

Abstract
P2X5 is a member of the P2X purinergic receptor family of ligand-gated cation channels, and has recently been shown to regulate inflammatory bone loss. Here, we report that P2X5 is a protective immune regulator during Listeria monocytogenes (Lm) infection, as P2X5-deficient mice exhibit increased bacterial loads in the spleen and liver, increased tissue damage, and early (within 3–6 days) susceptibility to systemic Lm infection. While P2X5-deficient mice experience normal monocyte recruitment in response to Lm, P2X5-deficient bone marrow-derived macrophages (BMMs) exhibit defective cytosolic killing of Lm. We further showed that P2X5 is required for Lm-induced inflammasome activation and IL-1β production, and that defective Lm killing in P2X5-deficient BMMs is substantially rescued by exogenous IL-1β or IL-18. Finally, in vitro BMM killing and in vivo Lm infection experiments employing either P2X7 deficiency or extracellular ATP depletion suggest that P2X5-dependent anti-Lm immunity is independent of the ATP-P2X7 inflammasome activation pathway. Together, our findings elucidate a novel and specific role for P2X5 as a critical mediator of protective immunity.
Keywords: P2X5, P2rx5, P2X7, BMMs, Listeria monocytogenes, inflammasome
Introduction
L. monocytogenes (Lm) is a gram-positive intracellular bacterium that can cause serious infections in immunocompromised individuals (1). Subsequent to phagocytosis, Lm escapes from the phagosomal compartment and gains access to the cytosol, where it grows rapidly within host cells (2). Therefore, the cytosolic innate immune system, including the inflammasome, is critical for host protection. Assembly of the inflammasome, a cytosolic complex of multiproteins, leads to cleavage and activation of caspase-1, which promotes maturation of the pro-inflammatory cytokines interleukin (IL)-1β and IL-18. Inflammasome formation is triggered by diverse stimuli that are encountered during infection, tissue damage or metabolic imbalances. Lm has been reported to activate the inflammasome, resulting in activation of caspase-1 (3–5). Caspase-1−/− mice exhibit early susceptibility to Lm, characterized by reduced IL-18 release and severely defective interferon-gamma (IFN-γ) production, suggesting the importance of caspase-1-mediated innate immunity in responses against Lm (6–9). Cytosolic Lm activates the NLRP3 inflammasome (4), and Lm total RNA can also stimulate the NLRP3 inflammasome through a variety of mechanisms (3). It has further been found that the Nlrc4 inflammasome contributes to IL-1β production in Lm-infected mouse cells (10). In another study, Sauer et al. demonstrated that bacteriolysis within the cytosol resulted in activation of the AIM2 inflammasome, implicating cytosolic survival and avoidance of cell autonomous defenses as important mechanisms of avoiding detection by the AIM2 inflammasome (11). NLRP6 inflammasome also can be activated by LTA, a molecule produced by gram-positive bacteria, leading to recruitment of pro-inflammatory caspases after Lm infection (12). Taken together, Lm has been demonstrated to engage various inflammasomes, and there are indications that Lm-triggered inflammasome activity is critical for immunity. However, it remains unclear by what upstream mechanism Lm-triggered inflammasome activation occurs in the context of protective immunity.
P2X receptors are ligand-gated, cation-selective channels with differential permeability to Na+, K+ and Ca2+. Of the seven known P2X receptors (P2X1–7), biological functions have been identified for P2X1, P2X2, P2X3, P2X4 and P2X7 (13). The P2X receptor best characterized for its roles in inflammation and immunity is P2X7, which is highly expressed by virtually all immune cells and is activated by extracellular ATP (14, 15). The discovery of the NALP1 (NLRP1) inflammasome and the identification of additional members of the family (namely NLRP3) enabled placement of P2X7 in a pathophysiological context, providing the molecular mechanism that couples P2X receptor activation to IL-1β processing (16, 17). As of now, P2X7 is understood as one of the most potent activators of NLRP3 inflammasome, capable of initiating caspase-1-mediated processing and release of the pro-inflammatory cytokines IL-1β and IL-18.
Much less is known about P2X5 in the immune system, with the exception of reported upregulation of P2X5 in CD34+ leukemic myeloid cell subpopulations (18) and in activated human T lymphocytes (19). We recently demonstrated a critical role for P2X5 in osteoclast maturation, with P2X5 deficiency resulting in protection against LPS-induced inflammatory bone loss in a manner that involved defects in both inflammasome activation and IL-1β production (20). Therefore, we speculated that P2X5 may also play a role in protective immune responses that are associated with inflammasome activity, such as the immune response to Lm infection. As we describe below, we found that P2X5 is a key factor in mounting proper innate immune responses against Lm infection in vivo, and that P2X5 is required specifically in BMMs for Lm induced-inflammasome activation.
Materials and Methods
Mice
P2X5-deficient (P2X5−/−) mice were generated on a C57BL/6J background using P2rx5−/− sperm obtained from the International Mouse Strain Resources (IMSR). P2X7-deficient (P2X7−/−) mice were purchased from Jackson Laboratory. P2X5−/−P2X7−/− mice were generated by crossing P2X5−/− and P2X7−/− mice. All mice were maintained and used in accordance with guidelines approved by the Institutional Animal Care and Use Committee (IACUC) at University of Pennsylvania.
Infection of mice
10403S wild-type (WT) Listeria monocytogenes were grown over night, re-inoculated until reaching an optical density at 600nm of 0.3–5 in BHI medium, and then washed in cold PBS. Mice were infected intravenously with 105 CFU Lm in PBS. In some experiments, mice were treated intraperitoneally with 20 U of apyrase (Millipore Sigma) the day before infection, 20 min before infection, and 6 hr after infection. Blood was collected and serum isolated for measuring lactate dehydrogenase (LDH) activity using a CytoTox 96 assay kit (Promega) according to the manufacturer’s protocol. Mice were sacrificed 2 and 4 days post-infection. Bacterial loads were determined by plating dilutions of tissue homogenates. For histology, livers and spleens were fixed with 4% paraformaldehyde. Fixed samples were embedded in paraffin, and sections were stained with hematoxylin and eosin (H&E) for microscopic analysis.
ELISA
IL-12p40, IFN-γ, CCL2, IL-1β and IL-18 protein levels were measured by ELISA (eBioscience) according to the manufacturer’s protocols. Assays were performed in triplicate for each independent experiment.
Ex vivo detection of TNF-α production
Splenocytes from mice infected for 0, 1 and 2 days were cultured with or without 108 bacteria/ml of heat-killed Listeria monocytogenes (HKLM) stimulation for 4 hr in the presence of brefeldin A (BFA), then surfaced stained for CD4, CD8a, CD11b, and finally fixed, permeabilized (Cytofix/Cytoperm, BD Pharmingen) and stained for intracellular TNF-α. Relevant cells were identified after gating from non-CD4, CD8 and CD11bint TNF-αhi lymphocytes/monocytes.
Mononuclear cell isolation and flow cytometry
Bone marrow cells were isolated by flushing femurs of mice with RPMI-1640 containing 2% FBS, and spleens were homogenized through 40 uM nylon mesh. Resulting cell suspensions were pelleted by centrifugation at 300x g. RBCs were lysed and washed three times and counted. Absolute cell numbers were calculated based on the percentage of monocytes from the total cell population acquired by flow cytometry. Single-cell suspensions were blocked with CD16/CD32 (2.4G2) and then stained variously with CD4 (RM4–5), CD8a (53–6.7), B220 (RA3–6B2), NK1.1 (PK136), CD11b (M1/70), Ly6C (HK1.4), Ly6G (1A8), Siglec-F (E50–2440). Live/Dead fixable dead cell stain kit (Invitrogen) was used to remove dead cells. Monocytes were gated as Ly6ChiCD11b+ from non-CD4, -CD8, -B220, -NK1.1, -Ly6G and -Siglec-F. All samples were acquired using a FACS LSRII (BD Biosciences) and analyzed using FlowJo software (Tree Star). All antibodies were purchased from BD Biosciences except anti-CD4 and anti-Ly6C (eBioscience).
