

Abstract
The Unfolded Protein Response (UPR) plays a key role in the adaptive response to loss of protein homeostasis within the endoplasmic reticulum (ER). The UPR has an adaptive function in protein homeostasis, however, sustained activation of the UPR due to hypoxia, nutrient deprivation, and increased demand for protein synthesis, alters the UPR program such that additional perturbation of ER homeostasis activates a pro-apoptotic program. Since ubiquitination followed by proteasomal degradation of misfolded proteins within the ER is a central mechanism for restoration of ER homeostasis, inhibitors of this pathway have proven to be valuable anti-cancer therapeutics. Ubiquitin activating enzyme 1(UAE1), activates ubiquitin for transfer to target proteins for proteasomal degradation in conjunction with E2 and E3 enzymes. Inhibition of UAE1 activity in response to TAK-243, leads to an accumulation of misfolded proteins within the ER, thereby aggravating ER stress, leading to DNA damage and arrest of cells in the G2/M phase of the cell cycle. Persistent drug treatment mediates a robust induction of apoptosis following a transient cell cycle arrest. These biological effects of TAK-243 were recapitulated in mouse models of PDAC demonstrating antitumor activity at a dose and schedule that did not exhibit obvious normal tissue toxicity. In vitro as well as studies in mouse models failed to show enhanced efficacy when TAK-243 was combined with ionizing radiation or gemcitabine, providing an impetus for future studies to identify agents that synergize with this class of agents for improved tumor control in PDAC.
Significance
The UAE1 inhibitor TAK-243, mediates activation of the unfolded protein response, accumulation of DNA breaks and apoptosis, providing a rationale for the use as a safe and efficacious anti-cancer therapeutic for PDAC.
Keywords: Pancreatic cancer, UAE1 Inhibitor, ER stress
Graphical abstract
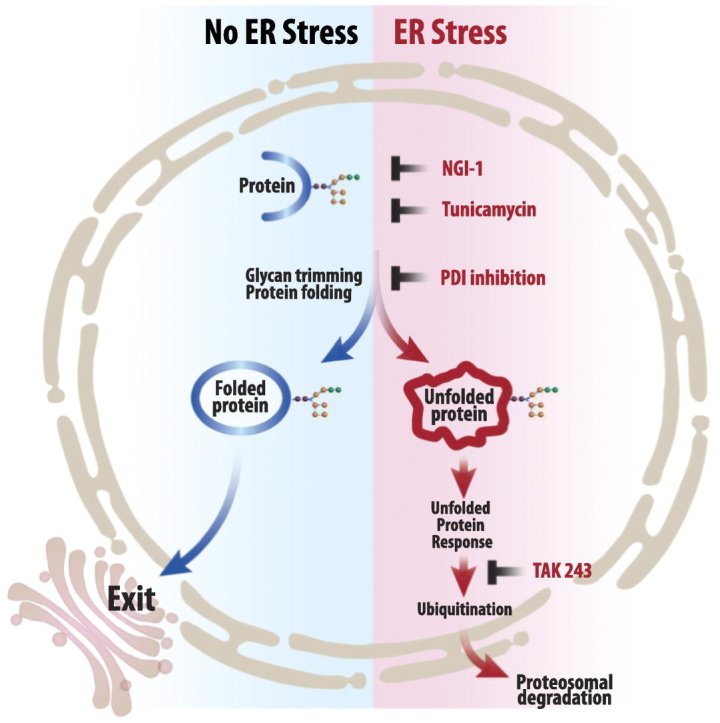
Highlights
-
•
Inhibition of Ubiquitin activating enzyme 1(UAE1) leads to an accumulation of misfolded proteins within the ER.
-
•
Persistent drug treatment mediates a robust induction of apoptosis in mouse models of Pancreatic Cancer demonstrating antitumor activity at a dose and schedule that did not exhibit obvious normal tissue toxicity.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a lethal disease with a five-year survival rate of 9.3%. Surgical resection at an early stage of the disease proves to be most effective, however, the asymptomatic nature of the disease leads to progression into advanced stages prior to diagnosis, when tumors metastasize to adjacent or distant tissue and organs [1,2]. Since tumor relapse is often seen in patients treated with only surgical intervention, adjuvant therapy with a modified FOLFIRINOX regimen has been adopted, leading to significantly longer survival than gemcitabine among patients with resected pancreatic cancer, at the expense of a higher incidence of toxic effects [[3], [4], [5], [6], [7]]. Targeted agents have shown benefit in patients bearing specific mutations (e.g. in the homologous recombination repair pathway, particularly BRCA) in several clinical trials [8,9], however, a majority of patients are not likely to benefit from these agents. This underscores a desperate need for novel therapeutic strategies that are effective and applicable for a majority of pancreatic cancer patients, irrespective of mutation status.
Solid tumors are constantly exposed to stress such as hypoxia, nutrient deprivation, as well as increased demand for protein synthesis. These stresses result in the accumulation of misfolded and unfolded protein within the endoplasmic reticulum (ER), a condition termed ER stress. In contrast to normal cells, cancer cells demonstrate a constant activation of the ER stress signaling pathway which contributes to their survival and adaptation to the tumor microenvironment [10]. ER stress activates the unfolded protein response (UPR) which includes inhibition of protein translation and transcriptional upregulation of chaperones that enhance the capacity for protein folding. Since accumulation of misfolded proteins within the ER harbors the risk of proteotoxicity, unfolded and misfolded proteins are retrotranslocated to the cytosol and ubiquitinated for proteasome-mediated degradation [11]. Post-translational ubiquitination of misfolded proteins is a requisite step for ER stress associated protein degradation (ERAD), involving the covalent conjugation of a ubiquitin moiety with a lysine residue of a target protein or another ubiquitin molecule (poly-ubiquitination). Ubiquitin activating enzyme (UAE) or E1, activates ubiquitin via an adenylate intermediate and catalyzes its transfer to an ubiquitin-conjugating enzyme (E2) and finally to the ubiquitin ligase (E3) that enables the conjugation of ubiquitin to the target substrate.
In cancer, the UPR and ERAD are an adaptive cellular response to the environment, and have been associated with oncogenic phenotypes including transformation and metastatic potential, in cell dormancy, genomic stability, angiogenesis as well as immunogenic tolerance [12]. Therapeutic targeting of proteostasis in cancer is increasingly being investigated as an opportunity to aggravate chronic ER stress within cancer cells by either inhibiting protein folding [11,13], inhibiting UPR signaling responses, as well as by inhibiting ERAD [14,15]. Bortezomib, a prototype for this approach, is a proteasome inhibitor and was approved in 2003 for the treatment of multiple myeloma and mantle cell lymphoma [16]. Inhibition of the 26S proteasome by the small molecule leads to the accumulation of misfolded proteins, thus aggravating ER stress, leading to proteotoxic conditions and the induction of apoptosis in cancer cells, while normal cells survive the insult due to their reserve capacity to adapt to the increased ER stress.
Prompted by this success, TAK-243, a first in class small molecule inhibitor of the UAE1, is being investigated as an anti-cancer therapeutic [[17], [18], [19]]. Since there is an urgent need to improve outcomes in patients with PDAC, the present study investigates the therapeutic potential of the agent in mouse models of the disease, the potential of combining it with standard of care therapies including radiation and gemcitabine as well as the mechanistic underpinnings of the agent's biological activity. We demonstrate that TAK-243 disrupts protein homeostasis through the inhibition of protein ubiquitination and thereby aggravating ER stress, leading to DNA damage and arrest of cells in G2/M phase of the cell cycle. Persistent drug treatment mediates a robust induction of apoptosis following a transient cell cycle arrest. These biological effects of TAK-243 were recapitulated in mouse models of PDAC and demonstrated antitumor activity at a dose and schedule that did not demonstrate obvious normal tissue toxicity. It should be noted that inhibition of UAE1 by TAK-243 may lead to cellular responses that are independent of the UPR, and a result of perturbation of a different cellular process dependent on UAE1 activity.
