

Abstract
Background
Few previous studies have comprehensively explored the level of DNA methylation and gene expression in ccRCC. The purpose of this study was to identify the key clear cell renal cell carcinoma- (ccRCC-) related DNA methylation-driven genes (MDG) and to build a prognostic model based on the level of DNA methylation.
Methods
RNA-seq transcriptome data and DNA methylation data were obtained from The Cancer Genome Atlas. Based on the MethylMix algorithm, we obtain ccRCC-related MDG. The univariate and multivariate Cox regression analyses were employed to investigate the correlation between patient overall survival and the methylation level of each MDG. Finally, a prognosis risk score was established based on a linear combination of the regression coefficient derived from the multivariate Cox regression model (β) multiplied with the methylation level of the gene.
Results
19 ccRCC-related MDG were identified. Three MDG (NCKAP1L, EVI2A, and BATF) were further screened and integrated into a prognostic risk score model, risk score = (3.710∗methylation level of NCKAP1L) + (−3.892∗methylation level of EVI2A) + (−3.907∗methylation level of BATF). The risk model was independent from conventional clinical characteristics as a prognostic factor for ccRCC (HR = 1.221, 95% confidence interval: 1.063–1.402, and P = 0.005). The joint survival analysis showed that the gene expression and methylation levels of the prognostic genes EVI2A and BATF were significantly related with prognosis.
Conclusion
This study provided an important bioinformatics foundation for in-depth studies of ccRCC DNA methylation.
1. Introduction
Clear cell renal cell carcinoma (ccRCC) is the most common pathological type of renal cancer [1, 2]. Approximately 350,000 new patients with RCC are confirmed globally every year, and RCC causes more than 140,000 deaths each year [3]. ccRCC has complex biological characteristics and is not sensitive to radiotherapy and chemotherapy; radical or partial nephrectomy is the main method for treating RCC [4, 5]. Although the surgical effect is definite, 20%-40% of patients experience local recurrence or distant metastasis after surgery [6], and the mortality rate is very high for advanced renal cell carcinoma [7]. In the last ten years, with the development of molecular biology, several ccRCC driver genes such as von Hippel–Lindau (VHL) and PBRM1 have been discovered [8, 9], and targeted drugs, such as tyrosine kinase inhibitors (TKIs) [10], have been developed to treat these advanced ccRCC patients; however, not all of these patients can benefit from these drugs. Therefore, the identification of new ccRCC-related driver genes and the establishment of risk models through bioinformatics analysis are very important for patient prognosis evaluation.
Epigenetic changes are thought to be closely related to cancer progression, and aberrant DNA methylation is one of the most crucial and routine epigenetic modifications [11]. DNA methylation is an epigenetic modification that plays a crucial role in regulating the growth, development, gene expression pattern, and stability of the genome without changing the DNA sequence [12, 13]. Many studies in recent years have found that abnormal DNA methylation is closely related to tumorigenesis, development, and cell canceration [14–16]. In particular, alterations in DNA methylation can supply crucial information for the early diagnosis and prognosis of cancer [17–20].
Few previous studies have comprehensively explored the level of DNA methylation and gene expression in ccRCC. The purpose of this study was to identify the key ccRCC-related methylation-driven genes and to build a prognostic model based on the level of DNA methylation.
2. Materials and Methods
2.1. Data Acquisition and Integrative Analysis
In this study, RNA-seq transcriptome data and DNA methylation data were obtained from The Cancer Genome Atlas (TCGA) website (https://portal.gdc.cancer.gov/repository). Of them, the DNA methylation data was using the Illumina Infinium HumanMethylation450 platform and 27 platform, and beta values, ranged from 0 to 1, were quantified to indicate the levels of DNA methylation. Based on R software and packages [21], we analyzed the above data to obtain differentially methylated genes and differentially expressed genes. In addition, clinicopathological data including survival status, survival time, age, sex, the International Society of Urological Pathology (ISUP) grade, and American Joint Committee on Cancer (AJCC) stage were obtained from TCGA. Based on the MethylMix package [22], we performed an analysis by combining the differentially methylated genes and differentially expressed genes. MethylMix is an algorithm for identifying the hypermethylation and hypomethylation of genes in diseases [23]. MethylMix is based on a β-mixture model to identify the methylation status and compares this status with the normal DNA methylation state. The correlation was computed between the gene methylation level and the gene expression level. Finally, the exported result of MethylMix is methylation-driven genes (MDG).
2.2. MDG Set Enrichment Analysis
Gene functional enrichment analyses were conducted to discover the main biological characteristics of the MDG, including Gene Ontology (GO) analyses with molecular function, biological process, and cellular component analyses [24]. In our study, the biological process category was selected for GO analysis through the enrichGO function in the clusterProfiler package (version 3.6) and the original database was acquired from the “org.Hs.eg.db” package [25].
2.3. Definition of the MDG-Related Prognostic Model
We randomly divided the tumor samples obtained from TCGA into two cohorts: a training cohort (244 patients, Table S1) and a validation cohort (242 patients, Table S2). We used the training cohort to build the Cox regression risk model, and the validation cohort was adopted to verify the performance of the model. First, we performed univariate Cox regression analysis to identify six methylation-driven genes related to overall survival in patients by considering P value < 0.01 as significant. Then, multivariate Cox regression analysis was used to build a prognostic risk model. The risk score was figured out using the regression coefficients (β) from the multivariate Cox regression model to weight the methylation values of the selected genes. Risk score = βgene (1) × methylation level of gene (1) + βgene (2) × methylation level of gene (2) + ⋯+βgene (n) × methylation level of gene (n). According to the median risk score, the training cohort was separated into the low-risk and high-risk groups. To assess the predictive power of the risk score, we performed receiver operating characteristic (ROC) curve analysis. The same method was adopted in the testing cohort to test the performance of the model.
