 3.
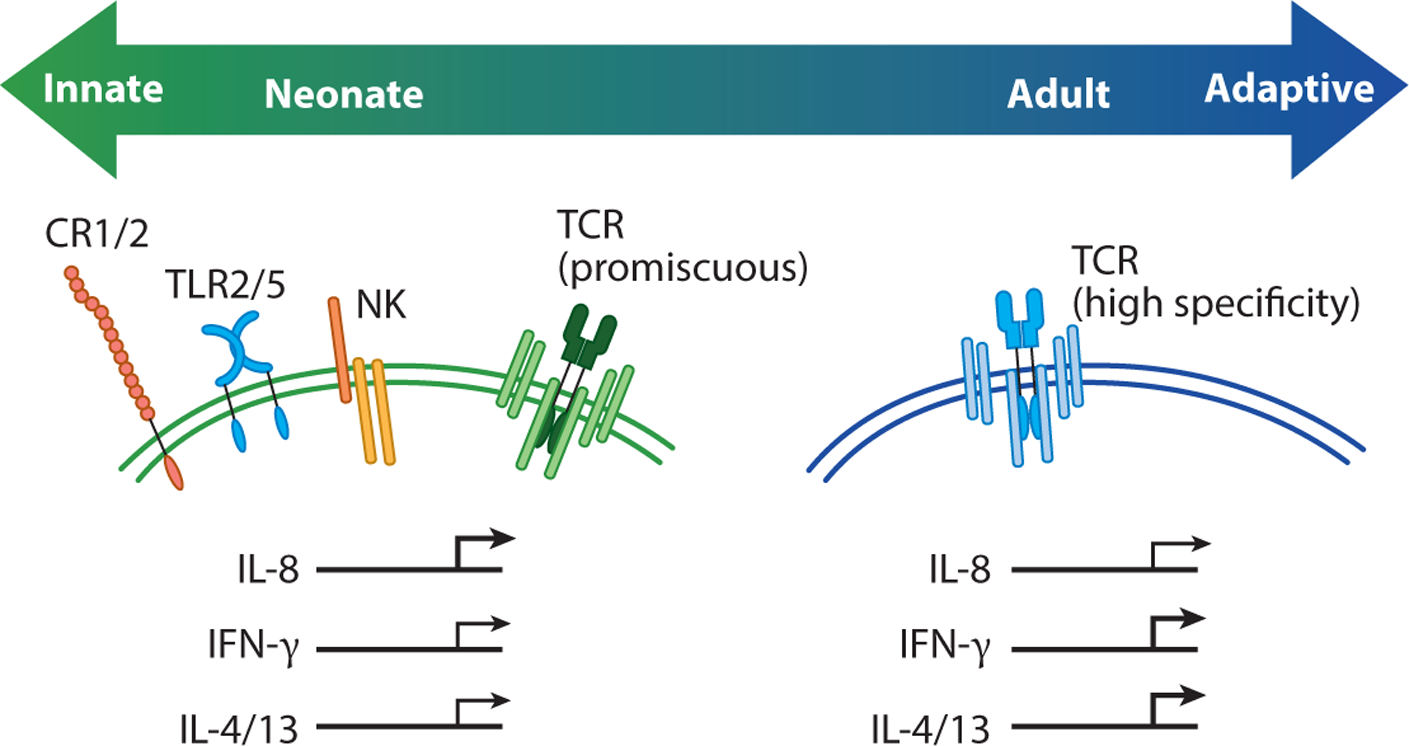
Neonatal T cells exhibit innate-like functions. This diagram shows how neonatal T cells express hardwired receptors and display functions that are more typically associated with innate and innate-like T cells. For example, naive neonatal T cells can express Toll-like receptors (TLRs), complement receptors, and natural killer (NK) cell receptors, as well as a more peptide-promiscuous T cell receptor (TCR). Following TCR stimulation, neonatal T cells also rapidly produce IL-8, which is consistent with their more broadly reactive nature. In contrast, adult T cells express a higher avidity TCR and produce more conventional cytokines (IFN-γ or IL-4 and IL-13) after clonal proliferation and differentiation.
Neonatal T cells may also rapidly deploy nonspecific defense mechanisms. In line with this idea, Galindo-Albarran et al. (82) found that naive CD8+ T cells (CD45RA+CD45RO−) in cord blood exhibit a unique gene expression profile and chromatin landscape, which are biased in favor of innate immune response genes. Among the transcripts that are upregulated in cord blood CD8+ T cells, many of them have known roles in the neutrophil activation response (e.g., antimicrobial peptides, chemokines, reactive oxygen species production) (82). These findings may help explain how neonatal CD8+ T cells are able to provide innate-like immune protection against extracellular pathogens in early life (85). Neonatal CD4+ T cells from human newborns have also been shown to express complement receptors (CR1 and CR2) and preferentially make IL-8 (CXCL8) after stimulation (12, 86–88). The discovery of IL-8 production by neonatal CD4+ T cells was particularly surprising because this chemokine is typically made by innate immune cells (e.g., macrophages) or structural cells (e.g., epithelial cells, endothelial cells) and plays an important role in the recruitment and activation of neutrophils and γδ T cells. The secretion of IL-8 by neonatal CD4+ T cells is elicited by TCR stimulation, though its production can be further enhanced by costimulation with flagellin (12). This attribute is reminiscent of other innate lymphocytes, such as marginal zone B cells, where signals generated from dual TLR and B cell receptor stimulation lead to greater amounts of antibodies (89). Lastly, ~25–30% of cord blood CD4+ T cells secrete IL-8 after stimulation (12), which likely relates to their recent thymic emigrant (RTE) status and previous amounts of homeostatic proliferation (86–88). Since levels of postthymic maturation and homeostatic proliferation differ among individuals, there is some hope that variability in IL-8 production by neonatal CD4+ T cells can be used to predict future clinical outcomes (90).
