

Abstract
This chapter reviews how allosteric (heterotrophic) effectors and natural mutations impact hemoglobin (Hb) primary physiological function of oxygen binding and transport. First, an introduction about the structure of Hb is provided, including the ensemble of tense and relaxed Hb states and the dynamic equilibrium of Hb multistate. This is followed by a brief review of Hb variants with altered Hb structure and oxygen binding properties. Finally, a review of different endogenous and exogenous allosteric effectors of Hb is presented with particular emphasis on the atomic interactions of synthetic ligands with altered allosteric function of Hb that could potentially be harnessed for the treatment of diseases.
Keywords: Hemoglobin, Allostery, Allosteric effectors, T state, Relaxed state, Oxygen affinty, X-ray crystallography, Hemoglobin variants
Introduction
Hemoglobin (Hb) is the most studied of the heme containing globulin proteins and yet is not fully understood. It was one of the first proteins to be studied by X-ray crystallography, and earned Max Perutz the Nobel Prize in Chemistry in 1962. The structural studies provided a plethora of data that offered glimpses of the magnificent molecular mechanisms behind Hb physiological functions. Hemoglobin is a polyfunctional molecule that is involved in several functions, such as catalytic (nitrite reductase, NO dioxygenase, monooxygenase, alkylhydroperoxidase, esterase, lipoxygenase); nitric oxide metabolism; metabolic reprogramming; pH regulation and maintaining redox balance (Kosmachevskaya and Topunov 2018). This chapter however, focuses on Hb primary function of oxygen transport and how mutations, endogenous or exogenous ligands or effectors affect Hb allostery.
Structure and Function of Hemoglobin
The primary function of Hb is to transport oxygen (O2) from the lung to tissues, binding and releasing O2 in a cooperative manner, as demonstrated by the oxygen equilibrium curve (OEC), which represents O2 saturation of Hb (SO2) at varying partial pressures of O2 (pO2) (Fig. 14.1). The pO2 at 50% SO2 (expressed as P50) measures the O2-affinity for Hb, which is about 26 mmHg for normal adult human Hb (HbA) (Fig. 14.1). Historically, Hb function has been explained in terms of equilibrium between two classical states: the tense (T) state (unliganded Hb) which exhibits low affinity for O2, and the relaxed (R) state (liganded Hb) which exhibits high affinity for O2, providing a structural basis for cooperative effects that facilitate the efficient uptake and release of O2 in vivo (Perutz 1972a, b; Perutz et al. 1998; Safo and Bruno 2011; Safo et al. 2011). The equilibrium between the T and R states is affected by endogenous heterotropic ligands, such as 2,3-bisphosphoglycerate (2,3-BGP), protons (H+), carbon dioxide (CO2), chloride (Cl−) or synthetic allosteric effectors that modulate Hb-O2 affinity, either by stabilizing the R state Hb (left-shift the OEC) (Fig. 14.1 colored red) or stabilizing the T state Hb (right-shift the OEC) (Fig. 14.1; cyan) (Perutz 1972a, b; Perutz et al. 1998; Safo and Bruno 2011; Safo et al. 2011).
Fig. 14.1.
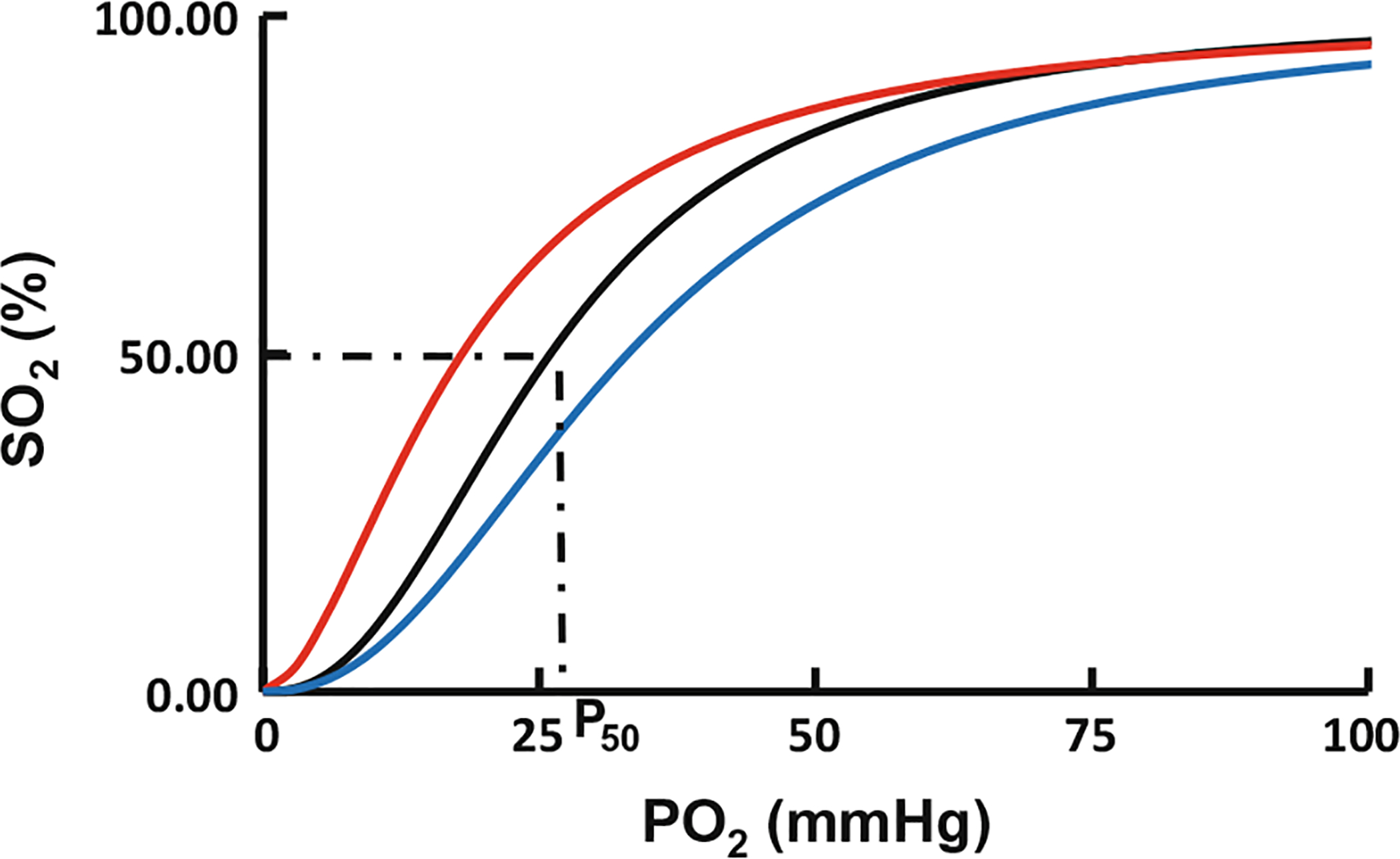
Oxygen equilibrium curve of Hb. The normal P50 value is indicated by dashed lines. The left-shift and right-shift in the curves are colored red and blue respectively
In most vertebrates, Hb is a tetramer, consisting of two α-subunits (α1 and α2) and two β-subunits (β1 and β2) that are structurally similar and about the same size (Fig. 14.2a). The two αβ dimers (named α1β1 and α2β2) are arranged around a 2-fold axis of symmetry resulting in a large central water cavity in the T or unliganded or deoxygenated structure and a narrower cavity in the R or liganded or oxygenated structure (Fig. 14.2b) (Fermi 1975; Safo and Bruno 2011; Safo et al. 2011). The α-and β-clefts are the two entry points into the central water cavity that are larger in T state structure than R state structure. The interdimer interface (α1β1–α2β2) of T state structure is also characterized by more salt-bridge/hydrogen-bond interactions than R state structure. (Fermi 1975; Safo and Bruno 2011; Safo et al. 2011).
Fig. 14.2.
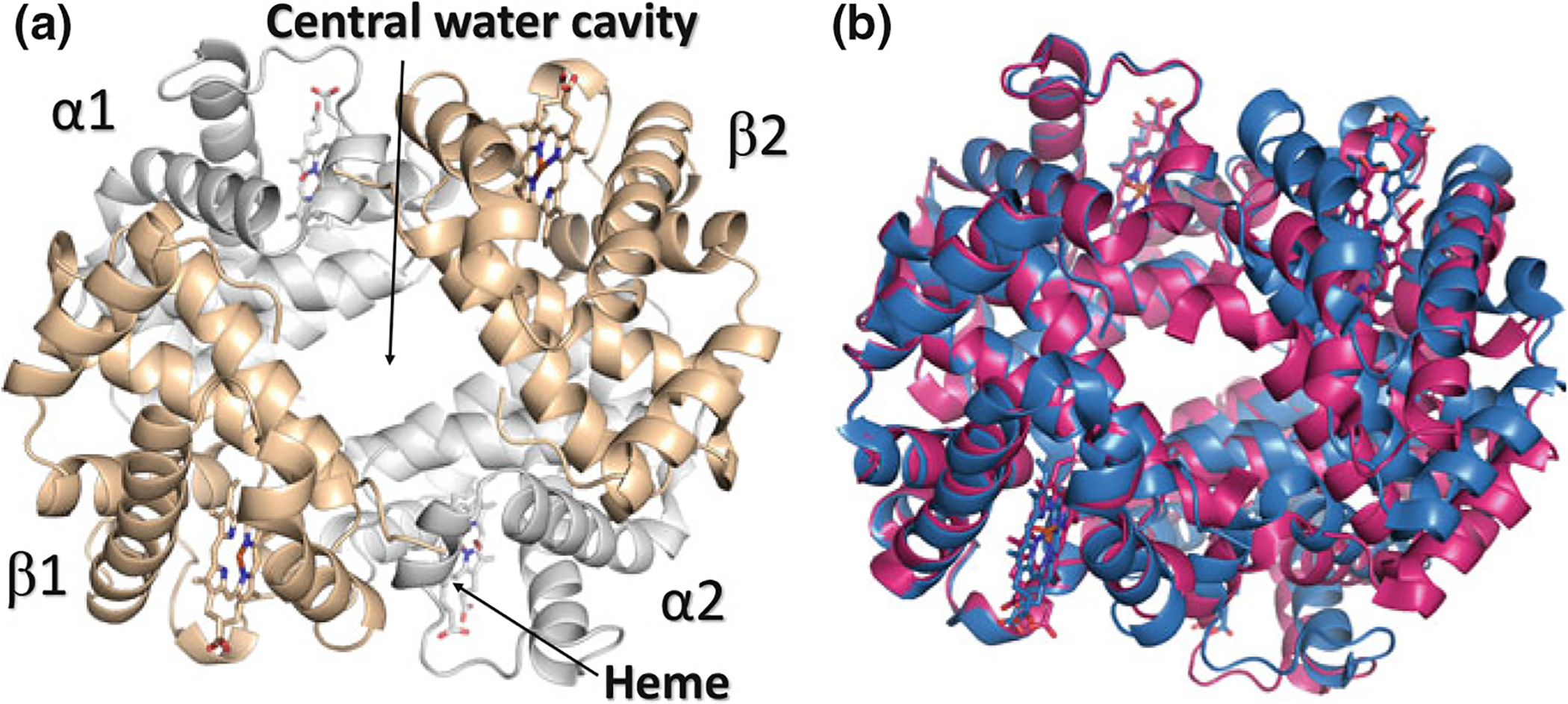
Crystal structure of hemoglobin. a Overall quaternary structure of Hb with the two α chains and β chains colored grey and tan, respectively. b Structure of oxygenated (R state) Hb (magenta) superimposed on the structure of deoxygenated (T state) Hb (blue). Note the larger central water cavity in the T structure
The α-subunits and β-subunits are formed of 7 and 8 helices, respectively named A–H that are joined by non-helical segments (referred to as corners). Each subunit has a binding pocket for heme formed by the E and F helices. The heme consists of a ferrous ion held in the center of a porphyrin and coordinated by the four nitrogen atoms of the porphyrin ring. The Fe is also covalently anchored to Hb at the heme proximal pocket by an imidazole of a histidine residue located on the F helix (known as the proximal histidine or His (F8)). This setup allows the Fe to bind O2 or other gasses at the distal pocket of the heme by a covalent bond to fulfill the octahedral coordination of six ligands. The O2 molecule binds in an “end-on bent” geometry where one oxygen atom binds to Fe, and the other protrudes at an angle. The imidazole of a histidine residue at the distal pocket (His E7) stabilizes the bound O2 through hydrogen-bond interaction. In the absence of oxygen (in deoxygenated Hb) at the α-cleft, a very weakly bonded water molecule fills the site, forming a distorted octahedron.
Other ligands, such as nitrite ion (NO2−), nitric oxide (NO), carbon monoxide (CO), cyanide (CN−), sulfur monoxide (SO), sulfide (S2−), may bind to the distal side of the heme and act as competitive inhibitors, affecting O2 binding. Some of these compounds bind with significantly higher affinity than oxygen, making these compounds highly toxic to humans. For example, Hb binding affinity for CO is as much as 240 times greater than O2, and a concentration of 0.1% CO in the air could lead to unconsciousness and eventually death (Winter and Miller 1976).
Ligand binding and unbinding events have far more ramifications, not just on the heme iron, but rather on the tertiary and the whole quaternary structure of Hb by inducing conformational changes to the globin E helix, CD and FG corners that extend to the heme environment (affecting the size of the distal pocket), central water cavity, α- and β-clefts, and salt-bridge/hydrogen-bond interactions across the α1β1–α2β2 (α1β2 or α2β1 or α1α2 or β1β2) dimer interface, subsequently, triggering cooperativity events that affect the T → R transition and giving rise to the phenomenon of allostery (Perutz 1972a, b; Baldwin and Chothia 1979; Paoli et al. 1996; Perutz et al. 1998; Safo and Bruno 2011; Safo et al. 2011).
Allosteric Models of Hemoglobin
Various allosteric models have been proposed to explain cooperative oxygen binding to hemoglobin. The earliest are the two-state Monod–Wyman–Changeux (MWC) and the Koshland–Némethy–Filmer (KNF) models. The MWC model assumes that, upon ligand binding, the T state switches to the R state without intermediate states (Monod et al. 1965), while the KNF model assumes one Hb conformation in the absence of ligand, which changes with each binding of ligand to the subunit that are sequentially transmitted to the rest of the subunits (Koshland et al. 1966). Following, Max Perutz proposed a stereochemical mechanism incorporating aspects of both the MWC and the KNF models to explain the cooperativity effect in Hb (Perutz 1972a, b; Perutz et al. 1998). According to this combined model, ligand binding to each subunit of the tetramer induces tertiary conformational changes that are transmitted to other subunits through direct communication between the α1 and β2 subunits; ultimately leading to a sequential increase in the affinity for the ligand at other heme sites and shifting the allosteric equilibrium from the T state towards the R state. The two-state model was based on a plethora of liganded and unliganded Hb crystal structures from different species, such as horse, human, and bovine (Muirhead and Perutz 1963; Perutz et al. 1968; Fermi 1975; Ladner et al. 1977). Liganded Hb structures are mostly co-crystallized with CO as the heme ligand since CO-liganded Hb is chemically stable, rendering it easier to manipulate and crystallize compared to the O2-liganded Hb. Nevertheless, the structures of O2- or CO-liganded Hb are very similar. Based on these crystal structures, Perutz partly attributed the low oxygen affinity in the T state to tension in the Fe–His (F8) bond, which restrains the Fe from moving into the porphyrin plane on ligand binding (Perutz 1972a, b; Perutz et al. 1998). This proposition was further supported by Paoli et al. (1996) who published for the first time the crystal structure of fully liganded Hb in T state (by introducing ligands to deoxygenated Hb crystals in the presence of T state stabilizing allosteric effector) demonstrating the rupture of the Fe–His(F8) bond in the α-subunits, which was cited as the reason behind uncoupling of the structural changes at the α-subunits from those at the β subunits. Barrick et al. (1997) also through site-directed mutagenesis studies noted that disrupting the Fe–His(F8) bond leads to a significant increase in ligand affinity, reduction in cooperativity, as well as slow down of the quaternary switch. However, the group also suggested that additional communication pathways exist (other than the Fe–His(F8) bond) that is responsible for the residual cooperativity observed (Barrick et al. 1997).
Perutz’s stereochemical model failed to account for the critical contributions of heterotropic ligands that can modulate the structure and function of Hb, and moreover, represented Hb as having only a binary state of either T or R without the existence of intermediary states. Several variations of Hb allosteric models were consequently put forward. Examples are the modified MWC Cooperon model of Brunori et al. (1986); the SK model of Szabo and Karplus (Szabo and Karplus 1972); the tertiary two-state (TTS) model of Henry and colleagues that describes distinct “tertiary t and r” within the T and R states conformations (Henry et al. 2002); and the global allostery model by Yonetani that also relates conformational changes to effector binding in both the T and R states and proposes that O2 affinity is dependent on heterotropic effector-induced tertiary structural changes (Yonetani et al. 2002; Yonetani and Tsuneshige 2003; Yonetani and Kanaori 2013). There are several excellent reviews of this topic, and the reader is referred to two such publications (Yonetani and Kanaori 2013; Gell 2018).
Multi States that Lie Along the T to R Transition
The early structural studies by Max Perutz and others (Muirhead and Perutz 1963; Perutz et al. 1968; Fermi 1975; Ladner et al. 1977) provided two classical end-state conformations of Hb during the T to R transition, but several lines of evidence drawn from functional, computational, structural, thermodynamic and spectroscopic experiments suggested discrete multi states along the T → R transition (Sawicki and Gibson 1976, 1978; Samaja et al. 1987; Schumacher et al. 1995; Jayaraman et al. 1995; Wilson et al. 1996; Perrella and Cera 1999; Yonetani and Tsuneshige 2003; Samuni et al. 2004; Song et al. 2008), which is expected to be due to the fact that proteins are flexible entities, and crystal structures can only provide a subset of the conformational ensembles present under physiological conditions. NMR and wide-angle X-ray scattering (WAXS), studies showed that the time-averaged solution structure of liganded Hb is unlikely to be identical to crystallized structures (Lukin et al. 2004; Gong et al. 2006; Sahu et al. 2007; Song et al. 2008; Makowski et al. 2011; Fan et al. 2013). Using laser photolysis and in the presence of the potent allosteric effector of Hb, inositol hexaphosphate (IHP), Sawicki and Gibson observed quaternary conformational changes in CO-liganded human Hb (Sawicki and Gibson 1976, 1978). Perrella and Cera (1999) also showed, using rapid quenching of the reaction between human Hb and CO, that different ligation intermediates exist with distinct conformations and oxygen affinities. Mozzarelli and collegues reported two distinct human Hb populations that had different oxygen-binding affinities (1000 and 100 times lower than the high-O2 affinity R state) and were non-cooperative (Mozzarelli et al. 1991). The former species was proposed to have T state like conformation, while the latter showed relaxed features that placed it closer to the R state.
Several crystal structures have also provided evidence of multi states along the T → R transition. A horse deoxygenated Hb trapped in the high-affinity relaxed state was reported to be a ligation intermediate of the R state (Wilson et al. 1996). Cross-linked human Hb, with trimesic acid exhibiting low oxygen affinity, was reported by Schumacher et al., in which the crystal structures have not yet reached the R state conformation, displaying several intermediate T and R features (Schumacher et al. 1995). Safo et al. (2002b) also showed that R state structure of CO-liganded human Hb with a phosphate molecule bound at the β-cleft exhibited subtle but significant tertiary T state features at the α1β2 interface. In the presence of heterotropic effectors, Yonetani and Tsuneshinge (2003) observed that liganded Hb exhibits T state constraint within an R-like quaternary structure.
Other studies have also shown the ligated T state structure with significant changes in the heme pockets, as well as changes at the α1β2 interface consistent with the presence of intermediate states along the T → R transition (Abraham et al. 1992a; Paoli et al. 1996; Song et al. 2008). An ensemble of related T-like quaternary structures induced by mutations in a cluster of residues at the αβ interface and centered at βTrp37 has been described by Arnone’s group (Kavanaugh et al. 2005). This cluster of residues was previously reported by Mozzarelli’s group to be the major region of the quaternary constraint (Noble et al. 2001).
Fully Liganded Hb Structure Trapped in a Tense Conformation
Several of the proposed allosteric models maintain the original MWC tenet that cooperative oxygen binding cannot occur in the absence of quaternary transition, consistent with the fact that a fully liganded T state hemoglobin heterotetramer, obtained from crystallizing ligated hemoglobin in solution, has never been reported. In a recent study, we reported the structure of such a uniquely fully liganded variant mammalian Hb (formed from Hb S by replacing adult α-globin with embryonic ζ-globin subunits), crystallized from CO-liganded Hb solution that remains trapped in a quaternary T state-like conformation (Safo et al. 2013, 2015). Hb inhibits polymerization of deoxygenated Hb S in vitro and ameliorates pathogenic features of sickle cell disease (SCD) in mouse models (He and Russell 2004a, b). The structure displayed a central water cavity, dimer interface and salt-bridge/hydrogen-bond interactions, β-cleft, etc. that are more typical for a tense conformation (Safo et al. 2013, 2015). It is clear that ligand binding to Hb is effected mostly by tertiary structural changes within the larger T- or R state structures, providing insights into the contributions of tertiary and quaternary structures to cooperative Hb-O2 ligand binding, as well as validating the hypothesis that Hb ligand affinity can be decoupled from overall quaternary structure (Henry et al. 2002; Yonetani et al. 2002; Yonetani and Tsuneshige 2003; Yonetani and Kanaori 2013). Moreover, the structure explains Hb antipolymer activities by favoring an alternate T state structure that is excluded from pathological deoxygenated Hb S polymers (Safo et al. 2015).
Multi Relaxed States Beyond the T → R Transition
Several studies by the Safo group and others have unveiled different relaxed Hb states that appear to lie beyond the classical T → R transition, suggesting that the relaxed state is not unique to the classical R state but is instead an ensemble of fully-liganded states exhibiting distinct quaternary conformations (Smith et al. 1991; Doyle et al. 1992; Silva et al. 1992; Janin and Wodak 1993; Smith and Simmons 1994; Srinivasan and Rose 1994; Schumacher et al. 1997; Fernandez et al. 2000; Mueser et al. 2000; Lukin et al. 2003; Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011; Abdulmalik et al. 2011). Examples of these states are the R2, RR2, R3, and RR3. Assigning these states structurally depends on different key parameters, such as rigid-body screw rotation (defined in terms of screw rotation angle, screw rotation translation, the direction of the screw rotation axis, and a point on the rotation axis) of the α1β1 dimer relative to the α2β2 dimer, interdimer salt-bridge/hydrogen-bond interactions, heme–heme distance, size of the distal heme pocket, size of the central water cavity, and size of the α-cleft and β-cleft (Baldwin and Chothia 1979; Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011).