Chemotaxis assay
Ly6Chi CD11b+ monocytes sorted on a FACS Aria II (BD Biosciences) were added to the upper chambers of 5um transwells (Corning) and 50ng/ml of CCL2 (Peprotech) was added to the lower chambers. The number of cells that migrated to the lower chamber was enumerated after 2hr.
In vitro killing assay
Bone marrow-derived macrophages (BMMs) were cultured for 7 days in alpha-MEM supplemented with10% fetal bovine serum (FBS) and recombinant M-CSF (30 ng/ml). 105 BMMs were plated in a 24 well plate 12–16 hr before stimulation. 10403S WT Lm, ΔactA mutant and LLO deleted (Δhly) mutant strains were grown to mid-log phase, washed with PBS, and added at a ratio of 10 to 1 cell (MOI 10). Nigericin (20 uM), IL-1β (100 ng/ml; Invivogen), or IL-18 (100 ng/ml; InvivoGen) were added 1hr before infection. 100 uM ATPγS, 10 U/ml apyrase, 100 uM ADPβS, 100 uM adenosine, and 10 U/ml adenosine deaminase were added 30 min before infection. All were purchased from Millpore Sigma. To measure phagocytosis, cells were lysed with 0.1% Triton X-100 for enumerating initial uptake for 30 min. Two hr post-infection, gentamycin was added to kill the extracellular bacteria, then two hr later, cells were lysed with 0.1% Triton X-100 and bacterial CFUs were enumerated by serial dilution.
Immunoblot analysis
BMMs were seeded at a density of 106 cells per well in 6-well plates and infected with bacteria. The cells were lysed in ice-cold radioimmunoprecipitation (RIPA) lysis buffer (Thermo Scientific) with protease and phosphatase inhibitor cocktail (Roche). Cell lysates were centrifuged to remove debris and quantified by Bradford assay. Equal amounts of lysates (2–50ug of protein) were fractionated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinyldifluoride (PVDF) membrane. The cleaved forms of caspase-1 and IL-1β were detected using anti-caspase-1 (Adipogen), anti-IL-1β (R&D Systems; AB-401-NA), with anti-β-actin (Sigma) used for loading control.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 6 program (GraphPad Software). Differences in mouse survival were assessed using the log-rank (Mantel-Cox) test. Statistical significance was defined as *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001.
Results
P2X5-deficient mice exhibit increased susceptibility to Lm infection
To examine the role of P2X5 in regulating host response to Lm, we infected WT and P2X5−/− mice by intravenous injection. During the early phase of infection (between days 3 and 6), P2X5−/− mice exhibited significant mortality, while all WT mice survived (Fig. 1a). The bacterial burdens in spleens and livers were determined at days 2 and 4, and P2X5−/− mice had 1–3 log-fold higher titers than WT mice in all tissues (Fig. 1b), confirming that P2X5 is required for bacterial clearance. Histopathological analyses revealed marked lymphocytolysis in P2X5−/− spleens, and more extensive coagulated necrosis with multifocal random microabscesses in P2X5−/− livers with increased LDH activity (Fig. 1c, d). These data indicate that P2X5 is required for host resistance to Lm infection.
Figure 1. P2X5-deficient mice exhibit increased susceptibility to Lm infection.
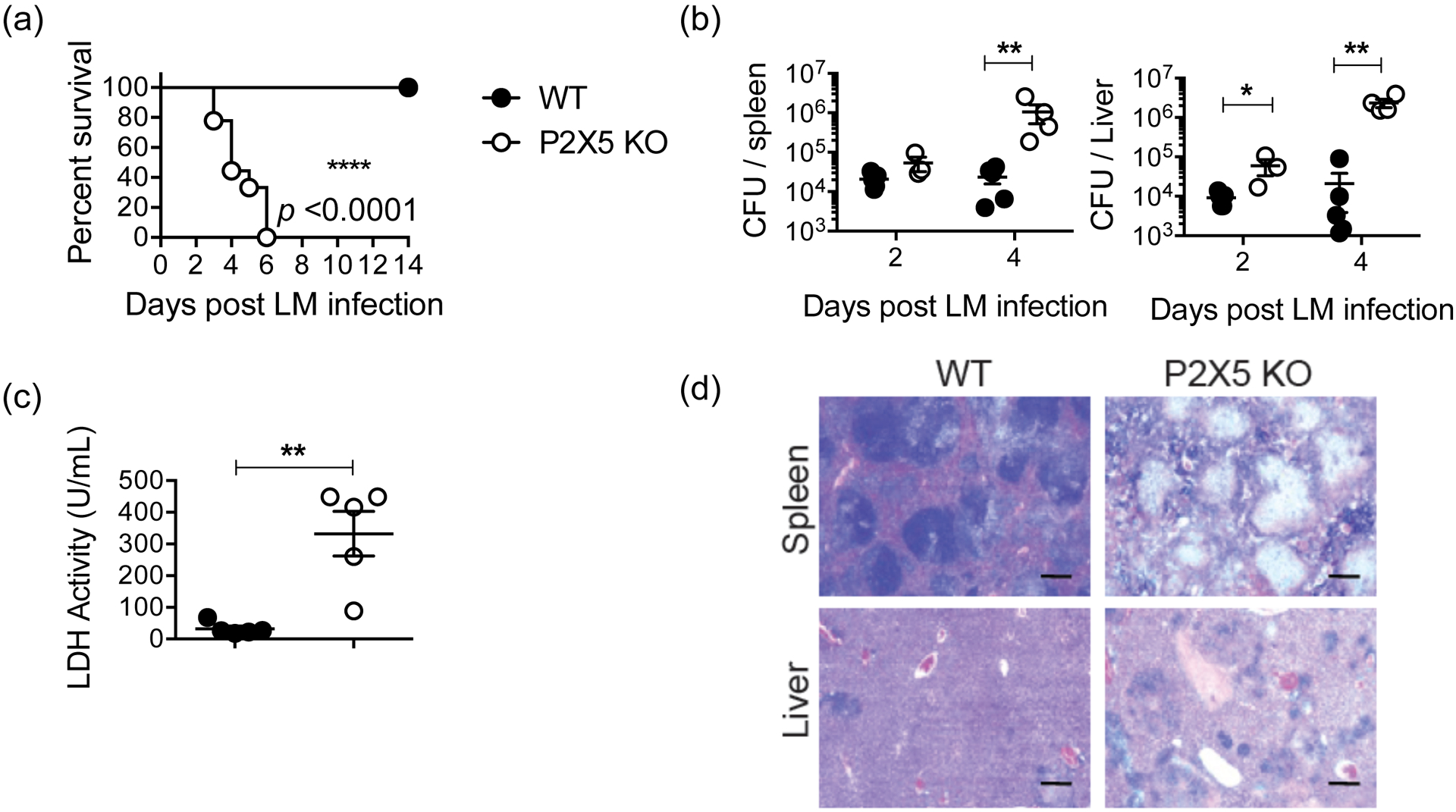
(a) WT and P2X5−/− mice were infected intravenously with 105 CFU of Lm and monitored for 14 days (n = 5–6 mice per group). Data are representative of five independent experiments. (b) Lm burden in the spleen (left) and liver (right) of WT and P2X5−/− mice at indicated days after infection. (c) Serum LDH activity at day 4 post-infection. (d) Histologic sections of spleen and liver from 4 days post-infection were stained with hematoxylin-eosin. Scale bar indicates 1 mm. (*p < 0.05, **p < 0.01 and ****p < 0.0001)
Lm-induced pro-inflammatory cytokine production in P2X5-deficient mice
Because P2X5-deficient mice exhibited early susceptibility to Lm infection, we began to investigate whether any critical elements of the anti-Lm immune response (2) require P2X5 for expression or function. In particular, early resistance to infection is attributed to the critical roles of myeloid differentiation factor 88 (MyD88)-dependent pro-inflammatory cytokine release (8), including TNF-α and IL-12, which promote production of IFN-γ (21). Studies with knockout mice show that both IFN-γ (22) and TNF-α (23) are essential for host defense against Lm. Therefore, we first measured the serum levels of pro-inflammatory cytokines upon Lm infection. Peak serum levels of IL-12 and IFN-γ appeared to be near normal in P2X5−/− mice, with IL-12p40 even slightly elevated (Fig 2a). Since TNF/iNOS-producing (Tip)-DCs have been reported as the predominant source of TNF-α during Lm infection (24), we measured TNF-α production by CD11b+ cells harvested from infected spleens, and found no significant defect in numbers of TNF-α-producing cells in P2X5−/− mice (Fig. 2b). Taken together, critical anti-Lm pro-inflammatory cytokines appear to be produced normally in the absence of P2X5, suggesting an alternative explanation for severe susceptibility to Lm.
