

Abstract
B cell receptor (BCR) recognizes foreign antigens to initiate humoral immunity that needs isotype-switched antibodies generated via class switch recombination (CSR); however, stimulating BCR in the absence of co-stimulation (e.g., CD40) does not induce CSR, thus, it remains elusive whether and how the BCR induces CSR mechanistically. Autoreactive B cells can maintain anergy via unresponsiveness of their BCRs to self-antigens. However, it remains unknown what molecule(s) restrict BCR signaling strength for licensing BCR-induced CSR and whether deficiency of such molecule(s) disrupts autoreactive B cell anergy and cause B cell-mediated diseases by modulating BCR signaling. Here, we employ mouse models to show that BCR’s capacity to induce CSR is restrained by B-cell intrinsic checkpoints, TRAF3 and TRAF2, whose deletion in B cells enables the BCR to induce CSR in the absence of co-stimulation. TRAF3-deficiency permits BCR-induced CSR by elevating BCR proximal signaling intensity. Furthermore, NF-κB2 is required for BCR-induced CSR in TRAF3-deficient B cells, but not for CD40-induced or LPS-induced CSR, suggesting that TRAF3 restricts NF-κB2 activation to specifically limit the BCR’s ability to induce CSR. TRAF3-deficiency also disrupts autoreactive B cell anergy by elevating calcium influx in response to BCR stimulation, leading to lymphoid organ disorders and autoimmune manifestations. We showed that TRAF3-deficiency-associated autoimmune phenotypes can be rectified by limiting BCR repertoires or attenuating BCR signaling strength. Thus, our studies highlight the importance of TRAF3-mediated restraint on BCR signaling strength for controlling CSR, B cell homeostasis and B cell-mediated disorders.
Keywords: B cell receptor, class switch recombination, activation-induced deaminase, tumor necrosis factor receptor-associated factor-3, B cell anergy and homeostasis
Introduction
To diversify antibody effector functions during pathogen infection or immunization, B cells switch from expressing IgM to IgA, IgG or IgE via class switch recombination (CSR) (1). During pathogen-mediated immune responses, the BCR and the co-receptors of B cells (e.g. CD40 or Toll-like receptors (TLR)) are both engaged. This pathogen-associated co-stimulation induces B cells to express activation-induced deaminase (AID) that initiates CSR to produce isotype-switched antibodies against pathogens (2, 3). However, signaling by the BCR alone does not lead to CSR (4, 5), although signaling by individual co-receptors such as CD40 can (2, 6, 7). Since the BCR recognizes antigens (Ag) to initiate humoral immunity and determines the specificity of isotype-switched antibody, it is counterintuitive that it cannot promote CSR on its own. Is it because there are negative regulatory mechanisms in play that prevent the BCR from inducing AID? Or simply because downstream signaling pathways activated by BCR engagement alone are not sufficient? These are fundamental knowledge gaps in the field of B-cell biology and immunology that have not been addressed before. Consequently, little is known regarding the specific role of BCR engagement in inducing CSR and AID expression, and what molecule(s) restrict BCR signaling strength required to induce CSR. Such a restriction on BCR’s function may be biologically important since autoreactive B-cells are present (8) and constantly encounter self-antigens.
Up to 70% newborn B-cells in bone marrow (BM) are autoreactive (8); a large fraction of them modify their BCR specificity by receptor editing or are eliminated by clonal deletion (9) while a portion of them can enter peripheral lymphoid tissues. To avoid antibody-mediated autoimmune diseases, these autoreactive B cells become anergic, namely, their BCR does not respond to further self-antigen stimulation (10). Thus, anergy maintains autoreactive B-cell tolerance and homeostasis (10). To maintain the anergic status of these B cells, the BCR signaling strength needs to be properly regulated by protein and lipid kinases or phosphatases, such as protein kinase Lyn, lipid kinase PI3K, protein phosphatase SHP-1 and PTPN22, lipid phosphatase PTEN and SHIP-1 (11). Genetic mutations of these enzymes may lead to autoimmune diseases by disrupting B-cell anergy (11). However, beyond the kinases and phosphatases, there is little knowledge about how the BCR signaling strength controls B-cell anergy and whether deficiency of other molecule(s) enhances BCR signaling to disrupt autoreactive B cell anergy and lead to B cell-mediated diseases.
Both TRAF2 and TRAF3 are signaling adaptors of the TNF receptor (TNFR) superfamily such as CD40 and B cell activating factor (BAFF) receptor (BAFFR) (12). TRAF3 and TRAF2 restrict the activation of nuclear factor kappa B2 (NF-κB2) induced by CD40, BAFFR and other TNFR members (13). In resting B cells, TRAF3 associates with NF-κB inducing kinase (NIK), while TRAF2 associates with cellular inhibitors of apoptosis protein 1 and 2 (cIAP1/2). Within this cytoplasmic complex, TRAF2 and TRAF3 heteromeric interaction allows cIAP1/2 to induce NIK polyubiquitination and degradation. Upon receptor (e.g., CD40 or BAFFR) clustering via ligand engagement, TRAF2/TRAF3 complex is recruited to the signalosome in membrane rafts where TRAF3 is degraded (14, 15), thereby releasing NIK and allowing its accumulation. NIK in turn phosphorylates inhibitory κB (IκB) kinase-α (IKKα). Activated IKKα phosphorylates NF-κB2 p100 and triggers p100 proteolytic cleavage into p52. NF-κB2 p52 then forms a heterodimer with RelB that translocates into nucleus and initiates target gene transcription. TRAF2 and TRAF3 were previously shown to negatively regulate BAFF-BAFFR signaling given that TRAF2 deletion rescued B cell survival defects in BAFF-deficient mice (16). Importantly, it has been previously shown that BCR signaling drives p100 transcription, while BAFFR signaling mediates the process of p100 into p52 (17, 18). However, it remains unclear whether and how BAFF and BCR signaling cooperate to induce CSR in the presence or absence of TRAF3.
We previously found that B cell-intrinsic TRAF2 is required while TRAF3 is dispensable for CD40-induced AID expression and CSR (6). As such, B cell-intrinsic TRAF2 deficiency impairs antibody responses against T cell-dependent (TD) Ag, whereas TRAF3 deficiency does not (6). However, we and others found that either TRAF2 or TRAF3 deletion in B cells enhances antibody responses against T-cell independent (TI) antigens (6, 19). We previously addressed the mechanism by which B cell-intrinsic TRAF2 and TRAF3 differentially regulate TD humoral immune response (6). However, it remains unknown why both B-cell-specific TRAF2 deficiency and TRAF3 deficiency promote humoral immune responses induced by TI Ags that can activate the BCR directly without T cell help.
Mutations or deletions of TRAF2 or TRAF3 are frequently found in human mature B cell lymphomas (12, 20, 21). Germline deletion of TRAF3 is neonatally lethal (22). B cell-specific deletion of TRAF2 or TRAF3 in mice results in abnormal B cell expansion including both marginal zone (MZ) and follicular (FO) B cells, lymphoid organ disorders, autoimmunity, and B cell lymphomas in late life (16, 19, 23). The expansion of MZ B cells in B-cell-specific TRAF2 deficient mice was independent of BAFF (16); however, it was not reported whether TRAF2/BAFF double conditional deficient mice still had increased B cell numbers in lymphoid organs as observed in B-cell-specific TRAF2 deficient mice (16). Previous studies also showed that B cell hyperplasia and splenomegaly phenotypes in B-cell-specific TRAF3 deficient mice were not dependent on BAFF signaling (19). Thus, it is still unclear which receptor signaling pathway(s) drives the pathogenesis caused by B cell-intrinsic deficiency of TRAF3, and the role of BCR signaling has not been explored in these pathogenic processes.
In the current study, we show that TRAF3 cooperates with TRAF2 to restrain BCR’s ability to induce CSR. We reveal that TRAF3-deficiency in B cells elevates BCR proximal signaling strength and constitutively activates NF-κB2, leading to BCR-induced CSR and disruption of autoreactive B cell anergy. Our studies provide new insight into negative regulatory mechanisms that ensure optimal humoral immunity while simultaneously maintaining B cell homeostasis and preventing autoimmunity by fine-tuning the BCR signaling intensity.
Materials and Methods
Mice, bone marrow transfer and chemical or Ag treatment
TRAF2flox/flox and TRAF3flox/flox mice were generated previously (19, 24). VDJ9/κ5 mice (25) were provided by Dr. Jason Cyster (University of California San Francisco, CA). HEL transgenic ML-5 mice (26) were provided by Dr. John C. Cambier (University of Colorado AMC, CO). Wild type C57BL/6 (B6), CD19Cre transgenic and NFκB2flox/flox mice were purchased from Jackson Laboratory. Cγ1Cre mice were described previously (27). B cell-specific TRAF2 and/or TRAF3 knockout (cKO) mice were generated by crossing TRAF2flox/flox and/or TRAF3flox/flox with CD19Cre, referred to as TRAF2-cKO or TRAF3-cKO throughout the text. We employed TRAF2flox/flox and/or TRAF3flox/flox as littermate control (LMC) for all experiments and acknowledge the caveat of lacking a CD19-Cre only control in some experiments. Cγ1Cre mice were crossed with TRAF3flox/flox mice to generate Cγ1Cre-TRAF3flox/flox mice used for Figure 2 only. VDJ9/κ5 mice were crossed with CD19Cre-TRAF3flox/flox mice to generate TRAF3-cKO-VDJ9/κ5 mice with HEL-specific BCR and B cell-specific TRAF3 deletion. TRAF2/TRAF3-DcKO mice were generated by crossing CD19Cre-TRAF3flox/flox mice with TRAF2flox/flox mice. B cell-specific TRAF3 and NF-κB2 double KO (TRAF3-NF-κB2-DcKO) mice were generated by crossing NF-κB2flox/flox with CD19Cre-TRAF3flox/flox mice. 6 to 12 weeks old mice were used for most experiments including CSR assay, western blot, signaling studies and proliferation assays. For Ibrutinib treatment, 20 days old mice were used. For kidney pathology analysis, 10-15 months old mice were used. Animal work was approved by the Institutional Animal Care and Use Committee of the University of Colorado Anschutz Medical Campus (Aurora, CO).
Figure 2. TRAF3 restricts BCR-induced CSR autonomously and TRAF2 and TRAF3 cooperatively inhibit BCR’s ability to induce CSR.
