

Abstract
Purpose
The use of chemotherapeutic agents to combat cancer is accompanied by high toxicity due to their inability to discriminate between cancer and normal cells. Therefore, cancer therapy research has focused on the targeted delivery of drugs to cancer cells. Here, we report an in vitro study of folate-poly(ethylene glycol)-poly(propylene succinate) nanoparticles (FA-PPSu-PEG-NPs) as a vehicle for targeted delivery of the anticancer drug paclitaxel in breast and cervical cancer cell lines.
Methods
Paclitaxel-loaded-FA-PPSu-PEG-NPs characterization was performed by in vitro drug release studies and cytotoxicity assays. The NPs cellular uptake and internalization mechanism were monitored by live-cell imaging in different cancer cell lines. Expression of folate receptor-α (FOLR1) was examined in these cell lines, and specific FOLR1-mediated entry of the FA-PPSu-PEG-NPs was investigated by free folic acid competition. Using inhibitors for other endocytic pathways, alternative, non-FOLR1 dependent routes for NPs uptake were also examined.
Results
Drug release experiments of Paclitaxel-loaded PPSu-PEG-NPs indicated a prolonged release of Paclitaxel over several days. Cytotoxicity of Paclitaxel-loaded PPSu-PEG-NPs was similar to free drug, as monitored in cancer cell lines. Live imaging of cells treated with either free Paclitaxel or Paclitaxel-loaded PPSu-PEG-NPs demonstrated tubulin-specific cell cycle arrest, with similar kinetics. Folate-conjugated NPs (FA-PPSu-PEG-NPs) targeted the FOLR1 receptor, as shown by free folic acid competition of the FA-PPSu-PEG-NPs cellular uptake in some of the cell lines tested. However, due to the differential expression of FOLR1 in the cancer cell lines, as well as the intrinsic differences between the different endocytic pathways utilized by different cell types, other mechanisms of nanoparticle cellular entry were also used, revealing that dynamin-dependent endocytosis and macropinocytosis pathways mediate, at least partially, cellular entry of the FA-PPSu-PEG NPs.
Conclusion
Our data provide evidence that Paclitaxel-loaded-FA-PPSu-PEG-NPs can be used for targeted delivery of the drug, FA-PPSu-PEG-NPs can be used as vehicles for other anticancer drugs and their cellular uptake is mediated through a combination of FOLR1 receptor-specific endocytosis, and macropinocytosis. The exploration of the different cellular uptake mechanisms could improve treatment efficacy or allow a decrease in dosage of anticancer drugs.
Keywords: Folate-PPSu-PEG copolymers, nanoparticles, drug delivery, Paclitaxel, targeted chemotherapy, breast cancer
Introduction
Breast Cancer is an eminent concern of public health, as it is the most common cancer in women worldwide, and the second most common cancer overall. It is the fifth most common cause of cancer-related deaths in women, and although less common in men, it may have a poorer outcome due to delayed diagnosis.1 The large majority of cases occur in less developed regions (statistics from 2018).2
The first-line therapy of solid tumors is based on surgery with supporting radiotherapy and/or chemotherapy. For metastasized tumors, chemotherapy is among the very few treatment options available. Paclitaxel, the most widely used anticancer drug, is mainly employed as second-line therapy in different types of cancer, including breast and cervical cancer.3,5 Paclitaxel targets microtubules and has been claimed as the most significant advance in chemotherapy during the last 30 years by National Cancer Institute (NCI).4,6 However, like all chemotherapy regimens, Paclitaxel treatment is accompanied by side effects, as it also affects normal cells.
Therefore, a main focus in cancer therapy in recent years has been the targeted delivery of the anti-tumor drugs to cancer cells, avoiding any harm to the normal cells. While systemic drug delivery depends mostly on drug characteristics and properties, most tumors act in their favor hindering effective drug delivery, through many mechanisms including increased cellular turnover, tissue density and drug resistance via active drug export.7,8
Nanosystems are under intense investigation as drug delivery systems, with the aim of achieving longer circulation, specific targeting, increased drug delivery, faster delivery to the tumor site, enhanced intracellular penetration and subsequently enhanced drug effect. In addition, nanocarriers aim to prevent the degradation of the drug, as well as to control the degree/rate of drug release, thereby allowing potential lower concentration and dosage frequency of the anticancer drug.7,8 Several nanocarriers have been used as drug delivery vehicles in the past decades, including liposomes, nanospheres, non-toxic biodegradable polymers, solid lipid nanoparticles NPs and inorganic-organic nanomaterials.9,13 The selective uptake of NPs by tumor cells can be enhanced by the addition of specific molecules on the nanoparticle surface (targeting moieties), recognized mainly by cancer cells.14
Recently, major progress has been made towards the development of functionalized nanocarriers with improved features, such as biocompatibility, biodegradability, and intracellular penetration. These nanocarriers may also be combined with ligands or small chemical compounds conferring directed cell targeting and stimuli responsiveness.8,9,15 Such nanocarriers include biocompatible and biodegradable polymers, with low degradation rates, such as poly(lactic acid) (PLA) and its copolymers with glycolic acid (PLGA), poly(e-caprolactone) (PCL) and poly(propylene succinate) (PPSu), a novel biocompatible polymer combining increased biodegradation and melting point close to the normal body temperature.16,19 These nanoparticles (NPs) demonstrate favorable pharmacokinetic and biodistribution profiles, for instance, pegylated PPSu NPs (PPSu-PEG-NPs) have decreased rate of removal from systemic circulation due to fast uptake by the reticuloendothelial system (RES),20,27 and show preferential accumulation in tumors due to the enhanced permeability and retention (EPR) effect. However, these NPs do not appear to show increased uptake by cancer cells.28
In this study, we examined the potential of PPSu-PEG-NPs as a new pharmaceutical form for Paclitaxel delivery. To this aim, we investigated the release profile and cytotoxicity of the drug-loaded biodegradable and biocompatible nanocarriers. As folic acid (FA) is reported to mediate tumor-specific recognition7,27,29 we also examined whether conjugation of FA to the PPSu-PEG-NPs (FA-PPSu-PEG-NPs) would specifically target the NPs to tumor cells. Moreover, fluorescent labeling of FA-PPSu-PEG-NPs with Rhodamine B allowed us to investigate FA-dependent and independent cellular internalization mechanism(s) in different cancer cell lines.
Materials and Methods
Materials
For the synthesis of conjugated polyester, succinic acid (99% pure), 1,3-propanediol (99% pure), tetrabutyl titanate (TBT), tripolyphenylphosphine (Ph3P), diethyl azodicarboxylate (DEAD), poly(ethylene glycol) (PEG) with molecular weight 2000, maleimide (MAL) and Sn(octanoate) (Sn(Oct)2) were purchased from Aldrich Chemical Co. Folic acid (FA), N-hydroxysuccinimide (NHS), triethylamine (TEA), dicyclohexylcarbodiimide (DCC) and Paclitaxel were also purchased from Aldrich Chemical Co. All other reagents were purchased from Analytical Grade.
Synthesis of Pegylated-Poly(Propylene Succinate) Copolymers Conjugated with Folic Acid
The copolymers were synthesized as described previously.9 Briefly, maleimide (MAL)-PEG was prepared, which reacted with PPSu to produce MAL-PPSu-PEG. A pre-determined amount of MAL–PEG–OH, PPSu and Sn(Oct)2 catalyst (0.1% amount of polyester in molar ratio) was dissolved in dry toluene in a flame-dried two neck flask equipped with a distillation set. The mixture was heated to 130°C and refluxed for 5 h. Subsequently, the solution was cooled to room temperature and precipitated into cold diethyl ether. The product (MAL–PEG–PPSu) was collected by filtration and dried in vacuum at room temperature.
In a second reaction, activated folic acid by N-hydroxysuccinimide (NHS) and dicyclohexylcarbodiimide (DCC) was used to prepare FA–PEG–PPSu copolymer. Briefly, 1g FA was dissolved in 50 mL dimethyl sulfoxide (DMSO). Then, NHS and DCC were added to the solution in a stoichiometric ratio of FA:NHS:DCC 1:2:1. The solution was stirred for 16 h at room temperature protected from light. Dicyclohexylurea, the by-product, was removed by filtration. The supernatant was added to glacial diethylether and the solid formed was filtrated and dried for 3 days at 30°C. The dried product was dissolved in DMSO and cysteamine- hydrochloride and 0.1 mL (TEA) were added. Subsequently, the solution was stirred for 24 h and the final product was recovered after pouring into diethylether. The modified-folic acid (FA-NHS) was washed thrice with purified water and dried at 30°C. In the second step, activated FA was reacted with MAL–PPSu-PEG. Briefly, 10 mg of FA-NHS were added to 20 mL of TEA and MAL-PPSu-PEG was dissolved in 10 mL of DMSO. The solutions were mixed and stirred for 24 h at room temperature. DMSO residue was removed via distillation in order to receive a yellow colored FA-PPSu-PEG polymer.
Synthesis of Pegylated-Poly(Propylene Succinate) Copolymers Conjugated with Rhodamine B Dye
The Rhodamine (Rho) conjugated PEG–PPSu was synthesized, as previously described.29 Briefly, activation of Rho was affected when 0.1 g of Rho was added into 10 mL DMSO and 0.06 mL TEA. After dissolving the dye, 51.6 mg of DCC and NHS were added. Magnetic stirring in the dark was continued for 24 h and the dicyclohexylurea produced was removed by filtration. DMSO and TEA were evaporated under vacuum and the activated Rho-NHS was dried under vacuum. The dried Rho-NHS was dissolved in 10 mL DMSO with 0.4 g of the PPSu-PEG and 0.02 mL TEA added in proportional amounts. The solution was stirred overnight and then dialyzed for 2 days. The final Rho-PPSu-PEG copolymer was collected by lyophilization.