Figure 2. Lm-induced pro-inflammatory cytokine production in P2X5-deficient mice.
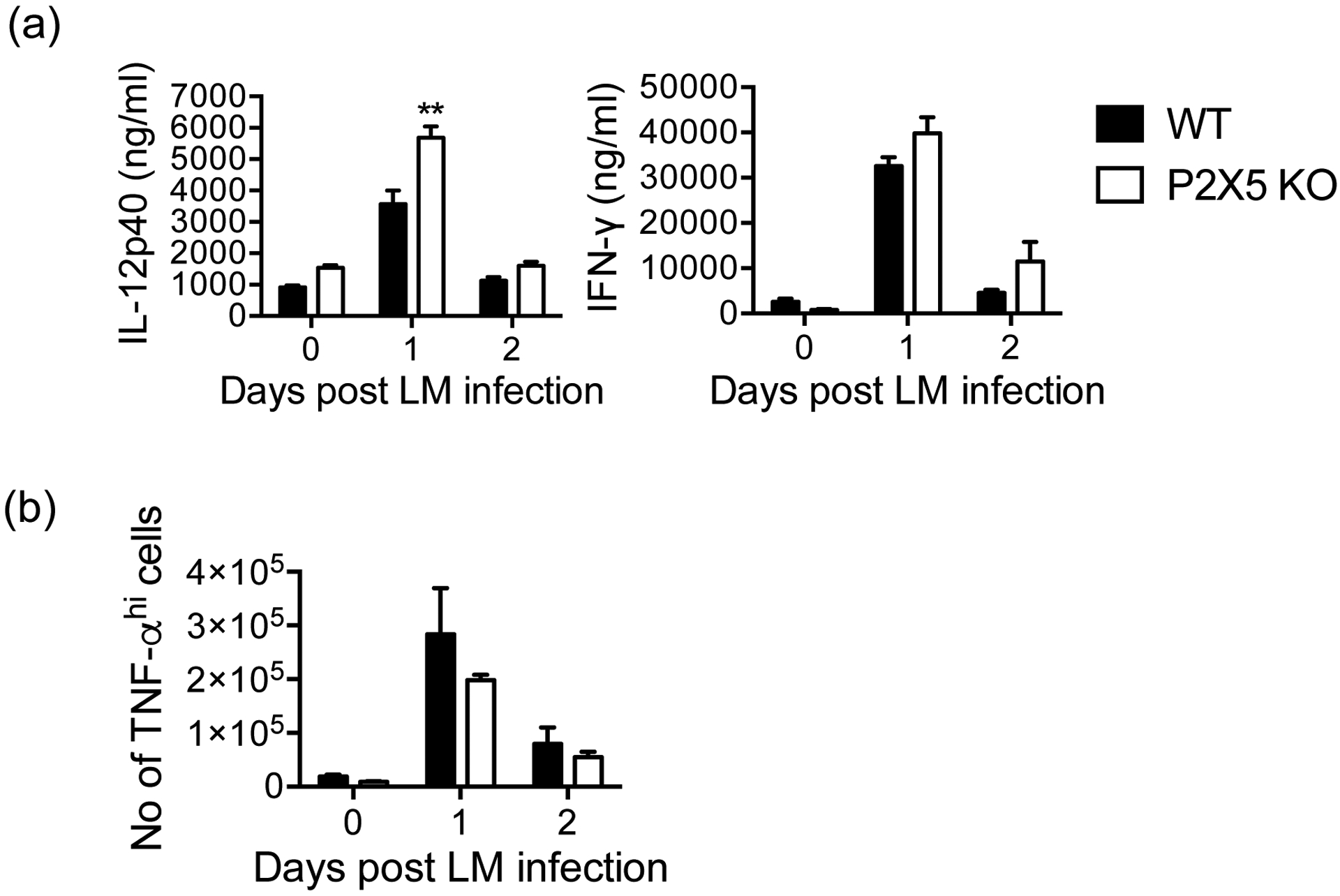
(a) IL-12p40 and IFN-γ were measured from collected serum at days 0, 1 and 2 post-Lm infection by ELISA. (b) Splenocytes from mice 0, 1 and 2 days post-Lm infection were cultured with or without HKLM stimulation for 4 hr in the presence of BFA and stained for intracellular TNF-α with surface staining for CD4, CD8a, and CD11b. Cell numbers are gated from non-CD4, CD8 and CD11bint TNF-αhi lymphocytes/monocytes. Data are representative of at least two independent experiments. (**p < 0.01)
Lm-induced monocyte migration in P2X5-deficient mice
The importance of monocyte recruitment in immune defense against Lm infection is well established (25), as shown by experiments that ablate signaling through CC-chemokine receptor (CCR2) (26, 27). Therefore, we investigated inflammatory monocyte recruitment during Lm infection and detected slightly higher peak serum levels of the CCR2 ligand CCL2/MCP-1 in P2X5−/− mice (Fig. 3a). We examined CCR2-mediated recruitment directly, but found no differences in the numbers of WT versus P2X5−/− sorted Ly6ChiCD11b+ monocytes that migrated in vitro in response to provision of recombinant CCL2 (Fig. 3b). Finally, we infected WT and P2X5−/− mice with Lm and examined recruitment of Ly6ChiCD11b+ inflammatory monocytes as part of the initial infiltrate from bone marrow to splenic white pulp. However, there were no detectable differences in monocyte migration into the tissues between WT and P2X5−/− mice (Fig. 3c), suggesting that P2X5 does not regulate resistance to Lm via monocyte migration.
Figure 3. Lm-induced monocyte migration in P2X5-deficient mice.