One possible explanation for why neonatal T cells are biased toward recognizing danger and inflammatory signals, while adult T cells appear to be more reliant on TCR signaling for activation, relates to the different environments present during neonatal and adult T cell development. Neonatal T cells develop in a relatively sterile environment but are later exposed to both commensal antigens (during microbiome colonization) and antigens from pathogens. Since there are no intrinsic molecular features of the microbes themselves to allow T cells (or TCRs) to discriminate commensals from pathogens, the extent of TCR recognition of these foreign antigens is not helpful in determining the necessary response. In this context, danger signals in the form of tissue damage (91, 92) may be more useful for discriminating between friend and foe. By contrast, adult T cells are exposed to commensal antigens during development and develop peripheral tolerance. For these cells, commensal tolerance is acquired during maturation, and thus recognition of the foreignness of antigens by the TCR is a more reliable indicator of recent and pathogenic infection. In this way, the changing bias toward inflammatory versus TCR signals could be viewed as a useful adaptation for the different types of antigens encountered by T cells at various stages of life. If we accept that neonatal T cells are not defective but rather more danger responsive and hyperfunctional compared to adult cells in responding to inflammatory stimuli, we may be able to develop more neonate-appropriate assays to better assess their functional abilities.
NEONATAL T CELLS ARE OPTIMALLY POSITIONED FOR RAPID RESPONSES
Some of the functions of neonatal T cells have likely been overlooked because past studies have focused on T cells in the blood and lymphoid tissue. However, there is now a gaining appreciation for the roles of neonatal T cells in peripheral organs. The current dogma for conventional T cell recirculation is that naive T cells are confined to the blood and lymphatics, whereas memory cells are able to recirculate throughout the peripheral organs (93, 94). Yet these rules do not appear to apply to T cells in fetal life. Using a sheep model, Mackay et al. (95, 96) elegantly showed that naive T cells circulate throughout extralymphoid tissues during normal development of the fetal immune system. Additional studies in mice have confirmed these findings and demonstrated that peripheral organs, such as the skin (97), lung (98), and liver (9), are selectively seeded by neonatal T cells in the first few weeks of life. Although the intestine was not examined in the aforementioned studies, other reports have shown that neonatal T cells express higher levels of gut homing receptors and preferentially traffic to the small intestine (99), placing them alongside other innate-like T cells, such as γδ T cells, intraepithelial lymphocytes, and MAIT cells. The propensity for neonatal T cells to localize in peripheral organs may suggest a role in organogenesis (100, 101) and help to explain why it is advantageous for them to express innate receptors in early life. In addition, the tolerance-prone neonatal T cells are ideally positioned to encounter a wide range of self-and commensal-derived antigens.
Do neonatal T cells exhibit distinct tissue distribution patterns in humans? Given the difficulty of sampling tissues from humans, our knowledge of how T cells populate peripheral organs has historically been sparse. However, key insights can be gleaned from a recent study by Farber’s group comparing the phenotypes of CD4+ and CD8+ T cells in tissues from pediatric (0–2 y of age) and adult (>15 y of age) organ donors (102). The authors found an elevated frequency of naive CD4+ and CD8+ T cells across all pediatric tissues, including the lung and intestine. Also, the pediatric tissues were comprised of a high frequency of Tregs (30–40%), which declined to a relatively low number in adulthood (1–10%). Interestingly, depletion of Tregs in infant samples allowed the remaining CD4+ and CD8+ T cells to undergo adult-like levels of proliferation and produce significantly more cytokines after stimulation. This finding is particularly notable because previous studies have compared the behavior of neonatal and adult T cells in bulk populations and reported that neonatal T cells are hypofunctional compared to adult T cells. However, the study by Thome et al. (102), as well as other studies in mice (103), indicate that rather than being intrinsically defective, neonatal T cells may simply be more suppressed than their adult counterparts. Such data illustrate not only how location shapes immune function but also the importance of comparing purified subsets of naive T cells from neonates and adults when studying their cell-intrinsic differences.
NEONATAL T CELLS ARE IMPAIRED AT FORMING MEMORY T CELLS—A USEFUL ADAPTATION?
Our understanding of the neonatal T cell response has evolved with the use of more refined models of infection. In the past, the strategy was to directly infect neonatal and adult mice and compare the numbers and function of T cells present in both groups at various times after infection. However, it was often unclear whether the altered behavior of neonatal T cells was due to a less diverse TCR repertoire, smaller numbers of precursor cells, or an altered priming environment. Thus, a number of groups have started using adoptive cotransfer experiments to control for age-related factors when identifying cell-intrinsic differences in the neonatal T cell response to infection. These studies suggest that the ability of neonatal T cells to rapidly proliferate and respond to innate signals may come at the cost of forming memory.
In the case of CD8+ T cells, Smith et al. (29) used an experimental strategy in which equal numbers of monoclonal CD8+ T cells from neonatal and adult donor TCR transgenic mice were transferred into the same host prior to systemic infection with vaccinia virus or a virulent strain of L. monocytogenes. Neonatal donor CD8+ T cells responded more quickly to infection and peaked sooner than adult donor cells but preferentially gave rise to short-lived effector cells and failed to transition into the long-lived memory pool. In contrast, the adult donor cells took longer to enter the proliferative response but preferentially became memory cells capable of responding to secondary infections. Thus, even when neonatal CD8+ T cells were placed in an adult environment and provided with all the signals that adult CD8+ T cells have during infection, they still rapidly proliferated and failed to form memory. Similarly, Siefker & Adkins (85) found that neonatal CD8+ T cells rapidly expand during the innate phase of the response to an extracellular bacterial enteropathogen (Yersinia enterocolitica), but they were not required for immune protection against secondary infection. Together, these papers suggest that the dominant function of neonatal CD8+ T cells is to provide an early innate-like response during primary infections.
