

Abstract
Chemical exchange saturation transfer (CEST) NMR experiments have emerged as a powerful tool for characterizing dynamics in proteins. We show here that the CEST approach can be extended to systems with symmetrical exchange, where the NMR signals of all exchanging species are severely broadened. To achieve this, multiquantum CEST (MQ-CEST) is introduced, where the CEST pulse is applied to a longitudinal multispin order density element and the CEST profiles are encoded onto nonbroadened nuclei. The MQ-CEST approach is demonstrated on the restricted rotation of guanidinium groups in arginine residues within proteins. These groups and their dynamics are essential for many enzymes and for noncovalent interactions through the formation of hydrogen bonds, salt-bridges, and π-stacking interactions, and their rate of rotation is highly indicative of the extent of interactions formed. The MQ-CEST method is successfully applied to guanidinium groups in the 19 kDa L99A mutant of T4 lysozyme.
A key strength of NMR spectroscopy is its ability to quantify the dynamics of molecules with atomic level resolution. Within biomolecules, conformational exchanges often occur on milli- to microsecond time scales, and these exchanges can be critical for function.1,2 A number of NMR-based approaches for characterizing exchange on these time scales now exist and provide important insights into conformations that are transiently populated, invisible to other high-resolution methods, and also broadened beyond detection in traditional NMR experiments.3−6 Although chemical exchange saturation transfer (CEST) methods have traditionally been used within the MRI field,7−9 CEST approaches have recently emerged as powerful tools also for studying biomolecular dynamics on a time scale from 20 to 200 s–1.10,11 In these experiments, first developed in the 1960s,12 saturation of magnetization by a weak pulse is transferred by exchange events within a network of exchanging conformers, and in particular, magnetization is transferred from invisible species to visible species in order to report on chemical shifts and rates of exchange.
However, for symmetrical exchange, where all the exchanging species are broadened in NMR spectra, a quantification of the exchanging system becomes challenging. This scenario is, for example, encountered for the rotational exchange about the Cζ–Nε bond in arginine side chains in proteins, Figure 1A. The importance of the arginine side chain for a range of protein functions, such as protein folding, catalysis, and noncovalent interactions, is well-established.13−15 It is the arginine guanidinium group, with its delocalized π system, that confers the functionality by allowing for a large range of interactions.16−18 Recently, it was shown that 13C-detection NMR provides an excellent tool to probe arginine side chains in proteins,19−22 and it was also shown that in favorable cases the rate of rotational exchange can be determined to provide a measure for the interactions formed by arginine guanidinium groups within proteins.23 Below, we present a multiquantum CEST (MQ-CEST) NMR experiment that is ideally suited to characterizing dynamics in symmetrically exchanging groups, and we apply this methodology to quantify the rotational dynamics of guanidinium groups in the side chains of arginine residues within proteins.
Figure 1.

(A) Illustration of an arginine side chain and the restricted rotation about the Cζ–Nε bond that causes the symmetrical exchange of the two 15Nη nuclei. (B) Example of a 15Nη–15Nη MQ-CEST profile, recorded for the R96 arginine in the L99A mutant of T4 lysozyme (T4L99A). The position of the two dips shows the 15Nη chemical shifts, and a least-squares fit to the profile provides the rate of exchange. (C) Example of a 13Cζ–15N correlation map for T4L99A that encodes the CEST profiles. (D) Pulse sequence derived for the MQ-CEST experiment applied to arginine side chains. The carrier frequencies are set to 7.15 ppm (1H), 157 ppm (13C), and 84 ppm (15N). Narrow and wide bars denote 90° and 180° pulses, respectively, with all pulses applied along x unless otherwise indicated. The bell shapes denote frequency-selective pulses, with the letter above showing selectivity and letter inside indicating the type: S refers to a Seduce shape,24 E to an EBURP shape, and R to a REBURP shape.25 Striped boxes indicate decoupling with the text indicating the scheme used. CW: continuous wave applied at CEST offset frequency. The delays are τa = 1/(4JHN)= 2.7 ms and τb = 1/(4JNC) = 12.5 ms. The phase cycle used is ϕ1 = x, −x; ϕ2 = 2(x), 2(−x); and ϕrec = x, −x, −x, x. Gradients are used to remove artifacts (full details in the caption to Figure S4).