2.4. Independence of the Risk Model from Traditional Clinical Features and Building a Nomogram
To verify whether the risk score was independent of other clinical variables in ccRCC patients (including age, sex, ISUP grade, and AJCC stage), the univariate and multivariate Cox regression analyses were performed on the entire TCGA cohort. According to results of multivariate Cox regression analysis, we constructed a nomogram with R package “rms” to assess the overall survival for ccRCC. The predictive performance was evaluated by the C index and calibration curve.
2.5. Joint Survival Analysis of the Gene Expression Levels and Methylation Levels of the MDG
To further identify the key genes related to the prognosis of ccRCC patients, based on the survival R package, a joint survival analysis was performed by combining the methylation levels of MDG with the corresponding gene expression levels.
2.6. Statistical Analysis
All statistical analyses were performed using R software. P < 0.05 was considered statistically significant. The MethylMix package was used to identify DNA methylation-driven genes. We performed the univariate and multivariate Cox regression analyses to determine whether the risk score had prognostic value independent of various clinicopathological characteristics. We conducted the log-rank test and Kaplan-Meier survival analysis to evaluate the predictive ability of the risk model.
3. Results
3.1. Data Analysis and Acquisition of DMG in ccRCC
All data were acquired from TCGA. The methylation data were downloaded from 542 cancer tissues and 357 noncancer tissues. In addition, the mRNA expression data were retrieved from 611 samples, consisting of 72 normal samples and 539 cancer samples. On the basis of the LIMMA software package, we analyzed the downloaded data to obtain differentially methylated genes and differentially expression genes. We performed an integrative analysis via the R package MethylMix, and the analysis required three datasets, including normal DNA methylation data, cancer DNA methylation data, and matched gene expression data (Tables S3, S4, and S5). ∣logFC | >0, P < 0.05, and ∣Cor | >0.3 were adopted for screening MDG. Finally, 19 MDG were obtained (Table 1). Heat maps of the ccRCC-related aberrant MDG are shown in Figure 1.
Table 1.
DNA methylation-driven genes in ccRCC.
| Gene | Normal me | Tumor me | logFC | P value | Cor | Cor P value |
|---|---|---|---|---|---|---|
| C1orf116 | 0.647716 | 0.896463 | 0.468882 | 3.76E-109 | -0.6038 | 4.66E-54 |
| ALDOC | 0.315874 | 0.201405 | -0.64925 | 1.32E-72 | -0.44678 | 2.04E-27 |
| NCKAP1L | 0.822054 | 0.656117 | -0.32528 | 1.06E-64 | -0.35805 | 1.66E-17 |
| EVI2A | 0.854583 | 0.71327 | -0.26077 | 1.95E-62 | -0.5227 | 1.45E-38 |
| CD2 | 0.74397 | 0.633319 | -0.23231 | 1.57E-54 | -0.46794 | 3.01E-30 |
| BATF | 0.696094 | 0.535991 | -0.37707 | 3.59E-48 | -0.43104 | 1.96E-25 |
| CD96 | 0.703644 | 0.600698 | -0.22821 | 2.19E-45 | -0.35773 | 1.79E-17 |
| RAB25 | 0.498379 | 0.687697 | 0.46453 | 2.40E-38 | -0.38485 | 3.44E-20 |
| IL20RB | 0.886919 | 0.731896 | -0.27716 | 1.13E-33 | -0.50225 | 2.79E-35 |
| TMEM173 | 0.514377 | 0.327888 | -0.64963 | 4.07E-30 | -0.36018 | 1.04E-17 |
| CTSZ | 0.478134 | 0.400661 | -0.25503 | 7.68E-30 | -0.32922 | 6.89E-15 |
| MASP1 | 0.600411 | 0.503547 | -0.25382 | 8.74E-26 | -0.40218 | 4.61E-22 |
| LAMA4 | 0.426073 | 0.346439 | -0.2985 | 4.99E-22 | -0.36743 | 2.04E-18 |
| LTA | 0.859977 | 0.795969 | -0.11159 | 1.31E-18 | -0.56031 | 3.21E-45 |
| SLC9B2 | 0.43967 | 0.392664 | -0.16312 | 3.45E-13 | -0.38856 | 1.40E-20 |
| FAM107A | 0.465871 | 0.414634 | -0.16809 | 1.77E-11 | -0.32415 | 1.86E-14 |
| SLC5A12 | 0.705801 | 0.634831 | -0.15289 | 1.16E-08 | -0.33355 | 2.90E-15 |
| AL592211. | 0.44045 | 0.575433 | 0.38567 | 0.002048 | -0.32193 | 2.87E-14 |
| LRMP | 0.534103 | 0.516217 | -0.04914 | 0.039959 | -0.33687 | 1.48E-15 |
Figure 1.
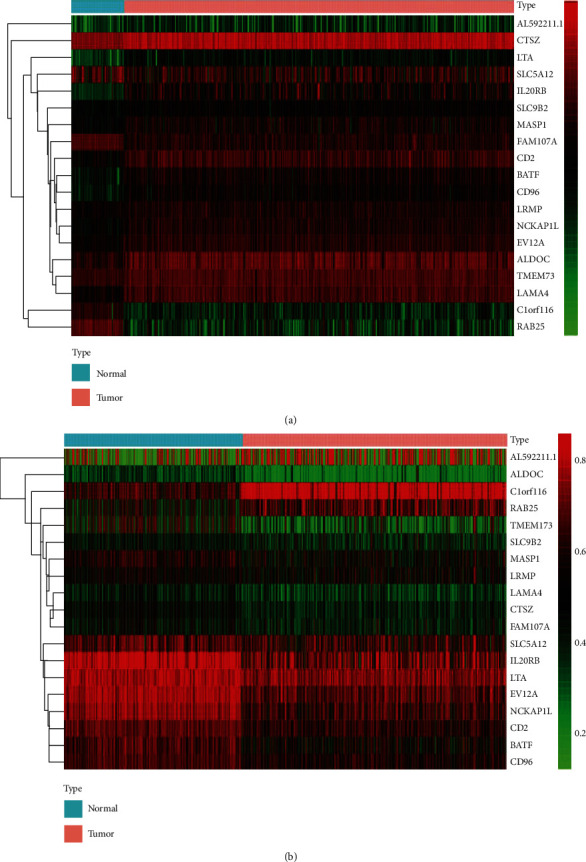
Heat maps of ccRCC-related aberrant methylation-driven genes. (a) The hierarchical clustering heat map of ccRCC-specific methylation-driven mRNAs. (b) The hierarchical clustering heat map of the methylation level of ccRCC-specific methylation-driven genes. In the figure, red represents highly methylated genes and green represents low methylated genes between ccRCC and normal tissues.
