

Abstract
Axonal and synaptic degeneration occur following nerve injury and during disease. Traumatic nerve injury results in rapid fragmentation of the distal axon and loss of synaptic terminals, in a process known as Wallerian degeneration (WD). Identifying and understanding factors that influence the rate of WD is of significant biological and clinical importance, as it will facilitate understanding of the mechanisms of neurodegeneration and identification of novel therapeutic targets. Here, we investigate levels of synaptic loss following nerve injury under a range of conditions, including during postnatal development, in a range of anatomically distinct muscles and in a mouse model of motor neuron disease. By utilising an ex vivo model of nerve injury, we show that synaptic withdrawal is slower during early postnatal development. Significantly more neuromuscular junctions remained fully innervated in the cranial nerve/muscle preparations analysed at P15 than at P25. Furthermore, we demonstrate variability in the level of synaptic withdrawal in response to injury in different muscles, with retraction being slower in abdominal preparations than in cranial muscles across all time points analysed. Importantly, differences between the cranial and thoracoabdominal musculature seen here are not consistent with differences in muscle vulnerability that have been previously reported in mouse models of the childhood motor neuron disease, spinal muscular atrophy (SMA), caused by depletion of survival motor neuron protein (Smn). To further investigate the relationship between synaptic degeneration in SMA and WD, we induced WD in preparations from the Smn2B/ − mouse model of SMA. In a disease‐resistant muscle (rostral band of levator auris longus), where there is minimal denervation, there was no change in the level of synaptic loss, which suggests that the process of synaptic withdrawal following injury is Smn‐independent. However, in a muscle with ongoing degeneration (transvs. abdominis), the level of synaptic loss significantly increased, with the percentage of denervated endplates increasing by 33% following injury, compared to disease alone. We therefore conclude that the presence of a die‐back can accelerate synaptic loss after injury in Smn2B/ − mice.
Keywords: axotomy, developmental regulation, die‐back, differential vulnerability, ex vivo, motor neuron disease, mouse model, neuromuscular disease, neuromuscular junction, peripheral neuropathy, postnatal development, spinal muscular atrophy, synaptic degeneration, Wallerian degeneration
Following traumatic nerve injury, distal synaptic terminals degenerate. Here we show that the rate of degeneration is slower during postnatal development and also varies significantly between anatomically distinct muscles. We further show that the presence of underlying synaptic pathology in a muscle accelerates the rate of synapse loss following injury. This data provides a valuable model system to investigate further the factors which affect synaptic stability and degeneration.

1. INTRODUCTION
Motor neurons are vulnerable to degeneration following physical or chemical injury and in a range of neurodegenerative diseases. Damaged axons and synapses degenerate in morphologically distinct ways depending on the nature of the original trauma. Following injury, spontaneous and rapid fragmentation occurs along the length of the injured axon in a characteristic process known as Wallerian degeneration (WD; Waller, 1851; Beirowski et al., 2004, 2005; Conforti et al., 2014). In WD, the cytoskeleton is disassembled and granular degeneration proceeds distal to the injury site (for review see Wang et al., 2012). This is morphologically distinct from degeneration that occurs during peripheral neuropathies, such as amyotrophic lateral sclerosis (ALS) and spinal muscular atrophy (SMA). In some forms of motor neuron disease, axons retrogradely retract away from the synaptic region and are described as ‘dying‐back’ (Cavanagh, 1964). An example of dying‐back pathology can be seen in the childhood motor neuron disease, SMA, where a lack of survival motor neuron (SMN) protein leads to the progressive loss of lower motor neurons (Lefebvre et al., 1995; Rodrigues et al., 1995). Despite morphological distinctions between WD and dying‐back pathology, their mechanistic differences or commonalities remain unclear. Understanding whether different morphologies also relate to different mechanisms would provide a powerful tool to better categorise different types of, neurodegeneration and potentially it will facilitate the identification of novel therapeutic targets.
Recent work has revealed that rates of axonal and synaptic degeneration are influenced by postnatal age. Significant differences in the response of neonatal vs. adult mice to peripheral nerve injury and hypoxic insult have been reported (Murray et al., 2011). In young adult mice (P25), tibial nerve lesion results in a loss of motor nerve terminals within the deep lumbrical (DL) muscles, with just 3% of motor endplates remaining innervated after 24 hr (Murray et al., 2011). Contrastingly, in mice aged less than 2 weeks of age, the rate of degeneration is much slower, with 86% of endplates remaining innervated after 24 hr (Murray et al., 2011). Synaptic loss in response to injury therefore appears to be developmentally regulated in the DL muscles. Neonatal, murine neuromuscular junctions (NMJs) also remain structurally intact following exposure to an ex vivo model of hypoxia reperfusion injury, with over 99% of endplates remaining fully occupied in P2 murine transvs. abdominis (TVA), compared with just 4% in adult mice (Murray et al., 2011).
Differential rates of degeneration have been reported between different motor neurons pools in humans and in mouse models of motor neuron disease (Bowerman et al., 2012). Transcriptional profiling of differentially vulnerable motor neurons has identified transcripts and pathways which can modify the rate of degeneration (Hedlund et al., 2010; Brockington et al., 2013; Kaplan et al., 2014; Comley et al., 2015; Murray et al., 2015; Kline et al., 2017; Boyd et al., 2017). Properties that are intrinsic to the motor neuron may therefore regulate the rate of axon degeneration. Understanding the properties of different groups of motor neurons should better inform us of the mechanisms of axon degeneration. Due to the limited numbers of muscle that have been studied in detail, it remains unclear whether there is any variability in the rate of synaptic loss that occurs following injury in different muscles. Determining how different pools of motor neurons respond to injury would provide a useful tool to investigate the factors that regulate WD, and how this relates to other types of axonal and synaptic degeneration.
It remains unclear whether developmental resistance to injury is unique to DL preparations. To investigate this, here we measured synaptic degeneration in cranial and thoracoabdominal musculature following nerve injury. This was achieved by using an ex vivo model of axon injury, where the nerve is transected, and the distal portion along with the musculature it innervates is removed and maintained in oxygenated solutions for 24 hr (Brown et al., 2015; Kline et al., 2019). We have applied this model system to cranial muscle/facial nerve and abdominal muscle/intercostal nerve preparation at different postnatal time points. We show that there is a developmental delay in synaptic withdrawal following injury in all cranial muscles. Interestingly, some muscles (such as adductor auris longus; AAL) showing significantly greater loss than other muscles of the same age (such as levator auris longus; LAL). Furthermore, synaptic degeneration was consistently reduced in the thoracoabdominal muscles compared to the cranial muscles. Together these data suggest that factors intrinsic to the muscle/motor neuron affect the degree of synaptic degeneration following injury. Comparison of the degree of synaptic loss following injury with the degree of synaptic loss in a mouse model of SMA, revealed strikingly different patterns. This suggests that the factors which affect the rate of synaptic degeneration following injury are not the same as those which affect the vulnerability of motor neurons to SMA. Finally, by exposing muscles from the mouse model of SMA to the ex vivo model of nerve injury, we show that the presence of SMA‐induced synaptic pathology significantly increases synaptic loss following injury.
