

Abstract
Liver fibrosis is an intrinsic repair process of chronic injury with excessive deposition of extracellular matrix. As an early stage of various liver diseases, liver fibrosis is a reversible pathological process. Therefore, if not being controlled in time, liver fibrosis will evolve into cirrhosis, liver failure, and liver cancer. It has been demonstrated that hepatic stellate cells (HSCs) play a crucial role in the formation of liver fibrosis. In particular, the activation of HSCs is a key step for liver fibrosis. Recent researches have suggested that autophagy and inflammasome have biological effect on HSC activation. Herein, we review current studies about the impact of autophagy and NOD-like receptors containing pyrin domain 3 (NLRP3) inflammasome on liver fibrosis and the underlying mechanisms.
1. Introduction
Liver fibrosis refers to a compensatory response of liver inflammation and damage caused by various pathogenic factors. Liver fibrosis has high morbidity and high mortality worldwide and is a common characteristic shared by many liver injuries, such as hepatitis B, autoimmune hepatitis, alcoholic liver disease, nonalcoholic steatohepatitis, and hepatocellular carcinoma [1]. After the emergence of steatosis and steatohepatitis, liver fibrosis will shape up gradually without timely cure. Liver fibrosis is characterized by excessive deposition of extracellular matrix (ECM), including collagen, glycoproteins, and proteoglycans, and severe fibrosis can lead to cirrhosis, portal hypertension, and even cancer [2]. It is well known that the activation of hepatic stellate cells (HSCs) plays a crucial role in the process of liver fibrosis. Upon stimulation by liver damage, the quiescent HSCs are activated and converted into myofibroblasts, which release a large amount of ECM, form scar tissues, and eventually lead to liver fibrosis [3, 4].
Autophagy is a process of degrading excess or damaged organelles or other components in the cell with the participation of lysosomes [5]. Autophagy occurs in all kinds of nucleated cells and is essential for cell survival, differentiation, growth, and homeostasis [6]. Therefore, dysregulated autophagy is involved in the development of various diseases, such as cancer, neurodegeneration, and diabetes [7]. It is no doubt that, as a physiological function of a cell, autophagy can also regulate the activity of HSCs. In the process of liver fibrosis, autophagy works as a double-edged sword. On the one hand, the upregulated autophagy can activate HSCs, promoting the development of fibrosis; on the other hand, excessive autophagy may inhibit the development of fibrosis [8, 9].
Inflammasome is a macromolecular protein complex, which is assembled by intracellular pattern recognition receptors. As an important component of the innate immunity, inflammasome promotes the maturation and secretion of proinflammatory factors [10]. Among all known inflammasomes, the NOD-like receptor family is the most characteristic, of which the NOD-like receptors containing pyrin domain 3 (NLRP3) inflammasome is a classic member [11]. Some researches found that NLRP3 inflammasome takes part in the development of sterile inflammation, which is induced by sterile cell death in the condition of mechanical trauma, ischemia, stress, and environmental cues [12]. In addition, sterile inflammation is regarded as a significant factor for fibrosis [13]. Thus, the NLRP3 inflammasome is associated with liver fibrosis to a great extent.
To sum up, autophagy and NLRP3 inflammasome are significant for liver fibrosis. The main purpose of this review is to systematically summarize the effects of autophagy and NLRP3 inflammasome on liver fibrosis and the underlying mechanisms.
2. The Overview of Liver Fibrosis
Liver fibrosis is a pathological state in which the ECM is secreted and deposited largely in response to liver injury, which disrupts the normal structure and function of the liver. There are many causes of liver fibrosis, such as viral or parasitic infections, cholestasis, metabolic diseases, and excessive intake of alcohol chronically [14]. Furthermore, these pathogenic factors induce liver fibrosis ultimately through inflammation and oxidative stress damage [15]. Age, alcohol intake, and gender are considered to be the three major factors affecting liver fibrosis, and about 33% of patients with liver fibrosis worldwide will progress to cirrhosis, if not being treated in time [16]. What is worse, about 85% of patients with cirrhosis will be diagnosed with hepatocellular carcinoma [17]. Liver fibrosis is regarded as a common pathological basis of numerous liver diseases. For instance, approximately 57% of patients with nonalcoholic steatohepatitis (NASH) are suffering from different degrees of liver fibrosis [18]. Liver fibrosis is a reversible pathological state, so the prevention and treatment of liver fibrosis are of great significance for prevention of various liver diseases.