3.2. MDG Set Enrichment Analysis in ccRCC
Gene Ontology analysis showed that MDG were mainly enriched in the regulation of interferon-gamma production, interferon-gamma production, regulation of the inflammatory response to antigenic stimuli, lymphocyte-mediated immunity, myeloid dendritic cell activation, regulation of lymphocyte-mediated immunity, negative regulation of interferon-gamma production, and negative regulation of lymphocyte-mediated immunity (P < 0.05, Figure 2).
Figure 2.
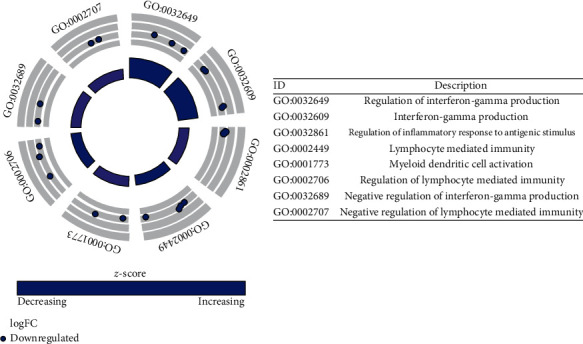
Functional enrichment analysis of methylation-driven mRNAs in ccRCC. The outer circle represents the expression (logFC) of methylation-driven mRNAs in each enriched GO (gene ontology) term: red dots on each GO term indicate upregulated methylation-driven mRNAs and blue dots indicate downregulated methylation-driven mRNAs. The inner circle indicates the significance of GO terms.
Above MDG set enrichment analysis revealed that MDG were significantly linked to inflammatory response regulation and immune regulation.
3.3. Construction of the MDG-Related Prognostic Model
The training cohort was used to construct the risk model. Firstly, we conducted a univariate Cox regression analysis of the methylation level of nineteen MDG. We identified six genes (NCKAP1L, EVI2A, BATF, CD96, IL20RB, and CTSZ) related to overall survival in the training cohort by considering P value < 0.01 as significant (Figure 3(a), P value < 0.01). To better predict the relationships between the methylation levels of the methylation-driven genes and overall survival of ccRCC, we further analyzed them by stepwise multivariate Cox regression analysis. Finally, three genes were selected to build a predictive model (Figure 3(b), P value < 0.05). The risk score was figured out using a linear combination of the methylation levels of the three selected MDG weighted by their specific regression coefficients (β). Risk score = (3.710∗methylation level of NCKAP1L) + (−3.892∗methylation level of EVI2A) + (−3.907∗methylation level of BATF). On the basis of the median risk score, the patients in the training cohort were classified as a high-risk group and a low-risk group. The risk score distribution of the patients according to the prognostic model is shown in Figure 4(a). Survival status scatter plots for the patients according to the prognostic model are shown in Figure 4(b), which shows that the high-risk subgroup contained a higher number of patients who died than the low-risk subgroup. We observed a significant difference in overall survival between the two groups (Figure 4(c), P < 0.0001), and the AUCs at three and five years were 0.719 and 0.689, respectively (Figures 4(d) and 4(e)).
Figure 3.
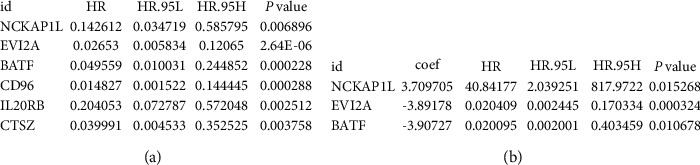
Further analysis of methylation-driven genes the in the training cohort. (a) Six methylation-driven genes selected through univariate Cox regression analysis. (b) Final multivariate prognostic model containing three survival-associated methylation-driven genes.
Figure 4.
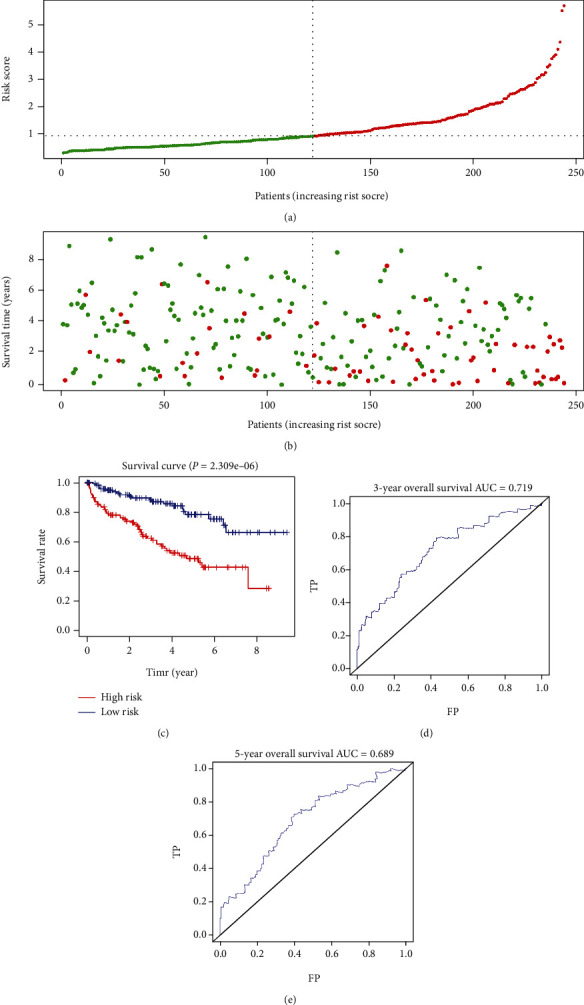
Prognostic analyses of the high-risk and low-risk patients in the training cohort. (a) Risk score distribution of patients in the prognostic model. (b) Survival status scatter plots for patients in the prognostic model (green dots: alive; red dots: death). (c) The Kaplan-Meier plot (high-risk vs. low-risk group) for the training cohort. (d, e) Receiver operating characteristic curves showed the predictive efficiency of the risk score for the training cohort. AUC: area under the curve; FP: false positive; TP: true positive.