2. METHODS
2.1. Mouse maintenance
All animal procedures were performed in accordance with the UK Home Office and institutional guidelines. C57BL/6/J MCos1, C57BL/6 Smn 2B/2B and Smn +/− mice were maintained as groups of less than four (or with parents/littermates) in individually ventilated cages under pathogen‐free conditions, with ad libitum access to food and water, on a 12:12 light/dark cycle within animal facilities at the University of Edinburgh. MCos1 mice were sacrificed between P15‐P25 and Smn2B/− mice were sacrificed at P18 (disease end‐stage) by an inhalational overdose of anaesthetic (isofluorane). Death was confirmed by exsanguination of the carotid artery. The Smn 2B/− mouse is a mouse model of the childhood motor neuron disease, spinal muscular atrophy that exhibits dying‐back pathology. The Smn2B allele consists of a three‐nucleotide substitution in the exon enhancer region within exon 7 of the murine Survival motor neuron (Smn) gene. The resultant mouse has reduced levels of full‐length, endogenous SMN protein which leads to progressive loss of lower motor neurons (DiDonato et al., 2001; Hammond et al., 2010; Bowerman et al., 2012). Onset of disease signs occurs at around P10, and, in our colonies, this model has a lifespan of around 21 days.
2.2. Ex vivo model of nerve injury
Cranial and abdominal nerve/muscle explants were dissected as described previously (Murray et al., 2010a, 2014) in Hepes‐buffered saline composed of NaCl (137 mM), KCl (5 mM), CaCl2·2H2O (2 mM), MgCl2·6H2O (1 mM), glucose (5.6 mM) and Hepes (5 mM) with pH corrected to 7.4. Nerves (facial or intercostal) were severed at a consistent point, and distal nerve portions with attached musculature were pinned to dental wax and suspended in oxygenated mammalian physiological saline containing: NaCl (120 mM), KCl (5 mM), CaCl2·2H2O (2 mM), MgCl2·6H2O (1 mM), NaH2PO4·2H2O (0.4 mM), NaHCO3 (23.8 mM), glucose (5.6 mM), gentamicin (50 mg/ml, Thermo Fisher) and kanamycin (100 mg/ml, Thermo Fisher) in a 30‐ml bijou tube. The bijou lid was modified, with two 0.8‐mm needles, one needle clipped short, above the buffer level, to act as a pressure valve. A length of 0.50 mm tubing (VWR) sufficient to reach the bottom of the receptacle was attached to the other needle to provide a constant supply of 95/5% O2/CO2 to equilibrate the saline, at a perfusion rate of approximately 5 ml/min. The bathing solution was equilibrated for approximately 30 min before adding the sample. The receptacle containing the nerve/muscle explant was then incubated in a 30°C water bath for 24 hr. Temperature was monitored during this period using a digital thermometer (Fisher Traceable Digital Thermometer with stainless steel probe).
2.3. Immunohistochemistry
After 24 hr incubation, preparations were fixed immediately in 4% paraformaldehyde (EMS) for 15 min. Muscles of interest were microdissected and permeabilised in 2% Triton‐X‐100‐PBS (Sigma; PBT) for 30 min on a rocking platform at room temperature. Blocking solution (4% bovine serum albumin [BSA]/1% PBT) was applied for 60 min followed by primary antibodies (SV2, 1:100; 2H3, 1:50, both from Developmental Studies Hybridoma Bank) in blocking solution, and left to incubate at 4°C on a rocking platform for 24 hr. Preparations were then washed three times for 5 min with PBS. Secondary antibody (anti‐mouse, Alexa Fluor 488, Stratech, 1:250 in 1×PBS) was added to each well and incubated in the dark at room temperature for a minimum of 4 hr. Muscles were washed three times in 1 × PBS for 5 min, and incubated with tetramethylrhodamine‐conjugated α‐bungarotoxin (TRITC‐BTX; Thermo Fisher, 1:250, 20 min). Muscles were washed and whole‐mounted on glass slides with Mowiol® (Calbiochem) and cover‐slipped. Slides were stored in the dark at 4°C.
2.4. Quantification and statistical analyses
Quantification of NMJ occupancy was undertaken by fluorescent microscopy at ×40 magnification. The investigator was blinded to the age, genotype and treatment of the muscle. A minimum of two to three fields of view (depending on muscle size and NMJ distribution) and 50 NMJs were sampled per muscle (average of 118 motor endplates per muscle). Fields of view were chosen at random from across the entire muscle using bungarotoxin staining only, to ensure a fair representation of the entire muscle and to reduce the chance of bias for innervated areas. A set of previously optimised and standardly used protocols for NMJ quantification were applied to quantify the percentage of fully occupied endplates (Murray et al., 2008a, 2011, 2013, 2015; Thomson et al., 2012; Sleigh et al., 2014a, 2014b; Brown et al., 2015; Comley et al., 2016; Kline et al., 2017; Courtney et al., 2019; Osman et al., 2019). In brief, fully occupied endplates were defined as complete apposition between the pre‐synaptic terminal and the motor endplate (see Figure 1a for example). This has been expressed as a percentage of the total number of endplates quantified for each muscle, with the remainder of endplates displaying synaptic withdrawal, appearing either only partially innervated (where only part of the AChR cluster is covered by a pre‐synaptic terminal) or vacant (where no pre‐synaptic staining is evident at the AChR cluster).
Figure 1.
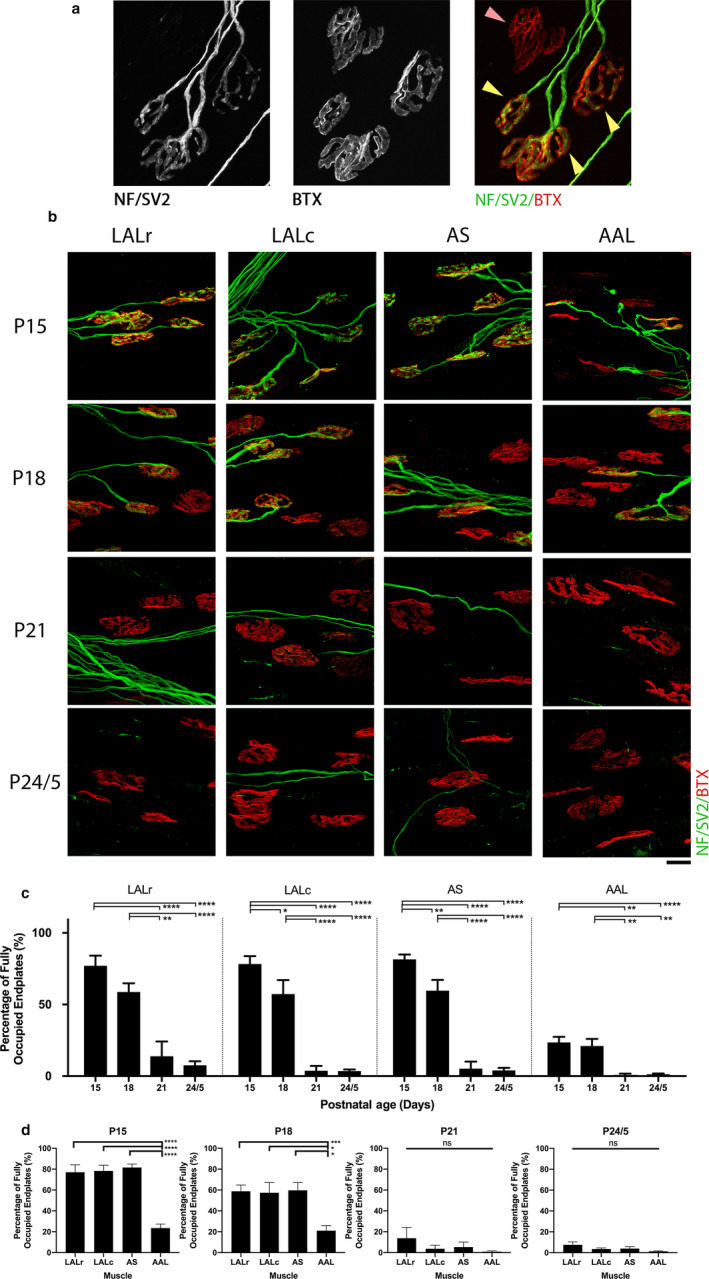