Material and methods
Cell lines and treatments
MiaPaCa-2 cells and Panc-1 cells were purchased from ATCC and the mouse pancreatic ductal adenocarcinoma cell line KPC2 (FVB) has been described previously [20]. All cells were maintained in DMEM (Corning) and 10% fetal bovine serum (Hyclone) and grown in 37 °C incubator with 5% CO2. All cell lines were routinely tested for mycoplasma (Lonza) and authenticated by short tandem repeat (STR) profiling at the University of Michigan Sequencing Core.
Apoptotic reporter cell lines were established by infecting pancreatic cell lines with a luciferase reporter for caspase-3 (Fig. 1A) [21]. Cells were dosed with CTEP agents: TAK-243 (MLN7243) (selleckchem, S8341), MLN4924 (pevonedistat or TAK-924) (selleckchem, S7109), VX-970 (M6620) (selleckchem, S7102), VX-984 (MedChemExpress, HY-19939S), AZD1775 (adavosertib), AZD2281 (olaparib) (selleckchem, S1060), BMN-673 (talazoparib) (selleckchem, S7048), ABT-888 (veliparib) (selleckchem, S1004) at 1 μM final concentration (in DMSO) for 4 h and luciferin (Promega) (in PBS) was added to each well at a final concentration at 150 μg/mL and luminescence was measured using a Perkin Elmer Envision microplate reader every 30 min for up to 36 h.
Fig. 1.

TAK-243 induces apoptosis in a dose and time dependent manner.
(A) A non-invasive reporter for apoptosis. Activated caspase-3 cleaves the DEVD sequence within the reporter to form a functioning luciferase enzyme that enzymatically releases photons of light in the presence of luciferin as substrate. (B) MiaPaCa-2 and Panc-1 cells with apoptosis reporter were dosed with CTEP compounds (1 μM) and luciferase activity was measured every 30 min to up to 19 h. (C) MiaPaCa-2, Panc-1 and KPC2 cells with apoptosis reporter in a 96-well plate were treated with TAK-243 at indicated concentration together with luciferin (150 μg/mL) and the plate was read every 30 min to up to 36 h. (D) MiaPaCa-2 and (E) Panc-1 cells were treated with TAK-243 at 50, 100, 200, 300 and 1000 nM or tunicamycin (5 μg/mL) for 4–24 h and the cells were lysed and assessed by immunoblotting for cleaved PARP.
Pancreatic cancer cells were infected with pLVX-XBP1-mNeonGreen (Addgene) and stable cells were established by puromycin selection. XBP1 reporter cells were plated onto glass bottom 6-well plate (Cellvis) and TAK-243 at 300 nM and 5 μg/mL tunicamycin (Sigma Aldrich) were added and images were taken every 30 min for 24 h on a Zeiss LSM800 confocal microscope.
Panc-1, MiaPaCa-2 and KPC2 cells were grown in 6-well culture dishes and treated with TAK-243 at 0 nM, 50 nM, 100 nM, 200 nM, 300 nM and 1 μM TAK-243 for 4, 8, 26, 24 h and cells were also treated with tunicamycin (Sigma Aldrich) at 5 μg/mL as indicated.
Western blotting
Immunoblotting was performed as previously described [13]. Briefly, whole cell lysates were collected in RIPA buffer, and 10–30 μg total protein was fractionated by SDS-PAGE and transferred to PVDF membranes (Millipore). Primary antibodies to BIP, ATF4, CHOP, RAD51, α-actin (all from Cell Signaling) and ubiquitin (Santa Cruz) were applied followed by horseradish peroxidase-conjugated goat antirabbit IgG (H + L) and goat anti-mouse (H + L) (Jackson ImmunoResearch) as secondary antibodies. Pierce™ ECL (Thermal Scientific) or ECL™ prime (GE Healthcare) were used as substrate.
Immunoprecipitation
For immunoaffinity purification of endogenous ubiquitinated RAD51, 10 cm tissue culture dish was washed with cold PBS and lysed using lysis buffer A (50 mM Tris-HCL (pH 7.5), 150 mM NaCl, 0.5 mM EDTA, 1% Triton X100) with fresh 1 mM NEM, protease and phosphatase inhibitors (Roche). 1 mg of protein was incubated overnight at 4 °C with anti-RAD51 antibody (Cell Signaling). Protein A sepharose beads (Pierce™)were added to these lysates and rotated at 40C for 2 h, washed 3 times with lysis buffer A and resuspended in 2× Laemmli buffer (Bio-Rad). The immunoprecipitates were divided into 2 parts; 90% of immunoprecipitates were probed for ubiquitin antibody whereas 10% of immunoprecipitates were probed with RAD51 antibody.
Quantitative polymerase chain reaction
MiaPaCa-2 cells were grown in 6-well dishes and TAK-243 was added at a final concentration at 300 nM for 0, 6, 8 and 10 h. Total RNA was extracted with Qiagen RNeasy kit and cDNA was synthesized using High-Capacity Reverse Transcription kit (Applied Biosystems). Quantitative PCR was performed using SYBR Green Master Mix (Bio-Rad) on a ViiA real-time PCR system (Applied Biosystems) with denaturation at 95 °C for 15 s; 55 °C for 30 s; 72 °C for 30 s. mRNA expression level was evaluated using the ΔΔ Ct method.
Cell cycle analysis
Cells were pelleted and re-suspended in 100 μL PBS and fixed in ice-old 70% ethanol and stained with propidium iodide (Sigma-Aldrich) at a final concentration of 20 μg/mL with RNase A (100 μg/mL from Invitrogen) for 30 min. Monitoring of cell cycle phase was performed on a Cytoflex flow cytometer (Beckman Coulter), and data was analyzed with FlowJo software (V10).
Comet assay
Double strand DNA damage was measured by neutral method with Trevigen CometAssay® kit according to manufacturer's instruction and Olive and colleague [22]. Briefly, cells were treated with TAK-243 (300 nM) for 8 and 24 h and 1 × 105 cells/mL in suspension was mixed with molten SeaPlaque™ Agarose at 1:10 (v/v, Lonza) and spread onto CometSlide™ (Trevigen), the slides were then submerged in lysis buffer (2% sarkosyl, 0.5 M Na2EDTA, 0.5 mg/mL proteinase K, pH 8.0) for overnight at 37 °C. Electrophoresis was performed in TBE buffer at 0.6 V/cm at 4 °C, and then the slides were immersed in 70% ethanol for 30 min and then stained with SYBR™ Green (Invitrogen). All images were taken with a fluorescence microscope and analyzed by Comet Assay IV software (Perceptive Instruments).
Clonogenic assay
Cells were treated with TAK-243 (300 nM) or PDI shRNA (72 h) and irradiated with 2, 4, 6, 8 Gy single dose with an IC-320 orthovoltage irradiator (Kimtron Medical). Cells were plated the following day at a clonogenic density with fresh medium for 7–14 days before fixing with 10% formalin (Sigma-Aldrich) and were stained with 0.1% crystal violet (Sigma-Aldrich). Radiation survival curves were normalized for drug toxicity, and the radiation enhancement ratio was calculated as the ratio of the mean inactivation dose (area under the cell survival curve) under control conditions divided by the mean inactivation dose after drug exposure. A value significantly greater than 1 indicates radiosensitization. Cytotoxicity in the absence of radiation treatment was calculated by normalizing the plating efficiencies of drug-treated cells to non–drug-treated cells [23].
Animal studies
A total of 1 × 106 MiaPaCa 2 cells were injected subcutaneously into 6 to 8 weeks old NCRNU sp/sp mice (Taconic) in 100 μL DMEM (Corning): matrigel (BD Bioscience, 1:1) suspension. Tumor size was monitored bi-weekly, and volume was calculated as (L × W × W)/2, where W is tumor width and L is tumor length. Mice were randomized into 4 groups when tumor volume reached around 100 mm3, and each group had 5 mice. TAK-243 was given at 12.5 mg/kg in 10% (2-hydroxypropyl)-β-cyclodextrin (Sigma Aldrich), twice per week via tail vein to one group; Gemcitabine was given 100 mg/kg, diluted in PBS, once per week via IP to one group, and one group of mice received both TAK-243 and Gemcitabine, the rest of the mice were given 10% (2-hydroxypropyl)-β-cyclodextrin for 3 weeks. All animal experiments were approved by the University of Michigan Committee on the Use and Care of Animals. Tumor tissues were harvested by the end of the treatment and fixed in 10% neutral-buffered formalin, sections were prepared and stained by University of Michigan Core services. Images were taken using an Olympus BX-84 and the number of positive cells was counted in three fields of view and analyzed using ImageJ software.