2.1. Potential Pathogenesis of Liver Fibrosis
2.1.1. The Role of Hepatic Stellate Cell Activation in Liver Fibrosis
Hepatic stellate cells (HSCs), known as fat-storing cells (FSCs), are mainly distributed in the hepatic sinusoidal space. The quiescent HSCs are rich in vitamin A, accounting for 8% to 14% of the total amount in hepatocytes. When stimulated by profibrotic factors, the quiescent HSCs are activated and transformed into myofibroblasts, storing and secreting large amounts of ECM [19–21]. ECM is mainly composed of α smooth muscle actin (α-SMA) and collagen, and as we mentioned before, excessive deposition of ECM could destroy the normal liver structure. At the same time, the activated HSCs could further proliferate and migrate, aggravating the process of liver fibrosis [22]. Except for ECM, HSCs also express matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs), which play important roles in the progression of liver fibrosis [23]. MMPs are defined as a group of extracellular endopeptidases consisting of 25 members [24, 25]. The family of MMPs is responsible for reversal and degradation of all components of ECM. Among the members of MMPs, some have the capacity to inhibit fibrosis, such as MMP-1, MMP-8, and MMP-13. Their upregulation can alleviate liver fibrosis and promote the proliferation of hepatocyte [26]. On the contrary, some MMP members such as MMP-12 and MMP-19 are fibrogenic. TIMPs are the endogenous inhibitors of MMPs. TIMPs are composed of TIMP-1, TIMP-2, TIMP-3, and TIMP-4, which bind to MMPs with 1 : 1 noncovalent stoichiometry [27]. In general, the unbalanced expression of MMPs and TIMPs after activation of HSCs may be an important contributor to the liver fibrosis progression. Some signals have been found about autophagy and NLRP3 inflammasome involved in HSC activation, such as the autophagic-CTSB-inflammasome axis [28] and P2X7R-NLRP3 inflammasome pathway [29]. Activation of HSCs is a crucial step in the initiation of liver fibrosis. There are numerous causes of HSC activation, among which inflammation and oxidative stress are two important aspects.
2.1.2. The Role of Oxidative Stress in Liver Fibrosis
Oxidation-antioxidant imbalance occurs when reactive oxygen species and free radicals accumulate excessively beyond the capacity of the antioxidant system and ultimately leads to oxidative stress [30]. Free radicals are by-products of cellular metabolism with high activity and reactivity, due to the unpaired electrons on the outermost layer. Free radicals are generated in various parts of the body by several stimulations, such as long-term stress, disease, and ultraviolet exposure [31, 32]. Reactive oxygen species (ROS) are chemically reactive molecules produced during oxygen-related reactions. When the liver is attacked by harmful stimulations, hepatocytes, Kupffer cells, and HSCs themselves will produce large amounts of ROS [33, 34], which react directly with intracellular lipids, proteins, and DNA, furtherly causing cell damage and death. Moreover, ROS can also activate HSCs through lipid peroxidation on the cell surface, leading to liver fibrosis. As found by Casini et al., followed by N-formyl-Met-Leu-phenylalanine (FMLP) stimulation, neutrophil-produced ROS induced HSC activation in the coculture model [35]. In addition, during oxidative stress, ROS can induce HSC activation by stimulating Kupffer cells and inflammatory cells to secrete profibrogenic factors, including transcription growth factor, cytokines, and chemokines. For instance, TGF-β1 is an acknowledged inducer for activating HSCs, which is released from Kupffer cells by excessive ROS [36]. Collectively, we can see that excessive oxidative stress is an important factor driving liver injury and fibrosis. Therefore, it is a significant strategy to attenuate oxidative stress for the treatment of liver fibrosis (Figure 1).
Figure 1.
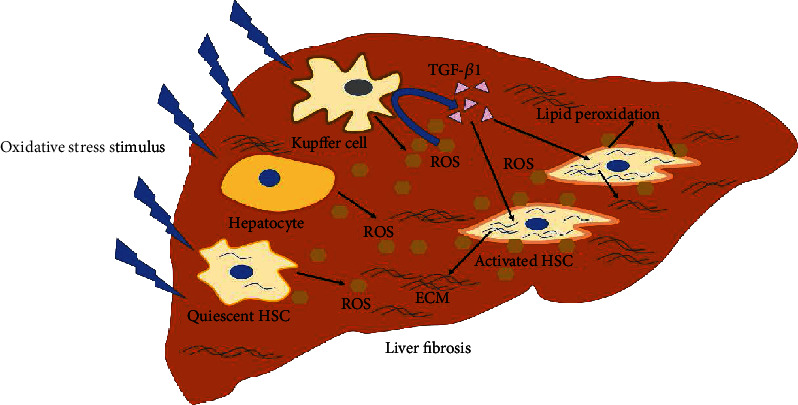
Oxidative stress plays an important role in HSC activation and liver fibrosis. In the liver, under impact of harmful stimulus, oxidative stress is induced and hepatocytes, Kupffer cells, and HSCs will produce mass of ROS. On the one hand, the ROS bond with cytomembrane of HSCs and cause its lipid peroxidation, leading to HSC activation. On the other hand, ROS will stimulate Kupffer cells to produce and release TGF-β1 in turn, which can induce HSC activation admittedly.