Functional Materials Characterization by Fourier-Transform Infrared Spectroscopy
FTIR spectra of the samples were taken with a FTIR–spectrometer (model FTIR–2000, Perkin Elmer, Dresden, Germany) using KBr disks (thickness of 500 μm). The spectra were recorded from 4000 to 400 cm–1 at a resolution of 2 cm–1 (64 co–added scans).
Preparation of Paclitaxel-Loaded Polyester Nanoparticles
The synthesized copolymers were used for nanoencapsulation of Paclitaxel, as previously described7,30,31 (Supplementary Figure 1A). An oil-in-water (o/w) emulsification and solvent evaporation technique was used for Paclitaxel nanoencapsulation in polyester matrices. In brief, 50 mg of polymer and 5mg of Paclitaxel were dissolved in 2 mL of dichloromethane. The polymer-drug solution was added to 6 mL of 12 mM sodium cholate aqueous solution and the mixture was sonicated for 1 min. Sodium cholate was added to prevent drug particle aggregation during solvent evaporation. As a result, drug-loaded polymer is dispersed in the form of nanoparticles. The emulsion formed was gently stirred until the evaporation of the organic solvent was completed. The obtained nanoparticles were purified by centrifugation (9000 rpm for 15 min) and reconstituted in deionized water. Any large polymer aggregates were removed by filtering the suspension through a 1.2 μm pore size microfilter. Nanoparticles were collected by lyophilization. The NPs used in the study are referred to as PPSu-PEG, PPSu-PEG-Paclitaxel and FA-PPSu-PEG-Rho having a core-shell structure as shown in Supplementary Figure 1B.
Characterization of Drug-Loaded Nanoparticles
Nanoparticle Yield, Drug Loading and Entrapment Efficiency
Drug content of the nanoparticles was determined by HPLC analysis using a Shimadzu HPLC (model LC-20AD). Supplementary Figure 2 displays the HPLC profiles of four standard Paclitaxel solutions.
Nanoparticles (3 mg) were added to 50mL of water/acrylonitrile (ACN) (50/50 v/v) and stirred with a magnetic stirrer until complete dissolution. A clear solution was obtained, which was filtered through a hydrophilic 0.45μm PVDF filter (since Paclitaxel is hydrophobic) and assayed for drug content by HPLC. The column used was a Eclipse XDB-C18, 5μm, 250 x 4.6 mm. The flow rate was 1 mL/min and the column temperature was 25°C. A diode array detector was used at 227 nm, and quantification of the API (Active Pharmaceutical Ingredient) was based on a calibration curve created by diluting with mobile phase a stock solution of 20 μg/mL Paclitaxel in water/ACN (50/50 v/v) to concentrations 20, 10, 5, 2.5, 1 and 0.5 μg/mL (Supplementary Figure 3). Nanoparticle yield, drug loading and drug entrapment efficiency were calculated from Equations (1)–(3), respectively:

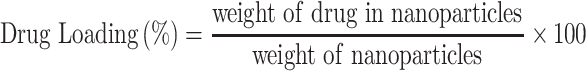 |
(2) |

Scanning Electron Microscopy (SEM)
Morphology of the prepared nanoparticles was examined with a Scanning Electron Microscope (JEOL, JMS–840). Samples were coated with carbon black to avoid charging under the electron beam. Operating conditions were: accelerating voltage 20 kV, probe current 45 nA, and counting time 60s.
Particle Size Distribution
Particle size distribution of the Paclitaxel/polyester nanoparticles was determined by Dynamic Light Scattering (DLS) using a Zetasizer Nano instrument (Malvern Instruments, Nano ZS, ZEN3600, UK) operating with a 532 nm laser. Nanoparticles were dispersed in distilled water at 1%w/v and kept under agitation at 37°C in a water bath. Particle size was measured at different time intervals after sample introduction into the dispersal medium. All measurements were performed in triplicate and the results were reported in terms of mean diameter ±SD.
In vitro Drug Release Studies
Lyophilized PPSu-PEG and PPSu-PEG-Paclitaxel NPs were suspended in PBS buffer (NaCl 137mM, KCl 2.7mM, Na2HPO4 10mM, K2HPO4 2mM) with pH 7.4 and handled as described above to ensure even distribution. Two rapid washes with PBS followed in order to remove any weakly bound drug remaining on the surface from the encapsulation reaction that could result in a burst effect.
The concentration of both nanoparticle types was calculated as polymer mass per volume (1.683 mg/mL) and the drug concentration was determined by the quantity of drug encapsulated in the polymer. Suspended NPs were incubated at 37ºC, with stirring at 100 rpm. At specific time intervals, the suspension was centrifuged (6000g for 10min) in order to separate the NPs (pellet) from the solution containing the released drug. A small sample volume (20μL) was removed from the supernatant of both PPSu-PEG-Paclitaxel and PPSu-PEG (used as control) at each time point and assayed in order to determine the amount of the released drug. The detection and quantification of the released drug was carried out using fluorescence spectroscopy (Infinite M1000, Tecan), with a suitable quartz cuvette, with the following parameters: excitation from 240nm, emissions scan from 280nm to 450nm, 150 flashes (400Hz) and 17nm bandwidth. The experiment was performed in triplicate.
Cell Lines and Cell Culture
Lyophilized NPs were suspended in PBS (as described in the relevant method). In order to avoid aggregate formation and uneven distribution of the NPs, suspension was followed by 30 min of sonication in a water bath (with intervals of ice cooling between every few sonication cycles) in order to enhance dispersion. Finally, NPs were diluted to the desired final concentration and added to the cell medium at 37°C. For each different type of synthesized NPs, the total mass of polymer was calculated and the final concentration used for each experiment was normalized to the concentration of the polymer (as mass of polymer per volume).
HeLa Kyoto, MCF7, MDA-MB-231 and T47D cells were obtained from ECACC. HeLa Kyoto stably expressing GFP-α-tubulin/mcherry-H2B cell line was a kind gift from Jan Ellenberg (EMBL, Heidelberg), has been authenticated by STR and has been used within 6 months of the time it was obtained. Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin (Invitrogen), at 37°C with 5% CO2 in a humidified incubator. For each experimental procedure the cell number was determined in duplicate using a haemocytometer.
RTCA Profiling
A real-time cell electronic sensing system, the xCELLigence Real-Time Cell Analysis (RTCA) system (ACEA Biosciences, Inc) was used for the dynamic measurement of Paclitaxel and PPSu-PEG-Paclitaxel NPs cytotoxicity. In principle, RTCA utilizes specialized microelectronic sensors, the 16-well E-Plates (Roche Applied Science, Indianapolis, IN), which are integrated with gold microelectrode arrays on a glass substrate in the bottom of the wells. Changes in cell status such as the cell number, the viability, the morphology, and the adherence are monitored and quantified by detecting sensor electrical impedance. Hence, electrode impedance is displayed as an arbitrary unit termed Cell Index (CI) and can be used to represent cell status. At the end of each experiment, the Cell Index curves can be normalized to the last time point before the addition of the compounds and they are expressed as normalized CI (NCI). Quantification via the RTCA Software allowed the plotting of sigmoidal dose–response curves and calculation of a half inhibitory concentration (IC50) 48 h after drug addition.
For our experiments, 2500 HeLa K or 10,000 MCF7 cells were added per well in the 16-well E-Plate. Cells were incubated for ~24h at 37°C, 5% CO2 at a final volume of 100 μL per well and the impedance of each well was monitored using the xCELLigence. Prior to addition, NPs were suspended in PBS (pH 7.4) followed by a wash step with PBS to remove any possible drug remaining on the surface from the encapsulation reaction, as described above. The concentration of the suspended NPs was calculated as mass per volume and the drug’s molarity was determined by the quantity of the encapsulated drug per mass contained in the final volume. Free Paclitaxel was diluted in DMSO (final DMSO concentration 0.1% v/v). After cell attachment, free Paclitaxel or PPSu-PEG-Paclitaxel were serially diluted and 50 μL were added to the culture at the relevant final concentrations. Cell attachment and proliferation were continuously monitored every 1 min for the first 3 h after drug addition, followed by 30 min intervals for 2 days in total. Assays were repeated at least in duplicate and wells with drug-free PPSu-PEG NPs, cell-free controls with the maximum concentration of NPs and cell-free controls with culture medium only were run in parallel. Untreated cells in normal culture medium (DMEM) and untreated cells in medium with 0.01% DMSO were also used as the appropriate controls.
Cell Proliferation Assay
For cell viability assays, the Premix WST-1 Cell Proliferation Assay System (Takara) was used. The kit enables measurement of cell proliferation and viability with a colorimetric assay, based on the cleavage of tetrazolium salts by mitochondrial dehydrogenase in viable cells. HeLa Kyoto, MCF7, MDA-MB-231 and T47D cells were seeded in a flat-bottomed 96-well plate at a density of 5000, 8500, 9000 and 10,000 viable cells per well, respectively, in 200 μL antibiotic-free DMEM (Invitrogen) and incubated overnight in a humidified atmosphere (37°C, 5% CO2) to allow cell attachment.