Statistical analysis
Statistical analysis of data was performed using GraphPad Prism v8.0. Statistical significance was determined by the 2-tailed unpaired Student t-test. P values are reported in the graphs. *, P < 0.05; **, P < 0.01; and ***, P < 0.001. n.s. denotes not significant. For clonogenic assay, cell survival curves were fit using the linear-quadratic equation, and the mean inactivation dose (MID), which is the linear area under the cell survival curve, was calculated. The radiation enhancement ratio was defined as the (MIDCTRL/MIDTREATMENT) so that a ratio > 1 indicates radiosensitization. Data are presented as the mean ± the standard error of at least three experiments.
Results
TAK-243 induces apoptosis in pancreatic cells
In an effort to identify effective anti-cancer agents for PDAC that can be rapidly moved to clinical applications, we selected agents from the Cancer Therapy Evaluation Program (CTEP) collection that target the DNA damage response of cancer cells so that these can be applied as radio- or chemo-sensitizers. Targeted agents including veliparib (PARP inhibitor), talazoparib (PARP inhibitor), and olaparib (PARP inhibitor), AZD1775 (Wee1 inhibitor), VX970 (ATR kinase inhibitor), VX984 (DNA-PK inhibitor), MLN4924 (NAE1 inhibitor) and TAK-243 (UAE1 inhibitor) were interrogated against live cell assay for Caspase 3/7 activation as a surrogate for the activation of the apoptotic cascade (Fig. 1A). TAK-243, a recently described UAE1 inhibitor [19], was identified as the most efficacious agent among the eight tested, and demonstrated a 25 and 30-fold activation of the caspase 3/7 reporter at 17.5 h in MiaPaCa-2 and Panc-1 cells respectively. The second most effective compound AZD1775, a WEE1 kinase inhibitor demonstrated a maximum of 5-fold increase in reporter activation compared to control (Fig. 1B). Since AZD1775 in combination with gemcitabine and radiation therapy has recently demonstrated substantially higher overall survival than prior results combining gemcitabine with radiation therapy, and was well tolerated [24], we concluded that TAK-243 warranted further investigation. A dose response study using TAK-243 showed a dose-dependent activation of the reporter in each of the human PDAC cell lines including a mouse PDAC line, KPC2. In each of the three cell lines, concentrations as low as 100 nM TAK-243 were able to induce detectable levels of apoptotic cell death within 24 h, with at least a 5-fold increase in reporter activation (Fig. 1C). In addition, Western blot analysis confirmed these findings biochemically through the detection of cleaved PARP, a marker for caspase 3 activation, in a dose and time dependent manner in each of the cell lines (Fig. 1D–E).
TAK-243 induces endoplasmic reticulum stress
UAE1 is a critical E1 component of the ubiquitin-proteasome system responsible for eliminating misfolded and unfolded proteins within the ER [25] by degradation (ERAD), and therefore plays a central role in proteostasis within the ER. We reasoned that inhibition of UAE1 would lead to accumulation of mis- and un-folded proteins and therefore induce ER stress and activate the unfolded protein response (UPR). We initially evaluated the activation of the UPR in response to TAK-243 in MiaPaCa-2 cells at the mRNA level using qPCR and observed an upregulation of transcripts for GRP78 as well as the spliced form of XBP1, key markers of the UPR [26,27]. A 1.3-fold increase (p < 0.05) after 2 h in both genes and a 3.5-fold (p < 0.01) and 53.4-fold (p < 0.001) increase at 6 h post-treatment of GRP78 and spliced XBP1 mRNA respectively was observed (Fig. 2A). To quantitatively and dynamically evaluate the UPR after UAE1 inhibition, we utilized a reporter construct that detects inositol-requiring enzyme 1 (IRE1)-alpha mediated splicing of X-box binding protein 1 (XBP1) [28] (Fig. 2B). Treatment of MiaPaCa-2 cells with TAK-243 (100 nM) led to a significant increase in GFP expression beginning 3 h (2-fold increase) and became saturated at approximately 16 h (4-fold increase) (Fig. 2C, D), whereas in Panc-1 cells, activation of IRE-1 became apparent at approximately 4 h (2-fold increase) and stabilized at 15 h (5.5-fold increase) upon TAK-243 treatment (Fig. 2C, E). We further confirmed these findings at the protein level wherein a robust, dose and time dependent accumulation of UPR responsive proteins: BiP, ATF4 and CHOP was observed after TAK-243 treatment in each of the PDAC cell lines tested (Fig. 2F–H). Activating transcription factor 4 (ATF4), an ER stress-induced transcription factor which mediates the expression of stress adaptive genes, was most readily detected as a differentially expressed protein upon TAK-243 treatment, even at doses that did not significantly induce apoptosis. However, under conditions of persistent (>12 h) ER stress or at high doses of the agent (>100 nM, Fig. 2F, G and H), a robust increase in ATF4 levels correlated with a large increase in caspase 3/7 activation (Fig. 1C). This is consistent with the duality of functions ascribed to ATF4 in cell adaptation and survival, while promoting cell death under persistent stress conditions [29].
Fig. 2.


TAK-243 activates the unfolded protein response.
(A) MiaPaCa-2 cells were treated with 300 nM TAK-243 for 1, 2, 4 and 6 h and total RNA was extracted for qRT-PCR of GRP78 and spliced XBP-1. Data is presented as mean ± SEM from three experiments, *, p < 0.05; **, p < 0.01; ***, p < 0.001. (B) IRE1 activity sensor expresses mNeonGreen when XBP-1 is spliced. Representative pictures of (C) MiaPaCa-2 and (D) Panc-1 (E) cells with spliced IRE1 reporter after TAK-243 or DMSO treatment at different time point. (E) Quantification of spliced XBP-1 fluorescence signal over surface area in MiaPaCa-2 and Panc-1 cells treated with 300 nM TAK-243, data is presented as mean ± SEM from three technical replicates. Immunoblotting of UPR markers: ATF-4, BIP and CHOP in (F) MiaPaCa-2, (G) Panc-1 and KPC2 (H) cells after TAK-243 or tunicamycin treatment at indicated dose and time. (I) Quantification of spliced XBP-1 fluorescence signal over surface area in MiaPaCa-2 cells treated with 300 nM TAK-243, BAP2, Tunicamycin, NGI-1 and PDI SiRNA. Data is presented as mean ± SEM from three technical replicates.
N-glycosylation and N-glycan trimming ensures that newly synthesized glycopolypeptides undergo proper folding, export and translocation within the ER [30]. Hence agents such as tunicamycin, which inhibit N-linked glycosylation, circumvent protein folding leading to activation of the UPR. Tunicamycin, an inhibitor of dolichyl-phosphate N-acetylglucosamine-phospho-transferase and a canonical activator of the UPR, when used as control in each of these studies, demonstrated an increase in BiP, ATF4 and CHOP protein levels (Fig. 2F–H), and led to the activation of caspase activity (Fig. 1D and E) although to a lesser extent compared to TAK-243, suggesting that these two compounds may activate the UPR in a distinct manner. As seen in Fig. 2F, and G, tunicamycin treatment elicited a UPR which was exemplified by an induction of BiP expression, a minor induction of ATF4 was observed in MiaPaCa-2 cells, however, this increase was dwarfed compared to what was observed in response to TAK-243. Conversely, the induction of BiP observed in response to tunicamycin treatment was greater compared to that observed in response to TAK-243. This differential response to ER stress was further investigated using the IRE-1α reporter, which demonstrated that activation of IRE-1α mediated RNA splicing peaked at 6 fold over background in response to TAK-243 at 35 h post-treatment. In contrast, using the same cell line, tunicamycin treatment resulted in peak activation at 20 h of 2.5 fold (Fig. 2H). To further corroborate this observation, we utilized a small molecule, NGI-1, which targets the oligosaccharyltransferase complex within the ER [31,32] and thereby inhibits the glycosylation machinery. NG-1 treatment resulted in a modest (1.8 fold) activation of the IRE1α reporter at 18 h post-treatment in MiaPaCa-2 cells. We next evaluated activation of the UPR in response to inhibition of protein disulfide isomerase (PDI) mediated protein folding activity, utilizing a small molecule inhibitor (BAP2) [11], as well as siRNA knockdown [13]. BAP2 mediated inhibition of PDI activity resulted in a robust (4 fold) activation of the reporter in both cell lines, while knockdown of PDI transcripts resulted in a 2.5-fold increase in the IRE1 reporter activity at 36 and 18 h, respectively in MiaPaCa-2 cells (Supplementary Fig. 1). These findings demonstrate that TAK-243 mediated UPR activation was robust and distinct from the canonical ER stress inducer tunicamycin in regards to downstream signaling effects.