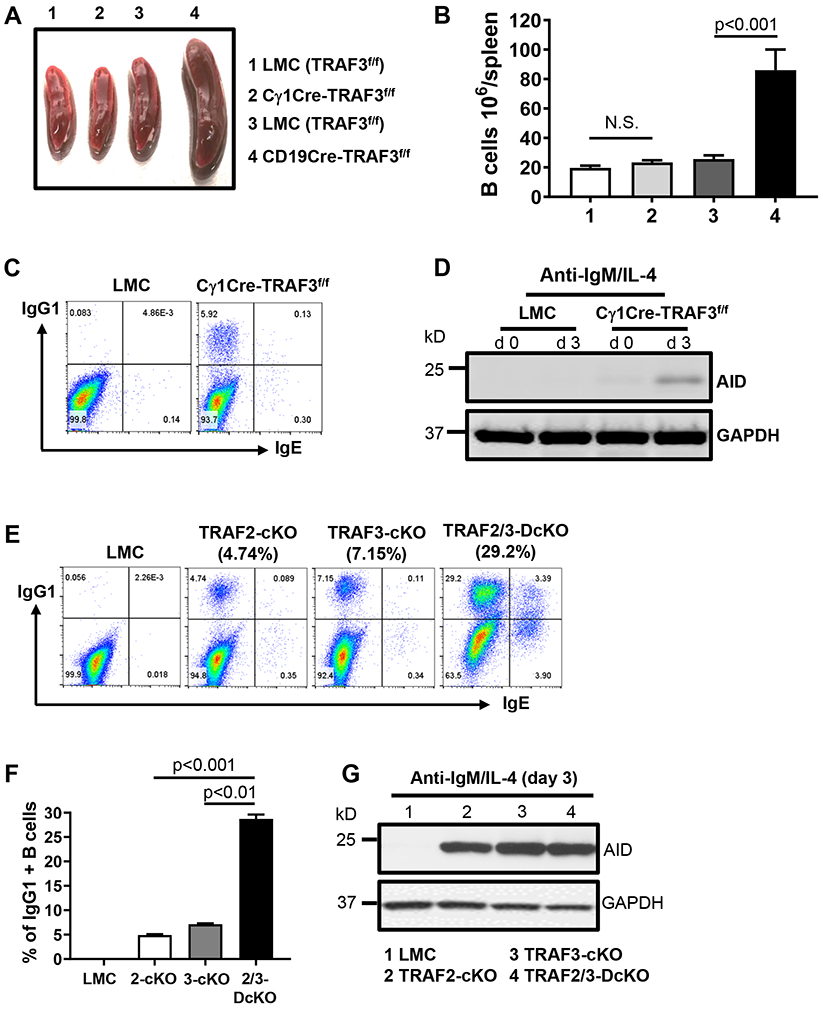
(A) Representative image of mouse spleens with indicated genotypes (group 1-4, n≥15/group, 8-12 weeks old). (B) Quantification of the number of splenic B cells in mice with indicated genotypes as labeled in (A) (n=5/group). (C) Representative flow data showing day4 IgG1 and IgE CSR induced by anti-IgM/IL-4 in indicated B cells. (D) Representative Western blot data showing AID protein expression induced by anti-IgM/IL-4 at day3 in indicated B cells. GAPDH as loading control. Data are representative of 3 to 4 independently repeated experiments. (E) Flow data showing day4 IgG1 and IgE CSR induced by anti-IgM/IL-4 in indicated B cells. (F) Quantification of IgG1+ B cell percentage from triplicates of one representative experiment. (G) Western blot showing AID protein expression induced by anti-IgM/IL-4 in indicated B cells. TRAF3-cKO, CD19Cre-Traf3f/f; TRAF2-cKO, CD19Cre-Traf2f/f; TRAF2/3-DcKO, CD19Cre-Traf2f/f/Traf3f/f. Data are representative of 3-6 independently repeated experiments.
BM transfer: BM cells were separately isolated from donors of VDJ9/κ5 or TRAF3-cKO-VDJ9/κ5 mice. Single cell suspension in PBS was prepared (5×106 cells/ml). Recipient ML-5 Tg mice were irradiated by two doses of 500 rad irradiation. 4 hours after second irradiation, 200 μl of BM cells (1×106) were injected via tail vein into recipient mice. 10-12 weeks after BM transfer, chimera mice were analyzed.
Ibrutinib in vivo treatment: Ibrutinib stock was made in DMSO at a concentration of 20 mg/ml. Before injection, the stock was diluted with DMSO by 5 times and further diluted with PBS by 4 times. Thus, 25% DMSO in PBS was used as vehicle control. 20 days old TRAF3-cKO mice were intraperitoneally injected with a dose of 6 mg Ibrutinib/kg body weight or the same volume of vehicle control every two days until mice become 3 months old; at this age, untreated TRAF3-cKO mice have significantly enlarged spleens.
Antibodies (Abs) and chemicals
All Abs used in the study were included in Supplemental Table 1. Chemicals were purchased from the following companies: PRT062607 (P505–15) from Selleck Chem (Houston, TX), Ibrutinib from Cayman (Ann Arbor, MI), Ionomycin from LC laboratories (Woburn, MA), U73122, and Lipopolysaccharides (LPS) (E.coli 0111: B4) from Sigma (St. Louis, MO). All chemicals were dissolved in H2O or DMSO. DMSO was titrated to determine its effect on B cell functions including B cell proliferation, CSR, AID expression and others. When DMSO was diluted more than 1:1000, it had no detectable effects on B cell functions. Since all chemicals were used at 1:5000 to 1:100,000 dilution from stocks, vehicle control was not included in assays when medium only was used as controls; otherwise, vehicle control was indicated. Mouse interleukin-4 (IL-4) was purchased from GenScript (Piscataway, NJ). BAFF was a gift provided by Dr. John Cambier’s lab (University of Colorado AMC).
ELISA for detection of secreted anti-HEL IgM
HEL Ag was dissolved in a carbonate buffer (pH 9.5) at 20μg/ml and coated on 96-well plates (Thermo scientific) for at least 12 hours at 4˚C. Coated plates were blocked with 200μl/well blocking buffer (2% BSA in PBS) for 2 hours at room temperature (RT). Culture media collected from stimulated B cells were first diluted at 1:5 with blocking buffer, then serially diluted to 1:15, 1:45, and 1:135. Diluted samples (60μl/well), in duplicate, were loaded onto plates and incubated at RT for 2 hours. Plates were washed with PBS-T. HEL-specific IgM was detected by HRP conjugated goat anti-mouse IgM. Plates were washed 6 times with PBS-T, and HRP substrate (1-Step Ultra TMB-ELISA, Thermo scientific) was added. Plates were incubated for 5-60 minutes to allow color development. The HRP substrate reaction was stopped with 2M H2SO4. OD value was read by Nanoquant infinite M200 (Switzerland) at wavelength 450nm. All plates were normalized to a reference serum sample. Culture medium serves as the negative control. The modified OD values for each sample were averaged between duplicate wells, subtracted by the OD of negative control and multiplied by the dilution factor to calculate the relative unit.
Measurement of calcium (Ca2+) flux
All steps were performed at room temperature. Single cell suspension of splenocytes was prepared and red blood cells were lysed with ACK buffer. Cells were washed twice with and re-suspended in 2% FBS RPMI1640 media at a concentration of 10×106 cells/ml. 2.5 μl Indo-1, AM (2 mM, I1223, Thermo Fisher Scientific) and 5 μl APC conjugated anti-B220 were added into 1 ml cell suspension described above. Samples were left at room temperature for 45 to 50 minutes, washed twice with and re-suspended in 2% FBS RPMI1640 (1 ml). Indo-1 bound or unbound intracellular Ca2+ was measured with BD LSRFortessa. Briefly, cells were collected for 30 seconds without stimulation to determine the basal Ca2+ level, then 5 μg F(ab)2 anti-mouse IgM or 0.5 μg HEL in 100 μl above medium was added to stimulate B cells that were continuously collected for additional 150 seconds to determine BCR activation-induced Ca2+ flux. Data were analyzed by FlowJo software.
Cell culture and flow cytometry
Spleens were harvested from mice of various genotypes. Naïve B cells were isolated with mouse B cell isolation EASY kit according to the manufacturer’s instructions (StemCell). Purified B cells (0.5×106/ml, 3 ml/well in 6-well plate) were stimulated in vitro with anti-CD40 (1μg/ml, Biolegend, clone HM40-3) plus IL-4 (10ng/ml, GenScript, Piscataway, NJ), LPS (2μg/ml) plus IL-4 (10ng/ml), BAFF (1μg/ml) plus IL-4 (10ng/ml), F(ab)2 fragment of goat anti-mouse IgM (10μg/ml) plus IL-4 (10ng/ml) or dextran conjugated goat anti-mouse IgD (10ng/ml) plus IL-4 (10ng/ml) in 10% FBS RPMI lymphocyte medium for various days (collectively, 2 days for mRNA of AID and β-actin, and Cγ1 GLT, 3 days for AID protein and 4 days for CSR) in a 5% CO2 incubator. For HEL-specific B cells, HEL (200ng/ml) plus IL-4 (10ng/ml) was used to stimulate these B cells. Activated B cells were examined by flow cytometry to detect the percentage of IgG1+ or IgE+ isotype-switched B cells. Intracellular IgE was detected as described previously (28). Proliferation assays were performed with the CellTrace™ CFSE or violet Cell Proliferation Kit for flow cytometry (Thermo Fisher Scientific) according to the manufacturer’s instructions.
For phosphor-BTK and phosphor-Syk flow, purified primary B cells were re-suspended in 1% FBS-RPMI medium at a concentration of 3×106 cells/ml and placed in 5% CO2 incubator for 30 minutes. Then, cells were aliquoted into flow tubes (300 μl/tube), stimulated with anti-IgM (10μg/ml) for 2 minutes, and immediately fixed with paraformaldehyde at a final concentration of 1.5% for 10 minutes at room temperature. Cells were spun down, re-suspended with ice-cold methanol (thoroughly vortex) and placed at 4℃ for 10 minutes. Cells were washed twice with 1% BSA in PBS and stained with anti-phosphor-BTK or -Syk for 30 minutes at room temperature. Flow cytometry was performed on BD LSRII, BD LSRFortessa, or FACSCalibur (BD Biosciences) platform. All Abs used for flow cytometry were included in Supplemental Table 1. Data were analyzed with Flow-Jo software.
