

Abstract
Objective
Fluzoparib (SHR3162) is a novel, potent poly(ADP-ribose) polymerases (PARP)1, 2 inhibitor that showed anti-tumor activity in xenograft models. We conducted a phase I, first-in-human, dose-escalation and expansion (D-Esc and D-Ex) trial in patients with advanced solid cancer.
Methods
This was a 3+3 phase I D-Esc trial with a 3-level D-Ex at 5 hospitals in China. Eligible patients for D-Esc had advanced solid tumors refractory to standard therapies, and D-Ex enrolled patients with ovarian cancer (OC). Fluzoparib was administered orally once or twice daily (bid) at 11 dose levels from 10 to 400 mg/d. Endpoints included dose-finding, safety, pharmacokinetics, and antitumor activity.
Results
Seventy-nine patients were enrolled from March, 2015 to January, 2018 [OC (47, 59.5%); breast cancer (BC) (16, 20.3%); colorectal cancer (8, 10.1%), other tumors (8, 10.1%)]; 48 patients were treated in the D-Esc arm and 31 in the D-Ex arm. The maximum tolerated dose (MTD) was 150 mg bid, with a half-life of 9.14 h. Grade 3/4 adverse events included anemia (7.6%) and neutropenia (5.1%). The objective response rate (ORR) was 30% (3/10) in patients with platinum-sensitive OC and 7.7% (1/13) in patients with BC. Among patients treated with fluzoparib ≥120 mg/d, median progression-free survival (mPFS) was 7.2 [95% confidence interval (95% CI), 1.8−9.3] months in OC, 9.3 (95% CI, 7.2−9.3) months in platinum-sensitive OC, and 3.5 (range, 2.0−28.0) months in BC. In patients with germline BC susceptibility gene mutation (gBRCAMut) (11/43 OC; 2/16 BC), mPFS was 8.9 months for OC (range, 1.0−23.2; 95% CI, 1.0−16.8) and 14 and 28 months for BC (those two patients both also had somaticBRCAMut).
Conclusions
The MTD of fluzoparib was 150 mg bid in advanced solid malignancies. Fluzoparib demonstrated single-agent antitumor activity in BC and OC, particularly in BRCAMut and platinum-sensitive OC.
Keywords: Phase I, PARP inhibitor (fluzoparib), solid tumor, pharmacokinetics, safety, antitumor activity
Introduction
The incidence of breast cancer (BC) is the highest in women. At present, the treatment of BC has tended to be standardized. Whether in the early or late stages, there are certain treatment rules to follow. However, there are still huge challenges in the treatment of advanced BC, and more new drugs are needed to improve the survival rate of BC (1). Poly(ADP-ribose) polymerases (PARPs) are important DNA repair enzymes. PARP inhibitors (PARPis) have shown promising efficacy in treatment and maintenance therapy of ovarian cancer (OC) (1,2). PARPis, including olaparib, rucaparib, niraparib and talazoparib, have been approved to treat OC and BC with germline breast cancer susceptibility gene mutation (gBRCAMut) (3-6). Olaparib was the first PARP inhibitor receiving USA Food and Drug Administration approval in 2014 to treat women with OC associated with a deleterious gBRCAMut(3,7). Rucaparib is also approved to treat women with germline or somatic BRCAMut or homologous recombination deficiencies (4,8). Niraparib, olaparib and rucaparib are approved for maintenance treatment of recurrent, platinum-sensitive OC, regardless of BRCAMut status (5,9,10), and olaparib and talazoparib are approved for the treatment of patients with HER2-negative metastatic BC with a gBRCAMut (11).
Fluzoparib (SHR3162) is an orally bioavailable PARP1, 2 inhibitor, with a half-maximum inhibitory concentration (IC50) of 2.0 nmol/L for PARP1, comparable to olaparib with an IC50 of 1.5 nmol/L. Fluzoparib inhibits tumor growth in both cell lines and MDA-MB-436 (BRCA1-deficient) xenograft models with loss of BRCA function. In particular, fluzoparib combined with paclitaxel and apatinib showed improved antitumor efficacy in a murine model compared with single paclitaxel or apatinib, without increased toxicity (12). On the basis of these pre-clinical results, we conducted the first-in-human, phase I, dose-escalation (D-Esc) trial of fluzoparib in patients with advanced solid malignancies, with a dose-expansion (D-Ex) cohort in patients with advanced OC. The study evaluated the safety, tolerability, maximum tolerated dose (MTD), and pharmacokinetic (PK) profile of fluzoparib, with a preliminary assessment of antitumor activity.
Materials and methods
Study design and procedures
This was a two-part, phase I, D-Esc and D-Ex study of fluzoparib led by the Fifth Medical Centre of Chinese PLA General Hospital and Peking University Cancer Hospital, with three additional centres (Tianjin Medical University Cancer Institute and Hospital; Sun Yat-sen University Cancer Centre; and the Comprehensive Cancer Centre of Drum Tower Hospital, the Affiliated Drum Tower Hospital of Nanjing University Medical School in China.
The study was approved by individual Ethics Committees at each site. All participants provided written informed consent before undergoing study-specific procedures. The study was conducted in accordance with the Declaration of Helsinki and the principles of good clinical practice. This trial was registered at ClinicalTrials.gov (NCT03509636).
Study design
D-Esc
A standard 3+3 design was used for the D-Esc arm with a starting fluzoparib dose of 10 mg/d by mouth once or twice daily (qd or bid). We increased doses at 100% fixed-dose increments to a dose of 160 mg/d. At fluzoparib doses >200 mg/d, dose increments were restricted to ≤50%. If a patient had a dose limiting toxicity (DLT) in the first cycle, cohorts were expanded to 6 patients. If 2 of 6 patients had DLTs in the first cycle, dose escalation ceased. The MTD was the highest dose at which at least one patient in a cohort of 6 had a DLT in the first cycle; and was deemed to be the recommended phase II dose unless a lower dose was found to have sufficient biological activity and drug exposure (biologic effective dose).
DLTs were assessed using National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 4.0 (13) and defined as treatment-related adverse events (AEs) that occurred in cycle one, including grade 4 anemia, thrombocytopenia or neutropenia; grade ≥3 neutropenia with fever ≥38.5 °C; grade ≥3 thrombocytopenia with clinically significant bleeding; and any grade ≥3 non-hematological AE, except for well-managed grade 3 nausea or vomiting. Patients who had DLTs in the first cycle held fluzoparib until toxicity improved to grade 1 or better, and then could restart therapy at the original or reduced dose. Study treatment was continued until disease progression, consent withdrawal, unacceptable toxic effects, or patients’ request to withdraw from the study.
D-Ex
Enrolment in the D-Ex arm proceeded after the MTD was determined. The absorption of fluzoparib based on PK data was used to determine qd or bid dosing. The D-Esc arm evaluated 3 dose groups, (the minimum effective dose, the MTD, and the dose in between). Eight to 10 participants with advanced OC were enrolled in each D-Ex group.
Evaluations
Each cycle was 28 d. Patients were assessed at baseline and on d 1, 3, 7, 15, 21 and 28 during cycle 1, on d 14 and 28 during cycle 2, and on d 28 of every subsequent treatment cycle. AEs were recorded from the first fluzoparib dose until 30 d after the last dose. Tumor responses were assessed by Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1 (14) after every 2 cycles by computed tomography (CT) or magnetic resonance imaging (MRI).
Collection of peripheral blood for detection of germline (g) BRCA1/2gene mutations (gBRCA1/2Mut) was required for all enrolled patients with OC or BC before starting study treatment. Testing was performed at the Beijing Genomics Institute. Additional plasma was collected for circulating tumor (ct) DNA detection as part of clinical routine practice for further detection of potential biomarkers. ctDNA detection was performed using next generation sequencing (NGS) by Huidu Shanghai Medical Sciences.