TAK-243 treatment leads to double strand breaks and a G2/M arrest
Cell cycle progression is strictly controlled by ubiquitin-mediated proteolysis of the key regulators including cyclins which are required for cyclin-dependent kinase activity [33]. We hence evaluated the consequence of UAE1 inhibition by TAK-243 on progression through the cell cycle, and identified an accumulation of cells in S and G2/M phases beginning at 6 h post-treatment, which eventually led to a predominantly G2/M arrest at 24 h (Fig. 3A), consistent with previous findings [18,19]. In comparison, induction of the UPR upon tunicamycin treatment led to a G1 arrest (Fig. 3A), further supporting our hypothesis that although TAK-243 and tunicamycin both activate the UPR, their impact on the cell cycle is distinct and could be attributed to a mechanism other than the activation of the UPR. Since the immediate cell cycle response of cells to TAK-243 was a delay in the S phase followed by a G2/M arrest, we reasoned that TAK-243 may alter DNA repair capacity, since ubiquitylation is required for the local enrichment of various protein complexes at sites of DNA damage to promote DNA repair [34]. Indeed, the COMET assay revealed persistent DNA damage upon TAK-243 treatment as early as 8 h after treatment, resulting in a 50% increase in tail moment at 24 h (Fig. 3B and C), consistent with previous findings [17,19,35].
Fig. 3.



TAK-243 induces cell cycle arrest, DNA damage and leads to a tumor growth delay.
(A) FACS analysis of DNA content in MiaPaCa-2 cells at 4, 6, 16, 24 h after DMSO, TAK-243 and tunicamycin treatment. (B) Representative images of comet assay from MiaPaCa-2 cells at indicated time points following 300 nM TAK-243 treatment. (C) Quantification of percentage of tail moment in MiaPaCa-2 cells after TAK-243 treatment, values are expressed as mean ± SEM for at least 100 counts from each condition. (D) MiaPaCa-2 tumor progression in control/DMSO, gemcitabine, TAK-243 and combination group, graph is plotted as mean ± SEM from at least 4 mouse. Representative images of immunohistochemistry on (E) ATF-4 and (F) Caspase-3 in tumors and their quantifications. Error bars represent S.E.M. *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.00005, ns: non-significant. (G) Quantification of ATF-4 and Caspase-3 expression and (H) Hematoxylin and eosin staining.
To leverage the observation that UAE1 inhibition leads to activation of the UPR, DNA damage, cell cycle arrest and apoptosis, we evaluated the therapeutic potential of TAK-243. As shown in Fig. 3D, mouse model studies to evaluate the safety and efficacy of TAK-243 (12.5 mg/kg, twice a week) demonstrated a significant delay in tumor growth of MiaPaCa-2 flank xenografts. This delay in tumor growth could be attributed to activation of the UPR (increase in ATF4, Fig. 3E) as well as activation of caspase 3 (Fig. 3F). Quantitative analysis of immunohistochemistry sections from multiple animals confirmed this finding (Fig. 3G). In addition, hematoxylin and eosin staining demonstrated elevated necrotic area after UAE inhibition (Fig. 3H), confirming that TAK-243 triggers ER stress and apoptosis, indicating the effectiveness of TAK-243 and hence its potential as a single agent for the treatment of PDAC.
Combination therapy for TAK-243
Although the above findings demonstrated safety and efficacy of TAK-243 in PDAC and at the same time provided in vivo confirmation for the mechanistic basis for drugs activity, we wanted to investigate if combination therapy with currently approved modalities may enhance the efficacy of the agent. We reasoned that the ability of TAK-243 to induce DNA damage could be leveraged to enhance the efficacy of DNA damage inducing modalities including radiation therapy [36,37], hence we evaluated the ability of TAK-243 to sensitize PDAC cells to radiation using clonogenic survival assays. However, these studies revealed that combination therapy using TAK-243 and ionizing radiation did not result in an enhanced cell killing at 100, 200 or 300 nM drug using MiaPaCa-2 or Panc-1(Fig. 4B and C). In contrast, inhibition of PDI, which also mediates ER stress and activation of the UPR [13], led to a potent radiosensitizing effect in MiaPaCa-2 cells as well as Panc-1 cells (enhancement ratios of 1.82 and 2.3 respectively, Fig. 4D and E). Enhancement of radiation sensitivity upon PDI inhibition has also been observed in glioblastoma cells previously [13] hence the lack of radiation sensitization upon TAK-243, a potent activator of the UPR, was surprising.
Fig. 4.

A lack of synergy when TAK-243 is combined with DNA damage inducing therapies.
Apoptotic signal in (A) MiaPaCa-2 and (B) Panc-1 cells treated with TAK-243 at indicated concentrations were radiated at 0, 2, 4, 6, 8 Gy and survival was assessed by plating cells 24 h post radiation. The values are expressed as mean ± SEM from three biological replicates. (C) MiaPaCa-2 and (D) Panc-1 cells with 72 h of PDI shRNA induction were radiated at 0, 2, 4, 6, 8 Gy and survival was assessed by plating cells 24 h post radiation, values are expressed as mean ± SEM from three biological replicates.
Since gemcitabine has been used for the management of patients with PDAC, we wanted to evaluate the efficacy of combination therapy using TAK-243 and gemcitabine. Using our caspase reporter, we observed that although TAK-243 as single agent mediated a potent dose dependent activation of apoptosis (Supplementary Fig. 2), inclusion of gemcitabine at 50 nM only modestly enhanced the induction of apoptosis in human (MiaPaCa-2 and Panc-1) and mouse (KPC2) PDAC models. Combination therapy compared to TAK-243 or gemcitabine alone, not only gave a small but significant increase in reporter activity, but also caused the peak activation to occur at earlier time points (Supplementary Fig. 2A) in the combination setting. Since these results suggested a superior potency of combining TAK-243 and gemcitabine, we then examined whether this could be recapitulated in vivo using MiaPaCa-2 cells in the flanks of nude mice. The combination therapy failed to show significant synergy in the mouse model (Supplementary Fig. 2B) confirming our observations using the caspase 3 reporter, that TAK-243 as a single agent exhibited efficacy, however, a lack of synergy between the TAK-243 and gemcitabine was demonstrated by a similar delay in tumor progression, although without additional systemic toxicity (Fig. 3D).
TAK-243 inhibits RAD51 ubiquitination and degradation
The consensus of current literature is that ER stress mediated activation of the UPR triggers activation of the ER-associated degradation pathway (ERAD) [38]. We and others have previously reported ubiquitination and proteasome mediated degradation of RAD51 in response to UPR activation [13,39,40], a key component of the homologous recombination mediated DNA repair machinery. However, we reasoned that inhibition of UAE1 by TAK-243 would abrogate ERAD, due to inability to ubiquitinate ERAD target proteins like RAD51. As shown in Fig. 5A, although TAK-243 led to a 15-fold increase in ATF4 levels, a 2-fold increase in RAD51 levels was observed in treated cells (Fig. 5A). In contrast, up-regulation of ATF4 and BiP protein levels in response to inhibition of protein glycosylation using tunicamycin or NGI-1 (Fig. 5B and C, respectively), as well as PDI inhibition (Fig. 5D), resulted in a 50% decrease in RAD51 levels. Restoration of the decreased RAD51 levels in these studies in the presence of the proteasome inhibitor MG132, recapitulated our previous work in glioblastoma, which demonstrate that activation of ERAD upon ER stress, leads to RAD51 degradation. In contrast, TAK-243 treated cells demonstrate elevated Rad51 levels, despite a robust activation of the UPR. The notion that RAD51 is ubiquitinated in response to activation of the UPR and ERAD was further elucidated using immunoprecipitation studies. As shown in Fig. 5E, treatment of cells with tunicamycin resulted in an increase in protein ubiquitination (in the presence or absence of MG132) as detected in whole cell extracts (input). Additionally, immunoprecipitation of RAD51 followed by Western blot analysis of the precipitate using a ubiquitin-specific antibody revealed that, compared to control cells or MG132 treated cells, RAD51 precipitated from tunicamycin treated cells (undergoing ERAD) demonstrated an increase in the ubiquitination form. MG132 treatment of tunicamycin treated cells further enhanced the ubiquitination of RAD51, presumably due to the inhibition of proteasomal degradation of the ubiquitinated RAD51 (Fig. 5E). Cells having activated UPR in response to PDI-inhibition also demonstrated an increase in global ubiquitination (Fig. 5F). In contrast, treatment of cells with TAK-243 (Fig. 5G) completely inhibited ubiquitination of total cellular proteins, even when MG132 was present. Immunoprecipitation of RAD51 from these samples confirmed that upon TAK-243 treatment, inhibition of UAE1 activity prevented the ubiquitination of RAD51 (Fig. 5G) despite a robust activation of the UPR.
