

Abstract
Background
Paragonimus spp. (lung flukes) are among the most injurious foodborne helminths, infecting ~23 million people and subjecting ~292 million to infection risk. Paragonimiasis is acquired from infected undercooked crustaceans and primarily affects the lungs but often causes lesions elsewhere including the brain. The disease is easily mistaken for tuberculosis owing to similar pulmonary symptoms, and accordingly, diagnostics are in demand.
Results
We assembled, annotated, and compared draft genomes of 4 prevalent and distinct Paragonimus species: Paragonimus miyazakii, Paragonimus westermani, Paragonimus kellicotti, and Paragonimus heterotremus. Genomes ranged from 697 to 923 Mb, included 12,072–12,853 genes, and were 71.6–90.1% complete according to BUSCO. Orthologous group analysis spanning 21 species (lung, liver, and blood flukes, additional platyhelminths, and hosts) provided insights into lung fluke biology. We identified 256 lung fluke–specific and conserved orthologous groups with consistent transcriptional adult-stage Paragonimus expression profiles and enriched for iron acquisition, immune modulation, and other parasite functions. Previously identified Paragonimus diagnostic antigens were matched to genes, providing an opportunity to optimize and ensure pan-Paragonimus reactivity for diagnostic assays.
Conclusions
This report provides advances in molecular understanding of Paragonimus and underpins future studies into the biology, evolution, and pathogenesis of Paragonimus and related foodborne flukes. We anticipate that these novel genomic and transcriptomic resources will be invaluable for future lung fluke research.
Keywords: lung flukes, Paragonimus, genomics, transcriptomics, diagnostics, paragonimiasis, infectious disease, trematodes
Background
The trematode genus Paragonimus, the lung flukes, is among the most injurious taxon of food-borne helminths. Approximately 23 million people are infected with lung flukes [1], an estimated 292 million people are at risk, mainly in eastern Asia [2], and billions of people live in areas where Paragonimus infections of animals are endemic. The life-cycle of Paragonimus species involves freshwater snails, crustacean intermediate hosts, and mammals in Asia, parts of Africa, and the Americas [3]. Human paragonimiasis is acquired by consuming raw or undercooked shrimp and crabs containing the metacercaria, which is the infective stage. Although primarily affecting the lungs, lesions can occur at other sites, including the brain, and pulmonary paragonimiasis is frequently mistaken for tuberculosis owing to similar respiratory symptoms [4].
Pathogenesis ensues because of the migration of the newly invading juveniles from the gut to the lungs and through not-infrequent ectopic migration to the brain, reproductive organs, and subcutaneous sites at the extremities, and because of toxins and other mediators released by the parasites during the larval migration [4, 5]. The presence of the flukes in the lung causes hemorrhage, inflammation with leukocytic infiltration, and necrosis of lung parenchyma that gradually proceeds to the development of fibrotic encapsulation except for a fistula from the evolving lesion to the respiratory tract. Eggs of the lung fluke exit the encapsulated lesion through the fistula to reach the sputum and/or feces of the host, where they pass to the external environment, accomplishing transmission of the parasite [6]. There are signs and symptoms that allow characterization of acute and chronic stages of paragonimiasis. In pulmonary paragonimiasis, for example, the most noticeable clinical symptom of an infected individual is a chronic cough with gelatinous, rusty brown, pneumonia-like, blood-streaked sputum [6]. Heavy work commonly induces hemoptysis. Pneumothorax, empyema from secondary bacterial infection, and pleural effusion might also be presented. When symptoms include only a chronic cough, the disease may be misinterpreted as chronic bronchitis and bronchiectasis or bronchial asthma. Pulmonary paragonimiasis is frequently confused with pulmonary tuberculosis [4]. The symptoms of extra-pulmonary paragonimiasis vary depending on the location of the fluke, including cerebral [5] and abdominal paragonimiasis [6].
Paragonimus is a large genus that includes >50 nominal species [7]. Seven of these species or species complexes of Paragonimus are known to infect humans [3]. This is also an ancient genus, thought to have originated before the breakup of Gondwana [8], but possibly also dispersing as colonists from the original East Asian clade, based on the distribution of host species [9]. To improve our understanding of pathogens across this genus at the molecular level, we have assembled, annotated, and compared draft genomes of 4 of these, 3 from Asia (Paragonimus westermani from Japan, Paragonimus herotremus, Paragonimus miyazakii) and 1 from North America (Paragonimus kellicotti). Among them, P. westermani is the best-known species causing pulmonary paragonimiasis. This name has been applied to a genetically and geographically diverse complex of lung fluke populations differing widely in biological features including infectivity to humans [10]. The complex extends from India and Sri Lanka eastwards to Siberia, Korea, and Japan, and southwards into Vietnam, Indonesia, and the Philippines. However, human infections are reported primarily from China, Korea, Japan, and the Philippines. Until this study, an Indian member of the P. westermani complex was the only lung fluke species for which a genome sequence was available [11]. Paragonimus heterotremus is the most common cause of pulmonary paragonimiasis in southern China, Lao PDR, Vietnam, northeastern India, and Thailand [6, 7]. Paragonimus miyazakii is a member of the Paragonimus skrjabini complex, to which Blair and co-workers accorded subspecific status [12]. Flukes of this complex tend not to mature in humans but frequently cause ectopic disease at diverse sites, including the brain. In North America, infection with P. kellicotti is primarily a disease of native, crayfish-eating mammals including the otter and mink. The occasional human infections can be severe, and thoracic involvement is typical [13, 14].
These 4 species represent a broad sampling of the phylogenetic diversity of the genus. Most of the known diversity, as revealed by DNA sequences from portions of the mitochondrial genome and the nuclear ribosomal genes, resides in Asia [15]. Analysis of the ITS2 marker by Blair et al. [15] indicates that each of the species sequenced occupies a distinct clade within the phylogenetic tree.
In addition to a greater understanding of the genome contents of this group of foodborne trematodes, the findings presented here provide new information to assist development of diagnostic tools and recognition of potential drug targets. The data and findings facilitate evolutionary, zoogeographical, and phylogenetic investigation of the genus Paragonimus and its host-parasite relationships through the comparative analysis of gene content relative to other sequenced platyhelminth and host species, and to known Paragonimus diagnostic antigen targets.
Data Description
Genomic sequence data were generated from DNA samples from 4 distinct Paragonimus species: 3 from Asia, P. miyazakii (Japan), P. heterotremus (LC strain, Vietnam), andParagonimus westermani (Japan), and 1 from North America, P. kellicotti (Missouri, USA). Illumina DNA sequencing produced short overlapping fragments and long insert size (3 and 8 kb) whole-genome shotgun libraries for all 4 spcies. Genome coverage per species is presented in Table 1. Owing to the higher fragmentation rate of the P. kellicotti assembly, long-read Pacific Biosciences (PacBio) reads were generated and used for assembly improvement (Table 2). To estimate the genetic divergence between geographically diverse samples, we compared our East Asian P. westermani sample from Japan with the previously published P. westermani genome from India by retrieving the Inidian genome from the previous study [11]. To facilitate gene annotation in the newly generated assemblies and to provide transcriptomic data for analysis, adult-stage RNA sequencing (RNA-seq) samples were also retrieved from our previous reports for P. westermani [16] and P. kellicotti [17]. We also collected adult-stage RNA samples for Illumina RNA-seq from young adult and adult samples for P. heterotremus, along with stages from the liver, peritoneal cavity, lung (adult), and pleural cavity for P. miyazakii.
Table 1:
Paragonimusspp. genome and RNA-seq data accessions
| Genome assemblies, annotations, and raw reads | |||
|---|---|---|---|
| Species | NCBI accession | Bioproject ID | Genome coverage (×) |
| Paragonimus miyazakii | JTDE00000000 | PRJNA245325 | 162 |
| Paragonimus heterotremus | LUCH00000000 | PRJNA284523 | 81 |
| Paragonimus kellicotti | LOND00000000 | PRJNA179523 | 77 (43*) |
| Paragonimus westermani | JTDF00000000 | PRJNA219632 | 152 |
| RNA-Seq dataset accessions | |||
| Species | NCBI accession | Bioproject ID | Body site or stage |
| Paragonimus miyazakii | SRX1100074 | PRJNA245325 | Pleural cavity |
| SRX1100062 | PRJNA245325 | Lung | |
| SRX1037170 | PRJNA245325 | Peritoneal cavity | |
| SRX1037172 | PRJNA245325 | Peritoneal cavity | |
| SRX1037171 | PRJNA245325 | Liver | |
| Paragonimus heterotremus | SRX3713099 | PRJNA284523 | Adult (technical rep 1) |
| SRX3713100 | PRJNA284523 | Adult (technical rep 2) | |
| SRX3713101 | PRJNA284523 | Young adult | |
| SRX3713102 | PRJNA284523 | Young adult | |
| Paragonimus kellicotti | SRX3718311 | PRJNA179523 | Adult |
| SRX3718310 | PRJNA179523 | Adult | |
| Paragonimus westermani | SRX1507710 | PRJNA219632 | Adult |
*Pacific Biosciences dataset coverage.
Table 2:
The draft genome of Paragonimus: assembly, size, and annotation characteristics
| Statistic | Paragonimus miyazakii | Paragonimus heterotremus | Paragonimus kellicotti | Paragonimus westermani (Japan) | Paragonimus westermani (India) |
|---|---|---|---|---|---|
| Assembly statistics | |||||
| Total genome length (Mb) | 915.8 | 841.2 | 696.5 | 923.3 | 922.8 |
| Number of contigs | 22,318 | 27,557 | 29,377 | 22,477 | 30,455 |
| Mean contig size (kb) | 41 | 30.5 | 23.7 | 41.1 | 30.3 |
| Median contig size (kb) | 15.1 | 9.3 | 10.2 | 17.2 | 4.8 |
| Maximum contig size (kb) | 919.8 | 715.6 | 826 | 829 | 809.4 |
| N50 length (kb) | 108.8 | 92.5 | 56.0 | 100.8 | 135.2 |
| N50 No. | 2320 | 2506 | 3316 | 2664 | 1943 |
| BUSCO completeness (303 genes, eukarota_odb9) | |||||
| Complete, single copy (%) | 84.5 | 82.5 | 70.3 | 88.78 | 76.90 |
| Complete, duplicated (%) | 1.3 | 0.0 | 1.3 | 1.32 | 2.31 |
| Fragmented (%) | 7.6 | 10.9 | 15.2 | 6.27 | 14.85 |
| Missing (%) | 6.6 | 6.6 | 13.2 | 3.63 | 5.94 |
| Gene statistics | |||||
| No. of genes | 12,652 | 12,490 | 12,853 | 12,072 | 12,771 |
| Mean gene length (kb) | 25.9 | 22.6 | 17.6 | 24.1 | 18.0 |
| Mean CDS length (kb) | 1.5 | 1.4 | 1.1 | 1.4 | 1.4 |
| Mean intron length (kb) | 4.2 | 4 | 3.6 | 4.2 | 4.0 |
| Mean No. exons per gene | 6.7 | 6.2 | 5.3 | 6.3 | 5.2 |
| Annotated (%) | |||||
| InterPro | 82 | 85 | 81 | 87 | 82 |
| KEGG | 40 | 41 | 34 | 43 | 43 |
CDS: coding sequence.