Participants
Eligibility for D-Esc included patients with advanced solid malignancies that were either refractory to standard therapies or for which no standard therapy exists, aged 18−70 years old, and with at least one measurable lesion according to RECIST, version 1.1 (14), a life expectancy of at least 12 weeks, an Eastern Cooperative Oncology Group (ECOG) performance scores of 0 or 1, and adequate hematologic, hepatic and renal functions. Eligibility for D-Ex was similar but included only patient with advanced OC with disease progression on or following standard therapy, preferably with platinum-sensitive disease. The following definitions of platinum responsiveness were used: platinum sensitive is recurrence ≥6 months after the last platinum treatment, platinum-resistant is recurrence <6 months after last platinum treatment and platinum-refractory is in progression during platinum treatment.
Patients were excluded if they had previously received a PARP inhibitor; they were unable to swallow or had gastrointestinal absorption dysfunction; they had residual grade ≥2 toxic effects from previous treatment; they were pregnant or breastfeeding; or they had uncontrolled medical disorders.
PK analysis
Plasma samples were assayed for fluzoparib concentrations using a validated high-performance liquid chromatograph-mass spectrometer (LC/MS-MS) detection method. Fluzoparib PK parameters following single and twice dosing were obtained using standard noncompartmental analysis methods in Phoenix WinNonlin7.0. Estimated PK parameters included peak concentration (Cmax); time to Cmax (Tmax); area under curve (AUC) from time 0 to time of last quantifiable concentration (AUC0−t), and AUC from time 0 extrapolated to infinity (AUC0−∞); Apparent total clearance of the drug (CL/F); apparent volume of distribution (Vz/F); and half-life. The bid PK parameters included minimum plasma concentration and CL/F at steady state (CLss/F).
Statistical analysis
The primary objective in the D-Esc arm of the study was to determine the DLT and MTD of oral fluzoparib; and the secondary objective included safety and PK profiles. For the D-Ex arm, efficacy parameters in OC were investigated based on RECIST by investigator assessment. The numbers and percentages of patients achieving a response or disease control were summarized and 95% confidence intervals (95% CIs) were calculated using the Clopper-Pearson method. Progression-free survival (PFS) was summarized using the Kaplan-Meier method. The data cut-off was March 1, 2019. SAS Analytics Software (Version 9.1; SAS Institute Inc., Cary, USA) was used for data analyses.
Results
Between March 3, 2015 and January 3, 2018, 79 patients with advanced solid tumors [OC: n=47 (59.5%); BC: n=16 (20.3%); colorectal cancer: n=8 (10.1%); other tumors: n=8 (10.1%)] were enrolled at 5 hospitals of China. The patients’ median age was 53 (range, 30−68) years old, 43 (54.4%) had ≥3 metastatic sites, and 48 (60.8%) had visceral disease (Table 1 ). All patients had received prior treatment, including 52 (65.8%) who received ≥3 prior lines of chemotherapy (including platinum-based therapies). Forty-eight patients were treated in the D-Esc arm and received fluzoparib at 11 dose levels from 10 to 400 mg/d. Thirty-one patients with OC were treated in the D-Ex arm at 3 dose levels of 80, 100 and 150 mg bid.
1. Demographic and baseline clinical characteristics (N=79).
| Parameter | Participants
[n (%)] |
| ECOG, Eastern Cooperative Oncology Group; BRCA, breast cancer susceptibility gene; OC, ovarian cancer; BC, breast cancer; *, chemotherapies were for advanced disease. | |
| Sex | |
| Female | 67 (84.8) |
| Male | 12 (15.2) |
| Age (year) [median (range)] | 53 (30−68) |
| ECOG scores | |
| 0 | 58 (73.4) |
| 1 | 21 (26.6) |
| Cancer type | |
| Ovarian | 47 (59.5) |
| Breast | 16 (20.3) |
| Colorectal | 8 (10.1) |
| Esophageal | 1 (1.3) |
| Fallopian tube | 1 (1.3) |
| Gastric | 4 (5.1) |
| Pancreatic | 2 (2.5) |
| BRCA status in patients with OC (n=43) | |
| BRCA mutation | 11 (25.6) |
| BRCA non-mutation | 31 (72.1) |
| Unknown | 1 (2.3) |
| BRCA status in patients with BC (n=16) | |
| BRCAmutation | 2 (12.5) |
| BRCA non-mutation | 14 (87.5) |
| Number of prior chemotherapies* (n=77) | |
| 1 prior chemotherapy | 9 (11.4) |
| 2 prior chemotherapy | 16 (20.3) |
| ≥3 prior chemotherapy | 52 (65.8) |
| Endocrine therapy for patients with BC (n=16) | |
| Yes | 9 (56.3) |
| No | 7 (43.7) |
| No. of metastasis site | |
| 1 | 12 (15.2) |
| 2 | 24 (30.4) |
| ≥3 | 43 (54.4) |
| Site of metastasis | |
| Visceral disease (liver, lung, spleen) | 48 (60.8) |
| No-visceral disease | 31 (39.2) |
Tolerability
Table 2 shows the numbers of patients per dose level, observed DLTs, dose reductions, and median treatment days for both D-Esc and D-Ex. DLTs in cycle 1 occurred in 1 of 6 patients at 10 mg/d fluzoparib and 2 of 4 patients at 400 mg/d (200 mg bid). The patient treated at 10 mg/d experienced a grade 3 lipase increase that was not considered related to study drug. Two of 4 patients treated at 400 mg/d experienced DLTs, including grade 3 asthenia, nausea and vomiting, and grade 3 abdominal pain, and 400 mg/d was deemed to be intolerable (Table 2 ). All DLTs resolved after temporary interruption of fluzoparib. No DLTs were observed in a group of 3 assessable patients treated at 300 mg/d (150 mg bid); and this dose was therefore determined to be the MTD. In the D-Ex arm (n=31), patients received fluzoparib at 80 mg (n=10), 100 mg (n=10), or 150 mg (n=11) bid. All three doses were well-tolerated.
2. Dose escalation schema, DLTs, dose reductions, and number of treatment days.
| Dose level | Patients (N=79) | DLTs in first cycle | Dose reduction (any cycle) (n) | No. of treatment days [median (range)] | |||
| Dose escalation (n=48) | Dose expansion (n=31) | Number | Description | ||||
| DLT, dose-limiting toxicity. | |||||||
| 10 mg qd | 6 | − | 0 | − | 0 | 46 (20−88) | |
| 10 mg bid | 4 | − | 0 | − | 0 | 88 (22−170) | |
| 20 mg bid | 3 | − | 0 | − | 0 | 106 (47−114) | |
| 40 mg bid | 3 | − | 0 | − | 0 | 57 (57−60) | |
| 120 mg qd | 8 | − | 0 | − | 0 | 112 (57−227) | |
| 60 mg bid | 8 | − | 0 | − | 1 | 86 (28−800+) | |
| 160 mg qd | 3 | − | 0 | − | 0 | 248 (44−393) | |
| 80 mg bid | 3 | 10 | 0 | − | 2 | 57 (28−632+) | |
| 100 mg bid | 3 | 10 | 0 | − | 0 | 257 (14−564) | |
| 150 mg bid | 3 | 11 | 0 | − | 3 | 56 (28−443+) | |
| 200 mg bid | 4 | − | 2 | 1 patient experienced grade 3 fatigue, nausea and vomiting and 1 patient experienced grade 3 abdominal pain | 3 | 66 (44−194) | |
Safety
All grades of treatment-related AEs occurring in at least 10% of participants are listed in Table 3 . The most common non-hematological AEs were fatigue (48.1%), nausea (34.2%), decreased appetite (29.1%), vomiting (17.7%) and weight loss (10.1%). The most common hematological AEs were anemia (53.2%), decreased neutrophil count (24.1%), and thrombocytopenia (17.7%). These AEs were mainly grade 1−2, and were manageable with supportive care and dose reduction. The most common grade 3−4 AEs were anemia (7.6%) and decreased neutrophil count (5.1%) at all groups (Table 3 ). Hematological AEs were uncomplicated and reversible.