In the multiquantum CEST (MQ-CEST) experiment, the CEST pulse is applied to a longitudinal multispin order spin-density element in order to quantify symmetrical exchange. The restricted rotation about the Cζ–Nε bond in arginine side chains, Figure 1A, corresponds to a symmetrical exchange of the two 15Nη nuclei. The MQ-CEST approach applied to a density operator that spans both of the 15Nη species is able to capture the rate of restricted rotation as well as the chemical shifts of the sometimes severely broadened 15Nη signals.
For the MQ-CEST approach applied to arginine guanidinium side chains, two equally sized dips are typically observed corresponding to the CEST pulse being resonant with one of the two 15Nη chemical shifts, Figure 1B and Figure S1. When the exchanging nuclei are in the so-called slow-exchange regime (kex ≪ |ω(15Nη1) – ω(15Nη2)| = |Δω|),26,27 two well-separated dips are visible in the MQ-CEST profile and the individual chemical shift of the two 15Nη nuclei can be directly identified from the center of the dips, Figure 1B and Figure S1. When the nuclei are in the intermediate chemical exchange regime (kex ≈ |Δω|), either because of fast rotation or because of a smaller chemical shift difference, the two dips coalesce into a single dip centered at the average of the two 15Nη chemical shifts. It is particularly near the intermediate chemical exchange regime that single-quantum 15Nη signals become severely broadened in NMR spectra.22 For the MQ-CEST approach, the CEST intensities are encoded onto 13Cζ–15Nε correlation maps that are unaffected by the rotational exchange. Therefore, as long as CEST offset frequencies are chosen to fully cover the CEST dip(s), the exchanging system can be characterized using the MQ-CEST approach, even for scenarios of intermediate exchange. When the rotation is in the fast-exchange regime (kex ≫ |Δω|), the width of the single CEST dip narrows.
Simulations were used to establish the range of parameters, where accurate exchange parameters, kex and Δω, can be derived using the MQ-CEST approach. In common with other CEST approaches, the MQ-CEST approach provides the most accurate parameters, when there is a substantial chemical shift difference between the two exchanging 15Nη species, which brings the system toward the slow-exchange regime. The simulations show that, for Δω ≥ 1 ppm, the MQ-CEST approach provides accurate exchange rates over the large range of kex from 5 to 1500 s–1, which covers almost the full range of possible arginine guanidinium rotation rates at 293 K. Generally, the larger the chemical shift differences are, Δω, the larger exchange rates kex are accessible, Figure S2, whereas accurate Δω values can be obtained for the full range of possible kex values from 5 to 1500 s–1, Figure S3, as long as Δω ≳ 0.5 ppm. For arginine side chains involved in very strong interactions and thus experiencing very slow rotational rates (<5 s–1), it is possible to set an upper bound for kex and accurately determine the two 15Nη chemical shifts from MQ-CEST experiments. In such cases of very slow exchange, the kex can be determined using the previous longitudinal exchange method,23 provided that the 13Cζ resonance is isolated. It is interesting to note that the range of chemical exchange rates, kex, accessible with the MQ-CEST approach is substantially larger than what is accessible from typical CEST experiments7,8 (20–200 s–1). This larger range of kex accessible with the MQ-CEST approach is mainly due to the symmetrical exchange with equal populations of the two exchanging sites. It is also important to note that with least-squares fitting (see Supporting Information) and detailed analysis of the MQ-CEST profiles it is possible to accurately extract the chemical shifts of the two 15Nη nuclei well beyond the range where these can be simply obtained from inspection of the MQ-CEST profiles.
Finally, the simulations show that it is highly desirable to collect data with different B1 CEST saturation field strengths and/or at different static magnetic field strengths, B0, Figures S2 and S3. It is interesting to note that MQ-CEST profiles with multiple B1 field strengths provide essentially as accurate kex rates and Δω as MQ-CEST profiles at multiple B0 fields. Recording MQ-CEST profiles at multiple static B0 fields, particularly at higher field strengths, gives access to higher rotational rates and smaller chemical shift differences, since the exchange is moved toward the slow-chemical exchange regime. A disadvantage of ultrahigh magnetic fields (B0 ≳ 19 T), however, is an increased 13Cζ transverse relaxation due to chemical shift anisotropy (CSA), which can reduce signal-to-noise in the resulting spectra.
