

Abstract
Oropharyngeal candidiasis (OPC) is an opportunistic infection of the oral mucosa caused by the commensal fungus C. albicans. IL-17 receptor signaling is essential to prevent OPC in mice and humans, but the individual roles of its ligands, IL-17A, IL-17F and IL-17AF, are less clear. A homozygous IL-17F deficiency in mice does not cause OPC susceptibility, whereas mice lacking IL-17A are moderately susceptible. In humans, a rare heterozygous mutation in IL-17F (IL-17F.S65L) was identified that causes chronic mucocutaneous candidiasis, suggesting the existence of essential antifungal pathways mediated by IL-17F and/or IL-17AF. To investigate the role of IL-17F and IL-17AF in more detail, we exploited this ‘experiment of nature’ by creating a mouse line bearing the homologous mutation in IL-17F (Ser65Leu) by CRISPR/Cas9. Unlike Il17f−/− mice that are resistant to OPC, Il17fS65L/S65L mice showed increased oral fungal burdens similar to Il17a−/− mice. In contrast to humans, however, disease was only evident in homozygous, not heterozygous, mutant mice. The mutation was linked to modestly impaired CXC chemokine expression and neutrophil recruitment to the infected tongue but not to alterations in oral antimicrobial peptide expression. These findings suggest mechanisms by which the enigmatic cytokine IL-17F contributes to host defense against fungi. Moreover, because these mice do not phenocopy Il17f−/− mice, they may provide a valuable tool to interrogate IL-17F and IL-17AF function in vivo in other settings.
Introduction
Fungal infections have a serious impact on human health, yet typically receive less attention than other pathogens (1). Candida albicans is a commensal fungus in humans, colonizing mucosal surfaces such as the oral cavity, vaginal tract and gut as well as skin. Although normally benign in healthy individuals, C. albicans can cause pathogenic infections such as oropharyngeal candidiasis (OPC, oral thrush) under settings of immunodeficiency. Immunity to C. albicans is highly dependent on CD4+ T cells, as up 95% of HIV+ patients suffer from recurrent OPC during progression to AIDS, correlating with T cell counts (2, 3).
Human T cell responses to C. albicans are dominated by Th17 cells (4–7). IL-17A (IL-17) is the signature cytokine of Th17 cells and is also expressed by various subsets of innate lymphocytes such as γ-T, NKT and ILC3 cells (8). IL-17 plays a critical role in antifungal immunity to C. albicans (9, 10). Studies in mice demonstrated the importance of IL-17 signaling against C. albicans oral infections, as mice lacking IL-17RA, IL-17RC or the signaling adaptor Act1 are all highly sensitive to systemic, oral and dermal candidiasis (11–15). Clinical data from cohorts of chronic mucocutaneous candidiasis disease (CMCD) patients confirmed that susceptibility to candidiasis is strongly associated with IL-17/Th17 deficiencies. Mutations in IL-17RA, IL-17RC or Act1 in humans, as in mice, cause susceptibility to mucocutaneous candidiasis (16–19). Clinical treatment with anti-IL-17A antibodies for autoimmunity is also linked to OPC (20). Similarly, STAT3, RORC, and STAT1 mutations are linked to reduced Th17 frequencies and CMCD (21–23).
The IL-17 family of cytokines is structurally distinct from other cytokine subclasses, composed of IL-17A, IL-17B, IL-17C, IL-17D, IL-17E (IL-25) and IL-17F (24). Among these, IL-17A shares the most homology with IL-17F at the amino acid level (56%) (25, 26). Both IL-17A and IL-17F form homodimers but can also form a heterodimer (IL-17AF) (27–29). All three isoforms signal through the IL-17RA:IL-17RC receptor complex, though the ligands have different binding affinities for the receptor, with IL-17A > IL-17AF > IL-17F (28, 30, 31). Recent data suggest that IL-17A may also signal through an IL17RA:IL-17RD receptor (32), and that IL-17C homodimer may also be competent for signaling (31, 33). Hence our understanding of the IL-17 cytokine family is still developing.
IL-17A and, to a lesser extent, IL-17F activate a program of inflammation in target cells, mainly fibroblasts and epithelial cell types (34, 35). IL-17A induces a characteristic gene signature that includes cytokines (IL-6, GM-CSF, G-CSF), chemokines (CXCL1, CXCL2, CXCL8, CCL2, CCL7, CCL20), matrix metalloproteinases (MMP1, 2, 3, 9, 13), transcriptional and post-transcriptional regulators (C/EBPs, IκBξ, Regnase-1, Arid5a) and antimicrobial peptides (AMPs) (β-defensins, S100A proteins, lipocalin 2) (36–40). Emerging studies also implicate IL-17A in metabolic and proliferation gene expression (41, 42). Though less well characterized, IL-17F induces a similar but not entirely overlapping panel of genes (43–46). Studies of the in vitro functions of the IL-17AF heterodimer are comparatively limited, but studies reported to date indicate a similar gene induction profile induced by IL-17AF compared to IL-17A and IL-17F (27, 28).
Despite their capacity to bind the same receptor, IL-17A and IL-17F exert distinct activities in vivo. Il17a−/− and Il17f−/− mice show differential susceptibilities to various diseases, both infectious and autoimmune (47, 48). This dichotomy is illustrated in OPC; whereas Il17a−/− or mice treated with IL-17A-neutralizing antibodies exhibit elevated susceptibility to OPC compared to immunocompetent control mice (49), Il17f−/− or mice treated with IL-17F neutralizing antibodies are fully resistant to C. albicans infection. However, there is evidence that IL-17F does participate in immunity to OPC, as dual blockade of IL-17A and IL-17F increases susceptibility to OPC over blockade of IL-17A alone (49, 50). In line with this, humans with autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED) caused by AIRE gene deficiencies have circulating auto-antibodies against both IL-17A and IL-17F, which is thought to underlie susceptibility to CMCD (51–53).
The physiological role of IL-17AF is still unclear. Although IL-17AF can be reliably detected (e.g., by sandwich ELISA), there are no commercial antibodies that block this isoform selectively or efficiently, and hence determining its specific role in vivo is challenging. Hints regarding IL-17F and IL-17AF function in came from an unusual cohort of rare CMCD patients that harbor a heterozygous serine-to-leucine mutation in the IL17F gene at position 65 (IL-17F.S65L) (16). The mutated residue in these individuals lies within the region of interaction of IL-17F with the IL-17 receptor. In vitro, this mutation impaired IL-17F binding to the receptor but had no apparent impact on the ability of IL-17F to dimerize with IL-17F or IL-17A. Cultured human fibroblasts treated with an IL-17F homodimer containing this mutation (IL-17FS65L/IL-17F or IL-17S65L/IL-17FS65L) or a mutant IL-17AF heterodimer (IL-17A/IL-17FS65L) showed strongly impaired signaling in vitro (16). Accordingly, the S65L mutation in human IL-17F appeared to be both a loss of function and a dominant negative mutation, causing functional blockade of IL-17F and IL-17AF. Importantly, IL-17A homodimers were still found in these patients yet were apparently insufficient to fully protect from candidiasis. These data thus implied that IL-17F and/or IL-17AF are significant contributors to the antifungal immune response in humans.
In this study, we created an IL-17F.S65L mouse strain using CRISPR/Cas9 technology to exploit this “experiment of nature” (16) in order to better understand the functions of IL-17F and IL-17AF in immune responses. There was no detectable disease susceptibility in mice heterozygous for the mutation, potentially suggesting differences between mouse and human IL-17F function in vivo. However Il17fS65L/S65L mice exhibited a similar susceptibility to OPC as Il17a−/− mice, which contrasted with the previously-known resistance of Il17f−/− mice to OPC (49). Thus, this mutation appears to be more than simply a loss-of-function mutation. The increased susceptibility of these mice to fungal infection was linked to impaired expression of CXC chemokines and concomitantly reduced neutrophil recruitment to the oral mucosa, but surprisingly not to expression of key antimicrobial peptides known to control OPC such as β-defensin-3. As these mice do not phenocopy Il17f−/− mice, they may provide a valuable tool to interrogate IL-17F and IL-17AF in vivo.
Methods
Generation of Il17fS65L/S65L knockin mice
Il17fS65L/S65L mice were created by CRISPR/Cas9 by the Transgenic and Gene Targeting (TGT) and Innovative Technologies Development (ITD) Core facilities in the Department of Immunology, University of Pittsburgh. Briefly, a S.py. Cas9 target sequence overlapping the Ser65 codon in the mature Il17f sequence (following signal peptide cleavage) was selected: GTTCCCCTCAGAGATCGCTG AGG. The protospacer adjacent motif (PAM) in bold was not included in the sgRNA. Cas9 mRNA and the sgRNA were produced as described (54, 55). A 127-mer oligonucleotide (Ultramer, IDT) was used as template for homology-directed repair (HDR): Il17f-S65L-HDRv3: 5’-CATCCTGCTTTACTTTTTATTTTTTTCCTTCAGCATCACTCGAGACCCCCACCGGTTCCCTCTAGAAATCGCTGAGGCCCAGTGCAGACACTCAGGCTGCATCAATGCCCAGGGTCAGGAAGACAGC-3’. The oligonucleotide contains a substitution to convert Serine 65 to Leucine, which contemporaneously disrupts an HPY188I restriction site (underlined) to facilitate genotyping. The sequence also contains an additional silent substitution, bringing a total of 4 mismatches (red, above) between the sgRNA target sequence and the designed allele, thus limiting further re-editing of the mutant allele by Cas9. C57BL/6J embryos were microinjected with the sgRNA (50 ng/μl), the HDR oligonucleotide (0.5 μM) and Cas9 mRNA (100 ng/μl). Embryos that developed to the 2-cell stage were transferred into pseudopregnant female surrogates. Sixteen founders were identified by PCR amplification of the target region (Forward: 3’-ATGGGAGAAACCCCGTTTTA-5’; reverse: 3’- TCCAACCTGAAGGAATTAGAACA-5’) followed by restriction digestion of the PCR product with HPY188I. The correct sequence was validated by Sanger sequencing of the PCR products following TOPO cloning. Of these, 5 founders were homozygous for the mutation (Supp. Fig. 2). Based on the CRISPOR website (56), there were only 2 potential off-target sequences with fewer than 4 mismatches in the mouse genome (Table 1). Among all founders analyzed, there were no mutations introduced at those sites, as determined by PCR and Sanger sequencing. The expanded line was backcrossed twice to C57BL/6J mice prior to colony expansion.