3. Common AEs (all grades, >10%; and 3−4 grade) assessed by investigator as related to treatment by dose.
| AEs | 10−80 mg/d (n=16) | 120 mg/d (n=16) | 160 mg/d (n=16) | 200 mg/d (n=13) | 300 mg/d (n=14) | 400 mg/d (n=4) | Total (N=79) | |||||||||||||
| All
grades |
3−4 grade | All
grades |
3−4 grade | All
grades |
3−4 grade | All
grades |
3−4 grade | All
grades |
3−4 grade | All
grades |
3−4 grade | All
grades |
3−4 grade | |||||||
| AE, adverse event; WBC, white blood cell; ALT, alanine aminotransferase; AST, aspartate aminotransferase; RBC, red blood cell. | ||||||||||||||||||||
| Anemia | 5 (31.3) | 1 (6.3) | 8 (50.0) | 2 (12.5) | 9 (56.3) | 1 (6.3) | 8 (61.5) | 1 (7.7) | 8 (57.1) | 0 | 4 (100) | 1 (25.0) | 42 (53.2) | 6 (7.6) | ||||||
| WBC count decreased | 7 (43.8) | 0 | 7 (43.8) | 3 (18.8) | 4 (25.0) | 0 | 5 (38.5) | 0 | 4 (28.6) | 0 | 3 (75.0) | 0 | 30 (38.0) | 3 (3.8) | ||||||
| Neutrophil count decreased | 4 (25.0) | 0 | 6 (37.5) | 3 (18.8) | 1 (6.3) | 0 | 5 (38.5) | 0 | 1 (7.1) | 0 | 2 (50.0) | 1 (25.0) | 19 (24.1) | 4 (5.1) | ||||||
| Platelet count decreased | 2 (12.5) | 0 | 2 (12.5) | 1 (6.3) | 3 (18.8) | 0 | 2 (15.4) | 1 (7.7) | 2 (14.3) | 0 | 3 (75.0) | 1 (25.0) | 14 (17.7) | 3 (3.8) | ||||||
| ALT increased | 3 (18.8) | 0 | 1 (6.3) | 0 | 4 (25.0) | 0 | 2 (15.4) | 0 | 2 (14.3) | 0 | 2 (50.0) | 0 | 14 (17.7) | 0 | ||||||
| AST increased | 2 (12.5) | 0 | 2 (12.5) | 0 | 4 (25.0) | 0 | 4 (30.8) | 0 | 2 (14.3) | 0 | 1 (25.0) | 0 | 15 (19.0) | 0 | ||||||
| Blood albumin decreased | 1 (6.3) | 0 | 3 (18.8) | 0 | 4 (25.0) | 0 | 4 (30.8) | 0 | 3 (21.4) | 0 | 1 (25.0) | 0 | 16 (20.3) | 0 | ||||||
| Blood triglycerides increased | 2 (12.5) | 0 | 11 (68.8) | 0 | 1 (6.3) | 0 | 4 (30.8) | 0 | 5 (35.7) | 0 | 0 | 0 | 23 (29.1) | 0 | ||||||
| Blood cholesterol increased | 2 (12.5) | 0 | 5 (31.3) | 0 | 1 (6.3) | 0 | 2 (15.4) | 0 | 4 (28.6) | 0 | 0 | 0 | 14 (17.7) | 0 | ||||||
| Blood potassium decreased | 1 (6.3) | 0 | 3 (18.8) | 0 | 4 (25.0) | 1 (6.3) | 0 | 0 | 0 | 0 | 0 | 0 | 8 (10.1) | 1 (1.3) | ||||||
| Blood calcium decreased | 2 (12.5) | 0 | 5 (31.3) | 0 | 4 (25.0) | 1 (6.3) | 1 (7.7) | 0 | 0 | 0 | 0 | 0 | 12 (15.2) | 1 (1.3) | ||||||
| Blood creatinine increased | 0 | 0 | 1 (6.3) | 0 | 1 (6.3) | 0 | 2 (15.4) | 0 | 4 (28.6) | 0 | 0 | 0 | 8 (10.1) | 0 | ||||||
| Lipase increased | 3 (18.8) | 1 (6.3) | 3 (18.8) | 0 | 3 (18.8) | 1 (6.3) | 1 (7.7) | 0 | 0 | 0 | 1 (25.0) | 1 (25.0) | 11 (13.9) | 3 (3.8) | ||||||
| Amylase increased | 0 | 0 | 2 (12.5) | 0 | 3 (18.8) | 0 | 1 (7.7) | 0 | 1 (7.1) | 0 | 1 (25.0) | 0 | 8 (10.1) | 0 | ||||||
| Proteinuria | 6 (37.5) | 1 (6.3) | 3 (18.8) | 0 | 4 (25.0) | 0 | 1 (7.7) | 0 | 1 (7.1) | 0 | 0 | 0 | 15 (19.0) | 1 (1.3) | ||||||
| WBC urine positive | 2 (12.5) | 0 | 2 (12.5) | 0 | 2 (12.5) | 0 | 1 (7.7) | 0 | 1 (7.1) | 0 | 0 | 0 | 8 (10.1) | 0 | ||||||
| RBC urine positive | 0 | 0 | 3 (18.8) | 0 | 1 (6.3) | 0 | 1 (7.7) | 0 | 3 (21.4) | 0 | 0 | 0 | 8 (10.1) | 0 | ||||||
| Nausea | 6 (37.5) | 0 | 4 (25.0) | 1 (6.3) | 7 (43.8) | 0 | 2 (15.4) | 0 | 6 (42.9) | 0 | 2 (50.0) | 1 (25.0) | 27 (34.2) | 2 (2.5) | ||||||
| Vomiting | 2 (12.5) | 0 | 2 (12.5) | 0 | 3 (18.8) | 0 | 2 (15.4) | 0 | 4 (28.6) | 1 (7.1) | 1 (25.0) | 1 (25.0) | 14 (17.7) | 2 (2.5) | ||||||
| Decreased appetite | 2 (12.5) | 0 | 5 (31.3) | 2 (12.5) | 8 (50.0) | 0 | 3 (23.1) | 0 | 3 (21.4) | 0 | 2 (50.0) | 0 | 23 (29.1) | 2 (2.5) | ||||||
| Diarrhea | 0 | 0 | 2 (12.5) | 0 | 3 (18.8) | 0 | 6 (46.2) | 1 (7.7) | 3 (21.4) | 1 (7.1) | 0 | 0 | 14 (17.7) | 2 (2.5) | ||||||
| Constipation | 0 | 0 | 2 (12.5) | 0 | 2 (12.5) | 0 | 3 (23.1) | 0 | 1 (7.1) | 0 | 0 | 0 | 8 (10.1) | 0 | ||||||
| Abdominal pain | 1 (6.3) | 0 | 3 (18.8) | 0 | 3 (18.8) | 1 (6.3) | 3 (23.1) | 0 | 6 (42.9) | 0 | 2 (50.0) | 2 (50.0) | 18 (22.8) | 3 (3.8) | ||||||
| Abdominal distension | 1 (6.3) | 1 (6.3) | 2 (12.5) | 0 | 5 (31.3) | 0 | 2 (15.4) | 0 | 3 (21.4) | 0 | 0 | 0 | 13 (16.5) | 1 (1.3) | ||||||
| Back pain | 1 (6.3) | 0 | 1 (6.3) | 0 | 3 (18.8) | 0 | 1 (7.7) | 0 | 3 (21.4) | 0 | 1 (25.0) | 0 | 10 (12.7) | 0 | ||||||
| Fatigue | 4 (25.0) | 0 | 7 (43.8) | 0 | 11 (68.8) | 2 (12.5) | 7 (53.8) | 1 (7.7) | 6 (42.9) | 0 | 3 (75.0) | 1 (25.0) | 38 (48.1) | 3 (3.8) | ||||||
| Weight decreased | 0 | 0 | 1 (6.3) | 0 | 3 (18.8) | 0 | 2 (15.4) | 0 | 0 | 0 | 2 (50.0) | 0 | 8 (10.1) | 0 | ||||||
Nine patients required dose reductions. Three patients discontinued therapy because of AEs, including 2 with fatigue (one during the first cycle and the other between the first and second cycle), and one with bowel obstruction (without clear evidence of disease progression). No treatment-related deaths occurred.