Biochemical assays and Western blotting
For detecting AID protein expression, purified B cells were treated by inhibitors or untreated, then stimulated with various stimuli for 3 days. Cells were harvested and lysed with lysis buffer (50mM Tris-base pH7.5, 150mM NaCl, 2mM EDTA, 2mM Na3O4V, 4mM NaF, 1% Triton-X100, 0.1% SDS, 0.5% Sodium deoxycholate) for 30 minutes on ice. Lysates were centrifuged at 12000 RPM for 10 minutes at 4˚C. Supernatants were collected for subsequent analysis. For signaling studies, freshly purified B cells were aliquoted into each tube (10×106 cells in 0.5 ml RPMI1640), treated with inhibitors or activators, then stimulated with 5μg anti-IgM for indicated times in a 5% CO2 incubator (37°C). After stimulation, cells were immediately cooled down with cold PBS on ice. Cell lysates were prepared as described above. Protein concentrations were determined with a BCA protein assay kit (Thermo scientific). 20μg of protein per sample was separated on SDS-PAGE (Bio-Rad, Hercules, CA) and transferred onto nitrocellulose membranes (Thermo scientific). Membranes were blocked and probed with specific Abs followed by HRP-conjugated anti-mouse or rabbit secondary Abs, respectively. Protein bands were read with ECL (Thermo scientific) on a G:Box Chemi-XX6 platform (Syngene, Frederick, MD) or exposed to Kodak BioMax MS Film.
Reverse Transcription (RT)-PCR
Semi-quantitative RT-PCR was performed as described previously (29). Purified B cells were stimulated as described above for 2 days. Total RNA was purified with TriPure (Roche, Indianapolis, IN). 2μg RNA per reaction was used for cDNA synthesis according to manufacturer’s instructions (Promega, Madison, WI). The cDNA was diluted as indicated in the figures. The primers and PCR reaction conditions were as follows. Forward primer for β-actin: 5'-TGGAATCCTGTGGCATCCATGAAAC-3'; Reverse primer for β-actin: 5'-TAAAACGCAGCTCAGTAACAGTCCG-3'. PCR conditions: 94°C 3 min, 94°C 1 min, 60°C 45s, 72°C 45s, 30 cycles, 72°C 10 min. Forward primer for IgG1 GLT (Iγ1): 5'-GGCCCTTCCAGATCTTTGAG-3'; Reverse primer for IgG1 GLT (Cγ1 exon1): 5'-CAGGGTCACCATGGAGTTAGTT-3'. PCR reaction conditions: 94°C 3 min, 94°C 1 min, 60°C 45s, 72°C 45s, 30 cycles, 72°C 10 min. Forward primer for AID: 5'-TTTCTTTACCAATTCAAAAATGTCCG-3'; Reverse primer for AID: 5'-TCAGCCTTGCGGTCCTCACA-3'. PCR reaction conditions: 94°C 3 min, 94°C 1 min, 60°C 1 min, 72°C 1 min, 39 cycles, 72°C 10 min. PCR products were separated by agarose gel electrophoresis. Agarose gels were imaged by G:Box Chemi-XX6 platform.
Statistical Analysis
All multiple group data comparison applied one-way or two-way ANOVA analysis and two group data comparison applied unpaired Student’s T test. Data were shown as mean±sem. P<0.0001 was defined very very very significant, p<0.001 as very very significant, p< 0.01 as very significant, p<0.05 as significant and p> 0.05 as not significant (N.S.).
Results
TRAF3-deficiency permits the BCR to induce CSR by anti-Ig or specific Ag stimulation
Prior studies using anti-Ig to mimic Ag-induced BCR activation showed that neither anti-IgM (4) nor anti-IgD (5) induced CSR in WT primary naïve B cells in the presence of interleukin-4 (IL-4), suggesting that the BCR is not able to induce CSR in the absence of co-stimulation. In contrast, CSR can be induced robustly in vitro by engaging CD40 or TLRs in the presence of cytokines (e.g., IL-4) (30, 31). Why engaging BCR cannot but engaging co-receptors (e.g., CD40) can induce CSR remains a long-lasting question to be addressed (2, 31). We and others have previously shown that TRAF3 deletion in B cells enhances antibody responses against TI Ags (6, 19) that activate the BCR directly without T cell help. Thus, we hypothesized that B cell-intrinsic TRAF3 may restrain BCR’s capacity to induce CSR.
To test whether TRAF3-deficiency in B cells enables the BCR to induce CSR, we stimulated naïve mature B cells from CD19Cre-Traf3f/f (TRAF3-cKO) or littermate control (LMC, Traf3f/f) mice with anti-IgM plus IL-4. Consistent with previous report (4), anti-IgM/IL-4 did not induce CSR in LMC B cells (Figure 1A, B). Strikingly, anti-IgM/IL-4 induced robust IgG1 switching in TRAF3-cKO B cells in a time- (Figure 1A, B) and dose- (Supplemental Figure 1A) dependent manner. Anti-IgM or IL-4 alone did not induce CSR in LMC or TRAF3-cKO B cells (Supplemental Figure 1B). Given that AID and germline transcripts (GLT) are two inducible factors uniquely required for CSR (1, 32), we next determined whether TRAF3-deficiency affected the expression of AID or the GLT of IgG1 region (Cγ1-GLT). Anti-IgM/IL-4 only induced the expression of AID protein and transcript in TRAF3-cKO but not in LMC B cells (Figure 1C, Supplemental Figure 1C). In contrast, anti-IgM/IL-4 induced the Cγ1 GLT in both LMC and TRAF3-cKO B cells (Supplemental Figure 1C). Thus, we conclude that TRAF3-deficiency enables the BCR to induce CSR by promoting AID transcription. In line with previous studies (5), anti-IgD/IL-4 did not induce CSR in LMC B cells; in contrast, anti-IgD/IL-4 induced robust CSR and AID expression in TRAF3-cKO B cells (Figure 1D, E).
Figure 1. TRAF3 restrains BCR’s capacity to induce AID and CSR.
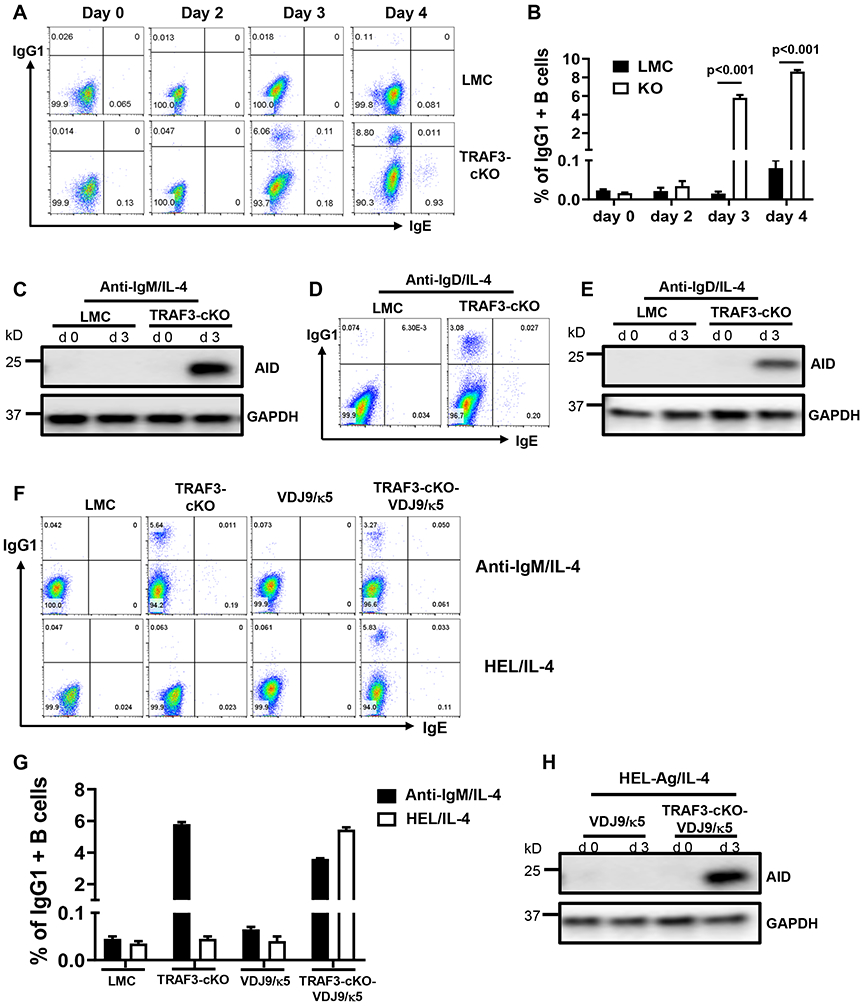
(A) Flow data showing IgG1 and IgE CSR kinetics induced by anti-IgM/IL-4 in indicated B cells. (B) Quantification of IgG1+ B cell percentage from triplicates of one representative experiment. (C) Western blot data showing AID protein expression induced by anti-IgM/IL-4 in indicated B cells. GAPDH as loading control. d0: day0 (unstimulated), d3: day3 (3 days after stimulation). (D) Flow data showing day4 IgG1 and IgE CSR induced by anti-IgD/IL-4. (E) Western blot data showing AID protein expression induced by anti-IgD/IL-4 in indicated B cells. (F) Flow data showing day4 IgG1 and IgE CSR induced by anti-IgM/IL-4 or HEL/IL-4 in indicated B cells. (G) Quantification of IgG1+ B cell percentage from triplicates of one representative experiment. (H) Western blot showing AID protein expression induced by HEL/IL-4 in indicated B cells. TRAF3-cKO, CD19Cre-Traf3f/f. Data are representative of 3-6 independently repeated experiments.
To test if engaging BCR by a specific Ag induces CSR in the absence of TRAF3, we crossed TRAF3-cKO mice with VDJ9/κ5 mice that harbor a unique knock-in (KI) BCR (25) specific for hen egg lysozyme (HEL) Ag (26) to generate TRAF3-cKO-VDJ9/κ5 mice. HEL/IL-4 induced CSR in TRAF3-cKO-VDJ9/κ5 B cells but not in various controls (Figure 1F, G). Consistently, HEL/IL-4 induced AID expression in TRAF3-cKO-VDJ9/κ5 but not in VDJ9/κ5 B cells (Figure 1H). Altogether, we conclude that TRAF3 functions as a checkpoint of BCR signaling to prevent AID expression and CSR.
TRAF3 restrains BCR’s capacity to induce CSR autonomously and TRAF2/TRAF3 double deficiency enhances BCR-induced CSR
CD19Cre-Traf3f/f mice have expanded MZ and FO B cell compartments (19). To exclude the possibility that TRAF3 restrains BCR-induced CSR via regulating B cell differentiation or development, we crossed Traf3f/f mice with Cγ1Cre mice in which Cre expression is driven by the Iγ1 promoter (27). Thus, Traf3f/f deletion only occurs upon B cell stimulation that turns on the Iγ1 promoter. Cγ1Cre-Traf3f/f mice have normal sized spleens (Figure 2A) and B cell numbers (Figure 2B) as well as MZ and FO B cell profiles (Supplemental Figure 1D). Engaging BCR still induced CSR and AID expression in Cγ1Cre-Traf3f/f B cells (Figure 2C, D). These data demonstrate that TRAF3 directly suppresses BCR’s capacity to induce AID and CSR.