Fig. 5.

TAK-243 mediated UPR is decoupled from ERAD.
MiaPaCa-2 cells were treated with (A) TAK-243 (300 nM), (B) tunicamycin (5 μg/mL), (C) NGI-1 (10 μM) with or without MG132 (10 μM) for 16, 12 and 6 h respectively, the cells were also transfected with (D) PDI siRNA for 48 h and MG132 was added for the last 16 h. Cell lysates were collected for immunoblotting for BIP, ATF4, RAD51, PDI and β-actin, and protein level was quantified and plotted as mean ± SEM from three biological replicates. MiaPaCa-2 cells were treated with (E) tunicamycin (5 μg/mL) with or without MG132 (10 μM) for 8 h and lysates were immunoprecipitated using RAD51 antibody, and the precipitates were immunoblotted with RAD51 and ubiquitin. (F) Cells were transfected with PDI siRNA and treated with or without with MG132 and total lysates were immunoblotted with ubiquitin and RAD51. (G) MiaPaCa-2 cells were treated with TAK-243 (300 nM) with or without MG132 (10 μM) for 6 h and lysates were immunoprecipitated as described above. Error bars represent S.E.M. *p < 0.05, **p < 0.005, ***p < 0.0005, ****p < 0.00005, ns: non-significant.
Discussion
Post-translational covalent attachment of the ubiquitin polypeptide (Ub) to specific protein substrates is required for efficient function of proteins as well as for protein turnover through the 26S proteasome. Ubiquitin-modification can lead to transcriptional or enzymatic activation, subcellular relocalization, intracellular trafficking, or degradation. Ubiquitination as well as additional ubiquitin-like post-translational modifications provide a regulatory node that maintains cellular homeostasis in normal development and in response to environmental insults. It is increasingly being appreciated that dysregulated ubiquitination contributes to the oncogenic phenotype by mediating abnormal cell proliferation and survival [41]. This post-translation modification of lysine residues on target substrate proteins involves activation, conjugation, and ligation by E1, E2, and E3 enzymes respectively. Although the E2 and E3 enzyme family is comprised of a large number of members (38 and >600, respectively), only two E1 family members exist in humans (UAE1 and UBA6). Of the two E1s, UAE1 charges a majority of cellular E2 ubiquitin conjugating enzymes [42].
The endoplasmic reticulum (ER) requires stringent protein homeostasis and contains elaborate signaling complexes to identify, ubiquitinate, and degrade misfolded proteins. ER proteostasis is critical for cell survival as exemplified by the endoplasmic reticulum associated degradation pathway (ERAD), wherein misfolded and unfolded proteins within the ER are degraded through the 26S proteasome upon ubiquitination. Perturbation of protein homeostasis within the ER leads to a rapid compensatory cellular response, through the UPR (unfolded protein response), to restore protein homeostasis. The tumor microenvironment perturbs protein homeostasis within cancer cells due to nutrient deprivation, hypoxia, and oxidative stress. Hence cancer cells experience chronic ER stress [12], which makes them vulnerable to additional proteostatic imbalance. The activation of oncogenes and the high proliferation rate also increases the demand on the protein synthesis machinery as well as the ER protein folding and quality control machinery, further contributing to increased ER stress in cancer cells compared to their normal counterparts [12]. Based on this rationale, many drugs have been developed that accentuate ER stress in cancer cells by targeting various processes within the UPR and ERAD [15]. Bortezomib, a first-in-class proteasome inhibitor, perturbs ER proteostasis, leading to cancer cell death, and has been approved for the treatment of specific cancers.
UAE1, a key ubiquitin activator required for ERAD, therefore provides a unique opportunity for the therapeutic intervention in cancer with the expectation that it would accentuate tumor cell ER stress. TAK-243, a first-in-class small molecule inhibitor of UAE1 [19], for which initial clinical safety studies are imminent (NCT02045095), has not been rigorously evaluated for efficacy in PDAC. The findings presented here provide compelling evidence for the potential for TAK-243 as a therapeutic in PDAC. Multiple human (MiaPaca-2 and Panc-1) as well as a mouse PDAC cell lines exhibit exquisite sensitivity to the agent, such that at doses that mediate an accumulation of unfolded proteins and activation of the UPR, lead to a robust induction of apoptosis. Administration of TAK-243 to mouse models of PDAC at doses that recapitulate the biological effects (activation of the UPR and apoptosis) observed in cultured cells, led to a tumor growth delay but did not exhibit any obvious normal tissue toxicity. This is in agreement with similar studies in AML [43], myeloma [35], B-cell lymphoma [18], squamous cell carcinoma, among others [44]. In agreement with the reported cellular response to TAK-243 in each of the above disease sites, our findings demonstrate that inhibition of protein ubiquitination leads to a dramatic induction of ER stress and activation of the UPR signaling pathway. At each dose tested, peak IRE1α (as determined using a non-invasive reporter), was observed prior to the induction of caspase activation (also evaluated using a live cell reporter). This is consistent with the observed peak in expression of ATF4, an important mediator of UPR-induced apoptosis [29]. In normal cells, translational upregulation of the ATF4 transcription factor in response to ER stress promotes the expression of adaptive genes, including those involved in amino acid transport and metabolism, protection from oxidative stress [45], as well as protein chaperones [46]. Distinct from its role in ER stress, ATF4 also regulates a wide range of genes including during osteogenesis [47], however, during long-term ER stress, an increase in ATF4 results in transcription of CCAAT-enhancer-binding protein homologous protein (CHOP), which is responsible for activating a transcriptional program that leads to initiation of the apoptotic cascade [48]. Under conditions of chronic ER stress (as experienced by cancer cells), accentuation of ER stress (e.g. in response to UAE1 inhibition) induces a change in the transcriptional program such that the ATF4-CHOP axis, leads to downregulation of the anti-apoptotic proteins like BCL2 and upregulation of pro-apoptotic proteins such as BIM, NOXA, and PUMA [29]. The differential response to ER stress of tumor cells compared to normal cells may be the basis for the therapeutic index of TAK-243 in mouse models.