Genomic raw reads, genome assemblies, genome annotations, and raw transcriptomic (RNA-Seq) fastq files were uploaded and are available for download from the NCBI SRA [18], with all accession numbers and relevant metadata provided in Table 1. Supplementary Table S1 provides, for each of the species, complete gene lists and gene expression levels for each of the RNA-Seq samples. All results of the genome-wide selection scan are provided in Supplementary Table S2. For each orthologous group (OG) identified, Supplementary Table S3 provides complete gene lists, counts of genes per species, and mean gene expression levels from each the Paragonimus transcriptome datasets described above. All relevant software versions, as well as commands specifying the parameters used, are presented in Supplementary Text S1.
Results and Discussion
Genome features
The sizes of the 4 newly generated Paragonimus genomes range from 697 to 923 Mb, containing between 12,072 and 12,853 genes. These draft genomes are estimated to be between 71.6% and 90.1% complete, according to the number of complete BUSCO eukaryote genes (single-copy or duplicate) [19], with the new P. westermani genome produced from a sample collected from Japan being more complete than the previously sequenced genome produced from a sample collected from India [11] (90.1% vs 70.2%, respectively; Table 2). Here, statements about P. westermani apply to the new Japanese genome unless otherwise stated. The total genome lengths of the Paragonimus spp. are larger than those of the Schistosomatidae and Opisthorchiidae but smaller than those of Fasciolidae. However, the total numbers of protein-coding genes are comparable (Table 2; complete gene lists for each species provided in Supplementary Table S1). Repetitive sequences occupy between 49% and 54% of the Paragonimus genomes (Fig. 1A). The repeat landscapes, depicting the relative abundance of repeat classes in the genome, vs the Kimura divergence from the consensus, revealed that P. kellicotti in particular has a significant number of copies of transposable elements (TEs) with high similarity to consensus (Kimura substitution level: 0–5), indicating recent and current TE activity (Fig. 1B). In a recent study [20], TE activity in the Fasciolidae was found to be low. TEs are potent sources of mutation that can rapidly create genetic variance, especially following genetic bottlenecks and environmental changes, providing bursts of allelic and phenotypic diversity upon which selection can act [21, 22]. Therefore, changes in TE activity, modulated by environmentally induced physiological or genomic stress, may have a major effect on adaptation of populations and species facing novel habitats and large environmental perturbations [23].
Figure 1:

Comparisons of the overall content of the assembled Paragonimus genome assemblies. Comparisons are based on (A) length (including statistics for other sequenced trematode genomes) and (B) repeat landscapes, measured using the Kimura substitution level, which indicates how much a repeat sequence has degenerated since its incorporation into the genome (i.e., how recently the repeat sequence was added). The high peak at the far left of P. kellicotti indicates a recent incorporation or active transposable element activity. LINE: long interspersed nuclear element; LTR: long terminal repeat.
Focusing on the gene content, P. kellicotti had the shortest mean total gene length among the species, and the lung flukes overall had similar gene lengths to other flukes, while platyhelminth species other than trematodes have shorter genes overall (Fig. 2A). The variability in gene lengths observed between species results from differences in both mean intron lengths (Fig. 2B) and the mean number of exons per gene (Fig. 2C), while the average coding sequence (CDS) lengths of the exons across all the platyhelminth species were similar to each other (Fig. 2D). Whereas there was species-to-species variability in gene lengths and exon counts, consistent patterns among the types of flukes were not apparent. Some of this variability may have arisen as a result of the variation in quality of the assemblies, but these differences were minimized by only using complete gene models with a start and stop codon identified in the same frame.
Figure 2:
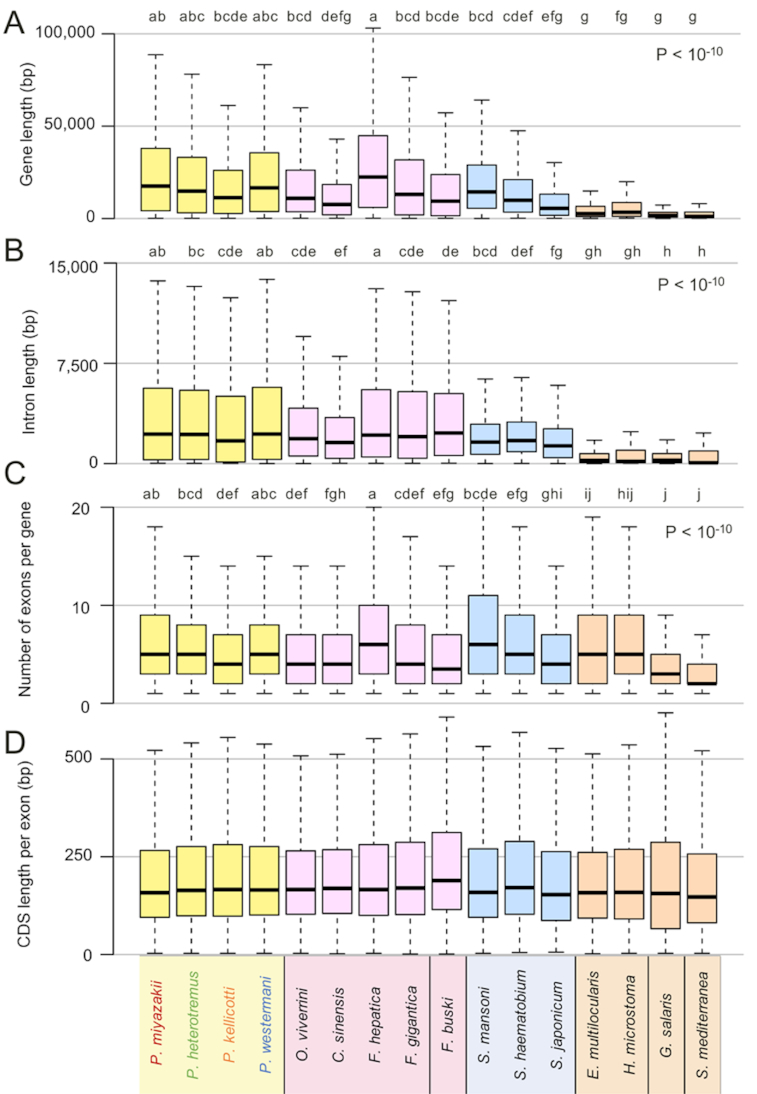
Comparison of genome annotation characteristics and attributes among several species of flatworms. Attributes characterized included (A) full gene lengths, including coding and noncoding sequences; (B) mean intron lengths per gene; (C) number of exons per gene; and (D) coding sequence (CDS) length per exon. P-values and letter groupings indicate significant differences among species, as calculated using ANOVA with Tukey HSD post hoc test (i.e., two species labeled the same letter in the group are not significantly different, P < 0.05). Boxes represent the interquartile ranges (IQRs) between the first and third quartiles, and the line inside the box represents the median value. Whiskers represent the lowest or highest values within values 1.5 times the IQR from the first or third quartiles.
Mitochondrial whole-genome–based clustering was performed for the 4 Paragonimus species plus some additional existing previously sequenced mitochondrial genome assemblies for P. ohirai and 4 for P. westermani (Fig. 3A). This indicated that our Japanese P. westermani sample clustered with the existing known P. westermani samples from eastern Asia and that all the other 3 newly sequenced species were distinct from P. ohirai.
Figure 3.
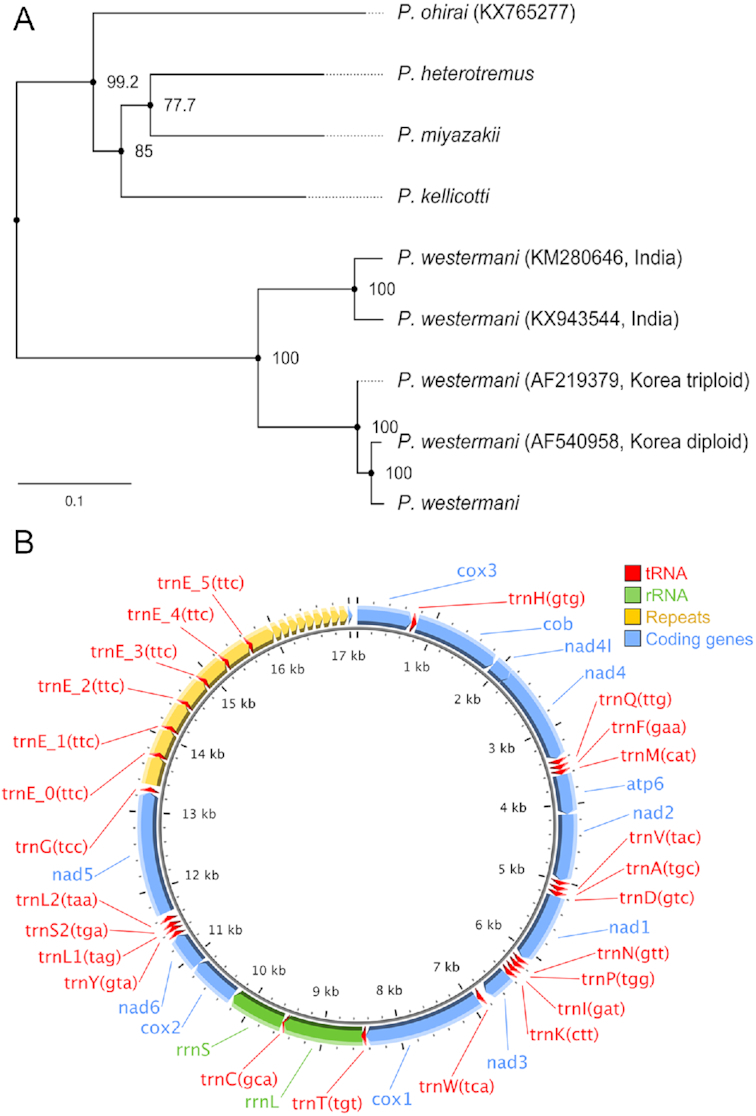
Clustering of Paragonimus species. (A) Mitochondrial whole-genome–based phylogeny, including previously sequenced Paragonimus mitochondrial genomes (with accessions indicated). The branch lengths represent nucleotide substitutions per site, and the numbers shown at nodes indicate SH-like approximate likelihood-ratio test (SH-aLRT) support values. (B) Paragonimus kellicotti mitogenome gene structure. rRNA: ribosomal RNA; tRNA: transfer RNA.