2.1.3. The Role of Inflammation in Liver Fibrosis
Inflammation is a defensive reaction to external invasion or damage, accompanied by congestion, exudation, and inflammatory cell infiltration [37]. When the liver suffers sustained damage, inflammatory cells and parenchymal cells secrete large amounts of cytokines and chemokines, such as interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α), transforming growth factor-β (TGF-β), and IL-6, which further cause the aggregation of inflammatory cells and amplify inflammatory response. Among these cytokines, IL-1β and TGF-β are closely related to liver fibrosis due to their capacity to activate HSCs [38, 39]. Moreover, TGF-β has been demonstrated to activate HSCs through the TGF-β/Smad pathway. In turn, the activated HSCs express TGF-β, thereby forming the inflammatory cascade [40]. Besides, inflammation-induced activation of the NF-κB signaling pathway is also associated with liver fibrosis. Specifically, after NF-κB is activated by proinflammatory factors, such as IL-1β and TNF-α, the IκB proteins are degraded as the IκB kinase complexes are activated. Then, the IκB proteins translocate to the nucleus and bind to the DNA binding sites, promoting fibrosis by regulating its targeting proinflammatory cytokines (IL-1, IL-2, IL-6, and TNF-α), adhesion factors (ICAM, VCAM, and E-selectin), proinflammatory enzymes (COX-2, iNOS), and growth factors (TGF-β) [41–43]. Simultaneously, activation of the NF-κB signaling pathway will further promote the accumulation of proinflammatory chemokines, leading to amplification of the NF-κB signaling pathway furtherly, along with HSC activation to a greater extent [38] (Figure 2).
Figure 2.
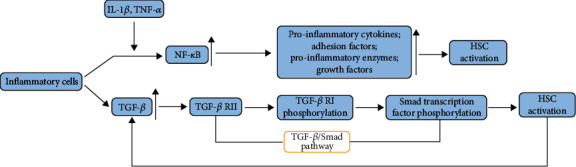
Inflammatory cells are involved in HSC activation. As shown in the picture, NF-κB pathway inflammatory cells of the liver can be activated through cytokines, such as IL-1β and TNF-α. After that, various profibrotic factors will be released and activate HSCs. Besides, the TGF-β, produced by inflammatory cells, can also activate HSCs via the TGF-β/Smad pathway.
3. The Role of NLRP3 Inflammasome and Autophagy in Liver Fibrosis
3.1. The Overview of NLRP3 Inflammasome
Inflammasome, as a biological macromolecule, is a component of innate immunity and plays a significant role in the host defense system and inflammatory signaling platform. It is responsible for detecting the damage factors during infection and tissue damage and activating the inflammatory response. NLRP3 inflammasome, composed of a nucleotide-binding oligomerization domain-like receptor 3 (NLRP3), an apoptosis-associated spot-like protein containing a caspase recruitment domain (ASC), and a serine protease caspase-1 [44], has remarkable effects on innate immunity and various inflammatory diseases, such as type 2 diabetes, atherosclerosis, and neurodegenerative diseases. In addition, neutrophils are involved in the sterile inflammatory response, and the NLRP3 inflammasome pathway plays an important role in the function of neutrophils [45]. Thus, it can be seen that NLRP3 inflammasome activation is associated with liver inflammation and fibrosis. The process of NLRP3 inflammasome activation is composed of two parts: primer phase (signal 1) and trigger phase (signal 2). Signal 1 is induced by LPS, and signal 2 is composed of damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), including intracellular ionic fluxes (Ca2+ influx and K+ efflux), ROS, and lysosomal damage. It has been reported that low concentration of K+ acts at the upstream signal of NLRP3 activation, and K+ efflux can be induced by ATP. In addition, mitochondrial Ca2+ overload may cause mitochondrial dysfunction and associated ROS generation [46].
3.2. The Activation of NLRP3 Inflammasome Promotes HSC Activation through Inflammatory Response
In the murine model of NASH, activation of NLRP3 inflammasome was indispensable for fibrotic response [44]. Moreover, several studies have shown that all components of NLRP3 inflammasome are present in HSCs, which regulate a variety of functions of HSCs and are associated with HSC activation [47]. After activation of NLRP3 inflammasome, pro-IL-18 and pro-IL-1β are cleaved by caspase-1 into matured IL-18 and IL-1β. As two downstream cytokines of NLRP3 inflammasome, IL-18 and IL-1β can promote the transformation of HSCs into mechanocytes [48, 49]. In addition, pyroptosis can be induced by caspase-1 followed by the activation of NLRP3 inflammasome in cells. Afterwards, the cytoplasmic contents, including IL-1β, IL-18, TGF-β, and connective tissue growth factor (CTGF), will be released into the extracellular environment, facilitating the development of liver fibrosis. However, fibrosis will be mitigated when hepatic inflammation was alleviated by the NLRP3 inhibitor [50, 51]. Besides, increased ROS in HSCs may also lead to cell activation via activating NLRP3 inflammasomes [52]. Therefore, NLRP3 inflammasomes play a potential role in the activation of HSCs and the development of liver fibrosis, but there are few relevant studies so far (Figure 3).
Figure 3.