Table 1.
Off target screening of Il17f S65L mutant mice
| Off Target 1a | Off Target 2a | |
|---|---|---|
| Score | 0.958618437118 | 0.455082125604 |
| Chromosome | 3 | 5 |
| Start | 131315589 | 141241627 |
| End | 131315611 | 141241649 |
| Strand | - | + |
| TargetPattern | GTTCCCCTCAGAGATCGCTGAGG | GTTCCCCTCAGAGATCGCTGAGG |
| Mismatch | 3 | 3 |
| Sequence | GTTCTGTTCAGAGATCGCTGTGG | GCTCCCCTCACAGAGCGCTGCGG |
| MismatchLoc | 5,6,7 | 2,11,15 |
| TargetNum | 1 | 1 |
| TargetStrand | + | + |
| Mmismatchln2 | 1 | 1 |
| Region | intergenic | exonic |
| GeneUCSCID | N/A | Uc009ail.2 |
| GeneSymbol | N/A | Sdk1 |
| Primer-forwardb | AGCTCCAATAAGGCCACACC | CACCTGGTGTCGTCCCAC |
| Primer-reverseb | CACGAGGACTAGACAGCGAC | CTCCGATGAGCAGCGAGC |
Two off-target sites with up to 3 bp mismatches were predicted in creation of Il17fS65L/S65L mice, located on chromosomes 3 and 5. Precise locations and sequences are shown.
PCR primers used to amplify and sequence these regions are indicated. See Methods section for details.
Other mice
Il17fThy1.1 reporter mice were previously described (57). Il17ra−/− mice were a gift from Amgen. Act1−/− mice were from U. Siebenlist, NIH. WT mice were from The Jackson Laboratory, Taconic Farms, or generated from breeding colonies. All mice were on the C57BL/6 background. Age matched mice (6–10 weeks) were used for experiments with both sexes. Animal use protocols were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Model of oropharyngeal candidiasis (OPC)
OPC was induced by sublingual inoculation with a cotton ball saturated in C. albicans (CAF2–1) for 75 min under anesthesia, as described (58). Tongue homogenates were prepared on a gentleMACS (Miltenyi Biotec), and fungal burdens determined by plating serial dilutions on YPD agar with Ampicillin. Anti-IL-17A or isotype control Abs (Bio X Cell) (200ug/ ul) were administered i.p. on days −1, 1, and 3 relative to C. albicans infection.
Flow cytometry
Tongues were digested with Collagenase IV (0.7mg/ml) in HBSS for 30 min. Cell suspensions were separated by Percoll gradient centrifugation. Abs were from eBioscience, BD Biosciences, and BioLegend, as described (59). Data were acquired on an LSR Fortessa and analyzed with FlowJo (Ashland, OR).
qPCR
Tongues were homogenized in in a Gentle MACS Dissociator and tongue RNA extracted using RNeasy kits (Qiagen). RNA from ST2 cells was extracted in RLT buffer (Qiagen). cDNA was generated using a SuperScript III First Strand Synthesis System (Invitrogen). Relative quantification of mRNA expression was determined by real-time PCR with SYBR green (Quanta BioSciences) normalized to Gapdh on the Applied Biosystems 7500 platform. Primers were from QuantiTect (Qiagen).
Cell culture and Th17 differentiation
The ST2 stromal cell line was cultured in α-minimum essential medium (α-MEM; Sigma-Aldrich, St. Louis MO) with L-glutamine, antibiotics, and 10% fetal bovine serum. 5×104 cells were seeded into 6 wells plates prior to cytokine stimulation. Recombinant IL-17F.S65L and IL-17F were synthesized by Bon Opus Biosciences (Millburn, NJ) by expression in Expi293 cells (ThermoFisher). Mouse TNFα was from Peprotech and used at 2 ng/ml (Rocky Hill, NJ).
Naïve splenic CD4+ T cells were purified by magnetic separation (Miltenyi Biotec). T cells were activated with α-CD28 (5 ug/ml; BioXCell) and plate-bound α-CD3 (clone 145-TC11, 5 ug/ml; BioXCell) in RPMI supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, 100 mg/ml streptomycin, 50 mM 2-β-mercaptoethanol, and sodium pyruvate) for 4 d with IL-1β (50 ng/ml), IL-6 (50 ng/ml), IL-23 (50 ng/ml), and TGFβ (5 ng/ml). Cytokines were from R&D Systems.
Statistics
Data were analyzed on Prism (Graphpad) using ANOVA or Student’s t test. Fungal burdens are presented as geometric mean analyzed by ANOVA with Mann-Whitney analysis. *P < 0.05; **< 0.01; *** < 0.001; **** < 0.0001.
Results
Murine IL-17F S65L is a loss-of-function mutation
The S65L residue in IL-17F that is mutated in humans with CMCD is conserved among many species, including mice (16). The human IL-17F.S65L cytokine has no signaling capacity in vitro (16). To determine whether the mouse orthologue of IL-17F.S65L functions similarly to its human counterpart, a His-tagged murine IL-17F.S65L and a corresponding wild type IL-17F control were expressed in Expi293 cells and purified from conditioned media on a nickel-charged affinity resin (Ni-NTA) (Fig. 1A). Recombinant IL-17F.S65L and IL-17F migrated according to their predicted dimeric sizes on non-reducing SDS-PAGE (Fig. 1B). The multiple bands in the reduced (R) and non-reduced (NR) gels are consistent with the expectation that these recombinant cytokines exist in multiple glycosylated and non-glycosylated forms (Fig. 1B, Suppl. Fig. 1).
Figure 1. Murine IL-17F.S65L is a loss-of-function mutation.

(A) Amino acid sequences of recombinant His-tagged WT IL-17F and the IL-17F.S65L mutant used in this study. The Ser65 residue is shown in red. (B) IL-17F and IL-17F.S65L were transfected into Expi293 cells. Culture medium was collected on day 5 post transfection purified by Ni-NTA. The eluted fraction was analyzed by SDS-PAGE in reducing (R) condition and non-reducing (NR) conditions. Size markers are indicated at left. (C, D) ST2 stromal cells were treated with the IL-17F (blue) and IL-17F.S65L (red) at increasing doses in the absence (C) or presence (D) of TNFα (2 ng/ml) for 3 h. Il6 or Cxcl1 mRNA was assessed by qPCR. Data is graphed as fold-change relative to untreated conditions ± SEM from 2 independent experiments, analyzed by ANOVA. **P < 0.01, *** < 0.001, **** < 0.0001.
To evaluate the signaling capability of murine IL-17F.S65L, murine ST2 stromal cells were treated with increasing concentrations of IL-17F.S65L or WT control IL-17F for 3 h. Two IL-17F-inducible genes, Il6 and Cxcl1, were assessed by qPCR as endpoints of signaling. As expected, control IL-17F induced Il6 and Cxcl1 in a dose-dependent manner (Fig. 1C). In contrast, IL-17F.S65L at most doses (6.25–50 ng/ml) did not detectably upregulate Il6 or Cxcl1. However, both mRNAs were slightly enhanced by IL-17F.S65L at a supraphysiological dose (100 ng/ml), but nonetheless showed significantly reduced activity compared to non-mutated IL-17F (Fig. 1C). Because IL-17F induces gene expression synergistically with other cytokines (43, 46), we evaluated IL-17F.S65L responsiveness in concert with a suboptimal dose of TNFα (2 ng/ml). Control IL-17F synergized with TNFα to induce Il6 and Cxcl1, but the IL-17F.S65L mutant did not synergistically enhance expression of these genes (Fig. 1D). Thus, analogous to the IL-17F.S65L mutation described CMCD patients, the mouse IL-17F.S65L homodimer appears to be a loss-of-function mutation.
Il17fS65L/S65L mice are modestly susceptible to OPC
To determine the function of the murine IL-17F.S65L mutation in vivo, we created an IL-17F.S65L mutant mouse strain by CRISPR/Cas9 technology in the C57BL/6 background. Sixteen founders were generated, of which 5 had a homozygous nucleotide replacement (Fig. 2A, Suppl. Fig. 2). None of the founder lines were found to have mutations in the either of the two predicted off-target sites, ascertained by PCR of genomic DNA and sequencing (Table 1, see Methods section for details). Similar to IL17a−/− and Il17f−/− mice (47), there were no obvious abnormalities in the health of these mice when maintained in SPF conditions, and they bred normally with Mendelian numbers of offspring.
Figure 2. Il17fS65L/S65L mice are modestly susceptible to OPC.