PK
Mean fluzoparib plasma concentration-time profiles after once and twice daily doses of fluzoparib were assayed. Fluzoparib PK parameters resulting from the analysis of the plasma concentration-time profiles are shown in Tables 4 −6 and Figure 1 . Fluzoparib demonstrated rapid absorption, with the maximum plasma concentration Cmax generally reached within 3 h after all evaluated doses and following both qd and bid dosing, with subsequent biphasic decrease and elimination. Cmax and exposure to the drug (measured by the AUC) were proportional to dose. With once daily dosing of 80, 100 and 150 mg, Cmaxwas approximately 2.53, 3.55 and 4.77 μg/mL, respectively, and AUC0−t was 42.1, 53.0 and 78.7 h·μg/mL, respectively. Steady-state plasma concentrations were reached by 15 d of daily dosing across all doses. In patients receiving fluzoparib at 80, 100 and 150 mg bid, Css,max at d 15 was 5.18, 5.94 and 8.45 μg/mL, respectively. Fluzoparib was well distributed into tissue compartments, and the estimated value of Vz/F was well in excess of the volume of the systemic circulatory space. Vz/F at 80, 100 and 150 mg once daily, were 33.3, 35.9 and 29.3 L, respectively; Vz/F at 80, 100 and 150 mg bid, were 35.8, 38.5 and 34.6 L, respectively. Plasma elimination followed biphasic kinetics with half-lives single dose (t1/2) was 12.3 h, 11.3 h and 10.2 h; bid was 11.4 h, 10.8 h and 9.14 h, respectively. Linear elimination across dose levels was apparent following both once and bid dosing as evidenced by parallel terminal phases of the log-linear profiles and similar CL/F estimates across dose levels. At equivalent total daily doses, AUC and Cmax for both the once-daily and bid dosing were similar on d 15; however, as expected, higher trough concentrations and much smaller peak-to-trough drug levels were achieved at steady state for bid dosing, supporting bid administration (Figure 1 , Table 4 ,5 ).
4. PK parameters and PARP inhibition following single fluzoparib dose.
| PK parameter | 80 mg (n=10) | 100 mg (n=13) | 150 mg (n=13) | ||||||||
| Mean | SD | CV (%) | Mean | SD | CV (%) | Mean | SD | CV (%) | |||
| PK, pharmacokinetic; PARP, poly(ADP-ribose) polymerases; Tmax, time to Cmax; Cmax, peak concentration; AUC0−t, area under the cueve from time 0 to last quantifiable concentration; AUC0−∞, AUC from time 0 extrapolated to infinity; λz, first-order rate constant of terminal phase; t1/2z, terminal half-life; MRT, mean retention time; Vz/F, apparent volume of distribution; CL/F, apparent total clearance of the drug; SD, standard deviation; CV, coefficient of variation. | |||||||||||
| Tmax (h) | 3.00 (1.50−4.00) | − | − | 3.00 (1.50−6.00) | − | − | 2.50 (1.00−6.00) | − | − | ||
| Cmax(μg/mL) | 2.53 | 0.67 | 26.7 | 3.55 | 1.52 | 42.9 | 4.77 | 1.21 | 25.4 | ||
| AUC0−t(h·μg/mL) | 42.1 | 15.8 | 37.5 | 53.0 | 31.8 | 59.9 | 78.7 | 30.6 | 39.0 | ||
| AUC0−∞ (h·μg/mL) | 46.5 | 20.7 | 44.5 | 58.2 | 36.5 | 62.7 | 82.8 | 33.9 | 40.9 | ||
| λz(1/h) | 0.0633 | 0.0214 | 33.8 | 0.0696 | 0.0260 | 37.4 | 0.0722 | 0.0191 | 26.5 | ||
| t1/2z(h) | 12.3 | 4.9 | 39.7 | 11.3 | 4.1 | 36.5 | 10.2 | 2.4 | 23.6 | ||
| MRT0−∞(h) | 17.8 | 6.8 | 38.1 | 16.3 | 6.3 | 38.8 | 15.1 | 3.2 | 21.3 | ||
| Vz/F (L) | 33.3 | 13.4 | 40.2 | 35.9 | 15.5 | 43.0 | 29.3 | 8.3 | 28.3 | ||
| CL/F (L/h) | 2.04 | 0.86 | 42.2 | 2.71 | 1.90 | 70.1 | 2.19 | 1.09 | 50.0 | ||
6. Clinical response rate assessed by RECIST v1.0 by cancer type in patients treated with fluzoparib.
| Variables | Ovarian cancer | Breast cancer | Colorectal cancer | Gastric cancer and other tumor types | Total |
| RECIST, Response Evaluation Criteria in Solid Tumor; CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease; ORR, objective response rate; DCR, disease control rate. | |||||
| Total subjects | 47 | 16 | 8 | 8 | 79 |
| Efficacy evaluable subjects | 37 | 13 | 8 | 7 | 65 |
| CR | 0 | 0 | 0 | 0 | 0 |
| PR | 3 | 1 | 0 | 0 | 4 |
| SD | 14 | 2 | 0 | 0 | 16 |
| PD | 20 | 10 | 8 | 7 | 45 |
| ORR based on total subjects (%) | 6.4 | 6.3 | 0 | 0 | 5.1 |
| DCR based on total subjects (%) | 36.2 | 18.8 | 0 | 0 | 25.3 |
| ORR based on evaluable subjects (%) | 8.1 | 7.7 | 0 | 0 | 6.2 |
| DCR based on evaluable subjects (%) | 45.9 | 23.1 | 0 | 0 | 30.8 |
1. Fluzoparib pharmacokinetic (PK) parameters. (A) Plasma concentration-time profiles for fluzoparib following a single dose fluzoparib at 10 mg (n=6), 80 mg (n=10), 100 mg (n=13), 120 mg (n=8), 150 mg (n=14), 160 mg (n=3) or 200 mg (n=4) (
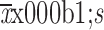 ); (B) Plasma concentration-time profiles on d 15 for fluzoparib following multiple dose fluzoparib at 10 mg bid (n=4), 20 mg bid (n=3), 40 mg bid (n=3), 60 mg bid (n=8), 80 mg bid (n=12), 100 mg bid (n=12), 150 mg bid (n=13) or 200 mg bid (n=3) (
); (B) Plasma concentration-time profiles on d 15 for fluzoparib following multiple dose fluzoparib at 10 mg bid (n=4), 20 mg bid (n=3), 40 mg bid (n=3), 60 mg bid (n=8), 80 mg bid (n=12), 100 mg bid (n=12), 150 mg bid (n=13) or 200 mg bid (n=3) (
 ); (C) Correlation analysis between Cmax and 7 different dose levels after multiple dose of fluzoparil (10−150 mg bid) at d 15; (D) Correlation analysis between AUC 0−10 h and 7 different dose levels after multiple dose of fluzoparil (0−150 mg bid) at d 15.