The pulse sequence derived for obtaining MQ-CEST profiles to characterize the symmetric exchange of arginine guanidinium groups in proteins is shown in Figure 1D. Briefly, equilibrium longitudinal magnetization residing on 1Hε, Hzε, is initially excited and transferred via an INEPT step between a and b to a 1Hε–15Nε longitudinal two-spin order density element, 2HzNzε. Using the magnetization on the 1Hε proton as the source brings two main advantages compared to methods where 13C magnetization is used as the source. First, the higher gyromagnetic ratio of 1H provides additional signal-to-noise, even though this is partly mitigated by the longer sequence. Second, 1H nuclei have substantially faster longitudinal R1 relaxation rates compared to 13C, which means that more scans can be acquired within a given time unit. Between b and c, the one-bond scalar coupling between 1Hε and 15Nε and between 15Nε and 13Cζ is evolved to generate the 13Cζ–15Nε longitudinal two-spin order element 2CzNzε, while concomitantly encoding the 15Nε chemical shift. The two selective 13C pulses between b and c ensure that 13Cδ–15Nε and 13Cζ–15Nε scalar couplings are refocused and evolved, respectively. Between c and d, a further INEPT is used to evolve 13Cζ–15Nε and 13Cζ–15Nη scalar couplings, yielding a density element proportional to 4CzNzηNz at point d. The MQ-CEST period between points d and e is carried out with the 15N carrier frequency being varied, providing the CEST intensities, I. A reference spectrum is also recorded without the CEST element (TCEST = 0 s), but including the gray block in Figure 1D. The reference spectrum provides I0 and the final MQ-CEST profiles are calculated as I/I0. The effects of scalar couplings between 15Nη and 1H are minimized through the application of high-power composite decoupling during the CEST period as described previously.28 As the 15N CEST pulse is applied to a 4CzζNzNzη density element, no decoupling is applied to 13C as this would deteriorate the signal. Instead, the effects of the 13Cζ–15Nη scalar couplings (∼20 Hz) are explicitly included in the analysis of the CEST profiles as described previously.21,29 It should be noted that several density elements are present during the 15N CEST pulse, including Zeeman order (4CzNzηNz), zero-quantum (e.g., 4CzζN+N–η), single-quantum (e.g., 4CzNzηN–), and double-quantum (e.g., 4CzζN+N+η) coherences. Whereas the double-quantum coherences are insensitive to the exchange,22 the zero-quantum coherence will report on the exchange process. Finally, after point e, the density element of interest is transferred to transverse in-phase 13Cζ magnetization for detection.
Several variations to the pulse sequence in Figure 1D have been developed (Figure S4). Of particular note is the 1H detected version (Figure S4B) in which magnetization, via additional INEPT steps, is transferred back to the 1Hε proton for detection. If relaxation is ignored, detecting 1Hε gives an 8-fold increase in signal-to-noise compared to 13C detection; however, in practice the additional delays required as well as exchange of 1Hε with the bulk solvent mean that this approach is only advantageous when the site in question has a local correlation time of less than approximately 10 ns (Figure S5). The guanidinium groups of greatest interest are often those that form interactions, making them more rigid, and so for the applications shown below on the 19 kDa T4L99A, the 13C detect sequence, Figure 1D, provides the best signal-to-noise.
In order to experimentally validate the MQ-CEST approach, experiments were carried out on a sample of free [13C6,15N4]-l-arginine at a high concentration (50 mM) and dissolved in a 50/50% vol mixture of H2O and MeOH under acidic conditions. With this sample, experiments can be carried out at temperatures below 0 °C, where the symmetrical exchange rate, kex, for free arginine is slow enough to be quantified by classical longitudinal exchange methods, such as zz-EXSY.30 This system therefore forms an ideal basis for validating and benchmarking the performance of the MQ-CEST approach.
The MQ-CEST profiles for free arginine measured at four temperatures between −15 and 2.4 °C are shown in Figure 2A. The chemical shifts of the 15Nη nuclei can be easily identified from the position of the dips at low temperatures, and the exchange rate can be obtained from a least-squares analysis at each temperature (see Supporting Information). The correlation between the obtained exchange rates, kex, from MQ-CEST and from longitudinal exchange is excellent, Figure 2C, validating the MQ-CEST approach. It is important to note that the longitudinal exchange approach is only applicable when the 15Nη nuclei give rise to diagonal and cross-peaks in single-quantum NMR spectra and these peaks can be accurately quantified. This is not the requirement for the MQ-CEST approach, since the CEST intensities are encoded onto the 13Cζ–15Nε cross-peaks.
Figure 2.