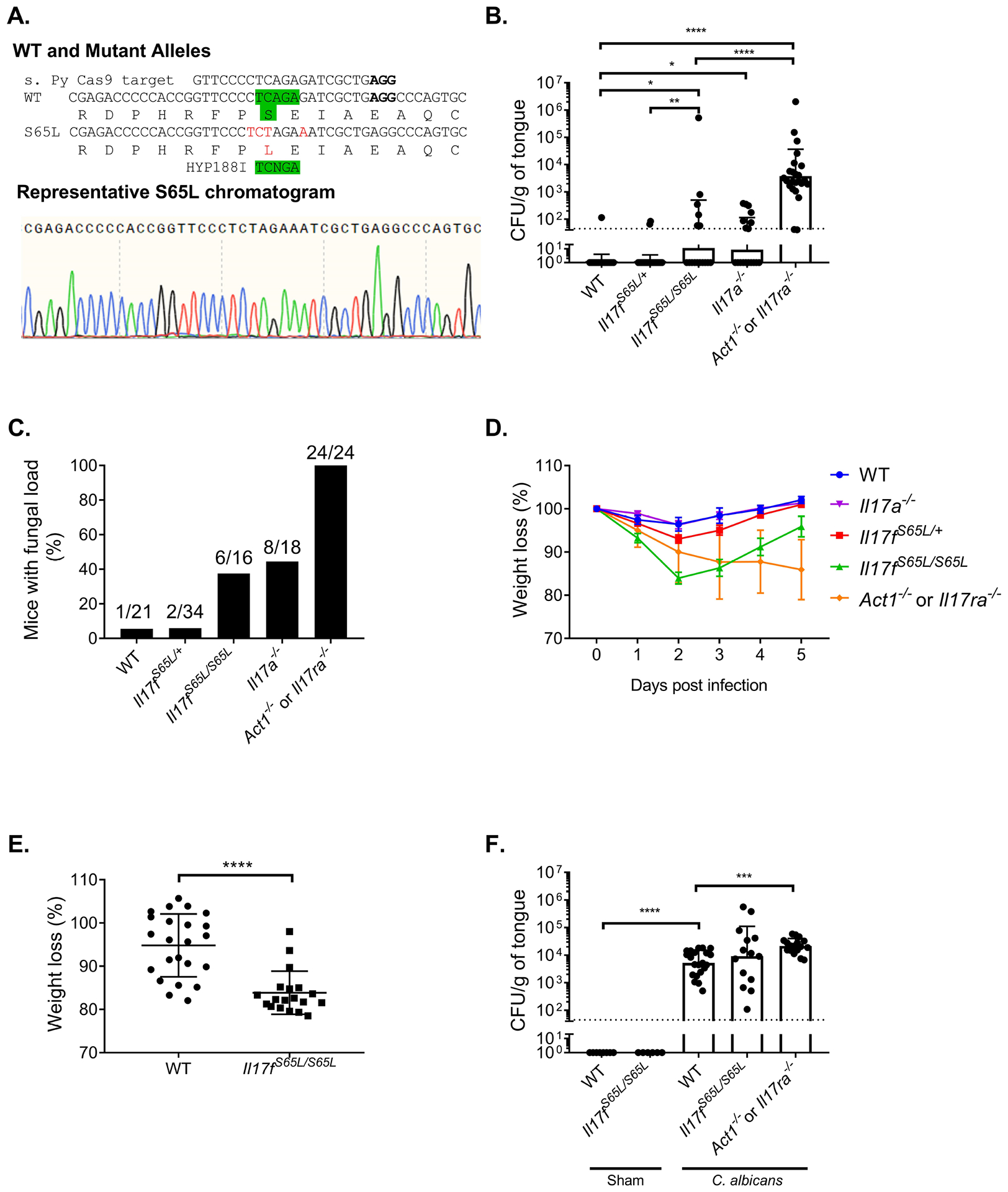
(A) Top: Schematic diagram of murine IL-17F.S65L substitution created by CRISPR/Cas9 including disruption of an endogenous HPY188I restriction site. Bottom: Representative chromatogram of genomic DNA sequencing from a representative founder mouse. (B) The indicated mice were infected sublingually with C. albicans or PBS (Sham). Fungal burdens were assessed by CFU enumeration on YPD/Amp on day 5 p.i. Graphs show geometric mean + SD. Dashed line indicates limit of detection (~30 CFU/g). Data were compiled from five 5 experiments. Each symbol represents one mouse. (C) Data from panel B is represented as percentage of mice with detectable fungal load in the tongue. Values above indicate number of mice with fungal burden/total. (D) Weight in the animals from panel B was assessed daily and percentage loss relative to day 0 is shown for each time point. (E) Weight loss of Il17fS65L/S65L and WT mice at day 2 p.i. from panel B. Graph shows mean ± SD. (F) Fungal burdens were assessed on day 2 p.i.. Data are compiled from 3 independent experiments. Data were analyzed by ANONVA or Student’s t-test, with Mann-Whitney analysis for fungal load analysis. *P < 0.05, ** < 0.01, *** < 0.001, **** < 0.0001.
In the one family of humans with this mutation identified, 5/7 affected individuals experienced CMCD as heterozygotes (no homozygote family members were described (16)). Based on this we predicted that mice that are homozygous or heterozygous for the IL17F.S65L mutation would be susceptible to mucosal candidiasis. To test this hypothesis, mice were subjected to OPC by a 75 minute sublingual exposure to C. albicans (strain CAF2–1) by standard methods (58, 60). In this model, WT mice typically clear C. albicans from the oral mucosa within 3 days and show no overt signs of illness such as prolonged weight loss (12, 61). We previously demonstrated that mice lacking IL-17RA or Act1 maintain a high fungal burden in the oral cavity after infection, which we typically measure at day 5 post infection (p.i.) (12, 14), Mice lacking IL-17A (Il17a−/− or given anti-IL-17A antibodies) have detectable fungal burdens, though consistently lower than Il17ra−/− or Act1−/− mice (49). In contrast, mice lacking IL-17F, either by knockout or with neutralizing antibodies, are fully resistant to OPC, at least within the detection limits of this system (49). Here, we observed that Il17fS65L/S65L mice had a significantly higher oral fungal burden compared to WT controls at 5 days p.i., at levels similar to Il17a−/− animals (Fig. 2B). Notably, however, the Il17f+/S65L heterozygous mice did not have elevated fungal loads compared to WT. Approximately 40% of the Il17fS65L/S65L and Il17a−/− mice still had a detectable fungal load at this time point, whereas the WT and the Il17/S65L/+ heterozygous mice almost all fully cleared the infection (Fig. 2C). Il17fS65L/S65L mice also lost slightly more weight than WT controls, which was most evident at day 2 p.i. (Fig. 2D, E). However, oral fungal burdens in Il17fS65L/S65L mice were not measurably different at day 2 (Fig. 2F). Collectively, these results indicate that IL-17F.S65L mutation contributes detectably, albeit modestly, to susceptibility to OPC, but only when the mutation is present on both alleles.
Il17fS65L/S65L mice show mildly impaired neutrophil recruitment during OPC
To understand the mechanisms by which the IL-17F.S65L mutation promotes susceptibility to OPC, we evaluated factors known to be critical for antifungal immunity mediated by IL-17R signaling (12, 15, 61). Neutrophils are vital for fungal clearance in OPC (62–64). We have observed that Il17ra−/− mice show impaired recruitment of neutrophils to the oral mucosa following OPC induction (12, 15, 59). IL-17F upregulates expression of neutrophil-attracting chemokines such as CXCL1 and CXCL2 (65–67), and both human and murine IL-17FS65L showed impaired induction of this chemokine in fibroblasts in vitro (Fig. 1, (16)). Consistent with this, Cxcl1 gene expression in the tongue was downregulated in Il17fS65L/S65L mice upon C. albicans oral infection, though not completely impaired. However, expression of Cxcl2 was similar between Il17fS65L/S65L mice and WT control (Fig. 3A). Flow cytometry revealed that the percentage of early neutrophil recruitment to the tongue measured at day 2 p.i. was partly, though by no means fully decreased in infected Il17fS65L/S65L mice compared to WT (45% versus 57%) (Fig. 3B). There was also a trend to reduced total numbers of neutrophils, though there was high variability in the number of cells recovered from the tongue (a common problem often encountered when isolating cells from this tissue (68, 69)). Thus, although not proven directly, impaired neutrophil recruitment in Il17fS65/S65L mice may help explain OPC susceptibility in these mice.
Figure 3. IL-17F.S65L mice exhibit impaired neutrophil recruitment during OPC.