); (C) Correlation analysis between Cmax and 7 different dose levels after multiple dose of fluzoparil (10−150 mg bid) at d 15; (D) Correlation analysis between AUC 0−10 h and 7 different dose levels after multiple dose of fluzoparil (0−150 mg bid) at d 15.

5. PK parameters and PARP inhibition following twice daily fluzoparib dosing.
| PK Parameter | 80 mg bid (n=10) | 100 mg bid (n=13) | 150 mg bid (n=13) | ||||||||
| Mean | SD | CV (%) | Mean | SD | CV (%) | Mean | SD | CV (%) | |||
| PK, pharmacokinetic; PARP, poly(ADP-ribose) polymerases; Tmax, time to peak concentration (Cmax); Css,max, Cmax at steady state; Css,min, minimum concentration at steady state; Css,avg, average steady-state concentration; AUC0–10 h, area under the curve from time 0 to10 h; λz, first-order rate constant of terminal phase; t1/2, terminal half-life; CLss/F, apparent total clearance of the drug at steady state; Vz/F, apparent volume of distribution; DF, coefficient of fluctuation; Rac_Cmax, accumulation in terms of Cmax; Rac_AUC, accumulation in terms of AUC; SD, standard deviation; CV, coefficient of variation. | |||||||||||
| Tmax(h) | 2.50 (1.00−3.00) | − | − | 3.00 (2.00−6.00) | − | − | 3.00 (1.00−4.00) | − | − | ||
| Css,max(μg/mL) | 5.18 | 1.68 | 32.4 | 5.94 | 2.99 | 50.3 | 8.45 | 1.83 | 21.6 | ||
| Css,min(μg/mL) | 2.69 | 1.07 | 39.8 | 3.15 | 1.96 | 62.4 | 4.06 | 1.05 | 25.8 | ||
| Css,avg (μg/mL) | 3.82 | 1.26 | 32.8 | 4.52 | 2.55 | 56.4 | 6.08 | 1.34 | 22.1 | ||
| AUC0−10 h(h·μg/mL) | 38.2 | 12.6 | 32.8 | 45.2 | 25.5 | 56.4 | 60.8 | 13.4 | 22.1 | ||
| λz(1/h) | 0.0706 | 0.0286 | 40.5 | 0.0763 | 0.0306 | 40.1 | 0.0800 | 0.0177 | 22.2 | ||
| t1/2(h) | 11.40 | 4.86 | 42.6 | 10.80 | 4.78 | 44.4 | 9.14 | 2.38 | 26.0 | ||
| CLss/F (L/h) | 2.31 | 0.76 | 32.9 | 2.96 | 1.69 | 57.0 | 2.58 | 0.57 | 22.0 | ||
| Vz/F (L) | 35.8 | 14.9 | 41.6 | 38.5 | 27.6 | 71.6 | 34.6 | 14.5 | 41.9 | ||
| DF (%) | 66.7 | 12.0 | 18.0 | 68.1 | 17.8 | 26.2 | 73.2 | 17.3 | 23.7 | ||
| Rac_Cmax | 2.09 | 0.52 | 24.7 | 1.73 | 0.41 | 23.4 | 1.76 | 0.33 | 18.7 | ||
| Rac_AUC | 2.23 | 0.51 | 22.8 | 2.08 | 0.39 | 18.5 | 1.86 | 0.35 | 18.7 | ||
Efficacy
Of the 65 patients whose disease was assessable for response by RECIST, 37 patients had OC, 13 patients had BC, 8 patients had colorectal cancer and 7 patients had gastric cancer or other tumor types (Table 6 ). The objective response rate (ORR) in patients with OC was 8.1%, including 3 partial response (PR), and 14 patients had stable disease (SD) lasting at least 24 weeks, for a disease control rate (DCR) of 45.9% at 24 weeks (Table 6 ). Among 43 patients with OC dosed at ≥120 mg/d, median PFS (mPFS) was 7.2 (95% CI, 1.8−9.3) months. Among the 11 efficacy-evaluable patients with platinum-sensitive OC dosed at ≥120 mg/d, ORR was 30% and mPFS was 9.3 (95% CI, 7.2−9.3) months (Figure 2 , Supplementary Table S1 ,2 ).
2. Efficacy-evaluable patients with OC treated with fluzoparib ≥120 mg/d. OC, ovarian cancer; PR, partial response; PFI, progression-free interval; #, progression-free survival censor (stop treatment without progression or death); +, breast cancer susceptibility genes (BRCA) mutation.

S1. ORR by platinum-sensitivity in ovarian cancer patients treated with fluzoparib ≥120 mg/d.
| Platinum-based therapy | End point | 120 mg qd | 60 mg bid | 160 mg qd | 80 mg bid | 100 mg bid | 150 mg bid | 200 mg bid | Total |
| ORR, objective response rate. | |||||||||
| First line platinum-sensitive and last course platinum-sensitive | Total subjects (N) | 1 | 1 | 0 | 1 | 6 | 2 | 0 | 11 |
| Efficacy evaluable subjects (n) | 1 | 1 | 0 | 1 | 5 | 2 | 0 | 10 | |
| ORR based on total subjects [n (%)] | 1 (100) | 1 (100) | 0 | 0 | 1 (16.7) | 0 | 0 | 3 (27.3) | |
| ORR based on efficacy evaluable subjects [n (%)] | 1 (100) | 1 (100) | 0 | 0 | 1 (20.0) | 0 | 0 | 3 (30.0) | |
| First line platinum-sensitive and last course platinum resistant | Total subjects (N) | 0 | 0 | 1 | 4 | 0 | 4 | 1 | 10 |
| Efficacy evaluable subjects (n) | 0 | 0 | 1 | 3 | 0 | 3 | 1 | 8 | |
| ORR based on total subjects [n (%)] | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| ORR based on efficacy evaluable subjects [n (%)] | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| First line platinum-sensitive and last course platinum refractory | Total subjects (N) | 0 | 1 | 0 | 2 | 4 | 2 | 0 | 9 |
| Efficacy evaluable subjects (n) | 0 | 1 | 0 | 2 | 4 | 2 | 0 | 9 | |
| ORR based on total subjects [n (%)] | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| ORR based on efficacy evaluable subjects [n (%)] | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| First line platinum-resistant or refractory | Total subjects (N) | 0 | 0 | 0 | 4 | 2 | 6 | 1 | 13 |
| Efficacy evaluable subjects (n) | 0 | 0 | 0 | 2 | 2 | 3 | 1 | 8 | |
| ORR based on total subjects [n (%)] | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| ORR based on efficacy evaluable subjects [n (%)] | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
S2. PFS by platinum-sensitivity in ovarian cancer patients treated with fluzoparib ≥120 mg/d.