Next, we determined whether BCR-induced CSR occurs differentially in MZ vs. FO B cells. We found that both MZ and FO B cells of TRAF3-cKO mice underwent BCR-induced CSR; furthermore, MZ B cells did not preferentially undergo BCR-induced CSR compared with FO B cells (Supplemental Figure 1E). In contrast, MZ B cells that underwent CSR appeared to acquire FO B cell phenotypes by upregulating CD23 (Supplemental Figure 1E), consistent with our previous studies of CD40 or LPS-induced CSR in MZ and FO B cells (6).
Notably, the level of BCR-induced CSR is much lower than that of CD40-induced CSR in TRAF3-cKO B cells (Supplemental Figure 1F) (6), suggesting that other checkpoint molecules might cooperatively inhibit BCR-induced CSR. We previously found that antibody responses against TI Ags were also elevated in TRAF2-cKO mice (CD19Cre-Traf2f/f) (6). We thus tested whether TRAF2 also restrained BCR-induced AID expression and CSR and found that anti-IgM/IL-4 induced CSR (Figure 2E, F) and AID expression (Figure 2G) in TRAF2-cKO B cells. TRAF2 and TRAF3 double deficiency synergistically promoted BCR-induced CSR, demonstrated by a much higher level of IgG1 and IgE CSR in TRAF2/3-DcKO (CD19Cre-Traf2f/f/Traf3f/f) than that of either single cKO B cells or both combined (Figure 2E, F). Hence, our data demonstrate that TRAF3 cooperates with TRAF2 to prohibit the BCR from inducing CSR, thereby providing a possible mechanistic explanation for increased antibody responses against TI Ags shown previously (6, 19).
TRAF3 deficiency amplifies BCR-induced proximal signaling intensity
We employed our newly established in vitro model to elucidate the signaling mechanisms by which the BCR induces AID expression and CSR. Spleen tyrosine kinase (Syk), Bruton tyrosine kinase (BTK) and phospholipase C gamma-2 (PLCγ2) are BCR proximal signaling elements (2). We found that engaging BCR induced a significantly higher level of phospho-BTK (Figure 3A, B) and phospho-Syk (Supplemental Figure 2A, B) as well as calcium (Ca2+) flux (Figure 3C, D) in TRAF3-cKO B cells than in controls, suggesting that TRAF3 deficiency permits the BCR to induce CSR by elevating BCR signaling strength. We found that TRAF3 deficiency in B cells did not affect the surface expression of IgM or the total protein expression of BTK and Syk (Supplemental Figure 2C, D).
Figure 3. TRAF3 deficiency increases the BCR proximal signaling intensity required for BCR-induced AID expression and CSR.
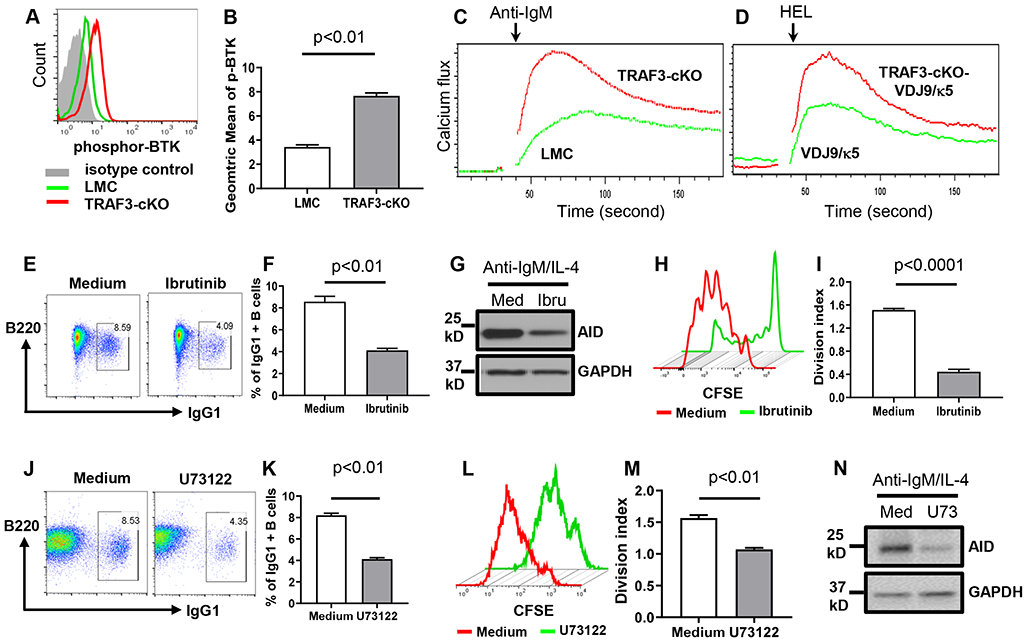
(A) Flow data showing phospho-BTK (p-BTK) induced by anti-IgM in indicated B cells. (B) Quantification of geometric mean of p-BTK intensity from duplicates of one representative experiment. (C and D) Flow data from duplicates of one experiment showing Ca2+ flux induced by anti-IgM (C) or HEL (D) in indicated B cells. (E to I) TRAF3-cKO B cells pre-treated with Ibrutinib (Ibru) (5nM) or medium (Med) then stimulated with anti-IgM/IL-4. (E) Flow data showing day4 IgG1 CSR. (F) Quantification of IgG1+ B cell percentage from triplicates of one representative experiment. (G) Western blot data showing AID protein expression at day3. (H) Proliferation pattern of TRAF3-cKO B cells as treated in (E). (I) Quantification of division index from triplicates of one representative experiment. (J to N) TRAF3-cKO B cells pretreated with U73122 (U73) (0.2 μM) or medium (Med) then stimulated with anti-IgM/IL-4. (J) Flow data showing day4 IgG1 CSR. (K) Quantification of IgG1+ B cell percentage from triplicates of one representative experiment. (L) Proliferation pattern of TRAF3-cKO B cells as treated in (J). (M) Quantification of division index from triplicates of one representative experiment. (N) Western blot showing AID protein expression at day3. TRAF3-cKO, CD19Cre-Traf3f/f. Data are representative of 3-6 independently repeated experiments.
To test whether TRAF3 deficiency has a differential effect on the BCR signaling of MZ or FO B cells, we purified mature naïve B cells including MZ and FO B cells from TRAF3-cKO mice. We stimulated the total purified B cells with anti-IgM, then examined the level of pBTK or pSyk in MZ B cells (CD23low) and FO B cells (CD23high) by flow cytometry. Our data showed that there was no difference in the level of pBTK or pSyk between MZ and FO B cells of TRAF3-cKO mice (Supplemental Figure 2E), demonstrating that changes in the BCR signaling are not a result of subpopulation skewing in TRAF3-cKO mice.
Next, we examined whether activation of these proximal signaling elements is required for the BCR to induce AID and CSR. BCR-induced CSR and AID expression were significantly inhibited by pretreating TRAF3-cKO B cells with a BTK inhibitor (Ibrutinib) (Figure 3E-G) or a Syk inhibitor (P505-15) (Supplemental Figure 2F, G). Both inhibitors also significantly inhibited B cell proliferation (Figure 3H, I, Supplemental Figure 2F), indicating that Syk and BTK are shared by BCR signaling to induce B cell proliferation and CSR. BCR-induced Ca2+ flux requires PLCγ2 (33). Pretreating TRAF3-cKO B cells with a PLCγ2 inhibitor (U73122) specifically blocked anti-IgM-induced but not ionomycin-induced Ca2+ flux (Supplemental Figure 2H). U73122 at the concentration that significantly reduced CSR (Figure 3J, K) also significantly inhibited B cell proliferation (Figure 3L, M), indicating that PLCγ2 signaling is required for both B cell proliferation and CSR. Inhibiting PLCγ2 also decreased BCR-induced AID expression (Figure 3N). However, inhibiting Syk or BTK did not impair CSR and AID expression induced by stimulating CD40 or TLR4 (Supplemental Figure 3A, B). These data indicate that the BCR employs a distinct signaling pathway from CD40 and TLRs to induce AID and CSR.
TRAF3-deficiency enables the processing of NF-κB2 precursor (p100) induced by BCR engagement into active NF-κB2 (p52)
We and others previously found that TRAF3-cKO B cells have elevated NF-κB2 activation (6, 16, 19). We thus examined whether TRAF3 deficiency influences NF-κB2 activation induced by stimulating BCR. Consistent with previous reports (6, 16, 19), resting TRAF3-cKO B cells expressed more active NF-κB2 p52 than resting LMC ones (Figure 4A, day 0). Engaging BCR with anti-IgM induced more precursor NF-κB2 p100 in LMC B cells but did not increase active NF-κB2 p52 compared to unstimulated counterpart (Figure 4A). These data suggest that BCR signaling activates NF-κB2 pathway via increasing NF-κB2 p100 expression; however, it cannot convert p100 into p52 in the presence of TRAF3, indicating that TRAF3 is a checkpoint for the BCR to activate NF-κB2 p100 downstream signaling. Engaging BCR did not further increase p52 in TRAF3-cKO B cells (Figure 4A), likely due to saturated NF-κB2 activation in the absence of TRAF3.
Figure 4. TRAF3 restricts the processing of NF-κB2 p100 induced by BCR engagement and an essential role of NF-κB2 in splenomegaly and B cell expansion caused by TRAF3-deficiency.