Neonatal CD4+ T cells have also been shown to be intrinsically defective at forming memory. Using a mouse model of influenza infection, Zens et al. (10) observed a reduced number of memory CD4+ T cells (and CD8+ T cells) in the lungs of mice infected as infants compared to those infected as adults. The reduction of neonatal memory CD4+ T cells in the lung was not due to a lack of responsiveness or proliferation but rather to an inherent propensity to become terminally differentiated and more rapidly lose their potential to transition into the long-lived tissue-resident memory pool. Importantly, adoptive transfer experiments demonstrated that the failure of neonatal CD4+ T cells to persist in the lung during the memory phase of infection is due to cell-intrinsic differences and is not a function of the infant environment. These findings are reminiscent of the cell-intrinsic differences observed in CD8+ T cells, suggesting that the propensity to quickly differentiate into short-lived effectors may be a general feature of neonatal T cells. From an evolutionary point of view, it is likely more beneficial for the neonate to mount a vigorous T cell response to infection than to develop immunological memory, since memory T cells are unimportant if the host fails to survive the initial infection. Also, since the neonatal T cell repertoire is extremely limited, likely it is of lesser value in the memory pool, as higher-avidity adult T cells will be made later in life, reducing the need for neonatal memory.
Compelling evidence suggests that similar principles apply to neonatal T cells in humans. For example, a study of infants experiencing viral respiratory tract infections reported an accumulation of more terminally differentiated CD8+ T cells (effector memory RA+ T cells, or TEMRAs) in the lungs of younger patients, whereas the less-differentiated tissue-resident memory CD8+ T cells were more prevalent in older children (104), potentially explaining why individuals are more susceptible to repeat infections with intracellular pathogens in early life (105). There is also evidence to suggest that infants are more susceptible to persistent infection (e.g., cytomegalovirus) because neonatal CD8+ T cells have an inherent propensity to become terminally differentiated and functionally exhausted (106).
In the future, it will be important to determine whether any routes of inoculation or priming conditions are favorable for the generation of neonatal memory T cells (see sidebar titled How Do Neonatal T Cells Perform in Clinical Settings?). Notably, vaccination of neonatal mice with an attenuated bacterium (ActA LM) (107) or lower doses of viral vectors (108, 109) resulted in more robust immune protection, suggesting that lower amounts of inflammation and antigen may be required for the optimal development of memory CD8+ T cells in early life. Also, immunization of neonatal mice with LAIV elicits more robust immune protection compared to IIV-vaccinated mice (10), which may relate to differences in the types of memory cells generated by different modes of immunization. Collectively, these studies indicate that careful consideration of the priming conditions and routes of inoculation will need to be applied to promote durable T cell immunity in early life.
CONCLUSION AND FUTURE PERSPECTIVES
Overwhelming evidence suggests that neonatal T cells can no longer be considered immature versions of adult cells. In fact, our understanding of the biology of neonatal T cells has likely been hampered by comparing neonatal T cells to their adult counterparts. If we instead consider the developmental biology of the immune system and compare neonatal T cells to other immune cells made during the same ontogenic window, their unique functions become clearer. Like other fetal-derived lymphocytes (B1 B cells, DETCs), neonatal T cells are a distinct developmental layer of T cells evolved to perform a specialized role for the host. They are a broadly reactive layer of T cells created with a preexisting effector-like state and an ability to migrate to peripheral tissues, which enables them to quickly develop into regulatory or effector cells, depending on the needs of the host. In this way, they resemble other fast-acting innate-like populations of lymphocytes made in early life and are therefore less similar to adult T cells, which exist in a more naive state in the lymphoid tissue and respond with slower kinetics.
Viewing neonatal T cells as a discrete ontogenic layer raises a number of critical questions. First, how do environmental (e.g., microbiome, diet) and genetic factors alter the persistence of neonatal T cells in adulthood, particularly in humans? A large number of studies have suggested that early-life microbial exposures can permanently program the offspring’s immune system and life-long disease risk (117–120), but whether this is accomplished by altering the developmental layering in the T cell compartment remains unclear. Second, can we predict infection outcomes and disease risk (allergies, autoimmunity) based upon the ratio of fetal-to adult-derived T cells present in the starting pool? Given that neonatal and adult T cells are long-lived and exhibit different cell-intrinsic properties, some of the interindividual variation is likely related to the layering variation of the T cell compartment. Third, can we purify or target certain subsets of naive T cells for specific therapeutic interventions? The fast-acting neonatal CD8+ T cells that persist into adulthood possess many features that could be highly desirable in the context of cancer immunotherapy. Understanding which developmental layers of T cells are associated with favorable or pathological outcomes will be essential to answering this question.
There has never been a more exciting time to study neonatal T cells. We now have state-of-the-art tools to examine their behavior in a more systematic and unbiased manner at a level never before possible, leaving the future open for discovery. These studies will broaden our fundamental understanding of neonatal T cells and potentially lead to new approaches for treating and preventing human disease.
WHEN IS A MOUSE NEONATAL?
In humans, the neonatal period extends from birth through the first month of life. In mice, the neonatal period is different, and there is often debate around finding equivalent stages of immune development in mice and humans. What we do know is that neonatal T cells in mice and humans are sculpted by the same mechanisms of immune development. Both are derived from fetal progenitors, comprised of more recent thymic emigrants, exhibit a less diverse TCR repertoire, and undergo more homeostatic proliferation. Mice have proven to be enormously helpful in providing key mechanistic insight into the biology of neonatal T cells. However, differences in the timing of key developmental events do exist between mice and humans and have led to the question of what age mice most closely resemble newborn humans. CD4+ and CD8+ T cells can be identified in humans at ~14 weeks gestation, for example, but sufficient numbers of peripheral T cells do not emerge in mice until 5–7 days after birth. Diversification of the TCR repertoire and the developmental switch in lymphopoiesis happen in the second trimester for humans, but they occur after birth in mice. Essentially, mice are born with a less developed immune system than humans, which is why most neonatal studies in mice are performed in 1-to 2-week-old pups.
DO NEONATAL T CELLS REPRESENT A DISTINCT LINEAGE OF LYMPHOCYTES?