Rigid-body screw rotation to quantify the allosteric movement between the T structure and the classical R structure was first reported by Baldwin and Chothia who found an ~14° rotation and ~1 Å translation of the α1β1 dimer relative to the α2β2 dimer (Baldwin and Chothia 1979). It was later used by several investigators to analyze the quaternary differences between several Hb states that were, in some instances, as significant or even larger than those between the T structure and the classical R structure (Silva et al. 1992; Mueser et al. 2000; Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). Here, we describe some of these structural differences with Figures using more recent high-resolution structures of the classical R (PDB ID: 2DN1), T (PDB ID: 2DN2), R2 (PDB ID: 1QXD or 1QXE) and R3 (PDB ID: 1YZI). Upon the transition from the T state to the classical R state, a sliding motion occurs between the β2-subunit and the opposite α1-subunit at a so called “switch region,” with a fulcrum, also at a so called “hinge region” (Baldwin and Chothia 1979; Silva et al. 1992; Lukin et al. 2003; Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). This motion places the β2FG corner residue β2His97 between α1Thr41 and α1Thr38 in the R structure (from its location between α1Pro44 and α1Thr41 in the T structure), where it forms a hydrogen-bond interaction with α1Thr38 (Fig. 14.3a). Moreover, the T → R transition leads to the narrowing of the central water cavity and the α- and β-clefts (Fig. 14.3b), as well as an increase in α1β2 iron–iron distance and a decrease in the β1β2 iron–iron-distance (Baldwin and Chothia 1979; Silva et al. 1992; Lukin et al. 2003; Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011).
Fig. 14.3.
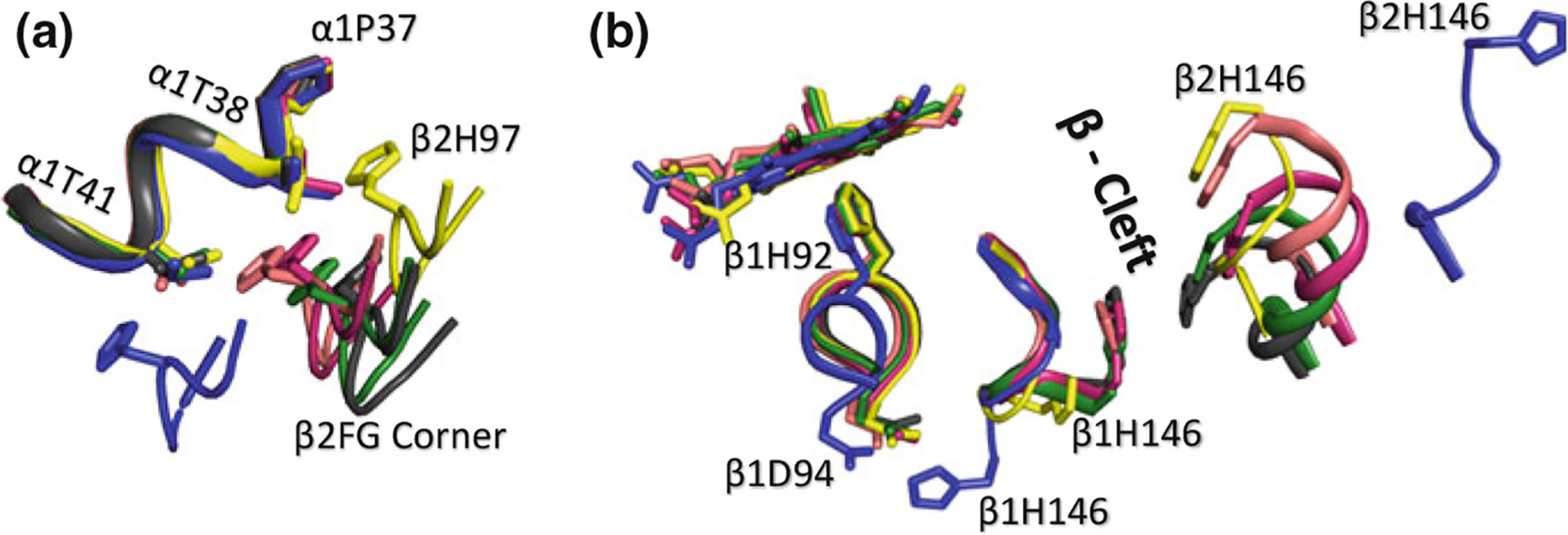
Superposed structures of T (blue), R (magenta), R3 (yellow), RR2 (green), R2 (black), and RR3 (salmon) on their α1β1 dimers. a Transitions between the different states lead to significant changes (sliding motion) at the α1β2 dimer interface switch regions. b Transitions from the T state to the relaxed states breaks a T state stabilizing salt-bridge interaction between βAsp94 and βHis146. In the R2 and RR2 structures β1His146 makes close contact with β2His146, while in the other relaxed structures, βHis146 becomes highly disordered. There is also a significant size decrease in the β-cleft of the relaxed structures compared to the T structure
Interestingly, the T → R2 transition (rotation/translation of ~23°/3.1 Å), and T → RR2 transition (~17°/2.6 Å) occur approximately in the same direction of the screw rotation axis of the T → R transition, in the order of T → R → RR2 → R2 (Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). It is interesting to note that the T → R2 transition was first proposed to lie along the T → R transition, with R as the end-state (Silva et al. 1992). However, as stated above, further analysis showed that R2 is not an intermediate but rather an end-state structure with the T → R2 transition first passing through the T → R transition and then the R RR2 transition (Janin and Wodak 1993; Schumacher et al. 1997; Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). The RR2 → R2 transition is characterized by a rotation/translation of ~6°/0.5 Å (Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). In both R2 and RR2 structures, the β2FG has further rotated perpendicularly from the R structure position, disengaging the hydrogen-bond interaction between β2His97 and α1Thr38 (Fig. 14.3a). This has led to widening of the central water cavity in these two structures, as well as bringing the two C-termini residues of βHis146 to engage in closer interactions, resulting in well-defined βHis146 positions (Fig. 14.3b) compared to the highly disordered βHis146 in the R structure (Silva et al. 1992; Lukin et al. 2003; Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). This observation, as will be discussed later, is significant due to the critical contribution of βHis146 to the Bohr effect (Silva et al. 1992).
Unlike the R, RR2 and R2 structures where β2His97 is located between α1Thr41 and α1Thr38, a rotation of β2FG corner in the R3 structure has further moved β2His97 away from the T state position, placing it between α1Thr38 and α1Pro37 (Fig. 14.3a), resulting in the smallest central water cavity and α-cleft (Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). Interestingly a complete closure of the β-cleft is observed in the R3 structure due to extensive hydrogen-bond interactions between residues from the opposite C-terminals of the HC segments (Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). The T → R3 and R → R3 transitions involve rotation/translation of ~22°/1.7 Å and ~10°/1.1 Å, respectively, and occur approximately in the same direction, with the R state mediating the T → R3 transition (Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011).
Another uniquely liganded structure, RR3 (PDB ID: 3D17) appears to lie in-between the R → R3 transition and in the order T → R → RR3 → R3 (Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). The transition from the RR3 to either R or R3 is ~6.2°/1.0 Å. Interestingly, the RR3 structure shows significant rotation of the distal βHis63(E7) out of the distal pocket, forming what is termed a His(E7) ligand channel to the bulk solvent (Fig. 14.4). A smaller rotation of His(E7) is also observed in the R3 structure (Fig. 14.4). The rotated position of the βHis63 in both R3 or RR3 is stabilized by a salt-bridge interaction between the imidazole side chain and the β-heme propionate (Safo and Abraham 2005; Jenkins et al. 2009). Of interest is that the closest distance between the βHis63 imidazole and the bound heme ligand are 6.6 Å and 4 Å in RR3 and R3 structures, respectively, compared to the ~3 Å observed in the classical R, R2, and RR2 structures, which allows stabilization of the bound ligand (Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). Overall, two trajectories have been proposed for the transition between the T and the relaxed states (Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). One trajectory suggests R2 to be an end-state with both R and RR2 lying on the T → R2 transition. The second shows R3 as another end-state with R and RR3 lying along the T → R3 transition (Fig. 14.5). Several other liganded Hb structures have also been reported, such as Gower II COHb (PDB ID: 1AJ9), bovine COHb (PDB ID: 1FSX, 1G08, and 1G09), and cross-linked forms of human COHb (PDB ID: 1HAB and 1HAC), that have distinct relaxed state quaternary structures with intermediate features between the R → RR2 and RR2 → R2 transitions (Schumacher et al. 1997; Fernandez et al. 2000; Mueser et al. 2000; Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011).
Fig. 14.4.
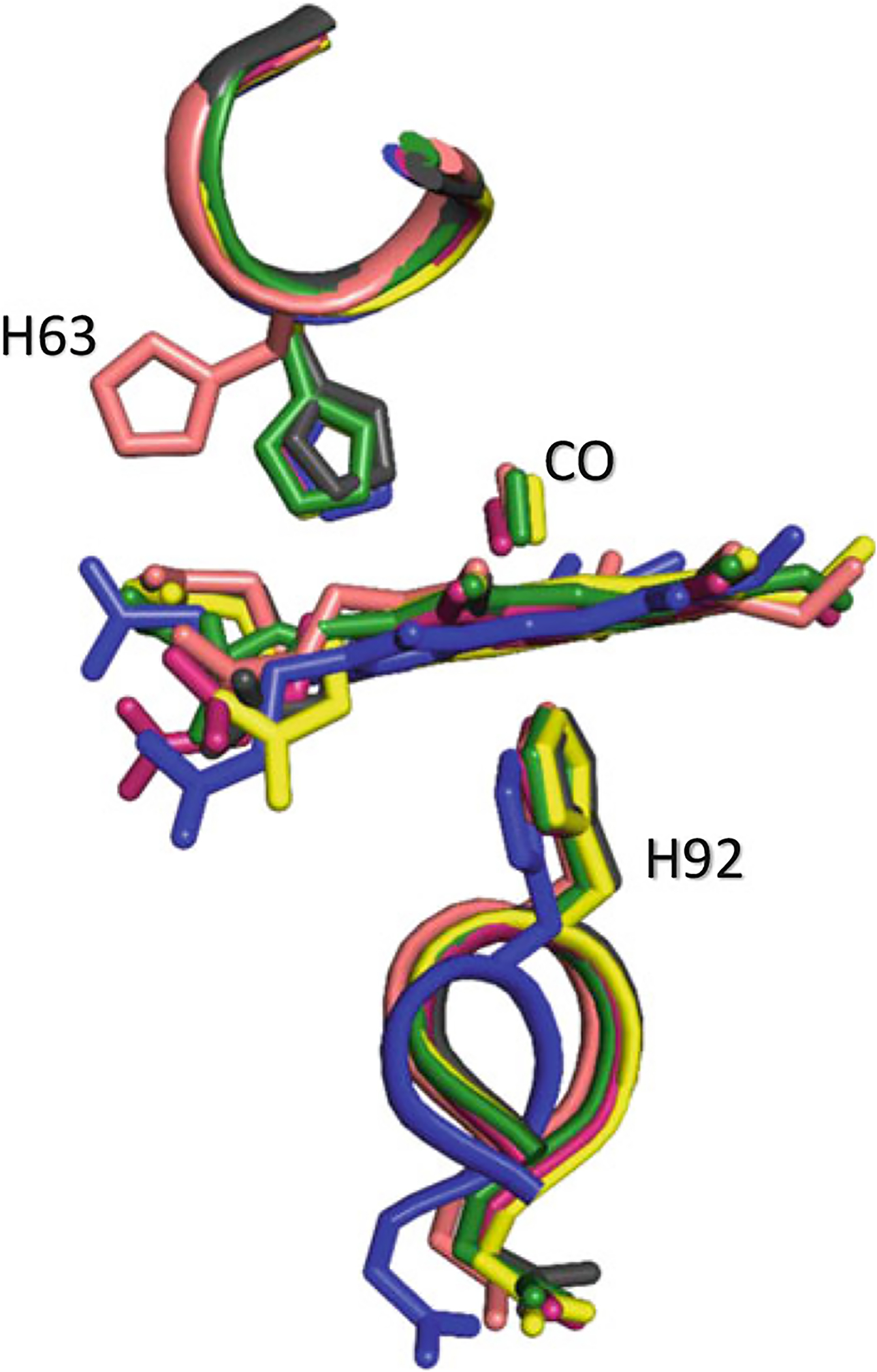
Superposed β heme structures of R (magenta), R3 (yellow), RR2 (green), R2 (black), and RR3 (salmon) showing the positions of βHis63. Note the rotation of βHis63 out of the distal pocket in the RR3 structure, creating a ligand channel to the bulk solvent, while in R, RR2, and R2 structures, βHis63 is still located in the pocket making hydrogen-bond interaction with the ligand. The R3 structure shows a partially opened ligand channel
Fig. 14.5.
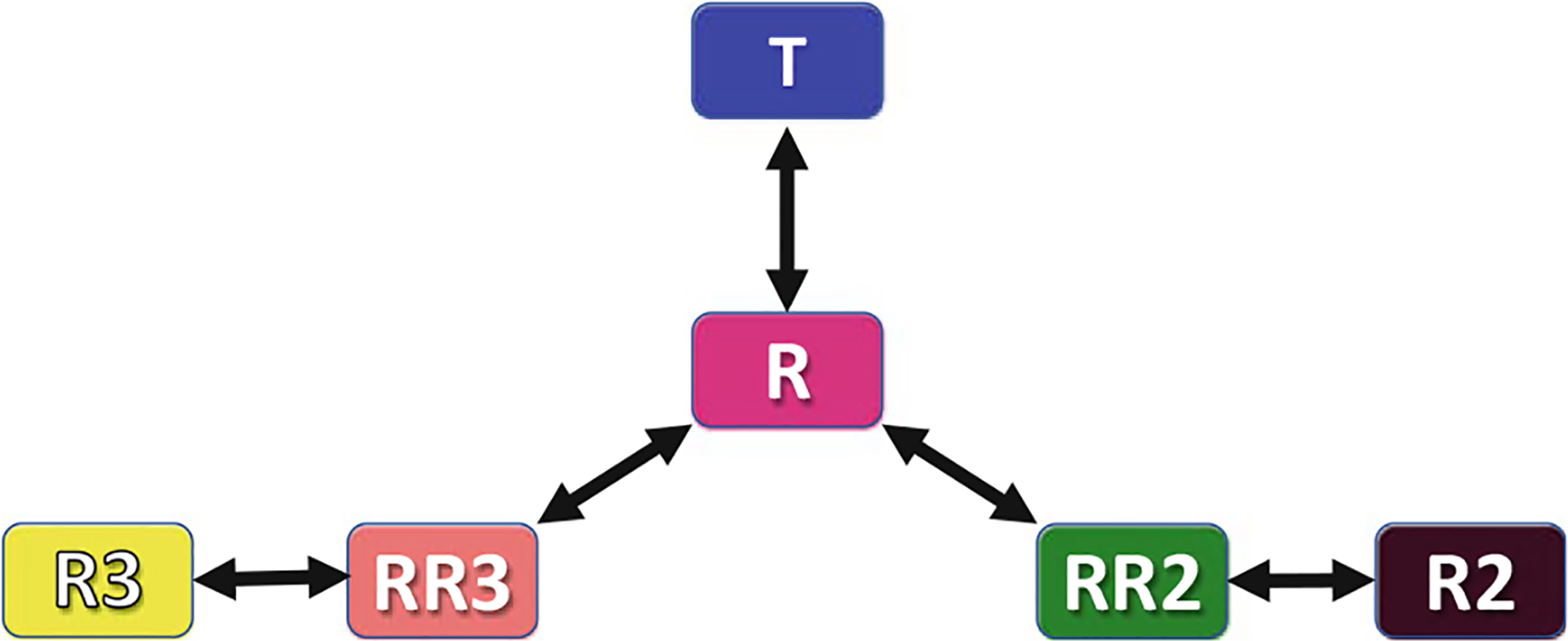
Schematic representation of the proposed allosteric pathway between the different Hb states
The R and R2 State Controversy
The discovery of the R2 structure in the early 1990s (Smith et al. 1991; Silva et al. 1992; Smith and Simmons 1994) initiated a controversy regarding the physiological relevance of the R2 or classical R structure, which took several years to resolve. The R structure was suggested to be an artifact and/or intermediate trapped between the T → R2 transition, and the R2 as the physiologically relaxed end-state (Srinivasan and Rose 1994; Schumacher et al. 1997). Other investigators, however suggested the R2 structure to be an intermediate between the T → R transition (Smith et al. 1991; Silva et al. 1992; Smith and Simmons 1994). Later studies and comprehensive analysis of the R and R2 structures, however, suggested that the R2 is not an intermediate, but rather an end-state relaxed structure (Doyle et al. 1992; Janin and Wodak 1993; Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011).
The argument for assigning the R2 structure as the physiologically relevant relaxed state, and R structure as an artifact was partly due to the former being crystallized with low-salt that mimics the in vivo environment, while the latter was crystallized with non-physiological high-salt condition. However, Safo and co-workers later crystallized the R2 structure using high-salt condition (Safo et al. 2004; Abdulmalik et al. 2011). Of interest is that the R2 crystals only form in high-salt when co-crystallized with antisickling aromatic aldehydes (Safo et al. 2004; Abdulmalik et al. 2011) usually at pH between 6.4 and 6.8. In the absence of the aldehyde, only R, RR2, RR3 or R3 or mixture of these crystals form with high-salt, and their ratio/formation appears to be a function of pH (Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). The R and R3 crystals predominate at high pH (>6.5) and low pH (<6.5), respectively, while the RR2 crystals typically appear at pH around 7 and not as abundant as the R and R3 crystals (Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). The RR3 crystal has only been observed once and appeared with R and R3 crystals at pH 6.4 (Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). It seems that various relaxed states do exist in dynamic equilibrium and their fraction and/or distribution appears to be pH-dependent with low energy barrier in changing one state to another. Consistently, an NMR study by Lukin et al. showed that liganded Hb exists as a mixture of R and R2, and a structure intermediate between the R and R2 (Lukin et al. 2003).
The existence of a multi-relaxed Hb states is appreciated even more when one considers crystal structures of liganded Hb in the presence of allosteric effectors. Aromatic aldehydes bind to the α-cleft of liganded Hb in the R2 form in a symmetry-related fashion that ties together the two α-subunits and stabilizes the relaxed state relative to the T state (Safo et al. 2004, 2011; Abdulmalik et al. 2011), which as will be discussed later is a potential pharmacologic strategy to treat sickle cell disease. As noted above, in high-salt liganded Hb without aromatic aldehyde crystallizes in R or RR2 or RR3 form depending on the pH, but in the presence of aromatic aldehyde appears to shift the equilibrium to the R2 form that crystallizes out. The binding pockets of the R, R3, and RR3 structures are sterically crowded, explaining the preferential binding of aromatic aldehyde to the R2 Hb (Safo et al. 2004; Abdulmalik et al. 2011). Gong et al. (2006) using NMR study reported that liganded Hb in the R2 form moves toward the R state in the presence of IHP. Unlike the aldehydes which bind at the α-cleft of R2 Hb, IHP binds at the β-cleft of classical R Hb (Song et al. 2008). It is interesting to note that the size of the β-cleft in the R structure is significantly larger than the other relaxed structures (Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011), suggesting preferential binding of IHP to the R structure, consistent with observed binding of phosphate at the β-cleft of R structure (Safo et al. 2002b). Thiols and imidazolylacryloyls seem to effect their antisickling activities in part by preferential binding to classical R and/or R3 structures and forming covalent interaction with βCys93 that lead to increase in Hb affinity for oxygen (Nakagawa et al. 2014, 2018; Omar et al. 2015).
Not clear is how the different liganded relaxed states fit in the overall scheme of Hb oxygen transport function. Based on the fact that the His(E7) ligand channel in the R, RR2, and R2 structures are closed, while R3 and RR3 are partially or fully opened, respectively, prompted Safo and colleagues to suggest that the R, R2, and RR2 conformations are involved in heme ligand transport, while the R3 and RR3 may be involved in ligand release (Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). Perutz has previously predicted such a rotation of His(E7) for access of ligand (Perutz 1989). Birukou et al. (2011) replaced the distal histidines, αHis58 and βHis63 with Trp in human Hb, which significantly reduced ligand access to the hemes, prompting the investigators to propose that over 90% of the ligand entry in human Hb is due to the His(E7) ligand channel. The dynamics of the His(E7) rotation is likely pH-dependent as relatively low pH appears to stabilize the open ligand channel conformation as observed in the RR3 structure, while high pH may favor closure of the channel (Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). Consistently, the rate constant for ligand binding to myoglobin increases with a decrease in pH (Traylor et al. 1983). Not obvious is whether His(E7) rotation out of the distal pocket in the T state is also pH-dependent since the pH in the lungs is not expected to be as low as in the tissues. Nevertheless, it is suggested that ligand may diffuse into the heme cavity, especially the α-heme through several hydrophobic pathways between the B, G, and H helices (Czerminski and Elber 1991), although not in an agreement by all investigators (Elber 2010).
Hemoglobin Variants with Altered Oxygen Affinity
Over 1000 naturally occurring hemoglobin variants have been identified, and although most are yet to be associated with any disease state, significant numbers are implicated in pathologies ranging from mild to severe, such as polycythemia or anemia, methemoglobinemia, cyanosis, tissue hypoxia and respiratory distress (Reissmann et al. 1961; Bonaventura and Riggs 1968; Schneider et al. 1975; Winslow and Charache 1975; Schoenborn 1976; Efremov et al. 1978; Arous et al. 1981; Dinçol et al. 1994; Kister et al. 1995; Borg et al. 1997; Marengo-Rowe 2006; Thom et al. 2013). We describe few examples of these variants that have mutations located at the surface of the protein, heme pocket, α1β2 or α1β1 interface, or the hydrophobic interior that alter hemoglobin structure and affect its oxygen binding properties. The most well-known variant is sickle Hb, which results from a single point mutation (βGlu6Val) in the β-globin chain of normal Hb to give sickle Hb (Bunn 1997). The mutation is located at the surface of the protein. Under hypoxia or when HbS is deoxygenated, the pathogenic βVal6 from one deoxygenated HbS molecule binds to a hydrophobic pocket on an adjacent deoxygenated HbS molecule, joining the molecules together to form insoluble polymers and giving rise to the classical sickle red blood cell (RBC) morphology (Eaton and Hofrichter 1990; Bunn 1997; Harrington et al. 1997; Ghatge et al. 2016). Sickled RBCs ultimately aggregate causing microvascular obstruction leading to SCD crisis (Belcher et al. 2003; Aliyu et al. 2008; De Franceschi 2009; Akinsheye and Klings 2010). Although the mutation does not directly affect the protein oxygen-binding property, a presumed elevation of 2,3-BPG in sickle red blood cells has been proposed to worsen the disease progression (Torrance et al. 1970; Grasso et al. 1975). Under physiological conditions, Hb releases 25–40% of O2 to tissues, aided by binding of 2,3-BPG to deoxygenated Hb. Unfortunately, in SCD, the repetitive deoxygenation-reoxygenation cycles with cell sickling lead to RBC membrane damage and hemolysis, reducing the life span to 10–20 days (from 90 to 120 days for normal RBCs), which manifests in chronic anemia (Zago and Bottura 1983; Connor et al. 1994), prompting compensatory elevation of 2,3-BPG in sickle RBC that decreases HbS affinity for O2 (P50 of 34 vs 26 mmHg in normal subjects) to unload more O2 to tissue (Torrance et al. 1970; Grasso et al. 1975). This counterproductive response, increases deoxygenated HbS concentration, worsening the HbS polymerization and thus the concomitant RBC sickling process. In vitro manipulation to reduce 2,3-BPG content has been suggested as a means to reduce the hypoxia-induced sickling (Poillon et al. 1986).