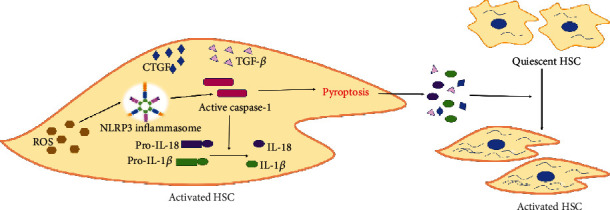
The activation of NLRP3 inflammasome is a key step of HSC activation. Lots of studies have testified that ROS is an activator of NLRP3 inflammasome. In HSCs, NLRP3 inflammasomes are activated by ROS and the procaspase-1 is transformed into activated caspase-1. Then, pro-IL-18 and pro-IL-1β were activated into mature IL-18 and IL-1β. Moreover, caspase-1 can also cause pyroptosis of HSCs. After pyroptosis, cytomembrane is ruptured and the cytoplasmic contents will be released into extracellular environment, such as IL-18, IL-1β, TGF-β, and CTGF, leading to HSC activation.
3.3. The Overview of Autophagy
Autophagy is the process of degrading harmful components in cells including misfolded proteins, damaged organelles, and excessive lipids, to maintain the cellular components and homeostasis [53]. Currently, three types of autophagy have been identified, including macroautophagy, microautophagy, and chaperone-mediated autophagy, among which macroautophagy is the most common form. This review will focus on macroautophagy (hereinafter referred to as autophagy). The general process of autophagy is to gradually encapsulate cytoplasmic components into vesicles of double-layer membranes to form autophagosomes, which then bind to lysosomes and degrade in lysosomes [54]. Autophagy is regulated by numerous autophagy-related genes (ATG), such as Beclin-1, ATG4, Light Chain 3 (LC3, ATG8), and p62. Of note, when autophagy is initiated, LC3I is lipidated to LC3II, which is involved in the formation of autophagosomes. The adaptor protein sequestosome 1 (p62) serves as substrates to LC3II and is degraded along with cargo. Therefore, LC3II and p62 are regarded as biomarkers of autophagic flux [55, 56]. In general, autophagy maintains at a basal level to facilitate cell survival. However, the autophagy level rises in circumstances of inflammation, oxidative stress, and nutrient deprivation. In turn, excessive autophagy will lead to autophagic cell death and is involved in infectious, inflammatory, and liver diseases [54, 57, 58].
3.4. Autophagy Upregulation Facilitates HSC Activation
Autophagy is a crucial manner for cells to adapt to various stress states. During the process of liver fibrosis, the involvement of autophagy in the mechanism of HSC activation has attracted increasing attention [59]. In the study of Li et al., on the one hand, autophagy could activate HSCs, increase the release of ECM, and aggravate the development of fibrosis; on the other hand, autophagy could also negatively modulate the activation of HSCs [58]. It has been demonstrated that autophagy can provide energy for HSC activation. A major feature of HSC activation is the release of lipid droplets containing retinol (vitamin A) and triglycerides, while autophagy can degrade lipid droplets in HSCs and hydrolyze the retinol into free fatty acids, which are further oxidized by mitochondria to produce ATP and provide energy for cell activation [60–62]. Inhibition of autophagy will reduce the degradation of lipid droplets, thereby preventing the activation of HSCs. Except that, during the autophagic process, lysosome-contained cathepsin B, an activator of NLRP3 inflammasome, will be released into cytoplasm [63], which can lead to NLRP3 inflammasome activation and final HSC activation.
3.5. Excessive Autophagy Inhibits Liver Fibrosis
On the contrary, several studies have suggested that autophagy can inhibit the progression of liver fibrosis. Lodder et al. have found that autophagy inhibits Kupffer cells from secreting cytokines such as IL-1β, thereby preventing HSC activation [64]. Then, Chen et al. have demonstrated that cells were damaged by excessive autophagy, resulting in HSC aging, followed by declining activity and reduced ECM secretion [9]. Therefore, autophagy may play a dual role in the process of liver fibrosis, and more evidence is needed (Figure 4).
Figure 4.
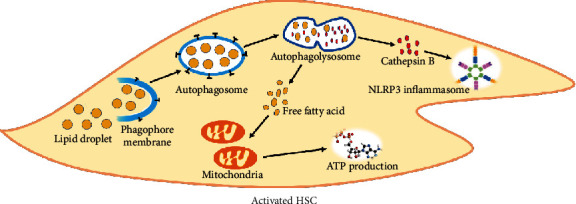
Autophagy participate in HSC activation. Autophagy is the normal physiological function of cells and maintain the balance of energy and substances. However, abnormal autophagy can also induce HSC activation. The consumption of lipid droplets is a character of HSC activation. Therefore, lipid droplets can be resolved into free fatty acid though autophagy. Then, mitochondria will produce ATP by consuming free fatty acid, providing energy for HSC activation. In addition, cathepsin B can be released into cytoplasm due to destruction of the lysosomal membrane. Cathepsin B has been demonstrated to be an inducer of NLRP3 inflammasome activation. So, this is another approach of activating HSCs by autophagy.