The following day, medium was discarded, and Paclitaxel or FA-PPSu-PEG-Paclitaxel serial dilutions in culture medium were added to cells in a total 200 μL volume per well, in triplicate. Paclitaxel is diluted in DMSO (Sigma) at 10 mM stock concentration, while the final working DMSO concentration is 0.01%. NPs were suspended in PBS buffer (pH 7.4), as described above, followed by a rapid wash with PBS. The concentration of the suspended NPs was calculated as mass per volume and drug molarity was determined by the quantity of the encapsulated drug per mass contained in the final volume. Untreated cells in normal culture medium (DMEM) and untreated cells in medium with 0.01% DMSO were used as the appropriate controls.
After 48 h of incubation with the NPs and/or the free drug, the medium was removed, cells were washed twice with 1xPBS, and 100 μL of DMEM w/o antibiotics containing 10 μL of Premix WST-1 reagent were added to each well and on an empty well (blank). Cells and WST-1 reagent were incubated in a humidified atmosphere (37°C, 5% CO2) for 1h. The absorbance of formazan was measured at 440 nm and at 660 nm as a reference wavelength with a plate reader (Infinite M1000, Tecan). Absorbance at 660 nm was subtracted from absorbance at 440 nm for each well. Absorbance of culture medium, in the absence of cells, is used as background control (blank) and untreated cells are considered as the control condition with 100% viability.
Interaction of Folate-Binding Protein with Folic Acid Conjugated NPs
Folate-Binding Protein (FBP), isolated from bovine milk was obtained from Sigma (F0504). 4 μg of FBP were mixed with either PPSu-PEG or FA-PPSu-PEG NPs (50 μg of polymer) in PBS solution, at pH 6.0 or pH 7.4 and mixture was incubated for 1 h at 25°C or 37°C. The mixture was then centrifuged (6000g for 10 min) to separate supernatant from pellet. The supernatant and the nanoparticle-containing pellet were loaded on a 12% SDS-PAGE gel in order to detect folate binding protein bound on the NPs or unbound, remaining in the supernatant. Quantification was performed using ImageJ 1.44n (National Institute of Health, USA) software.
Western Blot Analysis
Western blot analysis was performed as previously reported.32 Briefly, proteins were extracted from HeLa Kyoto, MCF7, MDA-MB-231, or T47D cells in RIPA buffer (150mM NaCl, 50mM Tris-HCl pH 8.0, 1% NP40 (v/v), supplemented with 10mM sodium orthovanadate, 10mM NaF and protease inhibitors). Protein concentrations were determined by the Bradford method with bovine serum albumin as standard and samples were analyzed in 12% SDS–PAGE according to standard procedures and transferred onto nitrocellulose membranes. Membranes were probed with polyclonal anti-α folate receptor (FOLR1) Antibody (Novus NBP1-32293) at 1/1000 dilution. Equal protein loading was verified by re-probing each membrane with an antibody against α-tubulin (DM1A 32293, Santa Cruz Biotechnology) in 1/1000 dilution. Binding was detected using horseradish peroxidase conjugated secondary antibody (Santa Cruz Biotechnology) and chemiluminescence was detected using ECL Western blotting substrates (Thermo Fisher Scientific, Rockford, IL, USA).
Live Cell Microscopy
For live cell microscopy, 3 x 104 HeLa Kyoto, 3 x 104 HeLa Kyoto GFP-α-tubulin/mcherry-H2B, 6 x 104 MCF7, 6 x 104 MDA-MB-231 or 8 x 104 T47D cells were plated on Labtek glass bottomed chambered dishes (Nunc) in complete DMEM and incubated overnight in a humidified atmosphere (37°C, 5% CO2) to allow cell attachment. The imaging was performed in a CO2-independent medium (041-95374, Life technologies), supplemented with 10% FBS, 1% L-Glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin (prior to imaging, cells were washed twice with the same imaging medium). In all cell lines (except HeLa Kyoto GFP-α-tubulin/mcherry-H2B), Hoechst 33342 was added to the cell medium (2 μg/mL), for nuclei staining.
Cells were imaged on a customized Andor Revolution Spinning Disk Confocal System built around a stand (IX81; Olympus) with a 20x air objective and a digital camera (Andor Ixon+885) (CIBIT Facility, MBG-DUTH) equipped with an incubator at 37°C. Image acquisition was performed with imaging software Andor IQ 2. The multi-dimensional time lapse experiments that are listed below were controlled by an autofocus pattern that coordinates the capturing of multi-field positions in a specified range of Z slices at repeated time intervals. Images were acquired as z stacks with selected optical sections every 3 μm through the entire cell volume every 6–10 min, according to experimental needs. Hoechst-treated cells and Rhodamine-labeled NPs were excited using 405nm and 561nm laser light, respectively.
Cell Cycle Arrest Monitoring
Stably transfected HeLa Kyoto GFP-α-tubulin/mcherry-H2B cells were treated with Paclitaxel-loaded PPSU-PEG NPs, containing 250nM of encapsulated Paclitaxel and ~5.5 μg/mL of polymers (calculated as described above) or 250nM of free Paclitaxel and monitored for 24 h post drug addition. The mcherry-H2B and GFP-α-tubulin expressed fluorescent proteins were excited at 561nm and 491nm laser light and detected at 625nm and at 525nm emission, respectively. Throughout the duration of the experiment, images were acquired every 10 min as z stacks, with selected optical sections of 6.24 μm (31.2 μm in 5 planes) through the entire cell volume. Stacked images were processed into uncompressed.avi files, and time-stamp is displayed at each timepoint on the upper right corner of each image, using ImageJ 1.44n (National Institute of Health, USA) software.
Cellular Uptake of FA-PPSu-PEG-Rho NPs
HeLa Kyoto, MCF7, MDA-MB-231 and T47D cells were incubated with FA-PPSu-PEG-Rho NPs, containing 11 μg/mL of polymer (designated as high-concentration). Cell nuclei were stained with Hoechst, as stated above. Images were acquired every 6 min for a total of 6.5 h as 15 planes of z stacks, 3 μm each.
Folic Acid Competition Experiments
HeLa Kyoto, MCF7, MDA-MB-231, and T47D cells were pre-incubated for 1 h with 3mM free folic acid (Sigma F7876) in imaging medium before nanoparticle addition. Following, FA-PPSU-PEG-Rho NPs were added at a final polymer concentration of 2.2 μg/mL (designated as low concentration), in the presence of free folic acid. NPs internalization was monitored for 4 h with live cell confocal microscopy as described above, and images were acquired every 10 min. NPs uptake followed folic acid treatment was compared to the uptake in the absence of folic acid.
NPs Internalization Experiments
Cells were pretreated for 1 h with either 80 μM Dynasore (3-Hydroxy-naphthalene-2-carboxylic acid (3,4-dihydroxy-benzylidene)-hydrazide hydrate) (Sigma D7693) or 50 μM EIPA (5-(N-Ethyl-N-isopropyl)amiloride) (Sigma A3085) in imaging medium. Following, FA-PPSu-PEG-Rho NPs were added (11μg/mL polymer) to the cell medium and cells were monitored for 2 h. The inhibitory effect of EIPA and Dynasore on nanoparticle internalization was compared to the uptake of untreated cells.
Quantification and Statistical Analysis
Image intensity analysis for data sets were performed with ImageJ 1.44n (National Institute of Health, USA) software. Integrated Rhodamine fluorescence intensity was quantified and divided by number of nuclei from average projection of z-stack images after the subtraction of integrate intensity of extracellular nanoparticle-formed aggregates. Intensity values were normalized against maximum values and therefore, expressed in arbitrary units. One-phase exponential association was used to estimate half-time of cellular uptake (Prism Version 5.0a software, San Diego, CA, USA).
Results and Discussion
Synthesis of Conjugated Copolymers and Paclitaxel Nanoencapsulation
The different copolymers were synthesized, characterized by FT-IR spectroscopy, and their successful modification along with the recorded spectra are presented in Figure 1A. The nanoparticles were produced by nanoencapsulation via an oil-in-water (o/w) emulsification and solvent evaporation technique, with a nanoparticle yield of about 72%. The prepared nanoparticles show a unimodal size distribution with diameters varying from 60 to 230 nm and a mean particle diameter of about 150–170 nm, as shown by DLS (Supplementary Figure 4). Nanoparticles of similar sizes were synthesized in our previous study, where Paclitaxel was nanoencapsulated using aliphatic polyesters.30
Figure 1.

FT-IR spectra and SEM micrographs of the synthesized NPs. (A) Regarding neat PPSu, a strong band at 1735 cm−1 corresponding to its carbonyl ester groups C=O and a broad band at 3433–3439 cm−1 assigned to –OH groups are observed. Additional characteristic peaks are recorded at 1454–1474 cm−1 (–CH–) and 1163–1167 cm−1 (C–O–C stretching). The FA- PEG-PPSu spectrum shows a strong band at 1726 cm−1, assigned to the carbonyl groups of polyester and a broad band at 3400–3500 cm−1 corresponding to the hydroxyl groups of PEG conjugated to the polymer. Several small peaks at 1608, 1686 and at 606 cm−1 indicate the successful conjugation of modified FA into PPSu-PEG, given that these three peaks exist only in FA.33 Similar peaks for the results are shown for PPSu-PEG-Rho copolymers. Rhodamine B has a characteristic peak at 2967 cm−1, due to the methylene groups of its aromatic rings, which is also recorded in conjugated copolymer, as well as a small peak at 1623 cm−1 (C=N stretching), indicating the successful conjugation of Rhodamine on PPSu-PEG copolymer. (B) SEM micrographs of FA-PPSu-PEG (left) and FA-PPSu-PEG encapsulated with Paclitaxel (right).