As noted above, the α1β1–α2β2 dimer interface (or α1β2 or α2β1 interface) is one of the most important structural determinant of the T → R transition (Baldwin and Chothia 1979; Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). It is, therefore, no surprise that several mutations affecting this interface have been reported to affect the oxygen affinity of Hb (Reissmann et al. 1961; Charache et al. 1966; Botha et al. 1966; Jones et al. 1967; Bunn et al. 1972; Lokich et al. 1973; Jensen et al. 1975). Hb Bassett (αAsp94Ala), with the αAla94 mutation located at the α1β2 interface is characterized by a markedly reduced oxygen affinity (P50 of cell free Hb at pH 7.0 = 22.0 mmHg compared with 10.5 mmHg in HbA) and low subunit cooperativity (n = 1.4 vs. 2.6 in HbA) (Abdulmalik et al. 2004; Safo et al. 2005). It is one of the few mutations where both the T and R structures of the mutant have been elucidated (Safo et al. 2005). The deoxygenated structure is characterized by two unique inter-dimer hydrogen-bond interactions (α1Tyr42–β2Asp99 and α1Asn97– β2Asp99) that are known to contribute to stabilization of T state Hb. Interestingly, the liganded R structure maintains these two hydrogen-bond interactions, in addition to losing a native R state stabilizing hydrogen-bond interaction between α1Asp94 and β2Asn102. These unique T state features in the R structure explain Hb Bassett low affinity for oxygen (Safo et al. 2005). Other low-O2 affinity Hb variants with mutation at the α1β2 interface include Hb Kansas (βAsn102Thr), Hb Saint Mande  (βAsn102Tyr), Hb Setif (αAsp94Tyr), Hb Capa (αAsp94Gly) and Hb Titusville (αAsp94Asn) (Bonaventura and Riggs 1968; Schneider et al. 1975; Arous et al. 1981; Dinçol et al. 1994; Huisman et al. 1996; Borg et al. 1997).
Hb Thionville, a variant with a αVal1Glu mutation has been reported to exhibit an increased stabilization relative to normal Hb (Vasseur et al. 1992). Structurally, this mutation results in the inhibition of the cleavage of the initiator methionine, which becomes acetylated. This results in several tertiary structural changes in the form of an extension of the α-chain N-terminus, the formation of new intra- and inter-subunit contacts in the α1α2 interface that stabilize the T state and ultimately result in decreased Hb–O2 binding affinity. However, the reduced oxygen affinity was not severe enough to produce any hematological abnormalities in the male carrier (Vasseur et al. 1992). In contrast, a similar mutation in the β chains of Hb (βVal1Glu; Hb Doha) resulted in significant anemia in a female carrier upon giving birth, who was otherwise healthy (Kamel et al. 1985). The introduction of a glutamic acid residue in Hb Doha also prevented the removal of the initiator methionine extending the N-terminus by one residue (Kamel et al. 1985). Unfortunately, we are not aware of any structural data for Hb Doha.
Several βAsp99 Hb variants, such as Hb Ypsilanti (βAsp99Tyr), Hb Radcliffe (βAsp99Ala), Hb Coimbra (βAsp99Glu) are known to exhibit increased affinity for O2 and decreased cooperativity (Jorge et al. 2018). These individuals are clinically characterized by erythrocytosis (Hardison et al. 1994, 2001; Riemer et al. 1998). The βAsp99 residue is located at the switch region of α1β2 dimer interface and forms unique hydrogen-bond interactions with α1Tyr42 and α1Asn97, stabilizing the T structure. Mutations of βAsp99 abrogate these hydrogen-bond interactions, shifting the allosteric equilibrium to the R state, and increasing Hb affinity for oxygen (Jorge et al. 2018).
Hb Rothschild is characterized by a mutation (βTrp37Arg) at the hinge region of α1β2 dimer interface, leading to significant structural perturbation at the α1β2 interface. Interestingly, this variant exhibits variable oxygen affinities, including both low and high affinity (Sharma et al. 1980; Nienhuis 1987). The T state structure showed a chloride ion bound at the α1β2 interface as a counterion to βArg37 (Kavanaugh et al. 1992, 2001). The chloride ion was suggested to affect the solution properties of this mutant, especially at varying chloride concentration that may explain the variations in oxygen binding properties (Kavanaugh et al. 1992, 2001).
Mutations in the heme pocket are also known to affect Hb oxygen binding properties (Thom et al. 2013). An example is Hb Kirklareli (αHis58Leu), which is associated with iron deficiency and increased CO binding (Bissé et al. 2017). The imidazole sidechain of αHis58 provides stability to the heme bound O2 through hydrogen-bond interactions, which is lost with αLeu58. Indeed, the crystal structure of Hb Kirklareli shows that the bound O2 is no longer stabilized, consistent with the observed increased autoxidation and loss of hemin ~200 times more rapidly than native α subunits (Bissé et al. 2017). Interestingly, Hb Kirklareli α subunit has an ~80,000-fold higher affinity for CO than O2, causing it to rapidly take up and retain carbon monoxide (Bissé et al. 2017).
Several Hb variants with mutations at the α1β1 (α2β2) interface have been identified, with some of these mutations causing significant alterations at the α1β1 interface that manifest in changes in Hb oxygen binding properties. Examples are Hb Philly (βTyr35Phe), which is characterized by increased oxygen affinity and decreased cooperativity, Hb Peterborough (βVal111Phe) and Hb Stanmore (βVal111Ala) also characterized by decreased oxygen affinities (Rieder et al. 1969; King et al. 1972; Como et al. 1991; Thom et al. 2013). These mutations cause hemolytic anemia and/or reticulocytosis (Thom et al. 2013).
Finally, mutations, deletions or insertions in the core nonpolar regions of Hb have been reported to produce unstable hemoglobins (Carrell et al. 1966; Dacie et al. 1967; Murari et al. 1977; Sakuragawa et al. 1984; Plaseska et al. 1991) that tend to undergo spontaneous oxidation causing precipitation and the formation of insoluble inclusions called Heinz bodies leading to hemolytic anemia.
Hemoglobin Modulators
Endogenous Modulators
2,3-Bisphosphoglycerate (2,3-BPG)
2,3-Bisphosphoglycerate (Fig. 14.6) plays a central role in hemoglobin allostery by reducing Hb–O2 affinity and allowing efficient tissue oxygenation (Arnone 1972; Perutz et al. 1986; Marden et al. 1990; Richard et al. 1993). Its effect on Hb oxygen affinity was first reported by Benesch and Benesch (1967). The atomic basis of its mechanism of action was elucidated with the crystal structure of Hb that showed 2,3-BPG preferentially bind to the β-cleft of deoxygenated Hb making interactions with βHis2, βLys82, βHis143, and βHis146, and linking together the two β-subunits to stabilize the T state (Fig. 14.7) (Arnone 1972; Richard et al. 1993). In contrast to the T structure, the ensemble relaxed structures have relatively smaller β-clefts explaining their lower affinity for 2,3-BPG, although liganded R state Hb with the largest β-cleft among the relaxed structures is known to bind 2,3-BPG, albeit at a lower affinity than deoxygenated Hb (Gupta et al. 1979). Crystallographic and NMR studies with phosphate and IHP (Fig. 14.6) also suggest that the R and RR2 structures are capable of binding 2,3-BPG at the β-cleft (Gupta et al. 1979; Safo et al. 2002b; Safo and Abraham 2005). It is quite obvious that the R3 structure, with its completely closed β-cleft due to elaborate β1–β2 inter-subunit hydrogen-bond interactions should preclude 2,3-BPG binding (Safo and Abraham 2005). As noted above 2,3-BPG has been proposed to be involved in the pathogenesis of sickle cell disease by its elevation in sickle RBC, which leads to further decrease in Hb affinity for oxygen to increase the concentration of the polymer forming deoxygenated HbS.
Fig. 14.6.
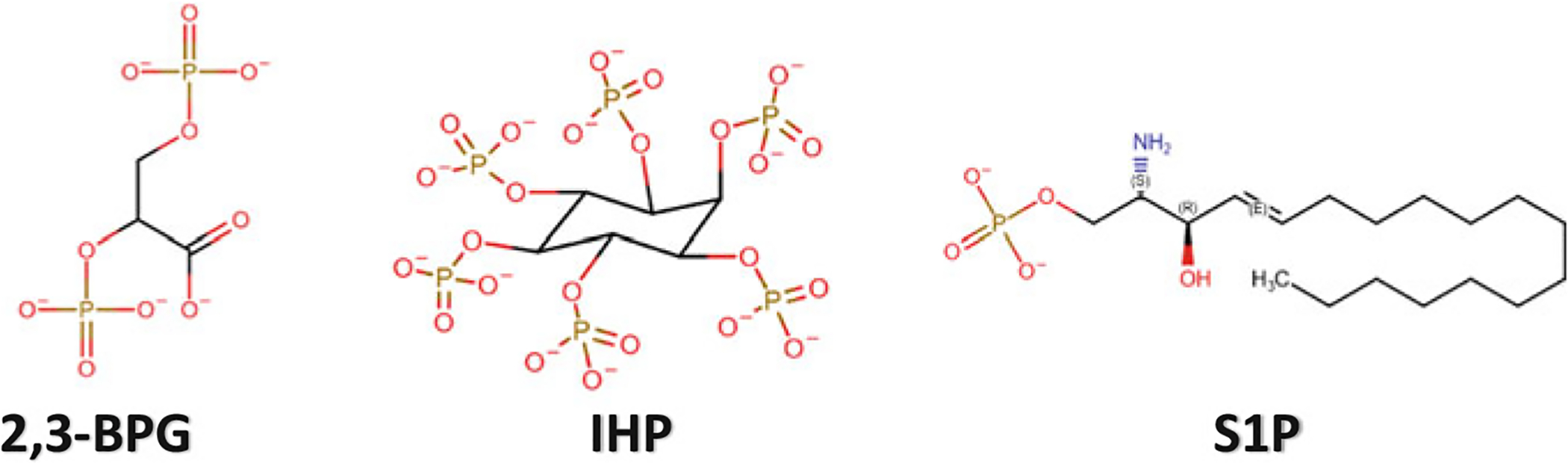
Chemical structures of IHP, 2,3-BPG and S1P
Fig. 14.7.
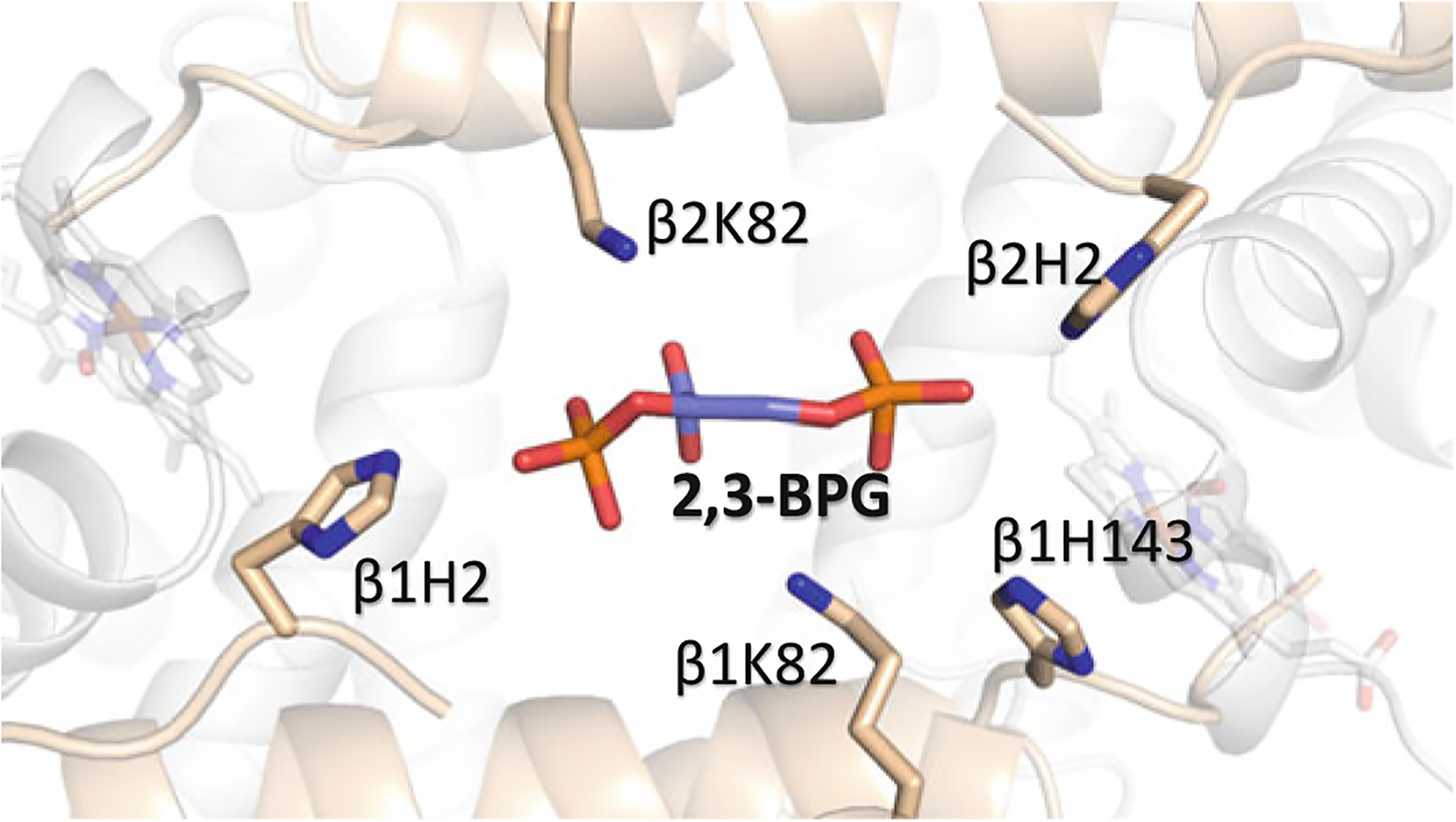
Binding of 2,3-BPG (purple) at the β-cleft of Hb. The α-subunits are colored in gray and the β-subunits are colored in tan
Sphingosine-1-Phosphate (S1P)
S1P (Fig. 14.6) is an important signaling molecule that is enriched in erythrocytes and is known to regulate diverse biological processes through activation of cell surface S1P receptors and/or by interaction with critical regulatory proteins within cells (Spiegel and Milstien 2003; Hänel et al. 2007; Ito et al. 2007). Interaction between S1P and hemoglobin was first uncovered by the Xia group after a metabolomic screen identified the pathological role of elevated S1P in SCD (Zhang et al. 2014). S1P at μM concentrations was found to regulate binding of deoxygenated Hb to the membrane protein Band 3 (cdB3) and ultimately resulting in metabolic reprogramming in SCD (Sun et al. 2017). The crystal structure of deoxygenated Hb in complex with S1P showed S1P to bind to the surface of the protein close to the heme pocket (making hydrophilic/hydrophobic interactions at the switch interface) (Fig. 14.8) (Sun et al. 2017). S1P only binds to the surface of Hb upon binding of 2,3-BPG at the β-cleft. The ternary complex leads to a significant conformational change that not only increase the T state character of the T structure but also sterically impede diffusion of diatomic ligands (O2) into the heme pocket (Sun et al. 2017). These structural changes have been proposed to in part explain the observed decrease in Hb–O2 affinity of HbS and the concomitant sickling of RBC (Sun et al. 2017). An interesting structural observation is that the last 3–4 carbon atoms of the bound S1P do not make any interaction with the Hb residue but hang out in the bulk solvent. These carbon atoms are hypothesized to mediate the hydrophobic interactions with cdB3, to also promote SCD pathogenesis (Sun et al. 2017). These findings add significant new insight to erythrocyte pathology and physiology, which paves the way for novel therapeutic interventions in SCD.
Fig. 14.8.
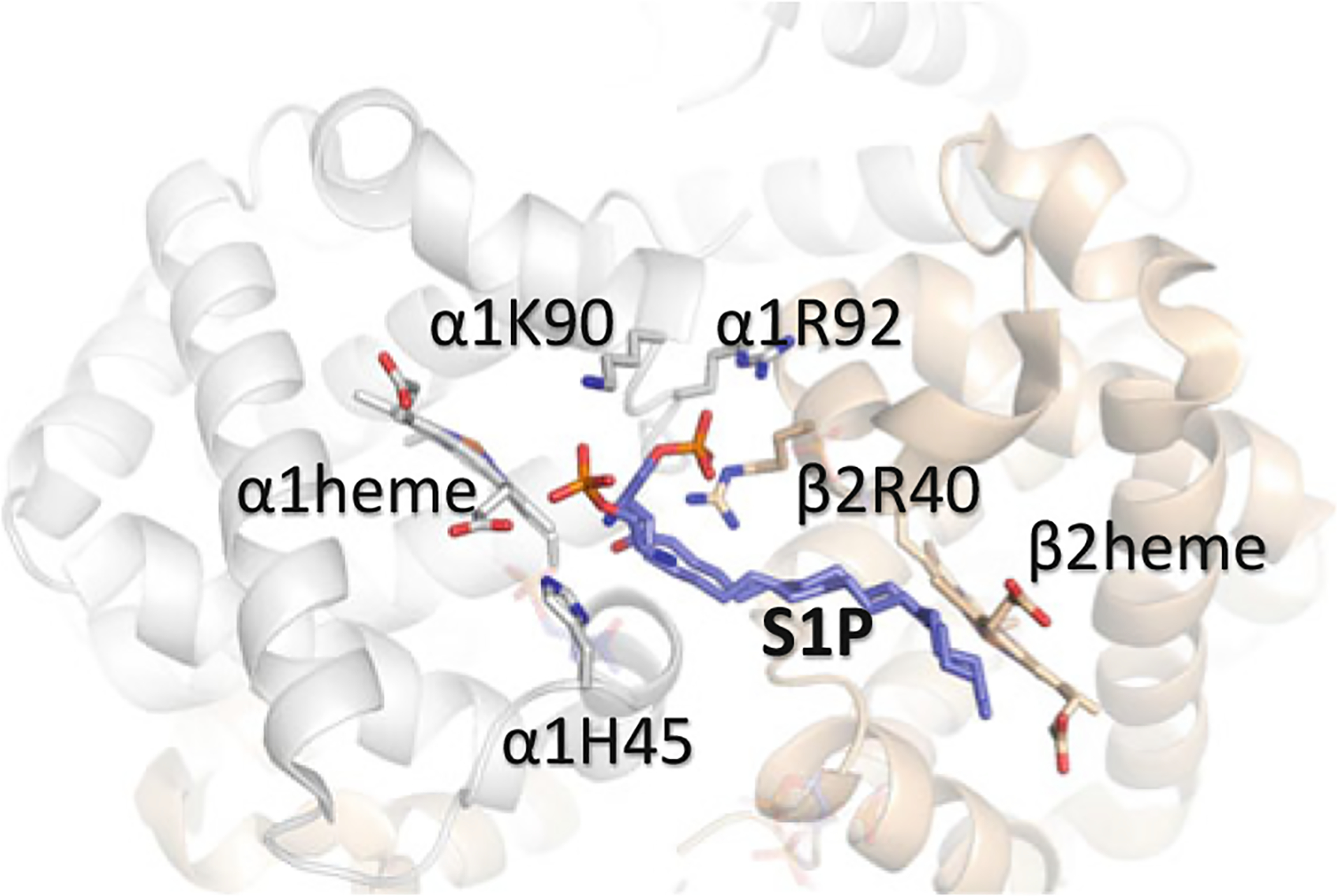
Binding of S1P (purple) on the surface of deoxygenated Hb. The α-subunits are colored in gray and the β-subunits are colored in tan. Note that S1P only binds to the surface of the protein when 2,3-BPG binds to the β-cleft
The Bohr Effect—Carbon Dioxide, Proton, and Chloride
For most mammalian hemoglobins, the heme affinity for ligands is dependent upon ambient pH (Bohr effect), due to tertiary structural perturbations (Shibayama and Saigo 2001; Yonetani et al. 2002; Yonetani and Tsuneshige 2003; Yonetani and Kanaori 2013), as well as the equilibrium between the quaternary T and R structures (Perutz 1972a, b; Perutz et al. 1998). In 1904, CO2 was discovered by Christian Bohr to lower the oxygen affinity of Hb, allowing efficient delivery of oxygen to tissues (Bohr et al. 1904). In the tissue, where CO2 concentration is high, its conversion to carbamate and/or bicarbonate releases hydrogen ions which in turn lowers the pH causing the oxygen affinity of Hb to decrease. Max Perutz and others have proposed a molecular basis for this proton-dependent Bohr effect that involves the C-terminal residues of both the α- and β-chains (αArg141 and βHis146, respectively) (Perutz 1976; O’Donnell et al. 1979; Mozzarelli et al. 1991; Kavanaugh et al. 1992; Perutz et al. 1993, 1994, 1998; Bettati and Mozzarelli 1997; Bettati et al. 1998; Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011). At high pH in deoxygenated Hb, βHis146 makes intra-subunit and inter-subunit salt-bridge interactions with βAsp94 (Fig. 14.3b) and αLys40, while αArg141 also participates in a separate inter-subunit salt-bridge interactions with αLys127 and αAsp126 (Fig. 14.9). These interactions stabilize the low-affinity T structure, consequently, facilitating O2 release. At low pH especially in the tissues, these salt-bridge interactions are broken (Figs. 14.3b and 14.9), which increases the mobility of both αArg141 and βHis146, facilitating the T → R transition, and consequently increasing Hb oxygen affinity. Other residues, such as αVal1, αHis122, βHis2, βLys82, βHi143, are also known to contribute to the Bohr effect through deoxygenation-linked proton binding (Perutz et al. 1969; Kilmartin et al. 1978; Perutz 1983; Lukin and Ho 2004; Berenbrink 2006).