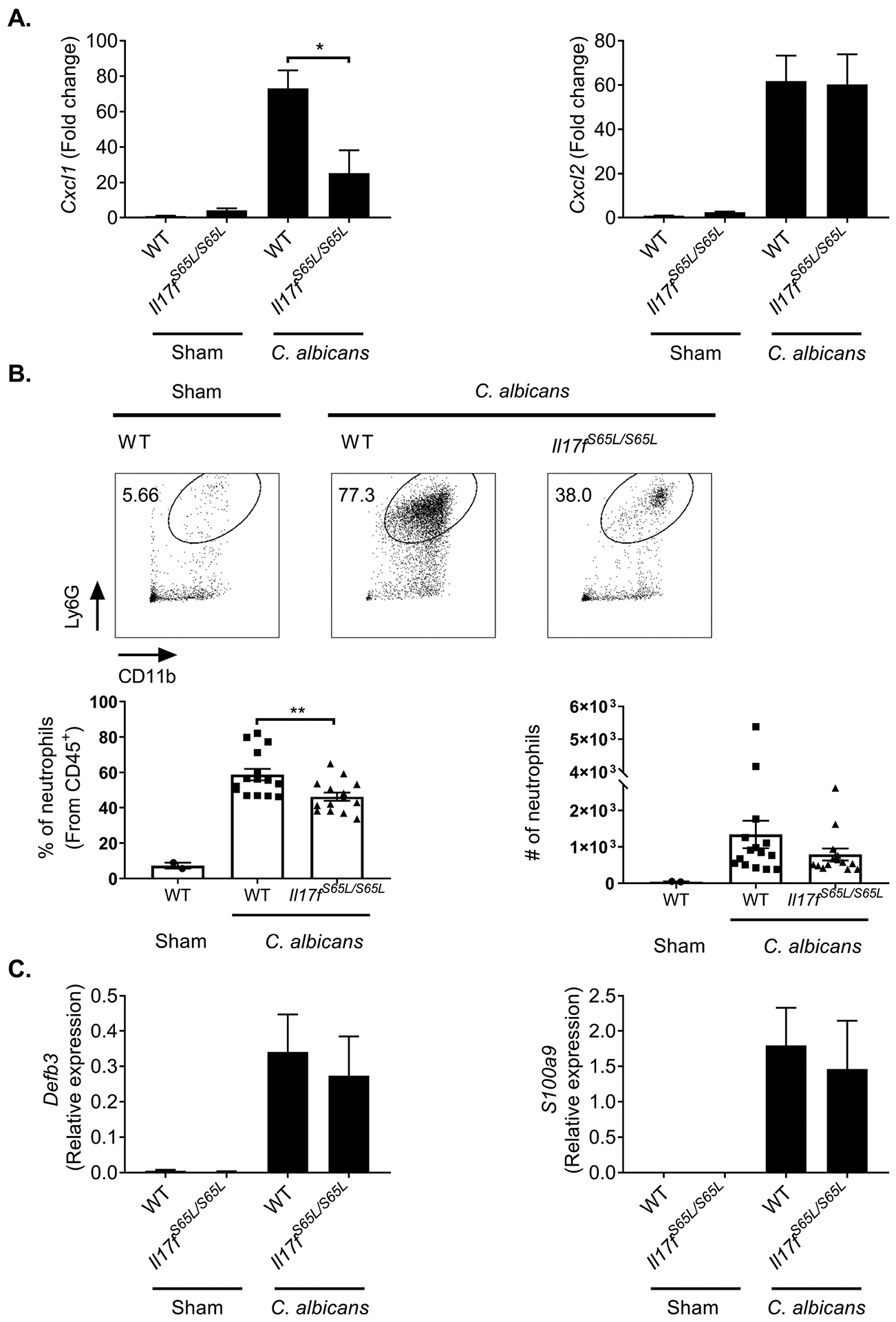
The indicated mice were infected sublingually with C. albicans or PBS (Sham). (A) Tongues were harvested at day 2 p.i.. Total mRNA from tongue homogenates was assessed by qPCR, normalized to Gapdh. Fold-change compared to Sham is indicated. Graphs show mean + SEM. Data were merged from 2 independent experiments. (B) Single cell suspensions from tongues harvested at day 2 p.i. were analyzed by flow cytometry. Cells were gated on the CD45+ live cell population and stained for CD11b and Ly6G. Top: representative FACS plots. Bottom: compiled results from 3 independent experiments showing percentages or total numbers of neutrophils in the CD45+ gate. Graph shows mean + SEM. The absolute neutrophil number is normalized to 5×104 total cells analyzed for each sample. (C) Total mRNA from tongue homogenates was assessed for Defb3 (encoding β defensin 3) and S100a9 by qPCR, normalized to Gapdh. Relative expression data are graphed as mean + SEM merged from 4 independent experiments. Data were analyzed by ANOVA. *P < 0.05, ** < 0.01.
AMPs such as β-defensins and calprotectin (S100A8/S100A9) have antifungal activity towards C. albicans (70–75). IL-17RA knockout mice show impaired AMP expression after C. albicans oral challenge (12, 61). Surprisingly, the induction of Defb3 and S100a9 in the oral mucosa of Il17fS65L/S65L mice was equivalent to WT upon oral C. albicans infection (Fig. 3C). Therefore, the increased disease susceptibility of OPC in Il17fS65L/S65L mice is apparently not due to inefficient AMP expression. Moreover, these data suggest that IL-17F and/or IL-17AF signaling may be more critical for the neutrophil response than for induction of AMPs.
Increased OPC in Il17fS65L/S65L mice is not due to impaired IL-17A
IL-17A has an antifungal activity in OPC (49, 76). To determine if IL-17A expression was impacted in Il17fS65L/S65L mice, we analyzed Il17a mRNA from tongue at day 2 p.i. Interestingly, Il17fS65L/S65L mice showed elevated Il17a as well as Il17f (Fig. 4A), arguing that the higher fungal susceptibility in Il17fS65L/S65L mice is likely not due to impaired Il17a expression; rather, the mildness of the disease susceptibility in these mice could be due to compensatory IL-17A levels. To determine whether the increased Il17a expression is directly caused by the IL-17F.S65L mutation or a result of fungus infection in the oral mucosa, we subjected splenic CD4+ T cells to in vitro differentiation for 4 days under Th17 conditions (IL-6, IL-23, TGFβ). IL-17A concentrations in supernatant were measured by ELISA. As shown, there was no significant difference in the amount of IL-17A produced by T cells obtained from Il17fS65L/S65L, Il17f−/− or WT mice (Fig. 4B). Thus, IL-17F.S65L does not directly influence IL-17A production from T cells.
Figure 4. The IL-17F.S65L mutation does not influence IL-17A production from T cells.

The indicated mice were infected sublingually with C. albicans or PBS (Sham). (A) At 2 days p.i., total mRNA was isolated from tongue and Il17a and Il17f measured by qPCR and normalized to Gapdh. Data show mean + SEM from 4 independent experiments. (B) Naïve CD4+ T cells from indicated mice were isolated from spleen and activated with anti-CD3 and anti-CD28 antibodies (5 ug/ml) under Th17 conditions (IL-1β, IL-6, IL-23 at 50 ng/ml and TGFβ at 5 ng/ml) for 4 days. IL-17A in supernatants was measured by ELISA. Data were analyzed by ANOVA. *P < 0.05.
To determine if the elevated IL-17A in the tongue could compensate for IL-17F signaling in Il17fS65L/S65L mice, we treated mice with neutralizing antibodies against IL-17A (76) during OPC induction. In WT mice, blockade of IL-17A caused increased fungal burdens and an increased percentage of mice with fungal loads, measured at day 5 p.i., as previously demonstrated (Fig. 5A, B) (49). However, IL-17A blockade in Il17fS65L/S65L mice did not further increase the oral fungal load compared to Il17fS65L/S65L isotype control mice or WT mice treated with α-IL-17A antibodies (Fig. 5A, B). Thus, in contrast to findings in Il17f−/− mice (49), IL-17A does not compensate for the IL-17F.S65L mutation during OPC.
Figure 5. IL-17A does not compensate for IL-17F signaling in Il17fS65L/S65L mice during OPC.

The indicated mice were infected sublingually with C. albicans or PBS (Sham). (A) Mice were injected i.p. with α-IL-17A neutralizing Abs or isotype control IgG (200ug) on days −1, 1, and 3 relative to the infection. CFU was determined on day 5 p.i.. Data were compiled from 2 independent experiments. (B) Data from panel A is represented as percentage of mice with detectable fungal loads. Values above indicate number of mice with fungal burden/total. *P ˂ 0.05 by ANOVA and Mann-Whitney analysis.
IL-17F is produced dominantly by γδ T cells
Unlike humans, mice do not harbor C. albicans as a commensal microbe. Multiple studies have verified that the initial response to oral infection with this organism during this first encounter in mice derive entirely from the innate immune compartment (50, 69, 77–80). The sources of IL-17A during acute OPC were previously shown to be dominantly from an unconventional, innate-acting TCRαβ+ cell population and γδ-T cells (69, 77). Group 3 innate lymphoid cells (ILC3s) were reported to produce IL-17A as well (50). Using Il17fThy1.1 reporter mice (57) we observed an increased level of Il17f-expressing cells 2 days after C. albicans infection (Fig. 6A), the time point at which Il17f mRNA expression peaks (49). γδ-T cells constituted the major Thy1.1+ population (64%), and TCRβ+ cells also comprised a significant portion of the Thy1.1+ cells (21%). A population of TCRγδ-negative TCRβ- negative cells (15.5%) that may be ILC3s was also observed (Fig. 6B).
Figure 6. IL-17F is dominantly produced by oral γδ-T cells during OPC.

Il17fThy1.1 reporter mice were subjected to OPC. At day 2 p.i., single cell suspensions from tongue were analyzed by flow cytometry. (A) Cells were stained for Thy1.1 (a reporter for IL-17F) and CD45 in the live lymphocyte gate. Left: representative FACS plots. Right: compiled results from 2 independent experiments. (B) Single cell suspensions from panel A were stained for TCRβ and TCRγδ in the CD45+ Thy1.1+ gate. Left: representative FACS plots. Right: compiled results from 2 independent experiments. Data were analyzed by ANOVA or Student’s t-test. ***P < 0.001, **** < 0.0001.