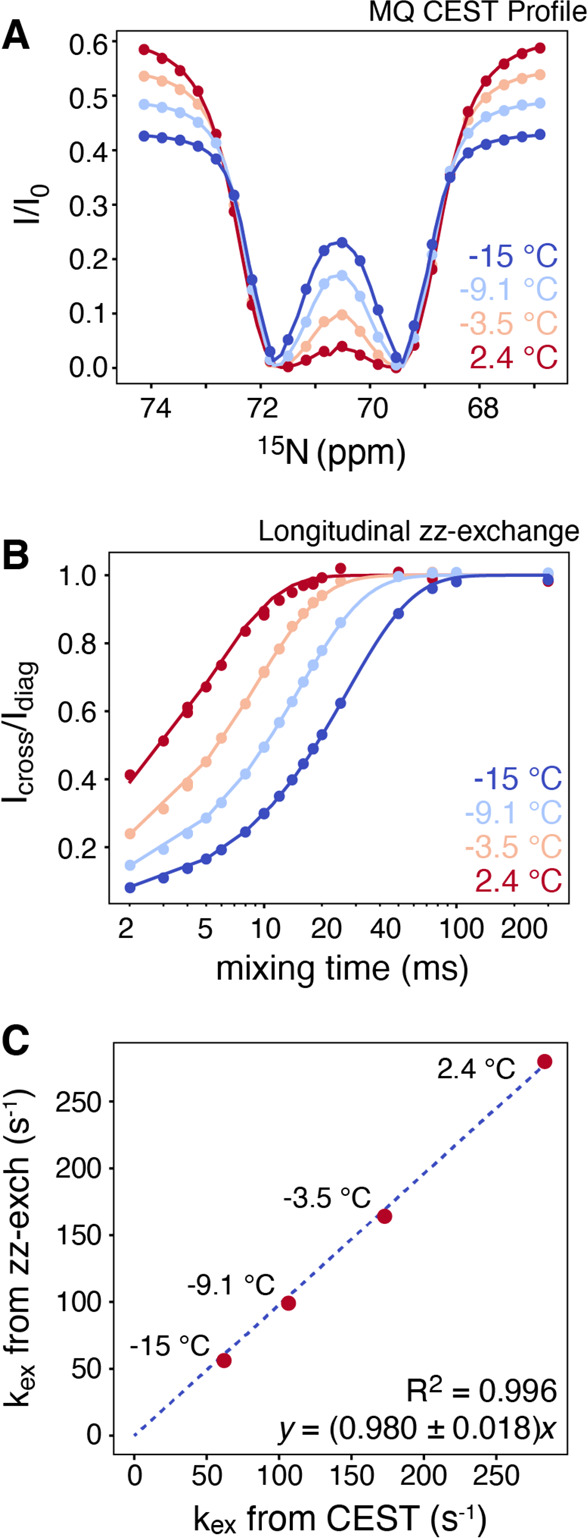
(A) MQ-CEST profiles recorded at four temperatures (−15 to 2.4 °C) on a 50 mM sample of [13C6, 15N4]-l-arginine dissolved in 50/50% vol H2O/MeOH at a static magnetic field of 14.1 T and using a 10 Hz B1 CEST saturation pulse for TCEST = 250 ms. (B) Corresponding longitudinal exchange, zz-EXSY, data recorded on the same sample. (C) Correlation plot of symmetrical exchange rates obtained from MQ-CEST (abscissa) and from longitudinal exchange (ordinate). The excellent linear correlation between the rates shows that accurate rotational exchange rates, kex, can be derived from the MQ-CEST experiment.
Having demonstrated the validity of the MQ-CEST approach, both theoretically and experimentally, for extracting the rate for symmetrical exchange of the arginine guanidinium group, we turned our attention to arginine side chains within the 19 kDa L99A mutant of T4 lysozyme (T4L99A). T4 lysozyme is a challenging test case since a large range of exchange rates spanning more than 3 orders of magnitude have been observed.23 Previously, D-evolution and longitudinal exchange were used to characterize the rotational dynamics of arginine guanidinium groups in T4L99A; however, these measurements rely on single-quantum 13Cζ–15Nη spectra, and only five of the 13 arginine residues in T4L99A could previously be characterized. On the contrary, the MQ-CEST approach relies on 13Cζ–15Nε (or 1Hε–15Nε) spectra, where well-separated cross-peaks are observed, e.g., Figure 1C. Thus, the MQ-CEST approach shows substantial improvements over the existing D-evolution method, since nearly every arginine residue can be resolved, resulting in an exchange rate for 11 out of the 13 arginine residues in T4L99A at 293 K. Four of these MQ-CEST profiles are shown in Figure 3, while all data is provided in Figure S6 and Table S2. For the arginine residues, where kex could previously be obtained from the D-evolution approach, there is an excellent agreement with the rates derived from the MQ-CEST profiles.23
Figure 3.