Fig. 14.9.
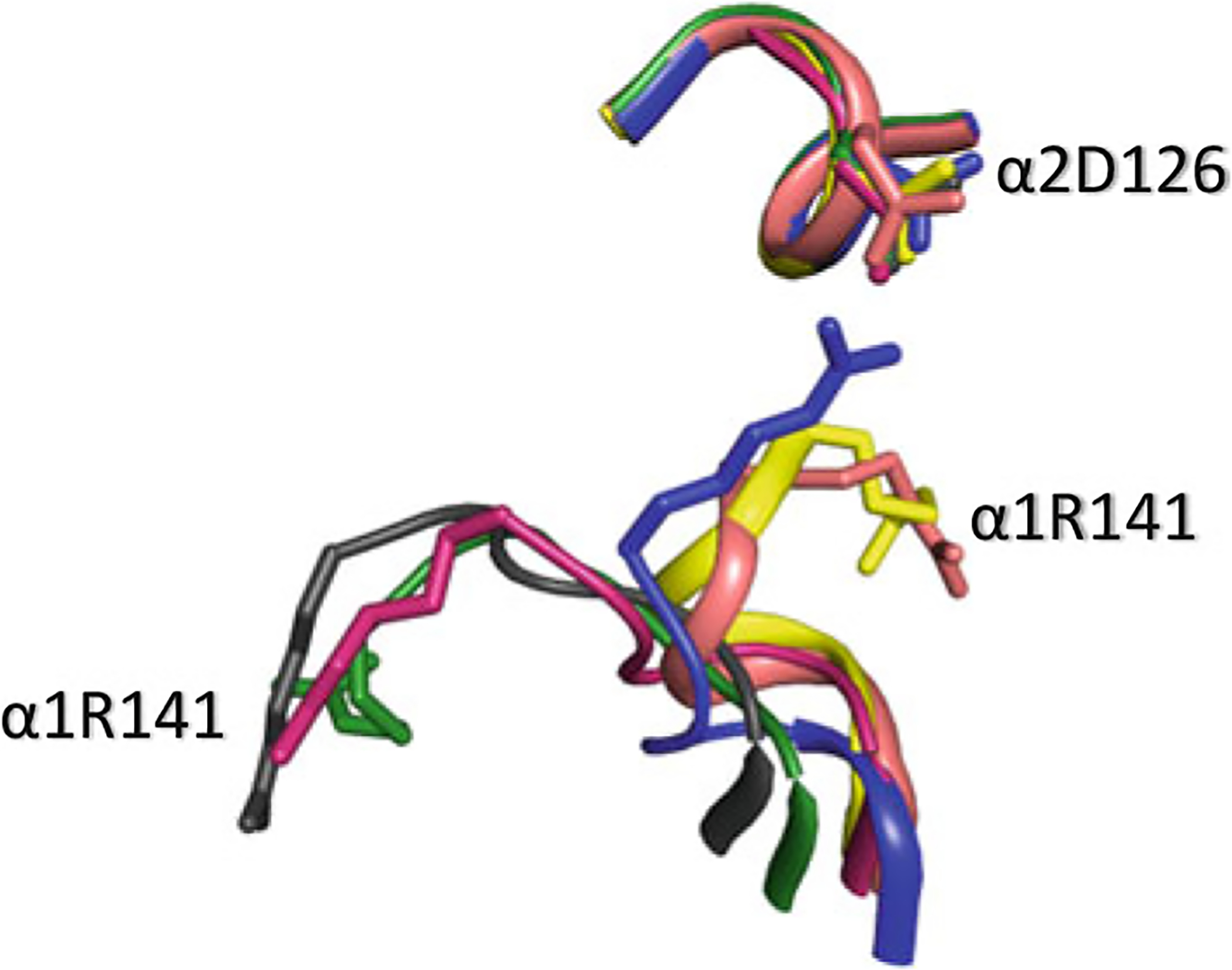
Superposed structures of T (blue), R (magenta), R3 (yellow), RR2 (green), R2 (black), and RR3 (salmon) on their α1β1 dimers. αArg141 participates in inter-subunit salt-bridge interaction with αAsp126 (as well as αLys127—not shown) in deoxygenated Hb stabilizing the low-affinity T state and facilitating O2 release. At higher pH, this interaction is broken increasing the mobility of αArg141, which facilitates the T → R transition, and increase Hb oxygen affinity
Excess positively charged residues, such as αVal1 and βLys82 in the central water cavity mutually repel each other, increasing the free energy of Hb (Bonaventura et al. 1994; Perutz et al. 1998). Chloride ions contribute to the Bohr effect by neutralizing these positive charges to stabilize Hb (Perutz et al. 1993, 1994; Fronticelli et al. 1994). More chloride ions are found in the larger T structure central water cavity when compared to the smaller R structure central water cavity, leading to greater stabilization of the T state with concomitant lowering of Hb oxygen affinity. Several Hb surface located histidines have also been suggested to contribute to the Bohr effect (Busch and Ho 1990; Sun et al. 1997). For a more detailed discussion of the Bohr effect, the reader is referred to a review article by Mairbäurl & Weber (2012).
Contribution to the Bohr effect by the various relaxed Hb structures may be different due to obvious tertiary and quaternary differences. In the R, R3 and RR3 structures, βHis146 is highly disordered, while in the RR2 and R2 structures, this residue is resolved due to close proximity of the two symmetry-related βHis146 residues that make interaction with each other (Fig. 14.3b) (Silva et al. 1992; Safo and Abraham 2005; Jenkins et al. 2009; Safo et al. 2011), prompting Arnone and co-workers to suggest that the contribution of βHis146 to the Bohr effect are different in the R and R2 states (Silva et al. 1992).
Exogenous Modulators
Modulators Shifting the Allosteric Equilibrium to the Low-O2 Affinity
A fundamental importance of Hb allostery is taking advantage of it to develop therapeutics for diseases. The discovery of 2,3-BPG prompted the search for synthetic Hb effectors for the treatment of ischemic-related diseases. One of the earliest compounds to be studied was inositol hexaphosphate (IHP) (Fig. 14.6), that binds similarly as 2,3-BPG to the β-cleft of deoxygenated Hb and was found to be 1000×more potent than 2,3-BPG in decreasing Hb affinity for oxygen (Yonetani et al. 2002). IHP has limited absorption profile for a useful therapeutic application (Stucker et al. 1985; Biolo et al. 2009). Nonetheless, it has been beneficial in investigating the allosteric properties of Hb. Analogs of IHP, such as myoinositol trispyrophosphate that are capable of crossing the membrane of erythrocytes, have been shown to enhance the exercise capacity in mice with severe heart failure (Biolo et al. 2009).
Propionates are a class of synthetic effectors that also induce low-O2 affinity by binding to Hb and stabilizing the T state. Examples of these compounds include the antilipidemic drug bezafibrate (BZF) and several of its urea derivatives (L35, L345, LR16) (Abraham et al. 1983a; Perutz and Poyart 1983; Lalezari et al. 1988, 1990; Shibayama et al. 2002; Yokoyama et al. 2006), RSR-13 and several of its derivatives (KDD3–138, RSR-40, RSR-4, TB-27, etc.) (Fig. 14.10); the latter group of compounds was discovered and studied by Abraham and co-workers (Randad et al. 1991; Wireko et al. 1991; Abraham et al. 1992b; Khandelwal et al. 1993; Phelps Grella et al. 2000; Safo et al. 2001, 2002a; Youssef et al. 2002). These compounds, unlike IHP or 2,3-BPG effect their allosteric activity in part by binding to the middle of the central water cavity of deoxygenated Hb. RSR-13 (aka Efaproxiral), the most well-known propionate was originally synthesized to mimic allosteric effects first seen with BZF, but with less protein binding in serum (Randad et al. 1991; Abraham et al. 1992b). The mode of atomic interactions of the propionates with deoxygenated Hb are very similar. X-ray crystal structures indicated that a pair of RSR-13 form noncovalent interactions with three subunits of the deoxygenated Hb tetramer within the central water cavity in a symmetry-related fashion (Fig. 14.11a), effectively stabilizing the T state from transitioning to the R state and thus reducing the affinity of Hb for O2 (_S1_Reference188Wireko et al. 1991; Abraham et al. 1992a; Safo et al. 2001). Each molecule of RSR-13 makes hydrogen-bond and hydrophobic interactions with two α-subunits and one β-subunit of the protein. Its specific interactions to the protein are illustrated in Fig. 14.11b (Safo et al. 2001). The allosteric effects of RSR-13 are additive to that of 2,3-BPG, since the compounds have distinct Hb binding sites (Laberge et al. 2005). RSR-13 has undergone preclinical testing in hypoxia- and ischemia-induced conditions, as well as late-stage human testing as an adjunct to enhance the O2-induced effects during whole-brain radiation (Abraham et al. 1992b; Khandelwal et al. 1993; Pagel et al. 1998; Woods et al. 1998; Kleinberg et al. 1999; Shaw et al. 2003).
Fig. 14.10.
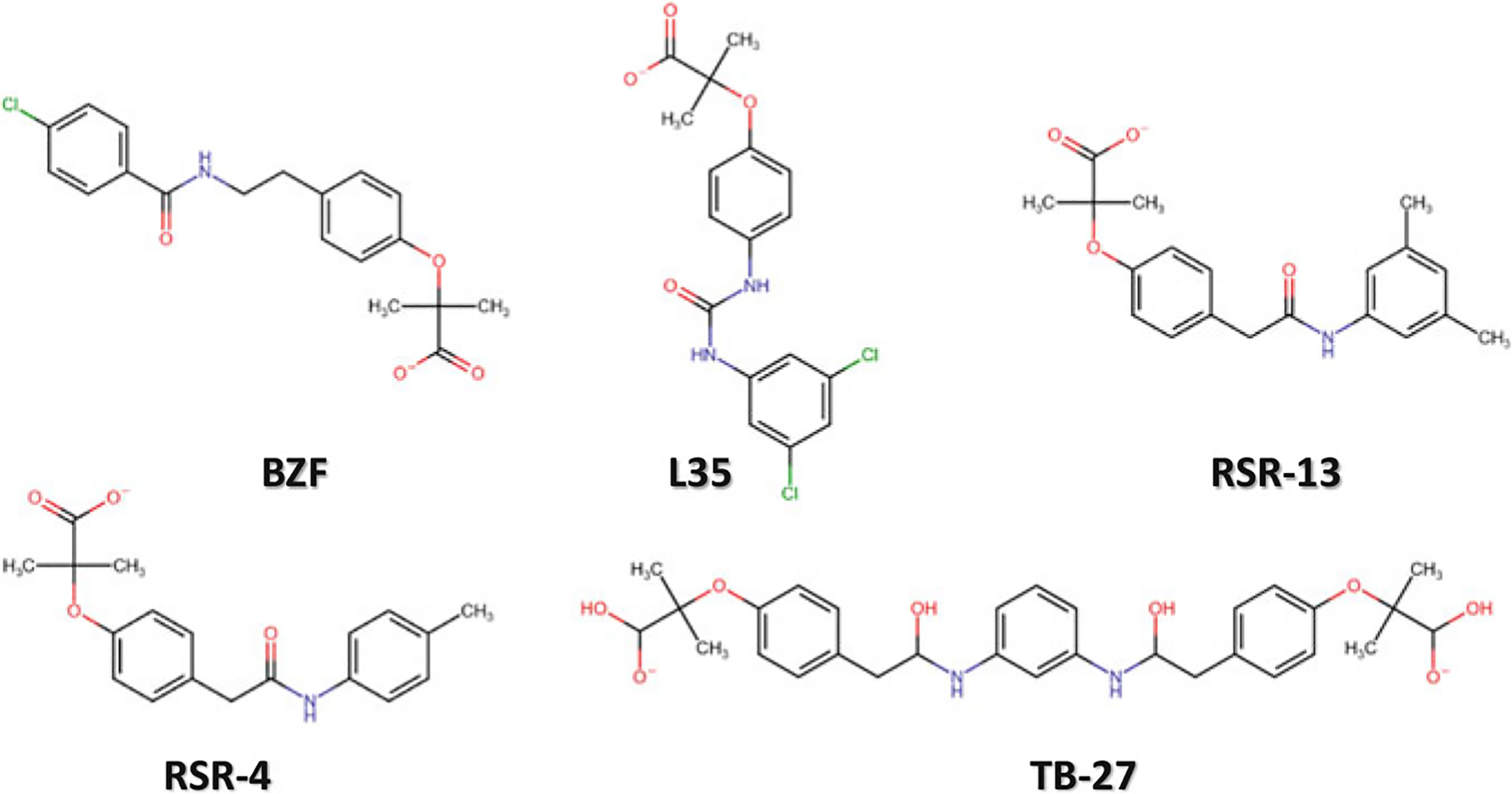
Chemical structures of BZF (and derivative L35) and RSR-13 and derivatives (RSR-4 and TB-27)
Fig. 14.11.
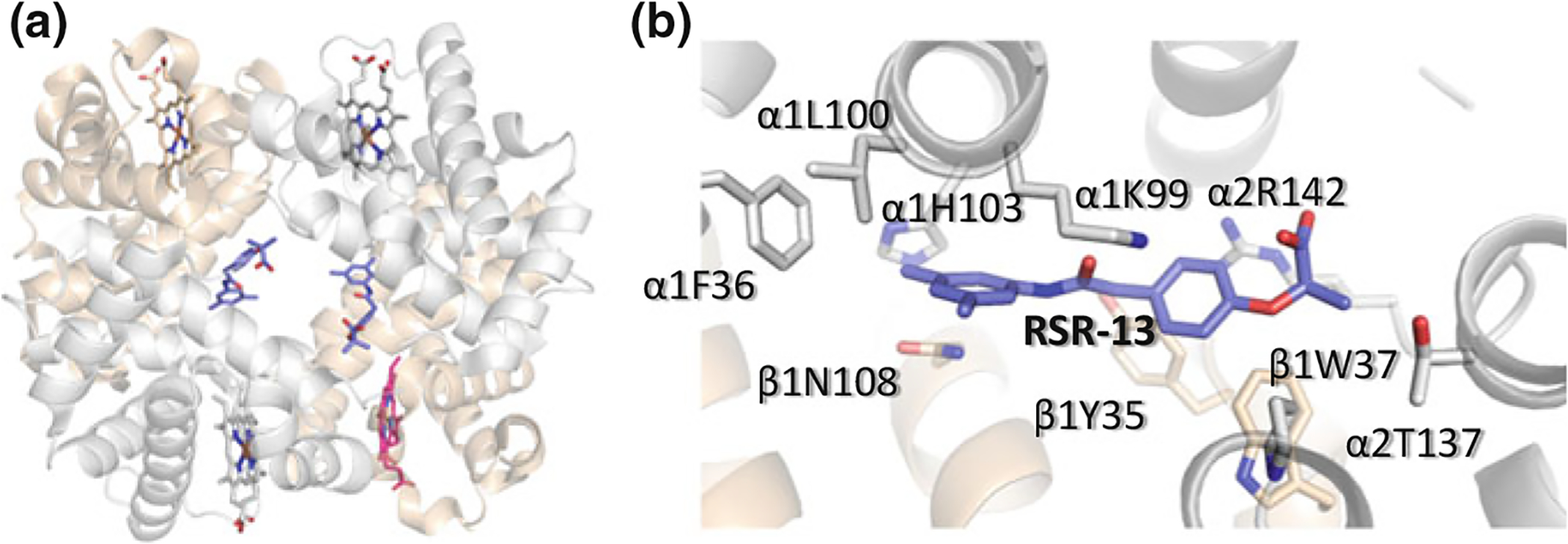
a Binding of a pair of RSR-13 (purple) at the central water cavity of deoxygenated (T state) Hb. b Detailed interactions between one of the RSR-13 molecules and the protein. The other molecule makes similar symmetry-related interactions. The two α-subunits are colored grey, while the β-subunit is colored tan
In addition to their interactions with deoxygenated Hb as described above for RSR-13, BZF and L35 have also been shown to bind to the central water cavity and/or surface of liganded Hb close to the α-heme (in the form of the classical R structure), which sterically impede ligand access to the heme (Shibayama et al. 2002; Chen et al. 2005; Yokoyama et al. 2006). Clearly, these effectors are capable of binding to both liganded and deoxygenated Hb, and their regulatory effect on Hb allostery appears to result from their interactions with both forms of Hb.
Safo’s group recently developed novel nitric oxide (NO)-releasing prodrugs of RSR-13 derivatives by attaching the NO-releasing moieties nitrooxyethyl, nitrooxypropyl, and 1-(pyrrolidin-1-yl)diazen-1-ium-1,2-diolate, respectively, to the carboxylate of RSR-13 (Xu et al. 2015). Crystallographic studies with the prodrugs showed RSR-13, the hydrolysis product of the prodrugs bound to the central water cavity of deoxygenated Hb (as described above) that explained these compounds ability to decrease Hb affinity for oxygen (Xu et al. 2015). Moreover, the released NO, was observed exclusively bound to the two α hemes, which was suggested to be due to RSR-13 decreasing the protein’s affinity for the ligand at the β hemes (Xu et al. 2015).
Aryloxyalkanoic Acids are another class of right-shifting compounds that bind non-covalently to Hb (Omar et al. 2016). The effect of these compounds on Hb’s oxygen affinity depends on the site of interaction. Aryloxyalkanoic acids that bind to the αTrp14 hydrophobic pocket of Hb have been shown to increase oxygen affinity, while those that bind to the central water cavity of the protein to stabilize the T state in most instances showed the opposite effect of decreasing the protein affinity for oxygen (Abraham et al. 1982, 1983b, 1984; Patwa et al. 1987; Mehanna and Abraham 1990).
Recently, a high-throughput screening campaign identified IRL-2500 (Fig. 14.12a), a synthetic peptide, that decreased the affinity of Hb for O2 (Goldstein et al. 2018). Structural studies with deoxygenated Hb showed this compound to overlap the 2,3-BPG binding site at the β-cleft, engaging in water-mediated and direct hydrogen-bond, as well as hydrophobic interactions with the β-cleft residues of βHis2, βLys82, βAsn139, and βHis143 (Fig. 14.12b), which provide additional interactions across the two β-subunit interface of deoxygenated Hb leading to further stabilization of the T state (Goldstein et al. 2018).
Fig. 14.12.
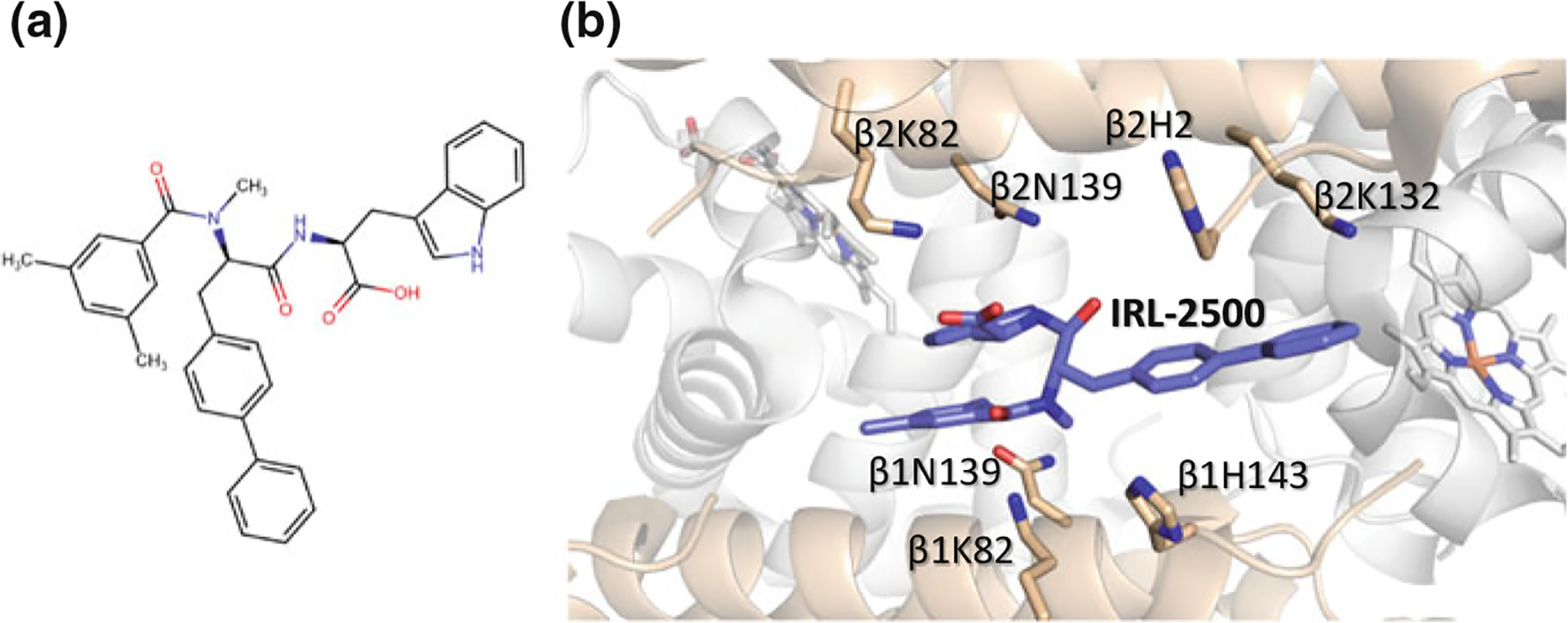
a Chemical structure of IRL-2500. b Binding of IRL-2500 (purple) at the β-cleft of Hb. The α-subunits are colored in gray and the β-subunits are colored in tan
Modulators Shifting the Allosteric Equilibrium to the High-O2 Affinity
Several synthetic and natural compounds have also been shown, in most instances to bind covalently to Hb to shift the allosteric equilibrium to the R state and increase the oxygen affinity of the protein. Some of these compounds have been clinically evaluated as antisickling agents for the treatment of sickle cell disease, as high-O2 affinity sickle Hb molecules are resistant to polymer formation.
Aromatic aldehydes (Fig. 14.13), the most well-studied effectors interact with Hb and increase the protein affinity for oxygen. The food flavoring agent vanillin was one of the earliest to be studied (Fig. 14.13) (Zaugg et al. 1977; Abraham et al. 1991). Although relatively non-toxic, its limited bioavailability and the large dose needed to increase Hb affinity for oxygen and elicit in vivo therapeutic antisickling effects were not clinically acceptable.