Combination chemotherapy is standard adjuvant treatment for patients with resected pancreatic cancer, because single-agent therapies have not impacted outcomes dramatically. Often, additional chemotherapy is given to these patients once recurrence occurs [4,24]. This prompted us to investigate the potential of combining TAK-243 with gemcitabine or radiation. The justification for these studies was based on the finding that TAK-243 treated cells exhibit an increase in DNA double strand breaks. Ubiquitylation not only facilitates the local enrichment of various protein complexes to sites of strand breaks to promote DNA repair, it also has important roles in determining which pathway is used to repair individual breaks. Hence it is not unexpected that TAK-243, despite abrogating Rad51 degradation, leads to unrepaired DNA damage [34]. Combination therapy including TAK-243 with DNA damage-inducing agents such as gemcitabine and radiation [36,49], should lead to synergistic cell killing, however, our in vitro studies failed to show any synergy, which was confirmed in mouse xenografts wherein TAK-243 was administered as single agent or in combination with gemcitabine. Since previous work from our laboratory as well as others [13,39,40] has shown synergy when ER stress inducing agents are combined with DNA damage inducing modalities, this result was unexpected. In an effort to explain these findings, we leveraged published work that associated ubiquitination and ERAD mediated degradation of RAD51 as an often observed phenomenon. Activation of the UPR in response to tunicamycin or PDI inhibition resulted in a decrease in RAD51 protein levels, which could be rescued in the presence of MG132, suggesting its ubiquitin-mediated proteasomal targeting. Indeed, analysis of ubiquitination of RAD51 immunoprecipitated from cells treated with each of these UPR activating insults, confirmed that an increase in ubiquitination of total cellular proteins and specifically Rad51 was detected, especially in the presence of MG132. In contrast, treatment with TAK-243, although led to a robust activation of the UPR as evidenced by IRE1α and ATF4 increase (but not BiP), resulted in an accumulation of RAD51 levels and a complete loss of ubiquitination of total cellular proteins as well as RAD51 itself. We hence propose ubiquitination and degradation of DNA repair proteins which includes RAD51 as a prototype, is critical for sensitization of cells to DNA damage inducing therapies when combined with agents that mediate ER stress, activation of the UPR and ERAD.
Targeting of pathways that maintain cellular protein homeostasis is now appreciated as a viable anti-cancer approach. Examples include Carfilzomib which is approved for clinical use, and MLN4924 (Pevonedistat, TAK-924) which is also undergoing clinical trials as combination therapy for many liquid and solid tumors. MLN4924 functions as a potent and selective inhibitor of NEDD8-activating enzyme (NAE). Within the neddylation pathway, NAE is a functional analog of the UAE1 (Ubiquitin-activating enzyme). MLN4924 is efficacious at doses that are safe and is being evaluated for use in combination with DNA damage inducing therapies such as radiation [[50], [51], [52], [53]]. Our in vivo findings clearly demonstrate that TAK-243 is able to activate the UPR pathway (as evidenced by ATF4 expression) in a tumor specific manner at doses that are safe. In addition, at these doses, a robust activation of caspase 3 was observed in response to UPR activation, which likely contributes to the tumor growth delay observed in mouse models. Inhibition of UAE1 by TAK-243 may lead to cellular responses that are independent of the UPR, and a result of perturbation of a different cellular process dependent on UAE1 activity. To address this possibility we perturbed cellular proteostasis through four distinct mechanisms (Tunicamycin, PDI-inhibition, NG-1 and TAK-243), thus providing some confidence that the observed phenotypes are related to the UPR. Clinical translation of the agent will require additional dose and schedule optimization studies as well as optimal combination therapy strategies. Our current findings of combining TAK-243 with DNA damaging agents including ionizing radiation and gemcitabine suggest that alternate strategies for combination therapy will need to be evaluated. Our current findings provide a detailed understanding of the cellular response to TAK-243 and provide potential biomarkers of drug-target engagement and efficacy using imaging and biochemical readouts, which will be invaluable for future translational studies for optimization of dose, schedule and combination therapies.
The following are the supplementary data related to this article.
MiaPaCa-2 and Panc-1 cells were treated with ER stress inducers: BAP2 (30 μM), tunicamycin (5 μg/mL), TAK-243 (300 nM), NGI-1 (20 μM), as well as PDI siRNA (Santa Cruz) and IRE1 activity was measured and recorded every 30 min for up to 24 h. Data is presented as mean ± SEM from 4 technical replicates.
(A) Fold change of cleaved Caspase-3 reporter in MiaPaCa-2, Panc-1 and KPC2 cells after gemcitabine and TAK-243 treatment at indicated doses and time, data is presented as mean ± SEM from three experiments. (B) MiaPaCa-2 cells tumor progression after vehicle control, TAK-243, gemcitabine and combination treatment, data is presented as mean tumor volume ± SEM (n ≥ 4).
MiaPaCa-2 cells were treated with PDI siRNA for 48 h and MG132 was added for the last 16 h. Cell lysates were collected for immunoblotting for BIP, RAD51, PDI and β-actin.
Financial support
This work was supported by the National Institutes of Health Grants: CA216449 (T.S.L., M.M. and A.R.), CA193690 (N.N. and A.R.) and P30 CA046592-29.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Acknowledgements
We would like to thank Drs. Meilan Liu, Chao Zhang, Qiang Zhang, Wenbin Ji (Department of Radiation Oncology) and Yin Wang (Department of Internal Medicine, Hematology and Oncology), for sharing their expertise on comet assay and live cell imaging, as well as Mr. Steven Kronenberg (Department of Radiation Oncology) for his help on preparing the figures. We also want to thank University of Michigan Biomedical Research Flow Cytometry Core, Advanced Genomics Core, Unit for Laboratory Animal Medicine and the In-Vivo Animal Core for their services.
Authors' contributions
Conception and design: A. Rehemtulla, M. Morgan, T. Lawrence and N. Neamati.
Development of methodology: M. Morgan, M. Ljungman, A. Delaney, Y. Liu, S. Awadia and S. Gonzalez.
Acquisition of data: A. Delaney, Y. Liu, S. Awadia, C. Engelke, S. Gonzalez, and M. Ljungman.
Analysis and interpretation of data: A. Rehemtulla, M. Morgan, M. Ljungman, A. Delaney, Y. Liu, S. Awadia and C. Engelke.
Preparation of manuscript: Y. Liu, A. Rehemtulla, S. Awadia and M. Morgan.
Administrative, technical, or material support: M. Sitto, H. Patel, A. Calcaterra, H. Lee, and J. Contessa.
References
- 1.Wagner M., Redaelli C., Lietz M., Seiler C.A., Friess H., Buchler M.W. Curative resection is the single most important factor determing outcome in patients with pancreatic adenocarcinoma. Brit J Surg. 2004;91:586–594. doi: 10.1002/bjs.4484. [DOI] [PubMed] [Google Scholar]
- 2.Winter J.M., Brennan M.F., Tang L.H., D'Angelica M.I., Dematteo R.P., Fong Y., Klimstra D.S., Jarnagin W.R., Allen P.J. Survival after resection of pancreatic adenocarcinoma: results from a single institution over three decades. Ann. Surg. Oncol. 2012;19:169–175. doi: 10.1245/s10434-011-1900-3. [DOI] [PubMed] [Google Scholar]