3.4. Testing of the Methylation-Driven Gene-Related Prognostic Model
To confirm the performance of the model, the testing cohort was analyzed. First, we used the selected methylation-driven genes (NCKAP1L, EVI2A, and BATF) to compute the risk score of each patient in the validation cohort. Similar results were observed in the testing cohort, and the risk score distribution of the patients according to the prognostic model is shown in Figure 5(a). Survival status scatter plots for the patients according to the prognostic model are shown in Figure 5(b). Significant survival differences were observed in the testing cohort (Figure 5(c), P < 0.0001). The AUCs at three and five years were 0.674 and 0.659, respectively (Figures 5(d) and 5(e)).
Figure 5.
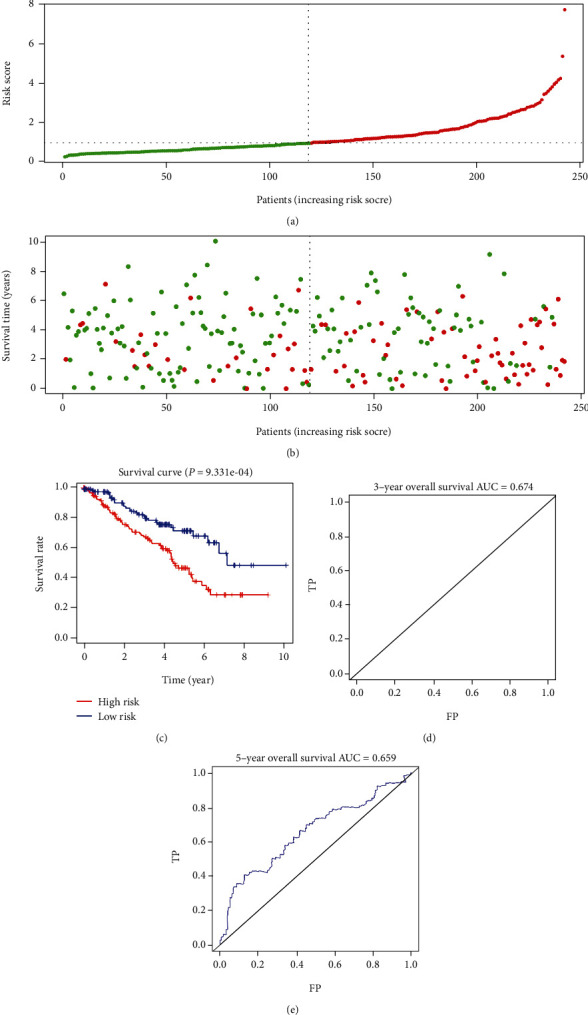
Prognostic analyses of the high-risk and low-risk patients in the testing cohort. (a) Risk score distribution of patients in the prognostic model. (b) Survival status scatter plots for patients in the prognostic model (green dots: alive; red dots: death). (c) The Kaplan-Meier plot (high-risk vs. low-risk group) for the testing cohort. (d, e) Receiver operating characteristic curves showed the predictive efficiency of the risk score for the testing cohort. AUC: area under the curve; FP: false positive; TP: true positive.
3.5. Independence of the Prognostic Model from Other Clinical Characteristics and Building a Nomogram Based on the Risk Score and Other Clinical Characteristics
We conducted the univariate and multivariate Cox regression analyses to determine whether the risk score was a prognostic factor of ccRCC independent from traditional clinical features (including age, sex, ISUP grade, and AJCC stage). The results revealed that the risk score was independent of conventional clinical features in the entire TCGA cohort (Figure 6), with a hazard ratio (HR) of 1.221 (95% confidence interval: 1.063–1.402; P = 0.005).
Figure 6.
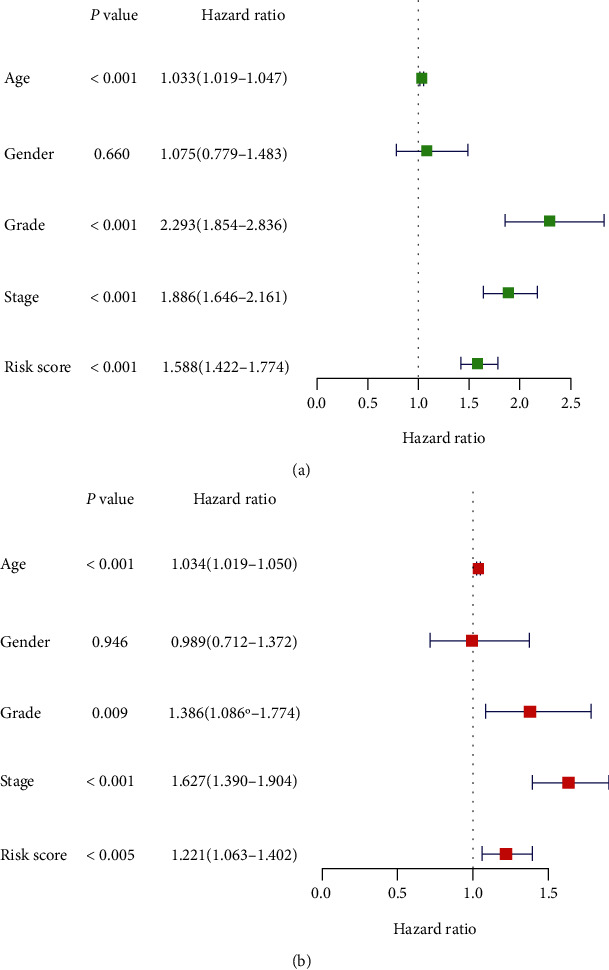
Univariate (a) and multivariate (b) Cox regression analyses of the entire TCGA cohort.