| Platinum-based therapy | End point | 120 mg qd | 60 mg bid | 160 mg qd | 80 mg bid | 100 mg bid | 150 mg bid | 200 mg bid | Total |
| PFS, progression-free survival; /, not applicable; −, not reached. | |||||||||
| First line platinum-sensitive and last course platinum-sensitive | Total subjects (N) | 1 | 1 | 0 | 1 | 6 | 2 | 0 | 11 |
| Efficacy evaluable subjects (n) | 1 | 1 | 0 | 1 | 5 | 2 | 0 | 10 | |
| mPFS (95% CI) based on total subjects (month) | 7.4 | 9.3 | / | − | − | − | / | 9.3 (7.2−9.3) | |
| mPFS (95% CI) based on efficacy evaluable subjects (month) | 7.4 | 9.3 | / | − | − | − | / | 9.3 (7.2−9.3) | |
| First line platinum-sensitive and last course platinum resistant | Total subjects (N) | 0 | 0 | 1 | 4 | 0 | 4 | 1 | 10 |
| Efficacy evaluable subjects (n) | 0 | 0 | 1 | 3 | 0 | 3 | 1 | 8 | |
| mPFS (95% CI) based on total subjects (month) | / | / | 8.3 | 3.6 | / | − | − | 3.6 (1.7−8.3) | |
| mPFS (95% CI) based on efficacy evaluable subjects (month) | / | / | 8.3 | 3.6 | / | − | − | 3.6 (1.7−8.3) | |
| First line platinum-sensitive and last course platinum refractory | Total subjects (N) | 0 | 1 | 0 | 2 | 4 | 2 | 0 | 9 |
| Efficacy evaluable subjects (n) | 0 | 1 | 0 | 2 | 4 | 2 | 0 | 9 | |
| mPFS (95% CI) based on total subjects (month) | / | − | / | − | − | 1.8 | / | 1.9 (1.7−1.9) | |
| mPFS (95% CI) based on efficacy evaluable subjects (month) | / | − | / | − | − | 1.8 | / | 1.9 (1.7−1.9) | |
| First line platinum-resistant or refractory | Total subjects (N) | 0 | 0 | 0 | 4 | 2 | 6 | 1 | 13 |
| Efficacy evaluable subjects (n) | 0 | 0 | 0 | 2 | 2 | 3 | 1 | 8 | |
| mPFS (95% CI) based on total subjects (month) | / | / | / | 1.8 | − | − | 1.7 | − | |
| mPFS (95% CI) based on efficacy evaluable subjects (month) | / | / | / | 1.8 | − | − | 1.7 | − | |
| Overall | Subjects (N) | 1 | 2 | 1 | 11 | 12 | 14 | 2 | 43 |
| Efficacy evaluable subjects (n) | 1 | 2 | 1 | 8 | 11 | 10 | 2 | 35 | |
| mPFS (95% CI) based on total subjects (month) | 7.4 | 9.3 | 6.3 | − | − | 1.9 | − | 7.2 (1.8−9.3) | |
| mPFS (95% CI) based on efficacy evaluable subjects (month) | 7.4 | 9.3 | 6.3 | − | − | 1.9 | − | 7.2 (1.8−9.3) | |
The ORR in patients with BC was 7.7% (1 PR), and 5 patients had SD for at least 8 weeks with 2 confirmed SD at 24 weeks. The DCR was 23.1% at 24 weeks (Table 6 ). Among BC patients dosed at ≥120 mg/d, PFS was 3.5 (range, 2.0−28.0) months, including one patient who is still on treatment at 28.0 months. As of the cut-off date (March 1, 2019), one patient with BC in the D-Esc arm and 3 patients with OC in the D-Ex arm continue on study therapy.
Of the patients with colorectal and gastric cancers or other tumor types, 6 patients had SD lasting 8 weeks, none confirmed at 24 weeks (Table 6 ).
Efficacy in patients with gBRCA1/2Mut
gBRCA1/2Mut was tested in 43 patients with OC and 16 patients with BC; 11 in 43 OC (7 gBRCA1Mut, 2 gBRCA2Mut, 2 both gBRCA1/2Mut) and 2 in 16 BC (one gBRCA2Mut, one both gBRCA1/2Mut) had a gBRCAMut. One of the two BC patients had a gBRCA2Mut that was classified as benign by American College of Medical Genetics (ACMG) Standards and Guidelines (15). Both BC patients with gBRCAMut also had plasma ctDNA analysis prior to fluzoparib treatment; both also had somatic (s) BRCAMut (Table 7 ). In 11 patients with OC and gBRCAMut, mPFS was 8.9 (range, 1.0−23.2; 95% CI, 1.0−16.8) months, with fluzoparib dosed at 80 mg bid in one, 100 mg bid in 5, and 150 mg bid in 5. For the 2 patients with BC with gBRCA1/2Mut and sBRCA1/2Mut, PFS was 28 months (fluzoparib 60 mg bid) and 14 months (fluzoparib 160 mg qd), respectively. As of the cut-off date of 3/1/2019, 4 patients continued on fluzoparib including one patient with BC (gBRCA2Mut and sBRCA1 60 mg bid, 28+ months) and 3 patients with OC (one gBRCA1/2Mut at 80 mg bid, 23+ months, one gBRCA1Mut and one with gBRCA2Mut both at 150 mg bid, 17+ and 16+ months PFS). The details of mutation status and efficacy in patients with gBRCAMut and sBRCAMut are shown in Figure 3 , Table 7 .
7. Site of mutation in germline (g) or somatic (s)BRCA .
| No. of patients | BRCAmutation type | Tumor type | Fluzoparib dose | Site of mutation | Clinically important | PFS
(month) |
| gBRCA, germline breast cancer susceptibility gene; OC, ovarian cancer; BC, breast cancer; PFS, progression-free survival; *, BRCA somatic mutation detected by next generation sequencing using plasma ctDNA; #, patients still on treatment. | ||||||
| 03002 | gBRCA1 | OC | 80 mg bid | c.5332+1delG | Pathogenic | 2.0 |
| 02041# | gBRCA1 | OC | 80 mg bid | c.5470_5477delATTGGGCA | Pathogenic | 23.2 |
| 01024 | gBRCA2 | OC | 100 mg bid | c.6405_6409delCTTAA | Pathogenic | 8.9 |
| 03004 | gBRCA1 | OC | 100 mg bid | c.2572C>T | Pathogenic | 12.6 |
| 01025 | gBRCA1 | OC | 100 mg bid | c.5035delC | Pathogenic | 2.0 |
| 02017 | gBRCA1 | OC | 120 mg qd | NM_007294.3:c.3756_3759delp.Leu 1252fs | Likely pathogenic | 9.3 |
| 03010 | gBRCA1 | OC | 150 mg bid | c.5470_5477delATTGGGCA | Pathogenic | 1.9 |
| gBRCA2 | c.3007C>G | Uncertain significance | ||||
| 03011# | gBRCA2 | OC | 150 mg bid | c.9117G>A | Pathogenic | 16.1 |
| 03009 | gBRCA1 | OC | 150 mg bid | c.5407-1G>A | Pathogenic | 1.0 |
| gBRCA2 | c.4599A>C, c.6325G>A | Likely benign | ||||
| 06001# | gBRCA1 | OC | 150 mg bid | c.66dupA | Pathogenic | 17.0 |
| 06005 | gBRCA1 | OC | 150 mg bid | c.5030_5033delCTAA | Pathogenic | 7.6 |
| 02021 | gBRCA1 | BC | 160 mg qd | rs3092994;c.4308T>C;c.3113A>G;c.2612C>T;c.2082C>T | Benign | 14.0 |
| gBRCA2 | c.7242A>G;c.7397T>C; c.4563A>G; | Benign | ||||
| gBRCA2 | c.4240delA | Pathogenic | ||||
| sBRCA2* | c.7230del | Pathogenic | ||||
| 02025# | gBRCA2 | BC | 60 mg bid | c.6513G>C;c.7397T>C | Benign | 28.0 |
| sBRCA1* | c.670+2T>A | Pathogenic | ||||
3. Germline breast cancer susceptibility gene mutation (gBRCAMut) related efficacy. OC, ovarian cancer; BC, breast cancer; all mutation in clinically important was pathogenic, unless patients No. 02017 was likely pathogenic, 02021 and 02025 were benign, but both had pathogenic somatic BRCAMut. According to American College of Medical Genetics (ACMG) Standards and Guidelines.