We generated a PacBio long-read–based mitochondrial assembly for P. kellicotti. The fully circularized complete genome was 17.3 kb in length, including a 3.7-kb non-coding repeat region between tRNAGly and cox3 (Fig. 3B). There are 7 copies of long repeats (378 bp) and 9.5 copies of short repeats (111 bp). The long repeats overlap with 6 copies of tRNAGlu. This structural organization of repeat sequences does not resemble those found in previous comparison of Paragonimus ohirai and P. westermani [11], where the non-coding region is partitioned by tRNAGlu into 2 parts.
Clustering based on nuclear genomes' single-member orthologous protein families (OPFs) of the 4 new lung flukes, 4 liver flukes, 3 blood flukes, 5 other platyhelminths, 4 host species, and a yeast outgroup was performed on the basis of the shared phylogeny among orthologous OPF groups. These findings mirrored the mitochondrial clustering results for the lung fluke species (Fig. 4), indicating that P. westermani is the earlier-diverging taxon, as previously suggested on the basis of ribosomal RNA [24].
Figure 4:
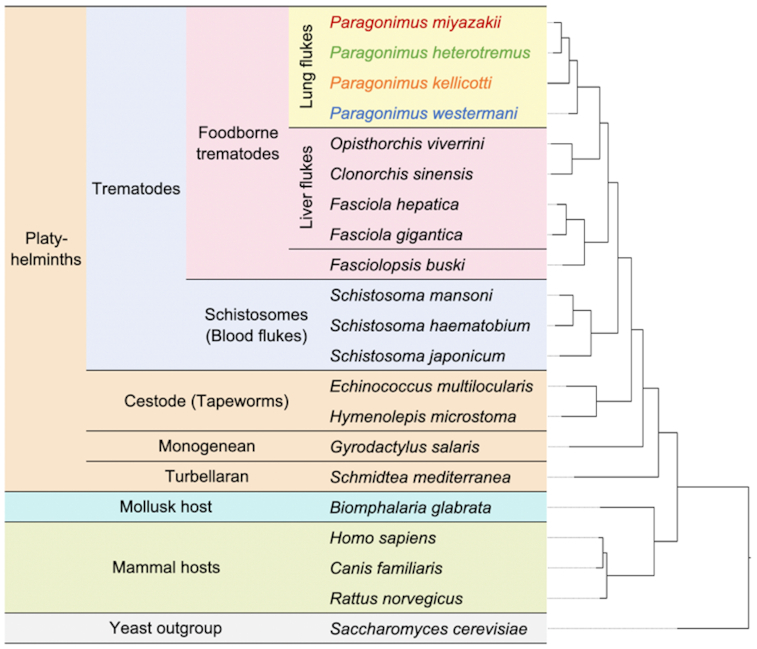
Species clustering based on single-member OPF sequences. A total of 262,720 genes (85% of all genes across the species) were assigned to 17,953 OPFs; 2,493 genes are in 326 species-specific OPFs.
Our P. westermani reference genome was assembled using samples collected from Japan (Amakusa, Kyushu). We compared the genomic sequences of our East Asian P. westermani to the recently published P. westermani genome from India (Changlang, Arunachal Pradesh) [11] to estimate the genetic divergence between geographically diverse samples. This analysis identified a mean nucleotide sequence identity of 87.6%.
Gene-family dynamics identify expanded functions distinguishing lung fluke species
We investigated large-scale differences in gene complements among families of digenetic trematodes (Fig. 5A) and modeled gene gain and loss while accounting for the phylogenetic history of species [25]. Gene families of interest that displayed pronounced differential expansion or contraction (Fig. 5B) included the papain-family cysteine proteases, cathepsins L, B, and F, dynein heavy chain, spectrin/dystrophin, heat shock 70 kDa protein, major vault protein, and multidrug resistance protein. Total protease and protease inhibitor counts are shown in Fig. 5C. Cathepsin F genes may have roles in nutrient digestion and remodeling of other physiologically active molecules, and Ahn et al. [26] reported differential expression of cathepsin F genes during development of P. westermani and showed that most are highly immunogenic. This flagged them as prospective diagnostic targets. The importance of cathepsin F for Paragonimus contrasts with its function in the fasciolids, where cathepsin L genes are expanded and are thought to play a more critical role in host invasion [20, 27].
Figure 5:
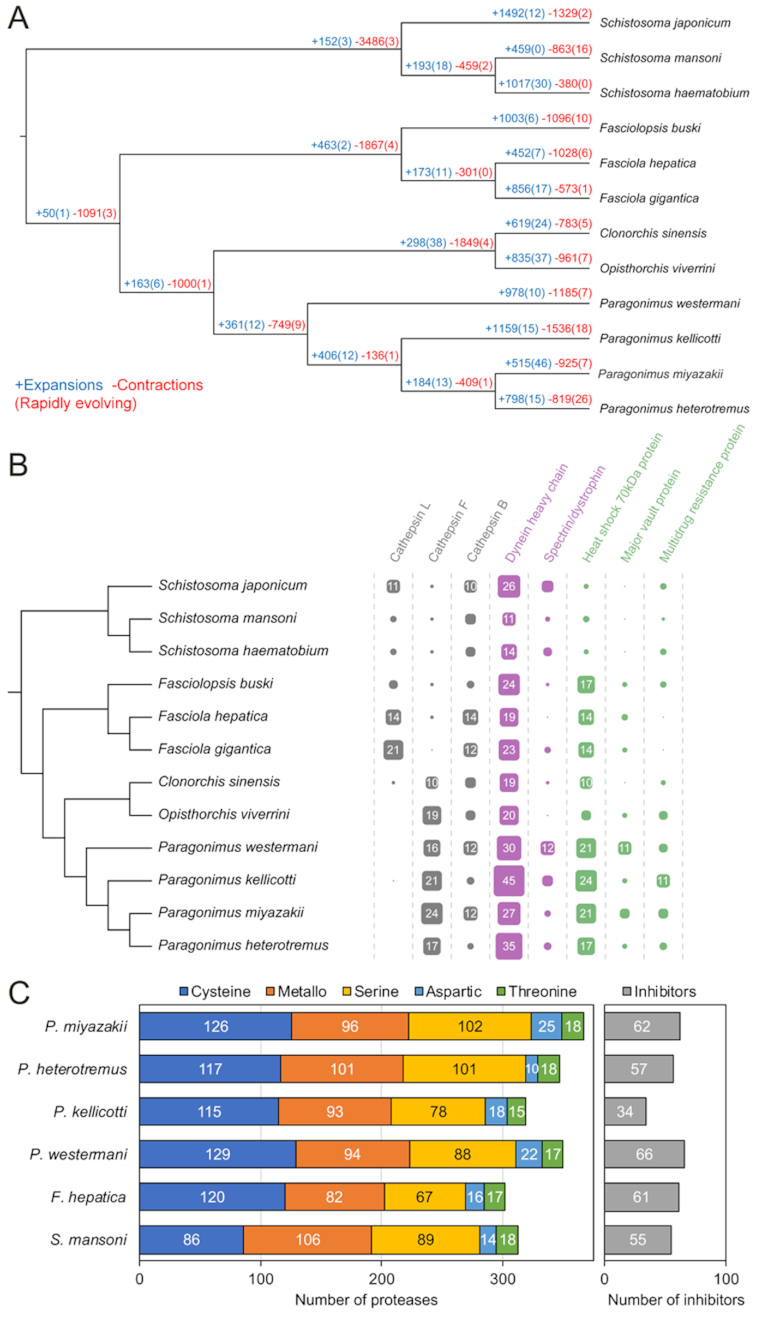
Gene-family dynamics among platyhelminth species. (A) Rapidly evolving families of interest are quantified at each stage of the phylogeny, including genes gained (blue) and lost (red) relative to other species. The numbers of rapidly evolving genes are indicated in parentheses. (B) Functionally annotated gene families of interest that displayed most pronounced differential expansions or contractions. (C) Overall protease and protease inhibitor abundance per species.
Differential expansion of cytoskeletal molecules is of interest in the context of tegument physiology [28]. Dynein is a microtubule motor protein, which transports intracellular cargo. Spectrin is an actin-binding protein, with a key role in maintenance of integrity of the plasma membrane. Dystrophin links microfilaments with extracellular matrix. The syncytial tegument of the surface of flatworms is a complex structure and a major adaptation to parasitism, and plays critical roles in nutrient uptake, immune response modulation and evasion, and other processes [28].
In Paragonimus spp., expanded gene families included heat shock proteins (HSPs), major vault proteins, and multidrug resistance proteins that play roles in maintaining cellular homeostasis under stress conditions. HSPs of flatworm parasites play a key role as molecular chaperones in the maintenance of protein homeostasis. They also are immunogenic and immunomodulatory. HSP is the most abundant family of proteins in the immature and mature egg of Schistosoma mansoni, and in the miracidium [29] and is highly abundant in the tegument of the adult schistosome [30]. In addition, HSP is abundant in the excretory/secretory products of the adult Schistosoma japonicum blood fluke [31]. HSP stimulates diverse immune cells, eliciting release of pro- and anti-inflammatory cytokines [32], and binds human low-density lipoprotein (the purpose of which is unknown but may be associated with transport of apoprotein B or in lipid trafficking [33]), and, given these properties, HSP represents a promising vaccine and diagnostic candidate [34]. Vaults, ribonucleoprotein complexes, are highly conserved in eukaryotes. Although their exact function remains unclear, it may be associated with multidrug resistance phenotypes and with signal transduction. In S. mansoni, up-regulation of major vault protein has been observed during the transition from cercaria to schistosomulum and in praziquantel-resistant adult worms [35]. ATP-binding cassette transporters (ABC transporters) are essential components of cellular physiological machinery, and some ABC transporters, including P-glycoproteins, pump toxins and xenobiotics out of the cell. Overexpression of P-glycoprotein has been reported in a praziquantel-resistant S. mansoni [36].
Tetraspanin sequence evolution in P. kellicotti
We searched for genes that evolved under positive selection in the 4 Paragonimus spp. based on the non-synonymous to synonymous substitution rate ratio (dN/dS). We conducted the branch-site test of positive selection to identify adaptive gene variants that became fixed in each species [37] (Supplementary Table S2). A tetraspanin from P. kellicotti (PKEL_00573) reached statistical significance after correction for multiple testing (dN/dS = 9.9, false discovery rate = 0.018). Tetraspanins are small integral proteins bearing 4 transmembrane domains, which form 2 extracellular loops [38]. In trematodes, they are major components of the tegument at the host-parasite interface [39], are highly immunogenic vaccine antigens [40, 41], and may play a role in immune evasion [42]. In the tetraspanin sequence of P. kellicotti, we detected 6 amino acid sites under positive selection (Fig. 6). Five of the 6 sites were predicted to be located within the extracellular loops believed to interact with the immune system of the host. A similar pattern of positive selection within regions that code for extracellular loops has been reported in tetraspanin-23 from African Schistosoma species [43].