Then, a nomogram predicting the overall survival in ccRCC was constructed based on the risk score and clinical characteristics (Figure 7(a)). The C index was 0.776. The calibration curve showed that the nomogram performed well (Figures 7(b) and 7(c)). Both the C index and the calibration curve suggested a good predictive performance.
Figure 7.
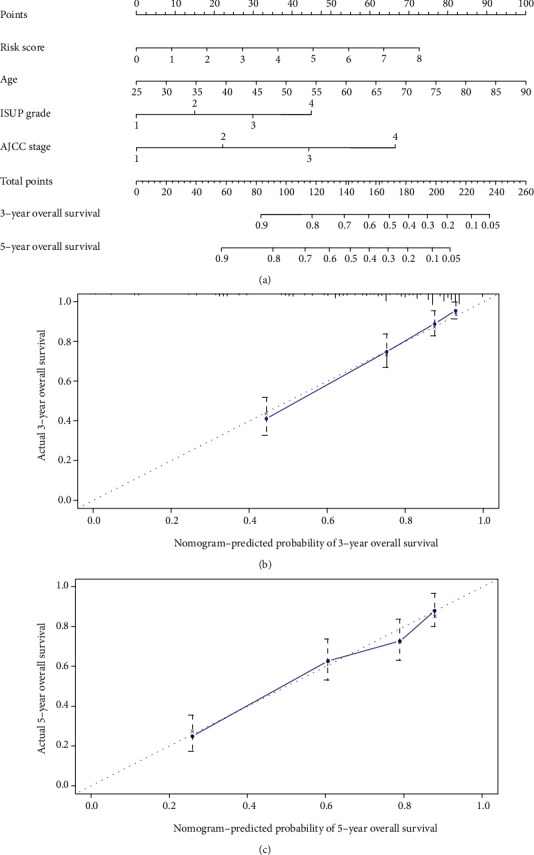
(a) Nomogram predicting 3- and 5-year overall survival for patients with ccRCC. The calibration curve for predicting patient survival at (b) 3 years and (c) 5 years in TCGA datasets. Nomogram-predicted probability of overall survival is plotted on the x-axis; actual overall survival is plotted on the y-axis.
3.6. MethylMix Model of Three Selected DNA MDG
The MethylMix model of the three selected DNA MDG is shown in Figure 8. The correlation between DNA methylation and gene expression is visualized in Figures 9(a)–9(c), Cor < −0.3.
Figure 8.
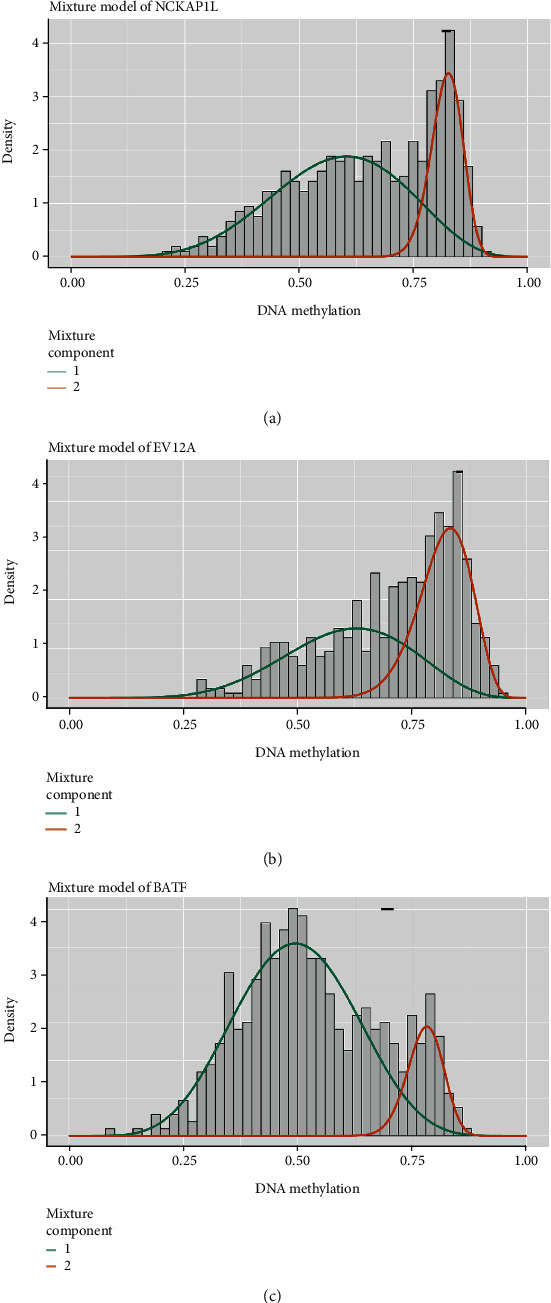
MethylMix model of three selected DNA methylation-driven genes. The distribution maps show the methylation states of methylated genes. The histogram represents the distribution of methylation in tumor samples. The horizontal black bar demonstrates the distribution of methylation in the normal samples. The distribution of the methylation degree can be clearly seen from (a–c).
Figure 9.