4. Autophagy and NLRP3 Inflammasome Activate HSCs via Hedgehog Signaling
Hedgehog (Hh) is a developmental regulator discovered firstly in modulating embryonic morphogenesis [65]. Besides, Hh signaling is also involved in wound healing in adult tissues [66]. The Hh signaling is activated when Hh ligands interact with receptor Patch (Ptch) on the membrane of Hh-responsive cells. After that, the nuclear transcription of the glioma-associated oncogene transcription factor (Gli) family is motivated, which regulates the expression of Hh target genes associated with cell growth and transformation [67]. In the liver, hepatocytes are the main producers of Hh ligands. After hepatocyte death, a mass of Hh ligands is released and stimulates Hh-responsive cells through liver sinusoids and bile canaliculi. It has been demonstrated that HSCs are Hh responsive and will be transformed into fibrogenic myofibroblasts (MFs). However, HSC activation can be inhibited by interfering Hh signaling [67, 68]. It is well-known that excessive autophagy and NLRP3 inflammasome activation can induce autophagic cell death and pyroptosis, respectively [69]. Therefore, if hepatocytes suffer autophagic cell death and pyroptosis, Hh ligands can be released and HSCs may be activated by Hh signaling, leading to liver fibrosis at last.
5. Conclusion and Future Perspectives
In summary, the process of liver fibrosis includes the activation of HSCs and the deposition of ECM. Moreover, autophagy and NLRP3 inflammasome have significant effects on the process of liver fibrosis. As the main downstream signaling of NLRP3 inflammasome, the IL-1 family pathway is a vital mediator of liver injury and fibrogenesis. However, the link between them is complicated, and there have been few studies about the combined effects of autophagy and NLRP3 inflammasome on HSC activation and liver fibrosis. In addition, the regulation of autophagy and NLRP3 inflammasome might be identified as the target for the treatment of liver fibrosis, and further clinical trials are required to verify it.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (NSFC, 81872566).
Conflicts of Interest
The authors declare that they have no conflicts of interest in the present study.
Authors' Contributions
Every author contributed to the writing and revision of the manuscript and accepted the final version. Ye Tao and Ningning Wang contributed equally to this work.
References
- 1.Feili X., Wu S., Ye W., Tu J., Lou L. MicroRNA-34a-5p inhibits liver fibrosis by regulating TGF-β1/Smad3 pathway in hepatic stellate cells. Cell Biology International. 2018;42(10):1370–1376. doi: 10.1002/cbin.11022. [DOI] [PubMed] [Google Scholar]
- 2.Jiang H., Zheng T., Duan T., Chen J., Song B. Non-invasive in vivo imaging grading of liver fibrosis. Journal of Clinical and Translational Hepatology. 2018;6(2):198–207. doi: 10.14218/jcth.2017.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fagone P., Mangano K., Mammana S., et al. Identification of novel targets for the diagnosis and treatment of liver fibrosis. International Journal of Molecular Medicine. 2015;36(3):747–752. doi: 10.3892/ijmm.2015.2264. [DOI] [PubMed] [Google Scholar]
- 4.Sui M., Jiang X., Chen J., Yang H., Zhu Y. Magnesium isoglycyrrhizinate ameliorates liver fibrosis and hepatic stellate cell activation by regulating ferroptosis signaling pathway. Biomedicine & Pharmacotherapy. 2018;106:125–133. doi: 10.1016/j.biopha.2018.06.060. [DOI] [PubMed] [Google Scholar]
- 5.Bolt A. M., Byrd R. M., Klimecki W. T. Autophagy is the predominant process induced by arsenite in human lymphoblastoid cell lines. Toxicology and Applied Pharmacology. 2010;244(3):366–373. doi: 10.1016/j.taap.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fang L., Zhou Y., Cao H., et al. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS One. 2013;8(4, article e60546) doi: 10.1371/journal.pone.0060546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bai J., Yao X., Jiang L., et al. Taurine protects against As2O3-induced autophagy in livers of rat offsprings through PPARγ pathway. Scientific Reports. 2016;6(1, article 27733) doi: 10.1038/srep27733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bai F., Huang Q., Nie J., et al. Trolline ameliorates liver fibrosis by inhibiting the NF-κB pathway, promoting hsc apoptosis and suppressing autophagy. Cellular Physiology and Biochemistry. 2017;44(2):436–446. doi: 10.1159/000485009. [DOI] [PubMed] [Google Scholar]
- 9.Chen W., Zhang Z., Yao Z., et al. Activation of autophagy is required for oroxylin a to alleviate carbon tetrachloride-induced liver fibrosis and hepatic stellate cell activation. International Immunopharmacology. 2018;56:148–155. doi: 10.1016/j.intimp.2018.01.029. [DOI] [PubMed] [Google Scholar]