Figure 6:
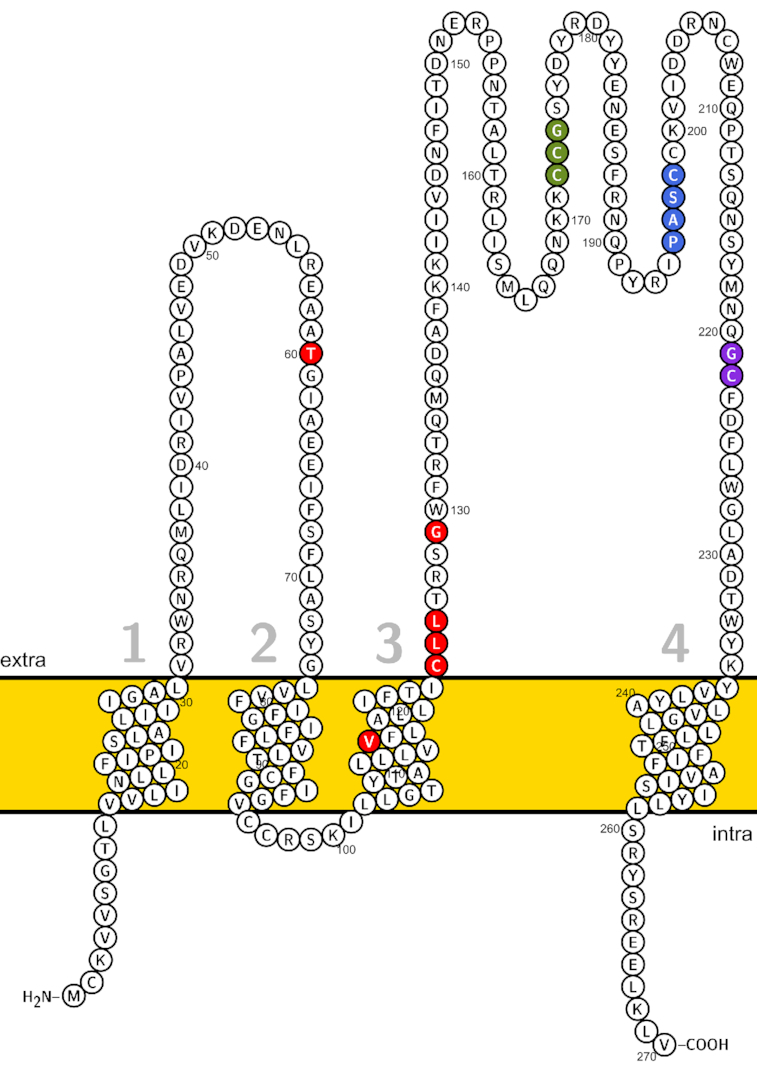
Predicted transmembrane helical topology of Paragonimus kellicotti tetraspanin (PKEL_00573). Amino acid sites under positive selection (red) and conserved motifs (CCG, PXSC, and GC motifs in green, blue and purple, respectively). The “PXSC” motif here is represented by the “PASC” sequence.
Gene phylogeny analysis identifies functions conserved and specific to fluke groups
We classified OGs on the basis of phylogenetic distribution of proteins from each of the 21 species (Fig. 4). Complete gene counts and lists per species and per OG are provided in Supplementary Table S3. These results were parsed to identify the OGs containing members among the platyhelminth species, and those that were conserved across all members of each group (lung, liver, and blood flukes and other platyhelminth species; Fig. 7A). This analysis identified 256 OGs that were conserved among, and exclusive to, the lung flukes (Fig. 7A and B). The lung fluke–conserved and –specific genes were significantly enriched for several gene ontology (GO) terms (Table 3; using P. miyazakii genes to test significance), most of which were related to peptidase activity (including serine proteases, which are involved in host tissue invasion, anticoagulation, and immune evasion [44]), as well as “iron binding” (which may be related to novel iron acquisition mechanisms from host tissue, which is not well understood in most metazoan parasites but has been described in schistosomes [45]). Lung (adult) stage RNA-Seq datasets were collected for each of the 4 lung fluke species (accessions in Table 1), and reads were mapped to each of their respective genomes. Based on the 1:1 gene orthologs (as defined by the previously described OG dataset), the orthologous genes across the lung flukes had consistent adult-stage gene expression levels, with Pearson correlations ranging from 0.72 to 0.85 (Fig. 8A and B).
Figure 7:

Orthologous Group (OG) distribution analysis. (A) OGs identified among groups of flukes. The OGs conserved in ≥1 of the species from each group are indicated in black, and the OGs conserved among all the species in the overlapping groups are indicated in red. (B) Counts of OGs among the 4 Paragonimus species, with Paragonimus-specific gene sets indicated in red. The 256 Paragonimus conserved-and-specific genes are highlighted in yellow. (C) Significant functional enrichment (Interpro domains) among the gene sets conserved among, and specific to, each major group of flukes (256, 758, and 270 OPFs in lung, liver, and blood flukes, respectively), relative to the functions in the complete gene sets.
Table 3:
“Molecular Function” Gene Ontology terms enriched among P. miyazakii genes that are conserved among and exclusive to lung flukes
| GO ID | GO term name | P value | No. conserved and specific | Total No. in genome |
|---|---|---|---|---|
| GO:0004175 | Endopeptidase activity | 5.2E−05 | 8 | 132 |
| GO:0008236 | Serine-type peptidase activity | 5.6E−05 | 6 | 67 |
| GO:0017171 | Serine hydrolase activity | 5.6E−05 | 6 | 67 |
| GO:0 004252 | Serine-type endopeptidase activity | 1.6E−04 | 5 | 51 |
| GO:00 70011 | Peptidase activity, acting on L-amino acid peptides | 6.1E−04 | 9 | 237 |
| GO:0 008233 | Peptidase activity | 8.7E−04 | 9 | 249 |
| GO:0 004568 | Chitinase activity | 2.1E−03 | 2 | 7 |
| GO:0 004190 | Aspartic-type endopeptidase activity | 1.1E−02 | 2 | 16 |
| GO:00 70001 | Aspartic-type peptidase activity | 1.1E−02 | 2 | 16 |
| GO:0 008199 | Ferric iron binding | 1.1E−02 | 2 | 16 |
Figure 8:

Analysis of gene expression data for species of lung flukes of the genus Paragonimus. (A) Comparison of adult-stage gene expression levels among 1:1 orthologs shared by P. westermani and P. miyazakii. Pearson correlation = 0.79. (B) Pearson correlation values between all lung fluke species for the adult-stage expression levels of all 1:1 orthologous genes.
Expansion of unique aspartic proteases (including those predicted to be retropepsins) and other peptidases in the lung flukes may be associated with digestion of ingested blood, given the key role of this category of hydrolases and their inhibitors in nutrition and digestion of hemoglobin by schistosomes, and indeed other blood-feeding worms including hookworms [46, 47]. Given that pulmonary hemorrhage and hemoptysis are cardinal signs of lung fluke infection, it can be anticipated that the lung flukes ingest host blood when localized at the ulcerous lesion induced in the pulmonary parenchyma by infection. Overall, protease counts across species were similar (Fig. 5C) although P. kellicotti had substantially fewer protease inhibitors compared to the other Paragonimus species (34 vs 57, 62, and 66), Fasciola hepatica (61), and S. mansoni (55). Protease inhibitors in flukes are thought to be important for creating a safe environment for the parasite inside the host by inhibiting and regulating protease activity and immunomodulation [91], so this may suggest a novel host interaction strategy by P. kellicotti.
Analysis of the adult-stage gene expression levels of the discrete protease classes (Fig. 9) did not identify substantial differences among the Paragonimus species, except for a lower expression of threonine proteases in P. kellicotti. During the adult stage, cysteine proteases in all Paragonimus species exhibited significantly higher expression overall compared to F. hepatica, but expression levels similar to those of S. mansoni. A previous study identified immunodominant excretory-secretory cysteine proteases of adult P. westermani involved in immune evasion [48], and another study identified critical roles for excretory-secretory cysteine proteases during tissue invasion by newly excysted metacercariae of P. westermani [49]. The rapid diversification and critical host-interaction functions of the proteases highlights their importance, both in terms of understanding Paragonimus biology and in terms of identifying targets for control.
Figure 9:
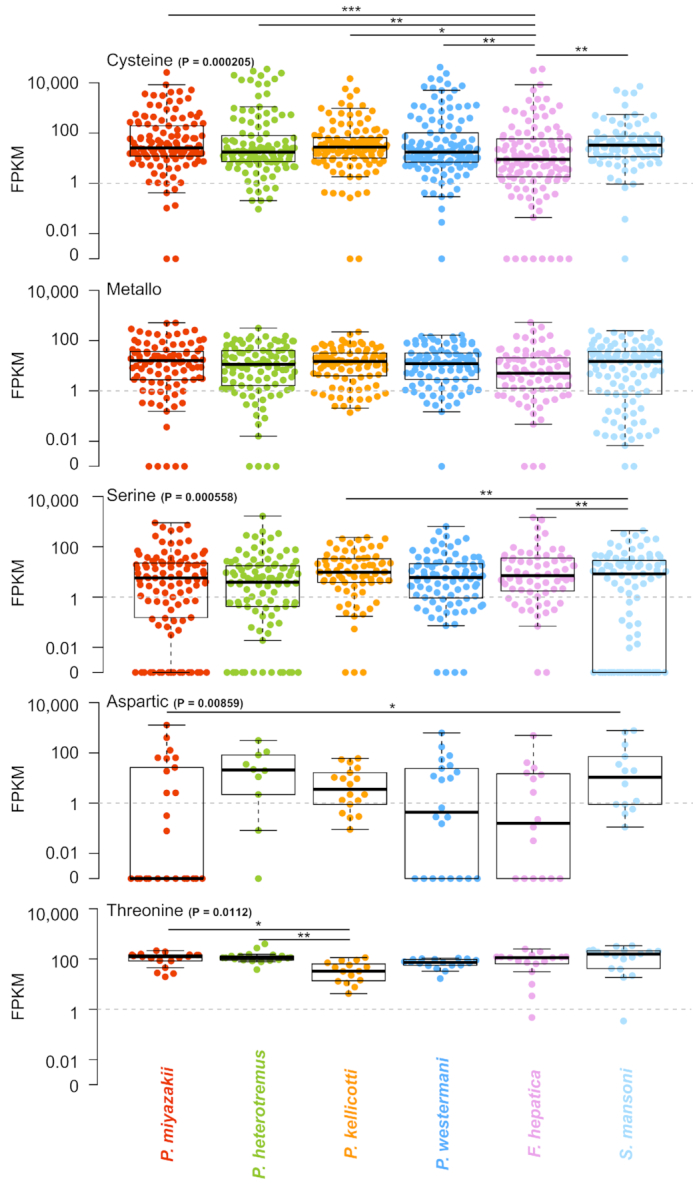
A comparison of adult-stage protease gene expression levels in the 4 Paragonimus species, F. hepatica, and S. mansoni. Boxes represent the interquartile ranges (IQRs) between the first and third quartiles, and the line inside the box represents the median value. Whiskers represent the lowest or highest values within values 1.5 times the IQR from the first or third quartiles. ANOVA P values are shown, and significant pairwise T-test comparisons are indicated with lines (* P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001).