Fig. 14.13.
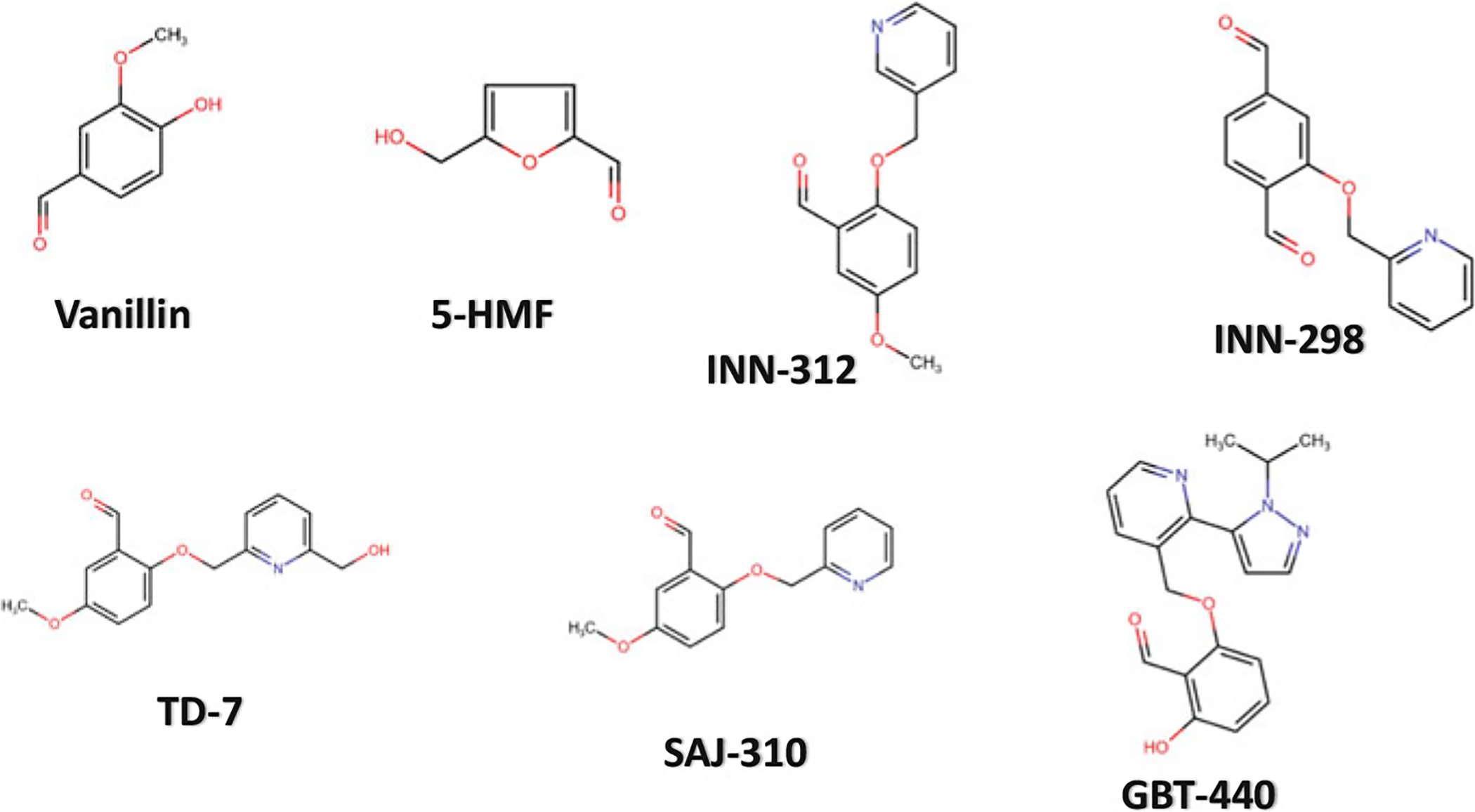
Chemical structures of high-O2 affinity antisickling aromatic aldehydes
Furfural and several of its analogs, such as 5-hydroxymethyl-2-furfural (5-HMF) (Fig. 14.13) (Safo et al. 2004; Abdulmalik et al. 2005; Xu et al. 2017) have also been shown to interact with Hb and increase its oxygen affinity. 5-HMF showed significant pharmacologic improvement over vanillin both in vitro and in vivo and was subsequently studied in phase II clinical trials for the treatment of SCD (Abdulmalik et al. 2005; Stern et al. 2012; Kato et al. 2013). The study was terminated due in part to in vivo bioavailability issue. Nonetheless, the study of 5-HMF helped trigger a dramatic increase in commercial interest in developing drugs for SCD that show a similar mechanism of action. It is important to note that prior to 5-HMF studies, the antisickling mechanism of action of aromatic aldehydes was suggested to be due to binding to deoxygenated Hb and destabilizing the T state and/or binding to liganded Hb and stabilizing the classical R state (Beddell et al. 1984; Abraham et al. 1991; Wireko and Abraham 1991). Safo and co-workers, however showed that mechanistically, 5-HMF, and for that matter, other antisickling aromatic aldehydes increase Hb affinity for O2 by binding to the R2 structure and not the T or classical R as proposed (Safo et al. 2004; Abdulmalik et al. 2005, 2011; Xu et al. 2017; Pagare et al. 2018; Deshpande et al. 2018). Two molecules of the aromatic aldehydes bind in a symmetry-related fashion at the α-cleft of the R2 structure, each molecule forming Schiff-base interaction between its aldehyde moiety and the N-terminal αVal1 nitrogen of the Hb, and through additional hydrogen-bond and/or hydrophobic interactions tie the two α-subunits together and restrict the transition to the T state (Fig. 14.14) (Safo et al. 2004; Abdulmalik et al. 2005, 2011; Xu et al. 2017; Pagare et al. 2018; Deshpande et al. 2018).
Fig. 14.14.
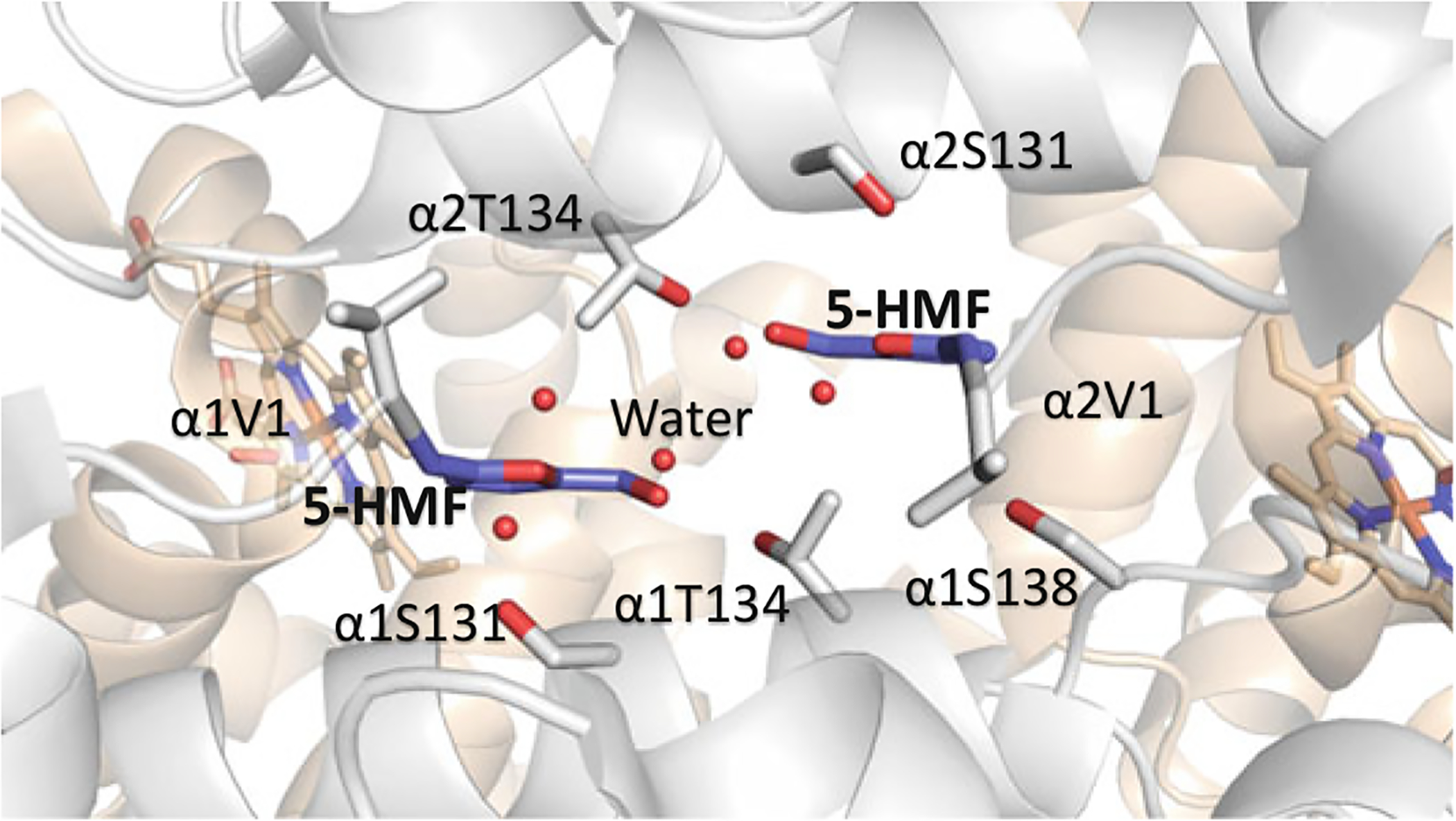
Binding of 5-HMF (purple) in a symmetry-related fashion at the α-cleft Hb. The α-subunits are colored in gray and the β-subunits are colored in tan. Water molecules are red spheres
To improve on vanillin and 5-HMF left-shifting potency and thus their anti-sickling activities, several derivatives of these lead compounds were developed. 5-HMF derivatives incorporate different substituents at the alcohol moiety of 5-HMF for additional interactions with the protein, which led to a significant increase in their allosteric activity, translating into 1.5–4.0-fold higher antisickling effects than 5-HMF (Xu et al. 2017).
Several groups have also embarked on structural modification of vanillin for more potent high-O2 affinity allosteric effectors. Some of these novel compounds include pyridyl derivatives of benzaldehydes that have been studied by Safo and colleagues, and shown to exhibit several-fold left-shifting potency over vanillin (Nnamani et al. 2008; Abdulmalik et al. 2011; Pagare et al. 2018; Deshpande et al. 2018). The crystal structures of some of these compounds, such as SAJ-310, INN-312, INN-298, and TD-7 (Fig. 14.13) complexed to Hb have been elucidated (Abdulmalik et al. 2011; Pagare et al. 2018; Deshpande et al. 2018) and show the compounds to expectedly bind similarly to the R2 structure as observed with 5-HMF. Of note is that the covalent bond between the aldehyde and the αVal1 N forced the molecules with meta-positioned methoxy-pyridine group, such as in INN-298 to direct down the central water cavity (Fig. 14.15), while those with ortho-positioned methoxypyridine, such as INN-312 or TD-7 is disposed toward the mouth of the α-cavity (Fig. 14.16) (Abdulmalik et al. 2011; Pagare et al. 2018; Deshpande et al. 2018). The latter group of compounds make interactions with the solvent-exposed αF-helix to exhibit a second antisickling mechanism of direct polymer destabilization that is independent of the primary mechanism of increasing HbS-O2 affinity (Pagare et al. 2018; Deshpande et al. 2018). The αF-helix has been shown to play an important role in polymer stabilization, and its perturbation is thought to destabilize the polymer (Safo et al. 2004, 2011; Abdulmalik et al. 2011). In line with this hypothesis, the Hb variant Stanleyville (αAsn78 → αLsy78) inhibits HbS polymerization (Benesch et al. 1979; Nagel et al. 1980; Rhoda et al. 1983). Four select representatives of these compounds, VZHE-039, PP-6, PP-10, and PP-14, are currently undergoing preclinical in vivo studies by Safo and co-workers (Safo, unreported studies).
Fig. 14.15.
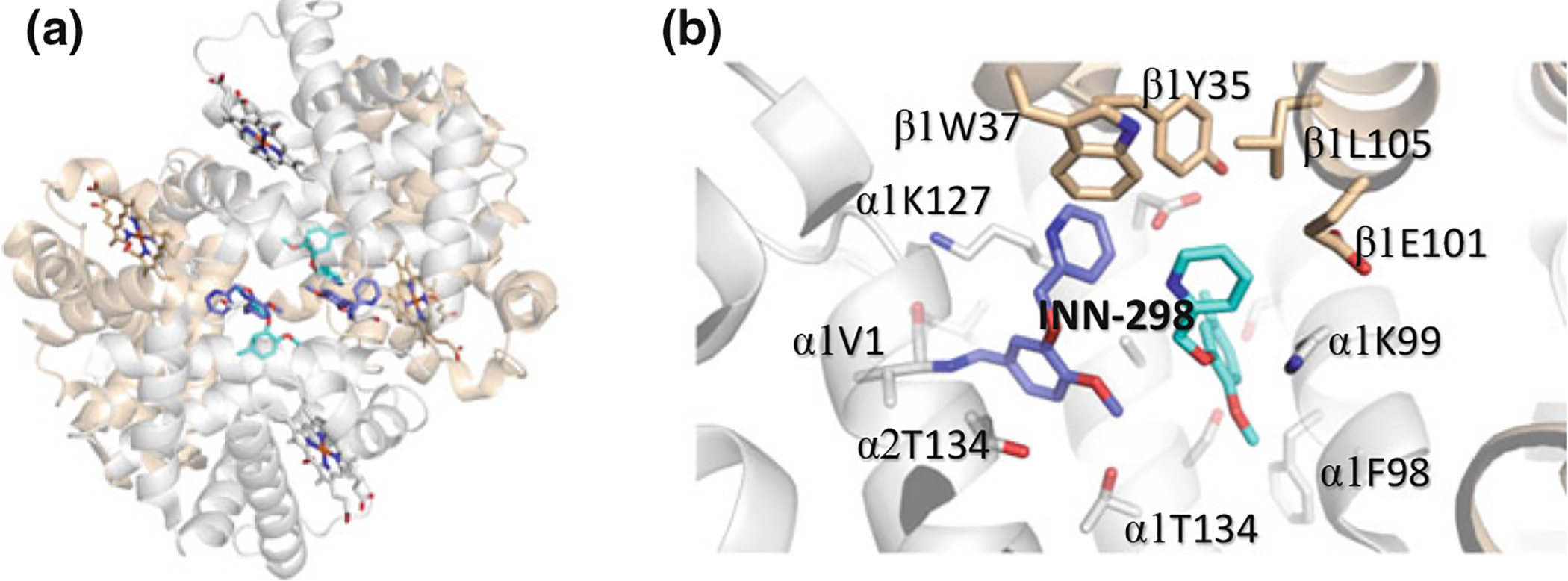
a Binding of INN-298 (purple and cyan) in a symmetry-related fashion at the α-cleft Hb. Unlike most aromatic aldehydes, four molecules of INN-298 bind per one Hb molecule. The molecules in purple make Schiff-base interactions with αVal1 nitrogen (primary), while the molecules in cyan (secondary) bind non-covalently and significantly weaker. b Detailed interactions of two of the INN-298 molecules (purple and cyan) with Hb. The meta-positioned methoxy-pyridine group of the primary bound INN-298 disposes further down the central water-cavity
Fig. 14.16.
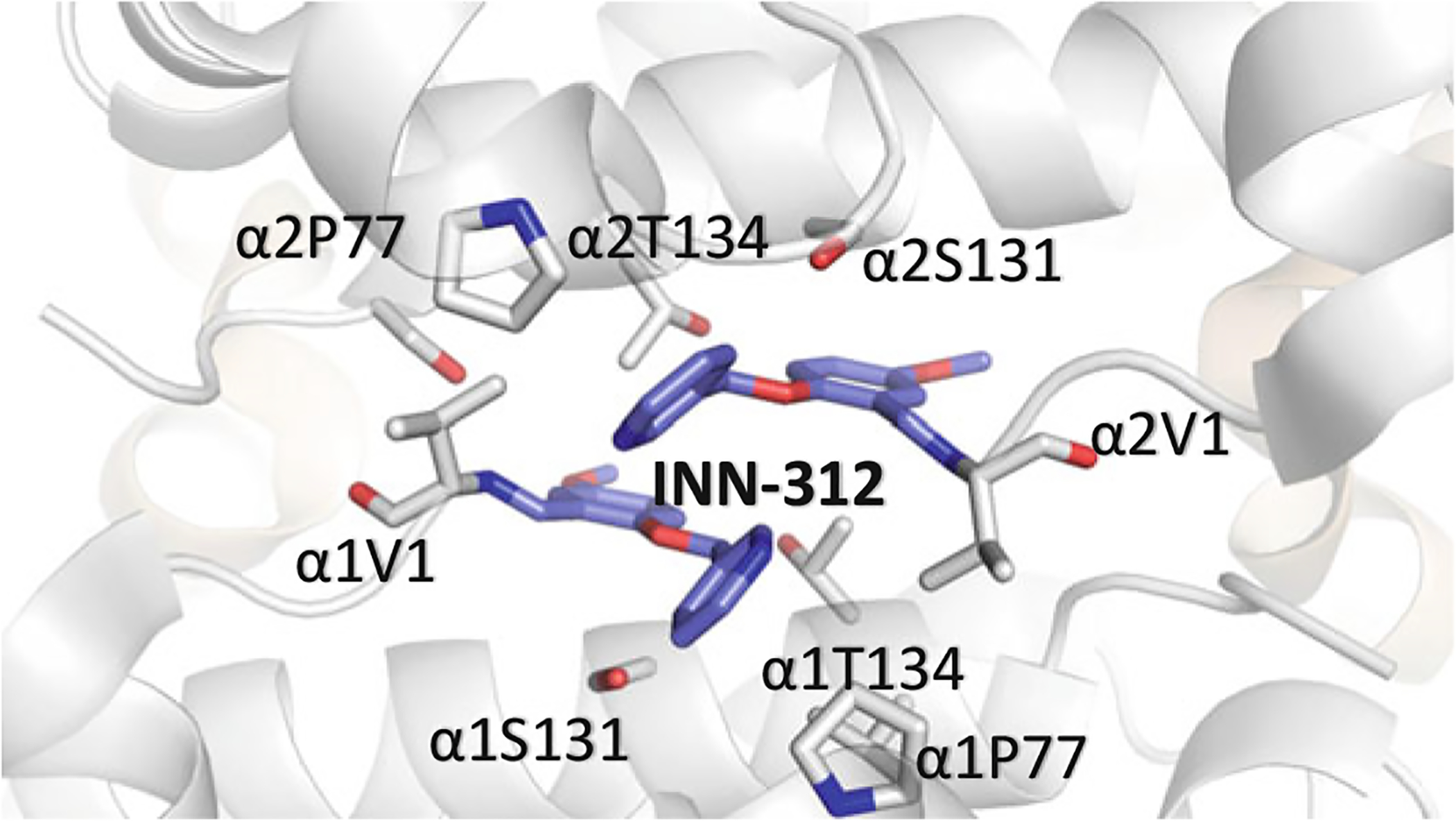
Binding of INN-312 (purple) in a symmetry-related fashion at the α-cleft of Hb. The α-subunits are colored in gray. The ortho-positioned methoxy-pyridine group disposes toward the surface of the Hb to make interaction with the αF-helix residue of Pro77
GBT-440 (Voxelotor; Fig. 14.13) is another synthetic aldehyde analog of vanillin developed by Global Blood Therapeutics, Inc. that also increases the oxygen affinity of Hb by stabilizing the relaxed state (Oksenberg et al. 2016; Metcalf et al. 2017). GBT-440 similarly binds to the N-terminal αVal1 of one of the α-chains of the R2 structure but because of the bulky pyrazole substituent, a second molecule is precluded from binding to the opposite α-chain resulting in a single GBT-440 molecule bound per Hb tetramer, which is in contrast with the observed 2:1 stoichiometry of other antisickling aromatic aldehydes (Fig. 14.17). GBT-440 is currently being studied in phase III clinical trials for the treatment of SCD (NCT03036813) (Oksenberg et al. 2016; Metcalf et al. 2017, p. 440; Vichinsky et al. 2019).
Fig. 14.17.