Heterogeneity is a hallmark feature of the adaptive immune system in vertebrates. For B cells, separate lineages in the starting population are made during distinct windows of development and differentiate along different pathways during infection. B1 B cells are generated from fetal progenitors and are preferentially found in newborn mice (17, 38). B2 B cells arise later in life and cannot be efficiently made from fetal progenitors. These two lineages of B cells are identified by their expression of CD5 (Ly-1) (16). Is a similar developmental architecture in place for conventional CD4+ and CD8+ T cells? In mice, both neonatal T cells and B1 B cells are derived from fetal HSCs and express broadly reactive lymphocyte receptors that are more germ line encoded (39, 40). Prior to stimulation, both B1 B cells and neonatal T cells express a distinct transcriptome and preferentially migrate to different compartments of the body than their adult counterparts (8, 25, 41–44). Neonatal T cells and B1 B cells also exhibit more immunoregulatory properties and preferentially respond during early stages of infection, while the B2 B cells and adult T cells exhibit slower kinetics and have an enhanced ability to persist into the memory phase (9, 29, 45). Finally, neonatal T cells persist into adulthood and retain their cell-intrinsic properties (9, 35, 37), similar to B1 B cells. If neonatal T cells have a separate developmental pathway, express different genes, and exhibit distinct functional properties, why are they not considered a separate lineage of cells, akin to B1 B cells? The answer may be that no surface marker or receptor currently exists to distinguish a neonatal CD4+ or CD8+ T cell from its adult counterpart. Identification of a unique marker for neonatal T cells would revolutionize the field in much the same way that identification of CD5 changed our perspective of B cells.
HOW DO NEONATAL T CELLS PERFORM IN CLINICAL SETTINGS?
The unique functional attributes of neonatal T cells have not gone unnoticed by cancer researchers. Treatment of aggressive forms of hematological malignancies often requires the reconstitution of the host immune system with hematopoietic stem cells. The more tolerant-prone and fast-acting neonatal T cells are ideally suited for cancer immunotherapy. Compared to transplantations with adult bone marrow, cord blood transplantations require less stringent HLA matching and have a decreased risk of graft-versus-host disease, likely due to protolerogenic cord-derived cells (110). Cord blood T cells have been shown to rapidly expand in the periphery and mediate better graft-antitumor responses than adult-derived T cells (110–112). The precursor frequency of T cells specific for some tumor-associated antigens (e.g., PR1) is also significantly higher in cord blood compared to adult peripheral blood mononuclear cells, which may be due to incomplete central tolerance in early life (113). In addition, cord blood T cells have been found to recognize a broad pool of unconventional cytomegalovirus (CMV) epitopes, which may prove useful for preventing reactivation of CMV after transplantation (114). Lastly, cord blood T cells undergo more vigorous proliferation in the presence of IL-7 and IL-15 (115), generating a large source of long-lived memory stem T cells, which are ideal candidates for adoptive immunotherapies (116). Better understanding of the biology of neonatal T cells may ultimately direct us to more effective therapeutic strategies for treating cancer in adults.
Footnotes
DISCLOSURE STATEMENT
The author is not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Billingham RE, Brent L, Medawar PB. 1953. Actively acquired tolerance of foreign cells. Nature 172:603–6 [DOI] [PubMed] [Google Scholar]
- 2.Gammon G, Dunn K, Shastri N, Oki A, Wilbur S, Sercarz EE. 1986. Neonatal T-cell tolerance to minimal immunogenic peptides is caused by clonal inactivation. Nature 319:413–15 [DOI] [PubMed] [Google Scholar]
- 3.Forsthuber T, Yip HC, Lehmann PV. 1996. Induction of TH1 and TH2 immunity in neonatal mice. Science 271:1728–30 [DOI] [PubMed] [Google Scholar]
- 4.Singh RR, Hahn BH, Sercarz EE. 1996. Neonatal peptide exposure can prime T cells and, upon subsequent immunization, induce their immune deviation: implications for antibody versus T cell-mediated autoimmunity. J. Exp. Med 183:1613–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen N, Field EH. 1995. Enhanced type 2 and diminished type 1 cytokines in neonatal tolerance. Transplantation 59:933–41 [DOI] [PubMed] [Google Scholar]
- 6.Prescott SL, Macaubas C, Smallacombe T, Holt BJ, Sly PD, Holt PG. 1999. Development of allergenspecific T-cell memory in atopic and normal children. Lancet 353:196–200 [DOI] [PubMed] [Google Scholar]
- 7.Romero R, Espinoza J, Goncalves LF, Kusanovic JP, Friel L, Hassan S. 2007. The role of inflammation and infection in preterm birth. Semin. Reprod. Med 25:21–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mold JE, Venkatasubrahmanyam S, Burt TD, Michaelsson J, Rivera JM, et al. 2010. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science 330:1695–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith NL, Patel RK, Reynaldi A, Grenier JK, Wang J, et al. 2018. Developmental origin governs CD8+ T cell fate decisions during infection. Cell 174:117–30.e14 [DOI] [PubMed] [Google Scholar]
- 10.Zens KD, Chen JK, Guyer RS, Wu FL, Cvetkovski F, et al. 2017. Reduced generation of lung tissue-resident memory T cells during infancy. J. Exp. Med 214:2915–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang G, Miyahara Y, Guo Z, Khattar M, Stepkowski SM, Chen W. 2010. “Default” generation of neonatal regulatory T cells. J. Immunol 185:71–78 [DOI] [PubMed] [Google Scholar]