Functional enrichment analysis among the lung, liver, and blood fluke conserved-and-exclusive OGs (Fig. 7C) indicated that each family of fluke has evolved a distinct set of aspartic peptidases, trematode eggshell synthesis genes, and saposin-like genes (which interact with lipids and are strongly immunogenic during fascioliasis [50]). The lung flukes, meanwhile, have uniquely expanded sets of serine proteases, as well as other gene families with functions including FAR1 DNA binding (a class of proteins that are important secreted host-interacting proteins in some parasitic nematodes [51]), fatty acid binding, and ferritin-like functions (intracellular proteins involved in iron metabolism, localized in vitelline follicles and eggs [52]).
Treatments, vaccine targets, and diagnostics
The World Health Organization currently recommends the use of praziquantel or, as a backup, triclabendazole for the treatment of paragonimiasis; both are highly effective for curing infections [53]. However, there are concerns about the development of resistance to these drugs; triclabendazole resistance of P. westermani was reported in a human case from Korea [54]. Furthermore, there is widespread resistance to triclabendazole in liver flukes in cattle in Australia and South America [55], and praziquantel resistance is anticipated in the future owing to its widespread use as a single treatment for schistosomiasis, a worrisome situation that has encouraged the search for novel drugs [56]. The comparative analysis presented here identifies valuable putative protein targets for drug development, including Paragonimus-specific proteins and trematode-conserved proteins that do not share orthology to human proteins. The protein annotation data available in Supplementary Table S1 also will enable prioritization including biological functional annotations [57, 58], protein weight and pi predictions [59], predictions of signal peptides and transmembrane domains [60] and cellular compartment localization [57], and sequence similarity matches to targets in the ChEMBL database [61]. This information can provide a starting point for future bioinformatic prioritization and drug testing.
Vaccination to prevent future infections would offer an attractive alternative to treatment, but development of vaccine protection against trematode infection has so far been unsuccessful and is unlikely to be practical for paragonimiasis in the near future [62]. However, the complete genome sequences and comparative analysis of the gene sets presented here provide valuable resources for future vaccine target development.
Pulmonary paragonimiasis is frequently mistaken for tuberculosis or pneumonia, and often patients do not shed eggs, which leads to false-positive diagnoses of other conditions such as malaria or pneumonia [4, 63, 64]. This highlights a pressing need for accurate, rapid, and affordable diagnostic approaches for paragonimiasis, a topic that has been the focus of numerous reports. We performed BLAST sequence similarity searches of previously identified Paragonimus diagnostic antigen targets among the 4 species (Fig. 10). These included (i) P. westermani and Paragonimus pseudoheterotremus cysteine proteases identified in 2 previous studies [65, 66] (matching to the same protein targets from both studies in P. heterotremus and P. kellicotti), 1 of which had high adult-stage expression levels in all 4 species [65]; (ii) 3 different tyrosine kinases (1 of which was identified in 2 different studies, in Clonorchis sinensis and in P. westermani [67, 68]), all of which had relatively low gene expression levels in adult stages; (iii) a previously unannotated P. heterotremus ELISA antigen [69] with low expression across life cycle stages, which we now annotate as a saposin protein (which we found to rapidly evolve among flukes [Fig. 7C] and which is strongly immunogenic in fascioliasis [50]); and (iv) eggshell proteins of P. westermani [70], for which we now provide full-length sequences. We observed that this gene was conserved across and specific to the lung flukes, with lower gene expression in the young adult stage (P. heterotremus) but higher expression in the adult stages of all species; (v) among serodiagnostic P. kellicotti antigens based on a transcriptome assembly and proteomic evidence [16], we identified the top 10 of the 25 prioritized transcripts that best matched between the transcript sequence and the newly annotated draft genome of P. kellicotti. Thereafter, the full-length gene sequence in P. kellicotti was used to query the other species. Several of these were highly expressed in the adult stage of all 4 species, including 1 that is fluke specific (PKEL_0 5597). However, not all of these had high sequence conservation across all species, with 2 only having weak hits in P. heterotremus (PKEL_00171 and PKEL_0 1872).
Figure 10:

Gene matches, expression level, and orthology for previously identified Paragonimus antigens. Top gene matches in each species (Diamond blastp) are shown, and the percent identity and percentage of the query sequence covered with the match are shown. Gene expression data correspond to the matched gene for each species, and orthology data indicate the conservation of the matched proteins according to the Orthologous Group analysis (dark grey = ortholog present in ≥1 species in group). *Query sequence was an amino acid sequence instead of a nucleotide sequence. **Of the top 25 P. kellicotti immunodominant antigen transcripts identified by McNulty and coworkers [17], the 10 best matches are presented (in terms of percent identity between the assembled transcript and the annotated gene). For the other 3 species, the BLAST searches were performed against the orthologous gene in P. kellicotti, not the original transcript sequence.
As a result of this newly developed genomic resource for the lung flukes, previously identified diagnostic targets were identified with full gene sequences across all 4 species. The complete gene sequences, conservation information, and transcriptomic gene expression data for these target proteins can allow for optimization of the targets for diagnostic testing that is effective on species spanning the genus (Fig. 10). This is noteworthy given the absence of a standardized, commercially available test for serodiagnosis for human paragonimiasis.
Conclusion
To substantially improve our understanding of the lung flukes at the molecular level, we sequenced, assembled, annotated, and compared draft genomes of 4 species of Paragonimus, 3 from Asia (P. miyazakii, P. westermani from Japan, P. heterotremus) and 1 from North America (P. kellicotti), thereby providing novel and valuable genomic resources across these important parasites for the first time. We have used these new resources to compare and analyze phylogenies, to identify gene sets and biological functions associated with parasitism in lung flukes, and to contribute a key resource for future investigation into host-parasite interactions for these poorly understood agents of neglected tropical disease. Our identification of previously prioritized Paragonimus diagnostic markers in each of the 4 lung fluke species revealed that the same protein targets were identified in multiple studies, and hence the availability of full gene sequences now should facilitate diagnostic assays aiming for reactivity across all species of lung fluke. Overall, the novel genomic and transcriptomic resources developed here will be invaluable for research on paragonimiasis, guiding experimental design and generation of novel hypotheses.
Methods
Parasite specimens
Samples of DNA and RNA of Paragonimus westermani were sourced in Japan. Paragonimus heterotremus (LC strain, Vietnam) were recovered from a cat experimentally infected with metacercariae from Lai Chau province, northern Vietnam (70% ethanol preserved; whole worm). Paragonimus miyazakii metacercariae were recovered from freshwater crabs (Geothelphusa dehaani), collected in Shizuoka Prefecture, central Japan [15], and were raised to adulthood in rats. DNA and RNA samples were prepared for each of the (pre-)adult flukes recovered from the lungs and from the pleural and peritoneal cavities of experimentally infected rats. Paragonimus kellicotti adult worms for genome sequencing were recovered from the lungs of Mongolian gerbils infected in the laboratory with metacercariae recovered from Missouri crayfish [71].
Genome sequencing, assembly, and annotation
DNA and RNA samples were collected from parasites of 4 distinct Paragonimus species: P. miyazakii (Japan), P. heterotremus (LC strain, Vietnam), P. kellicotti (Missouri, USA), and P. westermani (Japan). Illumina DNA sequencing produced fragments, 3- and 8-kb insert whole-genome shotgun libraries, and PacBio reads were generated for P. kellicotti. The sequences were generated on the Illumina platform and assembled using Allpaths_LG [72]. Scaffolding was improved using an in-house tool called Pygap (gap closure tool), the Pyramid assembler with Illumina paired reads to close gaps and extend contigs, and L_RNA_scaffolder [73], which uses transcript alignments to improve contiguity. For P. kellicotti, Nanocorr [74] was used to perform error correction on the PacBios data and PBJelly was used to fill gaps and improve the Illumina allpaths assembly using the PacBio reads [75]. The nuclear genomes were annotated using the MAKER pipeline v2.31.8 [76]. Repetitive elements were softmasked with RepeatMasker v4.0.6 using a species-specific repeat library created by RepeatModeler v1.0.8, RepBase repeat libraries [77], and a list of known transposable elements provided by MAKER [76]. RNA-seq reads were aligned to their respective genome assemblies and assembled using StringTie v1.2.4 [78] (P. miyazakii samples collected from stages in the liver, peritoneal cavity [2 replicates], lung [adult], and pleural cavity; P. heterotremus samples from adults and young adults [2 replicates]; P. westermani [16] and P. kellicotti [17] adult-stage transcriptomic reads were retrieved from published reports). The resulting alignments and transcript assemblies were used by BRAKER [79] and MAKER pipelines, respectively, as extrinsic evidence. In addition, messenger RNA (mRNA) and EST sequences for each species were retrieved from NCBI and were provided to MAKER as protein homology evidence along with protein sequences from UniRef100 [80] (Trematoda-specific, n = 205,161) and WormBase ParaSite WBPS7 [81]. Ab initio gene predictions from BRAKER v2 [79] and AUGUSTUS v3.2.2 (trained by BRAKER and run within MAKER) were refined using the transcript and protein evidence. Previously unpredicted exons and untranslated regions were added, and split models were merged. The best-supported gene models were chosen on the basis of annotation edit distance (AED) [82]. To reduce false-positive results, gene predictions without supporting evidence were excluded in the final annotation build, with the exception of those encoding Pfam domains, as detected by InterProScan v5.19 [57]. These Pfam encoding domains were rescued in order to improve the annotation accuracy overall by balancing sensitivity and specificity [76, 83]. Gene products were named using PANNZER2 [84] and sma3s v2 [85]. Table 1 provides details of database accessions for the genomes. The completeness of annotated gene sets was assessed using BUSCO v3.0, eukaryota_odb9 [19]. GO, KEGG, and protease annotations were performed using InterProScan v5.19 [57], GhostKOALA [58], and MEROPS [86], respectively. ExPASy was used to perform protein weight and pi predictions [59], SignalP was used to predict signal peptides and transmembrane domains [60], and gene product localization was predicted using the “cellular component” GO annotations provided by InterProScan [57].