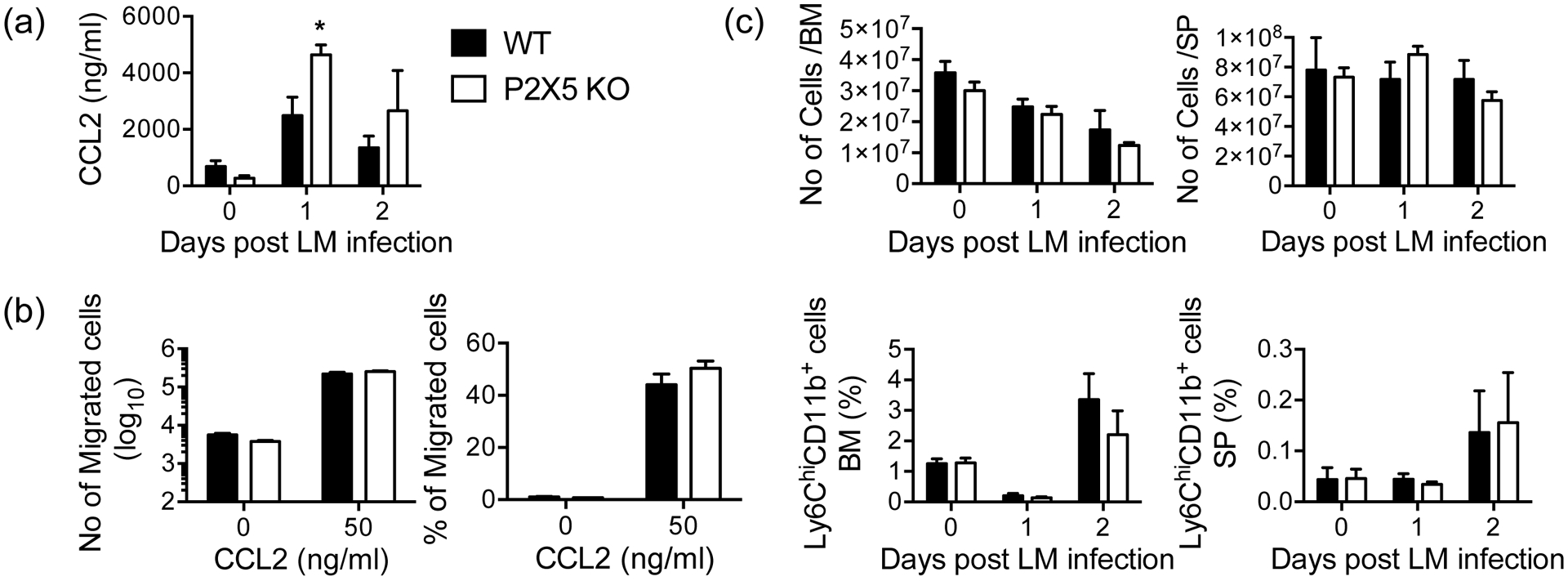
(a) Serum CCL2 was measured by ELISA. (b) Sorted Ly6Chi CD11b+ monocytes were added to top chambers of 5um transwells and the numbers of cells that migrated in response to 50 ng/ml of CCL2 to the lower chamber were enumerated. (c) Absolute numbers of bone marrow and spleen cells (upper), frequencies of Ly6Chi CD11b+ monocytes in the bone marrow and spleen (bottom). Data are representative of two independent experiments (n = 5 mice per group). (*p < 0.05)
Defective cytosolic Lm killing in P2X5-deficient macrophages
Roles for P2X receptors in macrophage function during pathophysiological conditions have previously been reported. P2X7 deficiency results in defective C. trachomatis (28) killing in macrophages, and P2X4 deficiency causes higher susceptibility in response to E. coli that is associated with uncontrolled bacterial killing in macrophages in vitro (29). The role of P2X receptors may depend on the type of bacterium and cell type studied. To specifically evaluate the relationship between Lm killing capacity by macrophages and P2X5 expression, we differentiated bone marrow-derived macrophages (BMMs) from WT and P2X5−/− mice and then performed in vitro infection with Lm, followed by quantification of remaining live bacteria that were taken up by BMMs. Killing capacity was found to be significantly reduced in P2X5−/− versus WT BMMs (Fig. 4). Decreased killing was not due to differences in either initial phagocytosis or intracellular growth, as similar numbers of colonies were counted within WT and P2X5−/− BMMs after 30 min incubation and growth curves for bacteria within WT and P2X5−/− BMMs remained similar throughout the entire course of infection (Supple FigS1). Further, levels of cell death exhibited by WT and P2X5−/− BMMs during Lm killing assays were similar (data not shown). To attempt to identify specific mechanisms of P2X5-dependent Lm killing, we also tested killing capacity using several Lm mutant strains, including LLO and actA mutants, and found that while the actA mutant resulted in a similar killing defect as WT Lm, LLO mutant killing was similarly reduced in both WT and P2X5−/− BMMs (Fig. 4). Upon entry of into the BMMs, Lm is initially contained within a phagocytic vacuole and listeriolysin O (LLO) is necessary for allowing Lm to escape into the cytosol (30). Once in the cytosol, Lm become motile by using actin tails generated by ActA, which allows Lm to spread to neighboring cells (31). Therefore, these mutants allow us to infer that the killing defect in the vacuole is P2X5-independent, while cytosolic killing is P2X5-dependent. In addition, since LLO is believed to be the primary Lm-associated molecule that triggers the inflammasome (32, 33), similarly defective LLO mutant killing in both WT and P2X5−/− BMMs suggests that P2X5 may be required for inflammasome-associated killing of Lm.
Figure 4. Defective cytosolic Lm killing in P2X5-deficient macrophages.