Synaptic stability following injury is developmentally regulated and non‐uniform in cranial muscles of the mouse. (a) Representative confocal micrographs showing example NMJs to exemplify criteria for occupancy quantification. NMJs are labelled with antibodies against neurofilament (NF, green) and synaptic vesicle protein 2 (SV2, green), and α‐bungarotoxin (BTX, red). The yellow arrowheads highlight endplates that would be quantified as ‘full’, whereas the pink arrowhead indicates a vacant endplate, where apposition between the pre‐synaptic terminal and the motor endplate has been lost (see Methods for further detail). (b) Representative confocal micrographs showing NMJs labelled with antibodies against NF (green) and SV2 (green), and BTX (red) from cranial muscles (levator auris longus rostral/caudal band [LALr/c], abductor auris longus [AAL], auricularis superior [AS]) that have been maintained ex vivo at 30°C for 24 hr. Postnatal age refers to the age of the animal at the time of dissection. Scale bar: 20 μm. (c) Bar charts showing quantification of the percentage of fully innervated motor endplates in cranial muscle bands between P15 and P24/5. Note that the percentage of fully occupied endplates decreases as postnatal age increases, supporting the finding that the response to injury is developmentally regulated. (d) Bar charts showing the percentage of fully occupied endplates in cranial muscles grouped by postnatal age at the time of ex vivo nerve injury. Note that there is a significant reduction in the percentage of fully occupied endplates in the AAL at both P15 and P18 time points in comparison with all other muscles. *p < .05, **p < .01, ***p < .001, ****p < .0001; one‐way ANOVA with Tukey’s correction. n = 10 at P15 and P24/5; n = 18/6 for the LALr/all other muscles at P18; and n = 4/5 at P21 for the AAL/all other muscles. Error bars represent mean ± SEM
All morphological analyses were collated into Microsoft excel before exporting to GraphPad prism 8 software for statistical analysis with n numbers referring to the number of muscles analysed. Statistical tests are detailed in the legend of the relevant graphs to which they refer. Data were tested for normality using a Shapiro–Wilk test. Data that were normally distributed were tested using one‐way analysis of variance (ANOVA) with post‐hoc Tukey correction. In groups that were not normally distributed, Kruskal–Wallis tests with Dunn’s post‐hoc analysis were applied. All data are presented as mean values ± standard error of the mean (SEM). Differences were considered significant when p < .05.
Micrographs are projections of Z‐image stacks collected on a Nikon A1R FLIM confocal microscope and were adjusted for overall image brightness and contrast only using imagej (downloadable from https://imagej.nih.gov/ij/) and Adobe photoshop CS6 software (licence purchased from https://www.adobe.com/uk/products/).
3. RESULTS
3.1. Synaptic loss following injury is developmentally regulated and non‐uniform in cranial muscles of the mouse
It is unclear whether developmental regulation of synaptic loss in response to injury is unique to the DL muscles or whether developmentally regulated delays in synaptic withdrawal also occurs in other muscle preparations. We therefore examined rates of synaptic withdrawal following injury during the critical time window of P15–P25, using an ex vivo model of nerve injury (see Section 2 for details). Nerve‐muscle preparations comprising facial nerve (CN VII) branches innervating the cranial musculature (comprising rostral and caudal bands of levator auris longus [LALr/LALc, respectively], abductor auris longus [AAL] and auricularis superior [AS]) were isolated and maintained in oxygenated physiological solution at 30°C for 24 hr. We hypothesised that synapse loss would be slower in younger mice and that the level of synaptic loss would differ between different muscles. We tested this by analysing levels of muscle innervation following 24‐hr exposure to the ex vivo model of nerve injury at different postnatal ages.
The percentage of fully innervated or ‘occupied’ endplates remaining in immunostained muscles from P15–P25 mice, 24 hr after ex vivo nerve injury were quantified (Figure 1a–d). Note that all ages refer to the age at dissection. The data show that postnatal age affected the degree of synapse loss in all preparations analysed (Figure 1c). For example, at P15, the LALr remained highly innervated, with 77.05 ± 7.05% (mean ± SEM) of endplates covered by motor nerve terminals. As expected, levels of innervation 24 hr after injury declined with age to 58.75 ± 6.06% innervation in preparations from mice aged P18, to 13.81 ± 10.4% aged P21, and 7.53 ± 2.84% aged P24/5. The LALc and AS muscles followed similar patterns. However, we found significantly more denervated endplates in AAL from the outset, with only 23.56 ± 3.85% innervation remaining at P15. This declined with mouse age to 21.02 ± 4.96% at P18, whereas less than 1.3% innervation remained at P21 and P24/5 (Figure 1d). As both AAL and LAL are homogeneously fast twitch muscles, these differences cannot be attributed to fibre type. This data suggests that there are other intrinsic properties of nerve or muscle that influence the rate of synaptic degeneration during postnatal development.
3.2. Levels of synaptic loss are uniformly reduced in thoracoabdominal muscles of the mouse
Data thus far suggest that developmental delays are present in a range of muscles. However, differences between the rates of synaptic withdrawal observed in the AAL at different time points, led us to ask whether there would be similar differences within thoracoabdominal musculature. To investigate the response to injury in these muscles, we examined the TVA and TS muscles 24 hr after ex vivo nerve injury using the same protocol as for cranial muscles. In contrast to cranial preparations, both the TVA and TS showed high levels of NMJ innervation across all time points analysed (Figure 2). There was no significant difference in the percentage of fully occupied endplates at any time point; in all cases, the muscle remained over 77% innervated (one‐way ANOVA with Tukey correction, p ≥ .09).
Figure 2.