Functional enrichment testing was performed using GOSTATS [87] for GO enrichment and negative binomial distribution tests for InterPro domain enrichment (minimum 3 annotated genes required for significant enrichment). Ribosomal RNAs and tRNAs were annotated using RNAmmer v1.2.1 [88] and tRNAscan-SE v1.23 [89], respectively. Genome characteristics and statistics including CDS, numbers and lengths of genes, exons and introns were defined using the longest complete mRNA (with start and stop codon) for each gene. Across the 4 species of Paragonimus, complete mRNAs were found for an average of 86.2% of all annotated genes.
Assembly of the mitochondrial genome of P. kellicotti was achieved using CANU [90] to align PacBio long reads, followed by error correction using Pilon [91].
MUMmer v4.0 [92] was used to estimate the level of genetic divergence between P. westermani samples from Japan and India. Nucmer was run first to generate genome alignments using draft assembly sequences. Dnadiff was then used to calculate the average sequence identity between the genomes considering only 1-to-1 alignments.
Transcriptome datasets and gene functional annotations
RNA-seq datasets were trimmed for adapters [93] and aligned [94] to their respective genome assemblies, and gene expression levels (FPKM) were quantified per gene per sample in each of the 4 species [95]. Interpro domains and GO terms [57], KEGG enzymes [58], and protease [86] annotations of the genes were used to identify putative functions of genes of interest and perform pathway enrichment [87]. All raw RNA-seq fastq files were uploaded to the NCBI SRA [18], and complete sample metadata and accession information are provided in Table 1. Supplementary Table S1 provides, for each of the species, complete gene lists and gene expression levels for each of the RNA-seq samples. Complete functional annotations for every gene are also provided for P. miyazakii in this table.
Repeat analysis
RepeatModeler v1.0.8 (with WU-BLAST as its search engine) was used to build, refine, and classify consensus models of putative interspersed repeats for each species. With the resulting repeat libraries, genomic sequences were screened using RepeatMasker v4.0.6 in “slow search” mode to generate a detailed annotation of the interspersed and simple repeats. Per-copy distances to consensus were calculated (Kimura 2-parameter model, excluding CpG sites) and were plotted as repeat landscapes where divergence distribution reflected the activity of TEs on a relative time scale per genome using the calcDivergenceFromAlign.pl and createRepeatLandscape.pl scripts included in the RepeatMasker package.
Gene family evolution
OGs of genes of 21 species were inferred with OrthoFinder v1.1.4 [96] using the longest isoform for each gene (Paragonimus genome source information in Table 1; worm gene sets were retrieved from WormBase ParaSite in June 2017 [81]; outgroup species gene sets were retrieved from Ensembl in June 2017 [97]). The CAFE method [25] was used to model gene gain and loss while accounting for the species' phylogenetic history based on an ultrametric species tree and the number of gene copies found in each species for each gene family. Birth-death (λ) parameters were estimated and the statistical significance of the observed family size differences among taxa were assessed. Results from OrthoFinder [96] were parsed to identify the OGs of interest based on conservation, including the lung fluke–conserved, liver fluke–conserved, and blood fluke–conserved OGs and gene sets per species. Supplementary Table S3 provides details of full OG counts per species and gene membership.
We used PosiGene [98] to search genome-wide for genes that evolved under positive selection based on the non-synonymous to synonymous substitution ratio. TMMOD [99] and Protter [100] were used for transmembrane helical topology prediction and visualization, respectively. We searched for genes that evolved under positive selection in the 4 Paragonimus spp. based on the non-synonymous to synonymous substitution rate ratio (dN/dS). We conducted the branch-site test of positive selection to identify adaptive gene variants that became fixed in each species [37].
Previously identified Paragonimus diagnostic antigen search
Nucleotide sequences (or, if unavailable, amino acid sequences) were retrieved from each of the cited publications (Fig. 10). Diamond blastx (nucleotides; v0.9.9.110) or Diamond blastp (amino acids; v0.9.9.110) were used to identify the top hit gene in each Paragonimus genome annotation (default settings). The best BLAST E-value was used to identify the top match, followed by top bit score, length, and percent ID in the case of ties. For the top 25 P. kellicotti immunodominant antigen transcripts identified in McNulty et al. 2014 [17], matches were identified between the assembled transcript and the annotated gene. For the other 3 species, the BLAST searches are performed against the identified P. kellicotti gene and not the original transcript sequence.
RNA-seq–based gene expression profiling
After adapter trimming using Trimmomatic v0.36 [93], RNA-seq reads were aligned to their respective genome assemblies using the STAR aligner [94] (2-pass mode, basic). All raw RNA-seq fastq files were uploaded to the NCBI SRA [18], and complete sample metadata and accession information are provided in Table 1. Read fragments (read pairs or single reads) were quantified per gene per sample using featureCounts (version 1.5.1) [95]. FPKM (fragments per kilobase of gene length per million reads mapped) normalization was also performed. Pearson correlation–based RNA-seq sample clustering was performed in R (using the hclust package, complete linkage).
Statistics
ANOVA analysis followed by Tukey HSD post hoc testing was performed to compare genome statistics and protease expression between species (Figs 2 and 9). Because comparisons for the genome statistics by t tests involved large numbers of values, which can falsely indicate positive statistical significance, a random selection of 100 values from each species was used (excluding the top and bottom 1% of data to avoid outliers). For Figure 2, Letter labels above the species indicate statistical groups; i.e., if 2 species share the same letter then they were not statistically significantly different. For Figure 3, individual pairwise significance values are indiciated since there were fewer differences between species. from each other.
Availability of Supporting Data and Materials
Genomic raw reads, genome assemblies, genome annotations, and raw transcriptomic (RNA-seq) fastq files were uploaded and are available for download from the NCBI SRA [18], with all accession numbers and relevant metadata provided in Table 1. Supplementary Table S1 provides, for each of the species, complete gene lists and gene expression levels for each of the RNA-seq samples. Other data further supporting this work are openly available in the GigaScience repository, GigaDB [101].
Additional Files
Supplementary Table S1: Gene expression and orthologous group data for each gene, for the 4 Paragonimus species: (A) P. miyazakii, (B) P. heterotremus, (C) P. kellicotti, (D) P. westermani (provided as a separate MS Excel database).
Supplementary Table S2: Genome-wide selection scan results for all Paragonimus species (provided as a separate MS Excel database).
Supplementary Table S3: Complete Orthologous Group (OG) counts per species, gene membership, and average Paragonimus gene expression levels per RNA-seq sample (provided as a separate MS Excel database).
Supplementary Text S1. Commands and parameters for analyses (provided as a separate MS Word file).
Rodrigo Baptista, Ph.D. -- 1/19/2020 Reviewed
Rodrigo Baptista, Ph.D. -- 4/13/2020 Reviewed
James Wasmuth -- 2/3/2020 Reviewed
James Wasmuth -- 4/21/2020 Reviewed
Abbreviations
ANOVA: analysis of variance; ATP: adenosine triphosphate; BLAST: Basic Local Alignment Search Tool; bp: base pairs; BUSCO: Benchmarking Universal Single-Copy Orthologs; CDS: coding sequence; ELISA: enzyme-linked immunosorbent assay; EST: expressed sequence tag; FPKM: fragments per kilobase of gene length per million reads mapped; GO: gene ontology; HSP: heat shock protein; kb: kilobase pairs; KEGG: Kyoto Encyclopedia of Genes and Genomes; LINE: long interspersed nuclear element; LTR: long terminal repeat; Mb: megabase pairs; mRNA: messenger RNA; NCBI: National Center for Biotechnology Information; NIH: National Institutes of Health; OG: Orthologous Group; OPF: orthologous protein family; PacBio: Pacific Biosciences; PDR: People's Democratic Republic; RNA-seq: RNA sequencing; SRA: Sequence Read Archive; TE: transposable element; tRNA: transfer RNA.
Competing Interests
The authors declare that they have no competing interests.
Funding
Sequencing of the genomes was supported by the “Sequencing the etiological agents of the Food-Borne Trematodiases” project (NIH—National Human Genome Research Institute award No. U54HG003079). Comparative genome analysis was funded by grants NIH—National Institute of Allergy and Infectious Diseases AI081803 and NIH—National Institute of General Medical Sciences GM097435 to M.M. Parasite material from Thailand was supported by Distinguished Research Professor Grant (W.M.), Thailand Research Fund (Grant No. DPG6280002).
Authors' Contributions
Conceptualization: M.M., P.J.B.
Formal analysis: B.A.R., Y.J.C., S.N.M., H.J., J.M.
Funding acquisition: P.J.B., M.M.
Methodology: P.J.B., P.U.F., D.B., M.M.
Resources: M.M., T.A., H.S., T.H.L., P.N.D., W.M., D.B., P.U.F.
Visualization: B.A.R., Y.J.C.
Writing—original draft: B.A.R., Y.J.C., M.M.
Writing—review and editing: D.B., P.J.B., P.U.F., M.M.
ACKNOWLEDGEMENTS
We gratefully acknowledge assistance provided by Xu Zhang and Kymberlie Pepin with genome assembly and annotation and by Rahul Tyagi for figure graphics. We thank Kurt Curtis for his help generating P. kellicotti parasite material.
Contributor Information
Bruce A Rosa, Department of Internal Medicine, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, MO 63110, USA.
Young-Jun Choi, Department of Internal Medicine, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, MO 63110, USA.
Samantha N McNulty, The McDonnell Genome Institute at Washington University, School of Medicine, 4444 Forest Park Ave, St. Louis, MO 63108, USA.
Hyeim Jung, Department of Internal Medicine, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, MO 63110, USA.
John Martin, Department of Internal Medicine, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, MO 63110, USA.
Takeshi Agatsuma, Department of Environmental Health Sciences, Kochi Medical School, Kohasu, Oko-cho 185-1, Nankoku, Kochi, 783-8505, Japan.
Hiromu Sugiyama, Laboratory of Helminthology, Department of Parasitology, National Institute of Infectious Diseases, 1-23-1 Toyama, Shinjuku-ku, Tokyo 162-8640, Japan.
Thanh Hoa Le, Department of Immunology, Institute of Biotechnology, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Cay Giay, Ha Noi 10307, Vietnam.