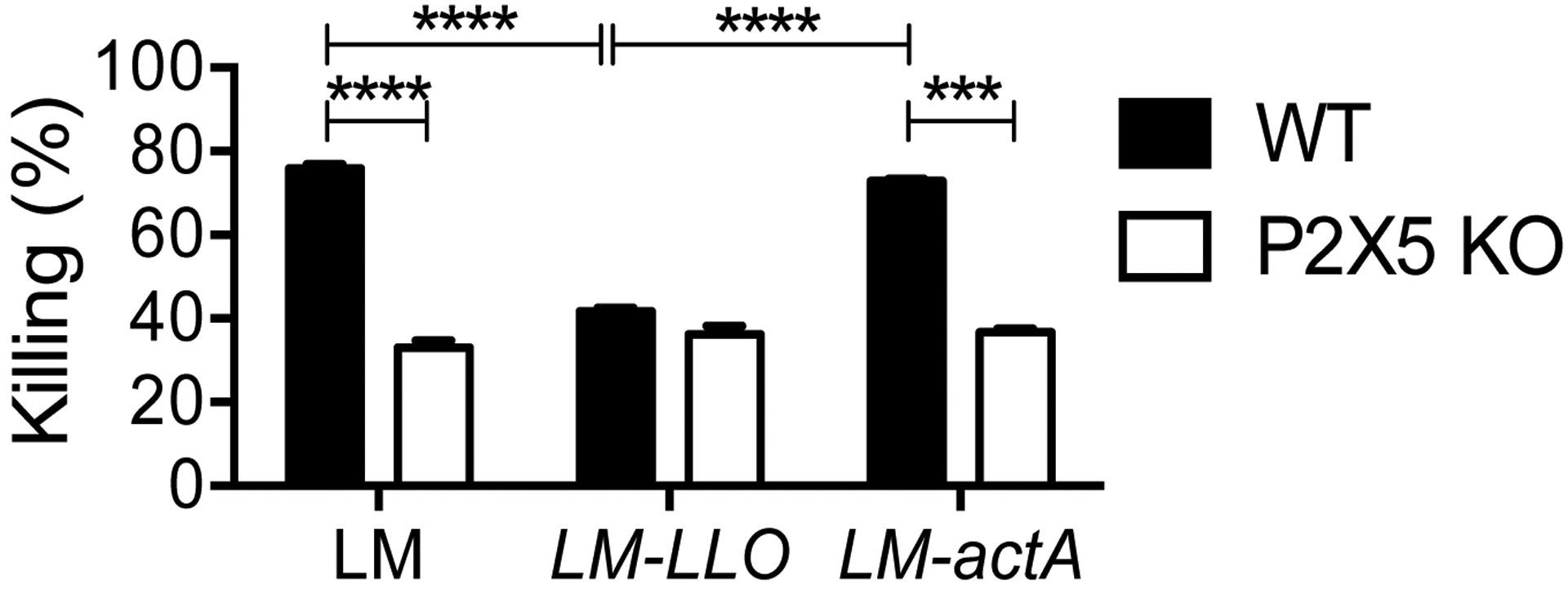
WT and P2X5−/− (BMMs) were infected in vitro with Lm, LLO and actA strains at MOI 10 for 30 min or 4 hr. BMM cell lysates were prepared and plated on BHI agar plates overnight and resulting Lm colonies were counted the next day. The percent of killing was calculated by comparing initial uptake after 30 min and remaining burden after 4 hr. (***p < 0.001 and ****p < 0.0001)
Defective inflammasome activation results in reduced Lm killing
Because the P2X5-dependent defect in Lm killing implicated the inflammasome, we next sought to determine whether P2X5 is required for Lm-induced inflammasome activation. In vitro infection of WT BMMs with Lm resulted in caspase-1 activation, as represented by cleavage of pro-caspase-1 to caspase-1p20, but P2X5−/− BMMs exhibited significant decreases in both caspase-1 activation and maturation of pro-IL-1β to the mature IL-1βp17 form (Fig. 5a). These defects were correlated with IL-1β protein levels measured in culture supernatants by ELISA (Fig. 5b), as well as with another inflammasome-dependent cytokine, IL-18, which also exhibited lower production in P2X5−/− BMMs (Fig. 5b). These results strongly suggest that P2X5 is required for Lm-triggered inflammasome activation in BMMs. To investigate whether P2X5-dependent Lm-triggered inflammasome activation is related to defective Lm killing in P2X5−/− BMMs, we treated WT and P2X5−/− BMMs with the K+ ionophore nigericin, a direct chemical activator of the inflammasome (34), in the context of an in vitro Lm killing assay. Nigericin-mediated rescue of Lm killing in P2X5−/− BMMs suggests both that inflammasome activation downstream of P2X5 remains intact, and that P2X5−/− BMMs capable of normal Lm killing if P2X5-dependent activation of the inflammasome is chemically bypassed (Fig. 5c). Finally, we showed that Lm killing by P2X5−/− BMMs was partially rescued by supplementing cultures with the inflammasome-dependent cytokines IL-1β or IL-18 for 1 hr prior to Lm infection (Fig. 5d). Together, these data suggest that BMM-expressed P2X5 is necessary for optimal inflammasome activation and production of the inflammasome-dependent cytokines IL-1β and IL-18, and that P2X5-dependent Lm killing requires inflammasome activation and production of IL-1β or IL-18.
Figure 5. Defective inflammasome activation results in reduced Lm killing.
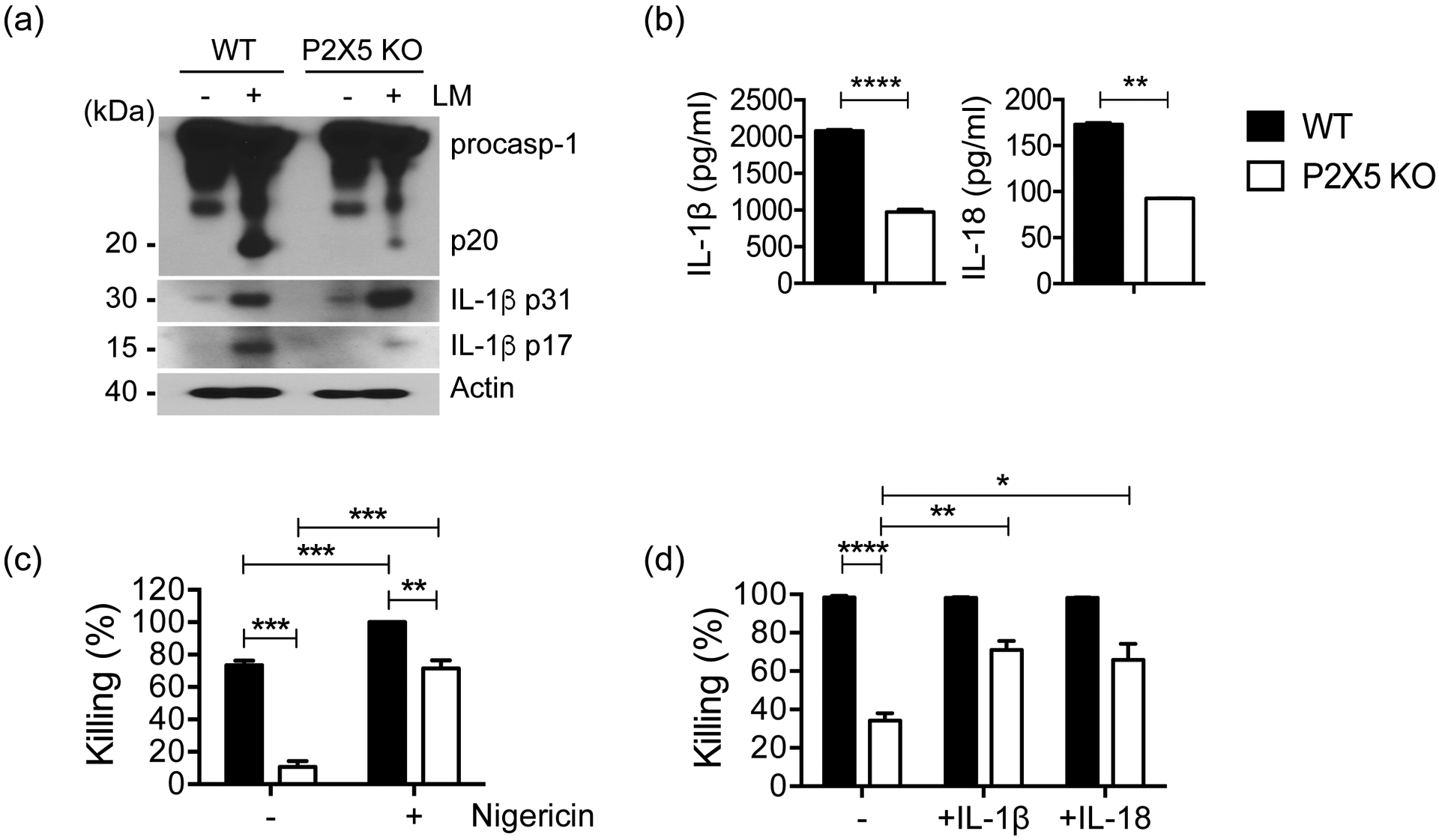
(a) The activation of caspase-1 and IL-1β processing in infected WT or P2X5−/− BMMs was analyzed using immunoblotting with anti-caspase-1 or anti-IL-1β Ab. (b) IL-1β and IL-18 secretion from infected BMMs into culture supernatants at 6 hr were measured by ELISA. (c) Killing assay with nigericin treatment of WT and P2X5−/− BMMs. (d) Killing assays with exogenous IL-1β or IL-18 to WT and P2X5−/− BMM cultures. Data are representative of at least two independent experiments. (*p < 0.05, **p < 0.01, ***p <0.001 and ****p < 0.0001).