Discussion
C. albicans asymptomatically colonizes healthy individuals and usually only causes mucocutaneous infections in immunocompromised individuals (2, 81). Deficits in IL-17 signaling or Th17 cell development are particularly linked to superficial C. albicans infections (21, 82). Although CMCD patients with null mutations in genes encoding IL-17RA, IL-17RC or the adaptor ACT1 have all been described, thus far no CMCD patients have been reported with a single IL-17A deficiency (22). Even anti-IL-17A biologics used to treat autoimmune disease cause only a modest increase in mucocutaneous Candida infections (20, 83).
In general, observations in mouse studies of OPC have been accurate predictors of the immune correlates in oral candidiasis, especially with regards to the IL-17 axis (12, 15, 84, 85). Mice treated with IL-17A neutralizing antibodies have lower fungal burdens compared to the Il17ra−/− mice during OPC (49, 76), which parallels the low percentage of patients who only experience mild C. albicans mucocutaneous infections during secukinumab treatment (20, 83, 86). Unlike humans (5, 87), laboratory mice do not harbor C. albicans as a commensal organism, and hence the acute OPC model system reflects events in the innate response (69, 77, 79, 88). Nonetheless, mice do generate potent and protective recall Th17 responses to C. albicans after a secondary encounter, which is similar to humans (78, 79, 89). Humans with STAT3 mutations experience CMC (21), and though STAT3 is not required in CD4+ cells to protect from OPC, we recently found that STAT3 is essential in oral epithelium (59, 77). Mice express different AMP proteins than humans, especially in saliva (90, 91). Of relevance to C. albicans infections, the AMP β-defensin 3 is essential to prevent OPC in mice (61), though has no direct orthologue in humans (92, 93).
Although humans with the IL-17F.S65L mutation were described only in a single kindred, the degree to which the phenotype of Il17fS65L/S65L knockin mouse is similar or different from humans is a matter of interpretation. Whereas mice with a complete IL-17F deficiency do not recapitulate the CMC phenotype found in humans with the IL-17F.S65L mutation, mice with the analogous IL-17F mutation do exhibit OPC in the homozygous state, pointing to similarities among species. Puel et al. reported that 70% of the individuals carrying the IL-17F.S65L mutation (5/7) had a confirmed diagnosis of mild CMCD (16). In contrast, almost all the Il17fS65L/+ mice fully cleared C. albicans from the mouth and were statistically indistinguishable from WT controls. However, susceptibility was seen in mice carrying the mutation on both alleles, contrasting with the lack of susceptibility previously documented in Il17f−/− mice (49). This observation could be interpreted to mean there is no dominant-negative activity of this mutation in mice. However, the CFU enumeration system used to quantify OPC probably underestimates the actual number of infectious fungi, since Candida albicans hyphae are not easily separable into single cells and hence each colony may represent more than a single organism (94). The finding that Il17fS65L/S65L mice showed fungal susceptibility that was closer to Il17a−/− mice than to Il17f−/− mice may imply there is some level of dominant-negative activity that OPC is not a sufficiently sensitive system to detect (49). Additinoally, there is one other report of a human IL-17F mutation, but unfortunately the nature of the mutation was not provided and so no functional inferences can be made (95).
Blocking IL-17A together with IL-17F increases susceptibility compared to blockade of IL-17A alone (49, 50), which is reminiscent of APECED (AIRE deficiency) where patients have neutralizing antibodies against multiple Th17 cytokines, including IL-17A and IL-17F (52, 53, 96). However, administration of IL-17A neutralizing antibodies (which also efficiently block IL-17AF (49)) in Il17fS65L/S65L mice did not further increase susceptibility to OPC, suggesting that residual IL-17A homodimers present in these animals do not provide additional detectable protection. Clearly there is more to learn about IL-17F and antifungal immunity.
Taken together, the above results are consistent with a protective role for the heterodimer IL-17AF in OPC, which has been challenging to determine due to a paucity of reagents to study the heterodimer. Studies in human fibroblasts showed that the IL-17F.S65L mutation impaired signaling of both the IL-17F homodimer and the IL-17AF heterodimer (16). The increased susceptibility to OPC in Il17fS65L/S65L mice could be due either to a contribution of IL-17F homodimer and/or the IL-17AF heterodimer. Nonetheless, anti-IL-17F neutralization did not cause an increase in fungal loads in WT animals (49), suggesting that a deficiency of the IL-17AF heterodimer rather than (or in addition to) IL-17F could be responsible for increased C. albicans infection in Il17fS65LS66L mice. A protective role of IL-17AF could also explain why IL-17A blockade failed to further promote a fungal burden in Il17fS65LS65L mice. If IL-17AF is indeed the primary effector cytokine among the three IL-17RA/IL-17RC receptor ligands, the increased susceptibility caused by anti-IL-17A treatment could be due to its capacity to block IL-17AF rather than the IL-17A homodimer, as generally assumed (49). Since Il17fS65L/S65L mice have a fully impaired IL-17AF signaling pathway, this could explain why anti-IL-17A antibody treatment did not lead to a higher fungal burden than isotype control Abs.
An unexpected observation made in these studies was that the IL-17F.S65L mutation affected CXCL1 mRNA expression and subsequent neutrophil recruitment, albeit modestly, yet had no detectable impact on expression of key antifungal AMPs, β-defensin 3 and S100A8/A9 (calprotectin). IL-17 is a potent regulator of the neutrophil axis, acting on target epithelial cells to induce chemokines that in turn recruit myeloid cells, especially CXCR2-expressing neutrophils (62). It is unclear if the moderate suppression on neutrophils observed here accounts for the susceptibility phenotype. In fact, not all studies of OPC have found that IL-17 regulates neutrophil infiltration during OPC (97); the reason for the discrepancy among labs is unclear, but could possibly relate to effects of different local oral microbiota (98, 99).
Although the IL-17RA/IL-17RC heterodimer is the canonical receptor complex thought to transduce signaling of IL-17A, IL-17F and IL-17AF (100, 101), recently several alternative configurations of the receptor have been proposed. IL-17RD was suggested to act in concert with IL-17RA to mediate IL-17A but not IL-17F signaling in keratinocytes to drive psoriasis-like skin inflammation (32). Thus far, the binding capacity of IL-17AF to an IL-17RA/RD receptor complex has not been characterized. In the OPC model, Il17rc−/− mice phenocopy Il17ra−/− mice in terms of fungal loads and other signs of disease (15), suggesting that IL-17RC is needed to mediate immunity. Il17rc−/− mice are also susceptible to dermal candidiasis (102). In agreement with results in mice, humans with IL17RC null mutations experience CMCD (18). Hence, it is unlikely that an IL-17RC-independent receptor complex mediates host-defense during mucocutaneous candidiasis.
A recent structural analysis of the IL-17RC subunit unexpectedly revealed that IL-17F may have the ability to signal through an IL-17RC homodimeric receptor (31), though this would contrast with our prior molecular studies showing that a forced dimer of murine IL-17RC is not signaling-competent (46). Interestingly, based on the human IL-17RC structure, the IL-17F.S65L mutation is predicted to cause steric hindrance that decreases its binding affinity to IL-17RC. However, the contacts of IL-17F to IL-17RA are less tight than to IL-17RC, so changes in binding affinity to this subunit would likely be less pronounced (31). Alignment of the IL-17RC residues that are proximal to IL-17F.S65 shows full conservation between the human and mouse receptors, arguing for strong overall structural conservation (Jean Michel Rondeau, personal communication). Putting this together, we speculate that the increased fungal burden in Il17fS65LS65L mice is caused by reduced interactions of IL-17F with the IL-17RC homodimeric receptor, rather than with the IL-17RA/IL-17RC heterodimer.
In summary, results from this new IL-17F.S65L mutant mouse strain in the murine OPC model are generally, though not completely, in line with data in CMCD patients with the IL-17F.S65L mutation. These data help reconcile the observations that, whereas a complete IL-17F deficiency does not cause candidiasis in mice, this naturally-occurring IL-17F mutation in humans does. The potential utility of this new mouse strain goes beyond studies of candidiasis. Because these mice show a different phenotype from Il17f−/− mice with respect to OPC, they could be valuable as an additional tool to interrogate the functions of IL-17F as well as the enigmatic IL-17AF heterodimer in other settings where these cytokines may participate, such as autoimmunity, cancer or other infectious disease settings.
Supplementary Material
Key findings:
A murine IL-17F.S65L orthologue has impaired signaling function in vitro
IL-17F.S65L knockin mice are susceptible to oral candidiasis, unlike Il17f−/− mice
Il17fS65L/S65L mice may be a broadly valuable tool to interrogate IL-17F function
Acknowledgements
We are grateful to Casey Weaver (University of Alabama) for Il17fThy1.1 reporter mice and Ulrich Siebenlist (NIH) for Act1−/− mice. Il17ra−/− mice were a kind gift from Amgen. We thank Chunming Bi of the University of Pittsburgh Transgenic and Gene Targeting for expert technical help. Drs. Jean Michel Rondeau and Frank Kolbinger (Novartis) and Yufang Shao (Bon Opus) provided valuable discussions. We also thank Drs. Mark Shlomchik, Jean-Laurent Casanova and Anne Puel for additional input.
Grant Funding: SLG was supported by NIH grants AI128991 and DE022550. DHK was supported by AR071720 and AR060744. R. Gordon was supported by T32-AI089443.