All nanoparticles have a spherical shape, as shown by SEM micrographs (Figure 1B, left), with calculated drug loading content ca 6.3% and entrapment efficiency ca 72%. Moreover, despite Paclitaxel encapsulation, the drug size remains almost unchanged (Figure 1B, right). These pegylated-aliphatic acid copolymer nanoparticles have a melting point near the natural body temperature7 and therefore may act as thermosensitive carriers.30
In vitro Drug Release from Paclitaxel-Loaded PPSu-PEG NPs
In order to investigate the prospective use of PPSu-PEG NPs as an anti-cancer drug delivery system, we initially assessed the release potential of the drug-loaded PPSu-PEG NPs under controlled conditions. The release profile of Paclitaxel from Paclitaxel-loaded PPSu-PEG NPs over a 14-day period (336 h) in PBS (pH 7.4) at 37°C (conditions simulating the physiological human body conditions) are shown in Figure 2A. The release profile is biphasic, with a rapid release during the first 24 h, followed by a prolonged release phase, reaching a plateau after 96 h. Approximately 66% of the loaded drug was released by 48 h (2 days) and 90% by 168 h (7 days) post the initial suspension of the NPs in solution. We mainly focused on the initial 48 h time window (Figure 2B), where increased release rate was observed, and thus performed subsequent cellular experiments (see below) over this 48 h time period. The observed release profile is typical for nanoparticle encapsulated drugs. It is in agreement with previous reports, particularly taking into account the hydrophobicity of the drug, the composition of the NPs in PEG and PPSu blocks and the physiochemical mechanisms that affect the nanoparticle stability and biodegradability.26,34,35
Figure 2.

Paclitaxel release profile from PPSu-PEG NPs. (A) Drug release profile from PPSu-PEG NPs over 14 days (336 h) in PBS is shown. The majority of the encapsulated drug (94.4%) is sustainably released. (B) Drug release profile of the PPSu-PEG NPs during the first 48 h in PBS in more detail. The initial burst effect of the non-encapsulated drug is also shown (36.9% of the theoretically loaded drug is washed out, after initial suspension).
In addition, an initial burst effect was observed within the first minutes of suspension that resulted in approximately 37% leakage of the theoretically loaded drug. The burst effect is mainly due to residual amounts of drug on the surface of nanoparticles, a common problem with the encapsulation process.36 Therefore, a wash step of the nanoparticles after their initial suspension in solution was performed in order to remove the weakly bound drug prior to cellular treatment. The effective NP-encapsulated Paclitaxel concentration was therefore re-calculated after NPs washing, monitoring the uncontrolled release of the non-encapsulated drug, and subtracting the amount of the washed drug. Our in vitro drug release experiments suggest that a significant proportion of the drug is released over a relatively long time (2 days, Figure 2) and therefore PPSu-PEG NPs are suitable as putative anti-cancer drug delivery carriers.
Real-Time Cytotoxicity of PPSu-PEG Nanoparticles
Next, we sought to investigate the cytotoxic capacity of Paclitaxel-loaded PPSu-PEG NPs. Two aspects were assessed: first, the possible toxic effect of empty PPSu-PEG NPs on physiological cell function and second, the evaluation of cytotoxic activity of the drug post release from the NPs compared to that of free Paclitaxel. The potential cytotoxicity of empty PPSu-PEG NPs was examined in two cell lines: the cervical cancer HeLa Kyoto (HeLa K) cell line, and the breast adenocarcinoma MCF7 cell line (Figure 3).
Figure 3.

Empty (drug-free) PPSu-PEG NPs are not toxic to cells. HeLa K and MCF7 cells were treated with PPSu-PEG NPs for 48 h. (A) Bar graph demonstrating PPSu-PEG NPs’ toxicity (containing 0–500 μg/mL of polymer) on HeLa K and MCF7 cells, as obtained from WST-1 cell proliferation assay. Graph represents percentage (%) of viable cells ± SEM of 3 experiments. (B) Graphs represent normalized cell index of i) HeLa K cells and ii) MCF7 cells treated with PPSu-PEG NPs (containing 11 μg/mL of polymer) for 48 h. Graphs were obtained from the RTCA assay. NCI demonstrates a dip at 24h, corresponding to the NPs’ addition, after which CI increases again in all cases.
First, cell toxicity of empty NPs was examined using the end-point WST-1 cell proliferation and cell viability assay. PPSu-PEG NPs were used in six different concentrations, containing 500, 100, 50, 10, 1, 0.1 μg/mL of polymers. Treatment of cells with PPSu-PEG NPs did not result in a significant loss of cell viability of either HeLa K or MCF7 cancer cells (Figure 3A).
For more accurate cell toxicity monitoring we also employed an impedance-based Real-Time Cell Analysis assay (RTCA) (xCELLigence). This is a high throughput non-invasive method that monitors continuously cell behaviour in real time, therefore providing more information than traditional end-point assays. Initially, we examined the Normalized Cell Index (NCI) responses of HeLa K and MCF7 cells incubated for 48 h with PPSu-PEG NPs containing 11 μg/mL polymers (Figure 3B). This concentration of polymer is designated as high concentration of NPs and corresponds to the re-calculated concentration of drug-loaded PPSu-PEG-NPs in order to contain 500 nM of Paclitaxel (see Figure 4). Cell-free control measurements showed that the PPSu-PEG NPs do not influence the impedance measurements. In agreement with the WST-1 assays drug-free PPSu-PEG NPs showed no toxic effect on either HeLa K or MCF7 cells; thus, further validating that PPSu-PEG NPs could be used as drug carriers.
Figure 4.

Monitoring of Paclitaxel-loaded PPSu-PEG NPs cell toxicity versus free Paclitaxel. HeLa K and MCF7 cells were treated with either Paclitaxel-loaded PPSu-PEG NPs, or free Paclitaxel and real-time monitored for 48 h after drug addition, using the RTCA assay. (A) Graphs representing normalized cell index of HeLa K cells treated with (i) Paclitaxel-loaded PPSu-PEG NPs (ii) free Paclitaxel, (B) Graphs representing normalized cell index of MCF7 cells treated with (i) Paclitaxel-loaded PPSu-PEG NPs, and (ii) free Paclitaxel. Cells were treated in duplicates with the indicated concentrations. (C–D) Dose response curves resulting from the RTCA assay for IC50 values of HeLa K (C) and MCF7 cells (D), treated with either Paclitaxel-loaded PPSu-PEG NPs (NPs-Paclitaxel) or free Paclitaxel.
Real-Time Cytotoxicity of Paclitaxel-Loaded PPSu-PEG NPs
We then tested the time-dependent cytotoxic effect of Paclitaxel-loaded PPSu-PEG NPs or free Paclitaxel on HeLa K and MCF7 cells using the RTCA assay (Figure 4A–D). A wide range of drug concentrations were used, corresponding to either 500, 250, 50, 25, 10 nM of free Paclitaxel, or 315, 157, 31.5, 15.7, and 6.4 nM (on a Paclitaxel basis) of washed Paclitaxel-loaded PPSu-PEG NPs. Cells with medium only (DMEM) served as untreated control (Figure 4A(i) and B(i)). Cells treated with DMSO (0.01%) also served as additional control (Figure 4A(ii) and B(ii)).
IC50 values of either free Paclitaxel or Paclitaxel-loaded PPSu-PEG NPs after treatment of HeLa K and MCF7 cells for 48 h were calculated. In HeLa K cells, the IC50 values of free Paclitaxel vs Paclitaxel-loaded PPSu-PEG NPs were 7.5 nM and 19.3 nM, respectively, while IC50 values for MCF7 cells were calculated as 19.2 nM and 27 nM, respectively (Figure 4C and D). Using the WST-1 cytotoxic assay we obtained similar results (data not shown).
Treatment with either NPs-Paclitaxel or free Paclitaxel resulted in similar time-dependent cell response profiles, with an initial steady decline of the cell index followed by recovery about 14–16 h post treatment, consistent with cells responding to antimitotic agents leading to mitotic arrest.37,38
We conclude that the mode of action of free Paclitaxel and Paclitaxel-loaded PPSu-PEG-NPs is similar. These results indicate that Paclitaxel-loaded PPSu-PEG-NPs are highly effective in mediating Paclitaxel-dependent cytotoxicity, despite the encapsulation of the drug.
Treatment with Paclitaxel-Loaded PPSu-PEG NPs Results in Tubulin-Specific Cell Arrest
To examine in more detail whether the encapsulation of Paclitaxel in PPSu-PEG NPs alters its effect on mitotic spindle assembly and mitotic progression, we performed live cell imaging using the HeLa K mcherry-H2B/GFP-α-tubulin cell line. This cell line stably expresses mcherry-tagged Histone H2B to visualize the DNA in the nucleus (and follow its fragmentation during cell arrest) and GFP-tagged α-tubulin, which is Paclitaxel’s cellular target. Thus, HeLa K mcherry-H2B/GFP-α-tubulin cells were treated with either Paclitaxel-loaded PPSu-PEG NPs containing 250 nM of encapsulated drug or 250 nM free Paclitaxel for ~24 h (Supplementary Videos S1–2, respectively). The effect of Paclitaxel-loaded PPSu-PEG NPs on cell cycle arrest (and its kinetics) was monitored for ~24 h at 37ºC using a spinning disc confocal microscope. Tracking 90–100 individual cells for 24 h post treatment showed similar results between Paclitaxel-loaded PPSu-PEG NPs or free Paclitaxel in terms of cell cycle arrest (Figure 5), mediated by tubulin stabilization; 24 h after Paclitaxel-loaded PPSu-PEG-NPs treatment 44.7% ± 0.5 of cells were arrested, while free Paclitaxel treatment resulted in 50.8% ± 4.5 of arrested cells, as seen by microtubule assemblies. The effect of either Paclitaxel-loaded PPSu-PEG-NPs or free Paclitaxel in cell arrest occurred with similar kinetics, with no significant difference. Moreover, in both cases DNA was fragmented, as expected in Paclitaxel induced arrest. As Paclitaxel is diluted in DMSO (0.5% v/v final concentration), we also tested whether DMSO alone results in cell cycle arrest, but cells behaved similarly to untreated control (Figure S5). These results are consistent with the cell cycle arrest observed in the RTCA assay and confirm our initial observations that PPSu-PEG NPs are suitable carriers for mediating Paclitaxel delivery, that the released drug remains active and that it affects the same cellular targets with the same kinetics as free Paclitaxel. However, the importance of using nanocarriers lies in the advantage of targeted delivery and therefore decreased toxicity at the organismal level.