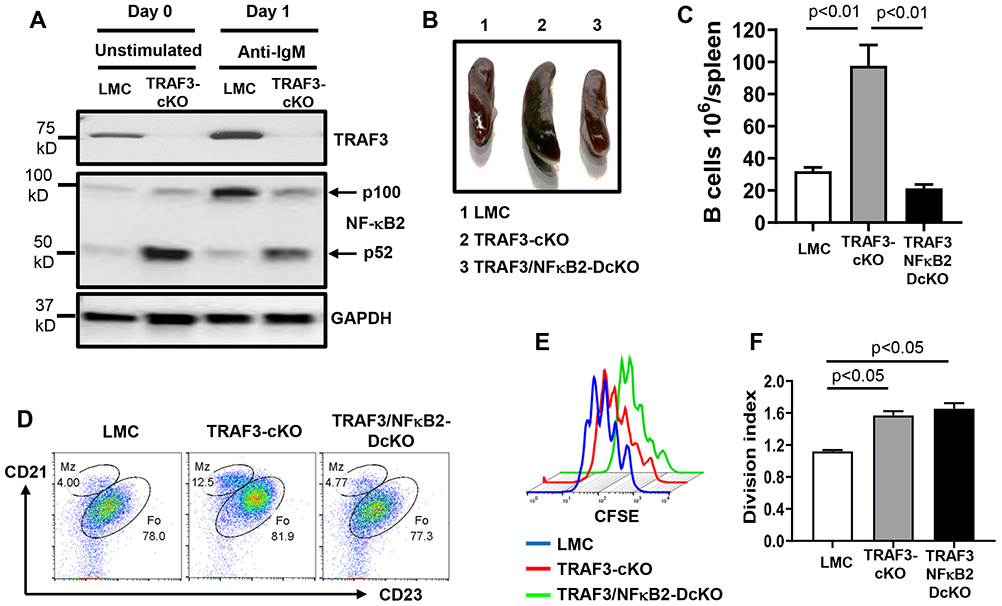
(A) Representative Western blot data showing precursor (p100) or active (p52) NF-κB2 and TRAF3 expression in indicated B cells that are either unstimulated (day 0) or stimulated (day 1) by anti-IgM/IL-4 for 1 day. GAPDH as loading control. (B) Representative image of mouse spleens with indicated genotypes (group 1-3, n=5/group, 8-12 weeks old). (C) Quantification of the number of splenic B cells in mice with indicated genotypes (n=5/group). (D) Representative flow data showing the profiles of MZ and FO B cells in mice with indicated genotypes (n=5/genotype). B cells were gated for B220+IgM+ double positive population (E) Proliferation pattern of B cells with indicated genotypes that were stimulated by anti-IgM/IL-4 for 4 days. (F) Quantification of division index from triplicates of one representative experiment in anti-IgM/IL-4 stimulated B cells with indicated genotypes. TRAF3-cKO, CD19Cre-Traf3f/f; TRAF3/NF-κB2-DcKO, CD19Cre-Traf3f/f-NF-κB2f/f. Data are representative of 3-6 independently repeated experiments.
NF-κB2 is required for splenomegaly, B cell expansion, and BCR-induced CSR caused by TRAF3-deficiency
To test whether NF-κB2 activation is required for phenotypes caused TRAF3-deficiency, we generated CD19Cre-Traf3f/f-NF-κB2f/f (TRAF3/NF-κB2-DcKO) mice. TRAF3/NF-κB2-DcKO mice had normal sized spleens (Figure 4B) and B cell numbers (Figure 4C) as well as a normal profile of MZ/FO B cells (Figure 4D), demonstrating an essential role of NF-κB2 in mediating B cell expansion and splenomegaly in TRAF3-cKO mice. TRAF3/NF-κB2-DcKO and TRAF3-cKO B cells proliferated equally well upon BCR stimulation, better than their LMC counterpart (Figure 4E, F), indicating that TRAF3-deficiency promotes BCR-induced proliferation, which is independent of NF-κB2.
BCR-induced CSR and AID expression were drastically reduced in TRAF3/NF-κB2-DcKO B cells (Figure 5A-C). GFP is a marker for NF-κB2 deletion and GFP+ B cells in TRAF3/NF-κB2-DcKO mice deleted NF-κB2 and TRAF3 and did not express AID (Figure 5D, E). We conclude that NF-κB2 is required for promoting BCR-induced AID expression and CSR. Intriguingly, NF-κB2-deficiency in TRAF3-cKO B cells did not significantly affect CD40-induced CSR and AID expression (Supplemental Figure 3C, D) or LPS-induced CSR (Supplemental Figure 3E), suggesting that TRAF3 restricts NF-κB2 activation to specifically limit the BCR’s ability to induce CSR.
Figure 5. NF-κB2 is required for BCR-induced CSR.
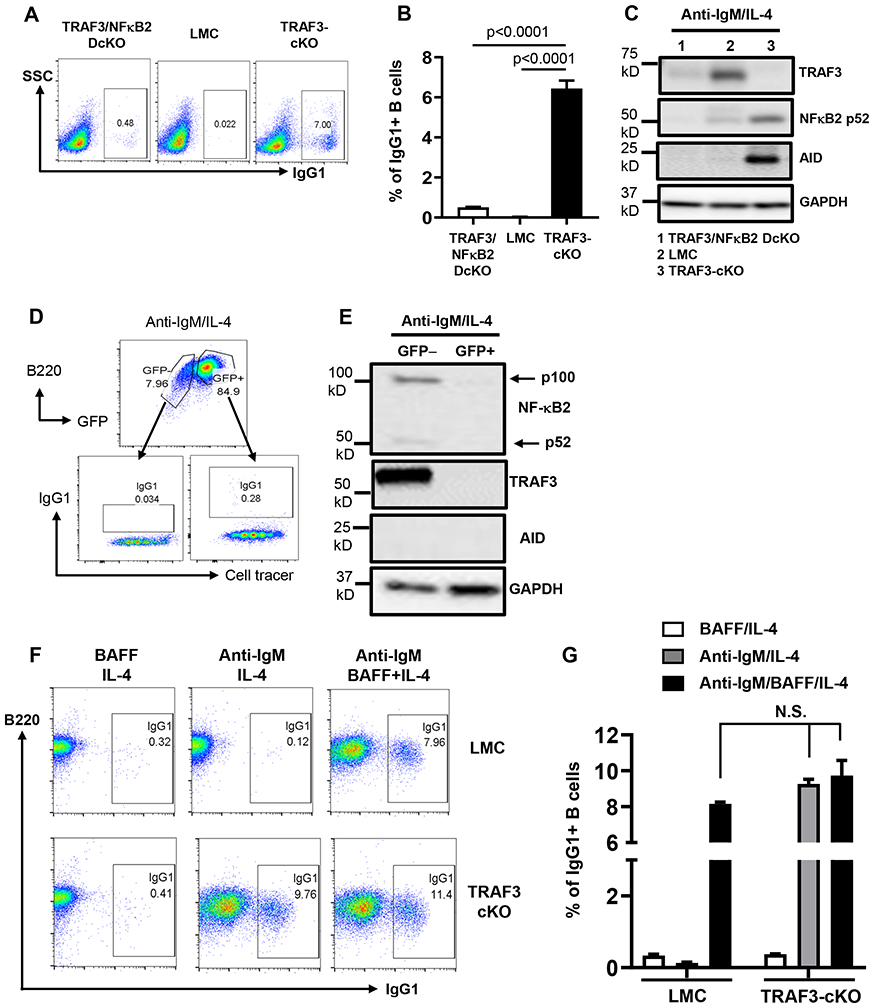
(A) Flow data showing day 4 IgG1 CSR induced by anti-IgM/IL-4 in indicated B cells. (B) Quantification of IgG1+ B cell percentage from triplicates of one representative experiment. (C) Western blot data showing indicated protein expression induced by anti-IgM/IL-4 at day3 in indicated B cells. (D) Representative flow data showing GFP expression (a marker for NF-κB2 deletion) in CD19Cre-TRAF3f/f/NF-κB2f/f B cells and day4 IgG1 CSR in GFP+ and GFP− B cells stimulated by anti-IgM/IL-4. Cell tracer indicates cell proliferation. (E) Representative Western blot data showing NF-κB2, TRAF3 and AID expression in CD19Cre-TRAF3f/f/NF-κB2f/f GFP− and GFP+ B cells stimulated with anti-IgM/IL-4 for 3 days. GAPDH as loading control. (F and G) The role of BAFF-BAFFR pathway in CSR of TRAF3-cKO B cells. (F) Representative flow data showing day 4 IgG1 CSR induced by indicated stimuli (IL4, 10 ng/ml; anti-IgM, 10 μg/ml; BAFF, 1 μg/ml). (G) Quantification of IgG1+ B cell percentage from triplicates of one representative experiment. N.S., not significant, Two-way ANOVA analysis between indicated groups. TRAF3-cKO, CD19Cre-Traf3f/f; TRAF3/NF-κB2-DcKO, CD19Cre-Traf3f/f-NF-κB2f/f. Data are representative of 3-6 independently repeated experiments.
Given that BAFF-BAFFR pathway can regulate NF-κB2 activation, we next examined BAFF’s role in CSR induction. Our data showed that BAFF/IL-4 stimulation induced a minimal level of IgG1 CSR in LMC B cells (Figure 5F, G). Importantly, BAFF/IL-4 stimulation did not increase the level of IgG1 CSR in TRAF3-cKO B cells (Figure 5F, G), suggesting that BAFFR pathway is not dysregulated in these KO B cells for CSR. In contrast, anti-IgM/IL-4 stimulation induced a robust level of CSR in TRAF3-cKO B cells but not in LMC B cells as shown above. BAFF markedly enhanced anti-IgM/IL-4-induced CSR in LMC B cells but not in TRAF3-cKO B cells (Figure 5F, G), suggesting that BAFF’s function in LMC B cells is to degrade TRAF3 during CSR. Taken together, these data demonstrate that TRAF3-deficiency enables BCR-induced CSR but has no effects on BAFF-induced CSR, and BCR-induced CSR in TRAF3-cKO B cells is not attributed to dysregulated BAFF-BAFFR pathway.
TRAF3 deficiency disrupts autoreactive B-cell anergy
Autoreactive anergic B cells do not generate Ca2+ flux when their BCR is being engaged with specific self-Ag or anti-IgM (34). Our studies revealed that engaging BCR by anti-IgM or specific HEL Ag induced an increased level of Ca2+ flux in the absence of TRAF3 (Figure 3C, D). Thus, we hypothesized that TRAF3 deficiency in B cells may disrupt anergy of autoreactive B cells. To test our hypothesis, we isolated BM cells from TRAF3-cKO-VDJ9/κ5 or VDJ9/κ5 mice and transferred them into irradiated ML-5-Tg recipients (Figure 6A). ML-5-Tg mice constitutively express HEL (26); thus, HEL becomes a self-Ag in the BM chimera. B cells from ML-5 chimeras receiving VDJ9/κ5 BM did not respond to HEL stimulation, evidenced by the absence of Ca2+ flux and reduced anti-HEL IgM (Figure 6B, C), suggesting anergy is maintained. However, B cells from ML-5 chimeras receiving TRAF3-cKO-VDJ9/κ5 BM responded to HEL stimulation almost as well as donor TRAF3-cKO-VDJ9/κ5 B cells (Figure 6B, C), suggesting anergy is disrupted. In addition, we examined the surface IgM expression in the donor and recipient B cells. Our data showed that the expression of surface IgM on B cells was downregulated in ML-5 chimeras receiving BM from VDJ9/κ5 mice compared with that in VDJ9/κ5 donor mice, consistent with anergy induction in these ML-5 chimeras (Figure 6D). In contrast, the expression of surface IgM on B cells was similar between ML-5 chimeras receiving BM from TRAF3-cKO-VDJ9/κ5 mice and TRAF3-cKO-VDJ9/κ5 donor mice (Figure 6E). These results show that IgM downregulation, a classic anergy phenotype, was not induced in these ML-5 chimeras that lack TRAF3 in B cells. These studies suggest that TRAF3 maintains B cell homeostasis and autoreactive B cell anergy by limiting BCR signaling strength induced by cognate Ag/BCR interaction. They also further indicate that the BCR in the absence of B-cell intrinsic TRAF3 is hypersensitive to Ag stimulation.