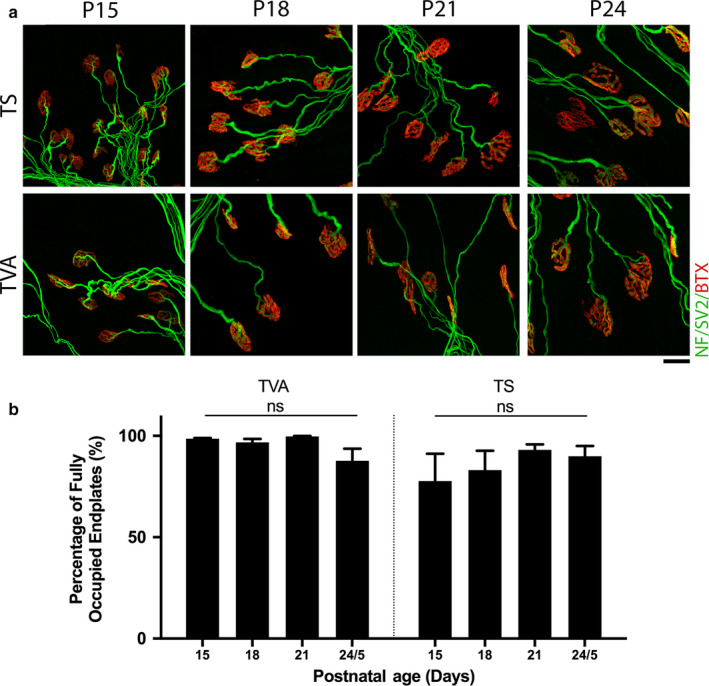
Minimal synaptic loss is observed following injury across all time points analysed in thoracoabdominal musculature. (a) Representative confocal micrographs showing NMJs labelled with antibodies against neurofilament (NF, green) and synaptic vesicle protein 2 (SV2, green), and α‐bungarotoxin (BTX, red) from transvs. abdominis [TVA] and triangularis sterni (TS) muscles which have been maintained ex vivo at 30°C for 24 hr. Postnatal age refers to the age of the animal at the time of dissection. Note that the majority of endplates remain fully occupied at all ages examined. Scale bar: 20 μm. (b) Bar charts showing quantification of the percentage of fully innervated motor endplates after ex vivo nerve injury in the TVA and TS at P15, P18, P21, and P24/5. The percentage of fully occupied endplates remains high across all time points and there is no significant difference within muscles at any time point. ns = not significant, p ≥ .09; one‐way ANOVA with Tukey’s correction. TVA, n = 6, 18, 4, and 6 muscles, at P15, 18, 21, and 24/5, respectively, and n = 6, 6, 6, and 4 in the TS at P15, 18, 21, and 24/5, respectively. Error bars represent mean ± SEM
Taken together, the data indicate that there are properties intrinsic to the muscle.., and/or motor neuron, that can influence the extent of synaptic withdrawal in response to injury during the postnatal period.
3.3. Patterns of differential syanptic stability following injury contrast with patterns of selective vulnerability in a mouse model of SMA
Variability in the levels of synaptic degeneration, in a given muscle, has previously been reported in mouse models of motor neuron disease. For instance, in the Smn2B/− mouse model of SMA, the TVA muscle is highly vulnerable and displays high levels of synaptic loss, whereas the LALr and AAL muscles are relatively spared, with little synaptic loss ‘even at late stages of disease’ (Murray et al., 2015). To investigate how synaptic stability differs between disease and injury, we compared levels of innervation remaining in muscles from end‐stage (P18) Smn2B/− mice in vivo with levels of innervation remaining in homologous muscles from wild‐type animals after ex vivo nerve injury. This provided insight into how synaptic responses differ following these distinct insults.
In line with previous work undertaken by Murray et al. (2015), here the LALr and AAL of Smn2B/− mice were largely spared from dying‐back neuropathy in vivo, with over 98% of motor endplates remaining fully innervated at P18 (Figure 3). Contrasting this, 24 hr after ex vivo nerve injury in P18 wild‐type mice, the LALr was 47.23 ± 2.58% innervated and the AAL only 21.02 ± 4.96% innervated (Figure 3). These cranial muscles therefore appear to exhibit more extensive synaptic loss following injury than in an SMA mouse model (Mann–Whitney U‐test, p ≤ .0007). Also in line with descriptions by Murray et al. (2015), in the Smn2B/− model, the TVA is vulnerable to dying‐back pathology in vivo, with 62.11 ± 3.78% innervation remaining at P18, compared with 99.54 ± 0.29% innervated in wild‐type age‐matched mice 24 hr after ex vivo nerve injury (Figure 3). This suggests that the TVA exhibits less extensive synaptic loss following injury than in an SMA mouse model (Mann‐Whitney U test, p = .0002). Thus, the factors responsible for determining relative synaptic stability following both nerve injury and SMA, appear to be distinct.
Figure 3.
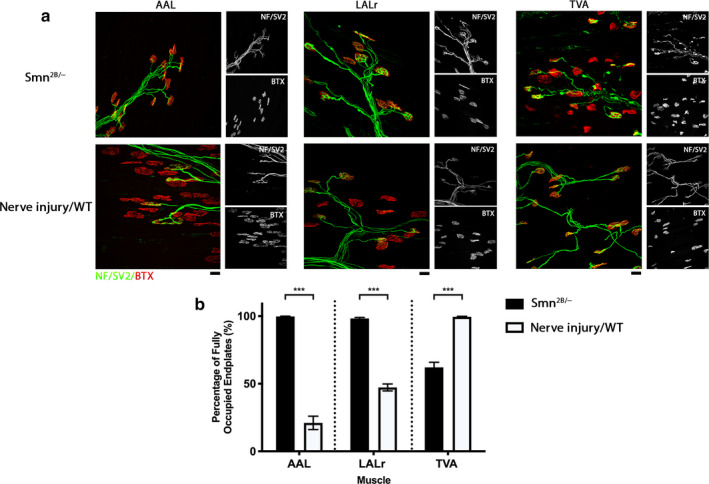
Synaptic stability is different in response to disease or injury conditions. (a) Representative confocal micrographs showing NMJs labelled with antibodies against neurofilament (NF, green) and synaptic vesicle protein 2 (SV2, green), and α‐bungarotoxin (BTX, red) showing innervation of abductor auris longus (AAL), levator auris longus rostral band (LALr), and transvs. abdominis (TVA) under disease conditions from Smn2B/− mice at P18 and following ex vivo nerve injury in wild‐type animals at P18. Nerve/muscle explants from the ‘nerve injury’ group were maintained ex vivo at 30°C for 24 hr. Scale bar: 20 μm. (b) Bar chart showing quantification of the percentage of fully innervated motor endplates in the AAL, LALr, and TVA after ex vivo nerve injury in WT vs. Smn2B/− mice. In the Smn2B/− mouse, the LALr and AAL cranial muscles are spared from degeneration in comparison with the TVA muscle. Following ex vivo nerve injury, LALr and AAL show high levels of synapse loss, whereas the TVA appears preserved. ns = not significant, ***p < .001; Mann–Whitney U‐test. Note that for the AAL, n = 9, 6; for the LALr, n = 8, 6; and for the TVA, n = 10, 6 muscles for Smn2B/− and nerve injury/WT groups, respectively. Error bars represent mean ± SEM
3.4. Reduced Smn levels do not influence the incidence of synaptic loss following injury in the LALr cranial muscle of the mouse
We next asked whether a reduction in Smn levels affects synaptic stability after injury. To address this, we isolated and maintained cranial nerve/muscle preparations from wild‐type mice and from Smn2B /− mice. In Smn2B/ − mice, cranial muscles are classified as selectively resistant, in that even at end‐stage of disease, there is no synapse loss evident (cf. Figure 3). Therefore, these muscles have reduced Smn levels but no resultant synaptic loss (Murray et al., 2015). We compared levels of denervation in the LALr under three conditions: LALr from wild type after ex vivo nerve injury; LALr from Smn2B/ −; and LALr from Smn2B/ − after ex vivo nerve injury.
In this disease‐resistant muscle, consistent with previous work by Murray et al. (2015), minimal denervation was found in vivo in Smn2B/ − mice, with over 93% of endplates remaining fully occupied. In wild‐type mice, ex vivo nerve injury‐induced extensive denervation, with 47.12 ± 4.08% of endplates remaining fully occupied. In muscles from Smn2B/ − mice subject to ex vivo nerve injury, we found that the level of synaptic loss was comparable to wild‐type levels, where 58.55 ± 3.528% of endplates remaining fully occupied (Unpaired t‐test with Welch’s correction, p = .087; Figure 4). We conclude that in the LALr, where synapse loss due to Smn deficiency is minimal, the synaptic loss caused by nerve injury is not altered. As Smn levels are reduced in this model, despite the lack of NMJ pathology, we conclude that synaptic withdrawal following ex vivo nerve injury is Smn‐independent.