Pham Ngoc Doanh, Institute of Ecology and Biological Resources, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Cay Giay, Ha Noi 10307, Vietnam; Graduate University of Science and Technology, Vietnam Academy of Science and Technology, 18 Hoang Quoc Viet, Cay Giay, Ha Noi 10307, Vietnam.
Wanchai Maleewong, Research and Diagnostic Center for Emerging Infectious Diseases, Khon Kaen University, 123 Moo 16 Mittraphap Rd., Nai-Muang, Muang District, Khon Kaen 40002, Thailand; Department of Parasitology, Faculty of Medicine, Khon Kaen University, 123 Moo 16 Mittraphap Rd., Nai-Muang, Muang District, Khon Kaen 40002, Thailand.
David Blair, College of Marine and Environmental Sciences, James Cook University, 1 James Cook Drive, Townsville, Queensland 4811, Australia.
Paul J Brindley, Departments of Microbiology, Immunology and Tropical Medicine, and Research Center for Neglected Diseases of Poverty, and Pathology School of Medicine & Health Sciences, George Washington University, Ross Hall 2300 Eye Street, NW, Washington, DC 20037, USA.
Peter U Fischer, Department of Internal Medicine, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, MO 63110, USA.
Makedonka Mitreva, Department of Internal Medicine, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, MO 63110, USA; The McDonnell Genome Institute at Washington University, School of Medicine, 4444 Forest Park Ave, St. Louis, MO 63108, USA.
References
- 1. Furst T, Keiser J, Utzinger J. Global burden of human food-borne trematodiasis: a systematic review and meta-analysis. Lancet Infect Dis. 2012;12(3):210–21. [DOI] [PubMed] [Google Scholar]
- 2. Utzinger J, Becker SL, Knopp S, et al. . Neglected tropical diseases: diagnosis, clinical management, treatment and control. Swiss Med Wkly. 2012;142:w13727. [DOI] [PubMed] [Google Scholar]
- 3. Blair D. Paragonimiasis. Adv Exp Med Biol. 2014;766:115–52. [DOI] [PubMed] [Google Scholar]
- 4. Furst T, Sayasone S, Odermatt P, et al. . Manifestation, diagnosis, and management of foodborne trematodiasis. BMJ. 2012;344:e4093. [DOI] [PubMed] [Google Scholar]
- 5. Lv S, Zhang Y, Steinmann P, et al. . Helminth infections of the central nervous system occurring in Southeast Asia and the Far East. Adv Parasitol. 2010;72:351–408. [DOI] [PubMed] [Google Scholar]
- 6. Sripa B, Kaewkes S, Intapan PM, et al. . Food-borne trematodiases in Southeast Asia epidemiology, pathology, clinical manifestation and control. Adv Parasitol. 2010;72:305–50. [DOI] [PubMed] [Google Scholar]
- 7. Blair D, Xu ZB, Agatsuma T. Paragonimiasis and the genus Paragonimus. Adv Parasitol. 1999;42:113–222. [DOI] [PubMed] [Google Scholar]
- 8. Blair D, Davis GM, Wu B. Evolutionary relationships between trematodes and snails emphasizing schistosomes and paragonimids. Parasitology. 2001;123:S229–S43. [DOI] [PubMed] [Google Scholar]
- 9. Attwood SW, Upatham ES, Meng XH, et al. . The phylogeography of Asian schistosoma (Trematoda: Schistosomatidae). Parasitology. 2002;125(Pt 2):99–112. [DOI] [PubMed] [Google Scholar]
- 10. Doanh NP, Tu AL, Bui TD, et al. . Molecular and morphological variation of Paragonimus westermani in Vietnam with records of new second intermediate crab hosts and a new locality in a northern province. Parasitology. 2016;143(12):1639–46. [DOI] [PubMed] [Google Scholar]
- 11. Oey H, Zakrzewski M, Narain K, et al. . Whole-genome sequence of the oriental lung fluke Paragonimus westermani. Gigascience. 2019;8(1):giy146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blair D, Chang Z, Chen M, et al. . Paragonimus skrjabini Chen, 1959 (Digenea: Paragonimidae) and related species in eastern Asia: a combined molecular and morphological approach to identification and taxonomy. Syst Parasitol. 2005;60(1):1–21. [DOI] [PubMed] [Google Scholar]
- 13. Lane MA, Marcos LA, Onen NF, et al. . Paragonimus kellicotti flukes in Missouri, USA. Emerg Infect Dis. 2012;18(8):1263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fischer PU, Weil GJ. North American paragonimiasis: epidemiology and diagnostic strategies. Expert Rev Anti Infect Ther. 2015;13(6):779–86. [DOI] [PubMed] [Google Scholar]
- 15. Blair D, Nawa Y, Mitreva M, et al. . Gene diversity and genetic variation in lung flukes (genus Paragonimus). Trans R Soc Trop Med Hyg. 2016;110(1):6–12. [DOI] [PubMed] [Google Scholar]
- 16. Li BW, McNulty SN, Rosa BA, et al. . Conservation and diversification of the transcriptomes of adult Paragonimus westermani and P. skrjabini. Parasit Vectors. 2016;9:497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McNulty SN, Fischer PU, Townsend RR, et al. . Systems biology studies of adult Paragonimus lung flukes facilitate the identification of immunodominant parasite antigens. PLoS Negl Trop Dis. 2014;8(10):e3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Leinonen R, Sugawara H, Shumway M, International Nucleotide Sequence Database Collaboration. The Sequence Read Archive. Nucleic Acids Res. 2011;39(Database issue):D19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Waterhouse RM, Seppey M, Simao FA, et al. . BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 2018;35(3):543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Choi YJ, Fontenla S, Fischer PU, et al. . Adaptive radiation of the flukes of the family Fasciolidae inferred from genome-wide comparisons of key species. Mol Biol Evol. 2020;37(1):84–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stapley J, Santure AW, Dennis SR. Transposable elements as agents of rapid adaptation may explain the genetic paradox of invasive species. Mol Ecol. 2015;24(9):2241–52. [DOI] [PubMed] [Google Scholar]
- 22. Schrader L, Schmitz J. The impact of transposable elements in adaptive evolution. Mol Ecol. 2019;28(6):1537–49. [DOI] [PubMed] [Google Scholar]
- 23. Chenais B, Caruso A, Hiard S, et al. . The impact of transposable elements on eukaryotic genomes: from genome size increase to genetic adaptation to stressful environments. Gene. 2012;509(1):7–15. [DOI] [PubMed] [Google Scholar]
- 24. Prasad PK, Tandon V, Biswal DK, et al. . Phylogenetic reconstruction using secondary structures and sequence motifs of ITS2 rDNA of Paragonimus westermani (Kerbert, 1878) Braun, 1899 (Digenea: Paragonimidae) and related species. BMC Genomics. 2009;10(Suppl 3):S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Han MV, Thomas GW, Lugo-Martinez J, et al. . Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol. 2013;30(8):1987–97. [DOI] [PubMed] [Google Scholar]
- 26. Ahn CS, Na BK, Chung DL, et al. . Expression characteristics and specific antibody reactivity of diverse cathepsin F members of Paragonimus westermani. Parasitol Int. 2015;64(1):37–42. [DOI] [PubMed] [Google Scholar]
- 27. McNulty SN, Tort JF, Rinaldi G, et al. . Genomes of Fasciola hepatica from the Americas reveal colonization with Neorickettsia endobacteria related to the agents of Potomac horse and human Sennetsu fevers. PLoS Genet. 2017;13(1):e1006537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jones MK, Gobert GN, Zhang L, et al. . The cytoskeleton and motor proteins of human schistosomes and their roles in surface maintenance and host-parasite interactions. Bioessays. 2004;26(7):752–65. [DOI] [PubMed] [Google Scholar]
- 29. Mathieson W, Wilson RA. A comparative proteomic study of the undeveloped and developed Schistosoma mansoniegg and its contents: the miracidium, hatch fluid and secretions. Int J Parasitol. 2010;40(5):617–28. [DOI] [PubMed] [Google Scholar]
- 30. Sotillo J, Pearson M, Becker L, et al. . A quantitative proteomic analysis of the tegumental proteins from Schistosoma mansoni schistosomula reveals novel potential therapeutic targets. Int J Parasitol. 2015;45(8):505–16. [DOI] [PubMed] [Google Scholar]
- 31. Liu F, Cui SJ, Hu W, et al. . Excretory/secretory proteome of the adult developmental stage of human blood fluke, Schistosoma japonicum. Mol Cell Proteomics. 2009;8(6):1236–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kolinski T, Marek-Trzonkowska N, Trzonkowski P, et al. . Heat shock proteins (HSPs) in the homeostasis of regulatory T cells (Tregs). Cent Eur J Immunol. 2016;41(3):317–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pereira AS, Cavalcanti MG, Zingali RB, et al. . Isoforms of Hsp70-binding human LDL in adult Schistosoma mansoniworms. Parasitol Res. 2015;114(3):1145–52. [DOI] [PubMed] [Google Scholar]
- 34. He S, Yang L, Lv Z, et al. . Molecular and functional characterization of a mortalin-like protein from Schistosoma japonicum (SjMLP/hsp70) as a member of the HSP70 family. Parasitol Res. 2010;107(4):955–66. [DOI] [PubMed] [Google Scholar]
- 35. Reis EV, Pereira RV, Gomes M, et al. . Characterisation of major vault protein during the life cycle of the human parasite Schistosoma mansoni. Parasitol Int. 2014;63(1):120–6. [DOI] [PubMed] [Google Scholar]
- 36. Messerli SM, Kasinathan RS, Morgan W, et al. . Schistosoma mansoni P-glycoprotein levels increase in response to praziquantel exposure and correlate with reduced praziquantel susceptibility. Mol Biochem Parasitol. 2009;167(1):54–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol Biol Evol. 2007;24(8):1586–91. [DOI] [PubMed] [Google Scholar]
- 38. Huang S, Yuan S, Dong M, et al. . The phylogenetic analysis of tetraspanins projects the evolution of cell-cell interactions from unicellular to multicellular organisms. Genomics. 2005;86(6):674–84. [DOI] [PubMed] [Google Scholar]
- 39. Chaiyadet S, Krueajampa W, Hipkaeo W, et al. . Suppression of mRNAs encoding CD63 family tetraspanins from the carcinogenic liver fluke Opisthorchis viverrini results in distinct tegument phenotypes. Sci Rep. 2017;7(1):14342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Krautz-Peterson G, Debatis M, Tremblay JM, et al. . Schistosoma mansoniinfection of mice, rats and humans elicits a strong antibody response to a limited number of reduction-sensitive epitopes on five major tegumental membrane proteins. PLoS Negl Trop Dis. 2017;11(1):e0005306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tran MH, Pearson MS, Bethony JM, et al. . Tetraspanins on the surface of Schistosoma mansoniare protective antigens against schistosomiasis. Nat Med. 2006;12(7):835–40. [DOI] [PubMed] [Google Scholar]
- 42. Wu C, Cai P, Chang Q, et al. . Mapping the binding between the tetraspanin molecule (Sjc23) ofSchistosoma japonicum and human non-immune IgG. PLoS One. 2011;6(4):e19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sealey KL, Kirk RS, Walker AJ, et al. . Adaptive radiation within the vaccine target tetraspanin-23 across nine Schistosoma species from Africa. Int J Parasitol. 2013;43(1):95–103. [DOI] [PubMed] [Google Scholar]