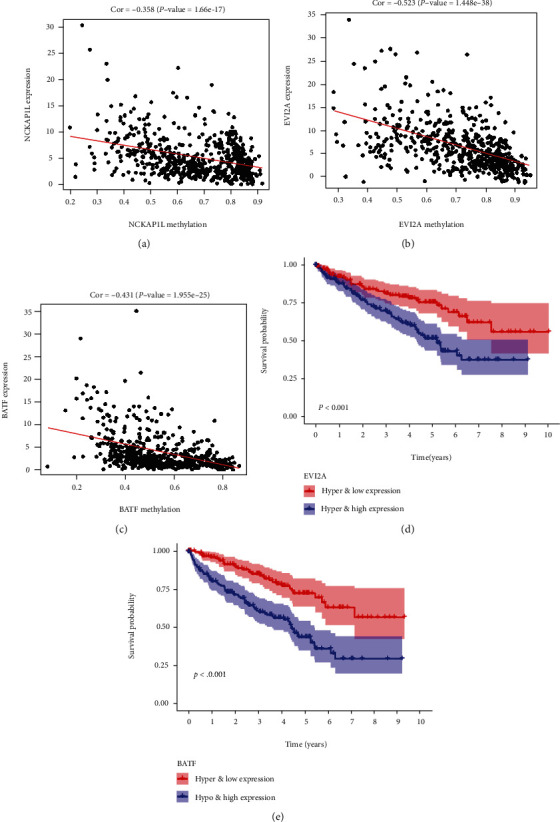
Correlation between DNA methylation and gene expression (a–c) and Kaplan-Meier survival curves for the integrative analysis of EVI2A and BATF (d, e).
3.7. Joint Prognostic Assessment of MDG in ccRCC
To further identify the key genes related to the prognosis of ccRCC patients, a joint survival analysis was conducted by combining the methylation levels of the MDG with the corresponding gene expression levels. The results are shown in Figures 9(d) and 9(e). The gene expression and methylation levels of the prognostic genes EVI2A and BATF were significantly related to prognosis.
4. Discussion
ccRCC is the most common pathological type of kidney cancer [26]. In-depth molecular pathogenesis research and the early detection of tumor prognostic biomarkers and specific driven-genes may have great significance for improving the prognosis of patients [27–29]. In recent years, with the deepening of epigenetic research, the epigenetic regulatory mechanisms of renal cancer, especially DNA methylation, are often used to predict the survival time of renal cancer, including DAB2IP [30], RCVRN [31], CRHBP [32], AR [33], and CDO1 [34]. However, the heterogeneity of ccRCC limits single-gene methylation in predicting ccRCC outcomes, so it is important to establish a multigene prediction model.
Related studies have shown that abnormal DNA MDG may cause transcriptional disorders, causing some gene expression mistakes and cell differentiation mistakes [35], and tumor suppressor genes and DNA repair genes were silenced due to hypermethylation. DNA methylation alterations at precancerous stages may determine tumor aggressiveness and patient prognosis [36]. Therefore, identifying abnormal DNA MDG can provide new insights for the risk assessment and prognosis of patients.
In this study, we identified key ccRCC-related MDG and constructed a prognostic model based on the level of DNA methylation. First, we screened nineteen methylation-driven genes by analyzing methylation and transcriptome data. Then, to discover the main biological characteristics of these methylation-driven genes, a series of gene functional enrichment analyses were performed. The gene functional enrichment analyses revealed that MDG were significantly linked to inflammatory response regulation and immune regulation (P < 0.05, Figure 2).
In addition, a risk score-based prognostic model was constructed for these methylation-driven genes, and the prediction ability of this model was validated in the testing dataset. Three methylation-driven genes (NCKAP1L, EVI2A, and BATF) were identified and used to construct a prognostic risk model for ccRCC. We performed the univariate and multivariate Cox regression analyses on the entire TCGA cohort to verify that the risk model was a prognostic factor for ccRCC independent from other clinicopathological data (age, sex, ISUP grade, and AJCC stage) (HR = 1.221, 95% confidence interval: 1.063–1.402, and P = 0.005). In addition, we constructed a nomogram to predict overall survival in patients with ccRCC. The C index and calibration curve indicated that the predictive performance of the nomogram was good. Finally, we performed joint survival analysis by combining the methylation levels of the methylation-driven genes with the corresponding gene expression levels. The prognostic genes EVI2A and BATF were significantly related to prognosis.
The gene EVI2A, as the human homolog of mouse genes, may be associated with other proteins in the membrane as a part of a cell surface receptor complex [37]; EVI2A has been shown to be an oncogene [38]; EVI2A is highly expressed in oral tongue squamous cell carcinoma [39] and osteosarcoma [40] and is a risk factor for cancer prognosis. In this study, we found that EVI2A is one of the MDG of ccRCC, and its hypomethylation leads to poor prognosis (Figure 9(d)).
The BATF gene acts a pivotal part in the development of different types of cancer, including colon cancer, lymphoma, and multiple myeloma [41–43]. In addition, Feng et al. found that BATF acted as an oncogene in non-small-cell lung cancer [44]. In this study, the joint survival analyses revealed that the BATF gene was closely related with poor prognosis in patients with ccRCC (Figure 9(e)).
There are some limitations in this study. The specific mechanisms of EVI2A and BATF in ccRCC were not investigated in the previous literature. Therefore, future studies may focus on the molecular mechanisms underlying the potential interactions of EVI2A and BATF in ccRCC.
5. Conclusion
In conclusion, we identified key ccRCC-related MDG and constructed a prognostic model based on the level of DNA methylation. In addition, we verified that the risk model was an independent prognostic factor for overall survival in ccRCC. Although further experimental verification is needed, this study provided an important bioinformatics foundation for in-depth studies of ccRCC DNA methylation.
Acknowledgments
This study was supported by the Beijing Hospital Clinical Research 121 Project (BJ-2018-090) and Chinese Academy of Medical Sciences Innovation Fund for Medical Sciences (2018-I2M-1-002).
Contributor Information
Ming Liu, Email: liumingbjh@126.com.
Jianye Wang, Email: wangjianyebjyy@126.com.
Data Availability
The data included in the current study was available in TCGA database (https://cancergenome.nih.gov/).
Ethical Approval
Because the dataset in the current study was downloaded from TCGA, and data acquiring and application complied with TCGA publication guidelines and data access policies, additional approval by an ethics committee and consent to participate were not needed.