- 10.Wu X., Zhang F., Xiong X., et al. Tetramethylpyrazine reduces inflammation in liver fibrosis and inhibits inflammatory cytokine expression in hepatic stellate cells by modulating nlrp3 inflammasome pathway. IUBMB Life. 2015;67(4):312–321. doi: 10.1002/iub.1348. [DOI] [PubMed] [Google Scholar]
- 11.Lim H., Min D. S., Park H., Kim H. P. Flavonoids interfere with nlrp3 inflammasome activation. Toxicology and Applied Pharmacology. 2018;355:93–102. doi: 10.1016/j.taap.2018.06.022. [DOI] [PubMed] [Google Scholar]
- 12.Feldman N., Rotter-Maskowitz A., Okun E. Damps as mediators of sterile inflammation in aging-related pathologies. Ageing Research Reviews. 2015;24, Part A:29–39. doi: 10.1016/j.arr.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 13.Kubes P., Mehal W. Z. Sterile inflammation in the liver. Gastroenterology. 2012;143(5):1158–1172. doi: 10.1053/j.gastro.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 14.Ma Z., Liu X., Dong H., et al. Sorafenib and praziquantel synergistically attenuate schistosoma japonicum-induced liver fibrosis in mice. Parasitology Research. 2018;117(9):2831–2839. doi: 10.1007/s00436-018-5972-x. [DOI] [PubMed] [Google Scholar]
- 15.Kabil S. L. Beneficial effects of cilostazol on liver injury induced by common bile duct ligation in rats: role of sirt1 signaling pathway. Clinical and Experimental Pharmacology & Physiology. 2018;45(12):1341–1350. doi: 10.1111/1440-1681.13004. [DOI] [PubMed] [Google Scholar]
- 16.Poynard T., Bedossa P., Opolon P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The Lancet. 1997;349(9055):825–832. doi: 10.1016/s0140-6736(96)07642-8. [DOI] [PubMed] [Google Scholar]
- 17.Lubecka K., Flower K., Beetch M., et al. Loci-specific differences in blood DNA methylation in hbv-negative populations at risk for hepatocellular carcinoma development. Epigenetics. 2018;13(6):605–626. doi: 10.1080/15592294.2018.1481706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hossain N., Afendy A., Stepanova M., et al. Independent predictors of fibrosis in patients with nonalcoholic fatty liver disease. Clinical Gastroenterology and Hepatology. 2009;7(11):1224–1229.e2. doi: 10.1016/j.cgh.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 19.Han X., Song J., Lian L.-H., et al. Ginsenoside 25-och3-ppd promotes activity of lxrs to ameliorate p2x7r-mediated nlrp3 inflammasome in the development of hepatic fibrosis. 2018;66:7023–7035. doi: 10.1021/acs.jafc.8b01982. [DOI] [PubMed] [Google Scholar]
- 20.Hong Y., Li S., Wang J., Li Y. In vitro inhibition of hepatic stellate cell activation by the autophagy- related lipid droplet protein ATG2A. Scientific Reports. 2018;8(1):p. 9232. doi: 10.1038/s41598-018-27686-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumar P., Smith T., Raeman R., et al. Periostin promotes liver fibrogenesis by activating lysyl oxidase in hepatic stellate cells. Journal of Biological Chemistry. 2018;293(33):12781–12792. doi: 10.1074/jbc.RA117.001601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He Q., Fu Y., Ding X., et al. High-mobility group box 1 induces endoplasmic reticulum stress and activates hepatic stellate cells. Laboratory Investigation. 2018;98(9):1200–1210. doi: 10.1038/s41374-018-0085-9. [DOI] [PubMed] [Google Scholar]
- 23.Benyon R. C., Arthur M. J. P. Extracellular matrix degradation and the role of hepatic stellate cells. Seminars in Liver Disease. 2001;21(3):373–384. doi: 10.1055/s-2001-17552. [DOI] [PubMed] [Google Scholar]
- 24.Ra H. J., Parks W. C. Control of matrix metalloproteinase catalytic activity. Matrix Biology. 2007;26(8):587–596. doi: 10.1016/j.matbio.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giannandrea M., Parks W. C. Diverse functions of matrix metalloproteinases during fibrosis. Disease Models & Mechanisms. 2014;7(2):193–203. doi: 10.1242/dmm.012062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robert S., Gicquel T., Victoni T., et al. Involvement of matrix metalloproteinases (mmps) and inflammasome pathway in molecular mechanisms of fibrosis. Bioscience Reports. 2016;36(4) doi: 10.1042/bsr20160107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Madtes D. K., Elston A. L., Kaback L. A., Clark J. G. Selective induction of tissue inhibitor of metalloproteinase-1 in bleomycin-induced pulmonary fibrosis. American Journal of Respiratory Cell and Molecular Biology. 2001;24(5):599–607. doi: 10.1165/ajrcmb.24.5.4192. [DOI] [PubMed] [Google Scholar]
- 28.Tao Y., Qiu T., Yao X., et al. Autophagic-ctsb-inflammasome axis modulates hepatic stellate cells activation in arsenic-induced liver fibrosis. Chemosphere. 2020;242, article 124959 doi: 10.1016/j.chemosphere.2019.124959. [DOI] [PubMed] [Google Scholar]
- 29.Jiang S., Zhang Y., Zheng J.-H., et al. Potentiation of hepatic stellate cell activation by extracellular atp is dependent on P2x7r-mediated NLRP3 inflammasome activation. Pharmacological Research. 2017;117:82–93. doi: 10.1016/j.phrs.2016.11.040. [DOI] [PubMed] [Google Scholar]