- 3.Conroy T., Desseigne F., Ychou M., Bouche O., Guimbaud R., Becouarn Y., Adenis A., Raoul J.L., Gourgou-Bourgade S., de la Fouchardiere C., Bennouna J., Bachet J.B., Khemissa-Akouz F., Pere-Verge D., Delbaldo C., Assenat E., Chauffert B., Michel P., Montoto-Grillot C., Ducreux M., Groupe Tumeurs Digestives of, U., and Intergroup, P FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011;364:1817–1825. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 4.Conroy T., Hammel P., Hebbar M., Ben Abdelghani M., Wei A.C., Raoul J.L., Chone L., Francois E., Artru P., Biagi J.J., Lecomte T., Assenat E., Faroux R., Ychou M., Volet J., Sauvanet A., Breysacher G., Di Fiore F., Cripps C., Kavan P., Texereau P., Bouhier-Leporrier K., Khemissa-Akouz F., Legoux J.L., Juzyna B., Gourgou S., O'Callaghan C.J., Jouffroy-Zeller C., Rat P., Malka D., Castan F., Bachet J.B., Canadian Cancer Trials G., the Unicancer, G. I. P. G. FOLFIRINOX or gemcitabine as adjuvant therapy for pancreatic cancer. N. Engl. J. Med. 2018;379:2395–2406. doi: 10.1056/NEJMoa1809775. [DOI] [PubMed] [Google Scholar]
- 5.Conroy T., Van Laethem J.L. Combination or single-agent chemotherapy as adjuvant treatment for pancreatic cancer? Lancet Oncol. 2019;20:336–337. doi: 10.1016/S1470-2045(19)30107-X. [DOI] [PubMed] [Google Scholar]
- 6.Kosuge T., Kiuchi T., Mukai K., Kakizoe T., Japanese Study Group of Adjuvant Therapy for Pancreatic, C A multicenter randomized controlled trial to evaluate the effect of adjuvant cisplatin and 5-fluorouracil therapy after curative resection in cases of pancreatic cancer. Jpn. J. Clin. Oncol. 2006;36:159–165. doi: 10.1093/jjco/hyi234. [DOI] [PubMed] [Google Scholar]
- 7.Neoptolemos J.P., Stocken D.D., Friess H., Bassi C., Dunn J.A., Hickey H., Beger H., Fernandez-Cruz L., Dervenis C., Lacaine F., Falconi M., Pederzoli P., Pap A., Spooner D., Kerr D.J., Buchler M.W., European Study Group for Pancreatic, C A randomized trial of chemoradiotherapy and chemotherapy after resection of pancreatic cancer. N. Engl. J. Med. 2004;350:1200–1210. doi: 10.1056/NEJMoa032295. [DOI] [PubMed] [Google Scholar]
- 8.Kindler H.L., Locker G.Y., Mann H., Golan T. POLO: a randomized phase III trial of olaparib tablets in patients with metastatic pancreatic cancer (mPC) and a germline BRCA1/2mutation (gBRCAm) who have not progressed following first-line chemotherapy. J. Clin. Oncol. 2017;33 [Google Scholar]
- 9.Xiong H.Q., Rosenberg A., LoBuglio A., Schmidt W., Wolff R.A., Deutsch J., Needle M., Abbruzzese J.L. Cetuximab, a monoclonal antibody targeting the epidermal growth factor receptor, in combination with gemcitabine for advanced pancreatic cancer: a multicenter phase II trial. J. Clin. Oncol. 2004;22:2610–2616. doi: 10.1200/JCO.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 10.Clarke H.J., Chambers J.E., Liniker E., Marciniak S.J. Endoplasmic reticulum stress in malignancy. Cancer Cell. 2014;25:563–573. doi: 10.1016/j.ccr.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 11.Xu S., Liu Y., Yang K., Wang H., Shergalis A., Kyani A., Bankhead A., 3rd, Tamura S., Yang S., Wang X., Wang C.C., Rehemtulla A., Ljungman M., Neamati N. Inhibition of protein disulfide isomerase in glioblastoma causes marked downregulation of DNA repair and DNA damage response genes. Theranostics. 2019;9:2282–2298. doi: 10.7150/thno.30621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang M., Kaufman R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer. 2014;14:581–597. doi: 10.1038/nrc3800. [DOI] [PubMed] [Google Scholar]
- 13.Liu Y., Ji W., Shergalis A., Xu J., Delaney A.M., Calcaterra A., Pal A., Ljungman M., Neamati N., Rehemtulla A. Activation of the unfolded protein response via inhibition of protein disulfide isomerase decreases the capacity for DNA repair to sensitize glioblastoma to radiotherapy. Cancer Res. 2019;79:2923–2932. doi: 10.1158/0008-5472.CAN-18-2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maurel M., McGrath E.P., Mnich K., Healy S., Chevet E., Samali A. Controlling the unfolded protein response-mediated life and death decisions in cancer. Semin. Cancer Biol. 2015;33:57–66. doi: 10.1016/j.semcancer.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 15.Schonthal A.H. Pharmacological targeting of endoplasmic reticulum stress signaling in cancer. Biochem. Pharmacol. 2013;85:653–666. doi: 10.1016/j.bcp.2012.09.012. [DOI] [PubMed] [Google Scholar]
- 16.Adams J., Kauffman M. Development of the proteasome inhibitor Velcade (Bortezomib) Cancer Investig. 2004;22:304–311. doi: 10.1081/cnv-120030218. [DOI] [PubMed] [Google Scholar]
- 17.Barghout S.H., Patel P.S., Wang X., Xu G.W., Kavanagh S., Halgas O., Zarabi S.F., Gronda M., Hurren R., Jeyaraju D.V., MacLean N., Brennan S., Hyer M.L., Berger A., Traore T., Milhollen M., Smith A.C., Minden M.D., Pai E.F., Hakem R., Schimmer A.D. Preclinical evaluation of the selective small-molecule UBA1 inhibitor, TAK-243, in acute myeloid leukemia. Leukemia. 2019;33:37–51. doi: 10.1038/s41375-018-0167-0. [DOI] [PubMed] [Google Scholar]
- 18.Best S., Hashiguchi T., Kittai A., Bruss N., Paiva C., Okada C., Liu T., Berger A., Danilov A.V. Targeting ubiquitin-activating enzyme induces ER stress-mediated apoptosis in B-cell lymphoma cells. Blood Adv. 2019;3:51–62. doi: 10.1182/bloodadvances.2018026880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hyer M.L., Milhollen M.A., Ciavarri J., Fleming P., Traore T., Sappal D., Huck J., Shi J., Gavin J., Brownell J., Yang Y., Stringer B., Griffin R., Bruzzese F., Soucy T., Duffy J., Rabino C., Riceberg J., Hoar K., Lublinsky A., Menon S., Sintchak M., Bump N., Pulukuri S.M., Langston S., Tirrell S., Kuranda M., Veiby P., Newcomb J., Li P., Wu J.T., Powe J., Dick L.R., Greenspan P., Galvin K., Manfredi M., Claiborne C., Amidon B.S., Bence N.F. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat. Med. 2018;24:186–193. doi: 10.1038/nm.4474. [DOI] [PubMed] [Google Scholar]
- 20.Boj S.F., Hwang C.I., Baker L.A., Chio I.I., Engle D.D., Corbo V., Jager M., Ponz-Sarvise M., Tiriac H., Spector M.S., Gracanin A., Oni T., Yu K.H., van Boxtel R., Huch M., Rivera K.D., Wilson J.P., Feigin M.E., Ohlund D., Handly-Santana A., Ardito-Abraham C.M., Ludwig M., Elyada E., Alagesan B., Biffi G., Yordanov G.N., Delcuze B., Creighton B., Wright K., Park Y., Morsink F.H., Molenaar I.Q., Borel Rinkes I.H., Cuppen E., Hao Y., Jin Y., Nijman I.J., Iacobuzio-Donahue C., Leach S.D., Pappin D.J., Hammell M., Klimstra D.S., Basturk O., Hruban R.H., Offerhaus G.J., Vries R.G., Clevers H., Tuveson D.A. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galban S., Jeon Y.H., Bowman B.M., Stevenson J., Sebolt K.A., Sharkey L.M., Lafferty M., Hoff B.A., Butler B.L., Wigdal S.S., Binkowski B.F., Otto P., Zimmerman K., Vidugiris G., Encell L.P., Fan F., Wood K.V., Galban C.J., Ross B.D., Rehemtulla A. Imaging proteolytic activity in live cells and animal models. PLoS One. 2013;8 doi: 10.1371/journal.pone.0066248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olive P.L., Banath J.P. The comet assay: a method to measure DNA damage in individual cells. Nat. Protoc. 2006;1:23–29. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 23.Morgan M.A., Meirovitz A., Davis M.A., Kollar L.E., Hassan M.C., Lawrence T.S. Radiotherapy combined with gemcitabine and oxaliplatin in pancreatic cancer cells. Transl. Oncol. 2008;1:36–43. doi: 10.1593/tlo.07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cuneo K.C., Morgan M.A., Sahai V., Schipper M.J., Parsels L.A., Parsels J.D., Devasia T., Al-Hawaray M., Cho C.S., Nathan H., Maybaum J., Zalupski M.M., Lawrence T.S. Dose escalation trial of the Wee1 inhibitor adavosertib (AZD1775) in combination with gemcitabine and radiation for patients with locally advanced pancreatic cancer. J. Clin. Oncol. 2019;37:2643–2650. doi: 10.1200/JCO.19.00730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruggiano A., Foresti O., Carvalho P. Quality control: ER-associated degradation: protein quality control and beyond. J. Cell Biol. 2014;204:869–879. doi: 10.1083/jcb.201312042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods. 2005;35:373–381. doi: 10.1016/j.ymeth.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 27.Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 28.Nougarede A., Tesniere C., Ylanko J., Rimokh R., Gillet G., Andrews D.W. Improved IRE1 and PERK pathway sensors for multiplex endoplasmic reticulum stress assay reveal stress response to nuclear dyes used for image segmentation. Assay Drug Dev Technol. 2018;16:350–360. doi: 10.1089/adt.2018.862. [DOI] [PubMed] [Google Scholar]