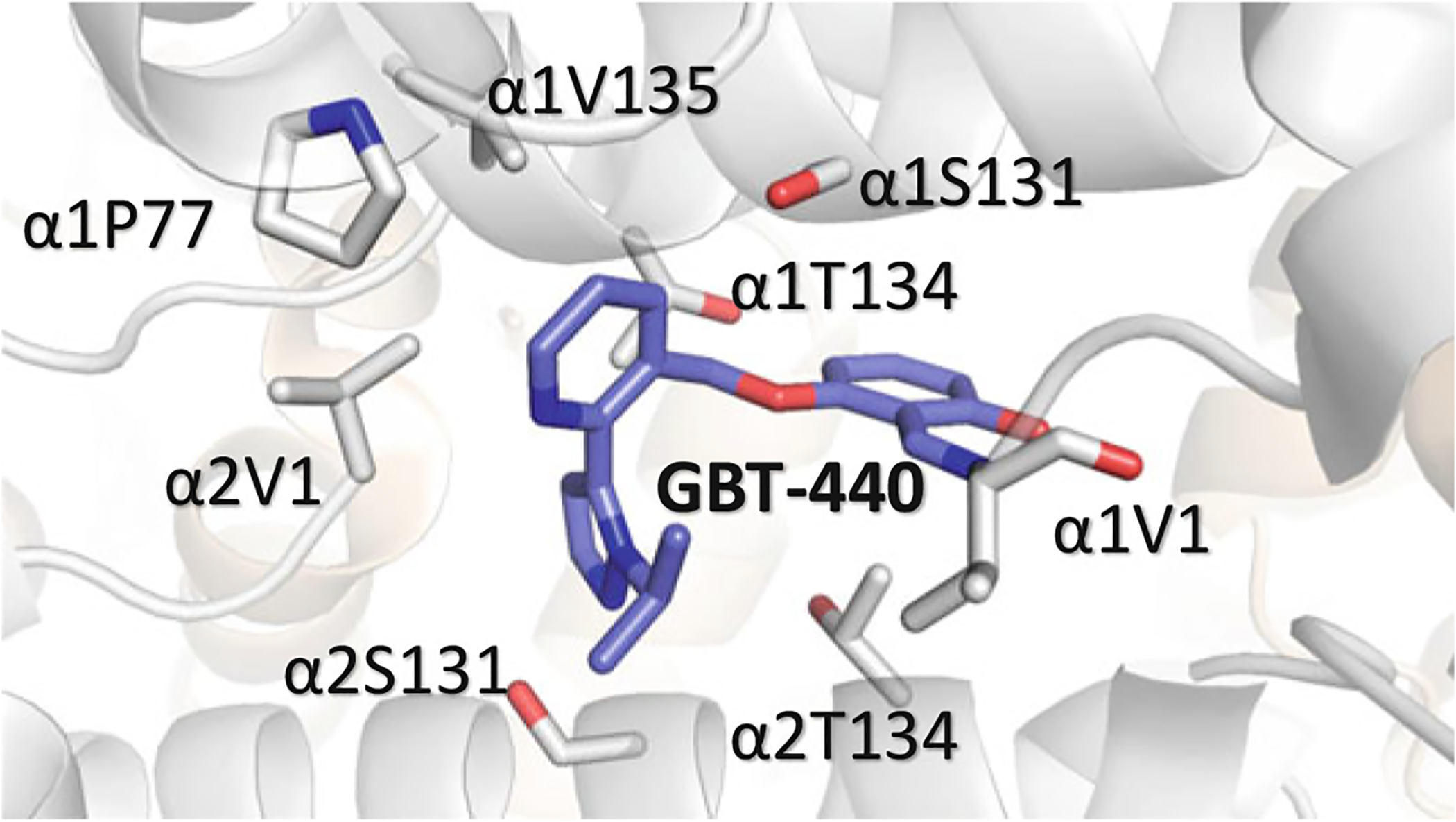
Binding of GBT-440 (purple) at the α-cleft Hb. The α-subunits are colored in gray and the β-subunits are colored in tan. Unlike other aldehyde effectors only one molecule of GBT-440 binds per one Hb molecule
High-throughput screening of a small molecule library has identified thiol-containing effectors of hemoglobin, TD-1, and TD-3 (Fig. 14.18) (Nakagawa et al. 2014, 2018). These compounds, unlike the aromatic aldehydes, act via covalent disulfide bond formation with βCys93 of deoxygenated Hb that inhibits a T state salt-bridge formation between βAsp94 and βHis146 shifting the equilibrium to the relaxed state and increasing Hb affinity for oxygen (Fig. 14.19a) (Nakagawa et al. 2014, 2018). The compounds also bind to the R and/or R3 structures that stabilize the relaxed state, contributing to the increase in Hb oxygen affinity (Fig. 14.19b) (Nakagawa et al. 2014, 2018).
Fig. 14.18.
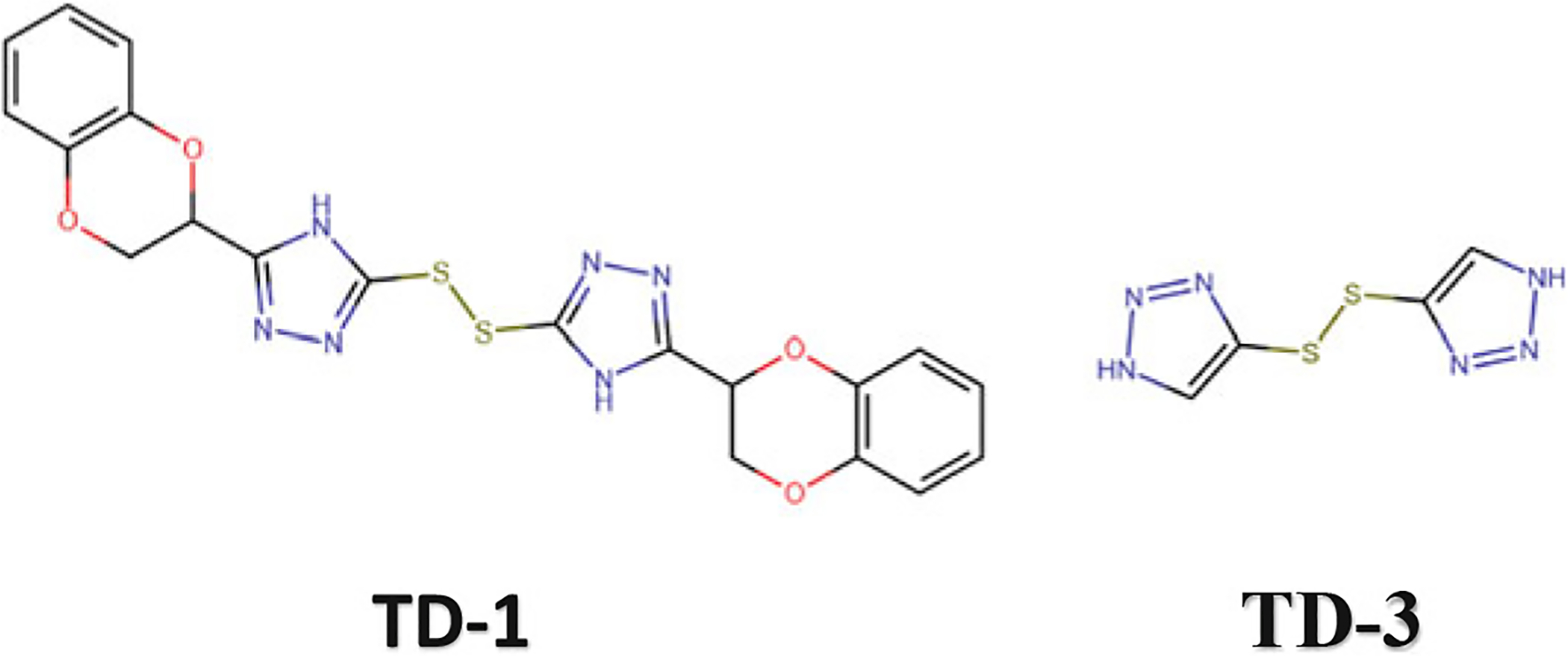
Chemical structures of antisickling thiol-containing agents
Fig. 14.19.
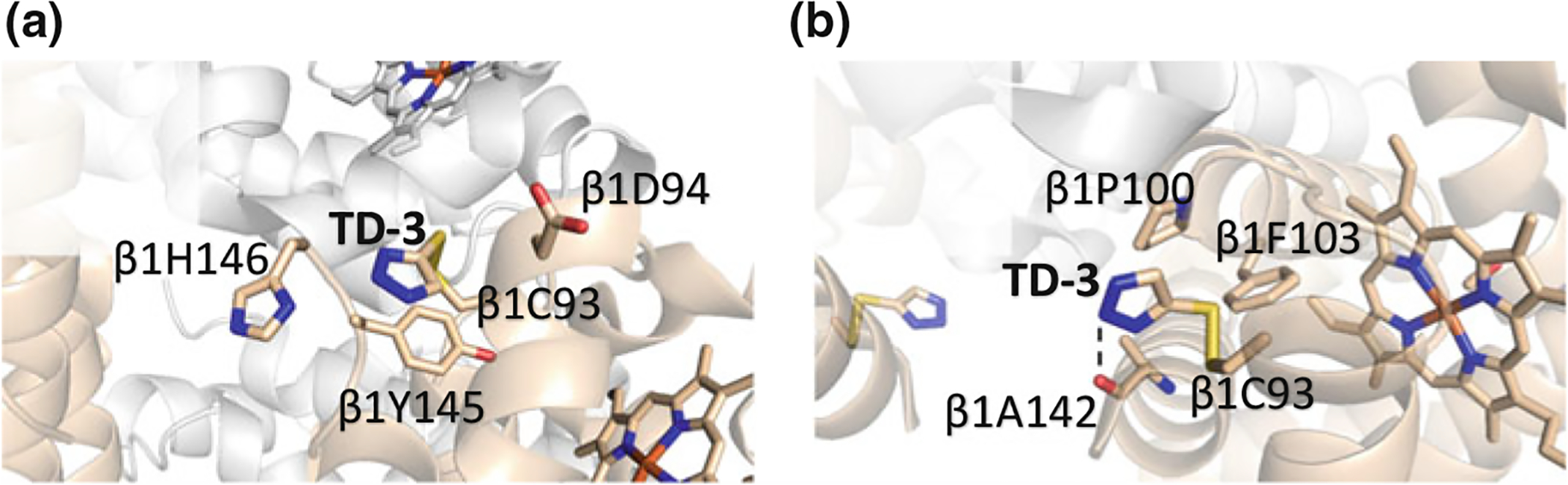
Crystal structure of TD-3 in complex with T and R structures, where TD-3 forms disulfide bond with βCys93 sulfur atom. Hb α and β subunits are shown as grey and tan, respectively. a The binding of TD-3 with βCys93 in deoxygenated Hb leads to disruption of the T state stabilizing salt-bridge interaction between βAsp94 and βHis146. b The binding of TD-3 in CO-liganded Hb (R structure) prevents possible interaction between βAsp94 and βHis146 that is required to shift the allosteric transition to the T state
Allosteric Effectors that Target the Same Site but Induce Opposite Equilibrium Shifts
Above, we described several aromatic aldehydes that increase the oxygen affinity of Hb by forming Schiff-base interaction with Hb αVal1 nitrogen at the α-cleft. However, not all αVal1 Schiff-base adducts with aromatic aldehydes result in increasing Hb affinity for oxygen. Abraham’s group tested several mono-aldehyde-acid effectors of Hb, such as 5-FSA, 2-BF, and 2-PEF (Fig. 14.20), that were hypothesized to produce a high-O2 affinity Hb (Abraham et al. 1995). Unexpectedly, these compounds showed the opposite effect by reducing Hb affinity for oxygen, despite forming Schiff-base interaction with the Hb αVal1 nitrogen. Structural analysis showed that because of the carboxylate substituent on the benzene ring (relative to the aldehyde), these compounds preferentially bind to the α-cleft of deoxygenated Hb, where they form Schiff-base interaction with αVal1 nitrogen, as well as an inter-subunit salt-bridge interaction between the carboxylate and the guanidinium group of the opposite αArg141 of Hb, providing additional constraints to the T state (Fig. 14.21) (Abraham et al. 1995). The R2 structure does not have αArg141 at the correct position to make such salt-bridge interaction with the carboxylate of the compounds, while R, RR2 and R3 structures sterically preclude binding at their respective α-clefts. Removal of the carboxylate from the compounds abrogates the αArg141 interaction in deoxygenated T structure, resulting in these molecules preferentially binding to liganded Hb in the form of R2 structure as observed with vanillin and several of its analogs (Abdulmalik et al. 2011; Pagare et al. 2018; Deshpande et al. 2018). Nonetheless, these non-carboxylate aromatic aldehydes still bind to deoxygenated Hb, albeit significantly weaker because of lack of strong inter-subunit interactions (Safo et al. 2004; Abdulmalik et al. 2011). Moreover, binding to deoxygenated Hb does not appear to contribute to the stability of the T state but rather further destabilizes it by replacing a T state stabilizing chloride ion in the central water cavity (Safo et al. 2004).
Fig. 14.20.
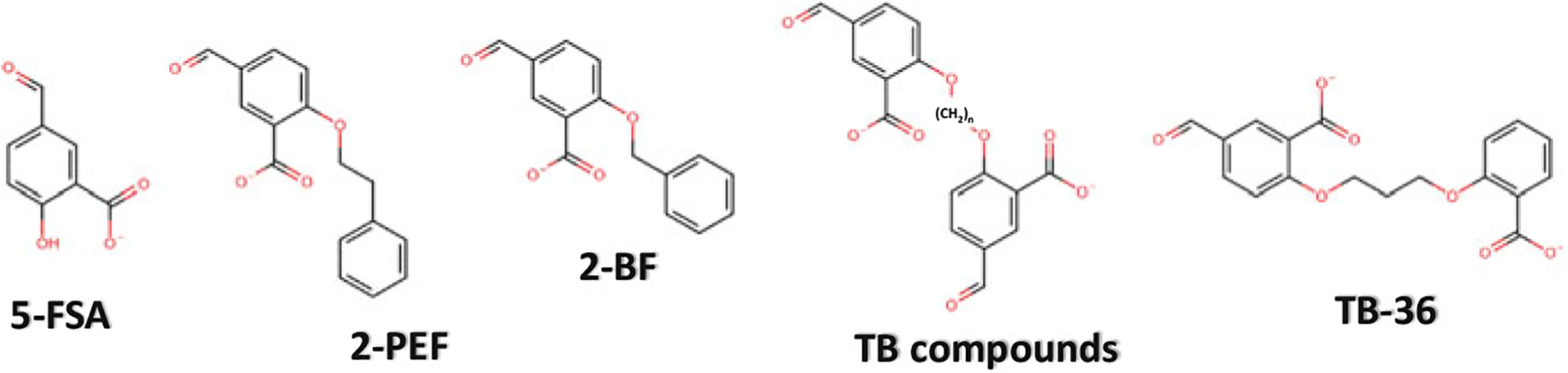
Chemical structures of right-shifting (low-O2 affinity) aromatic aldehydes
Fig. 14.21.
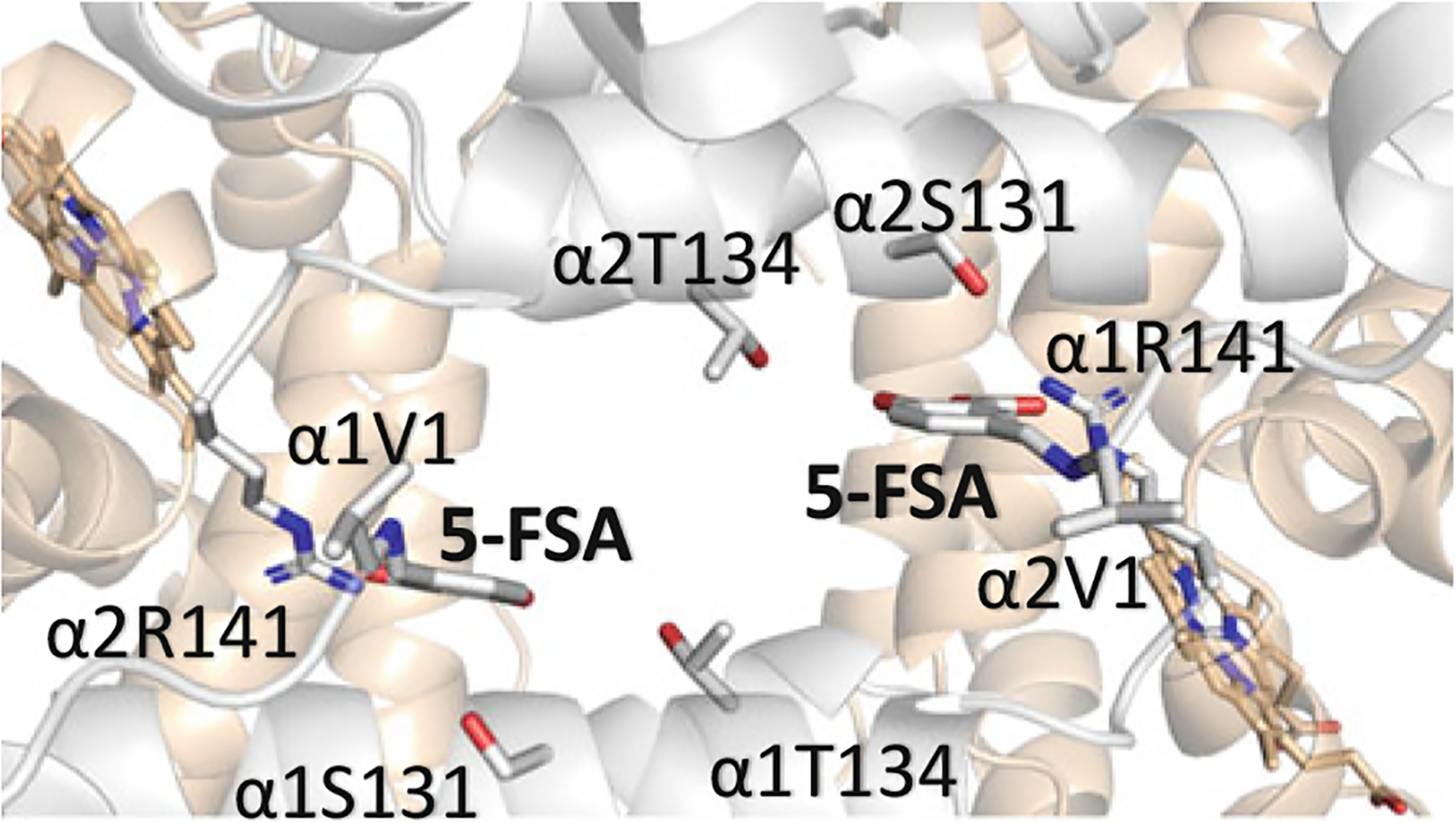
(A) Crystal structures of deoxygenated Hb in complex with the mono-aldehyde-acid molecule of 5-FSA
The crystallographic result with the mono-aldehyde-acid compounds prompted Abraham et al. to design di-aldehyde-acid derivatives by adding a second aromatic aldehyde at the para position of the first aldehyde, such as TB compounds (Fig. 14.20), which was expected to form a second Schiff-base adduct with αLys99 of the opposite α-subunit to further constrain the T structure (Boyiri et al. 1995). Different classes of these compounds were synthesized that showed several-fold increase in reducing Hb affinity for oxygen compared to the mono-aldehyde analogs (Boyiri et al. 1995). Crystallographic studies with the di-aldehyde compounds showed a pair of compounds bound to deoxygenated Hb as designed (Boyiri et al. 1995). The first aldehyde made a Schiff-base interaction with α1Val1 nitrogen, while the second aldehyde made an inter-subunit Schiff-base interaction with the amine of the α2Lys99. The meta-carboxylate moiety engaged in an inter-subunit salt-bridge interaction with α2Arg141 similar to the parent mono-aldehdye-acid compounds. Overall, these interactions provided additional stabilization to the T state, further reducing the oxygen affinity of Hb. It is worth noting that effectors with the shortest bridges exhibited the most potent effect, suggesting that, the tighter the two α-subunits are held together, the higher the degree of constraint on the T-structure. Such covalent cross-linkers of Hb subunits, with further optimization of cell permeability, have the potential for being used as cell-free Hb-based blood substitutes.
Concluding Remarks
Hb is a fascinating molecule that never ceases to amaze scientists. It helped to shape our understanding of one of the most complex theories in molecular biology, which is allostery, although it is yet to be fully unravelled. The remarkable advances in structural biology techniques, such as cryo-electron microscopy and time-resolved spectroscopic methods will, hopefully, provide more glimpses into the phenomenon of Hb allostery. In this chapter, we provided an overview of the molecular structure of Hb, relating to its primary function of oxygen transport. We discussed the concept of allostery and the different models that were proposed over the years to explain this phenomenon. It is clear that the oxygen carrying function of Hb involves an ensemble of states, although their specific functions, especially the relaxed states are yet to be fully understood. Finally, we provided an overview of the molecular mechanisms of how different Hb variants, as well as effectors modulate Hb oxygen binding property, and the effort being made to harness the latter for therapeutics. These allosteric effectors, depending on their mode of interactions with T or relaxed Hb, can decrease or increase Hb affinity for oxygen, the later useful for treating sickle cell disease and the former for hypoxia- and ischemia-induced conditions. Of interest is that the α-cleft of T and R2 structures of Hb act as sinks for aromatic aldehydes, and preferential binding to the T or R2 α-cleft can have profound effect on Hb oxygen affinity, given rise to a new understanding that an allosteric effector can bind to the same site but produce opposite allosteric effects. These studies have uncovered crucial roles played by several Hb residues, such as αVal1, αLys99, αArg141, G helix, F helix residues in modulating Hb allostery.
Footnotes
Conflict-of-Interest Disclosure Virginia Commonwealth University and Martin K. Safo have patents related to several aromatic aldehydes mentioned in the chapter.
Contributor Information
Mostafa H. Ahmed, Department of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University, Richmond, VA 23219, USA
Mohini S. Ghatge, Department of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University, Richmond, VA 23219, USA Institute for Structural Biology, Drug Discovery and Development, Virginia Commonwealth University, Richmond, VA 23219, USA.
Martin K. Safo, Department of Medicinal Chemistry, School of Pharmacy, Virginia Commonwealth University, Richmond, VA 23219, USA Institute for Structural Biology, Drug Discovery and Development, Virginia Commonwealth University, Richmond, VA 23219, USA.