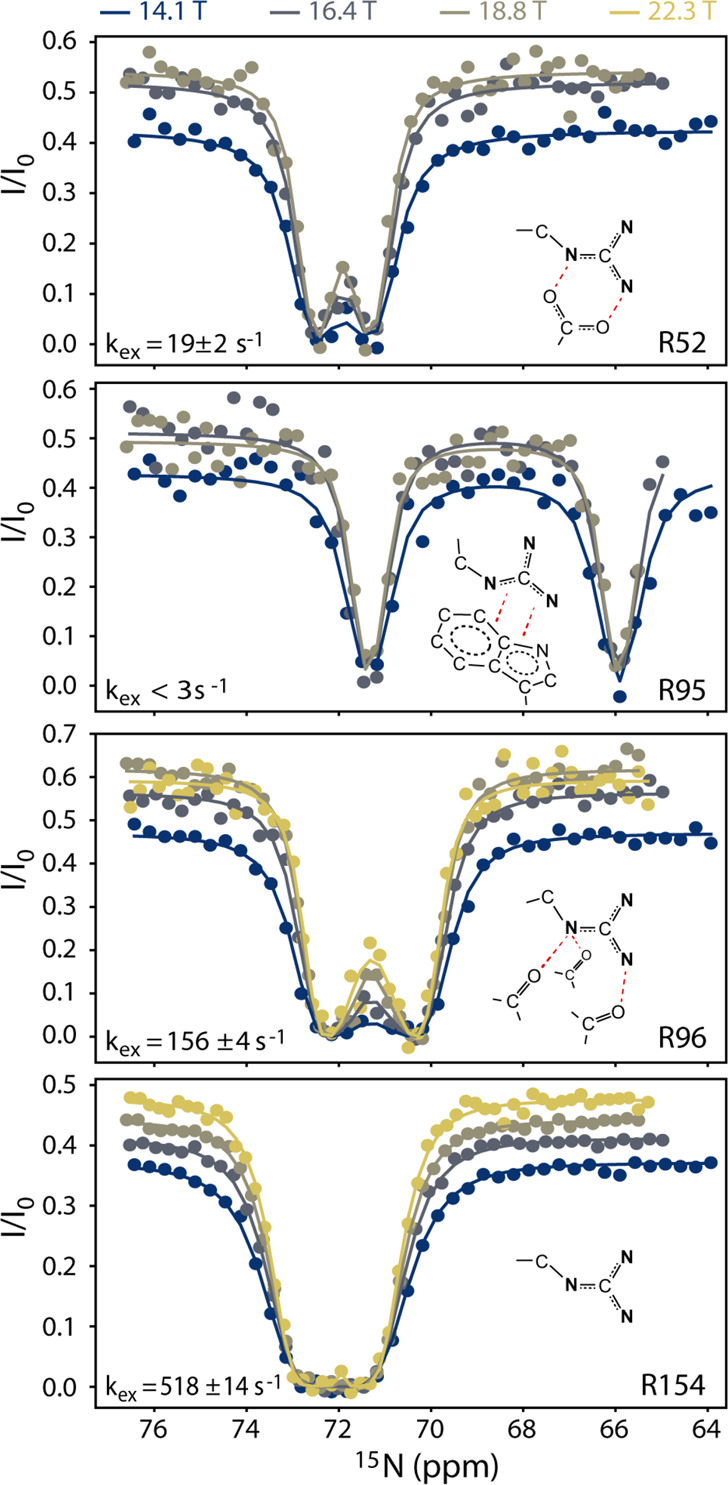
MQ-CEST profiles for arginine residues R52, R95, R96, and R154 of T4L99A. All data shown is collected at 293 K at static magnetic fields, B0, between 14.1 and 22.3 T. For a given arginine guanidinium group, all data is analyzed simultaneously to give the rate of symmetrical exchange, kex, and the chemical shift of the two 15Nη nuclei.
The obtained kex rates confirm that the rate of rotation is a very good indicator of interactions formed by a particular arginine side chain within the protein environment. For example, from the crystal structure of T4L99A,31 various interactions are observed for R52, R95, and R96, Figure 3, and these residues show a large range of rates, albeit all substantially slower than free arginine. The guanidinium group that shows the slowest rotational rate is in R95 that forms cation−π and π–π interactions with the large aromatic system of tryptophan W126.