- 12.Gibbons D, Fleming P, Virasami A, Michel ML, Sebire NJ, et al. 2014. Interleukin-8 (CXCL8) production is a signatory T cell effector function of human newborn infants. Nat. Med 20:1206–10 [DOI] [PubMed] [Google Scholar]
- 13.Wissink EM, Smith NL, Spektor R, Rudd BD, Grimson A. 2015. MicroRNAs and their targets are differentially regulated in adult and neonatal mouse CD8+ T cells. Genetics 201:1017–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu HR, Hsu TY, Huang HC, Kuo HC, Li SC, et al. 2016. Comparison of the functional microRNA expression in immune cell subsets of neonates and adults. Front. Immunol 7:615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palin AC, Ramachandran V, Acharya S, Lewis DB. 2013. Human neonatal naive CD4+ T cells have enhanced activation-dependent signaling regulated by the microRNA miR-181a. J. Immunol 190:2682–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayakawa K, Hardy RR, Herzenberg LA, Herzenberg LA. 1985. Progenitors for Ly-1 B cells are distinct from progenitors for other B cells. J. Exp. Med 161:1554–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kantor AB, Stall AM, Adams S, Herzenberg LA, Herzenberg LA. 1992. Differential development of progenitor activity for three B-cell lineages. PNAS 89:3320–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Havran WL, Allison JP. 1988. Developmentally ordered appearance of thymocytes expressing different T-cell antigen receptors. Nature 335:443–45 [DOI] [PubMed] [Google Scholar]
- 19.Ikuta K, Kina T, MacNeil I, Uchida N, Peault B, et al. 1990. A developmental switch in thymic lymphocyte maturation potential occurs at the level of hematopoietic stem cells. Cell 62:863–74 [DOI] [PubMed] [Google Scholar]
- 20.Herzenberg LA, Herzenberg LA. 1989. Toward a layered immune system. Cell 59:953–54 [DOI] [PubMed] [Google Scholar]
- 21.Jotereau F, Heuze F, Salomon-Vie V, Gascan H. 1987. Cell kinetics in the fetal mouse thymus: precursor cell input, proliferation, and emigration. J. Immunol 138:1026–30 [PubMed] [Google Scholar]
- 22.Douagi I, Andre I, Ferraz JC, Cumano A. 2000. Characterization of T cell precursor activity in the murine fetal thymus: evidence for an input of T cell precursors between days 12 and 14 of gestation. Eur. J. Immunol 30:2201–10 [DOI] [PubMed] [Google Scholar]
- 23.Adkins B. 1991. Developmental regulation of the intrathymic T cell precursor population. J. Immunol 146:1387–93 [PubMed] [Google Scholar]
- 24.Kim I, Saunders TL, Morrison SJ. 2007. Sox17 dependence distinguishes the transcriptional regulation of fetal from adult hematopoietic stem cells. Cell 130:470–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Wissink EM, Watson NB, Smith NL, Grimson A, Rudd BD. 2016. Fetal and adult progenitors give rise to unique populations of CD8+ T cells. Blood 128:3073–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adkins B. 2003. Peripheral CD4+ lymphocytes derived from fetal versus adult thymic precursors differ phenotypically and functionally. J. Immunol 171:5157–64 [DOI] [PubMed] [Google Scholar]
- 27.Hebel K, Weinert S, Kuropka B, Knolle J, Kosak B, et al. 2014. CD4+ T cells from human neonates and infants are poised spontaneously to run a nonclassical IL-4 program. J. Immunol 192:5160–70 [DOI] [PubMed] [Google Scholar]
- 28.Webster RB, Rodriguez Y, Klimecki WT, Vercelli D. 2007. The human IL-13 locus in neonatal CD4+ T cells is refractory to the acquisition of a repressive chromatin architecture. J. Biol. Chem 282:700–9 [DOI] [PubMed] [Google Scholar]
- 29.Smith NL, Wissink E, Wang J, Pinello JF, Davenport MP, et al. 2014. Rapid proliferation and differentiation impairs the development of memory CD8+ T cells in early life. J. Immunol 193:177–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reynaldi A, Smith NL, Schlub TE, Venturi V, Rudd BD, Davenport MP. 2016. Modeling the dynamics of neonatal CD8+ T-cell responses. Immunol. Cell Biol 94:838–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCarron MJ, Reen DJ. 2010. Neonatal CD8+ T-cell differentiation is dependent on interleukin-12. Hum. Immunol 71:1172–79 [DOI] [PubMed] [Google Scholar]
- 32.Marchant A, Goetghebuer T, Ota MO, Wolfe I, Ceesay SJ, et al. 1999. Newborns develop a Th1-type immune response to Mycobacterium bovis bacillus Calmette-Guerin vaccination. J. Immunol 163:2249–55 [PubMed] [Google Scholar]
- 33.Mascart F, Verscheure V, Malfroot A, Hainaut M, Pierard D, et al. 2003. Bordetella pertussis infection in 2-month-old infants promotes type 1 T cell responses. J. Immunol 170:1504–9 [DOI] [PubMed] [Google Scholar]
- 34.Reibke R, Garbi N, Ganss R, Hammerling GJ, Arnold B, Oelert T. 2006. CD8+ regulatory T cells generated by neonatal recognition of peripheral self-antigen. PNAS 103:15142–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D. 2015. Immune tolerance: Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 348:589–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mold JE, Michaelsson J, Burt TD, Muench MO, Beckerman KP, et al. 2008. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science 322:1562–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reynaldi A, Smith NL, Schlub TE, Tabilas C, Venturi V, et al. 2019. Fate mapping reveals the age structure of the peripheral T cell compartment. PNAS 116:3974–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Montecino-Rodriguez E, Leathers H, Dorshkind K. 2006. Identification of a B-1 B cell-specified progenitor. Nat. Immunol 7:293–301 [DOI] [PubMed] [Google Scholar]
- 39.Berland R, Wortis HH. 2002. Origins and functions of B-1 cells with notes on the role of CD5. Annu. Rev. Immunol 20:253–300 [DOI] [PubMed] [Google Scholar]
- 40.Bogue M, Candeias S, Benoist C, Mathis D. 1991. A special repertoire of alpha:beta T cells in neonatal mice. EMBO J. 10:3647–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hardy RR, Hayakawa K. 2015. Perspectives on fetal derived CD5+ B1 B cells. Eur. J. Immunol 45:2978–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li YS, Zhou Y, Tang L, Shinton SA, Hayakawa K, Hardy RR. 2015. A developmental switch between fetal and adult B lymphopoiesis. Ann. N. Y. Acad. Sci 1362:8–15 [DOI] [PubMed] [Google Scholar]