Figure 6. TRAF3 deficiency disrupts autoreactive B cell anergy.
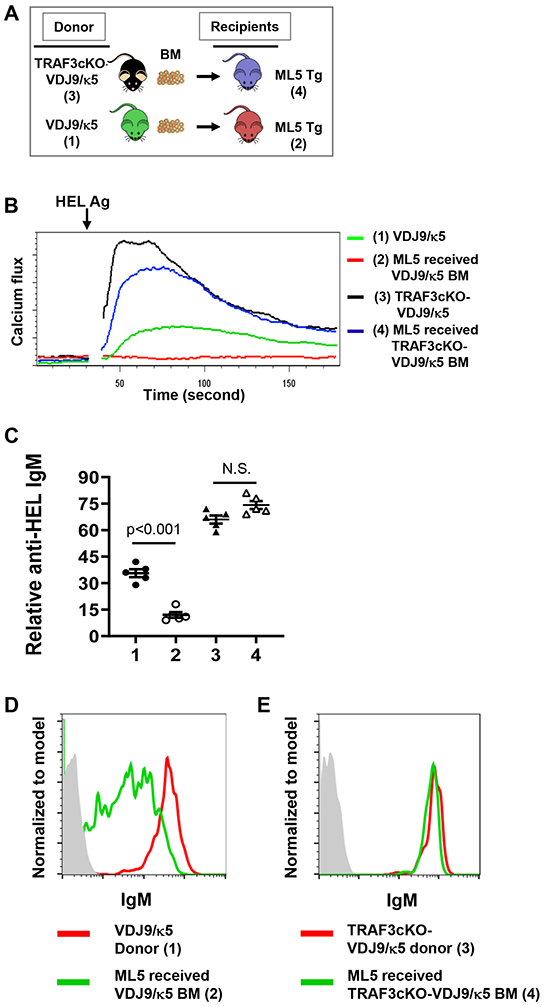
(A) Schematics of BM transfer experiments. (B) Representative flow data showing Ca2+ flux induced by HEL Ag in indicated B cells (group 1-4, n=5/group). (C) Relative level of secreted anti-HEL IgM induced by HEL/IL-4 in indicated B cell culture supernatant (n=5/group). ID of each group as labeled in (B). (D and E) Representative flow data showing surface IgM expression in the B cells of indicated donor and recipient mice. B cells were gated on B220+ populations. TRAF3-cKO, CD19Cre-Traf3f/f. Data are representative of 3 independently repeated experiments.
Limiting BCR repertoires or attenuating BCR signaling strength rectified lymphoid organ disorders and autoimmunity caused by TRAF3-deficiency
Based on our data, we hypothesized that the B cell expansion, splenomegaly and glomerulonephritis in TRAF3-cKO mice (19) may be driven by the elevated BCR signaling of autoreactive B cells. To test our hypothesis, we generated the TRAF3-cKO-VDJ9/κ5 mice in which an endogenous diverse BCR repertoire recognizing various self and non-self-Ags is replaced by a single VDJ9/κ5 BCR recognizing HEL Ag (25). In sharp contrast to TRAF3-cKO mice that had abnormal B cell expansion and splenomegaly, TRAF3-cKO-VDJ9/κ5 mice had normal sized spleens as well as normal numbers of MZ and FO B cells, compared to those of LMC or VDJ9/κ5 mice (Figure 7A-C, Supplemental Figure 3F). TRAF3-cKO-VDJ9/κ5 mice completely lacked lymphocyte infiltration in kidney or glomerulonephritis (Figure 7D). Because TRAF3-cKO-VDJ9/κ5 B cells can only recognize HEL that is absent in these mice, these B cells cannot receive stimulatory signals from their BCR. We infer from these data that introducing a non-autoreactive BCR abrogates the abnormal expansion of B cells and reduces the severity of autoimmunity.
Figure 7. Lymphoid organ disorder and autoimmune manifestations caused by TRAF3-deficiency are rectified by limiting BCR repertoires or attenuating BCR signaling strength.
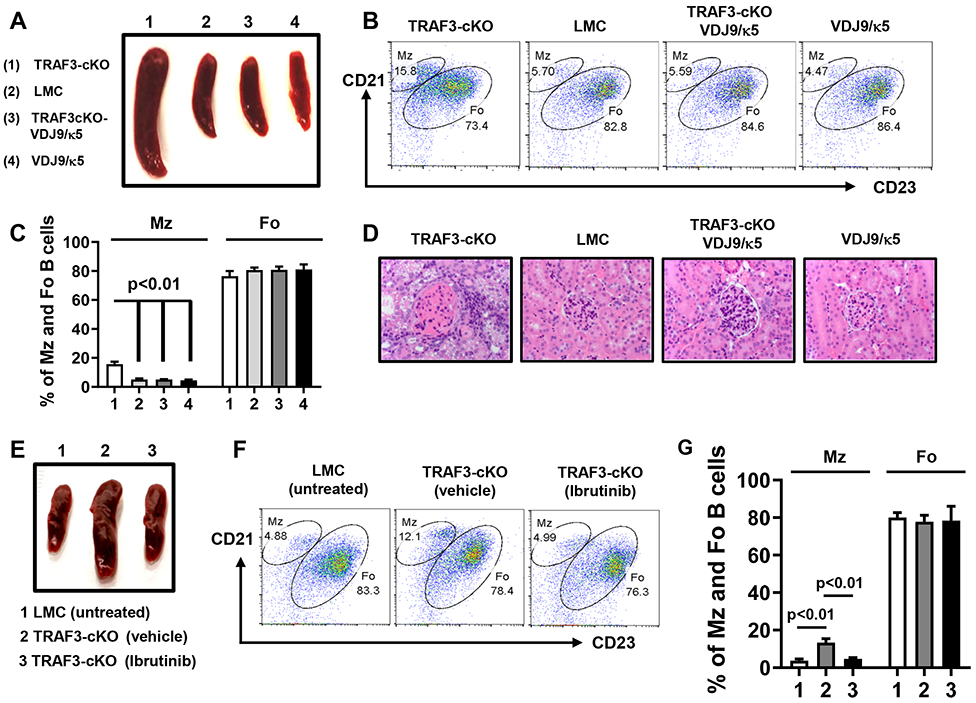
(A to D) TRAF3-cKO mice were crossed with VDJ9/κ5 mice to generate TRAF3-cKO-VDJ9/κ5 mice. (A) Representative image of mouse spleens with indicated genotypes and indicated number of mice examined in total: (1) TRAF3-cKO (n=24); (2) LMC (n=63); (3) TRAF3-cKO-VDJ9/κ5 (n=5); and (4) VDJ9/κ5 (n=10) (8-12 weeks old). (B) Flow data showing the profiles of MZ and FO B cells in mice with indicated genotypes. B cells were gated for B220+IgM+ double positive population. (C) Quantification of the percentage of MZ and FO B cells in mice with indicated genotypes as labeled in (A) (n=5/group). (D) Representative H&E staining of kidney samples. Magnification 40×. (E) Representative image of mouse spleens with indicated genotype and treatment (group 1-3, n=5/group, 12 weeks old). (F) Representative flow data showing the profiles of MZ and FO B cells. B cells were gated for B220+IgM+ double positive population. (G) Quantification of the percentage of MZ and FO B cells in mice with indicated genotype and treatment as labeled in (E) (n=5/group). TRAF3-cKO, CD19Cre-Traf3f/f. Data are representative of 3-6 independently repeated experiments.
Because replacing BCR repertoire is not practicable for therapeutic purpose, we next tested whether attenuating BCR proximal signaling strength by chemicals can prevent splenomegaly and abnormal B cell expansion in TRAF3-cKO mice. Ibrutinib is a drug that specifically targets BTK, a BCR proximal signaling element, and used in clinic to treat B cell lymphomas. As shown above, Ibrutinib strongly inhibits BCR-induced CSR and AID expression (Figure 3E-G). Ibrutinib-treated TRAF3-cKO mice had normal sized spleens and MZ and FO B cell profiles (Figure 7E-G) as well as total B cell numbers (Supplemental Figure 3G). In contrast, vehicle-treated TRAF3-cKO mice exhibited aforementioned abnormalities (Figure 7E-G, Supplemental Figure 3G). Our data show that attenuating BCR signaling strength prevents lymphoid organ disorders caused by TRAF3-deficiency.
Discussion
Our studies address a long-lasting question in the field of B cell biology and immunology, i.e., why BCR alone cannot induce AID and CSR as co-receptor CD40 does (2, 31). We uncovered new biological functions of TRAF3 in controlling BCR-induced CSR and autoreactive B-cell anergy by restricting BCR signaling strength. We found that: (1) BCR’s capacity to induce AID and CSR is restrained by TRAF2 and TRAF3; (2) TRAF3 limits BCR proximal signaling strength by reducing phosphorylation of Syk and BTK kinases as well as PLCγ2-dependent Ca2+ flux; (3) TRAF3 deficiency resulted in elevated BCR signaling intensity and constitutively active NF-κB2, both of which are required for enabling the BCR to induce CSR; (4) TRAF3-deficiency breaks autoreactive B-cell anergy; (5) Lymphoid organ disorders and autoimmune manifestations caused by TRAF3-deficiency can be rectified by limiting BCR repertoires or attenuating BCR proximal signaling strength by Ibrutinib, a BTK inhibitor. We suggest that when BCR signaling strength is elevated to a level that is sufficient to induce AID and CSR, it may break autoreactive B cell tolerance and disrupt B cell homeostasis. These results may lead to a new conceptual view, which explains how signaling components of the BCR and co-receptor pathways ensure optimal humoral immunity while simultaneously maintain B cell homeostasis and prevent malignancy by fine-tuning the BCR signaling intensity.
Based on our findings, we propose a model to explain why engaging BCR alone cannot induce CSR in the absence of co-stimulatory signals such as ligands for CD40 or BAFFR (Figure 8). Normally, BCR-induced CSR does not occur because the signaling strength for the BCR to do so is potentially dangerous, and if the BCR signaling strength reaches this level, it may break B-cell anergy, disrupt B-cell homeostasis or cause B-cell malignancy. Thus, a high threshold is set by B cell-intrinsic checkpoints such as TRAF2 and TRAF3; only after removal of the checkpoint(s) such as deleting TRAF2 or TRAF3, the BCR is able to induce CSR. Once TRAF3 is deleted, BCR-induced NF-κB2 precursor p100 will be processed into active NF-κB2 p52, which is required for BCR-induced CSR (Figure 8). We predict that there are additional B cell-intrinsic checkpoints that remain to be identified in this context.