Figure 4.
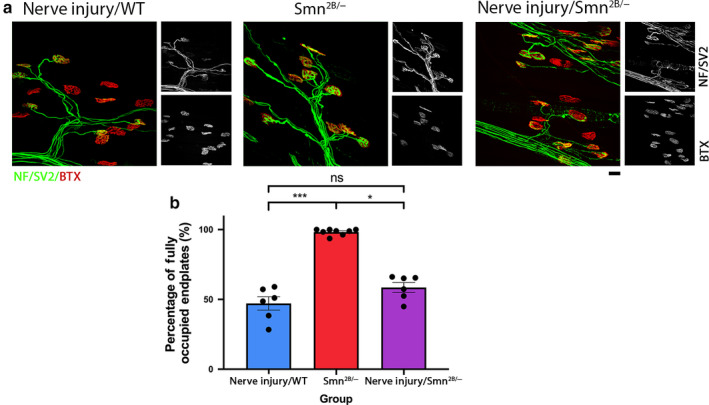
Reduced Smn levels have no effect on the level of synaptic degeneration following injury in the LALr cranial muscle of the mouse. (a) Representative confocal micrographs showing NMJs labelled with antibodies against neurofilament (NF, green) and synaptic vesicle protein 2 (SV2, green), and α‐bungarotoxin (BTX, red) from levator auris longus rostral band (LALr) in wild‐type animals after ex vivo nerve injury (nerve injury/WT), in Smn2B/− mice and in Smn2B/− mice after ex vivo nerve injury (nerve injury/Smn2B/−). In nerve injury/WT and nerve injury/Smn2B/− conditions, LALr muscles were maintained ex vivo at 30°C for 24 hr. Scale bar: 20 μm. (b) Bar charts showing quantification of the percentage of fully occupied endplates in LALr in nerve injury/WT, Smn2B/−, and nerve injury/Smn2B/− conditions. The percentage of fully occupied endplates after nerve injury in Smn2B/− mice is not significantly different than after nerve injury in wild‐type animals. *p < .05, ***p < .001, ns = not significant. For all comparisons with the Smn2B/− group, Kruskal–Wallis with Dunn’s correction was used to account for non‐normally distributed data; otherwise a one‐way ANOVA with Tukey’s correction was used. Note that n = 6, 8, 6 for the nerve injury/WT, Smn2B/− and nerve injury/Smn2B/− groups, respectively. Error bars: mean ± SEM
3.5. The presence of dying‐back pathology increases the level of synaptic loss following injury in the TVA muscle of the mouse
To investigate the relationship between synaptic loss that occurs in injury vs. disease, we also investigated whether there was any alteration in the level of synaptic loss following injury in the presence of dying‐back pathology. Unlike the LALr, the TVA muscle of the Smn2B/ − mouse model exhibits high levels of synaptic pathology, including denervation, pre‐synaptic swelling, and post‐synaptic shrinkage at end‐stages of disease (P18). We compared levels of denervation in the TVA under three conditions at P18: TVA from wild type after ex vivo nerve injury; TVA from Smn2B/ −; and TVA from Smn2B/ − after ex vivo nerve injury.
Ex vivo nerve injury of the TVA muscle from Smn2B/ − mice resulted in a significant reduction in the percentage of fully occupied endplates, to just 8.05 ± 6.23% in comparison with wild‐type TVA with ex vivo nerve injury (98.53 ± 0.29% of endplates fully innervated, Kruskal–Wallis with Dunn’s correction, p < .0001) and to Smn2B/ − in vivo (41.11 ± 3.98% of endplates fully innervated, Unpaired t‐test with Welch’s correction, p = .0018) in isolation (Figure 5). Overall, as we have already established that SMN reduction alone is not sufficient to increase levels of synaptic loss, it follows that the increase in synapse loss observed here is induced by the presence of disease‐induced synaptic pathology.
Figure 5.
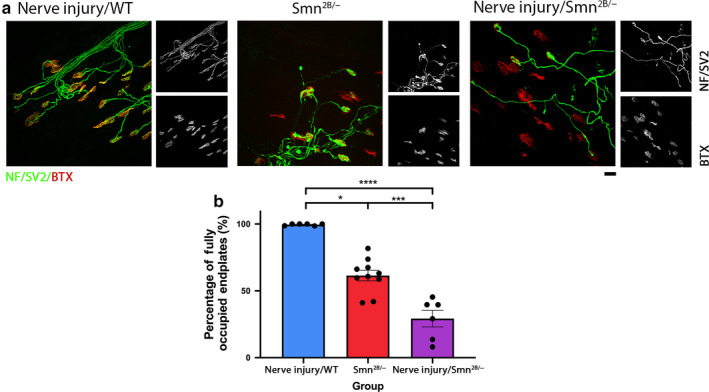
Presence of synaptic pathology in the Smn2B/− mouse significantly amplifies the level of synaptic loss induced by injury in the TVA muscle. (a) Representative confocal micrographs showing NMJs labelled with antibodies against neurofilament (NF, green) and synaptic vesicle protein 2 (SV2, green), and α‐bungarotoxin (BTX, red) from transvs. abdominis (TVA) at P18 in wild‐type animals after ex vivo nerve injury (nerve injury/WT), in the Smn2B/− mice, and in Smn2B/− after ex vivo nerve injury (nerve injury/Smn2B/) conditions, respectively. In ex vivo conditions, TVA muscles were maintained ex vivo at 30°C for 24 hr. P18 refers to the postnatal age of the animal at the time of dissection. Scale bar: 20 μm. (b) Bar charts showing quantification of the percentage of fully occupied endplates in the TVA in nerve injury/WT, Smn2B/−, and nerve injury/Smn2B/− conditions. The percentage of fully occupied endplates following injury in Smn2B/− mice was significantly increased in comparison with either wild‐type injury or Smn2B/− conditions in isolation. *p < .05, **p < .01, ****p < .0001. For all comparisons with the nerve injury/WT group, Kruskal–Wallis with Dunn’s correction was used to account for non‐normally distributed data, otherwise a one‐way ANOVA with Tukey’s correction was used. Note that n = 6, 10, 6 muscles for the nerve injury/WT, Smn2B/−, and nerve injury/Smn2B/− groups, respectively. Error bars: mean ± SEM
4. DISCUSSION
In this study, we have applied an ex vivo assay to investigate the effect of traumatic nerve injury in a range of muscles that have not been examined in this context previously. We have demonstrated that postnatal maturity influences the synaptic stability following injury in the cranial muscles. We have also shown that levels of synaptic loss are non‐uniform in these muscles, with a significant increase in the rate of synaptic withdrawal in response to injury in the AAL at younger ages when compared with other cranial muscles. We also found that high levels of innervation persisted in the thoracoabdominal muscles after injury across P15–P25. This suggests that muscle and/or motor neuron intrinsic properties affect synaptic stability following injury. Comparison of the patterns of relative synaptic stability following injury to the patterns of synaptic vulnerability in a mouse model of SMA further suggests that the factors which affect synaptic stability following injury are distinct from those in peripheral neuropathies such as SMA. By exposing a muscle from a mouse model of SMA that is resistant to dying‐back pathology with the ex vivo nerve injury assay, we have shown that synaptic loss in response to injury is SMN‐independent. Furthermore, exposure of a muscle with ongoing dying‐back pathology to the ex vivo nerve injury assay has demonstrated that the presence of dying‐back pathology accelerates the rate of synaptic loss in response to injury.