Conflicts of Interest
The authors declared that they have no conflict of interest.
Authors' Contributions
MH and JX carried out the extraction of data and interpretation of results and wrote the initial manuscript. MH and HH performed the data analyses. ML and JW provided feedback on the manuscript and contributed to the study idea. All authors read and approved the final manuscript. Maolin Hu and Jiangling Xie contributed equally to this work.
Supplementary Materials
Table S1 a training cohort. Table S2 a validation cohort. Table S3 normal DNA methylation data. Table S4 cancer DNA methylation data. Table S5 matched gene expression data.
References
- 1.Jonasch E., Gao J., Rathmell W. K. Renal cell carcinoma. BMJ. 2014;349, article g4797 doi: 10.1136/bmj.g4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shuch B., Amin A., Armstrong A. J., et al. Understanding pathologic variants of renal cell carcinoma: distilling therapeutic opportunities from biologic complexity. European Urology. 2015;67(1):85–97. doi: 10.1016/j.eururo.2014.04.029. [DOI] [PubMed] [Google Scholar]
- 3.Capitanio U., Montorsi F. Renal cancer. The Lancet. 2016;387(10021):894–906. doi: 10.1016/S0140-6736(15)00046-X. [DOI] [PubMed] [Google Scholar]
- 4.MacLennan S., Imamura M., Lapitan M. C., et al. Systematic review of oncological outcomes following surgical management of localised renal cancer. European Urology. 2012;61(5):972–993. doi: 10.1016/j.eururo.2012.02.039. [DOI] [PubMed] [Google Scholar]
- 5.Bazzi W. M., Sjoberg D. D., Feuerstein M. A., et al. Long-term survival rates after resection for locally advanced kidney cancer: Memorial Sloan Kettering Cancer Center 1989 to 2012 experience. The Journal of Urology. 2015;193(6):1911–1917. doi: 10.1016/j.juro.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teng J., Gao Y., Chen M., et al. Prognostic value of clinical and pathological factors for surgically treated localized clear cell renal cell carcinoma. Chinese Medical Journal. 2014;127(9):1640–1644. [PubMed] [Google Scholar]
- 7.Miller K. D., Nogueira L., Mariotto A. B., et al. Cancer treatment and survivorship statistics, 2019. CA: A Cancer Journal for Clinicians. 2019;69(5):363–385. doi: 10.3322/caac.21565. [DOI] [PubMed] [Google Scholar]
- 8.Shen C., Kaelin W. G., Jr. The VHL/HIF axis in clear cell renal carcinoma. Seminars in Cancer Biology. 2013;23(1):18–25. doi: 10.1016/j.semcancer.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z., Peng S., Guo L., et al. Prognostic and clinicopathological value of PBRM1 expression in renal cell carcinoma. Clinica Chimica Acta. 2018;486:9–17. doi: 10.1016/j.cca.2018.07.014. [DOI] [PubMed] [Google Scholar]
- 10.Atkins M. B., Tannir N. M. Current and emerging therapies for first-line treatment of metastatic clear cell renal cell carcinoma. Cancer Treatment Reviews. 2018;70:127–137. doi: 10.1016/j.ctrv.2018.07.009. [DOI] [PubMed] [Google Scholar]
- 11.Zheng X., Zhang N., Wu H. J., Wu H. Estimating and accounting for tumor purity in the analysis of DNA methylation data from cancer studies. Genome Biology. 2017;18(1):p. 17. doi: 10.1186/s13059-016-1143-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernstein B. E., Meissner A., Lander E. S. The mammalian epigenome. Cell. 2007;128(4):669–681. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 13.Pogribny I. P., Beland F. A. DNA hypomethylation in the origin and pathogenesis of human diseases. Cellular and Molecular Life Sciences. 2009;66(14):2249–2261. doi: 10.1007/s00018-009-0015-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang C., Zhao N., Yuan L., Liu X. Computational detection of breast cancer invasiveness with DNA methylation biomarkers. Cell. 2020;9(2):p. 326. doi: 10.3390/cells9020326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Z., Yin J., Zhou W., et al. Complex impact of DNA methylation on transcriptional dysregulation across 22 human cancer types. Nucleic Acids Research. 2020;48(5):2287–2302. doi: 10.1093/nar/gkaa041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El‐Zein M., Cheishvili D., Gotlieb W., et al. Genome-wide DNA methylation profiling identifies two novel genes in cervical neoplasia. International Journal of Cancer. 2020 doi: 10.1002/ijc.32880. [DOI] [PubMed] [Google Scholar]
- 17.Feng H., Zhang Z., Qing X., Wang X., Liang C., Liu D. Promoter methylation of APC and RAR-β genes as prognostic markers in non-small cell lung cancer (NSCLC) Experimental and Molecular Pathology. 2016;100(1):109–113. doi: 10.1016/j.yexmp.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 18.Zhang S., Wang Y., Gu Y., et al. Specific breast cancer prognosis-subtype distinctions based on DNA methylation patterns. Molecular Oncology. 2018;12(7):1047–1060. doi: 10.1002/1878-0261.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hao X., Luo H., Krawczyk M., et al. DNA methylation markers for diagnosis and prognosis of common cancers. Proceedings of the National Academy of Sciences of the United States of America. 2017;114(28):7414–7419. doi: 10.1073/pnas.1703577114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao J., Le Wang D. K., Hu G., Wei B. Construction of novel DNA methylation-based prognostic model to predict survival in glioblastoma. Journal of Computational Biology. 2020;27(5):718–728. doi: 10.1089/cmb.2019.0125. [DOI] [PubMed] [Google Scholar]