P2X5-dependent anti-Lm immunity is independent of extracellular ATP-mediated signaling
P2X7, which is highly expressed on various immune cells, including BMMs, is the P2X family member most associated with immune regulation, and has been implicated in responses to bacterial infections (35, 36). It is therefore reasonable to investigate whether P2X5-dependent anti-Lm immunity is linked to P2X7 function. To examine the role of P2X7 in regulating host response to Lm, and how P2X7 expression might affect P2X5-dependent anti-Lm immunity, we infected WT, P2X7−/−, and P2X5−/−P2X7−/− mice by intravenous injection. While P2X5−/− and P2X5−/−P2X7−/− mice exhibited significant mortality (between days 3 and 5), all WT and P2X7−/− mice survived (Fig. 6a), suggesting that P2X7 is dispensable for early systemic immunity to Lm. Investigating in vitro killing of Lm, we found that unlike P2X5−/− BMMs, Lm killing capacity by P2X7−/− BMMs was normal (Fig. 6b). These results led us to investigate whether control of Lm requires the presence of the established P2X/P2X7 purinergic receptor agonist, extracellular ATP. To do so, we treated WT and P2X5−/− mice before and after Lm infection with the ATP-hydrolyzing agent apyrase and found that apyrase treatment caused no increase in mortality of WT mice (Fig. 6c). More surprisingly, apyrase treatment resulted in complete rescue of Lm-infected P2X5−/− mice (Fig. 6c), suggesting that ATP-mediated signaling is not required for early anti-Lm immunity, but even possibly that apyrase-induced production of ATP metabolites can compensate for loss of P2X5-dependent anti-Lm function. To investigate the role of extracellular ATP and ATP metabolites on Lm killing by BMMs, we performed in vitro killing assays using WT and P2X5−/− BMMs in the presence of apyrase, extracellular ATP and ATP metabolites, and found, consistent with our in vivo infection findings, that apyrase rescues defective Lm killing by P2X5−/− BMMs (Fig. 6d). By contrast, treatment with a non-hydrolyzable form of ATP (ATPγS) had no effect on either WT or P2X5−/− killing, or any significant effect on apyrase-mediated rescue (Fig. 6d). Consistent with our speculation, however, addition of ATP metabolites ADPβS or adenosine combined with deaminase (ADA) did induce partial rescue of defective P2X5−/− killing (Fig. 6d). Together, these results suggest that P2X5 does not utilize conventional ATP-triggered signaling to effect anti-Lm immunity.
Figure 6. P2X5-dependent anti-Lm immunity is independent of extracellular ATP-mediated signaling.
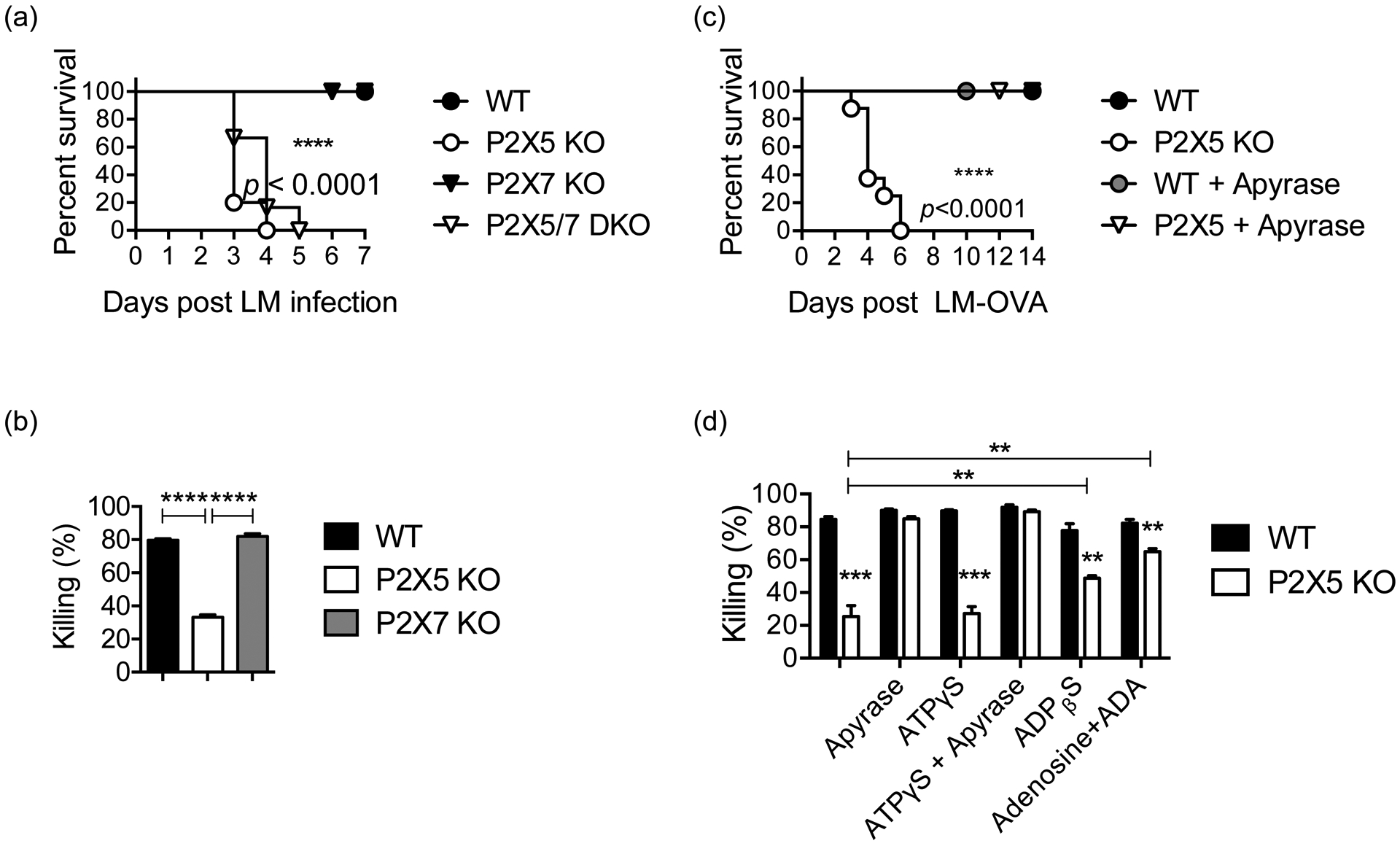
(a) WT, P2X5−/−, P2X7−/− and P2X5/7 DKO mice were infected intravenously with 105 CFU of Lm and monitored for 7 days (n = 5–6 mice per group) (b) Killing assays employing WT, P2X5−/− and P2X7−/− BMMs were performed with Lm infection at MOI 10 for 30 min or 4 hr. (c) WT and P2X5−/− were treated with 20 U apyrase intraperitoneally the day before infection, 20 min after infection, and 6hr after infection, and then monitored for 14 days (n = 5–6 mice per group). (d) Killing assays employing WT and P2X5−/− BMMs treated with either medium only, apyrase, ATPγS, apyrase combined with ATPγS, ADPβS, or adenosine combined with ADA were performed with Lm infection at MOI 10 for 30 min or 4 hr. Data are representative of at least two independent experiments. (**p < 0.01, ***p < 0.001 and ****p < 0.0001).