4.1. Synaptic stability following injury is governed by properties intrinsic to the muscle and/or motor neuron
We have shown that synaptic withdrawal in response to injury can be influenced by developmental age. Across all four cranial muscles analysed, we observed an increase in the incidence of denervated endplates with increasing postnatal age. These findings are in line with previous work that has shown that following either peripheral nerve injury or hypoxic insult, resistance is gradually lost across the first few weeks of life as the muscle transitions to adult levels of vulnerability (Murray et al., 2011). Furthermore, some muscles, like the AAL, reach adult levels of degeneration at an earlier time point, whereas others, such as the TVA, remain comparatively stable following injury, even at P25 (Murray et al., 2011). This suggests that postnatal maturity as well as properties specific to the muscle and/or motor neuron, are capable of regulating synaptic degeneration following insult. Discerning these mechanisms would be of great value and would contribute significantly to our understanding of the mechanisms regulating synaptic stability and vulnerability to injury and disease.
Indeed, various genetic mutations have been shown to alter the rate of WD in animal models, and have massively increased our understanding of the mechanisms of WD. This includes Wlds and Sterile Alpha and TIR motif‐containing 1 (SARM1) knockout (Lunn et al., 1989; Ribchester et al., 1995; Mack et al., 2001; Wang et al., 2001; Gillingwater et al., 2002, 2004, 2006; Coleman, 2005; Coleman and Freeman, 2010; Gilley and Coleman, 2010; Osterloh et al., 2012; Di Stefano et al., 2015; Gilley et al., 2015). Furthermore, differences in synaptic vulnerability in motor neuron disease have been exploited to investigate the underlying factors that may be responsible making motor neurons vulnerable in motor neuron disease. Transcriptional profiling of differentially vulnerable motor neurons has helped identify transcripts and pathways which can modify the rate of degeneration (Hedlund et al., 2010; Brockington et al., 2013; Kaplan et al., 2014; Comley et al., 2015; Murray et al., 2015; Boyd et al., 2017; Kline et al., 2017). We have recently used this approach to identify alpha‐synuclein and stathmin 1 as phenotypic modifiers of pathology in motor neuron diseases. Importantly, when levels of alpha‐synuclein or stathmin 1 were increased, disease severity was reduced and NMJ pathology ameliorated (Kline et al., 2017; Villalon et al., 2019). Identification of motor neurons that are differentially affected during axon degeneration therefore serves as a useful tool to identify factors that can modify axon degeneration in both disease and injury.
Here we have shown that muscle and/or motor neuron‐specific properties are another factor that can influence the rate of synaptic loss, with responses varying between muscles. This is unlikely to be attributed to muscle fibre type. For example, both LAL and AAL are predominantly fast twitch muscles, but they display a marked difference in synaptic stability following injury (Erzen et al., 2000; Murray et al., 2008a, 2010a, 2010b). In wild‐type rats and in Wlds mice, the rate of synaptic loss has also been shown to be dependent on nerve stump length, with failure of neuromuscular transmission occurring more rapidly when nerve stump length is shorter (Slater, 1966; Ribchester et al., 1995). However, the differences we have reported here cannot be attributed to differences in nerve stump length, as stump length is the same for all of the cranial muscles.
A promising avenue which may explain the differing synaptic stability following injury in different muscles at different postnatal ages, is differences in mitochondrial bioenergetics. In a recent study, we have profiled the nerve and muscle proteome in tibial nerve/lumbrical muscles between P12 and P24, and report a consistent up‐regulation in proteins involved with mitochondrial bioenergetics in both muscle and nerve. Interestingly, this same profile has been observed in the tibialis anterior muscle, and during postnatal development of the heart (Puente et al., 2014; Kim et al., 2019). Importantly, we further showed that inhibition of complex I of the mitochondrial respiratory chain could slow axon degeneration, whereas pharmacological activation of oxidative phosphorylation accelerated synaptic degeneration (Kline et al., 2019). It will now be important to investigate mitochondrial bioenergetics in the range of ages and muscles investigated in this study, to determine whether this can explain the differences in synaptic stability observed. This will help identify factors responsible for conferring rate differences and will provide insight into the mechanisms of synaptic degeneration, and aid in the identification of targets to manipulate the rate of degeneration.
4.2. Mechanisms of synaptic loss following injury vs. dying‐back neuropathy
Following traumatic injury in the Smn2B /− mouse model of SMA, we assessed the influence on the level of synaptic loss. Differential vulnerability is a known feature in this disease model, therefore we investigated both a die‐back‐resistant (LALr) and die‐back‐vulnerable (TVA) muscle (Murray et al., 2015). As synaptic withdrawal after injury was amplified in the presence of a die‐back, we conclude that the presence of dying‐back pathology can increase synaptic loss in following injury.
Our results are perhaps initially surprising based on previous work where the presence of dying‐back pathology reduced synaptic loss and axon fragmentation following nerve injury. In Wasted mice, which display a dying‐back neuromuscular phenotype, translation elongation factor eEF1A2 expression is required to prevent dying‐back pathology at the NMJ, whereas loss of this factor inhibits the initiation and progression of WD (Murray et al., 2008b). Although differing requirements for eEF1A2 expression may represent a mechanistic distinction between injury‐induced degeneration and dying‐back pathology, this does not exclude the possible occurrence of convergence in degenerative pathways further downstream.
Commonalities between WD and dying‐back neuropathies have been reported in animal models of motor neuron disease. For example, Wlds has been found to be protective in some motor neuron disease and peripheral neuropathy models, suggesting that there may be some convergence between degenerative mechanisms. Similarly, in mice with progressive motor neuronopathy, Wlds expression also delays dying‐back pathology (Ferri et al., 2003). Delays have also been reported in animal models of Parkinson’s and demyelinating diseases (Conforti et al., 2014). Several similarities have also been highlighted at the cellular level between WD and dying‐back pathology, including poor axonal transport, mitochondrial dysfunction, and an increase in intra‐axonal calcium (for review see Coleman, 2005; Conforti et al., 2014). Our recent proteomic work in mice has identified alterations in mitochondrial bioenergetics that coincide with delays in the change in level of synaptic loss in response to injury during postnatal development in the hindlimb (Kline et al., 2019). Targeting bioenergetic pathways by enhancing mitochondrial bioenergetics has also been found to rescue motor axon defects in a model of SMA (Boyd et al., 2017). Alterations in mitochondrial dynamics may therefore represent a promising, and perhaps uniting factor in WD and dying‐back neuropathy. Overall, the identification of common factors capable of influencing the rate of neurodegeneration under different physiological conditions could aid in the identification of targets that are common across multiple diseases.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
AUTHOR CONTRIBUTIONS
A. J. Mole: Design of experiments in Figures 1, 2, 3, 4, 5, acquisition, analysis, and interpretation of data for Figures 1, 2, 3, 4, 5; drafting of manuscript. S. Bell: Acquisition and processing of data for Figures 4 and 5, approval of manuscript. A .K. Thomson: Design of experiments for Figures 1 and 2, optimisation of experimental systems, critical reading and approval of manuscript. K. N. Dissanayake: Concept and design of Figures 1 and 2, approval of manuscript. R. R. Ribchester: Concept and design, critical reading and approval of manuscript. L. M. Murray: Concept and design of study, critical revision of the manuscript and approval of the manuscript.