Figure 5.

The effect of Paclitaxel-loaded PPSu-PEG NPs on tubulin. HeLa K cells stably expressing GFP-α-tubulin/mcherry-histone H2B were imaged for 24 h with 10-minute intervals, after the addition of 250 nM free Paclitaxel or encapsulated in PPSu-PEG NPs, and the number of cell cycle arrested cells were counted. Graph demonstrates the % of arrested cells by either Paclitaxel or Paclitaxel-loaded PPSu-PEG NPs, as measured during 24 h of live cell monitoring. At least 100 cells were imaged, per condition per experiment.
Real-Time Cellular Uptake of FA-Conjugated PPSu-PEG NPs
To expand the potential drug delivery applications of PPSu-PEG NPs, we set out to explore whether functionalization of these particles provides beneficial cell targeting properties. Conjugation of nanocarriers with folic acid (FA) provides a specific tag which has been reported to mediate tumor-specific recognition.27,41 We sought to investigate whether conjugation of FA to the PPSu-PEG NPs would affect the NPs’ cellular internalization. In order to enable real-time microscopic visualization, Rhodamine B was covalently bound to FA-conjugated PPSu-PEG NPs,7 resulting in the FA-PPSu-PEG-Rho NPs.
First, we tested internalization efficiency and kinetics of FA-conjugated NPs in three breast and one cervical cancer cell lines (MCF7, T47D, MDA-MB-231 and HeLa K, respectively). The different breast cancer cell lines represent diverse phenotypes observed in human breast cancer.39,40 Cellular entry of FA-PPSu-PEG-Rho NPs was monitored using live cell imaging for 4 h post NPs’ addition to the cell medium. For these experiments, we used high NPs concentration (11 μg/mL of polymer) to allow their tracking within the shortest possible time. Figure 6A depicts the cellular uptake of fluorescent NPs in all four cancer cell lines, as measured by the relative fluorescence intensity within the cell cytoplasm, over a period of 4 h. Quantification of the cellular uptake and the time required for cell entry showed that more NPs enter the MDA-MB-231 cells and with the highest rate (t1/2: 51.6 min), followed by MCF7 (t1/2: 66.1 min) and T47D (t1/2: 80.2 min) (Figure 6B). HeLa K cells can internalize less NPs compared to the three breast cancer cell lines tested (Figure 6A), although with a much faster rate (t1/2: 22.9 min) (Figure 6B). Figure 6C shows representative images recorded at 6, 30, 60 and 120 min after addition of NPs in the cell medium. In all four cell lines, a small amount of NPs enter the cells within the first 6 min, while a significant amount is internalized by 120 min. It should be noted that when using the high concentration of NPs, formation of extracellular aggregates is observed gradually over time. This is because the NPs are in excess compared to the cells, and the remaining NPs that are not internalized gradually tend to aggregate.
Figure 6.

FA-PPSu-PEG-Rho NPs cell internalization efficiency and kinetics. HeLa K, T47D, MCF7 and MDA-MB-231 (designated as MDA) cells were incubated with FA-PPSu-PEG-Rho NPs in high concentration (11 μg/mL of polymer) and monitored for 4 h. (A) Graph demonstrating the relative uptake of FA-PPSu-PEG-Rho NPs from HeLa K, T47D, MCF7 and MDA-MB-231, shown as normalized intensity (a.u.) over time, with maximum fluorescence of MDA-MB-231 at 1. (B) Table showing the time required for ½ of the total NPs amount (t1/2 uptake) to enter the cell. (C) Representative pictures showing cellular internalization of FA-PPSu-PEG-Rho nanoparticles, in the four different cell lines, at 6min, 30min, 60min and 120min after addition of the NPs in the medium. Images were captured during live cell imaging confocal microscopy. Cell nuclei are stained with Hoechst 33342 and shown in blue, Rhodamine labeled NPs are shown in red. Scale bar denotes 5 μm.
To conclude, FA-PPSu-PEG-Rho NPs are successfully internalized into all four cell lines used and are mainly localized in the cell cytoplasm. The entry of the NPs within the different cell lines occurs at a different concentration and with a different speed, indicating that these features may be important for their evaluation for in vivo applications, having an impact on both the uptake efficiency and cytotoxicity induction.
Folic-Acid Functionalized PPSu-PEG NPs are Specific Molecular Binders
Since the FA-PPSu-PEG-Rho NPs show different uptake efficiency within the different cell lines, we investigated whether the FA-specific properties of the functionalized NPs were contributing to these differences.
We first examined the binding capacity of the conjugated FA-PPSu-PEG-Rho particles to Folate Binding Protein (FBP) using an in vitro pull-down assay (Figure 7). The FBP interaction with FA-PPSu-PEG NPs was examined under neutral pH, similar to the physiological pH of normal cells, but also in more acidic pH, to mimic the sporadically acidic microenvironment present in cancer cells and tissues.42,43 37°C was selected to resemble normal human body temperature, while a lower temperature, 25°C, was also applied to test possible thermal effects on FBP at 37°C. Non-conjugated control PPSu-PEG NPs (Figure 7A) or FA-conjugated PPSu-PEG (Figure 7B) were incubated with 4 μg of FBP for 1 h, in PBS, at 25°C or 37°C, and at two different pH conditions (pH 6.0 and 7.4). After incubation, NPs were separated from the solution and the presence of pulled-down FBP was determined by SDS-PAGE. As shown in Figure 7A, FBP appears mainly as a ~26 kDa band (monomeric form of protein) with a small fraction at ~35kDa (dimeric form). In all pull-downs with PPSu-PEG NPs, the total amount of FBP protein is detected only in the supernatant (Figure 7A). However, FBP is specifically pulled-down by FA-PPSu-PEG NPs under all conditions tested (Figure 7B). The quantification of bound FBP upon interaction with FA-PPSu-PEG NPs (Figure 7C) suggests a higher affinity at pH 6.0 than pH 7.4, which is well documented elsewhere; FBP exhibits multiple isoelectric points in the pH range 7–8, is more hydrophobic at pH values close to its pI, while at pH lower than its pI it shows higher water solubility and increased affinity for folic acid.44 Structurally, the ligand’s binding induces a conformational change to the protein with a tendency to self-association into more stable and soluble complexes.45,46 Thus, our data provide strong evidence of specific FA-mediated recognition of FA-PPSu-PEG NPs by the Folate-Binding Protein.
Figure 7.

In vitro interaction of Folate-Binding Protein with FA-PPSu-PEG NPs. 4 μg of FBP were incubated with non-conjugated control PPSu-PEG NPs (A) or FA-conjugated PPSu-PEG (B) (both containing 50 μg of polymer) for 1 h, in PBS, at 25°C or 37°C, and at two different pH conditions (pH 6.0 and 7.4). SDS PAGE analysis of FBP interaction with PPSu-PEG and FA-PPSu-PEG NPs is shown. Lanes are marked as p (pelleted NPs) or s (supernatant). Protein marker with the relevant band sizes is also shown. (C) Densitometric quantification of FBP pulled-down amount upon interaction with FA-PPSu-PEG NPs. Numbers 1–8 correspond to referred lanes in Figure 7B.
Free Folic Acid Competes with FA-PPSu-PEG-Rho NPs for FOLR1 Binding
As FA-PPSu-PEG NPs are specifically recognized by the Folate-Binding Protein and readily enter MCF7, MDA-MB-231, T47D and HeLa K cells, we further explored the possible mechanism(s) of NPs internalization. First, we questioned whether internalization occurs via the conjugated Folic Acid (FA) on the NPs’ surface binding to the Ffolate receptor or via another path.47,50 Increased expression of the folate receptor-α (FOLR1) has been associated with the development and progression of many cancer types, including breast and ovarian cancer, and therefore would make a good potential targeted delivery site.51,53
To examine whether the FA-conjugated NPs uptake is indeed mediated via the FOLR1, cells were treated with excess of free Folic Acid (FA) prior to their exposure to FA-PPSu-PEG-Rho NPs. We hypothesized that excess of FA will compete for binding to folate receptors, thus delaying, or inhibiting internalization of FA-NPs. All four different cell lines (HeLa K, T47D, MCF7 and MDA-MB-231) were incubated with 3mM of free folic acid for 1 h prior to the addition of NPs. A lower polymer concentration was used in this experiment (2.2 μg/mL of polymer compared to 11 μg/mL in Figure 6) in order to avoid fast saturation of NPs inside cells and to facilitate the kinetic analysis. FA-PPSu-PEG-Rho NPs’ uptake was monitored for 4 h in all four cell lines in the presence or absence of free FA in the cell medium (Figure 8). Normalized fluorescence intensity in arbitrary units (A.U.) for each cell line is calculated as fold of its own maximum intensity at 240 minutes after NPs’ addition. The effect of FA on NPs’ internalization was monitored and expressed as fold-change, relative to fluorescence values measured upon NPs addition. We noted that the amount of NPs entering the cell was not significantly altered in the presence or absence of free FA in the case of HeLa K cells (Figure 8A, first row and B), and the MDA-MB-231 cells (Figure 8A 4th row, and E). However, NPs’ uptake was decreased by 59% in T47D cells in the presence of free FA as compared to its absence (Figure 8A 2nd row, and C). A decrease in the cellular uptake of the NPs by 39% was also observed for MCF7 cells (Figure 8A 3rd row, and D).