Discussion
In our previous studies, we demonstrated a relationship between P2X5 and inflammation by showing the regulation of inflammasome activity in inflammatory bone loss (20, 37). In this study, we sought to identify the activation pathway in Lm-induced inflammatory conditions. We clearly showed the role of P2X5 in controlling Lm infection in vivo and elucidated a mechanism for control of intracellular Lm bacteria in BMMs in vitro, whereby P2X5 specifically increases Lm killing but not phagocytosis. In addition, we provide evidence that BMM-expressed P2X5 functions to activate caspase-1 and IL-1β and IL-18 maturation in response to Lm in association with optimal bacterial clearance. Of note, the levels of IL-1β and IL-18 in the serum of Lm-infected P2X5−/− mice was found not to be reduced compared to infected control mice (Supple FigS2). Therefore, consistent with previous reports that neither IL-1β nor IL-18 are required for early resistance to Lm (38, 39), our findings showing that rescue of defective Lm killing by P2X5−/− BMMs via addition of exogenous IL-1β or IL-18 may be indicative of a local rather than systemic defect. It is also possible that increased systemic IL-18 production in Lm-infected P2X5−/− mice might be due to inflammatory factors released due to uncontrolled Lm infection.
Caspase-1 activation is controlled by the inflammasome, a multiprotein complex formed by NLR proteins and the adaptor ASC (41). NLRP3, a critical inflammasome component, is activated by microbial stimuli such as Lm. One of the favored models of NLRP3 inflammasome activation is that P2X7-dependent pore formation by pannexin-1 allows extracellular NLRP3 agonists to enter the cytosol and directly engage NLRP3 (42). However, it has also been reported that P2X7 receptor is differentially required for caspase-1 activation induced by intracellular and extracellular bacteria (43). Activation of inflammasomes by intracellular bacteria, including Lm, proceeds normally in the absence of P2X7 receptor-mediated cytoplasmic K+ perturbations. Consistent with this model, we found that unlike P2X5−/− mice, P2X7−/− mice are resistant to Lm infection, and further that P2X7−/− BMMs exhibit normal Lm killing. Because P2X7 is highly expressed on BMMs, as well as other immune cells, the fact that it is dispensable in the same context where P2X5 is required suggests distinct functional activity that is unique to each receptor. Interestingly, we found that early immunity to Lm infection and BMM killing of Lm did not require the presence of the established P2X/P2X7 agonist extracellular ATP. While ATP metabolites may be capable of overcoming P2X5-associated defects, it remains unclear what Lm-associated factors are necessary for triggering P2X5-dependent immunity. Regardless, our findings suggest that P2X5 is required to induce Lm-triggered inflammasome activation in BMMs and is the first example of an upstream activating receptor of caspase-1 that is specifically required for anti-Lm immunity.
Numerous factors identified as being critically important to early anti-Lm immunity, including MyD88 (7, 8), caspase-1 (6), and IFNγR (21, 22), are associated with production and/or function of IFN-γ. In addition, monocyte recruitment, which is linked to CCR2 expression is a critical function in early anti-Lm immunity (25). However, while P2X5−/− mice exhibit severe susceptibility to Lm, they produce normal levels of IFN-γ and exhibit normal monocyte recruitment. These observations are notable in comparison to the phenotype of caspase-1−/− mice, which show somewhat similar susceptibility to Lm as P2X5−/− mice, but which also exhibit a severe defect in serum IFN-γ production (6). These observations suggest first that there may be other Lm-triggered receptors that activate caspase-1 in vivo, possibly expressed on cells other than macrophages, that are responsible for other functions (e.g., IFN-γ and IL-18 production) in the context of Lm infection. Second, given that P2X5−/− mice show early susceptibility to Lm, even though they exhibit apparently normal IFN-γ production and monocyte recruitment, suggests that IFN-γ production and monocyte recruitment together are not sufficient for early resistance to Lm, and that P2X5-dependent responses may highlight a novel requisite component of early anti-Lm immunity. Future studies focused on cell-specific expression of P2X5 will be important for better understanding how this little characterized purinergic receptor plays a critical role in anti-Lm immunity. Finally, we believe the findings we have presented here are important for elucidating not only how P2X receptors regulate immune responses, but also how anti-bacterial immunity relies on various specialized mechanisms of purinergic signaling under different pathophysiologic conditions.
Supplementary Material
Key Points.
P2X5 is required for early protective immunity to Listeria monocytogenes (Lm)
Macrophages use P2X5 for Lm-induced inflammasome activation and Lm killing
P2X5-dependent anti-Lm functions are independent of both P2X7 and extracellular ATP
Acknowledgements
We thank Dr Sunny Shin, Dr Hyunsoo Kim and Dr Noriko Takegahara at University of Pennsylvania for helpful discussion and critical reading of the manuscript.
This work was supported in part by NIH grant AI125284 (to Y.C.).
Abbreviations used in this article:
- BMMs
bone marrow-derived macrophages
- Lm
Listeria monocytogenes
- WT
wild-type
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Gellin BG, and Broome CV. 1989. Listeriosis. JAMA 261: 1313–1320. [PubMed] [Google Scholar]
- 2.Pamer EG 2004. Immune responses to Listeria monocytogenes. Nat Rev Immunol 4: 812–823. [DOI] [PubMed] [Google Scholar]
- 3.Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, Bertin J, Coyle A, Grant EP, Akira S, and Nunez G. 2006. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440: 233–236. [DOI] [PubMed] [Google Scholar]
- 4.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, and Dixit VM. 2006. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440: 228–232. [DOI] [PubMed] [Google Scholar]
- 5.Martinon F, Petrilli V, Mayor A, Tardivel A, and Tschopp J. 2006. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440: 237–241. [DOI] [PubMed] [Google Scholar]
- 6.Tsuji NM, Tsutsui H, Seki E, Kuida K, Okamura H, Nakanishi K, and Flavell RA. 2004. Roles of caspase-1 in Listeria infection in mice. Int Immunol 16: 335–343. [DOI] [PubMed] [Google Scholar]
- 7.Edelson BT, and Unanue ER. 2002. MyD88-dependent but Toll-like receptor 2-independent innate immunity to Listeria: no role for either in macrophage listericidal activity. J Immunol 169: 3869–3875. [DOI] [PubMed] [Google Scholar]
- 8.Seki E, Tsutsui H, Tsuji NM, Hayashi N, Adachi K, Nakano H, Futatsugi-Yumikura S, Takeuchi O, Hoshino K, Akira S, Fujimoto J, and Nakanishi K. 2002. Critical roles of myeloid differentiation factor 88-dependent proinflammatory cytokine release in early phase clearance of Listeria monocytogenes in mice. J Immunol 169: 3863–3868. [DOI] [PubMed] [Google Scholar]
- 9.Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M and Dixit VM. 2011. Non-canonical inflammasome activation targets caspase-11. Nature 479: 117–121. [DOI] [PubMed] [Google Scholar]