- 43.Zhou Y, Li YS, Bandi SR, Tang L, Shinton SA, et al. 2015. Lin28b promotes fetal B lymphopoiesis through the transcription factor Arid3a. J. Exp. Med 212:569–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yuan J, Nguyen CK, Liu X, Kanellopoulou C, Muljo SA. 2012. Lin28b reprograms adult bone marrow hematopoietic progenitors to mediate fetal-like lymphopoiesis. Science 335:1195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Montecino-Rodriguez E, Dorshkind K. 2006. New perspectives in B-1 B cell development and function. Trends Immunol 27:428–33 [DOI] [PubMed] [Google Scholar]
- 46.Bedoui S, Gebhardt T, Gasteiger G, Kastenmuller W. 2016. Parallels and differences between innate and adaptive lymphocytes. Nat. Immunol 17:490–94 [DOI] [PubMed] [Google Scholar]
- 47.Wells AC, Daniels KA, Angelou CC, Fagerberg E, Burnside AS, et al. 2017. Modulation of let-7 miRNAs controls the differentiation of effector CD8 T cells. eLife 6:e26398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thornton JE, Gregory RI. 2012. How does Lin28 let-7 control development and disease? Trends Cell Biol. 22:474–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu H, Shyh-Chang N, Segre AV, Shinoda G, Shah SP, et al. 2011. The Lin28/let-7 axis regulates glucose metabolism. Cell 147:81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tabilas C, Wang J, Xiajoing L, Locasale JW, Smith NL, Rudd BD. 2019. Cutting edge: Elevated glycolytic metabolism limits the formation of memory CD8+ T cells in early life. J. Immunol 203:2571–76 10.4049/jimmunol.1900426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bronevetsky Y, Burt TD, McCune JM. 2016. Lin28b regulates fetal regulatory T cell differentiation through modulation of TGF-β signaling. J. Immunol 197:4344–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li QJ, Chau J, Ebert PJ, Sylvester G, Min H, et al. 2007. miR-181a is an intrinsic modulator of T cell sensitivity and selection. Cell 129:147–61 [DOI] [PubMed] [Google Scholar]
- 53.Li G, Yu M, Lee WW, Tsang M, Krishnan E, et al. 2012. Decline in miR-181a expression with age impairs T cell receptor sensitivity by increasing DUSP6 activity. Nat. Med 18:1518–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schietinger A, Delrow JJ, Basom RS, Blattman JN, Greenberg PD. 2012. Rescued tolerant CD8 T cells are preprogrammed to reestablish the tolerant state. Science 335:723–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cabaniols JP, Fazilleau N, Casrouge A, Kourilsky P, Kanellopoulos JM. 2001. Most α/β T cell receptor diversity is due to terminal deoxynucleotidyl transferase. J. Exp. Med 194:1385–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gilfillan S, Dierich A, Lemeur M, Benoist C, Mathis D. 1993. Mice lacking TdT: mature animals with an immature lymphocyte repertoire. Science 261:1175–78 [DOI] [PubMed] [Google Scholar]
- 57.Bogue M, Gilfillan S, Benoist C, Mathis D. 1992. Regulation of N-region diversity in antigen receptors through thymocyte differentiation and thymus ontogeny. PNAS 89:11011–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feeney AJ. 1991. Junctional sequences of fetal T cell receptor beta chains have few N regions. J. Exp. Med 174:115–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schelonka RL, Raaphorst FM, Infante D, Kraig E, Teale JM, Infante AJ. 1998. T cell receptor repertoire diversity and clonal expansion in human neonates. Pediatr. Res 43:396–402 [DOI] [PubMed] [Google Scholar]
- 60.Dong M, Artusa P, Kelly SA, Fournier M, Baldwin TA, et al. 2017. Alterations in the thymic selection threshold skew the self-reactivity of the TCR repertoire in neonates. J. Immunol 199:965–73 [DOI] [PubMed] [Google Scholar]
- 61.Mandl JN, Monteiro JP, Vrisekoop N, Germain RN. 2013. T cell-positive selection uses self-ligand binding strength to optimize repertoire recognition of foreign antigens. Immunity 38:263–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.White JT, Cross EW, Burchill MA, Danhorn T, McCarter MD, et al. 2016. Virtual memory T cells develop and mediate bystander protective immunity in an IL-15-dependent manner. Nat. Commun 7:11291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cho JH, Kim HO, Surh CD, Sprent J. 2010. T cell receptor-dependent regulation of lipid rafts controls naive CD8+ T cell homeostasis. Immunity 32:214–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fulton RB, Hamilton SE, Xing Y, Best JA, Goldrath AW, et al. 2015. The TCR’s sensitivity to self peptide-MHC dictates the ability of naive CD8+ T cells to respond to foreign antigens. Nat. Immunol 16:107–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baumgarth N. 2011. The double life of a B-1 cell: Self-reactivity selects for protective effector functions. Nat. Rev. Immunol 11:34–46 [DOI] [PubMed] [Google Scholar]
- 66.Carey AJ, Hope JL, Mueller YM, Fike AJ, Kumova OK, et al. 2017. Public clonotypes and convergent recombination characterize the naive CD8+ T-cell receptor repertoire of extremely preterm neonates. Front. Immunol 8:1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Venturi V, Nzingha K, Amos TG, Charles WC, Dekhtiarenko I, et al. 2016. The neonatal CD8+ T cell repertoire rapidly diversifies during persistent viral infection. J. Immunol 196:1604–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rudd BD, Venturi V, Smith NL, Nzingha K, Goldberg EL, et al. 2013. Acute neonatal infections ‘lock-in’ a suboptimal CD8+ T cell repertoire with impaired recall responses. PLOS Pathog. 9:e1003572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rudd BD, Venturi V, Davenport MP, Nikolich-Zugich J. 2011. Evolution of the antigen-specific CD8+ TCR repertoire across the life span: evidence for clonal homogenization of the old TCR repertoire. J. Immunol 186:2056–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ruckwardt TJ, Malloy AM, Gostick E, Price DA, Dash P, et al. 2011. Neonatal CD8 T-cell hierarchy is distinct from adults and is influenced by intrinsic T cell properties in respiratory syncytial virus infected mice. PLOS Pathog. 7:e1002377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gilfillan S, Bachmann M, Trembleau S, Adorini L, Kalinke U, et al. 1995. Efficient immune responses in mice lacking N-region diversity. Eur. J. Immunol 25:3115–22 [DOI] [PubMed] [Google Scholar]