Figure 8.

Effect of free FA on FA-PPSu-PEG-Rho NPs cellular uptake. HeLa K, T47D, MCF7 and MDA-MB-231 cells were pre-incubated for 1 h with 3mM free folic acid, followed FA-PPSU-PEG-Rhodamine nanoparticle addition at a low concentration (2.2 μg/mL polymer). NPs internalization was monitored for 4 h, and images were acquired every 10 min. (A) Representative images of HeLa K, T47D, MCF7 and MDA-MB-231 cell lines, upon addition of the FA-PPSu-PEG-Rho NPs (control), or 60 and 240min post NPs addition, in the presence (+FA) or absence of free folic acid (-FA). Cell nuclei were stained with Hoechst 33342 and shown in blue, whereas Rhodamine-labeled NPs are shown in red. Scale bar denotes 5 μm. (B–E) Graphs demonstrating cellular uptake of the FA-PPSu-PEG-Rho NPs for the four cell lines in the presence (+FA) or absence of FA. Relative fluorescence intensity is shown in Arbitrary Units (A.U) and fluorescence for each cell line is calculated as fold of its own maximum intensity at 240 minutes after NPs’ addition. The effect of FA on NPs’ internalization was monitored for 4 h and expressed as fold-change, relative to fluorescence values measured upon NPs addition. The fluorescent intensity of at least 100 cells per cell line was measured. For each cell line, nanoparticles uptake after FA treatment was plotted against the uptake of NPs per se. (F) SDS PAGE analysis showing the expression of FOLR1 protein in the four different cell lines: HeLa K, T47D, MCF7 and MDA-MB-231. α-tubulin serves as a loading control.
We also examined the expression of the folate receptor-α (FOLR1) receptor in all four cell lines, since existing data are controversial (https://www.proteinatlas.org/ENSG00000110195-FOLR1/cell).54,58 Western blotting analysis showed high protein levels of FOLR1 in T47D and MCF7 cells, while HeLa K cells had detectable but lower levels of the receptor (Figure 8F). However, no FOLR1 expression was detected in MDA-MB-231 cells (Figure 8F). The increased levels of FOLR1 expression in T47D and MCF7cells corroborate well with the observed reduction of the FA-NPs uptake in these cell lines, in the presence of free Folic Acid (Figure 8C and D). Moreover, although HeLa K cells demonstrate low levels of FOLR1 expression, there is no significant inhibition of NPs uptake upon addition of free Folic Acid in the cell medium, suggesting that the NPs enter these cells via alternative internalization routes. Similarly, MDA-MB-231 cells, despite the absence of FOLR1 expression, internalize FA-PPSu-PEG-Rho NPs at a high concentration and at a high rate (see also Figure 6), suggesting the presence of other FOLR1-independent internalization mechanisms.
FOLR1-Independent Cellular Uptake of FA- PPSu-PEG-Rho NPs
In all cell lines tested FA- PPSu-PEG-Rho NPs cellular uptake was observed, even in the absence of the FOLR1 receptor in some cell lines (MDA-MB-231), or in the presence of competitive free FA in the cell lines that express FOLR1 (T47D, MCF7, and HeLa K). These observations suggest that other cellular entry mechanisms play a role in NPs’ uptake.
To understand the potential involvement of additional mechanisms in NPs internalization, we investigated the role of dynamin-dependent endocytosis and macropinocytosis, using live cell imaging. Two small molecules known to inhibit distinct mechanisms of cellular uptake were used: Dynasore, which inhibits dynamin-dependent endocytosis59 and EIPA, a selective blocker of the Na+/H+ anti-port, which inhibits macropinocytosis.60 Integrated fluorescence intensity data from internalized NPs were obtained using single-cell analysis from time-lapsed confocal images. Since Dynasore and EIPA exert their maximum inhibitory action within a 1–2 h time window (depending on the cell type), the effect of either of the inhibitors on NPs’ internalization was monitored for 2 h and expressed as fold-change, relative to fluorescence values measured upon NPs addition.
As shown in Figure 9, both inhibitors’ maximum effect occurred at 120 min. EIPA reduced cellular uptake of FA-PPSu-PEG-Rho in all four cell lines, EIPA reduced NPs entry by 56% in HeLa K, by 91% in T47D cells, by 58% in MCF7 and by 94% in MDA-MB-231 cells (Figure 9A, C, E and G, respectively). Dynasore decreased cellular uptake by 74% in HeLa K, 60% in T47D cells, 40% in MCF7, while it had no significant effect on MDA-MB-231 cells (Figure 9A, C, E and G). In general, EIPA affected NPs internalization more dramatically than Dynasore in all cell lines tested, with the exception of HeLa K, where the opposite effect was observed (Figure 9B).
Figure 9.

Endocytosis pathways used for NPs cellular internalization in one cervical and three breast cancer cell lines. HeLa K, T47D, MCF7 and MDA-MB-231 cells were pretreated for 1 h with either 80 μM Dynasore or 50 μM EIPA followed by FA-PPSu-PEG-Rho NPs addition at a high concentration (11μg/mL polymer), and cells were monitored for 2 h. Graphs representing the real-time effect of Dynasore and EIPA on NPs internalization for 120 min post NPs addition are shown in (A) HeLa K (C) T47D (E) MCF7 and (G) MDA-MB-231 cells. Representative confocal images of NPs’ uptake in (B) HeLa K (D) T47D (F) MCF7 and (H) MDA-MB-231 cells at 120 min post treatment with NPs in the presence or absence of Dynasore or EIPA. Scale bar denotes 5μm.
The cervical cell line HeLa K expresses low amounts of FOLR1 receptor (Figure 8F) is largely affected by the Dynasore inhibitor, and to a lesser extent by EIPA (Figure 9A and B), suggesting that it utilizes mainly the dynamin-dependent endocytosis (but not through the FOLR1 receptor) and to a lesser degree the macropinocytosis pathway for NPs uptake; yet the relative amount of NPs uptake is the least compared to the three breast cancer cell lines tested, although the rate of internalization is the highest of all tested cell lines (Figure 6A and B).
In contrast, in MDA-MB-231 cells that do not express FOLR1 receptor (Figure 8F), EIPA treatment dramatically inhibits NPs’ uptake (Figure 9G), while Dynasore has no effect, suggesting that these cells do not internalize the NPs via the dynamin-dependent endocytosis, but primarily through macropinocytosis. MDA-MB-231 cells demonstrate the highest amount of NPs uptake, and exhibit a relative fast rate (t1/2) of NPs' uptake (Figure 6A and B).
T47D and MCF7 cells show an “intermediate” amount of NPs uptake, with a relative slow rate of internalization compared to MDA-MB-231 and HeLa K (Figure 6A and B), and they seem to utilize the FOLR1-dependent route, the dynamin-dependent endocytosis, as well as the macropinocytosis pathway (Figure 8C and D; Figure 9C and E).
Apart from the fact that the different cell lines exhibit different active endocytosis mechanisms, the size of the nanoparticles may have also affected the internalization mechanism utilized. The relatively broad size distribution of the FA-PPSu-PEG-Rho nanoparticles (60–230 nm) may have played a role on the parallel different mechanisms of their cellular uptake. Thus, the existence of non-FOLR-1 dependent endocytosis routes with T47D, MCF7 and HeLa K cells expressing FOLR-1 (Figure 8F and Figure 9A,C and E) may partly be explained by the possible different uptake mechanism of the relatively small versus the relatively large FA-PPSu-PEG-Rho NPs. The larger FA-PPSu-PEG-Rho NPs (those with diameters higher than 200 nm) could have followed a caveolae-mediated endocytosis mechanism, as the results obtained with the uptake of FA-decorated methoxy-poly(ethylene glycol)-b-polycaprolactone NPs by the human retinal pigment epithelium cell line would suggest.61 On the other hand, the relatively broad size distribution of the FA-PPSu-PEG-Rho NPs apparently did not exert any significant influence on the mechanism of their uptake by the MDA-MB-231 cells, which did not express the FOLR-1, as the endocytosis of the NPs in these cells followed almost exclusively a macropinocytosis pathway.
Conclusion
Our results strongly suggest that in different cell lines with differential FOLR1 expression, FA-targeting is acting complementary to other parallel, but not mutually exclusive, processes of cellular entry, such as dynamin-dependent endocytosis and macropinocytosis pathways. The “use” of alternative pathways or the use of more than one pathway does not correspond to cell aggressiveness, since MDA-MB-231, the most aggressive breast cancer cells, do not possess neither FOLR1 on their cell surface, nor do they utilize dynamin-dependent endocytosis for NPs internalization. On the other hand, the least aggressive cell line, MCF7, although it utilizes all three tested internalization pathways, still resulted in a lower amount of NPs internalized within the first 240 min, as compared to MDA-MB-231.