In order to further assess the robustness of the MQ-CEST analysis for the extraction of exchange rates, the relaxation rates, R(4CzζNyNyη) and R1(Cz), were measured experimentally for T4L99A. First, the experimentally measured relaxation rates were compared to the corresponding rates obtained from an analysis of the MQ-CEST profiles, where those rates were allowed to vary, however, independent of the static magnetic field (Figure S7). Second, two-dimensional χ2(kex, R2) grid plots were generated, Figure S8, to quantify the influence of the relaxation rates on the derived kex. For nuclei with slower exchange rates, accurate transverse 15Nη relaxation rates can be obtained. In cases of faster rotational exchange rates, the exchange rate is uncorrelated with the transverse relaxation rate, Figure S8, meaning that the transverse relaxation rate can be safely fixed to a sensible value in the fitting process. In all cases, accurate rotational exchange rates, kex, can be obtained (Figure S9).
In summary, we have described a multiquantum CEST NMR experiment, which is ideally suited for characterizing the rate of symmetrical exchange and the chemical shifts of the involved nuclei. The MQ-CEST approach was applied to quantify the rotation of guanidinium groups in arginine side chains in proteins, and it is shown that the MQ-CEST approach can accurately provide the rate of exchange over a very large range of time scales and also report on sites that previously remained undetected. The results confirm that the symmetrical rotational rate of arginine guanidinium groups within a protein environment is an insightful parameter reporting on the strength of interactions formed by the group. It is anticipated that the MQ-CEST methodology presented here will be generally applicable to quantify symmetric exchange over a large range of time scales and in many sites.
Experimental Methods
Sample preparations are described in the Supporting Information. All spectra were processed using NMRPipe32 and visualized with NMRFAM-Sparky.33 Peak intensities were quantified using FuDa.34 MQ-CEST profiles were simulated and analyzed using an in-house program that numerically propagates the Bloch–McConnell equations.29 A detailed description is provided in the Supporting Information.
Acknowledgments
Karola Gerecht is acknowledged for producing the sample of L99A T4L used in this study. The BBSRC (BB/R000255/1), the Wellcome Trust (ref 101569/z/13/z), and the EPSRC are acknowledged for supporting the biomolecular NMR facility at University College London. Access to ultra-high-field NMR spectrometers was supported by the Francis Crick Institute through provision of access to the MRC Biomedical NMR Centre and by the University of Oxford Wellcome Institutional Strategic Support Fund, the John Fell Fund, as well as the Edward Penley Abraham Cephalosporin Fund, and the Engineering and Physical Sciences Research Council (EP/R029849/1). The Francis Crick Institute receives its core funding from Cancer Research UK (FC001029), the UK Medical Research Council (FC001029), and the Wellcome Trust (FC001029). This research is supported by the Leverhulme Trust (RPG-2016-268).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.0c01322.
Samples of arginine and T4L99A sample preparations; a detailed list of experimental conditions for all NMR experiments, Table S1; detailed description of the NMR and computational methods; supporting tables with parameters derived and pulse sequences for the experiments performed;. supplementary Figures S1–S9 showing simulated MQ-CEST data; detailed NMR pulse sequence diagrams including for the 1H-detected sequence; a comparison of the sensitivity of 13C and 1H detected MQ-CEST spectra; all experimental data for T4L99A; and an error analysis of fitted parameters for T4L99A and analysis of the effects of relaxation rates on the results (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Karplus M.; Kuriyan J. Molecular Dynamics and Protein Function. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 6679–6685. 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzler-Wildman K.; Kern D. Dynamic Personalities of Proteins. Nature 2007, 450, 964–972. 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- Alderson T. R.; Kay L. E. Unveiling Invisible Protein States with NMR Spectroscopy. Curr. Opin. Struct. Biol. 2020, 60, 39–49. 10.1016/j.sbi.2019.10.008. [DOI] [PubMed] [Google Scholar]