- 72.Gavin MA, Bevan MJ. 1995. Increased peptide promiscuity provides a rationale for the lack of N regions in the neonatal T cell repertoire. Immunity 3:793–800 [DOI] [PubMed] [Google Scholar]
- 73.Conde C, Weller S, Gilfillan S, Marcellin L, Martin T, Pasquali JL. 1998. Terminal deoxynucleotidyl transferase deficiency reduces the incidence of autoimmune nephritis in (New Zealand Black × New Zealand White)F1 mice. J. Immunol 161:7023–30 [PubMed] [Google Scholar]
- 74.Feeney AJ, Lawson BR, Kono DH, Theofilopoulos AN. 2001. Terminal deoxynucleotidyl transferase deficiency decreases autoimmune disease in MRL-Faslpr mice. J. Immunol 167:3486–93 [DOI] [PubMed] [Google Scholar]
- 75.Robey IF, Peterson M, Horwitz MS, Kono DH, Stratmann T, et al. 2004. Terminal deoxynucleotidyltransferase deficiency decreases autoimmune disease in diabetes-prone nonobese diabetic mice and lupus-prone MRL-Faslpr mice. J. Immunol 172:4624–29 [DOI] [PubMed] [Google Scholar]
- 76.Houston EG Jr., Fink PJ. 2009. MHC drives TCR repertoire shaping, but not maturation, in recent thymic emigrants. J. Immunol 183:7244–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Matsutani T, Ohmori T, Ogata M, Soga H, Kasahara S, et al. 2007. Comparison of CDR3 length among thymocyte subpopulations: impacts of MHC and BV segment on the CDR3 shortening. Mol. Immunol 44:2378–87 [DOI] [PubMed] [Google Scholar]
- 78.He Q, Morillon YM 2nd, Spidale NA, Kroger CJ, Liu B, et al. 2013. Thymic development of autoreactive T cells in NOD mice is regulated in an age-dependent manner. J. Immunol 191:5858–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gilfillan S, Benoist C, Mathis D. 1995. Mice lacking terminal deoxynucleotidyl transferase: adult mice with a fetal antigen receptor repertoire. Immunol. Rev 148:201–19 [DOI] [PubMed] [Google Scholar]
- 80.Halkias J, Rackaityte E, Hillman SL, Aran D, Mendoza VF, et al. 2019. CD161 contributes to prenatal immune suppression of IFNγ-producing PLZF+ T cells. J. Clin. Investig 130:3562–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McCarron M, Reen DJ. 2009. Activated human neonatal CD8+ T cells are subject to immunomodulation by direct TLR2 or TLR5 stimulation. J. Immunol 182:55–62 [DOI] [PubMed] [Google Scholar]
- 82.Galindo-Albarran AO, Lopez-Portales OH, Gutierrez-Reyna DY, Rodriguez-Jorge O, Sanchez-Villanueva JA, et al. 2016. CD8+ T cells from human neonates are biased toward an innate immune response. Cell Rep. 17:2151–60 [DOI] [PubMed] [Google Scholar]
- 83.Komai-Koma M, Jones L, Ogg GS, Xu D, Liew FY. 2004. TLR2 is expressed on activated T cells as a costimulatory receptor. PNAS 101:3029–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sinnott BD, Park B, Boer MC, Lewinsohn DA, Lancioni CL. 2016. Direct TLR-2 costimulation unmasks the proinflammatory potential of neonatal CD4+ T cells. J. Immunol 197:68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Siefker DT, Adkins B. 2016. Rapid CD8+ function is critical for protection of neonatal mice from an extracellular bacterial enteropathogen. Front. Pediatr 4:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van den Broek T, Delemarre EM, Janssen WJ, Nievelstein RA, Broen JC, et al. 2016. Neonatal thymectomy reveals differentiation and plasticity within human naive T cells. J. Clin. Investig 126:1126–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Pekalski ML, Garcia AR, Ferreira RC, Rainbow DB, Smyth DJ, et al. 2017. Neonatal and adult recent thymic emigrants produce IL-8 and express complement receptors CR1 and CR2. JCI Insight 2:93739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Das A, Rouault-Pierre K, Kamdar S, Gomez-Tourino I, Wood K, et al. 2017. Adaptive from innate: Human IFN-γ+CD4+ T cells can arise directly from CXCL8-producing recent thymic emigrants in babies and adults. J. Immunol 199:1696–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cerutti A, Cols M, Puga I. 2013. Marginal zone B cells: virtues of innate-like antibody-producing lymphocytes. Nat. Rev. Immunol 13:118–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scheible KM, Emo J, Laniewski N, Baran AM, Peterson DR, et al. 2018. T cell developmental arrest in former premature infants increases risk of respiratory morbidity later in infancy. JCI Insight 3:96724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Matzinger P. 1994. Tolerance, danger, and the extended family. Annu. Rev. Immunol 12:991–1045 [DOI] [PubMed] [Google Scholar]
- 92.Janeway CA Jr. 1989. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb. Symp. Quant. Biol 54(Part 1):1–13 [DOI] [PubMed] [Google Scholar]
- 93.Hunter MC, Teijeira A, Halin C. 2016. T cell trafficking through lymphatic vessels. Front. Immunol 7:613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Masopust D, Schenkel JM. 2013. The integration of T cell migration, differentiation and function. Nat. Rev. Immunol 13:309–20 [DOI] [PubMed] [Google Scholar]