- 10.Sauer JD, Pereyre S, Archer KA, Burke TP, Hanson B, Lauer P, and Portnoy DA. 2011. Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proc Natl Acad Sci U S A 108: 12419–12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sauer JD, Witte CE, Zemansky J, Hanson B, Lauer P, and Portnoy DA. 2010. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 7: 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hara H, Seregin SS, Yang D, Fukase K, Chamaillard M, Alnemri ES, Inohara N, Chen GY, and Nunez G. 2018. The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell 175: 1651–1664 e1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.North RA 2002. Molecular physiology of P2X receptors. Physiol Rev 82: 1013–1067. [DOI] [PubMed] [Google Scholar]
- 14.Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, and Falzoni S. 2017. The P2X7 Receptor in Infection and Inflammation. Immunity 47: 15–31. [DOI] [PubMed] [Google Scholar]
- 15.Adinolfi E, Giuliani AL, De Marchi E, Pegoraro A, Orioli E, and Di Virgilio F. 2018. The P2X7 receptor: A main player in inflammation. Biochem Pharmacol 151: 234–244. [DOI] [PubMed] [Google Scholar]
- 16.Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, Panther E, and Di Virgilio F. 2006. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol 176: 3877–3883. [DOI] [PubMed] [Google Scholar]
- 17.Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, and Rubartelli A. 2008. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A 105: 8067–8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Norde WJ, Overes IM, Maas F, Fredrix H, Vos JC, Kester MG, van der Voort R, Jedema I, Falkenburg JH, Schattenberg AV, de Witte TM, and Dolstra H. 2009. Myeloid leukemic progenitor cells can be specifically targeted by minor histocompatibility antigen LRH-1-reactive cytotoxic T cells. Blood 113: 2312–2323. [DOI] [PubMed] [Google Scholar]
- 19.Abramowski P, Ogrodowczyk C, Martin R, and Pongs O. 2014. A truncation variant of the cation channel P2RX5 is upregulated during T cell activation. PLoS One 9: e104692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim H, Walsh MC, Takegahara N, Middleton SA, Shin HI, Kim J, and Choi Y. 2017. The purinergic receptor P2X5 regulates inflammasome activity and hyper-multinucleation of murine osteoclasts. Sci Rep 7: 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tripp CS, Wolf SF, and Unanue ER. 1993. Interleukin 12 and tumor necrosis factor alpha are costimulators of interferon gamma production by natural killer cells in severe combined immunodeficiency mice with listeriosis, and interleukin 10 is a physiologic antagonist. Proc Natl Acad Sci U S A 90: 3725–3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harty JT, and Bevan MJ. 1995. Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity 3: 109–117. [DOI] [PubMed] [Google Scholar]
- 23.Pfeffer K, Matsuyama T, Kundig TM, Wakeham A, Kishihara K, Shahinian A, Wiegmann K, Ohashi PS, Kronke M, and Mak TW. 1993. Mice deficient for the 55 kd tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell 73: 457–467. [DOI] [PubMed] [Google Scholar]
- 24.Serbina NV, Salazar-Mather TP, Biron CA, Kuziel WA, and Pamer EG. 2003. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity 19: 59–70. [DOI] [PubMed] [Google Scholar]
- 25.Serbina NV, and Pamer EG. 2006. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 7: 311–317. [DOI] [PubMed] [Google Scholar]
- 26.Rosen H, Gordon S, and North RJ. 1989. Exacerbation of murine listeriosis by a monoclonal antibody specific for the type 3 complement receptor of myelomonocytic cells. Absence of monocytes at infective foci allows Listeria to multiply in nonphagocytic cells. J Exp Med 170: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurihara T, Warr G, Loy J, and Bravo R. 1997. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J Exp Med 186: 1757–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coutinho-Silva R, Stahl L, Raymond MN, Jungas T, Verbeke P, Burnstock G, Darville T, and Ojcius DM. 2003. Inhibition of chlamydial infectious activity due to P2X7R-dependent phospholipase D activation. Immunity 19: 403–412. [DOI] [PubMed] [Google Scholar]
- 29.Csoka B, Nemeth ZH, Szabo I, Davies DL, Varga ZV, Paloczi J, Falzoni S, Di Virgilio F, Muramatsu R, Yamashita T, Pacher P, and Hasko G. 2018. Macrophage P2X4 receptors augment bacterial killing and protect against sepsis. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamon MA, Ribet D, Stavru F, and Cossart P. 2012. Listeriolysin O: the Swiss army knife of Listeria. Trends Microbiol 20: 360–368. [DOI] [PubMed] [Google Scholar]
- 31.Kocks C, Gouin E, Tabouret M, Berche P, Ohayon H, and Cossart P. 1992. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 68: 521–531. [DOI] [PubMed] [Google Scholar]
- 32.Meixenberger K, Pache F, Eitel J, Schmeck B, Hippenstiel S, Slevogt H, N’Guessan P, Witzenrath M, Netea MG, Chakraborty T, Suttorp N, and Opitz B. 2010. Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J Immunol 184: 922–930. [DOI] [PubMed] [Google Scholar]
- 33.Hara H, Tsuchiya K, Nomura T, Kawamura I, Shoma S, and Mitsuyama M. 2008. Dependency of caspase-1 activation induced in macrophages by Listeria monocytogenes on cytolysin, listeriolysin O, after evasion from phagosome into the cytoplasm. J Immunol 180: 7859–7868. [DOI] [PubMed] [Google Scholar]
- 34.Katsnelson MA, Rucker LG, Russo HM, and Dubyak GR. 2015. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J Immunol 194: 3937–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coutinho-Silva R,JL Rerfecttini PM Persechini ADautry-Varsat and DM Ojcius. 2001. Modulation of P2Z/P2X(7) receptor activity in macrophages infected with Chlamydia psittaci. Am J Physiol Cell Physiol 280(1): C81–C89. [DOI] [PubMed] [Google Scholar]
- 36.Kusner DJ. and Adams J. 2000. ATP-induced killing of virulent Mycobacterium tuberculosis with human macrophages requires phospholipase D. J Immunol 164(1): 379–388. [DOI] [PubMed] [Google Scholar]
- 37.Csoka B, Nemeth ZH, Toro G, Idzko M, Zech A, Koscso B, Spolarics Z, Antonioli L, Cseri K, Erdelyi K, Pacher P and Hasko G. 2019. Extracellular ATP protects against sepsis through macrophage P2X7 purinergic receptors by enhancing intracellular bacterial killing. The FASEB J 29: 3626–3637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng H, Fletcher D, Kozak W, Jiang M, Hofmann K, Conn C, Soszynski D, Grabiec C, Trumbauer M, Shaw A, Kostura M, Stevens K, Rosen H, North R, Che H, Tocci M, Kluger M and Can Der Plog L. 1995. Resistence to fever induction and impaired acute-phase response in interleukine-1β-deficient mice. Immunity 3:9–19. [DOI] [PubMed] [Google Scholar]
- 39.Lochner M, Kastenmuller K, Neuenhahn M, Weighardt H, Busch DH, Reindl W and Forster I. 2008. Decreased susceptibility of mice to infection with Listeria monocytogenes in the absence of interleukin-18. Infect. Immun. 76(9): 3881–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim H, Kajikawa T, Walsh MC, Takegahara N, Jeong YH, Hajishengallis G, and Choi Y. 2018. The purinergic receptor P2X5 contributes to bone loss in experimental periodontitis. BMB Rep 51: 468–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Franchi L, Eigenbrod T, Munoz-Planillo R, and Nunez G. 2009. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10: 241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanneganti TD, Lamkanfi M, Kim YG, Chen G, Park JH, Franchi L, Vandenabeele P, and Nunez G. 2007. Pannexin-1-mediated recognition of bacterial molecules activates the cryopyrin inflammasome independent of Toll-like receptor signaling. Immunity 26: 433–443. [DOI] [PubMed] [Google Scholar]
- 43.Franchi L, Kanneganti TD, Dubyak GR, and Nunez G. 2007. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem 282: 18810–18818. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.