- 44. Yang Y, Wen Y, Cai YN, et al. . Serine proteases of parasitic helminths. Korean J Parasitol. 2015;53(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Glanfield A, McManus DP, Anderson GJ, et al. . Pumping iron: a potential target for novel therapeutics against schistosomes. Trends Parasitol. 2007;23(12):583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Brindley PJ, Kalinna BH, Wong JY, et al. . Proteolysis of human hemoglobin by schistosome cathepsin D. Mol Biochem Parasitol. 2001;112(1):103–12. [DOI] [PubMed] [Google Scholar]
- 47. Williamson AL, Brindley PJ, Abbenante G, et al. . Cleavage of hemoglobin by hookworm cathepsin D aspartic proteases and its potential contribution to host specificity. FASEB J. 2002;16(11):1458–60. [DOI] [PubMed] [Google Scholar]
- 48. Lee EG, Na BK, Bae YA, et al. . Identification of immunodominant excretory-secretory cysteine proteases of adult Paragonimus westermani by proteome analysis. Proteomics. 2006;6(4):1290–300. [DOI] [PubMed] [Google Scholar]
- 49. Na BK, Kim SH, Lee EG, et al. . Critical roles for excretory-secretory cysteine proteases during tissue invasion of Paragonimus westermani newly excysted metacercariae. Cell Microbiol. 2006;8(6):1034–46. [DOI] [PubMed] [Google Scholar]
- 50. Caban-Hernandez K, Espino AM. Differential expression and localization of saposin-like protein 2 of Fasciola hepatica. Acta Trop. 2013;128(3):591–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Basavaraju SV, Zhan B, Kennedy MW, et al. . Ac-FAR-1, a 20 kDa fatty acid- and retinol-binding protein secreted by adult Ancylostoma caninumhookworms: gene transcription pattern, ligand binding properties and structural characterisation. Mol Biochem Parasitol. 2003;126(1):63–71. [DOI] [PubMed] [Google Scholar]
- 52. Jones MK, McManus DP, Sivadorai P, et al. . Tracking the fate of iron in early development of human blood flukes. Int J Biochem Cell Biol. 2007;39(9):1646–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. World Health Organization . Schistosomiasis: Strategy - Control and preventive chemotherapy, 2019. https://www.who.int/schistosomiasis/strategy/en/ .Accessed on August 25, 2019. [Google Scholar]
- 54. Kyung SY, Cho YK, Kim YJ, et al. . A paragonimiasis patient with allergic reaction to praziquantel and resistance to triclabendazole: successful treatment after desensitization to praziquantel. Korean J Parasitol. 2011;49(1):73–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kelley JM, Elliott TP, Beddoe T, et al. . Current threat of triclabendazole resistance in Fasciola hepatica. Trends Parasitol. 2016;32(6):458–69. [DOI] [PubMed] [Google Scholar]
- 56. Mader P, Rennar GA, Ventura AMP, et al. . Chemotherapy for fighting schistosomiasis: past, present and future. ChemMedChem. 2018;13(22):2374–89. [DOI] [PubMed] [Google Scholar]
- 57. Jones P, Binns D, Chang HY, et al. . InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30(9):1236–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol. 2016;428(4):726–31. [DOI] [PubMed] [Google Scholar]
- 59. Artimo P, Jonnalagedda M, Arnold K, et al. . ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012;40(Web Server issue):W597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Almagro Armenteros JJ, Tsirigos KD, Sonderby CK, et al. . SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol. 2019;37(4):420–3. [DOI] [PubMed] [Google Scholar]
- 61. Mendez D, Gaulton A, Bento AP, et al. . ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019;47(D1):D930–D40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stutzer C, Richards SA, Ferreira M, et al. . Metazoan parasite vaccines: present status and future prospects. Front Cell Infect Microbiol. 2018;8:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Radzikowska E, Chabowski M, Bestry I. Tuberculosis mimicry. Eur Respir J. 2006;27(3):652 . [DOI] [PubMed] [Google Scholar]
- 64. Eapen S, Espinal E, Firstenberg M. Delayed diagnosis of paragonimiasis in Southeast Asian immigrants: a need for global awareness. Int J Acad Med. 2018;4(2):173–7. [Google Scholar]
- 65. Yang SH, Park JO, Lee JH, et al. . Cloning and characterization of a new cysteine proteinase secreted by Paragonimus westermani adult worms. Am J Trop Med Hyg. 2004;71(1):87–92. [PubMed] [Google Scholar]
- 66. Yoonuan T, Nuamtanong S, Dekumyoy P, et al. . Molecular and immunological characterization of cathepsin L-like cysteine protease of Paragonimus pseudoheterotremus. Parasitol Res. 2016;115(12):4457–70. [DOI] [PubMed] [Google Scholar]
- 67. Kim SH, Bae YA. Lineage-specific expansion and loss of tyrosinase genes across platyhelminths and their induction profiles in the carcinogenic oriental liver fluke, Clonorchis sinensis. Parasitology. 2017;144(10):1316–27. [DOI] [PubMed] [Google Scholar]
- 68. Bae YA, Kim SH, Ahn CS, et al. . Molecular and biochemical characterization of Paragonimus westermani tyrosinase. Parasitology. 2015;142(6):807–15. [DOI] [PubMed] [Google Scholar]
- 69. Pothong K, Komalamisra C, Kalambaheti T, et al. . ELISA based on a recombinantParagonimus heterotremus protein for serodiagnosis of human paragonimiasis in Thailand. Parasit Vectors. 2018;11(1):322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bae YA, Kim SH, Cai GB, et al. . Differential expression of Paragonimus westermani eggshell proteins during the developmental stages. Int J Parasitol. 2007;37(3–4):295–305. [DOI] [PubMed] [Google Scholar]
- 71. Fischer PU, Curtis KC, Marcos LA, et al. . Molecular characterization of the North American lung fluke Paragonimus kellicotti in Missouri and its development in Mongolian gerbils. Am J Trop Med Hyg. 2011;84(6):1005–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gnerre S, Maccallum I, Przybylski D, et al. . High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc Natl Acad Sci U S A. 2011;108(4):1513–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Xue W, Li JT, Zhu YP, et al. . L_RNA_scaffolder: scaffolding genomes with transcripts. BMC Genomics. 2013;14:604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Goodwin S, Gurtowski J, Ethe-Sayers S, et al. . Oxford Nanopore sequencing, hybrid error correction, and de novo assembly of a eukaryotic genome. Genome Res. 2015;25(11):1750–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. English AC, Richards S, Han Y, et al. . Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PLoS One. 2012;7(11):e47768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Holt C, Yandell M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics. 2011;12:491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bao W, Kojima KK, Kohany O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 2015;6:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Pertea M, Pertea GM, Antonescu CM, et al. . StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hoff KJ, Lange S, Lomsadze A, et al. . BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics. 2016;32(5):767–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. The UniProt Consortium . UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2017;45(D1):D158–D69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Howe KL, Bolt BJ, Shafie M, et al. . WormBase ParaSite - a comprehensive resource for helminth genomics. Mol Biochem Parasitol. 2017;215:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Eilbeck K, Moore B, Holt C, et al. . Quantitative measures for the management and comparison of annotated genomes. BMC Bioinformatics. 2009;10:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Campbell MS, Law M, Holt C, et al. . MAKER-P: a tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiol. 2014;164(2):513–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Koskinen P, Törönen P, Nokso-Koivisto J, et al. . PANNZER: high-throughput functional annotation of uncharacterized proteins in an error-prone environment. Bioinformatics. 2015;31(10):1544–52. [DOI] [PubMed] [Google Scholar]
- 85. Casimiro-Soriguer CS, Munoz-Merida A, Perez-Pulido AJ. Sma3s: a universal tool for easy functional annotation of proteomes and transcriptomes. Proteomics. 2017;17(12), doi: 10.1002/pmic.201700071. [DOI] [PubMed] [Google Scholar]
- 86. Rawlings ND, Barrett AJ, Finn R. Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2016;44(D1):D343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23(2):257–8. [DOI] [PubMed] [Google Scholar]
- 88. Lagesen K, Hallin P, Rodland EA, et al. . RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Koren S, Walenz BP, Berlin K, et al. . Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27(5):722–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Walker BJ, Abeel T, Shea T, et al. . Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Marcais G, Delcher AL, Phillippy AM, et al. . MUMmer4: a fast and versatile genome alignment system. PLoS Comput Biol. 2018;14(1):e1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dobin A, Davis CA, Schlesinger F, et al. . STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30. [DOI] [PubMed] [Google Scholar]
- 96. Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Zerbino DR, Achuthan P, Akanni W, et al. . Ensembl 2018. Nucleic Acids Res. 2018;46(D1):D754–D61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Sahm A, Bens M, Platzer M, et al. . PosiGene: automated and easy-to-use pipeline for genome-wide detection of positively selected genes. Nucleic Acids Res. 2017;45(11):e100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kahsay RY, Gao G, Liao L. An improved hidden Markov model for transmembrane protein detection and topology prediction and its applications to complete genomes. Bioinformatics. 2005;21(9):1853–8. [DOI] [PubMed] [Google Scholar]
- 100. Omasits U, Ahrens CH, Muller S, et al. . Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics. 2014;30(6):884–6. [DOI] [PubMed] [Google Scholar]
- 101. Rosa BA, Choi YJ, McNulty SN, et al.. Supporting data for “Comparative genomics and transcriptomics of 4 Paragonimus species provide insights into lung fluke parasitism and pathogenesis.”. GigaScience Database. 2020. 10.5524/100757. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Rosa BA, Choi YJ, McNulty SN, et al.. Supporting data for “Comparative genomics and transcriptomics of 4 Paragonimus species provide insights into lung fluke parasitism and pathogenesis.”. GigaScience Database. 2020. 10.5524/100757. [DOI] [PMC free article] [PubMed]
Supplementary Materials
Rodrigo Baptista, Ph.D. -- 1/19/2020 Reviewed
Rodrigo Baptista, Ph.D. -- 4/13/2020 Reviewed
James Wasmuth -- 2/3/2020 Reviewed
James Wasmuth -- 4/21/2020 Reviewed