Abbreviations
- AMP
antimicrobial peptide
- APECED
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy
- CMCD
chromic mucocutaneous candidiasis disease
- ILC
innate lymphoid cell
- OPC
oropharyngeal candidiasis
References
- 1.Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, and White TC. 2012. Hidden killers: human fungal infections. Sci Transl Med 4: 165rv113. [DOI] [PubMed] [Google Scholar]
- 2.Fidel PL Jr. 2011. Candida-Host Interactions in HIV Disease: Implications for Oropharyngeal Candidiasis. Adv Dent Res 23: 45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Challacombe SJ, and Naglik JR. 2006. The effects of HIV infection on oral mucosal immunity. Adv Dent Res 19: 29–35. [DOI] [PubMed] [Google Scholar]
- 4.Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, and Napolitani G. 2007. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol 8: 639–646. [DOI] [PubMed] [Google Scholar]
- 5.Bacher P, Hohnstein T, Beerbaum E, Rocker M, Blango MG, Kaufmann S, Rohmel J, Eschenhagen P, Grehn C, Seidel K, Rickerts V, Lozza L, Stervbo U, Nienen M, Babel N, Milleck J, Assenmacher M, Cornely OA, Ziegler M, Wisplinghoff H, Heine G, Worm M, Siegmund B, Maul J, Creutz P, Tabeling C, Ruwwe-Glosenkamp C, Sander LE, Knosalla C, Brunke S, Hube B, Kniemeyer O, Brakhage AA, Schwarz C, and Scheffold A. 2019. Human Anti-fungal Th17 Immunity and Pathology Rely on Cross-Reactivity against Candida albicans. Cell 176: 1340–1355 e1315. [DOI] [PubMed] [Google Scholar]
- 6.Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, Monticelli S, Lanzavecchia A, and Sallusto F. 2012. Pathogen-induced human T(H)17 cells produce IFN-gamma or IL-10 and are regulated by IL-1β. Nature 484: 514–518. [DOI] [PubMed] [Google Scholar]
- 7.van de Veerdonk FL, Marijnissen R, Joosten LA, Kullberg BJ, Drenth JP, Netea MG, and van der Meer JW. 2010. Milder clinical hyperimmunoglobulin E syndrome phenotype is associated with partial interleukin-17 deficiency. Clin Exp Immunol 159: 57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGeachy MJ, Cua DJ, and Gaffen SL. 2019. The IL-17 Family of Cytokines in Health and Disease. Immunity 50: 892–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conti HR, and Gaffen SL. 2015. IL-17-Mediated Immunity to the Opportunistic Fungal Pathogen Candida albicans. J Immunol 195: 780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drummond RA, and Lionakis MS. 2018. Organ-specific mechanisms linking innate and adaptive antifungal immunity. Semin Cell Dev Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang W, Na L, Fidel PL, and Schwarzenberger P. 2004. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis 190: 624–631. [DOI] [PubMed] [Google Scholar]
- 12.Conti H, Shen F, Nayyar N, Stocum E, JN S, Lindemann M, Ho A, Hai J, Yu J, Jung J, Filler S, Masso-Welch P, Edgerton M, and Gaffen S. 2009. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med 206: 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kagami S, Rizzo HL, Kurtz SE, Miller LS, and Blauvelt A. 2010. IL-23 and IL-17A, but not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans. J Immunol 185: 5453–5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferreira MC, Whibley N, Mamo AJ, Siebenlist U, Chan YR, and Gaffen SL. 2014. Interleukin-17-induced protein lipocalin 2 is dispensable for immunity to oral candidiasis. Infect Immun 82: 1030–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ho A, Shen F, Conti H, Patel N, Childs E, Peterson A, Hernandez-Santos N, Kolls J, Kane L, Ouyang W, and Gaffen S. 2010. IL-17RC is required for immune signaling via an extended SEF/IL-17R signaling domain in the cytoplasmic tail. J. Immunol 185: 1063–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puel A, Cypowji S, Bustamante J, Wright J, Liu L, Lim H, Migaud M, Israel L, Chrabieh M, Audry M, Gumbleton M, Toulon A, Bodemer C, El-Baghdadi J, Whitters M, Paradis T, Brooks J, Collins M, Wolfman N, Al-Muhsen S, Galicchio M, Abel L, Picard C, and Casanova J-L. 2011. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science 332: 65–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boisson B, Wang C, Pedergnana V, Wu L, Cypowyj S, Rybojad M, Belkadi A, Picard C, Abel L, Fieschi C, Puel A, Li X, and Casanova J-L. 2013. A biallelic ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity 39: 676–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ling Y, Cypowyj S, Aytekin C, Galicchio M, Camcioglu Y, Nepesov S, Ikinciogullari A, Dogu F, Belkadi A, Levy R, Migaud M, Boisson B, Bolze A, Itan Y, Goudin N, Cottineau J, Picard C, Abel L, Bustamante J, Casanova JL, and Puel A. 2015. Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J Exp Med 212: 619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levy R, Okada S, Beziat V, Moriya K, Liu C, Chai LY, Migaud M, Hauck F, Al Ali A, Cyrus C, Vatte C, Patiroglu T, Unal E, Ferneiny M, Hyakuna N, Nepesov S, Oleastro M, Ikinciogullari A, Dogu F, Asano T, Ohara O, Yun L, Della Mina E, Bronnimann D, Itan Y, Gothe F, Bustamante J, Boisson-Dupuis S, Tahuil N, Aytekin C, Salhi A, Al Muhsen S, Kobayashi M, Toubiana J, Abel L, Li X, Camcioglu Y, Celmeli F, Klein C, AlKhater SA, Casanova JL, and Puel A. 2016. Genetic, immunological, and clinical features of patients with bacterial and fungal infections due to inherited IL-17RA deficiency. Proc Natl Acad Sci U S A 113: E8277–E8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saunte DM, Mrowietz U, Puig L, and Zachariae C. 2017. Candida infections in psoriasis and psoriatic arthritis patients treated with IL-17 inhibitors and their practical management. Br J Dermatol. 177:47–62 [DOI] [PubMed] [Google Scholar]
- 21.Milner J, and Holland S. 2013. The cup runneth over: lessons from the ever-expanding pool of primary immunodeficiency diseases. Nat Rev Immunol 13: 635–648. [DOI] [PubMed] [Google Scholar]
- 22.Li J, Vinh DC, Casanova JL, and Puel A. 2017. Inborn errors of immunity underlying fungal diseases in otherwise healthy individuals. Curr Opin Microbiol 40: 46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, Alzahrani M, Al-Muhsen S, Halwani R, Ma CS, Wong N, Soudais C, Henderson LA, Marzouqa H, Shamma J, Gonzalez M, Martinez-Barricarte R, Okada C, Avery DT, Latorre D, Deswarte C, Jabot-Hanin F, Torrado E, Fountain J, Belkadi A, Itan Y, Boisson B, Migaud M, Arlehamn CS, Sette A, Breton S, McCluskey J, Rossjohn J, de Villartay JP, Moshous D, Hambleton S, Latour S, Arkwright PD, Picard C, Lantz O, Engelhard D, Kobayashi M, Abel L, Cooper AM, Notarangelo LD, Boisson-Dupuis S, Puel A, Sallusto F, Bustamante J, Tangye SG, and Casanova JL. 2015. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science 349: 606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monin L, and Gaffen SL. 2017. Interleukin 17 Family Cytokines: Signaling Mechanisms, Biological Activities, and Therapeutic Implications. Cold Spring Harb Perspect Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolls JK, and Linden A. 2004. Interleukin-17 family members and inflammation. Immunity 21: 467–476. [DOI] [PubMed] [Google Scholar]
- 26.Starnes T, Robertson MJ, Sledge G, Kelich S, Nakshatri H, Broxmeyer HE, and Hromas R. 2001. Cutting edge: IL-17F, a novel cytokine selectively expressed in activated T cells and monocytes, regulates angiogenesis and endothelial cell cytokine production. J Immunol 167: 4137–4140. [DOI] [PubMed] [Google Scholar]
- 27.Chang SH, and Dong C. 2007. A novel heterodimeric cytokine consisting of IL-17 and IL-17F regulates inflammatory responses. Cell Res 17: 435–440. [DOI] [PubMed] [Google Scholar]
- 28.Wright JF, Bennett F, Li B, Brooks J, Luxenberg DP, Whitters MJ, Tomkinson KN, Fitz LJ, Wolfman NM, Collins M, Dunussi-Joannopoulos K, Chatterjee-Kishore M, and Carreno BM. 2008. The human IL-17F/IL-17A heterodimeric cytokine signals through the IL-17RA/IL-17RC receptor complex. J Immunol 181: 2799–2805. [DOI] [PubMed] [Google Scholar]
- 29.Wright JF, Guo Y, Quazi A, Luxenberg DP, Bennett F, Ross JF, Qiu Y, Whitters MJ, Tomkinson KN, Dunussi-Joannopoulos K, Carreno BM, Collins M, and Wolfman NM. 2007. Identification of an Interleukin 17F/17A Heterodimer in Activated Human CD4+ T Cells. J Biol Chem 282: 13447–13455. [DOI] [PubMed] [Google Scholar]
- 30.Goepfert A, Lehmann S, Wirth E, and Rondeau JM. 2017. The human IL-17A/F heterodimer: a two-faced cytokine with unique receptor recognition properties. Sci Rep 7: 8906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goepfert A, Lehmann S, Blank J, Kolbinger F, and Rondeau JM. 2020. Structural Analysis Reveals that the Cytokine IL-17F Forms a Homodimeric Complex with Receptor IL-17RC to Drive IL-17RA-Independent Signaling. Immunity 52: 499–512. [DOI] [PubMed] [Google Scholar]