- 21.Ritchie M. E., Phipson B., Wu D., et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research. 2015;43(7, article e47) doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gevaert O. MethylMix: an R package for identifying DNA methylation-driven genes. Bioinformatics (Oxford, England) 2015;31(11):1839–1841. doi: 10.1093/bioinformatics/btv020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gevaert O., Tibshirani R., Plevritis S. K. Pancancer analysis of DNA methylation-driven genes using MethylMix. Genome Biology. 2015;16(1):p. 17. doi: 10.1186/s13059-014-0579-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gene Ontology Consortium. The Gene Ontology (GO) project in 2006. Nucleic Acids Research. 2006;34(90001):D322–D326. doi: 10.1093/nar/gkj021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu G., Wang L. G., Han Y., He Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen F., Zhang Y., Senbabaoglu Y., et al. Multilevel genomics-based taxonomy of renal cell carcinoma. Cell Reports. 2016;14(10):2476–2489. doi: 10.1016/j.celrep.2016.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li R., Yang Y. E., Yin Y. H., Zhang M. Y., Li H., Qu Y. Q. Methylation and transcriptome analysis reveal lung adenocarcinoma-specific diagnostic biomarkers. Journal of Translational Medicine. 2019;17(1):p. 324. doi: 10.1186/s12967-019-2068-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanai Y. Genome-wide DNA methylation profiles in precancerous conditions and cancers. Cancer Science. 2010;101(1):36–45. doi: 10.1111/j.1349-7006.2009.01383.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arai E., Kanai Y. Genetic and epigenetic alterations during renal carcinogenesis. International Journal of Clinical and Experimental Pathology. 2010;4(1):58–73. [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Z. R., Wei J. H., Zhou J. C., et al. Validation of DAB2IP methylation and its relative significance in predicting outcome in renal cell carcinoma. Oncotarget. 2016;7(21):31508–31519. doi: 10.18632/oncotarget.8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Golovastova M. O., Tsoy L. V., Bocharnikova A. V., et al. The cancer-retina antigen recoverin as a potential biomarker for renal tumors. Tumour Biology. 2016;37(7):9899–9907. doi: 10.1007/s13277-016-4885-5. [DOI] [PubMed] [Google Scholar]
- 32.Tezval H., Dubrowinskaja N., Peters I., et al. Tumor specific epigenetic silencing of corticotropin releasing hormone -binding protein in renal cell carcinoma: association of hypermethylation and metastasis. PLoS One. 2016;11(10, article e0163873) doi: 10.1371/journal.pone.0163873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao H., Leppert J. T., Peehl D. M. A protective role for androgen receptor in clear cell renal cell carcinoma based on mining TCGA data. PLoS One. 2016;11(1, article e0146505) doi: 10.1371/journal.pone.0146505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Deckers I. A. G., Schouten L. J., Van Neste L., et al. Promoter methylation of CDO1 identifies clear-cell renal cell cancer patients with poor survival outcome. Clinical Cancer Research. 2015;21(15):3492–3500. doi: 10.1158/1078-0432.CCR-14-2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morris M. R., Ricketts C. J., Gentle D., et al. Genome-wide methylation analysis identifies epigenetically inactivated candidate tumour suppressor genes in renal cell carcinoma. Oncogene. 2011;30(12):1390–1401. doi: 10.1038/onc.2010.525. [DOI] [PubMed] [Google Scholar]
- 36.Arai E., Chiku S., Mori T., et al. Single-CpG-resolution methylome analysis identifies clinicopathologically aggressive CpG island methylator phenotype clear cell renal cell carcinomas. Carcinogenesis. 2012;33(8):1487–1493. doi: 10.1093/carcin/bgs177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cawthon R. M., O'Connell P., Buchberg A. M., et al. Identification and characterization of transcripts from the neurofibromatosis 1 region: the sequence and genomic structure of EVI2 and mapping of other transcripts. Genomics. 1990;7(4):555–565. doi: 10.1016/0888-7543(90)90199-5. [DOI] [PubMed] [Google Scholar]
- 38.Cawthon R. M., Andersen L. B., Buchberg A. M., et al. cDNA sequence and genomic structure of EVI2B, a gene lying within an intron of the neurofibromatosis type 1 gene. Genomics. 1991;9(3):446–460. doi: 10.1016/0888-7543(91)90410-G. [DOI] [PubMed] [Google Scholar]
- 39.Qiu Z., Sun W., Gao S., et al. A 16-gene signature predicting prognosis of patients with oral tongue squamous cell carcinoma. PeerJ. 2017;5, article e4062 doi: 10.7717/peerj.4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li S., Yang F., Yang Y.-K., Zhou Y. Increased expression of ecotropic viral integration site 2A indicates a poor prognosis and promotes osteosarcoma evolution through activating MEK/ERK pathway. Journal of Receptor and Signal Transduction Research. 2019;39(4):368–372. doi: 10.1080/10799893.2019.1669182. [DOI] [PubMed] [Google Scholar]
- 41.Dai L., Cui X., Zhang X., et al. SARI inhibits angiogenesis and tumour growth of human colon cancer through directly targeting ceruloplasmin. Nature Communications. 2016;7(1, article 11996) doi: 10.1038/ncomms11996. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Schleussner N., Merkel O., Costanza M., et al. The AP-1-BATF and -BATF3 module is essential for growth, survival and TH17/ILC3 skewing of anaplastic large cell lymphoma. Leukemia. 2018;32(9):1994–2007. doi: 10.1038/s41375-018-0045-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gil M., Pak H. K., Park S. J., et al. Engagement of CD99 reduces AP-1 activity by inducing BATF in the human multiple myeloma cell line RPMI8226. Immune Network. 2015;15(5):260–267. doi: 10.4110/in.2015.15.5.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng Y., Pan L., Zhang B., Huang H., Ma H. BATF acts as an oncogene in non-small cell lung cancer. Oncology Letters. 2020;19(1):205–210. doi: 10.3892/ol.2019.11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 a training cohort. Table S2 a validation cohort. Table S3 normal DNA methylation data. Table S4 cancer DNA methylation data. Table S5 matched gene expression data.
Data Availability Statement
The data included in the current study was available in TCGA database (https://cancergenome.nih.gov/).