References
- Abdulmalik O, Safo MK, Lerner NB et al. (2004) Characterization of hemoglobin bassett (alpha94Asp → Ala), a variant with very low oxygen affinity. Am J Hematol 77:268–276. 10.1002/ajh.20184 [DOI] [PubMed] [Google Scholar]
- Abdulmalik O, Safo MK, Chen Q et al. (2005) 5-hydroxymethyl-2-furfural modifies intracellular sickle haemoglobin and inhibits sickling of red blood cells. Br J Haematol 128:552–561. 10.1111/j.1365-2141.2004.05332.x [DOI] [PubMed] [Google Scholar]
- Abdulmalik O, Ghatge MS, Musayev FN et al. (2011) Crystallographic analysis of human hemoglobin elucidates the structural basis of the potent and dual antisickling activity of pyridyl derivatives of vanillin Corrigendum. Acta Crystallogr D Biol Crystallogr 67:1076 10.1107/S0907444911045860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham DJ, Mehanna AS, Williams FL (1982) Design, synthesis, and testing of potential antisickling agents. 1. halogenated benzyloxy and phenoxy acids. J Med Chem 25:1015–1017. 10.1021/jm00351a002 [DOI] [PubMed] [Google Scholar]
- Abraham DJ, Perutz MF, Phillips SE (1983a) Physiological and x-ray studies of potential antisickling agents. Proc Natl Acad Sci USA 80:324–328. 10.1073/pnas.80.2.324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham DJ, Perutz MF, Phillips SE (1983b) Physiological and x-ray studies of potential antisickling agents. PNAS 80:324–328. 10.1073/pnas.80.2.324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham DJ, Kennedy PE, Mehanna AS et al. (1984) Design, synthesis, and testing of potential antisickling agents. 4. structure-activity relationships of benzyloxy and phenoxy acids. J Med Chem 27:967–978. 10.1021/jm00374a006 [DOI] [PubMed] [Google Scholar]
- Abraham DJ, Mehanna AS, Wireko FC et al. (1991) Vanillin, a potential agent for the treatment of sickle cell anemia. Blood 77:1334–1341 [PubMed] [Google Scholar]
- Abraham DJ, Peascoe RA, Randad RS, Panikker J (1992a) X-ray diffraction study of di and tetraligated T-state hemoglobin from high salt crystals. J Mol Biol 227:480–492. 10.1016/0022-2836(92)90902-V [DOI] [PubMed] [Google Scholar]
- Abraham DJ, Wireko FC, Randad RS et al. (1992b) Allosteric modifiers of hemoglobin: 2-[4-[[(3,5-disubstituted anilino)carbonyl]methyl]phenoxy]-2-methylpropionic acid derivatives that lower the oxygen affinity of hemoglobin in red cell suspensions, in whole blood, and in vivo in rats. Biochemistry 31:9141–9149. 10.1021/bi00153a005 [DOI] [PubMed] [Google Scholar]
- Abraham DJ, Safo MK, Boyiri T et al. (1995) How allosteric effectors can bind to the same protein residue and produce opposite shifts in the allosteric equilibrium. Biochemistry 34:15006–15020. 10.1021/bi00046a007 [DOI] [PubMed] [Google Scholar]
- Akinsheye I, Klings ES (2010) Sickle cell anemia and vascular dysfunction: the nitric oxide connection. J Cell Physiol 224:620–625. 10.1002/jcp.22195 [DOI] [PubMed] [Google Scholar]
- Aliyu ZY, Gordeuk V, Sachdev V et al. (2008) Prevalence and risk factors for pulmonary artery systolic hypertension among sickle cell disease patients in Nigeria. Am J Hematol 83:485–490. 10.1002/ajh.21162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnone A (1972) X-ray diffraction study of binding of 2,3-diphosphoglycerate to human deoxyhaemoglobin. Nature 237:146–149. 10.1038/237146a0 [DOI] [PubMed] [Google Scholar]
- Arous N, Braconnier F, Thillet J et al. (1981) Hemoglobin Saint Mandé beta 102 (G4) asn replaced by tyr: a new low oxygen affinity variant. FEBS Lett 126:114–116. 10.1016/0014-5793(81)81046-0 [DOI] [PubMed] [Google Scholar]
- Baldwin J, Chothia C (1979) Haemoglobin: The structural changes related to ligand binding and its allosteric mechanism. J Mol Biol 129:175–220. 10.1016/0022-2836(79)90277-8 [DOI] [PubMed] [Google Scholar]
- Barrick D, Ho NT, Simplaceanu V et al. (1997) A test of the role of the proximal histidines in the Perutz model for cooperativity in haemoglobin. Nat Struct Biol 4:78–83 [DOI] [PubMed] [Google Scholar]
- Beddell CR, Goodford PJ, Kneen G et al. (1984) Substituted benzaldehydes designed to increase the oxygen affinity of human haemoglobin and inhibit the sickling of sickle erythrocytes. Br J Pharmacol 82:397–407. 10.1111/j.1476-5381.1984.tb10775.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcher JD, Bryant CJ, Nguyen J et al. (2003) Transgenic sickle mice have vascular inflammation. Blood 101:3953–3959. 10.1182/blood-2002-10-3313 [DOI] [PubMed] [Google Scholar]
- Benesch R, Benesch RE (1967) The effect of organic phosphates from the human erythrocyte on the allosteric properties of hemoglobin. Biochem Biophys Res Commun 26:162–167. 10.1016/0006-291X(67)90228-8 [DOI] [PubMed] [Google Scholar]
- Benesch RE, Kwong S, Edalji R, Benesch R (1979) Alpha chain mutations with opposite effects on the gelation of hemoglobin S. J Biol Chem 254:8169–8172 [PubMed] [Google Scholar]
- Berenbrink M (2006) Evolution of vertebrate haemoglobins: Histidine side chains, specific buffer value and Bohr effect. Respir Physiol Neurobiol 154:165–184. 10.1016/j.resp.2006.01.002 [DOI] [PubMed] [Google Scholar]
- Bettati S, Mozzarelli A (1997) T state hemoglobin binds oxygen noncooperatively with allosteric effects of protons, inositol hexaphosphate, and chloride. J Biol Chem 272:32050–32055. 10.1074/jbc.272.51.32050 [DOI] [PubMed] [Google Scholar]
- Bettati S, Mozzarelli A, Perutz MF (1998) Allosteric mechanism of haemoglobin: rupture of salt-bridges raises the oxygen affinity of the T-structure. J Mol Biol 281:581–585. 10.1006/jmbi.1998.1983 [DOI] [PubMed] [Google Scholar]
- Biolo A, Greferath R, Siwik DA et al. (2009) Enhanced exercise capacity in mice with severe heart failure treated with an allosteric effector of hemoglobin, myo-inositol trispyrophosphate. PNAS 106:1926–1929. 10.1073/pnas.0812381106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukou I, Soman J, Olson JS (2011) Blocking the gate to ligand entry in human hemoglobin. J Biol Chem 286:10515–10529. 10.1074/jbc.M110.176271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bissé E, Schaeffer-Reiss C, Van Dorsselaer A et al. (2017) Hemoglobin Kirklareli (α H58L), a new variant associated with iron deficiency and increased CO binding. J Biol Chem 292:2542–2555. 10.1074/jbc.M116.764274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohr C, Hasselbalch K, Krogh A (1904) Ueber einen in biologischer Beziehung wichtigen Einfluss, den die Kohlensäurespannung des Blutes auf dessen Sauerstoffbindung übt1. Skandinavisches Archiv Für Physiologie 16:402–412. 10.1111/j.1748-1716.1904.tb01382.x [DOI] [Google Scholar]
- Bonaventura J, Riggs A (1968) Hemoglobin Kansas, a human hemoglobin with a neutral amino acid substitution and an abnormal oxygen equilibrium. J Biol Chem 243:980–991 [PubMed] [Google Scholar]
- Bonaventura C, Arumugam M, Cashon R et al. (1994) Chloride masks effects of opposing positive charges in Hb A and Hb Hinsdale (β139 Asn → Lys) that can modulate cooperativity as well as oxygen affinity. J Mol Biol 239:561–568. 10.1006/jmbi.1994.1395 [DOI] [PubMed] [Google Scholar]
- Borg I, Valentino M, Fiorini A, Felice AE (1997) Hb Setif [alpha 94(G1)Asp → Tyr] in Malta. Hemoglobin 21:91–96 [DOI] [PubMed] [Google Scholar]
- Botha MC, Beale D, Isaacs WA, Lehmann H (1966) Hemoglobin J Cape Town-alpha-2 92 arginine replaced by glutamine beta-2. Nature 212:792–795. 10.1038/212792a0 [DOI] [PubMed] [Google Scholar]
- Boyiri T, Safo MK, Danso-Danquah RE et al. (1995) Bisaldehyde allosteric effectors as molecular ratchets and probes. Biochemistry 34:15021–15036. 10.1021/bi00046a008 [DOI] [PubMed] [Google Scholar]
- Brunori M, Coletta M, Di Cera E (1986) A cooperative model for ligand binding to biological macromolecules as applied to oxygen carriers. Biophys Chem 23:215–222. 10.1016/0301-4622(86)85006-2 [DOI] [PubMed] [Google Scholar]
- Bunn HF (1997) Pathogenesis and treatment of sickle cell disease. N Engl J Med 337:762–769. 10.1056/NEJM199709113371107 [DOI] [PubMed] [Google Scholar]
- Bunn HF, Bradley TB, Davis WE et al. (1972) Structural and functional studies on hemoglobin Bethesda (alpha2beta2 145His), a varient associated with compensatory erythrocytosis. J Clin Invest 51:2299–2309. 10.1172/JCI107040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch MR, Ho C (1990) Effects of anions on the molecular basis of the Bohr effect of hemoglobin. Biophys Chem 37:313–322. 10.1016/0301-4622(90)88031-M [DOI] [PubMed] [Google Scholar]
- Carrell RW, Lehmann H, Hutchison HE (1966) Haemoglobin Köln (beta-98 valine–methionine): an unstable protein causing inclusion-body anaemia. Nature 210:915–916. 10.1038/210915a0 [DOI] [PubMed] [Google Scholar]
- Charache S, Weatherall DJ, Clegg JB (1966) Polycythemia associated with a hemoglobinopathy. J Clin Invest 45:813–822. 10.1172/JCI105397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Lalezari I, Nagel RL, Hirsch RE (2005) Liganded hemoglobin structural perturbations by the allosteric effector L35. Biophys J 88:2057–2067. 10.1529/biophysj.104.046136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Como PF, Wylie BR, Trent RJ et al. (1991) A new unstable and low oxygen affinity hemoglobin variant: Hb Stanmore [beta 111(G13)Val–Ala]. Hemoglobin 15:53–65 [DOI] [PubMed] [Google Scholar]
- Connor J, Pak CC, Schroit AJ (1994) Exposure of phosphatidylserine in the outer leaflet of human red blood cells. relationship to cell density, cell age, and clearance by mononuclear cells. J Biol Chem 269:2399–2404 [PubMed] [Google Scholar]
- Czerminski R, Elber R (1991) Computational studies of ligand diffusion in globins: I. Leghemoglobin. Proteins: Struct Funct Bioinform 10:70–80. 10.1002/prot.340100107 [DOI] [PubMed] [Google Scholar]
- Dacie JV, Shinton NK, Gaffney PJ, Lehmann H (1967) Haemoglobin Hammersmith (beta-42 (CDI) Phe replaced by ser). Nature 216:663–665. 10.1038/216663a0 [DOI] [PubMed] [Google Scholar]
- De Franceschi L (2009) Pathophisiology of sickle cell disease and new drugs for the treatment. Mediterr J Hematol Infect Dis 1:e2009024 10.4084/MJHID.2009.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande TM, Pagare PP, Ghatge MS et al. (2018) Rational modification of vanillin derivatives to stereospecifically destabilize sickle hemoglobin polymer formation. Acta Crystallogr D Struct Biol 74:956–964. 10.1107/S2059798318009919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinçol G, Dinçol K, Erdem S et al. (1994) Hb Capa or alpha (2)94(G1)Asp → Gly beta 2, a mildly unstable variant with an A → G (GAC → GGC) mutation in codon 94 of the alpha 1-globin gene. Hemoglobin 18:57–60 [DOI] [PubMed] [Google Scholar]
- Doyle ML, Lew G, Turner GJ et al. (1992) Regulation of oxygen affinity by quaternary enhancement: does hemoglobin ypsilanti represent an allosteric intermediate? Proteins: Struct Funct Bioinform 14:351–362. 10.1002/prot.340140304 [DOI] [PubMed] [Google Scholar]
- Eaton WA, Hofrichter J (1990) Sickle cell hemoglobin polymerization. Adv Protein Chem 40:63–279 [DOI] [PubMed] [Google Scholar]
- Efremov GD, Stojmirovic E, Lam HL et al. (1978) HB Beth Israel (beta 102 [G4] Asn replaced by Ser) observed in a Yugoslavian teenager. Hemoglobin 2:75–77. 10.3109/03630267808999192 [DOI] [PubMed] [Google Scholar]
- Elber R (2010) Ligand diffusion in globins: simulations versus experiment. Curr Opin Struct Biol 20:162–167. 10.1016/j.sbi.2010.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J-S, Zheng Y, Choy W-Y et al. (2013) Solution structure and dynamics of human hemoglobin in the carbonmonoxy form. Biochemistry 52:5809–5820. 10.1021/bi4005683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fermi G (1975) Three-dimensional fourier synthesis of human deoxyhaemoglobin at 2–5 a resolution: refinement of the atomic model. J Mol Biol 97:237–256. 10.1016/s0022-2836(75)80037-4 [DOI] [PubMed] [Google Scholar]
- Fernandez EJ, Abad-Zapatero C, Olsen KW (2000) Crystal structure of Lysβ182-Lysβ282 crosslinked hemoglobin: A possible allosteric intermediate 11 Edited by K. Nagai. J Mol Biol 296:1245–1256. 10.1006/jmbi.2000.3525 [DOI] [PubMed] [Google Scholar]
- Fronticelli C, Pechik I, Brinigar WS et al. (1994) Chloride ion independence of the Bohr effect in a mutant human hemoglobin beta (V1M + H2deleted). J Biol Chem 269:23965–23969 [PubMed] [Google Scholar]
- Gell DA (2018) Structure and function of haemoglobins. Blood Cells Mol Dis 70:13–42. 10.1016/j.bcmd.2017.10.006 [DOI] [PubMed] [Google Scholar]
- Ghatge MS, Ahmed MH, Omar ASM et al. (2016) Crystal structure of carbonmonoxy sickle hemoglobin in R-state conformation. J Struct Biol 194:446–450. 10.1016/j.jsb.2016.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein SR, Liu C, Safo MK et al. (2018) Design, synthesis, and biological evaluation of allosteric effectors that enhance CO release from carboxyhemoglobin. ACS Med Chem Lett 9:714–718. 10.1021/acsmedchemlett.8b00166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Q, Simplaceanu V, Lukin JA et al. (2006) Quaternary structure of carbonmonoxyhemoglobins in solution: structural changes induced by the allosteric effector inositol hexaphosphate. Biochemistry 45:5140–5148. 10.1021/bi052424h [DOI] [PubMed] [Google Scholar]
- Grasso JA, Sullivan AL, Sullivan LW (1975) Ultrastructural studies of the bone marrow in sickle cell anaemia. II. the morphology of erythropoietic cells and their response to deoxygenation in vitro. Br J Haematol 31:381–389. 10.1111/j.1365-2141.1975.tb00869.x [DOI] [PubMed] [Google Scholar]
- Gupta RK, Benovic JL, Rose ZB (1979) Location of the allosteric site for 2,3-bisphosphoglycerate on human oxy- and deoxyhemoglobin as observed by magnetic resonance spectroscopy. J Biol Chem 254:8250–8255 [PubMed] [Google Scholar]
- Hänel P, Andréani P, Gräler MH (2007) Erythrocytes store and release sphingosine 1-phosphate in blood. FASEB J 21:1202–1209. 10.1096/fj.06-7433com [DOI] [PubMed] [Google Scholar]
- Hardison R, Chao KM, Schwartz S et al. (1994) Globin gene server: a prototype E-mail database server featuring extensive multiple alignments and data compilation for electronic genetic analysis. Genomics 21:344–353. 10.1006/geno.1994.1275 [DOI] [PubMed] [Google Scholar]
- Hardison RC, Chui DH, Riemer C et al. (2001) Databases of human hemoglobin variants and other resources at the globin gene server. Hemoglobin 25:183–193 [DOI] [PubMed] [Google Scholar]
- Harrington DJ, Adachi K, Royer WE (1997) The high resolution crystal structure of deoxyhemoglobin S. J Mol Biol 272:398–407. 10.1006/jmbi.1997.1253 [DOI] [PubMed] [Google Scholar]
- He Z, Russell JE (2004a) Effect of zeta-globin substitution on the O2-transport properties of Hb S in vitro and in vivo. Biochem Biophys Res Commun 325:1376–1382. 10.1016/j.bbrc.2004.10.180 [DOI] [PubMed] [Google Scholar]
- He Z, Russell JE (2004b) Antisickling effects of an endogenous human alpha-like globin. Nat Med 10:365–367. 10.1038/nm1022 [DOI] [PubMed] [Google Scholar]
- Henry ER, Bettati S, Hofrichter J, Eaton WA (2002) A tertiary two-state allosteric model for hemoglobin. Biophys Chem 98:149–164. 10.1016/S0301-4622(02)00091-1 [DOI] [PubMed] [Google Scholar]
- Huisman THJ, Carver MFH, Efremov G (1996) A syllabus of human hemoglobin variants. The Sickle Cell Anemia Foundation, Augusta, GA, USA [Google Scholar]
- Ito K, Anada Y, Tani M et al. (2007) Lack of sphingosine 1-phosphate-degrading enzymes in erythrocytes. Biochem Biophys Res Commun 357:212–217. 10.1016/j.bbrc.2007.03.123 [DOI] [PubMed] [Google Scholar]
- Janin J, Wodak SJ (1993) The quaternary structure of carbonmonoxy hemoglobin ypsilanti. Proteins: Struct Funct Bioinform 15:1–4. 10.1002/prot.340150102 [DOI] [PubMed] [Google Scholar]
- Jayaraman V, Rodgers KR, Mukerji I, Spiro TG (1995) Hemoglobin allostery: resonance Raman spectroscopy of kinetic intermediates. Science 269:1843–1848. 10.1126/science.7569921 [DOI] [PubMed] [Google Scholar]
- Jenkins JD, Musayev FN, Danso-Danquah R et al. (2009) Structure of relaxed-state human hemoglobin: insight into ligand uptake, transport and release. Acta Cryst D 65:41–48. 10.1107/S0907444908037256 [DOI] [PubMed] [Google Scholar]
- Jensen M, Oski FA, Nathan DG, Bunn HF (1975) Hemoglobin Syracuse (alpha2beta2–143(H21)His leads to Pro), a new high-affinity variant detected by special electrophoretic methods. observations on the auto-oxidation of normal and variant hemoglobins. J Clin Invest 55:469–477. 10.1172/JCI107953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RT, Osgood EE, Brimhall B, Koler RD (1967) Hemoglobin Yakina. I. clinical and biochemical studies. J Clin Invest 46:1840–1847. 10.1172/JCI105674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorge SE, Bringas M, Petruk AA et al. (2018) Understanding the molecular basis of the high oxygen affinity variant human hemoglobin Coimbra. Arch Biochem Biophys 637:73–78. 10.1016/j.abb.2017.11.010 [DOI] [PubMed] [Google Scholar]
- Kamel K, el-Najjar A, Webber BB et al. (1985) Hb Doha or alpha 2 beta 2[X-N-Met-1(NA1)Val–Glu]; a new beta-chain abnormal hemoglobin observed in a Qatari female. Biochim Biophys Acta 831:257–260. 10.1016/0167-4838(85)90043-3 [DOI] [PubMed] [Google Scholar]
- Kato GJ, Lawrence MP, Mendelsohn LG et al. (2013) Phase 1 clinical trial of the candidate anti-sickling agent Aes-103 In adults with sickle cell anemia. Blood 122:1009–1009 [Google Scholar]
- Kavanaugh JS, Rogers PH, Case DA, Arnone A (1992) High-resolution x-ray study of deoxyhemoglobin Rothschild 37.beta. Trp.fwdarw. Arg: a mutation that creates an intersubunit chloride-binding site. Biochemistry 31:4111–4121. 10.1021/bi00131a030 [DOI] [PubMed] [Google Scholar]
- Kavanaugh JS, Weydert JA, Rogers PH et al. (2001) Site-directed mutations of human hemoglobin at residue 35beta: a residue at the intersection of the alpha1beta1, alpha1beta2, and alpha1alpha2 interfaces. Protein Sci 10:1847–1855. 10.1110/ps.16401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh JS, Rogers PH, Arnone A (2005) Crystallographic evidence for a new ensemble of ligand-induced allosteric transitions in hemoglobin: the T-to-T(high) quaternary transitions. Biochemistry 44:6101–6121. 10.1021/bi047813a [DOI] [PubMed] [Google Scholar]
- Khandelwal SR, Randad RS, Lin PS et al. (1993) Enhanced oxygenation in vivo by allosteric inhibitors of hemoglobin saturation. Am J Physiol 265:H1450–H1453. 10.1152/ajpheart.1993.265.4.H1450 [DOI] [PubMed] [Google Scholar]
- Kilmartin JV, Imai K, Jones RT et al. (1978) Role of Bohr group salt bridges in cooperativity in hemoglobin. Biochim Biophys Acta 534:15–25. 10.1016/0005-2795(78)90471-3 [DOI] [PubMed] [Google Scholar]
- King MA, Wiltshire BG, Lehmann H, Morimoto H (1972) An unstable haemoglobin with reduced oxygen affinity: haemoglobin Peterborough, 3 (GI3) Valine lead to Phenylalanine, its interaction with normal haemoglobin and with haemoglobin Lepore. Br J Haematol 22:125–134. 10.1111/j.1365-2141.1972.tb08794.x [DOI] [PubMed] [Google Scholar]
- Kister J, Kiger L, Francina A et al. (1995) Hemoglobin Roanne [alpha 94(G1) Asp → Glu]: a variant of the alpha 1 beta 2 interface with an unexpected high oxygen affinity. Biochim Biophys Acta 1246:34–38. 10.1016/0167-4838(94)00190-r [DOI] [PubMed] [Google Scholar]
- Kleinberg L, Grossman SA, Piantadosi S et al. (1999) Phase I trial to determine the safety, pharmacodynamics, and pharmacokinetics of RSR13, a novel radioenhancer, in newly diagnosed glioblastoma multiforme. J Clin Oncol 17:2593–2603. 10.1200/JCO.1999.17.8.2593 [DOI] [PubMed] [Google Scholar]
- Koshland DE, Némethy G, Filmer D (1966) Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 5:365–385. 10.1021/bi00865a047 [DOI] [PubMed] [Google Scholar]
- Kosmachevskaya OV, Topunov AF (2018) Alternate and additional functions of erythrocyte hemoglobin. Biochem Mosc 83:1575–1593. 10.1134/S0006297918120155 [DOI] [PubMed] [Google Scholar]
- Laberge M, Kövesi I, Yonetani T, Fidy J (2005) R-state hemoglobin bound to heterotropic effectors: models of the DPG, IHP and RSR13 binding sites. FEBS Lett 579:627–632. 10.1016/j.febslet.2004.12.033 [DOI] [PubMed] [Google Scholar]
- Ladner RC, Heidner EJ, Perutz MF (1977) The structure of horse methaemoglobin at 2–0 A resolution. J Mol Biol 114:385–414. 10.1016/0022-2836(77)90256-x [DOI] [PubMed] [Google Scholar]
- Lalezari I, Rahbar S, Lalezari P et al. (1988) LR16, a compound with potent effects on the oxygen affinity of hemoglobin, on blood cholesterol, and on low density lipoprotein. PNAS 85:6117–6121. 10.1073/pnas.85.16.6117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalezari I, Lalezari P, Poyart C et al. (1990) New effectors of human hemoglobin: structure and function. Biochemistry 29:1515–1523. 10.1021/bi00458a024 [DOI] [PubMed] [Google Scholar]