Figure 8. Signaling mechanisms of BCR-induced CSR.
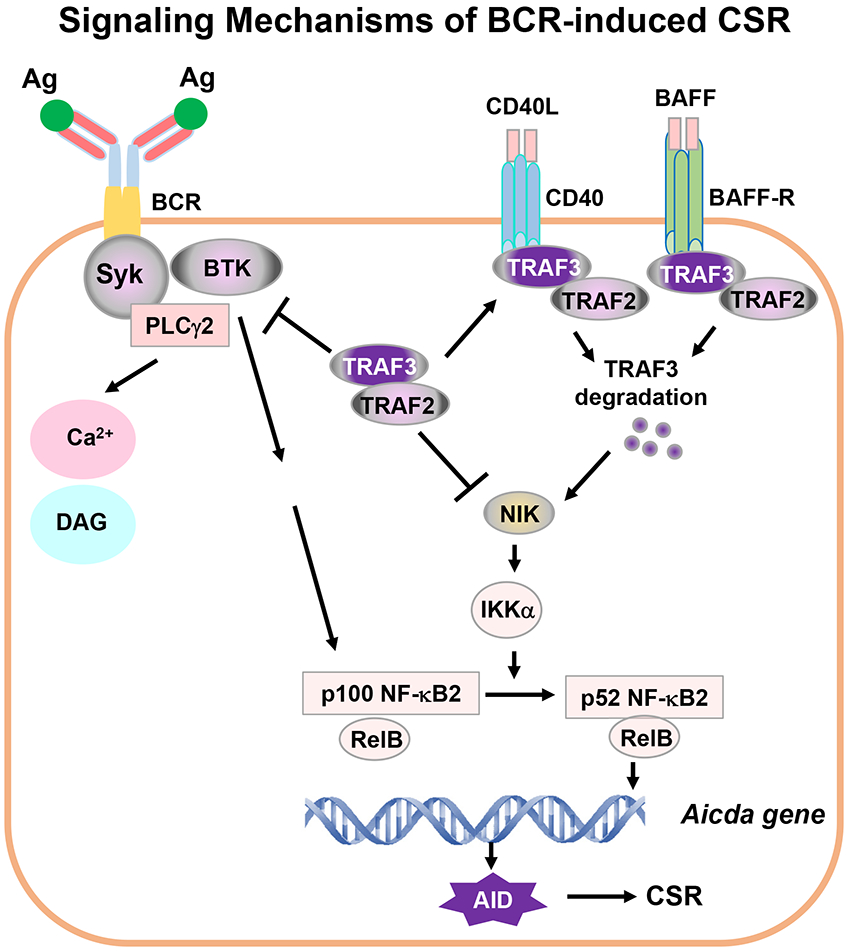
Ag stimulation of BCR activates proximal signaling elements, Syk, BTK and PLCγ2, whose activation triggers Ca2+ flux. Syk/BTK/PLCγ2 complex may activate additional unknown signaling pathway(s) to promote NF-κB2 precursor p100 synthesis. TRAF2 and TRAF3 block NIK activity. Thus, Syk/BTK/PLCγ2 complex cannot activate signaling components downstream of NF-κB2 p100, i.e., cannot activate NF-κB2 transcription factor to initiate AID expression. Removal of TRAF3 and/or TRAF2 leads to constitutively active NF-κB2 p52 via NIK and IKKα pathway. Active NF-κB2 complexes act together with additional factors to initiate AID transcription. AID protein targets Igh locus to induce CSR. During infection or immunization, CD40/CD40L or BAFF/BAFFR interaction recruits TRAF3/TRAF2 to membrane lipid rafts, which eventually causes TRAF3 degradation, a physiological situation of TRAF3-deficiency. TRAF3 degradation results in NIK and NF-κB2 complex activation. NF-κB2 activation is specifically required for the BCR to induce CSR. We propose that one critical function of co-stimulatory signals is to degrade TRAF3 to allow NF-κB2-dependent BCR-induced CSR that is essential for in vivo antibody responses. Of note, TRAF3 not only restricts Syk, BTK and PLCγ2 hyperactivation upon Ag stimulation but also blocks NF-κB2 activation, which may be especially important for maintaining autoreactive B-cell anergy.
Transient TRAF3-deficiency can occur during normal humoral immune responses against TD Ag. During pathogen infection or immunization, T cells or other innate immune cells can be activated that provide CD40 ligand (CD40L) or the ligand for BAFFR to activate B cells, induce B cell differentiation as well as promote germinal center (GC) formation (35, 36). CD40L/CD40 or BAFF/BAFFR interaction induces TRAF3 degradation in B cells (15, 16, 37) (Figure 8). Once TRAF3 is degraded, NIK will be released and allowed to accumulate in cytoplasm to activate IKKα. IKKα then phosphorylates NF-κB2 p100 whose expression can be induced by engaging BCR as shown previously (17, 18) and in the current study. Phosphorylated p100 converts into active p52 by ubiquitination-mediated proteolysis, thereby enabling the BCR to induce CSR (Figure 8). Consistently, we showed that BAFF/IL-4/anti-IgM induced a robust level of IgG1 CSR, whereas neither BAFF/IL-4 nor anti-IgM/IL-4 did so in LMC B cells. Of note, we found that BAFF/IL-4 stimulation only induced a minimal level of IgG1 CSR in LMC B cells, much lower than that reported previously (38). This discrepancy might be due to the vast difference in IL-4 concentration used previously (50μg/ml) (38). Importantly, we showed that BAFF did not enhance anti-IgM/IL-4-induced CSR in TRAF3-cKO B cells, suggesting that BAFF’s function is to degrade TRAF3 in WT B cells. Once foreign Ags disappear, T cells and innate immune cells will cease providing CD40L or BAFF and B cell-intrinsic TRAF3 expression will recover; consequently, the BCR cannot induce AID expression to initiate CSR anymore. Thus, our study explains how the BCR and co-receptors (e.g., BAFFR) cooperate to induce CSR.
We identified the NF-κB2 as an essential transcription factor for the BCR to induce AID and CSR. NF-κB1 activation is important for CSR induced by co-receptors including CD40 and TLR4 as inhibiting NF-κB1 activation significantly decreased CSR induced by these receptors (5, 6). In contrast, NF-κB2 activation is specifically required for the BCR signaling to induce CSR but not CD40 or TLR4 (Figure 5, Supplemental Figure 3). Our results are supported by previous studies showing that p52, the active NF-κB2, is not required for CD40-induced CSR in vitro but is essential for in vivo antibody responses during Ag immunization (39). Previously, it was suggested that p52 promotes antibody responses possibly by supporting GC formation (39). Our current studies suggest that p52 can also directly promote antibody responses by enabling the BCR to induce CSR. In this regard, the promoter region of AID contains NF-κB2 binding motifs (2, 30), an evolutionary evidence supporting the importance of NF-κB2-dependent BCR-induced CSR.
The physiological role of AID is to induce SHM and CSR at Ig loci (3). However, even endogenous AID can target non-Ig genes such as Bcl-6 (40, 41) or c-myc oncogene (42). If dysregulated, AID can target non-Ig genes genome-wide to induce point mutations or DNA double strand breaks (DSBs) that may cause chromosomal translocations, thereby contributing to B cell lymphomagenesis (43-45). Our study showed that NF-κB2 activation is essential for the BCR to induce AID expression; moreover, active NF-κB2 is also required for driving B cell expansion and lymphoid organ disorders caused by TRAF3-deficiency. Taken together, elevated BCR proximal signaling and constitutive NF-κB2 activation may lead to survival and proliferation of B cells, which, together with abnormally induced AID expression, would significantly increase the likelihood of tumorigenesis.
We observed a more robust CSR level in TRAF2/3-DcKO B cells, although these DcKO B cells did not express more AID protein. CSR efficiency can be regulated not only by AID protein level but also by other mechanisms such as phosphorylation status of the AID protein, nuclear translocation of the AID protein and target sequence accessibility as well as Ig gene germline transcription (32, 46, 47). For instance, double deficiency of TRAF2 and TRAF3 may promote AID phosphorylation to enhance AID activity or facilitate the access of AID to target sequences, namely, Sγ1 and Sε regions. It is also possible that double deficiency of TRAF2 and TRAF3 may enhance Cγ1 and Cε germline transcription.
Inhibitors of BTK, PLCγ2, and Syk not only reduced BCR-induced CSR but also inhibited proliferation in TRAF3-cKO B cells. CSR in response to other stimuli is known to be linked to proliferation, and our data suggest that this linkage between CSR and proliferation also occurs in BCR-induced CSR. An interesting question is − does signaling through the Syk/BTK/PLCγ2 pathway directly promote CSR? Or does it promote proliferation which subsequently facilitates CSR induced by other signaling pathways? We predict that both possibilities are likely, for example, Syk signaling is required for the BCR-induced expression of precursor NF-κB2 p100 (18, 48). In the absence of TRAF3, the BCR-induced p100 can be constitutively processed into active NF-κB2 p52 that is essential for BCR-induced CSR as shown in the current study (Figure 5). Thus, Syk signaling not only affects B cell proliferation but also may directly promote CSR by inducing p100 expression. On the other hand, it is likely that these BCR proximal signaling elements just promote B cell proliferation, and the proliferating state of B cells allows CSR to be induced by other signaling pathways, such as NF-κB2 in the absence of TRAF3. To further address these questions, future studies are needed that could uncouple CSR and proliferation.
Supplementary Material
Key Points: (1) We identify checkpoints restricting the BCR-induced class switch recombination. (2) TRAF3 maintains B cell homeostasis by fine-tuning the BCR signaling strength.
Acknowledgments:
We thank Dr. Jason Cyster (University of California San Francisco, San Francisco, CA), Dr. Douglas Mann (Washington University, St. Louis, MO), and Dr. John C. Cambier (University of Colorado, Aurora, CO) for generously providing VDJ9/κ5 mice, TRAF2flox/flox mice, ML-5-Tg mice, respectively. We thank Nicholas Rotello Kuri and Yonatan Kramer for technical help. We apologize to those whose work was not cited due to length restrictions.