- 32.Su Y, Huang J, Zhao X, Lu H, Wang W, Yang XO, Shi Y, Wang X, Lai Y, and Dong C. 2019. Interleukin-17 receptor D constitutes an alternative receptor for interleukin-17A important in psoriasis-like skin inflammation. Sci Immunol 4:eaau9657. [DOI] [PubMed] [Google Scholar]
- 33.De Luca A, Pariano M, Cellini B, Costantini C, Villella VR, Jose SS, Palmieri M, Borghi M, Galosi C, Paolicelli G, Maiuri L, Fric J, and Zelante T. 2017. The IL-17F/IL-17RC Axis Promotes Respiratory Allergy in the Proximal Airways. Cell Rep 20: 1667–1680. [DOI] [PubMed] [Google Scholar]
- 34.Veldoen M 2017. Interleukin 17 is a chief orchestrator of immunity. Nat Immunol 18: 612–621. [DOI] [PubMed] [Google Scholar]
- 35.Li X, Bechara R, Zhao J, McGeachy MJ, and Gaffen SL. 2019. Interleukin 17 receptor-based signaling and implications for disease. Nature Immunology 20: 1594–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Onishi R, and Gaffen SL. 2010. IL-17 and its Target Genes: Mechanisms of IL-17 Function in Disease. Immunology 129: 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Amatya N, Garg AV, and Gaffen SL. 2017. IL-17 Signaling: The Yin and the Yang. Trends Immunol 38: 310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kao CY, Chen Y, Thai P, Wachi S, Huang F, Kim C, Harper RW, and Wu R. 2004. IL-17 markedly up-regulates β-defensin-2 expression in human airway epithelium via JAK and NF-κB signaling pathways. J Immunol 173: 3482–3491. [DOI] [PubMed] [Google Scholar]
- 39.Kao CY, Kim C, Huang F, and Wu R. 2008. Requirements for two proximal NF-κB binding sites and IκB-ξ in IL-17A-induced human β-defensin 2 expression by conducting airway epithelium. J Biol Chem 283: 15309–15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Velichko S, Zhou X, Zhu L, Anderson JD, Wu R, and Chen Y. 2016. A Novel Nuclear Function for the Interleukin-17 Signaling Adaptor Protein Act1. PLoS One 11: e0163323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Majumder S, Amatya N, Revu S, Jawale C, Wu D, Rittenhouse N, Menk A, Kupul S, Du F, Raphael I, Bhattacharjee A, Siebenlist U, Hand TW, Delgoffe GM, Poholek AC, Gaffen SL, Biswas P , and Mcgeachy M. 2019. IL-17 metabolically reprograms activated fibroblastic reticular cells for proliferation and survival. Nat Immunol 20: 534–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kohlgruber A, Gal-Oz S, LaMarche N, Shimazaki M, Duquette D, Nguyen H, Mina A, Paras T, Tavakkoli A, von Adrian U, Banks A, Shay T, Brenner M, and Lynch L. 2018. Gammadelta T cells producing interleukin-17A regulate adipose regulatory T cell homeostasis and thermogenesis. Nat Immunol 19: 464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zrioual S, Ecochard R, Tournadre A, Lenief V, Cazalis MA, and Miossec P. 2009. Genome-wide comparison between IL-17A- and IL-17F-induced effects in human rheumatoid arthritis synoviocytes. J Immunol 182: 3112–3120. [DOI] [PubMed] [Google Scholar]
- 44.Hot A, Zrioual S, Toh ML, Lenief V, and Miossec P. 2011. IL-17A- versus IL-17F-induced intracellular signal transduction pathways and modulation by IL-17RA and IL-17RC RNA interference in rheumatoid synoviocytes. Ann Rheum Dis 70: 341–348. [DOI] [PubMed] [Google Scholar]
- 45.Bertelsen T, Ljungberg C, Boye Kjellerup R, Iversen L, and Johansen C. 2017. IL-17F regulates psoriasis-associated genes through IκBξ. Exp Dermatol 26: 234–241. [DOI] [PubMed] [Google Scholar]
- 46.Onishi R, Park S, Hanel W, Maitra A, and Gaffen S. 2010. The SEFIR is not enough: An extended region downstream of the Interleukin-17RA SEFIR domain is required for IL-17-dependent signal transduction. J Biol Chem 285: 32751–32759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, and Iwakura Y. 2009. Differential roles of interleukin-17A and −17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30: 108–119. [DOI] [PubMed] [Google Scholar]
- 48.Iwakura Y, Ishigame H, Saijo S, and Nakae S. 2011. Functional specialization of interleukin-17 family members. Immunity 34: 149–162. [DOI] [PubMed] [Google Scholar]
- 49.Whibley N, Tritto E, Traggiai E, Kolbinger F, Moulin P, Brees D, Coleman BM, Mamo A, Garg A, Jaycox JR, Siebenlist U, Kammueller M, and Gaffen SL. 2016. Antibody blockade of IL-17-family cytokines in immunity to acute murine oral mucosal candidiasis. J Leukoc Biol 99: 1153–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gladiator A, Wangler N, Trautwein-Weidner K, and Leibundgut-Landmann S. 2013. Cutting Edge: IL-17-Secreting Innate Lymphoid Cells Are Essential for Host Defense against Fungal Infection. J Immunol 190: 521–525. [DOI] [PubMed] [Google Scholar]
- 51.Browne SK, and Holland SM. 2010. Anti-cytokine autoantibodies explain some chronic mucocutaneous candidiasis. Immunol Cell Biol 88: 614–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, Ersvaer E, Perheentupa J, Erichsen MM, Bratanic N, Meloni A, Cetani F, Perniola R, Ergun-Longmire B, Maclaren N, Krohn KJ, Pura M, Schalke B, Strobel P, Leite MI, Battelino T, Husebye ES, Peterson P, Willcox N, and Meager A. 2010. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J Exp Med 207: 299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Puel A, Doffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, Cobat A, Ouachee-Chardin M, Toulon A, Bustamante J, Al-Muhsen S, Al-Owain M, Arkwright PD, Costigan C, McConnell V, Cant AJ, Abinun M, Polak M, Bougneres PF, Kumararatne D, Marodi L, Nahum A, Roifman C, Blanche S, Fischer A, Bodemer C, Abel L, Lilic D, and Casanova JL. 2010. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med 207: 291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lear TB, McKelvey AC, Evankovich JW, Rajbhandari S, Coon TA, Dunn SR, Londino JD, McVerry BJ, Zhang Y, Valenzi E, Burton CL, Gordon R, Gingras S, Lockwood KC, Jurczak MJ, Lafyatis R, Shlomchik MJ, Liu Y, and Chen BB. 2019. KIAA0317 regulates pulmonary inflammation through SOCS2 degradation. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pelletier S, Gingras S, and Green DR. 2015. Mouse genome engineering via CRISPR-Cas9 for study of immune function. Immunity 42: 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Concordet JP, and Haeussler M. 2018. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res 46: W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, and Weaver CT. 2009. Late developmental plasticity in the T helper 17 lineage. Immunity 30: 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Solis NV, and Filler SG. 2012. Mouse model of oropharyngeal candidiasis. Nat Protoc 7: 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aggor FEY, Break T, Trevejo-Nunez G, Whibley N, Coleman BM, Bailey R, Kaplan DH, Naglik JR, Shan W, Shetty A, McCracken C, Durum SK, Biswas P , Bruno VM, Kolls J , Lionakis MS, and Gaffen SL. 2020. Oral epithelial IL-22/STAT3 signaling licenses IL-17-mediated immunity to oral mucosal candidiasis Sci Immunol in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Conti HR, Huppler AR, Whibley N, and Gaffen SL. 2014. Animal models for candidiasis. Curr Protoc Immunol 105: 19 16 11–19 16 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Conti H, Bruno V, Childs E, Daugherty S, Hunter J, Mengesha B, Saevig D, Hendricks M, Coleman BM, Brane L, Solis NV, Cruz JA, Verma A, Garg A, Hise AG, Naglik J, Naglik JR, Filler SG, Kolls JK, Sinha S, and Gaffen SL. 2016. IL-17RA signaling in oral epithelium is critical for protection against oropharyngeal candidiasis. Cell Host Microbe 20: 606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huppler AR, Conti HR, Hernandez-Santos N, PS B, Darville T, and Gaffen SL. 2014. Role of neutrophils in IL-17-dependent immunity to mucosal candidiasis. J Immunol 192: 1745–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Altmeier S, Toska A, Sparber F , Teijeira A, Halin C, and LeibundGut-Landmann S. 2016. IL-1 Coordinates the Neutrophil Response to C. albicans in the Oral Mucosa. PLoS Pathog 12: e1005882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Desai JV, and Lionakis MS. 2018. The role of neutrophils in host defense against invasive fungal infections. Curr Clin Microbiol Rep 5: 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Riedel JH, Paust HJ, Krohn S, Turner JE, Kluger MA, Steinmetz OM, Krebs CF, Stahl RA, and Panzer U. 2016. IL-17F Promotes Tissue Injury in Autoimmune Kidney Diseases. J Am Soc Nephrol 27: 3666–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oda N, Canelos PB, Essayan DM, Plunkett BA, Myers AC, and Huang SK. 2005. Interleukin-17F induces pulmonary neutrophilia and amplifies antigen-induced allergic response. Am J Respir Crit Care Med 171: 12–18. [DOI] [PubMed] [Google Scholar]