In the present study, we examined a novel pegylated-aliphatic acid copolymer with a melting point near the natural body temperature7 and therefore may act as a thermosensitive carrier.30 For the first time, these copolymers were conjugated with folic acid to facilitate targeted delivery of Paclitaxel, a very efficient chemotherapeutic drug used for breast and cervical cancer treatment. Thus, these NPs could be used as effective nanocarriers for several anti-cancer drugs. The release profile of the encapsulated drug, the NPs’ toxicity, as well as their effectiveness in drug-induced cell cycle arrest and cell death was investigated and was similar to free Paclitaxel. In addition, targeting of drug-loaded, biocompatible and biodegradable FA-PPSu-PEG NPs7 to the FOLR1 receptor, which is present in a wide range of cancer cell membranes, would retain Paclitaxel’s efficiency in killing cancer cells, but, importantly, could result in reduced toxicity to normal cells. Interestingly, our experiments showed that although NPs are decorated with FA, they manage to enter cells, even in the absence of FOLR1 receptor on the cell surface, as in the case of MDA-MB-231 breast cancer cell line. In addition, MDA-MB-231 exhibit the highest amount of NPs’ uptake with a fast rate, utilizing almost exclusively the macropinocytosis pathway. Thus, as shown in this study, cells seem to take advantage of alternative internalization mechanisms, independent of FOLR1 expression. Moreover, these differential internalization mechanisms seem to be cell-type specific. Therefore, the effective utilization of different cellular entry pathways, in the presence or absence of FOLR1 should be re-evaluated in order to enter the era of personalized medicine and individuals should be tested for different cellular uptake mechanisms before their tailor-cut anti-cancer treatment.
Acknowledgments
This work was supported by ”Nanobilon”, National Action Range “Sinergasia”-Act I OP “Competitiveness and Entrepreneurship” (EPAN-II). We are grateful to Dr S. Boukouvala, for technical assistance. We also thank the CiBIT microscopy facility at the Department of MBG, Democritus University of Thrace and Dr Andreas Girod for assistance with the microscopy experiments.
Disclosure
Dr Avgi Tsolou report grants from Ministry of Education and Religious Affairs, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
- 1.Ghoncheh M, Pournamdar Z, Salehiniya H. Incidence and mortality and epidemiology of breast cancer in the world. Asian Pac J Cancer Prev. 2016;17(S3):43–46. doi: 10.7314/APJCP.2016.17.S3.43 [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. doi: 10.3322/caac.20107 [DOI] [PubMed] [Google Scholar]
- 3.Kitagawa R, Katsumata N, Shibata T, et al. Paclitaxel plus carboplatin versus paclitaxel plus cisplatin in metastatic or recurrent cervical cancer: the open-label randomized phase III trial JCOG0505. J Clin Oncol. 2015;33(19):2129–2135. doi: 10.1200/JCO.2014.58.4391 [DOI] [PubMed] [Google Scholar]
- 4.Singla AK, Garg A, Aggarwal D. Paclitaxel and its formulations. Int J Pharm. 2002;235(1–2):179–192. doi: 10.1016/S0378-5173(01)00986-3 [DOI] [PubMed] [Google Scholar]
- 5.Spencer CM, Paclitaxel FD. A review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in the treatment of cancer. Drugs. 1994;48(5):794–847. doi: 10.2165/00003495-199448050-00009 [DOI] [PubMed] [Google Scholar]
- 6.Sofias AM, Dunne M, Storm G, Allen C. The battle of “nano” paclitaxel. Adv Drug Deliv Rev. 2017;122:20–30. doi: 10.1016/j.addr.2017.02.003 [DOI] [PubMed] [Google Scholar]
- 7.Siafaka P, Betsiou M, Tsolou A, et al. Synthesis of folate-pegylated polyester nanoparticles encapsulating ixabepilone for targeting folate receptor overexpressing breast cancer cells. J Mater Sci Mater Med. 2015;26(12):275. doi: 10.1007/s10856-015-5609-x [DOI] [PubMed] [Google Scholar]
- 8.Torchilin V. Multifunctional and stimuli-sensitive pharmaceutical nanocarriers. Eur J Pharm Biopharm. 2009;71(3):431–444. doi: 10.1016/j.ejpb.2008.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siafaka PI, Ustundag Okur N, Karavas E, Bikiaris DN. Surface modified multifunctional and stimuli responsive nanoparticles for drug targeting: current status and uses. Int J Mol Sci. 2016;17(9):1440. doi: 10.3390/ijms17091440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang T, Choi MK, Cui FD, et al. Preparation and evaluation of paclitaxel-loaded PEGylated immunoliposome. J Control Release. 2007;120(3):169–177. doi: 10.1016/j.jconrel.2007.05.011 [DOI] [PubMed] [Google Scholar]
- 11.Liggins RT, Burt HM. Paclitaxel-loaded poly(L-lactic acid) microspheres 3: blending low and high molecular weight polymers to control morphology and drug release. Int J Pharm. 2004;282(1–2):61–71. doi: 10.1016/j.ijpharm.2004.05.026 [DOI] [PubMed] [Google Scholar]
- 12.Zhang Z, Feng SS. The drug encapsulation efficiency, in vitro drug release, cellular uptake and cytotoxicity of paclitaxel-loaded poly(lactide)-tocopheryl polyethylene glycol succinate nanoparticles. Biomaterials. 2006;27(21):4025–4033. doi: 10.1016/j.biomaterials.2006.03.006 [DOI] [PubMed] [Google Scholar]
- 13.Huh KM, Min HS, Lee SC, Lee HJ, Kim S, Park K. A new hydrotropic block copolymer micelle system for aqueous solubilization of paclitaxel. J Control Release. 2008;126(2):122–129. doi: 10.1016/j.jconrel.2007.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parveen S, Misra R, Sahoo SK. Nanoparticles: a boon to drug delivery, therapeutics, diagnostics and imaging. Nanomedicine. 2012;8(2):147–166. doi: 10.1016/j.nano.2011.05.016 [DOI] [PubMed] [Google Scholar]
- 15.Karimi M, Ghasemi A, Sahandi Zangabad P, et al. Smart micro/nanoparticles in stimulus-responsive drug/gene delivery systems. Chem Soc Rev. 2016;45(5):1457–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bikiaris D, Karavelidis V, Karavas E. Effectiveness of various drug carriers in controlled release formulations of raloxifene HCl prepared by melt mixing. Curr Drug Deliv. 2009;6(5):425–436. doi: 10.2174/156720109789941632 [DOI] [PubMed] [Google Scholar]
- 17.Bikiaris D, Karavelidis V, Karavas E. Novel biodegradable polyesters. Synthesis and application as drug carriers for the preparation of raloxifene HCl loaded nanoparticles. Molecules. 2009;14(7):2410–2430. doi: 10.3390/molecules14072410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bikiaris D, Papageorgiou G, Achilias D. Synthesis and comparative biodegradability studies of three poly(alkylene succinate)s. Polym Degrad Stabil. 2006;91(1):31–43. [Google Scholar]
- 19.Papageorgiou G, Bikiaris D. Crystallization and melting behavior of three biodegradable poly(alkylene succinates). A comparative study. Polym. 2005;46(26):12081–92. [Google Scholar]
- 20.Chan P, Kurisawa M, Chung JE, Yang YY. Synthesis and characterization of chitosan-g-poly(ethylene glycol)-folate as a non-viral carrier for tumor-targeted gene delivery. Biomaterials. 2007;28(3):540–549. doi: 10.1016/j.biomaterials.2006.08.046 [DOI] [PubMed] [Google Scholar]
- 21.Couvreur P, Vauthier C. Nanotechnology: intelligent design to treat complex disease. Pharm Res. 2006;23(7):1417–1450. doi: 10.1007/s11095-006-0284-8 [DOI] [PubMed] [Google Scholar]
- 22.Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6(9):688–701. doi: 10.1038/nrc1958 [DOI] [PubMed] [Google Scholar]
- 23.Chan P, Kurisawa M, Eun Chung J, Yang Y-Y. Synthesis and characterization of chitosan-g-poly(ethylene glycol)-folate as a non-viral carrier for tumor-targeted gene delivery. Biomaterials. 2007;28(3):540–9. [DOI] [PubMed] [Google Scholar]
- 24.Kyriakopoulou S, Matthaiolampakis G, Papadimitriou S, Karavas E, Bikiaris D, Avgoustaκis K. Ppsu-PEG copolymers and their application in the preparation of cisplatin-loaded nanoparticles. Curr Nanosci. 2011;7(4):503–9. [Google Scholar]
- 25.Tong R, Cheng J. Anticancer polymeric nanomedicines. Polym Rev. 2007;47(3):345–381. doi: 10.1080/15583720701455079 [DOI] [Google Scholar]
- 26.Vassiliou AA, Papadimitriou SA, Bikiaris DN, Mattheolabakis G, Avgoustakis K. Facile synthesis of polyester-PEG triblock copolymers and preparation of amphiphilic nanoparticles as drug carriers. J Control Release. 2010;148(3):388–395. doi: 10.1016/j.jconrel.2010.09.017 [DOI] [PubMed] [Google Scholar]