This work was supported by University of Colorado School of Medicine and Cancer Center startup funds to J.H.W., Cancer League of Colorado, R21-CA184707, R21-AI110777, R01-CA166325, R21-AI133110, R01-CA229174 and R01-CA249940 to J.H.W., and a fund from American Cancer Society (ACS IRG #16-184-56) to Z.C. X.G.W. was supported by an AAI Careers in Immunology Fellowship. R.A.W. is supported by a NIH F31 fellowship (F31DE027854). S.M.Y.C. is supported by a NIH T32 fellowship (T32 AI007405). The sponsors or funders have no role in the design and conduct of the study, in the collection, analysis, and interpretation of the data, and in the preparation, review, or approval of the manuscript.
Footnotes
Competing interests: All authors have declared that no conflict of interest exists.
Data and materials availability: all data is available in the manuscript or the Supplemental materials.
References
- 1.Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, Vuong B, Wang J, Phan RT, Datta A, Manis J, and Alt FW. 2007. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Adv Immunol 94: 157–214. [DOI] [PubMed] [Google Scholar]
- 2.Chen Z, and Wang JH. 2019. Signaling control of antibody isotype switching. Adv Immunol 141: 105–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, and Honjo T. 2000. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102: 553–563. [DOI] [PubMed] [Google Scholar]
- 4.Heltemes-Harris LM, Gearhart PJ, Ghosh P, and Longo DL. 2008. Activation-induced deaminase-mediated class switch recombination is blocked by anti-IgM signaling in a phosphatidylinositol 3-kinase-dependent fashion. Mol Immunol 45: 1799–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pone EJ, Zhang J, Mai T, White CA, Li G, Sakakura JK, Patel PJ, Al-Qahtani A, Zan H, Xu Z, and Casali P. 2012. BCR-signalling synergizes with TLR-signalling for induction of AID and immunoglobulin class-switching through the non-canonical NF-kappaB pathway. Nature communications 3: 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woolaver RA, Wang X, Dollin Y, Xie P, Wang JH, and Chen Z. 2018. TRAF2 Deficiency in B Cells Impairs CD40-Induced Isotype Switching That Can Be Rescued by Restoring NF-kappaB1 Activation. J Immunol 201: 3421–3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zan H, and Casali P. 2013. Regulation of Aicda expression and AID activity. Autoimmunity 46: 83–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, and Nussenzweig MC. 2003. Predominant autoantibody production by early human B cell precursors. Science 301: 1374–1377. [DOI] [PubMed] [Google Scholar]
- 9.Pelanda R, and Torres RM. 2006. Receptor editing for better or for worse. Current opinion in immunology 18: 184–190. [DOI] [PubMed] [Google Scholar]
- 10.Yarkoni Y, Getahun A, and Cambier JC. 2010. Molecular underpinning of B-cell anergy. Immunological reviews 237: 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franks SE, and Cambier JC. 2018. Putting on the Brakes: Regulatory Kinases and Phosphatases Maintaining B Cell Anergy. Frontiers in immunology 9: 665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin WW, Hostager BS, and Bishop GA. 2015. TRAF3, ubiquitination, and B-lymphocyte regulation. Immunological reviews 266: 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Kooten C 2000. Immune regulation by CD40-CD40-l interactions - 2; Y2K update. Front Biosci 5: D880–693. [DOI] [PubMed] [Google Scholar]
- 14.Hostager BS, Haxhinasto SA, Rowland SL, and Bishop GA. 2003. Tumor necrosis factor receptor-associated factor 2 (TRAF2)-deficient B lymphocytes reveal novel roles for TRAF2 in CD40 signaling. J Biol Chem 278: 45382–45390. [DOI] [PubMed] [Google Scholar]
- 15.Liao G, Zhang M, Harhaj EW, and Sun SC. 2004. Regulation of the NF-kappaB-inducing kinase by tumor necrosis factor receptor-associated factor 3-induced degradation. J Biol Chem 279: 26243–26250. [DOI] [PubMed] [Google Scholar]
- 16.Gardam S, Sierro F, Basten A, Mackay F, and Brink R. 2008. TRAF2 and TRAF3 signal adapters act cooperatively to control the maturation and survival signals delivered to B cells by the BAFF receptor. Immunity 28: 391–401. [DOI] [PubMed] [Google Scholar]
- 17.Cancro MP 2009. Signalling crosstalk in B cells: managing worth and need. Nat Rev Immunol 9: 657–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stadanlick JE, Kaileh M, Karnell FG, Scholz JL, Miller JP, Quinn WJ 3rd, Brezski RJ, Treml LS, Jordan KA, Monroe JG, Sen R, and Cancro MP. 2008. Tonic B cell antigen receptor signals supply an NF-kappaB substrate for prosurvival BLyS signaling. Nat Immunol 9: 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie P, Stunz LL, Larison KD, Yang B, and Bishop GA. 2007. Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity 27: 253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu S, Jin J, Gokhale S, Lu AM, Shan H, Feng J, and Xie P. 2018. Genetic Alterations of TRAF Proteins in Human Cancers. Frontiers in immunology 9: 2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore CR, Edwards SK, and Xie P. 2015. Targeting TRAF3 Downstream Signaling Pathways in B cell Neoplasms. J Cancer Sci Ther 7: 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Y, Cheng G, and Baltimore D. 1996. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity 5: 407–415. [DOI] [PubMed] [Google Scholar]
- 23.Moore CR, Liu Y, Shao C, Covey LR, Morse HC 3rd, and Xie P. 2012. Specific deletion of TRAF3 in B lymphocytes leads to B-lymphoma development in mice. Leukemia 26: 1122–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grech AP, Amesbury M, Chan T, Gardam S, Basten A, and Brink R. 2004. TRAF2 differentially regulates the canonical and noncanonical pathways of NF-kappaB activation in mature B cells. Immunity 21: 629–642. [DOI] [PubMed] [Google Scholar]
- 25.Allen CD, Okada T, Tang HL, and Cyster JG. 2007. Imaging of germinal center selection events during affinity maturation. Science 315: 528–531. [DOI] [PubMed] [Google Scholar]
- 26.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, and et al. 1988. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 334: 676–682. [DOI] [PubMed] [Google Scholar]
- 27.Casola S, Cattoretti G, Uyttersprot N, Koralov SB, Seagal J, Hao Z, Waisman A, Egert A, Ghitza D, and Rajewsky K. 2006. Tracking germinal center B cells expressing germ-line immunoglobulin gamma1 transcripts by conditional gene targeting. Proc Natl Acad Sci U S A 103: 7396–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z, Getahun A, Chen X, Dollin Y, Cambier JC, and Wang JH. 2015. Imbalanced PTEN and PI3K Signaling Impairs Class Switch Recombination. J Immunol 195: 5461–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Z, Ranganath S, Viboolsittiseri SS, Eder MD, Chen X, Elos MT, Yuan S, Hansen E, and Wang JH. 2014. AID-initiated DNA lesions are differentially processed in distinct B cell populations. J Immunol 193: 5545–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu Z, Zan H, Pone EJ, Mai T, and Casali P. 2012. Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nat Rev Immunol 12: 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stavnezer J, and Schrader CE. 2014. IgH chain class switch recombination: mechanism and regulation. J Immunol 193: 5370–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vaidyanathan B, Yen WF, Pucella JN, and Chaudhuri J. 2014. AIDing Chromatin and Transcription-Coupled Orchestration of Immunoglobulin Class-Switch Recombination. Frontiers in immunology 5: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hashimoto A, Takeda K, Inaba M, Sekimata M, Kaisho T, Ikehara S, Homma Y, Akira S, and Kurosaki T. 2000. Cutting edge: essential role of phospholipase C-gamma 2 in B cell development and function. J Immunol 165: 1738–1742. [DOI] [PubMed] [Google Scholar]
- 34.Browne CD, Del Nagro CJ, Cato MH, Dengler HS, and Rickert RC. 2009. Suppression of phosphatidylinositol 3,4,5-trisphosphate production is a key determinant of B cell anergy. Immunity 31: 749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foy TM, Laman JD, Ledbetter JA, Aruffo A, Claassen E, and Noelle RJ. 1994. gp39-CD40 interactions are essential for germinal center formation and the development of B cell memory. J Exp Med 180: 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalled SL 2006. Impact of the BAFF/BR3 axis on B cell survival, germinal center maintenance and antibody production. Semin Immunol 18: 290–296. [DOI] [PubMed] [Google Scholar]
- 37.Hacker H, Tseng PH, and Karin M. 2011. Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol 11: 457–468. [DOI] [PubMed] [Google Scholar]
- 38.Castigli E, Wilson SA, Scott S, Dedeoglu F, Xu S, Lam KP, Bram RJ, Jabara H, and Geha RS. 2005. TACI and BAFF-R mediate isotype switching in B cells. J Exp Med 201: 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caamano JH, Rizzo CA, Durham SK, Barton DS, Raventos-Suarez C, Snapper CM, and Bravo R. 1998. Nuclear factor (NF)-kappa B2 (p100/p52) is required for normal splenic microarchitecture and B cell-mediated immune responses. J Exp Med 187: 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Z, Viboolsittiseri SS, O'Connor BP, and Wang JH. 2012. Target DNA sequence directly regulates the frequency of activation-induced deaminase-dependent mutations. J Immunol 189: 3970–3982. [DOI] [PubMed] [Google Scholar]
- 41.Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, and Schatz DG. 2008. Two levels of protection for the B cell genome during somatic hypermutation. Nature 451: 841–845. [DOI] [PubMed] [Google Scholar]
- 42.Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, Honjo T, Nussenzweig A, and Nussenzweig MC. 2004. AID is required for c-myc/IgH chromosome translocations in vivo. Cell 118: 431–438. [DOI] [PubMed] [Google Scholar]
- 43.Chen Z, and Wang JH. 2014. Generation and repair of AID-initiated DNA lesions in B lymphocytes. Frontiers of medicine 8: 201–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, McBride KM, Klein IA, Stone G, Eisenreich TR, Ried T, Nussenzweig A, and Nussenzweig MC. 2009. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Molecular cell 36: 631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gu X, Booth CJ, Liu Z, and Strout MP. 2016. AID-associated DNA repair pathways regulate malignant transformation in a murine model of BCL6-driven diffuse large B-cell lymphoma. Blood 127: 102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matthews AJ, Zheng S, DiMenna LJ, and Chaudhuri J. 2014. Regulation of immunoglobulin class-switch recombination: choreography of noncoding transcription, targeted DNA deamination, and long-range DNA repair. Adv Immunol 122: 1–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vuong BQ, and Chaudhuri J. 2012. Combinatorial mechanisms regulating AID-dependent DNA deamination: interacting proteins and post-translational modifications. Semin Immunol 24: 264–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaileh M, Vazquez E, MacFarlane A. W. t., Campbell K, Kurosaki T, Siebenlist U, and Sen R. 2016. mTOR-Dependent and Independent Survival Signaling by PI3K in B Lymphocytes. PLoS One 11: e0146955. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.