- 67.Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, and Dong C. 2008. Regulation of inflammatory responses by IL-17F. J Exp Med 205: 1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pandiyan P, Bhaskaran N, Zhang Y, and Weinberg A. 2014. Isolation of T cells from mouse oral tissues. Biol Proced Online 16: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Verma A, Richardson J, Zhou C, Coleman BM, Moyes D, Ho J, Huppler AR, Ramani K, McGeachy MJ, Mufazalov IA, Waisman A, Kane LP, Biswas P , Hube B, Naglik J, and Gaffen SL. 2017. Oral epithelial cells orchestrate innate Type 17 responses to Candida albicans through the virulence factor Candidalysin. Sci Immunol 2: eeam8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yano J, Noverr MC, and Fidel PL Jr. 2012. Cytokines in the host response to Candida vaginitis: Identifying a role for non-classical immune mediators, S100 alarmins. Cytokine 58: 118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sroussi HY, Kohler GA, Agabian N, Villines D, and Palefsky JM. 2009. Substitution of methionine 63 or 83 in S100A9 and cysteine 42 in S100A8 abrogate the antifungal activities of S100A8/A9: potential role for oxidative regulation. FEMS Immunol Med Microbiol 55: 55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tomalka J, Azodi E, Narra HP, Patel K, O’Neill S, Cardwell C, Hall BA, Wilson JM, and Hise AG. 2015. β-Defensin 1 plays a role in acute mucosal defense against Candida albicans. J Immunol 194: 1788–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schröder JM, Wang JM, Howard OMZ, and Oppenheim JJ. 1999. b-Defensins: Linking innate immunity and adaptive immunity through dendritic and T cell CCR6. Science 286: 525–528. [DOI] [PubMed] [Google Scholar]
- 74.Conti H, Baker O, Freeman A, Jang W, Li R, Holland S, Edgerton M, and Gaffen S. 2011. New mechanism of oral immunity to mucosal candidiasis in hyper-IgE syndrome. Mucosal Immunol 4: 448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feng Z, Jiang B, Chandra J, Ghannoum M, Nelson S, and Weinberg A. 2005. Human β-defensins: differential activity against candidal species and regulation by Candida albicans. J Dent Res 84: 445–450. [DOI] [PubMed] [Google Scholar]
- 76.Shen F, Verma AH, Volk A, Jones B, Coleman BM, Loza MJ, Malaviya R, Moore B, Weinstock D, Elloso MM, Gaffen SL, and Ort T. 2019. Combined Blockade of TNF-α and IL-17A Alleviates Progression of Collagen-Induced Arthritis without Causing Serious Infections in Mice. J Immunol 202: 2017–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Conti H, Peterson A, Huppler A, Brane L, Hernández-Santos N, Whibley N, Garg A, Simpson-Abelson M, Gibson G, Mamo A, Osborne L, Bishu S, Ghilardi N, Siebenlist U, Watkins S, Artis D, McGeachy M, and Gaffen S. 2014. Oral-resident ‘natural’ Th17 cells and γδ-T cells control opportunistic Candida albicans infections. J Exp Med 211: 2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hernández-Santos N, Huppler AR, Peterson AC, Khader SA, KC M, and Gaffen SL. 2013. Th17 cells confer long term adaptive immunity to oral mucosal Candida albicans infections. Mucosal Immunol 6: 900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bär E, Gladiator A, Bastidas S, Roschitzki B, Acha-Orbea H, Oxenius A, and LeibundGut-Landmann S. 2012. A novel Th cell epitope of Candida albicans mediates protection from fungal infection. J Immunol 188: 5636–5643. [DOI] [PubMed] [Google Scholar]
- 80.Sparber F, Dolowschiak T, Mertens S, Lauener L, Clausen BE, Joller N, Stoitzner P, Tussiwand R, and LeibundGut-Landmann S. 2018. Langerin+ DCs regulate innate IL-17 production in the oral mucosa during Candida albicans-mediated infection. PLoS Pathog 14: e1007069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Moyes DL, and Naglik JR. 2011. Mucosal immunity and Candida albicans infection. Clin Dev Immunol 2011: 346307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lionakis MS, and Levitz SM. 2018. Host Control of Fungal Infections: Lessons from Basic Studies and Human Cohorts. Annu Rev Immunol 36: 157–191. [DOI] [PubMed] [Google Scholar]
- 83.Deodhar A, Mease PJ, McInnes IB, Baraliakos X, Reich K, Blauvelt A, Leonardi C, Porter B, Das Gupta A, Widmer A, Pricop L, and Fox T. 2019. Long-term safety of secukinumab in patients with moderate-to-severe plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis: integrated pooled clinical trial and post-marketing surveillance data. Arthritis Res Ther 21: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Farah C, Hu Y, Riminton S, and Ashman R. 2006. Distinct roles for interleukin-12p40 and tumour necrosis factor in resistance to oral candidiasis defined by gene targeting. Oral Microbiol Immunol 21: 252–255. [DOI] [PubMed] [Google Scholar]
- 85.Goupil M, Cousineau-Cote V, Aumont F, Senechal S, Gaboury L, Hanna Z, Jolicoeur P, and de Repentigny L. 2014. Defective IL-17- and IL-22-dependent mucosal host response to Candida albicans determines susceptibility to oral candidiasis in mice expressing the HIV-1 transgene. BMC Immunol 15: 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.van de Kerkhof PC, Griffiths CE, Reich K, Leonardi CL, Blauvelt A, Tsai TF, Gong Y, Huang J, Papavassilis C, and Fox T. 2016. Secukinumab long-term safety experience: A pooled analysis of 10 phase II and III clinical studies in patients with moderate to severe plaque psoriasis. J Am Acad Dermatol 75: 83–98 e84. [DOI] [PubMed] [Google Scholar]
- 87.Naglik JR, Richardson JP, and Moyes DL. 2014. Candida albicans pathogenicity and epithelial immunity. PLoS Pathog 10: e1004257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Verma AH, Zafar H, Ponde NO, Hepworth OW, Sihra D, Aggor FEY, Ainscough JS, Ho J, Richardson JP, Coleman BM, Hube B, Stacey M, McGeachy MJ, Naglik JR, Gaffen SL, and Moyes DL. 2018. IL-36 and IL-1/IL-17 Drive Immunity to Oral Candidiasis via Parallel Mechanisms. J Immunol 201: 627–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shao TY, Ang WXG, Jiang TT, Huang FS, Andersen H, Kinder JM, Pham G, Burg AR, Ruff B, Gonzalez T, Khurana Hershey GK, Haslam DB, and Way SS. 2019. Commensal Candida albicans Positively Calibrates Systemic Th17 Immunological Responses. Cell Host Microbe 25: 404–417 e406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Salvatori O, Puri S, Tati S, and Edgerton M. 2016. Innate Immunity and Saliva in Candida albicans-mediated Oral Diseases. J Dent Res 95: 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Vylkova S, Li XS, Berner JC, and Edgerton M. 2006. Distinct antifungal mechanisms: β-defensins require Candida albicans Ssa1 protein, while Trk1p mediates activity of cysteine-free cationic peptides. Antimicrob Agents Chemother 50: 324–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Joly S, Organ CC, Johnson GK, McCray PB Jr., and Guthmiller JM. 2005. Correlation between β-defensin expression and induction profiles in gingival keratinocytes. Mol Immunol 42: 1073–1084. [DOI] [PubMed] [Google Scholar]
- 93.Ganz T 2003. Defensins: Antimicrobial peptides of innate immunity. Nat Rev Immunol 3: 710–720. [DOI] [PubMed] [Google Scholar]
- 94.Graham CE, Cruz MR, Garsin DA, and Lorenz MC. 2017. Enterococcus faecalis bacteriocin EntV inhibits hyphal morphogenesis, biofilm formation, and virulence of Candida albicans. Proc Natl Acad Sci U S A 114: 4507–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bader O, Weig M, Gross U, Schon M, Mempel M, and Buhl T. 2012. A 32-Year-Old Man With Ulcerative Mucositis, Skin Lesions, and Nail Dystrophy. Clin Infect Dis 54: 1035–1036. [DOI] [PubMed] [Google Scholar]
- 96.Browne SK, and Holland SM. 2010. Immunodeficiency secondary to anticytokine autoantibodies. Curr Opin Allergy Clin Immunol 10: 534–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Trautwein-Weidner K, Gladiator A, Nur S, Diethelm P, and LeibundGut-Landmann S. 2015. IL-17-mediated antifungal defense in the oral mucosa is independent of neutrophils. Mucosal Immunol 8: 221–231. [DOI] [PubMed] [Google Scholar]
- 98.Pandiyan P, Bhaskaran N, Zou M, Schneider E, Jayaraman S, and Huehn J. 2019. Microbiome Dependent Regulation of Tregs and Th17 Cells in Mucosa. Front Immunol 10: 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bhaskaran N, Quigley C, Paw C, Butala S, Schneider E, and Pandiyan P. 2018. Role of Short Chain Fatty Acids in Controlling Tregs and Immunopathology During Mucosal Infection. Front Microbiol 9: 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ho A, and Gaffen SL. 2010. IL-17RC: A partner in IL-17 signaling and beyond. Semin Immunopathol 32: 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, Tocker J, and Peschon JJ. 2006. Cutting Edge: Interleukin-17 signals through a heteromeric receptor complex. J. Immunol 177: 36–39. [DOI] [PubMed] [Google Scholar]
- 102.Conti HR, Whibley N, Coleman B, Garg A, Jaycox J, and Gaffen S. 2015. Signaling through IL-17C/IL-17RE is dispensable for immunity to systemic, oral and cutaneous candidiasis. PLoS One 10: e0122807. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.