Discussion
This phase I trial was designed to test the safety and PK of fluzoparib monotherapy, with a preliminary evaluation of efficacy. In addition, the trial included a dose expansion in patients with OC to further assess efficacy in a sensitive population. A standard 3+3 dose escalation design was used in our study. Because this is the first clinical study used in humans, it was reasonable to choose a lower starting dose (10 mg/d) and increased it at a lower dose level. Dose escalation continued until dose-limiting toxicities were observed in >33% of participants, we totally completed 11 cohorts in the dose escalation part, each cohort takes at least two months. Enrolment in the dose expansion part proceeded after MTD was determined. The dose expansion arm evaluated 31 patients, taking about 5 months.
Fluzoparib is a potent oral PARP1, 2 inhibitor that has catalytic activity equivalent to that of olaparib (16). It induces DNA double-strand break accumulation, G2/M arrest and subsequent apoptosis in HR-deficient cells (12,17,18). This first-in-human study demonstrating that fluzoparib monotherapy is active in OC, especially in platinum-sensitive tumors, and has antitumor activity in BC.
Fluzoparib was well tolerated (Table 3 ). Hematologic AEs (all grade) included anemia (53.2%), thrombocytopenia (17.7%) and neutropenia (24.1%); non-hematologic AEs included fatigue (48.1%), vomiting (17.7%), nausea (34.2%) and decreased appetite (29.1%) and were mild in severity. Grade 3−4 AEs were infrequent and included anemia (7.6%) and neutropenia (5.1%). The AEs were primarily managed with drug interruption and/or dose reduction and otherwise routine medical interventions. The fluzoparib AE profile is comparable to other approved PARPis (3,6,19-21). In the phase III olaparib study in OC, AEs (at grades 1/2 and 3/4, respectively) for olaparib at 300 mg bid were nausea (73% and 3%), anemia (24% and 18%), neutropenia (14% and 5%), and abdominal pain (22% and 3%) (9). In our study, 18 patients (22.8%) had abdominal pain. For rucaparib in Study 10 and ARIEL2, rates for abdominal pain, nausea and anemia were 31.6%, 76.9% and 43.8%, respectively, and grade 3/4 anemia was 24.9% (4). In the quality of life analysis evaluating talazoparib in BC (6), fatigue was the most common side effect at 36.6%. In this phase I trial with heavily pre-treated patients, fatigue was reported in 48.1%. Although our study and these studies have a relatively small sample size, they are still comparable.
Fluzoparib demonstrated favourable PK properties with rapid absorption, and dose-proportional increases in total exposure (AUC) among a dose range of 80, 100 and 150 mg bid. Steady state was reached approximately 2 weeks after initiating dosing. At the recommended dose of 150 mg bid, fluzoparib plasma concentrations were maintained above 4 μg/mL, suggesting that systemic concentrations of fluzoparib were adequate to inhibit PARP activity. The PK effects of food on fluzoparib demonstrated that fasting compared to post-prandial administration extended the peak concentration of fluzoparib from 3 h to 6 h; peak concentration decreased by 19.8%, and the AUC did not change significantly. In view of this, it is recommended that fluzoparib be administered following meals.
The elimination half-life of fluzoparib is 12.3, 11.3 and 10.2 h with a single oral dose of 80 mg, 100 mg and 150 mg, respectively, and was 11.4, 10.8 and 9.14 h with 80 mg, 100 mg and 150 mg given bid. These results support bid administration.
Fluzoparib demonstrated the promising antitumor activity in patients with platinum-sensitive OC and heavily pre-treated BC. Single-agent ORR in patients with platinum-sensitive OC was 30% and mPFS in patients with OC with a gBRCAMut was 8.9 months, comparing favourably to the efficacy of olaparib, niraparib and rucaparib in larger studies of patients with BRCAMut OC (1-3,5,22).
Among the 13 efficacy-evaluable patients with BC treated with fluzoparib, ORR was 7.7% and DCR was 23.1%. Two patients with heavily pre-treated gBRCAMut BC achieved prolonged PFS at 14 months and >28 months. These data add to larger studies in g BRCAMut BC showing significant PFS benefit with olaparib or talazoparib (11,19,23-25).
According to ACMG Standards and Guidelines (15), the germline mutation in one of these 2 patients with BC is classified as benign. Therefore, we further evaluated plasma ctDNA by NGS in these 2 patients and found an additional sBRCAMutin both patients (Table 7 ), suggesting that patients with somatic pathogenic mutations in BRCA might benefit from PARP inhibitors. One review reported similar clinical benefit from PARP inhibitors in patients with both germline and somatic BRCA-mutated OC (26). In our study one patient had both pathogenic gBRCAMut and sBRCAMut with PFS of 14 months. The other patient had a benign gBRCAMut and a pathogenicsBRCAMut with PFS over than 28 months. Previous reports had shown that the rate of sBRCAMut in BC has variable prevalence. Winteret al.reported deleterious gBRCAMut in 9% and sBRCAMut in 3% in patients with BC (27). Our previous study showed that gBRCAMut (rs80350973) had a high prevalence in Chinese patients with triple negative BC (7.2%), suggesting the potential for different prevalence of specific mutations in the Chinese population (28). Indeed, ongoing studies are evaluating the efficacy of PARP inhibitors in patients with gBRCAMut, sBRCAMut and those with defects in homologous recombination. Due to the limited number of patients in our study, efficacy must be confirmed in a larger study. Inclusion of patients with either germline and/or somatic mutations in BRCA should be considered.
Further clinical testing of fluzoparib is ongoing in multiple tumor types, including a phase Ib trial in platinum-sensitive, recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer with BRCAMut (NCT03509636), a phase I trial to evaluate the safety and efficacy of fluzoparib in combination with apatinib in patients with OC or BC (NCT03075462), a phase I study of fluzoparib in combination with apatinib and paclitaxel in patients with gastric cancer (NCT03026881), and a phase III trial of fluzoparib maintenance therapy in patients with OC after response to platinum chemotherapy (NCT03863860).
PARP inhibitors have demonstrated clinically important efficacy in patients with gBRCAMut OC and BRCAMut (both germline and somatic) BC and are part of the therapeutic strategy to treat these tumors (18,29). Fluzoparib has the potential to add to this therapeutic armamentarium.
Conclusions
In this phase I study, fluzoparib demonstrated a tolerable safety profile similar to that reported for other approved PARP inhibitors. PK profiling supports bid dosing at an MTD of 150 mg bid. Fluzoparib demonstrated single-agent antitumor activity in BC and OC, especially in those with platinum-sensitive OC and BRCAMut. The strengths of this study demonstrated that there’s no 3/4 grade gastrointestinal disorders, alanine aminotransferase (ALT)/aspartate aminotransferase (AST) elevation observed in RP2D; favorable PK properties, being rapidly absorbed, showed much higher exposure in steady state. The limitation of this data is the small study size, and only 11 OC and 2 BC patients had BRCAMut. There was no detection of homologous recombination deficiency.