- 27.van Vlerken LE, Amiji MM. Multi-functional polymeric nanoparticles for tumour-targeted drug delivery. Expert Opin Drug Deliv. 2006;3(2):205–216. doi: 10.1517/17425247.3.2.205 [DOI] [PubMed] [Google Scholar]
- 28.Choi CH, Alabi CA, Webster P, Davis ME. Mechanism of active targeting in solid tumors with transferrin-containing gold nanoparticles. Proc Natl Acad Sci U S A. 2010;107(3):1235–1240. doi: 10.1073/pnas.0914140107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu X, Yan L, Xiao H, Li X, Jing X. Application of microwave-assisted click chemistry in the preparation of functionalized copolymers for drug conjugation. J Appl Polym Sci. 2013;127(5):3365–3373. doi: 10.1002/app.37662 [DOI] [Google Scholar]
- 30.Karavelidis V, Bikiaris D, Avgoustakis K. New thermosensitive nanoparticles prepared by biocompatible pegylated aliphatic polyester block copolymers for local cancer treatment. J Pharm Pharmacol. 2015;67(2):215–230. doi: 10.1111/jphp.12337 [DOI] [PubMed] [Google Scholar]
- 31.Gryparis EC, Mattheolabakis G, Bikiaris D, Avgoustakis K. Effect of conditions of preparation on the size and encapsulation properties of PLGA-mPEG nanoparticles of cisplatin. Drug Deliv. 2007;14(6):371–380. doi: 10.1080/10717540701202937 [DOI] [PubMed] [Google Scholar]
- 32.Tsolou A, Nelson G, Trachana V, et al. The 19S proteasome subunit Rpn7 stabilizes DNA damage foci upon genotoxic insult. IUBMB Life. 2012;64(5):432–442. doi: 10.1002/iub.1018 [DOI] [PubMed] [Google Scholar]
- 33.Anderson KE, Stevenson BR, Rogers JA. Folic acid-PEO-labeled liposomes to improve gastrointestinal absorption of encapsulated agents. J Control Release. 1999;60(2–3):189–198. doi: 10.1016/S0168-3659(99)00072-3 [DOI] [PubMed] [Google Scholar]
- 34.Karavelidis V, Karavas E, Giliopoulos D, Papadimitriou S, Bikiaris D. Evaluating the effects of crystallinity in new biocompatible polyester nanocarriers on drug release behavior. Int J Nanomedicine. 2011;6:3021–3032. doi: 10.2147/IJN.S26016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Filippousi M, Papadimitriou SA, Bikiaris DN, et al. Novel core-shell magnetic nanoparticles for Taxol encapsulation in biodegradable and biocompatible block copolymers: preparation, characterization and release properties. Int J Pharm. 2013;448(1):221–230. doi: 10.1016/j.ijpharm.2013.03.025 [DOI] [PubMed] [Google Scholar]
- 36.Brazel CS, Huang X. The cost of optimal drug delivery: reducing and preventing the burst effect in matrix systems In: Carrier-Based Drug Delivery. Vol. 879 American Chemical Society; 2004:267–282. [Google Scholar]
- 37.Ke N, Wang X, xu X, Abassi Y. The xCELLigence System for Real-Time and Label-Free Monitoring of Cell Viability. Vol. 7402011. [DOI] [PubMed] [Google Scholar]
- 38.Abassi YA, Xi B, Zhang W, et al. Kinetic cell-based morphological screening: prediction of mechanism of compound action and off-target effects. Chem Biol. 2009;16(7):712–723. doi: 10.1016/j.chembiol.2009.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lacroix M, Leclercq G. Relevance of breast cancer cell lines as models for breast tumours: an update. Breast Cancer Res Treat. 2004;83(3):249–289. [DOI] [PubMed] [Google Scholar]
- 40.Holliday DL, Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011;13(4):215. doi: 10.1186/bcr2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang S, Luo Y, Zeng S, et al. Dodecanol-poly(D,L-lactic acid)-b-poly (ethylene glycol)-folate (Dol-PLA-PEG-FA) nanoparticles: evaluation of cell cytotoxicity and selecting capability in vitro. Colloids Surf B Biointerfaces. 2013;102:130–135. doi: 10.1016/j.colsurfb.2012.07.030 [DOI] [PubMed] [Google Scholar]
- 42.Swietach P, Vaughan-Jones RD, Harris AL, Hulikova A. The chemistry, physiology and pathology of pH in cancer. Philos Trans R Soc Lond B Biol Sci. 2014;369(1638):20130099. doi: 10.1098/rstb.2013.0099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4(11):891–899. doi: 10.1038/nrc1478 [DOI] [PubMed] [Google Scholar]
- 44.Holm J, Hansen SI, Hoier-Madsen M. Ionic charge, hydrophobicity and tryptophan fluorescence of the folate binding protein isolated from cow’s milk. Biosci Rep. 2001;21(3):305–313. doi: 10.1023/A:1013234231960 [DOI] [PubMed] [Google Scholar]
- 45.Holm J, Lawaetz AJ, Hansen SI. Ligand binding induces a sharp decrease in hydrophobicity of folate binding protein assessed by 1-anilinonaphthalene-8-sulphonate which suppresses self-association of the hydrophobic apo-protein. Biochem Biophys Res Commun. 2012;425(1):19–24. doi: 10.1016/j.bbrc.2012.07.036 [DOI] [PubMed] [Google Scholar]
- 46.Kaarsholm NC, Kolstrup AM, Danielsen SE, Holm J, Hansen SI. Ligand-induced conformation change in folate-binding protein. Biochem J. 1993;292(Pt 3):921–925. doi: 10.1042/bj2920921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamen BA, Wang MT, Streckfuss AJ, Peryea X, Anderson RG. Delivery of folates to the cytoplasm of MA104 cells is mediated by a surface membrane receptor that recycles. J Biol Chem. 1988;263(27):13602–13609. [PubMed] [Google Scholar]
- 48.Rothberg KG, Ying YS, Kolhouse JF, Kamen BA, Anderson RG. The glycophospholipid-linked folate receptor internalizes folate without entering the clathrin-coated pit endocytic pathway. J Cell Biol. 1990;110(3):637–649. doi: 10.1083/jcb.110.3.637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lewis CM, Smith AK, Kamen BA. Receptor-mediated folate uptake is positively regulated by disruption of the actin cytoskeleton. Cancer Res. 1998;58(14):2952–2956. [PubMed] [Google Scholar]
- 50.Sabharanjak S, Mayor S. Folate receptor endocytosis and trafficking. Adv Drug Deliv Rev. 2004;56(8):1099–1109. doi: 10.1016/j.addr.2004.01.010 [DOI] [PubMed] [Google Scholar]
- 51.Campbell IG, Jones TA, Foulkes WD, Trowsdale J. Folate-binding protein is a marker for ovarian cancer. Cancer Res. 1991;51(19):5329–5338. [PubMed] [Google Scholar]
- 52.Knutson KL, Krco CJ, Erskine CL, et al. T-cell immunity to the folate receptor alpha is prevalent in women with breast or ovarian cancer. J Clin Oncol. 2006;24(26):4254–4261. doi: 10.1200/JCO.2006.05.9311 [DOI] [PubMed] [Google Scholar]
- 53.Kelemen LE. The role of folate receptor alpha in cancer development, progression and treatment: cause, consequence or innocent bystander? Int J Cancer. 2006;119(2):243–250. doi: 10.1002/ijc.21712 [DOI] [PubMed] [Google Scholar]
- 54.Jhaveri MS, Rait AS, Chung KN, Trepel JB, Chang EH. Antisense oligonucleotides targeted to the human alpha folate receptor inhibit breast cancer cell growth and sensitize the cells to doxorubicin treatment. Mol Cancer Ther. 2004;3(12):1505–1512. [PubMed] [Google Scholar]
- 55.Brooks SC, Locke ER, Soule HD. Estrogen receptor in a human cell line (MCF-7) from breast carcinoma. J Biol Chem. 1973;248(17):6251–6253. [PubMed] [Google Scholar]
- 56.Masters JR. HeLa cells 50 years on: the good, the bad and the ugly. Nat Rev Cancer. 2002;2(4):315–319. doi: 10.1038/nrc775 [DOI] [PubMed] [Google Scholar]
- 57.Meier R, Henning TD, Boddington S, et al. Breast cancers: MR imaging of folate-receptor expression with the folate-specific nanoparticle P1133. Radiology. 2010;255(2):527–535. doi: 10.1148/radiol.10090050 [DOI] [PubMed] [Google Scholar]
- 58.Matherly LH, Gangjee A. Discovery of novel antifolate inhibitors of de novo purine nucleotide biosynthesis with selectivity for high affinity folate receptors and the proton-coupled folate transporter over the reduced folate carrier for cellular entry In: Jackman ALC, editor. Targeted Drug Strategies for Cancer and Inflammation. Boston, MA: Springer; 2011:119–134. [Google Scholar]
- 59.Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, Kirchhausen T. Dynasore, a cell-permeable inhibitor of dynamin. Dev Cell. 2006;10(6):839–850. doi: 10.1016/j.devcel.2006.04.002 [DOI] [PubMed] [Google Scholar]
- 60.Fretz M, Jin J, Conibere R, et al. Effects of Na+/H+ exchanger inhibitors on subcellular localisation of endocytic organelles and intracellular dynamics of protein transduction domains HIV-TAT peptide and octaarginine. J Control Release. 2006;116(2):247–254. doi: 10.1016/j.jconrel.2006.07.009 [DOI] [PubMed] [Google Scholar]
- 61.Langston Suen WL, Chau Y. Size-dependent internalisation of folate-decorated nanoparticles via the pathways of clathrin and caveolae-mediated endocytosis in ARPE-19 cells. J Pharm Pharmacol. 2014;66(4):564–573. doi: 10.1111/jphp.12134 [DOI] [PubMed] [Google Scholar]