ACKNOWLEDGEMENTS
This work was funded by grants from the Muscular Dystrophy Association (MDA) to L.M.M. (grant number 417757), the Motor Neuron Disease Association (MNDA) to R.R.R. (Ref. 838‐791), and the Institute Strategic Support Fund (ISSF3) from the University of Edinburgh supported by the Wellcome Trust (IS3‐R2.23) to L.M.M. A.J.M. is a recipient of a studentship from the Anatomical Society.
Mole AJ, Bell S, Thomson AK, Dissanayake KN, Ribchester RR, Murray LM. Synaptic withdrawal following nerve injury is influenced by postnatal maturity, muscle‐specific properties, and the presence of underlying pathology in mice. J. Anat. 2020;237:263–274. 10.1111/joa.13187
DATA AVAILABILITY STATEMENT
The raw data that support the findings of this study are available from the corresponding author (L.M.M.) upon reasonable request.
REFERENCES
- Beirowski, B. , Berek, L. , Adalbert, R. , Wagner, D. , Grumme, D.S. , Addicks, K. , et al. (2004) Quantitative and qualitative analysis of Wallerian degeneration using restricted axonal labelling in YFP‐H mice. Journal of Neuroscience Methods, 134, 23–35. [DOI] [PubMed] [Google Scholar]
- Beirowski, B. , Adalbert, R. , Wagner, D. , Grumme, D.S. , Addicks, K. , Ribchester, R.R. , et al. (2005) The progressive nature of Wallerian degeneration in wild‐type and slow Wallerian degeneration (WldS) nerves. BMC Neuroscience, 6, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman, M. , Murray, L.M. , Beauvais, A. , Pinheiro, B. and Kothary, R. (2012) A critical Smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromuscular Disorders, 22, 263–276. [DOI] [PubMed] [Google Scholar]
- Boyd, P.J. , Tu, W.Y. , Shorrock, H.K. , Groen, E.J.N. , Carter, R.N. , Powis, R.A. , et al. (2017) Bioenergetic status modulates motor neuron vulnerability and pathogenesis in a zebrafish model of spinal muscular atrophy. PLoS Genetics, 13, e1006744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockington, A. , Ning, K. , Heath, P.R. , Wood, E. , Kirby, J. , Fusi, N. , et al. (2013) Unravelling the enigma of selective vulnerability in neurodegeneration: Motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathologica, 125, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, R. , Hynes‐Allen, A. , Swan, A.J. , Dissanayake, K.N. , Gillingwater, T.H. and Ribchester, R.R. (2015) Activity‐dependent degeneration of axotomized neuromuscular synapses in Wld S mice. Neuroscience, 290, 300–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanagh, J.B. (1964) The significance of the ‘dying back’ process in experimental and human neurological disease. International Review of Experimental Pathology, 3, 219–267. [PubMed] [Google Scholar]
- Coleman, M. (2005) Axon degeneration mechanisms: commonality amid diversity. Nature Reviews Neuroscience, 6, 889–898. [DOI] [PubMed] [Google Scholar]
- Coleman, M.P. and Freeman, M.R. (2010) Wallerian degeneration, Wld(s), and Nmnat. Annual Review of Neuroscience, 33, 245–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comley, L. , Allodi, I. , Nichterwitz, S. , Nizzardo, M. , Simone, C. , Corti, S. , et al. (2015) Motor neurons with differential vulnerability to degeneration show distinct protein signatures in health and ALS. Neuroscience, 291, 216–229. [DOI] [PubMed] [Google Scholar]
- Comley, L.H. , Nijssen, J. , Frost‐Nylen, J. and Hedlund, E. (2016) Cross‐disease comparison of amyotrophic lateral sclerosis and spinal muscular atrophy reveals conservation of selective vulnerability but differential neuromuscular junction pathology. Journal of Comparative Neurology, 524, 1424–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti, L. , Gilley, J. and Coleman, M.P. (2014) Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nature Reviews Neuroscience, 15, 394–409. [DOI] [PubMed] [Google Scholar]
- Courtney, N.L. , Mole, A.J. , Thomson, A.K. and Murray, L.M. (2019) Reduced P53 levels ameliorate neuromuscular junction loss without affecting motor neuron pathology in a mouse model of spinal muscular atrophy. Cell Death & Disease, 10, 515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Stefano, M. , Nascimento‐Ferreira, I. , Orsomando, G. , Mori, V. , Gilley, J. , Brown, R. , et al. (2015) A rise in NAD precursor nicotinamide mononucleotide (NMN) after injury promotes axon degeneration. Cell Death and Differentiation, 22, 731–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiDonato, C.J. , Lorson, C.L. , De Repentigny, Y. , Simard, L. , Chartrand, C. , Androphy, E.J. , et al. (2001) Regulation of murine survival motor neuron (Smn) protein levels by modifying Smn exon 7 splicing. Human Molecular Genetics, 10, 2727–2736. [DOI] [PubMed] [Google Scholar]
- Erzen, I. , Cvetko, E. , Obreza, S. and Angaut‐Petit, D. (2000) Fiber types in the mouse levator auris longus muscle: a convenient preparation to study muscle and nerve plasticity. Journal of Neuroscience Research, 59, 692–697. [DOI] [PubMed] [Google Scholar]
- Ferri, A. , Sanes, J.R. , Coleman, M.P. , Cunningham, J.M. and Kato, A.C. (2003) Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Current Biology, 13, 669–673. [DOI] [PubMed] [Google Scholar]
- Gilley, J. and Coleman, M.P. (2010) Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biology, 8, e1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley, J. , Orsomando, G. , Nascimento‐Ferreira, I. and Coleman, M.P. (2015) Absence of SARM1 rescues development and survival of NMNAT2‐deficient axons. Cell Reports, 10, 1974–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillingwater, T.H. , Haley, J.E. , Ribchester, R.R. and Horsburgh, K. (2004) Neuroprotection after transient global cerebral ischemia in Wld(s) mutant mice. Journal of Cerebral Blood Flow and Metabolism, 24, 62–66. [DOI] [PubMed] [Google Scholar]
- Gillingwater, T.H. , Ingham, C.A. , Parry, K.E. , Wright, A.K. , Haley, J.E. , Wishart, T.M. , et al. (2006) Delayed synaptic degeneration in the CNS of Wlds mice after cortical lesion. Brain, 129, 1546–1556. [DOI] [PubMed] [Google Scholar]
- Gillingwater, T.H. , Thomson, D. , Mack, T.G. , Soffin, E.M. , Mattison, R.J. , Coleman, M.P. ,, et al. (2002) Age‐dependent synapse withdrawal at axotomised neuromuscular junctions in Wld(s) mutant and Ube4b/Nmnat transgenic mice. Journal of Physiology, 543, 739–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, S.M. , Gogliotti, R.G. , Rao, V. , Beauvais, A. , Kothary, R. and DiDonato, C.J. (2010) Mouse survival motor neuron alleles that mimic SMN2 splicing and are inducible rescue embryonic lethality early in development but not late. PLoS ONE, 5, e15887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedlund, E. , Karlsson, M. , Osborn, T. , Ludwig, W. and Isacson, O. (2010) Global gene expression profiling of somatic motor neuron populations with different vulnerability identify molecules and pathways of degeneration and protection. Brain, 133, 2313–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan, A. , Spiller, K.J. , Towne, C. , Kanning, K.C. , Choe, G.T. , Geber, A. , et al. (2014) Neuronal matrix metalloproteinase‐9 is a determinant of selective neurodegeneration. Neuron, 81, 333–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. , Yang, D.S. , Katti, P. and