- 30.Nencini C., Giorgi G., Micheli L. Protective effect of silymarin on oxidative stress in rat brain. Phytomedicine. 2007;14(2-3):129–135. doi: 10.1016/j.phymed.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 31.Giordano F. J. Oxygen, oxidative stress, hypoxia, and heart failure. The Journal of Clinical Investigation. 2005;115(3):500–508. doi: 10.1172/jci24408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ezhilarasan D. Oxidative stress is bane in chronic liver diseases: clinical and experimental perspective. Arab Journal of Gastroenterology. 2018;19(2):56–64. doi: 10.1016/j.ajg.2018.03.002. [DOI] [PubMed] [Google Scholar]
- 33.Urtasun R., Nieto N. Células estrelladas hepáticas y estrés oxidativo. Revista Española de Enfermedades Digestivas. 2007;99(4) doi: 10.4321/s1130-01082007000400007. [DOI] [PubMed] [Google Scholar]
- 34.Svegliati-Baroni G., Saccomanno S., van Goor H., Jansen P., Benedetti A., Moshage H. Involvement of reactive oxygen species and nitric oxide radicals in activation and proliferation of rat hepatic stellate cells. Liver. 2001;21(1):1–12. doi: 10.1034/j.1600-0676.2001.210101.x. [DOI] [PubMed] [Google Scholar]
- 35.Casini A., Ceni E., Salzano R., et al. Neutrophil-derived superoxide anion induces lipid peroxidation and stimulates collagen synthesis in human hepatic stellate cells: role of nitric oxide. Hepatology. 1997;25(2):361–367. doi: 10.1053/jhep.1997.v25.pm0009021948. [DOI] [PubMed] [Google Scholar]
- 36.Sanchez-Valle V., Chavez-Tapia N. C., Uribe M., Mendez-Sanchez N. Role of oxidative stress and molecular changes in liver fibrosis: a review. Current Medicinal Chemistry. 2012;19(28):4850–4860. doi: 10.2174/092986712803341520. [DOI] [PubMed] [Google Scholar]
- 37.Marzouk B., Marzouk Z., Haloui E., Fenina N., Bouraoui A., Aouni M. Screening of analgesic and anti-inflammatory activities of Citrullus colocynthis from southern Tunisia. Journal of Ethnopharmacology. 2010;128(1):15–19. doi: 10.1016/j.jep.2009.11.027. [DOI] [PubMed] [Google Scholar]
- 38.Wang W., Wang X., Zhang X. S., Liang C. Z. Cryptotanshinone attenuates oxidative stress and inflammation through the regulation of nrf-2 and NF-κB in mice with unilateral ureteral obstruction. Basic & Clinical Pharmacology & Toxicology. 2018;123(6):714–720. doi: 10.1111/bcpt.13091. [DOI] [PubMed] [Google Scholar]
- 39.Amicone L., Marchetti A. Microenvironment and tumor cells: two targets for new molecular therapies of hepatocellular carcinoma. Transl Gastroenterol Hepatol. 2018;3:p. 24. doi: 10.21037/tgh.2018.04.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Derynck R., Zhang Y. E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425(6958):577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 41.Zandi E., Rothwarf D. M., Delhase M., Hayakawa M., Karin M. The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell. 1997;91(2):243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 42.Liu M., Ning X., Li R., et al. Signalling pathways involved in hypoxia-induced renal fibrosis. Journal of Cellular and Molecular Medicine. 2017;21(7):1248–1259. doi: 10.1111/jcmm.13060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grande M. T., Perez-Barriocanal F., Lopez-Novoa J. M. Role of inflammation in túbulo-interstitial damage associated to obstructive nephropathy. Journal of Inflammation. 2010;7(1):p. 19. doi: 10.1186/1476-9255-7-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wree A., McGeough M. D., Pena C. A., et al. NLRP3 inflammasome activation is required for fibrosis development in nafld. Journal of Molecular Medicine. 2014;92(10):1069–1082. doi: 10.1007/s00109-014-1170-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mankan A. K., Dau T., Jenne D., Hornung V. The NLRP3/ASC/Caspase-1 axis regulates il-1β processing in neutrophils. European Journal of Immunology. 2012;42(3):710–715. doi: 10.1002/eji.201141921. [DOI] [PubMed] [Google Scholar]
- 46.He Y., Hara H., Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends in Biochemical Sciences. 2016;41(12):1012–1021. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watanabe A., Sohail M. A., Gomes D. A., et al. Inflammasome-mediated regulation of hepatic stellate cells. American Journal of Physiology Gastrointestinal and Liver Physiology. 2009;296(6):G1248–G1257. doi: 10.1152/ajpgi.90223.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reiter F. P., Wimmer R., Wottke L., et al. Role of interleukin-1 and its antagonism of hepatic stellate cell proliferation and liver fibrosis in the abcb4−/− mouse model. World Journal of Hepatology. 2016;8(8):401–410. doi: 10.4254/wjh.v8.i8.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yaping Z., Ying W., Luqin D., Ning T., Xuemei A., Xixian Y. Mechanism of interleukin-1β-induced proliferation in rat hepatic stellate cells from different levels of signal transduction. APMIS. 2014;122(5):392–398. doi: 10.1111/apm.12155. [DOI] [PubMed] [Google Scholar]