- Baldwin A. J.; Kay L. E. NMR Spectroscopy Brings Invisible Protein States into Focus. Nat. Chem. Biol. 2009, 5, 808–814. 10.1038/nchembio.238. [DOI] [PubMed] [Google Scholar]
- Vallurupalli P.; Hansen D. F.; Kay L. E. Structures of Invisible, Excited Protein States by Relaxation Dispersion NMR Spectroscopy. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 11766–11771. 10.1073/pnas.0804221105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauerwein A. C.; Hansen D. F.. Relaxation Dispersion NMR Spectroscopy. In Protein NMR: Modern Techniques and Biomedical Applications; Springer: Boston, MA, 2015; pp 75–132. [Google Scholar]
- Jones K. M.; Pollard A. C.; Pagel M. D. Clinical Applications of Chemical Exchange Saturation Transfer (CEST) MRI. J. Magn. Reson. Imaging 2018, 47, 11–27. 10.1002/jmri.25838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling W.; Eliav U.; Navon G.; Jerschow A. Chemical Exchange Saturation Transfer by Intermolecular Double-Quantum Coherence. J. Magn. Reson. 2008, 194, 29–32. 10.1016/j.jmr.2008.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.; Lee J.; Leninger M.; Windschuh J.; Traaseth N. J.; Jerschow A. Magnetization Transfer in Liposome and Proteoliposome Samples That Mimic the Protein and Lipid Composition of Myelin. NMR Biomed. 2019, 32, e4097 10.1002/nbm.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallurupalli P.; Bouvignies G.; Kay L. E. Studying “Invisible” Excited Protein States in Slow Exchange with a Major State Conformation. J. Am. Chem. Soc. 2012, 134, 8148–8161. 10.1021/ja3001419. [DOI] [PubMed] [Google Scholar]
- Vallurupalli P.; Sekhar A.; Yuwen T.; Kay L. E. Probing Conformational Dynamics in Biomolecules via Chemical Exchange Saturation Transfer: A Primer. J. Biomol. NMR 2017, 67, 243–271. 10.1007/s10858-017-0099-4. [DOI] [PubMed] [Google Scholar]
- Forsén S.; Hoffman R. A. Study of Moderately Rapid Chemical Exchange Reactions by Means of Nuclear Magnetic Double Resonance. J. Chem. Phys. 1963, 39, 2892–2901. 10.1063/1.1734121. [DOI] [Google Scholar]
- Borders C. L.; Broadwater J. A.; Bekeny P. A.; Salmon J. E.; Lee A. S.; Eldridge A. M.; Pett V. B. A Structural Role for Arginine in Proteins: Multiple Hydrogen Bonds to Backbone Carbonyl Oxygens. Protein Sci. 1994, 3, 541–548. 10.1002/pro.5560030402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascal S. M.; Yamazaki T.; Singer A. U.; Kay L. E.; Forman-Kay J. D. Structural and Dynamic Characterization of the Phosphotyrosine Binding Region of a Src Homology 2 Domain-Phosphopeptide Complex by NMR Relaxation, Proton Exchange, and Chemical Shift Approaches. Biochemistry 1995, 34, 11353–11362. 10.1021/bi00036a008. [DOI] [PubMed] [Google Scholar]
- Friedt J.; Leavens F. M. V.; Mercier E.; Wieden H. J.; Kothe U. An Arginine-Aspartate Network in the Active Site of Bacterial TruB Is Critical for Catalyzing Pseudouridine Formation. Nucleic Acids Res. 2014, 42, 3857–3870. 10.1093/nar/gkt1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong P. A.; Vernon R. M.; Forman-Kay J. D. RGG/RG Motif Regions in RNA Binding and Phase Separation. J. Mol. Biol. 2018, 430, 4650–4665. 10.1016/j.jmb.2018.06.014. [DOI] [PubMed] [Google Scholar]
- Goldschen-Ohm M. P.; Wagner D. A.; Jones M. V. Three Arginines in the GABAA Receptor Binding Pocket Have Distinct Roles in the Formation and Stability of Agonist- versus Antagonist-Bound Complexes. Mol. Pharmacol. 2011, 80, 647–656. 10.1124/mol.111.072033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley P. B.; Golovin A. Cation-π Interactions in Protein-Protein Interfaces. Proteins: Struct., Funct., Genet. 2005, 59, 231–239. 10.1002/prot.20417. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y.; Oktaviani N. A.; Yonezawa K.; Kamikubo H.; Mulder F. A. A. Unambiguous Determination of Protein Arginine Ionization States in Solution by NMR Spectroscopy. Angew. Chem., Int. Ed. 2017, 56, 239–242. 10.1002/anie.201609605. [DOI] [PubMed] [Google Scholar]