- 29.Wortel I.M.N., van der Meer L.T., Kilberg M.S., van Leeuwen F.N. Surviving stress: modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol. Metab. 2017;28:794–806. doi: 10.1016/j.tem.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Helenius A., Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 31.Baro M., Lopez Sambrooks C., Quijano A., Saltzman W.M., Contessa J. Oligosaccharyltransferase inhibition reduces receptor tyrosine kinase activation and enhances glioma radiosensitivity. Clin. Cancer Res. 2019;25:784–795. doi: 10.1158/1078-0432.CCR-18-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lopez-Sambrooks C., Shrimal S., Khodier C., Flaherty D.P., Rinis N., Charest J.C., Gao N., Zhao P., Wells L., Lewis T.A., Lehrman M.A., Gilmore R., Golden J.E., Contessa J.N. Oligosaccharyltransferase inhibition induces senescence in RTK-driven tumor cells. Nat. Chem. Biol. 2016;12:1023–1030. doi: 10.1038/nchembio.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakayama K.I., Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 34.Schwertman P., Bekker-Jensen S., Mailand N. Regulation of DNA double-strand break repair by ubiquitin and ubiquitin-like modifiers. Nat. Rev. Mol. Cell Biol. 2016;17:379–394. doi: 10.1038/nrm.2016.58. [DOI] [PubMed] [Google Scholar]
- 35.Zhuang J., Shirazi F., Singh R.K., Kuiatse I., Wang H., Lee H.C., Berkova Z., Berger A., Hyer M., Chattopadhyay N., Syed S., Shi J.Q., Yu J., Shinde V., Tirrell S., Jones R.J., Wang Z., Davis R.E., Orlowski R.Z. Ubiquitin-activating enzyme inhibition induces an unfolded protein response and overcomes drug resistance in myeloma. Blood. 2019;133:1572–1584. doi: 10.1182/blood-2018-06-859686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morgan M.A., Lawrence T.S. Molecular pathways: overcoming radiation resistance by targeting DNA damage response pathways. Clin. Cancer Res. 2015;21:2898–2904. doi: 10.1158/1078-0432.CCR-13-3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reichert Z.R., Wahl D.R., Morgan M.A. Translation of targeted radiation sensitizers into clinical trials. Semin. Radiat. Oncol. 2016;26:261–270. doi: 10.1016/j.semradonc.2016.06.001. [DOI] [PubMed] [Google Scholar]
- 38.Travers K.J., Patil C.K., Wodicka L., Lockhart D.J., Weissman J.S., Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 39.Pinkham K., Park D.J., Hashemiaghdam A., Kirov A.B., Adam I., Rosiak K., da Hora C.C., Teng J., Cheah P.S., Carvalho L., Ganguli-Indra G., Kelly A., Indra A.K., Badr C.E. Stearoyl CoA desaturase is essential for regulation of endoplasmic reticulum homeostasis and tumor growth in glioblastoma cancer stem cells. Stem Cell Reports. 2019;12:712–727. doi: 10.1016/j.stemcr.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamamori T., Meike S., Nagane M., Yasui H., Inanami O. ER stress suppresses DNA double-strand break repair and sensitizes tumor cells to ionizing radiation by stimulating proteasomal degradation of Rad51. FEBS Lett. 2013;587:3348–3353. doi: 10.1016/j.febslet.2013.08.030. [DOI] [PubMed] [Google Scholar]
- 41.Song L., Luo Z.Q. Post-translational regulation of ubiquitin signaling. J. Cell Biol. 2019;218:1776–1786. doi: 10.1083/jcb.201902074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jin J., Li X., Gygi S.P., Harper J.W. Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature. 2007;447:1135–1138. doi: 10.1038/nature05902. [DOI] [PubMed] [Google Scholar]
- 43.Barghout S.H., Schimmer A.D. The ubiquitin-activating enzyme, UBA1, as a novel therapeutic target for AML. Oncotarget. 2018;9:34198–34199. doi: 10.18632/oncotarget.26153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McHugh A., Fernandes K., South A.P., Mellerio J.E., Salas-Alanis J.C., Proby C.M., Leigh I.M., Saville M.K. Preclinical comparison of proteasome and ubiquitin E1 enzyme inhibitors in cutaneous squamous cell carcinoma: the identification of mechanisms of differential sensitivity. Oncotarget. 2018;9:20265–20281. doi: 10.18632/oncotarget.24750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harding H.P., Zhang Y., Zeng H., Novoa I., Lu P.D., Calfon M., Sadri N., Yun C., Popko B., Paules R., Stojdl D.F., Bell J.C., Hettmann T., Leiden J.M., Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 46.Novoa I., Zhang Y., Zeng H., Jungreis R., Harding H.P., Ron D. Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 2003;22:1180–1187. doi: 10.1093/emboj/cdg112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang X., Matsuda K., Bialek P., Jacquot S., Masuoka H.C., Schinke T., Li L., Brancorsini S., Sassone-Corsi P., Townes T.M., Hanauer A., Karsenty G. ATF4 is a substrate of RSK2 and an essential regulator of osteoblast biology; implication for Coffin-Lowry Syndrome. Cell. 2004;117:387–398. doi: 10.1016/s0092-8674(04)00344-7. [DOI] [PubMed] [Google Scholar]
- 48.Nishitoh H. CHOP is a multifunctional transcription factor in the ER stress response. J. Biochem. 2012;151:217–219. doi: 10.1093/jb/mvr143. [DOI] [PubMed] [Google Scholar]
- 49.Morgan M.A., Parsels L.A., Maybaum J., Lawrence T.S. Improving the efficacy of chemoradiation with targeted agents. Cancer Discov. 2014;4:280–291. doi: 10.1158/2159-8290.CD-13-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brownell J.E., Sintchak M.D., Gavin J.M., Liao H., Bruzzese F.J., Bump N.J., Soucy T.A., Milhollen M.A., Yang X., Burkhardt A.L., Ma J., Loke H.K., Lingaraj T., Wu D., Hamman K.B., Spelman J.J., Cullis C.A., Langston S.P., Vyskocil S., Sells T.B., Mallender W.D., Visiers I., Li P., Claiborne C.F., Rolfe M., Bolen J.B., Dick L.R. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol. Cell. 2010;37:102–111. doi: 10.1016/j.molcel.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 51.Jia L., Li H., Sun Y. Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia. 2011;13:561–569. doi: 10.1593/neo.11420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Milhollen M.A., Traore T., Adams-Duffy J., Thomas M.P., Berger A.J., Dang L., Dick L.R., Garnsey J.J., Koenig E., Langston S.P., Manfredi M., Narayanan U., Rolfe M., Staudt L.M., Soucy T.A., Yu J., Zhang J., Bolen J.B., Smith P.G. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-{kappa}B-dependent lymphoma. Blood. 2010;116:1515–1523. doi: 10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- 53.Wei D., Li H., Yu J., Sebolt J.T., Zhao L., Lawrence T.S., Smith P.G., Morgan M.A., Sun Y. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 2012;72:282–293. doi: 10.1158/0008-5472.CAN-11-2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MiaPaCa-2 and Panc-1 cells were treated with ER stress inducers: BAP2 (30 μM), tunicamycin (5 μg/mL), TAK-243 (300 nM), NGI-1 (20 μM), as well as PDI siRNA (Santa Cruz) and IRE1 activity was measured and recorded every 30 min for up to 24 h. Data is presented as mean ± SEM from 4 technical replicates.
(A) Fold change of cleaved Caspase-3 reporter in MiaPaCa-2, Panc-1 and KPC2 cells after gemcitabine and TAK-243 treatment at indicated doses and time, data is presented as mean ± SEM from three experiments. (B) MiaPaCa-2 cells tumor progression after vehicle control, TAK-243, gemcitabine and combination treatment, data is presented as mean tumor volume ± SEM (n ≥ 4).
MiaPaCa-2 cells were treated with PDI siRNA for 48 h and MG132 was added for the last 16 h. Cell lysates were collected for immunoblotting for BIP, RAD51, PDI and β-actin.