- 50.Mridha A. R., Wree A., Robertson A. A. B., et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental nash in mice. Journal of Hepatology. 2017;66(5):1037–1046. doi: 10.1016/j.jhep.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miao E. A., Rajan J. V., Aderem A. Caspase-1-induced pyroptotic cell death. Immunological Reviews. 2011;243(1):206–214. doi: 10.1111/j.1600-065X.2011.01044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cai S. M., Yang R. Q., Li Y., et al. Angiotensin-(1-7) improves liver fibrosis by regulating the nlrp3 inflammasome via redox balance modulation. Antioxidants & Redox Signaling. 2016;24(14):795–812. doi: 10.1089/ars.2015.6498. [DOI] [PubMed] [Google Scholar]
- 53.Mizushima N., Levine B., Cuervo A. M., Klionsky D. J. Autophagy fights disease through cellular self-digestion. Nature. 2008;451(7182):1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu B., Tan M., Cai W., Wang B., He P., Zhang X. Arsenic trioxide induces autophagic cell death in osteosarcoma cells via the ROS-TFEB signaling pathway. Biochemical and Biophysical Research Communications. 2018;496(1):167–175. doi: 10.1016/j.bbrc.2018.01.018. [DOI] [PubMed] [Google Scholar]
- 55.Guimei M., Eladl M. A., Ranade A. V., Manzoor S. Autophagy related markers (Beclin-1 and Atg4b) are strongly expressed in wilms' tumor and correlate with favorable histology. Histology and Histopathology. 2019;34(1):47–56. doi: 10.14670/HH-18-023. [DOI] [PubMed] [Google Scholar]
- 56.Klionsky D. J., Abdelmohsen K., Abe A., et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Booth L. A., Tavallai S., Hamed H. A., Cruickshanks N., Dent P. The role of cell signalling in the crosstalk between autophagy and apoptosis. Cellular Signalling. 2014;26(3):549–555. doi: 10.1016/j.cellsig.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J., Zeng C., Zheng B., et al. Hmgb1-induced autophagy facilitates hepatic stellate cells activation: a new pathway in liver fibrosis. Clinical Science (London, England) 2018;132(15):1645–1667. doi: 10.1042/cs20180177. [DOI] [PubMed] [Google Scholar]
- 59.Shao Y. Z., Zhao H. J., Wang Y., et al. The apoptosis in arsenic-induced oxidative stress is associated with autophagy in the testis tissues of chicken. Poultry Science. 2018;97(9):3248–3257. doi: 10.3382/ps/pey156. [DOI] [PubMed] [Google Scholar]
- 60.Park S., Kim S., Kim M. J., et al. Golga2 loss causes fibrosis with autophagy in the mouse lung and liver. Biochemical and Biophysical Research Communications. 2018;495(1):594–600. doi: 10.1016/j.bbrc.2017.11.049. [DOI] [PubMed] [Google Scholar]
- 61.Wang B., Yang H., Fan Y., et al. 3-Methyladenine ameliorates liver fibrosis through autophagy regulated by the NF-κB signaling pathways on hepatic stellate cell. Oncotarget. 2017;8(64):107603–107611. doi: 10.18632/oncotarget.22539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hernández-Gea V., Friedman S. L. Autophagy fuels tissue fibrogenesis. Autophagy. 2014;8(5):849–850. doi: 10.4161/auto.19947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qiu T., Pei P., Yao X., et al. Taurine attenuates arsenic-induced pyroptosis and nonalcoholic steatohepatitis by inhibiting the autophagic-inflammasomal pathway. Cell Death & Disease. 2018;9(10):p. 946. doi: 10.1038/s41419-018-1004-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lodder J., Denaes T., Chobert M. N., et al. Macrophage autophagy protects against liver fibrosis in mice. Autophagy. 2015;11(8):1280–1292. doi: 10.1080/15548627.2015.1058473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu F. X., Guan K. L. The hippo pathway: regulators and regulations. Genes & Development. 2013;27(4):355–371. doi: 10.1101/gad.210773.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gao L., Zhang Z., Zhang P., Yu M., Yang T. Role of canonical hedgehog signaling pathway in liver. International Journal of Biological Sciences. 2018;14(12):1636–1644. doi: 10.7150/ijbs.28089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yan J., Huang H., Liu Z., et al. Hedgehog signaling pathway regulates hexavalent chromium-induced liver fibrosis by activation of hepatic stellate cells. Toxicology Letters. 2020;320:1–8. doi: 10.1016/j.toxlet.2019.11.017. [DOI] [PubMed] [Google Scholar]
- 68.Kumar V., Dong Y., Kumar V., Almawash S., Mahato R. I. The use of micelles to deliver potential hedgehog pathway inhibitor for the treatment of liver fibrosis. Theranostics. 2019;9(25):7537–7555. doi: 10.7150/thno.38913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wree A., Eguchi A., McGeough M. D., et al. Nlrp3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014;59(3):898–910. doi: 10.1002/hep.26592. [DOI] [PMC free article] [PubMed] [Google Scholar]