- 95.Mackay CR, Marston WL, Dudler L. 1990. Naive and memory T cells show distinct pathways of lymphocyte recirculation. J. Exp. Med 171:801–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mackay CR, Kimpton WG, Brandon MR, Cahill RN. 1988. Lymphocyte subsets show marked differences in their distribution between blood and the afferent and efferent lymph of peripheral lymph nodes. J. Exp. Med 167:1755–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Scharschmidt TC, Vasquez KS, Truong HA, Gearty SV, Pauli ML, et al. 2015. A wave of regulatory T cells into neonatal skin mediates tolerance to commensal microbes. Immunity 43:1011–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Alferink J, Tafuri A, Vestweber D, Hallmann R, Hammerling GJ, Arnold B. 1998. Control of neonatal tolerance to tissue antigens by peripheral T cell trafficking. Science 282:1338–41 [DOI] [PubMed] [Google Scholar]
- 99.Staton TL, Habtezion A, Winslow MM, Sato T, Love PE, Butcher EC. 2006. CD8+ recent thymic emigrants home to and efficiently repopulate the small intestine epithelium. Nat. Immunol 7:482–88 [DOI] [PubMed] [Google Scholar]
- 100.Crespo M, Martinez DG, Cerissi A, Rivera-Reyes B, Bernstein HB, et al. 2012. Neonatal T-cell maturation and homing receptor responses to Toll-like receptor ligands differ from those of adult naive T cells: relationship to prematurity. Pediatr. Res 71:136–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shi J, Wei PK. 2016. Interleukin-8: a potent promoter of angiogenesis in gastric cancer. Oncol. Lett 11:1043–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Thome JJ, Bickham KL, Ohmura Y, Kubota M, Matsuoka N, et al. 2016. Early-life compartmentalization of human T cell differentiation and regulatory function in mucosal and lymphoid tissues. Nat. Med 22:72–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fernandez MA, Puttur FK, Wang YM, Howden W, Alexander SI, Jones CA. 2008. T regulatory cells contribute to the attenuated primary CD8+ and CD4+ T cell responses to herpes simplex virus type 2 in neonatal mice. J. Immunol 180:1556–64 [DOI] [PubMed] [Google Scholar]
- 104.Connors TJ, Baird JS, Yopes MC, Zens KD, Pethe K, et al. 2018. Developmental regulation of effector and resident memory T cell generation during pediatric viral respiratory tract infection. J. Immunol 201:432–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chang J, Braciale TJ. 2002. Respiratory syncytial virus infection suppresses lung CD8+ T-cell effector activity and peripheral CD8+ T-cell memory in the respiratory tract. Nat. Med 8:54–60 [DOI] [PubMed] [Google Scholar]
- 106.Huygens A, Lecomte S, Tackoen M, Olislagers V, Delmarcelle Y, et al. 2015. Functional exhaustion limits CD4+ and CD8+ T-cell responses to congenital cytomegalovirus infection. J. Infect. Dis 212:484–94 [DOI] [PubMed] [Google Scholar]
- 107.Kollmann TR, Reikie B, Blimkie D, Way SS, Hajjar AM, et al. 2007. Induction of protective immunity to Listeria monocytogenes in neonates. J. Immunol 178:3695–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fadel SA, Cowell LG, Cao S, Ozaki DA, Kepler TB, et al. 2006. Neonate-primed CD8+ memory cells rival adult-primed memory cells in antigen-driven expansion and anti-viral protection. Int. Immunol 18:249–57 [DOI] [PubMed] [Google Scholar]
- 109.Sarzotti M, Robbins DS, Hoffman PM. 1996. Induction of protective CTL responses in newborn mice by a murine retrovirus. Science 271:1726–28 [DOI] [PubMed] [Google Scholar]
- 110.Hiwarkar P, Hubank M, Qasim W, Chiesa R, Gilmour KC, et al. 2017. Cord blood transplantation recapitulates fetal ontogeny with a distinct molecular signature that supports CD4+ T-cell reconstitution. Blood Adv. 1:2206–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hiwarkar P, Qasim W, Ricciardelli I, Gilmour K, Quezada S, et al. 2015. Cord blood T cells mediate enhanced antitumor effects compared with adult peripheral blood T cells. Blood 126:2882–91 [DOI] [PubMed] [Google Scholar]
- 112.Lee YS, Kim TS, Kim DK. 2011. T lymphocytes derived from human cord blood provide effective antitumor immunotherapy against a human tumor. BMC Cancer 11:225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.St. John LS, Wan L, He H, Garber HR, Clise-Dwyer K, et al. 2016. PR1-specific cytotoxic T lymphocytes are relatively frequent in umbilical cord blood and can be effectively expanded to target myeloid leukemia. Cytotherapy 18:995–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hanley PJ, Cruz CR, Savoldo B, Leen AM, Stanojevic M, et al. 2009. Functionally active virus-specific T cells that target CMV, adenovirus, and EBV can be expanded from naive T-cell populations in cord blood and will target a range of viral epitopes. Blood 114:1958–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Schonland SO, Zimmer JK, Lopez-Benitez CM, Widmann T, Ramin KD, et al. 2003. Homeostatic control of T-cell generation in neonates. Blood 102:1428–34 [DOI] [PubMed] [Google Scholar]
- 116.Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, et al. 2013. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 121:573–84 [DOI] [PubMed] [Google Scholar]
- 117.Schaub B, Liu J, Hoppler S, Schleich I, Huehn J, et al. 2009. Maternal farm exposure modulates neonatal immune mechanisms through regulatory T cells. J. Allergy Clin. Immunol 123:774–82.e5 [DOI] [PubMed] [Google Scholar]
- 118.Risnes KR, Belanger K, Murk W, Bracken MB. 2011. Antibiotic exposure by 6 months and asthma and allergy at 6 years: findings in a cohort of 1,401 US children. Am. J. Epidemiol 173:310–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shaw SY, Blanchard JF, Bernstein CN. 2010. Association between the use of antibiotics in the first year of life and pediatric inflammatory bowel disease. Am. J. Gastroenterol 105:2687–92 [DOI] [PubMed] [Google Scholar]
- 120.Kronman MP, Zaoutis TE, Haynes K, Feng R, Coffin SE. 2012. Antibiotic exposure and IBD development among children: a population-based cohort study. Pediatrics 130:e794–803 [DOI] [PMC free article] [PubMed] [Google Scholar]