Glancy, B. (2019) Protein composition of the muscle mitochondrial reticulum during postnatal development. Journal of Physiology, 597, 2707–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline, R.A. , Dissanayake, K.N. , Hurtado, M.L. , Martínez, N.W. , Ahl, A. , Mole, A.J. , et al. (2019) Altered mitochondrial bioenergetics are responsible for the delay in Wallerian degeneration observed in neonatal mice. Neurobiology of Diseases, 130, 104496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline, R.A. , Kaifer, K.A. , Osman, E.Y. , Carella, F. , Tiberi, A. , Ross, J. , et al. (2017) Comparison of independent screens on differentially vulnerable motor neurons reveals alpha‐synuclein as a common modifier in motor neuron diseases. PLoS Genetics, 13, e1006680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre, S. , Burglen, L. , Reboullet, S. , Clermont, O. , Burlet, P. , Viollet, L. , et al. (1995) Identification and characterization of a spinal muscular atrophy‐determining gene. Cell, 80, 155–165. [DOI] [PubMed] [Google Scholar]
- Lunn, E.R. , Perry, V.H. , Brown, M.C. , Rosen, H. and Gordon, S. (1989) Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. European Journal of Neuroscience, 1, 27–33. [DOI] [PubMed] [Google Scholar]
- Mack, T.G. , Reiner, M. , Beirowski, B. , Mi, W. , Emanuelli, M. , Wagner, D. , et al. (2001) Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nature Neuroscience, 4, 1199–1206. [DOI] [PubMed] [Google Scholar]
- Murray, L.M. , Comley, L.H. , Thomson, D. , Parkinson, N. , Talbot, K. and Gillingwater, T.H. (2008a) Selective vulnerability of motor neurons and dissociation of pre‐ and post‐synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Human Molecular Genetics, 17, 949–962. [DOI] [PubMed] [Google Scholar]
- Murray, L.M. , Thomson, D. , Conklin, A. , Wishart, T.M. and Gillingwater, T.H. (2008b) Loss of translation elongation factor (eEF1A2) expression in vivo differentiates between Wallerian degeneration and dying‐back neuronal pathology. Journal of Anatomy, 213, 633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, L.M. , Gillingwater, T.H. and Parson, S.H. (2010a) Using mouse cranial muscles to investigate neuromuscular pathology in vivo. Neuromuscular Disorders, 20, 740–743. [DOI] [PubMed] [Google Scholar]
- Murray, L.M. , Lee, S. , Baumer, D. , Parson, S.H. , Talbot, K. and Gillingwater, T.H. (2010b) Pre‐symptomatic development of lower motor neuron connectivity in a mouse model of severe spinal muscular atrophy. Human Molecular Genetics, 19, 420–433. [DOI] [PubMed] [Google Scholar]
- Murray, L.M. , Comley, L.H. , Gillingwater, T.H. and Parson, S.H. (2011) The response of neuromuscular junctions to injury is developmentally regulated. FASEB Journal, 25, 1306–1313. [DOI] [PubMed] [Google Scholar]
- Murray, L.M. , Beauvais, A. , Bhanot, K. and Kothary, R. (2013) Defects in neuromuscular junction remodelling in the Smn(2B/‐) mouse model of spinal muscular atrophy. Neurobiology of Diseases, 49, 57–67. [DOI] [PubMed] [Google Scholar]
- Murray, L. , Gillingwater, T.H. and Kothary, R. (2014) Dissection of the transvs. abdominis muscle for whole‐mount neuromuscular junction analysis. Journal of Visualized Experiments, 83, e51162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, L.M. , Beauvais, A. , Gibeault, S. , Courtney, N.L. and Kothary, R. (2015) Transcriptional profiling of differentially vulnerable motor neurons at pre‐symptomatic stage in the Smn (2b/‐) mouse model of spinal muscular atrophy. Acta Neuropathologica Communications, 3, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman, E.Y. , Rietz, A. , Kline, R.A. , Cherry, J.J. , Hodgetts, K.J. , Lorson, C.l. , et al. (2019) Intraperitoneal delivery of a novel drug‐like compound improves disease severity in severe and intermediate mouse models of Spinal Muscular Atrophy. Scientific Reports, 9, 1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterloh, J.M. , Yang, J. , Rooney, T.M. , Fox, A.N. , Adalbert, R. , Powell, E.H. , et al. (2012) dSarm/Sarm1 is required for activation of an injury‐induced axon death pathway. Science, 337, 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente, B.N. , Kimura, W. , Muralidhar, S.A. , Moon, J. , Amatruda, J.F. , Phelps, K.L. , et al. (2014) The oxygen‐rich postnatal environment induces cardiomyocyte cell‐cycle arrest through DNA damage response. Cell, 157, 565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribchester, R.R. , Tsao, J.W. , Barry, J.A. , Asgari‐Jirhandeh, N. , Perry, V.H. and Brown, M.C. (1995) Persistence of neuromuscular junctions after axotomy in mice with slow Wallerian degeneration (C57BL/WldS). European Journal of Neuroscience, 7, 1641–1650. [DOI] [PubMed] [Google Scholar]
- Rodrigues, N.R. , Owen, N. , Talbot, K. , Ignatius, J. , Dubowitz, V. and Davies, K.E. (1995) Deletions in the survival motor neuron gene on 5q13 in autosomal recessive spinal muscular atrophy. Human Molecular Genetics, 4, 631–634. [DOI] [PubMed] [Google Scholar]
- Slater, C.R. (1966) Time course of failure of neuromuscular transmission after motor nerve section. Nature, 209, 305–306. [DOI] [PubMed] [Google Scholar]
- Sleigh, J.N. , Burgess, R.W. , Gillingwater, T.H. and Cader, M.Z. (2014a) Morphological analysis of neuromuscular junction development and degeneration in rodent lumbrical muscles. Journal of Neuroscience Methods, 227, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleigh, J.N. , Grice, S.J. , Burgess, R.W. , Talbot, K. and Cader, M.Z. (2014b) Neuromuscular junction maturation defects precede impaired lower motor neuron connectivity in Charcot‐Marie‐Tooth type 2D mice. Human Molecular Genetics, 23, 2639–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson, S.R. , Nahon, J.E. , Mutsaers, C.A. , Thomson, D. , Hamilton, G. , Parson, S.H. , et al. (2012) Morphological characteristics of motor neurons do not determine their relative susceptibility to degeneration in a mouse model of severe spinal muscular atrophy. PLoS ONE, 7, e52605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalon, E. , Kline, R.A. , Smith, C.E. , Lorson, Z.C. , Osman, E.Y. , O'Day, S. , et al. (2019) AAV9‐Stathmin1 gene delivery improves disease phenotype in an intermediate mouse model of Spinal Muscular Atrophy. Human Molecular Genetics, 28(22), 3742–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waller, A. (1851) Experiments on the section of the glosso‐pharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Edinburgh Medical and Surgical Journal, 76, 369–376. [PMC free article] [PubMed] [Google Scholar]
- Wang, M.S. , Fang, G. , Culver, D.G. , Davis, A.A. , Rich, M.M. and Glass, J.D. (2001) The WldS protein protects against axonal degeneration: A model of gene therapy for peripheral neuropathy. Annals of Neurology, 50, 773–779. [DOI] [PubMed] [Google Scholar]
- Wang, J.T. , Medress, Z.A. and Barres, B.A. (2012) Axon degeneration: Molecular mechanisms of a self‐destruction pathway. Journal of Cell Biology, 196, 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data that support the findings of this study are available from the corresponding author (L.M.M.) upon reasonable request.