- Werbeck N. D.; Kirkpatrick J.; Hansen D. F. Probing Arginine Side-Chains and Their Dynamics with Carbon-Detected NMR Spectroscopy: Application to the 42 KDa Human Histone Deacetylase 8 at High PH. Angew. Chem., Int. Ed. 2013, 52, 3145–3147. 10.1002/anie.201209385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard R. B.; Hansen D. F. Characterising Side Chains in Large Proteins by Protonless 13 C-Detected NMR Spectroscopy. Nat. Commun. 2019, 10, 1747. 10.1038/s41467-019-09743-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie H. W.; Hansen D. F. A 13C-Detected 15N Double-Quantum NMR Experiment to Probe Arginine Side-Chain Guanidinium 15Nη Chemical Shifts. J. Biomol. NMR 2017, 69, 123–132. 10.1007/s10858-017-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerecht K.; Figueiredo A. M.; Hansen D. F. Determining Rotational Dynamics of the Guanidino Group of Arginine Side Chains in Proteins by Carbon-Detected NMR. Chem. Commun. 2017, 53, 10062–10065. 10.1039/C7CC04821A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy M. A.; Mueller L. Selective Shaped Pulse Decoupling in NMR: Homonuclear [13C]Carbonyl Decoupling. J. Am. Chem. Soc. 1992, 114, 2108–2112. 10.1021/ja00032a026. [DOI] [Google Scholar]
- Geen H.; Freeman R. Band-Selective Radiofrequency Pulses. J. Magn. Reson. 1991, 93, 93–141. 10.1016/0022-2364(91)90034-Q. [DOI] [Google Scholar]
- McConnell H. M. Reaction Rates by Nuclear Magnetic Resonance. J. Chem. Phys. 1958, 28, 430–431. 10.1063/1.1744152. [DOI] [Google Scholar]
- Hansen D. F.; Led J. J. Implications of Using Approximate Bloch-McConnell Equations in NMR Analyses of Chemically Exchanging Systems: Application to the Electron Self-Exchange of Plastocyanin. J. Magn. Reson. 2003, 163, 215–227. 10.1016/S1090-7807(03)00062-4. [DOI] [PubMed] [Google Scholar]
- Chakrabarti K. S.; Ban D.; Pratihar S.; Reddy J. G.; Becker S.; Griesinger C.; Lee D. High-Power 1H Composite Pulse Decoupling Provides Artifact Free Exchange-Mediated Saturation Transfer (EST) Experiments. J. Magn. Reson. 2016, 269, 65–69. 10.1016/j.jmr.2016.05.013. [DOI] [PubMed] [Google Scholar]
- Vallurupalli P.; Bouvignies G.; Kay L. E. A Computational Study of the Effects of 13c-13c Scalar Couplings on 13C CEST NMR Spectra: Towards Studies on a Uniformly 13C-Labeled Protein. ChemBioChem 2013, 14, 1709–1713. 10.1002/cbic.201300230. [DOI] [PubMed] [Google Scholar]
- Farrow N. A.; Zhang O.; Forman-Kay J. D.; Kay L. E. A Heteronuclear Correlation Experiment for Simultaneous Determination of 15N Longitudinal Decay and Chemical Exchange Rates of Systems in Slow Equilibrium. J. Biomol. NMR 1994, 4, 727–734. 10.1007/BF00404280. [DOI] [PubMed] [Google Scholar]
- Liu L.; Baase W. A.; Matthews B. W. Halogenated Benzenes Bound within a Non-Polar Cavity in T4 Lysozyme Provide Examples of I···S and I···Se Halogen-Bonding. J. Mol. Biol. 2009, 385, 595–605. 10.1016/j.jmb.2008.10.086. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. NMRPipe: A Multidimensional Spectral Processing System Based on UNIX Pipes. J. Biomol. NMR 1995, 6, 277–293. 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Lee W.; Tonelli M.; Markley J. L. NMRFAM-SPARKY: Enhanced Software for Biomolecular NMR Spectroscopy. Bioinformatics 2015, 31, 1325–1327. 10.1093/bioinformatics/btu830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen D. F.; Yang D.; Feng H.; Zhou Z.; Wiesner S.; Bai Y.; Kay L. E. An Exchange-Free Measure of 15N Transverse Relaxation: An NMR Spectroscopy Application to the Study of a Folding Intermediate with Pervasive Chemical Exchange. J. Am. Chem. Soc. 2007, 129, 11468–11479. 10.1021/ja072717t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.