Acknowledgements
The authors wish to acknowledge the patients, investigators and participating institutions for their support and endorsement.
Footnote
Conflicts of Interest: This trial received support for sponsored research to the Peking University Cancer Hospital from Jiangsu Hengrui Medicine Co., Ltd. Dr. Li reports research support to the Peking University Cancer Hospital from Pfizer, Roche and Lilly, and being a speaker invited by Novartis, Pfizer, Roche, Lilly and AstraZeneca. Dr. Rugo reports research support to the University of California San Francisco from Pfizer, Merck, Novartis, Lilly, Macrogenics, Roche, OBI, Odonate, Eisai and Daichi, as well as travel support from Pfizer, Novartis, Roche and Mylan. Quanren Wang and Guangze Li are employees of Hengrui Medicine Co., Ltd. No other disclosures were declared.
Contributor Information
Huiping Li, Email: huipingli2012@hotmail.com.
Jianming Xu, Email: jmxu2003@yahoo.com.
References
- 1.National Health Commission of the People’s Republic of China Chinese guidelines for diagnosis and treatment of breast cancer 2018 (English version) Chin J Cancer Res. 2019;31:259–77. doi: 10.21147/j.issn.1000-9604.2019.02.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ledermann J, Harter P, Gourley C, et al Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366:1382–92. doi: 10.1056/NEJMoa1105535. [DOI] [PubMed] [Google Scholar]
- 3.Domchek SM, Aghajanian C, Shapira-Frommer R, et al Efficacy and safety of olaparib monotherapy in germline BRCA1/2 mutation carriers with advanced ovarian cancer and three or more lines of prior therapy. Gynecol Oncol. 2016;140:199–203. doi: 10.1016/j.ygyno.2015.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oza AM, Tinker AV, Oaknin A, et al Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high-grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: Integrated analysis of data from Study 10 and ARIEL2. Gynecol Oncol. 2017;147:267–75. doi: 10.1016/j.ygyno.2017.08.022. [DOI] [PubMed] [Google Scholar]
- 5.Mirza MR, Monk BJ, Herrstedt J, et al Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375:2154–64. doi: 10.1056/NEJMoa1611310. [DOI] [PubMed] [Google Scholar]
- 6.Ettl J, Quek RGW, Lee KH, et al Quality of life with talazoparib versus physician’s choice of chemotherapy in patients with advanced breast cancer and germline BRCA1/2 mutation: patient-reported outcomes from the EMBRACA phase III trial. Ann Oncol. 2018;29:1939–47. doi: 10.1093/annonc/mdy257. [DOI] [PubMed] [Google Scholar]
- 7.Robson M, Im SA, Senkus E, et al Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377:523–33. doi: 10.1056/NEJMoa1706450. [DOI] [PubMed] [Google Scholar]
- 8.Drew Y, Ledermann J, Hall G, et al Phase 2 multicentre trial investigating intermittent and continuous dosing schedules of the poly(ADP-ribose) polymerase inhibitor rucaparib in germline BRCA mutation carriers with advanced ovarian and breast cancer. Br J Cancer. 2016;114:723–30. doi: 10.1038/bjc.2016.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pujade-Lauraine E, Ledermann JA, Selle F, et al Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017;18:1274–84. doi: 10.1016/S1470-2045(17)30469-2. [DOI] [PubMed] [Google Scholar]
- 10.Coleman RL, Oza AM, Lorusso D, et al Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:1949–61. doi: 10.1016/S0140-6736(17)32440-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Litton JK, Rugo HS, Ettl J, et al Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379:753–63. doi: 10.1056/NEJMoa1802905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Yang C, Xie C, et al Pharmacologic characterization of fluzoparib, a novel poly(ADP-ribose) polymerase inhibitor undergoing clinical trials. Cancer Sci. 2019;110:1064–75. doi: 10.1111/cas.13947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE). Available online: https://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm#ctc_40
- 14.Schwartz LH, Litière S, de Vries E, et al RECIST 1.1-Update and clarification: From the RECIST committee. Eur J Cancer. 2016;62:132–7. doi: 10.1016/j.ejca.2016.03.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards S, Aziz N, Bale S, et al Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bochum S, Berger S, Martens UM. In: Martens U. (eds) Small Molecules in Oncology. Recent Results in Cancer Research, vol 211. Berlin: Springer, Cham, 2018.
- 17.Bundred N, Gardovskis J, Jaskiewicz J, et al Evaluation of the pharmacodynamics and pharmacokinetics of the PARP inhibitor olaparib: a phase I multicentre trial in patients scheduled for elective breast cancer surgery. Invest New Drugs. 2013;31:949–58. doi: 10.1007/s10637-012-9922-7. [DOI] [PubMed] [Google Scholar]
- 18.Hopkins TA, Ainsworth WB, Ellis PA, et al PARP1 trapping by PARP inhibitors drives cytotoxicity in both cancer cells and healthy bone marrow. Mol Cancer Res. 2019;17:409–19. doi: 10.1158/1541-7786.MCR-18-0138. [DOI] [PubMed] [Google Scholar]
- 19.Tutt A, Robson M, Garber JE, et al Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–44. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 20.Somlo G, Frankel PH, Arun BK, et al Efficacy of the PARP inhibitor veliparib with carboplatin or as a single agent in patients with germline BRCA1- or BRCA2-associated metastatic breast cancer: California Cancer Consortium Trial NCT01149083. Clin Cancer Res. 2017;23:4066–76. doi: 10.1158/1078-0432.CCR-16-2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamel D, Gray C, Walia JS, et al PARP inhibitor drugs in the treatment of breast, ovarian, prostate and pancreatic cancers: An update of clinical trials. Curr Drug Targets. 2018;19:21–37. doi: 10.2174/1389450118666170711151518. [DOI] [PubMed] [Google Scholar]
- 22.Swisher EM, Lin KK, Oza AM, et al Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017;18:75–87. doi: 10.1016/S1470-2045(16)30559-9. [DOI] [PubMed] [Google Scholar]
- 23.McCann EK Advances in the use of PARP inhibitors for BRCA1/2-associated breast cancer: talazoparib. Future Oncol. 2019;15:1707–15. doi: 10.2217/fon-2018-0751. [DOI] [PubMed] [Google Scholar]
- 24.Robson ME, Tung N, Conte P, et al OlympiAD final overall survival and tolerability results: Olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann Oncol. 2019;30:558–66. doi: 10.1093/annonc/mdz012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garber HR, Litton JK Integrating poly(ADP-ribose) polymerase (PARP) inhibitors in the treatment of early breast cancer. Curr Opin Oncol. 2019;31:247–55. doi: 10.1097/CCO.0000000000000516. [DOI] [PubMed] [Google Scholar]
- 26.Faraoni I, Graziani G Role of BRCA mutations in cancer treatment with poly(ADP-ribose) polymerase (PARP) inhibitors. Cancers (Basel) 2018;10:487. doi: 10.3390/cancers10120487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Winter C, Nilsson MP, Olsson E, et al Targeted sequencing of BRCA1 and BRCA2 across a large unselected breast cancer cohort suggests that one-third of mutations are somatic. Ann Oncol. 2016;27:1532–8. doi: 10.1093/annonc/mdw209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu X, Li H, Shao B, et al Identification of recurrent BRCA1 mutation and its clinical relevance in Chinese Triple-negative breast cancer cohort. Cancer Med. 2017;6:547–54. doi: 10.1002/cam4.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beniey M, Haque T, Hassan S Translating the role of PARP inhibitors in triple-negative breast cancer. Oncoscience. 2019;6:287–8. doi: 10.18632/oncoscience.474. [DOI] [PMC free article] [PubMed] [Google Scholar]