- Lokich JJ, Moloney WC, Bunn HF et al. (1973) Hemoglobin brigham (alpha2Abeta2100 Pro–Leu). Hemoglobin variant associated with familial erythrocytosis. J Clin Invest 52:2060–2067. 10.1172/JCI107390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukin JA, Ho C (2004) The structure–function relationship of hemoglobin in solution at atomic resolution. Chem Rev 104:1219–1230. 10.1021/cr940325w [DOI] [PubMed] [Google Scholar]
- Lukin JA, Kontaxis G, Simplaceanu V et al. (2003) Quaternary structure of hemoglobin in solution. PNAS 100:517–520. 10.1073/pnas.232715799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukin JA, Kontaxis G, Simplaceanu V et al. (2004) Backbone resonance assignments of human adult hemoglobin in the carbonmonoxy form. J Biomol NMR 28:203–204. 10.1023/B:JNMR.0000013816.64039.6f [DOI] [PubMed] [Google Scholar]
- Mairbäurl H, Weber RE (2012) Oxygen transport by hemoglobin. Compr Physiol 2:1463–1489. 10.1002/cphy.c080113 [DOI] [PubMed] [Google Scholar]
- Makowski L, Bardhan J, Gore D et al. (2011) WAXS studies of the structural diversity of hemoglobin in solution. J Mol Biol 408:909–921. 10.1016/j.jmb.2011.02.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marden MC, Bohn B, Kister J, Poyart C (1990) Effectors of hemoglobin. separation of allosteric and affinity factors. Biophys J 57:397–403. 10.1016/S0006-3495(90)82556-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marengo-Rowe AJ (2006) Structure-function relations of human hemoglobins. Proc (Bayl Univ Med Cent) 19:239–245. 10.1080/08998280.2006.11928171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehanna AS, Abraham DJ (1990) Comparison of crystal and solution hemoglobin binding of selected antigelling agents and allosteric modifiers. Biochemistry 29:3944–3952. 10.1021/bi00468a022 [DOI] [PubMed] [Google Scholar]
- Metcalf B, Chuang C, Dufu K et al. (2017) Discovery of GBT440, an orally bioavailable R-state stabilizer of sickle cell hemoglobin. ACS Med Chem Lett 8:321–326. 10.1021/acsmedchemlett.6b00491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux JP (1965) On the nature of allosteric transitions: a plausible model. J Mol Biol 12:88–118. 10.1016/s0022-2836(65)80285-6 [DOI] [PubMed] [Google Scholar]
- Mozzarelli A, Rivetti C, Rossi GL et al. (1991) Crystals of haemoglobin with the T quaternary structure bind oxygen noncooperatively with no Bohr effect. Nature 351:416–419. 10.1038/351416a0 [DOI] [PubMed] [Google Scholar]
- Mueser TC, Rogers PH, Arnone A (2000) Interface sliding as illustrated by the multiple quaternary structures of liganded hemoglobin. Biochemistry 39:15353–15364. 10.1021/bi0012944 [DOI] [PubMed] [Google Scholar]
- Muirhead H, Perutz MF (1963) Structure of hæemoglobin: a three-dimensional Fourier synthesis of reduced human haemoglobin at 5.5 Å resolution. Nature 199:633–638. 10.1038/199633a0 [DOI] [PubMed] [Google Scholar]
- Murari J, Smith LL, Wilson JB et al. (1977) Some properties of hemoglobin Gun Hill. Hemoglobin 1:267–282 [DOI] [PubMed] [Google Scholar]
- Nagel RL, Johnson J, Bookchin RM et al. (1980) Beta-chain contact sites in the haemoglobin S polymer. Nature 283:832–834. 10.1038/283832a0 [DOI] [PubMed] [Google Scholar]
- Nakagawa A, Lui FE, Wassaf D et al. (2014) Identification of a small molecule that increases hemoglobin oxygen affinity and reduces SS erythrocyte sickling. ACS Chem Biol 9:2318–2325. 10.1021/cb500230b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa A, Ferrari M, Schleifer G et al. (2018) A triazole disulfide compound increases the affinity of hemoglobin for oxygen and reduces the sickling of human sickle cells. Mol Pharm 15:1954–1963. 10.1021/acs.molpharmaceut.8b00108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nienhuis AW (1987) Hemoglobin: Molecular, genetic and clinical aspects: By H. F. Bunn and B.G. Forget. Philadelphia: W. B. Saunders Company. (1986). 690 pp. $99.00. Cell 48:731 10.1016/0092-8674(87)90069-9 [DOI] [Google Scholar]
- Nnamani IN, Joshi GS, Danso-Danquah R et al. (2008) Pyridyl derivatives of benzaldehyde as potential antisickling agents. Chem Biodivers 5:1762–1769. 10.1002/cbdv.200890165 [DOI] [PubMed] [Google Scholar]
- Noble RW, Hui HL, Kwiatkowski LD et al. (2001) Mutational effects at the subunit interfaces of human hemoglobin: evidence for a unique sensitivity of the T quaternary state to changes in the hinge region of the alpha 1 beta 2 interface. Biochemistry 40:12357–12368. 10.1021/bi010988p [DOI] [PubMed] [Google Scholar]
- O’Donnell S, Mandaro R, Schuster TM, Arnone A (1979) X-ray diffraction and solution studies of specifically carbamylated human hemoglobin A. evidence for the location of a proton- and oxygen-linked chloride binding site at valine 1 alpha. J Biol Chem 254:12204–12208 [PubMed] [Google Scholar]
- Oksenberg D, Dufu K, Patel MP et al. (2016) GBT440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half-life in a murine model of sickle cell disease. Br J Haematol 175:141–153. 10.1111/bjh.14214 [DOI] [PubMed] [Google Scholar]
- Omar AM, Mahran MA, Ghatge MS et al. (2015) Identification of a novel class of covalent modifiers of hemoglobin as potential antisickling agents. Org Biomol Chem 13:6353–6370. 10.1039/c5ob00367a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omar AM, Mahran MA, Ghatge MS et al. (2016) Aryloxyalkanoic acids as non-covalent modifiers of the allosteric properties of hemoglobin. Molecules 21 10.3390/molecules21081057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagare PP, Ghatge MS, Musayev FN et al. (2018) Rational design of pyridyl derivatives of vanillin for the treatment of sickle cell disease. Bioorg Med Chem 26:2530–2538. 10.1016/j.bmc.2018.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagel PS, Hettrick DA, Montgomery MW et al. (1998) RSR13, a synthetic allosteric modifier of hemoglobin, enhances recovery of stunned myocardium in dogs. Adv Exp Med Biol 454:527–531. 10.1007/978-1-4615-4863-8_63 [DOI] [PubMed] [Google Scholar]
- Paoli M, Liddington R, Tame J et al. (1996) Crystal structure of T state haemoglobin with oxygen bound at all four haems. J Mol Biol 256:775–792. 10.1006/jmbi.1996.0124 [DOI] [PubMed] [Google Scholar]
- Patwa DC, Abraham DJ, Hung TC (1987) Design, synthesis, and testing of potential antisickling agents. 6. Rheologic studies with active phenoxy and benzyloxy acids. Blood Cells 12:589–601 [PubMed] [Google Scholar]
- Perrella M, Cera ED (1999) CO ligation intermediates and the mechanism of hemoglobin cooperativity. J Biol Chem 274:2605–2608. 10.1074/jbc.274.5.2605 [DOI] [PubMed] [Google Scholar]
- Perutz MF (1972a) Stereochemical mechanism of cooperative effects in haemoglobin. Biochimie 54:587–588. 10.1016/s0300-9084(72)80142-1 [DOI] [PubMed] [Google Scholar]
- Perutz MF (1972b) Nature of haem-haem interaction. Nature 237:495–499. 10.1038/237495a0 [DOI] [PubMed] [Google Scholar]
- Perutz MF (1976) Structure and mechanism of haemoglobin. Br Med Bull 32:195–208. 10.1093/oxfordjournals.bmb.a071363 [DOI] [PubMed] [Google Scholar]
- Perutz MF (1983) Species adaptation in a protein molecule. Mol Biol Evol 1:1–28. 10.1093/oxfordjournals.molbev.a040299 [DOI] [PubMed] [Google Scholar]
- Perutz MF (1989) Myoglobin and haemoglobin: role of distal residues in reactions with haem ligands. Trends Biochem Sci 14:42–44. 10.1016/0968-0004(89)90039-X [DOI] [PubMed] [Google Scholar]
- Perutz MF, Poyart C (1983) Bezafibrate lowers oxygen affinity of haemoglobin. The Lancet 322:881–882. 10.1016/S0140-6736(83)90870-X [DOI] [PubMed] [Google Scholar]
- Perutz MF, Muirhead H, Cox JM, Goaman LC (1968) Three-dimensional Fourier synthesis of horse oxyhaemoglobin at 2.8 A resolution: the atomic model. Nature 219:131–139. 10.1038/219131a0 [DOI] [PubMed] [Google Scholar]
- Perutz MF, Muirhead H, Mazzarella L et al. (1969) Identification of residues responsible for the alkaline Bohr effect in haemoglobin. Nature 222:1240–1243. 10.1038/2221240a0 [DOI] [PubMed] [Google Scholar]
- Perutz MF, Giulio Fermi, Abraham DJ et al. (1986) Hemoglobin as a receptor of drugs and peptides: x-ray studies of the stereochemistry of binding. J Am Chem Soc 108:1064–1078. 10.1021/ja00265a036 [DOI] [Google Scholar]
- Perutz MF, Fermi G, Poyart C et al. (1993) A novel allosteric mechanism in haemoglobin: structure of bovine deoxyhaemoglobin, absence of specific chloride-binding sites and origin of the chloride-linked Bohr effect in bovine and human haemoglobin. J Mol Biol 233:536–545. 10.1006/jmbi.1993.1530 [DOI] [PubMed] [Google Scholar]
- Perutz MF, Shih DT-b., Williamson D (1994) The chloride effect in human haemoglobin. a new kind of allosteric mechanism. J Mol Biol 239:555–560. 10.1006/jmbi.1994.1394 [DOI] [PubMed] [Google Scholar]
- Perutz MF, Wilkinson AJ, Paoli M, Dodson GG (1998) The stereochemical mechanism of the cooperative effects in hemoglobin revisited. Annu Rev Biophys Biomol Struct 27:1–34. 10.1146/annurev.biophys.27.1.1 [DOI] [PubMed] [Google Scholar]
- Phelps Grella M, Danso-Danquah R, Safo MK et al. (2000) Synthesis and structure-activity relationships of chiral allosteric modifiers of hemoglobin. J Med Chem 43:4726–4737. 10.1021/jm000199q [DOI] [PubMed] [Google Scholar]
- Plaseska D, Dimovski AJ, Wilson JB et al. (1991) Hemoglobin Montreal: a new variant with an extended beta chain due to a deletion of Asp, Gly, Leu at positions 73, 74, and 75, and an insertion of Ala, Arg, Cys, Gln at the same location. Blood 77:178–181 [PubMed] [Google Scholar]
- Poillon WN, Kim BC, Welty EV, Walder JA (1986) The effect of 2,3-diphosphoglycerate on the solubility of deoxyhemoglobin S. Arch Biochem Biophys 249:301–305. 10.1016/0003-9861(86)90006-8 [DOI] [PubMed] [Google Scholar]
- Randad RS, Mahran MA, Mehanna AS, Abraham DJ (1991) Allosteric modifiers of hemoglobin. 1. Design, synthesis, testing, and structure-allosteric activity relationship of novel hemoglobin oxygen affinity decreasing agents. J Med Chem 34:752–757. 10.1021/jm00106a041 [DOI] [PubMed] [Google Scholar]
- Reissmann KR, Ruth WE, Nomura T (1961) A human hemoglobin with lowered oxygen affinity and impaired heme-heme interactions. J Clin Invest 40:1826–1833. 10.1172/JCI104406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoda MD, Martin J, Blouquit Y et al. (1983) Sickle cell hemoglobin fiber formation strongly inhibited by the Stanleyville II mutation (alpha 78 Asn leads to Lys). Biochem Biophys Res Commun 111:8–13. 10.1016/s0006-291x(83)80109-0 [DOI] [PubMed] [Google Scholar]
- Richard V, Dodson GG, Mauguen Y (1993) Human deoxyhaemoglobin-2,3-diphosphoglycerate complex low-salt structure at 2.5 A resolution. J Mol Biol 233:270–274. 10.1006/jmbi.1993.1505 [DOI] [PubMed] [Google Scholar]
- Rieder RF, Oski FA, Clegg JB (1969) Hemoglobin Philly (beta 35 tyrosine phenylalanine): studies in the molecular pathology of hemoglobin. J Clin Invest 48:1627–1642. 10.1172/JCI106128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemer C, ElSherbini A, Stojanovic N et al. (1998) A database of experimental results on globin gene expression. Genomics 53:325–337. 10.1006/geno.1998.5524 [DOI] [PubMed] [Google Scholar]
- Safo MK, Abraham DJ (2005) The enigma of the liganded hemoglobin end state: a novel quaternary structure of human carbonmonoxy hemoglobin. Biochemistry 44:8347–8359. 10.1021/bi050412q [DOI] [PubMed] [Google Scholar]
- Safo MK, Bruno S (2011) Allosteric effectors of hemoglobin: past, present and future. In: Chemistry and biochemistry of oxygen therapeutics. Wiley, Ltd, pp 285–300 [Google Scholar]
- Safo MK, Moure CM, Burnett JC et al. (2001) High–resolution crystal structure of deoxy hemoglobin complexed with a potent allosteric effector. Protein Sci 10:951–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safo MK, Boyiri T, Burnett JC et al. (2002a) X-ray crystallographic analyses of symmetrical allosteric effectors of hemoglobin: compounds designed to link primary and secondary binding sites. Acta Crystallogr D Biol Crystallogr 58:634–644. 10.1107/s0907444902002627 [DOI] [PubMed] [Google Scholar]
- Safo MK, Burnett JC, Musayev FN et al. (2002b) Structure of human carbonmonoxyhemoglobin at2.16 Å: a snapshot of the allosteric transition. Acta Crystallographica Section D 58:2031–2037. 10.1107/S0907444902015809 [DOI] [PubMed] [Google Scholar]
- Safo MK, Abdulmalik O, Danso-Danquah R et al. (2004) Structural basis for the potent antisickling effect of a novel class of five-membered heterocyclic aldehydic compounds. J Med Chem 47:4665–4676. 10.1021/jm0498001 [DOI] [PubMed] [Google Scholar]
- Safo MK, Abdulmalik O, Lin HR et al. (2005) Structures of R- and T-state hemoglobin Bassett: elucidating the structural basis for the low oxygen affinity of a mutant hemoglobin. Acta Crystallogr D Biol Crystallogr 61:156–162. 10.1107/S0907444904030501 [DOI] [PubMed] [Google Scholar]
- Safo MK, Ahmed MH, Ghatge MS, Boyiri T (2011) Hemoglobin-ligand binding: understanding Hb function and allostery on atomic level. Biochim Biophys Acta 1814:797–809. 10.1016/j.bbapap.2011.02.013 [DOI] [PubMed] [Google Scholar]
- Safo MK, Ko T-P, Abdulmalik O et al. (2013) Structure of fully liganded Hb ζ2β2s trapped in a tense conformation. Acta Crystallogr D Biol Crystallogr 69:2061–2071. 10.1107/S0907444913019197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safo MK, Ko T-P, Schreiter ER, Russell JE (2015) Structural basis for the antipolymer activity of Hb ζ2β2s trapped in a tense conformation. J Mol Struct 1099:99–107. 10.1016/j.molstruc.2015.06.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahu SC, Simplaceanu V, Gong Q et al. (2007) Insights into the solution structure of human deoxyhemoglobin in the absence and presence of an allosteric effector. Biochemistry 46:9973–9980. 10.1021/bi700935z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuragawa M, Ohba Y, Miyaji T et al. (1984) A Japanese boy with hemolytic anemia due to an unstable hemoglobin (Hb Bristol). Nippon Ketsueki Gakkai Zasshi 47:896–902 [PubMed] [Google Scholar]
- Samaja M, Rovida E, Niggeler M et al. (1987) The dissociation of carbon monoxide from hemoglobin intermediate. J Biol Chem 262:4528–4533 [PubMed] [Google Scholar]
- Samuni U, Dantsker D, Juszczak LJ et al. (2004) Spectroscopic and functional characterization of T state hemoglobin conformations encapsulated in silica gels. Biochemistry 43:13674–13682. 10.1021/bi048531d [DOI] [PubMed] [Google Scholar]
- Sawicki CA, Gibson QH (1976) Quaternary conformational changes in human hemoglobin studied by laser photolysis of carboxyhemoglobin. J Biol Chem 251:1533–1542 [PubMed] [Google Scholar]
- Sawicki CA, Gibson QH (1978) The relation between carbon monoxide binding and the conformational change of hemoglobin. Biophys J 24:21–33. 10.1016/S0006-3495(78)85328-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider RG, Atkins RJ, Hosty TS et al. (1975) Haemoglobin Titusville: alpha94 Asp replaced by Asn. a new haemoglobin with a lowered affinity for oxygen. Biochim Biophys Acta 400:365–373 [PubMed] [Google Scholar]
- Schoenborn BP (1976) Dichloromethane as an antisickling agent in sickle cell hemoglobin. Proc Natl Acad Sci USA 73:4195–4199. 10.1073/pnas.73.11.4195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher MA, Dixon MM, Kluger R et al. (1995) Allosteric transition intermediates modelled by crosslinked haemoglobins. Nature 375:84–87. 10.1038/375084a0 [DOI] [PubMed] [Google Scholar]
- Schumacher MA, Zheleznova EE, Poundstone KS et al. (1997) Allosteric intermediates indicate R2 is the liganded hemoglobin end state. Proc Natl Acad Sci USA 94:7841–7844. 10.1073/pnas.94.15.7841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma VS, Newton GL, Ranney HM et al. (1980) Hemoglobin Rothschild (beta 37(C3)Trp replaced by Arg): a high/low affinity hemoglobin mutant. J Mol Biol 144:267–280. 10.1016/0022-2836(80)90090-x [DOI] [PubMed] [Google Scholar]
- Shaw E, Scott C, Suh J et al. (2003) RSR13 plus cranial radiation therapy in patients with brain metastases: comparison with the radiation therapy oncology group recursive partitioning analysis brain metastases database. J Clin Oncol 21:2364–2371. 10.1200/JCO.2003.08.116 [DOI] [PubMed] [Google Scholar]
- Shibayama N, Saigo S (2001) Direct observation of two distinct affinity conformations in the T state human deoxyhemoglobin. FEBS Lett 492:50–53. 10.1016/s0014-5793(01)02225-6 [DOI] [PubMed] [Google Scholar]
- Shibayama N, Miura S, Tame JRH et al. (2002) Crystal structure of horse carbonmonoxyhemoglobinbezafibrate complex at 1.55-A resolution. a novel allosteric binding site in R-state hemoglobin. J Biol Chem 277:38791–38796. 10.1074/jbc.M205461200 [DOI] [PubMed] [Google Scholar]
- Silva MM, Rogers PH, Arnone A (1992) A third quaternary structure of human hemoglobin A at1.7-A resolution. J Biol Chem 267:17248–17256 [PubMed] [Google Scholar]
- Smith FR, Simmons KC (1994) Cyanomet human hemoglobin crystallized under physiological conditions exhibits the Y quaternary structure. Proteins 18:295–300. 10.1002/prot.340180310 [DOI] [PubMed] [Google Scholar]
- Smith FR, Lattman EE, Carter CW (1991) The mutation beta 99 Asp-Tyr stabilizes Y-a new, composite quaternary state of human hemoglobin. Proteins 10:81–91. 10.1002/prot.340100202 [DOI] [PubMed] [Google Scholar]
- Song X, Simplaceanu V, Ho NT, Ho C (2008) Effector-induced structural fluctuation regulates the ligand affinity of an allosteric protein: binding of inositol hexaphosphate has distinct dynamic consequences for the T and R states of hemoglobin. Biochemistry 47:4907–4915. 10.1021/bi7023699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S, Milstien S (2003) Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev MolCell Biol 4:397–407. 10.1038/nrm1103 [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Rose GD (1994) The T-to-R transformation in hemoglobin: a reevaluation. PNAS 91:11113–11117. 10.1073/pnas.91.23.11113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern W, Mathews D, McKew J et al. (2012) A phase 1, first-in-man, dose-response study of Aes-103 (5-HMF), an anti-sickling, allosteric modifier of hemoglobin oxygen affinity in healthy norman volunteers. Blood 120:3210–3210 [Google Scholar]
- Stucker O, Laurent D, Duvelleroy M et al. (1985) Incorporation of inositol hexaphosphate in stored erythrocytes: effect on tissue oxygenation. Life Support Syst 3(Suppl 1):458–461 [PubMed] [Google Scholar]
- Sun DP, Zou M, Ho NT, Ho C (1997) Contribution of surface histidyl residues in the alpha-chain to the Bohr effect of human normal adult hemoglobin: roles of global electrostatic effects. Biochemistry 36:6663–6673. 10.1021/bi963121d [DOI] [PubMed] [Google Scholar]
- Sun K, D’Alessandro A, Ahmed MH et al. (2017) Structural and functional insight of sphingosine 1-phosphate-mediated pathogenic metabolic reprogramming in sickle cell disease. Sci Rep 7:15281 10.1038/s41598-017-13667-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo A, Karplus M (1972) A mathematical model for structure-function relations in hemoglobin. J Mol Biol 72:163–197. 10.1016/0022-2836(72)90077-0 [DOI] [PubMed] [Google Scholar]
- Thom CS, Dickson CF, Gell DA, Weiss MJ (2013) Hemoglobin variants: biochemical properties and clinical correlates. Cold Spring Harb Perspect Med 3:a011858 10.1101/cshperspect.a011858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrance J, Jacobs P, Restrepo A et al. (1970) Intraerythrocytic adaptation to anemia. N Engl J Med 283:165–169. 10.1056/NEJM197007232830402 [DOI] [PubMed] [Google Scholar]
- Traylor TG, Deardurff LA, Coletta M et al. (1983) Reactivity of ferrous heme proteins at low pH. JBiol Chem 258:12147–12148 [PubMed] [Google Scholar]
- Vasseur C, Blouquit Y, Kister J et al. (1992) Hemoglobin Thionville. An alpha-chain variant with a substitution of a glutamate for valine at NA-1 and having an acetylated methionine NH2 terminus. J Biol Chem 267:12682–12691 [PubMed] [Google Scholar]
- Vichinsky E, Hoppe CC, Ataga KI et al. (2019) A phase 3 randomized trial of Voxelotor in sickle cell disease. N Engl J Med 381:509–519. 10.1056/NEJMoa1903212 [DOI] [PubMed] [Google Scholar]
- Wilson J, Phillips K, Luisi B (1996) The crystal structure of horse deoxyhaemoglobin trapped in the high-affinity (R) state. J Mol Biol 264:743–756. 10.1006/jmbi.1996.0674 [DOI] [PubMed] [Google Scholar]
- Winslow RM, Charache S (1975) Hemoglobin Richmond. Subunit dissociation and oxygen equilibrium properties. J Biol Chem 250:6939–6942 [PubMed] [Google Scholar]
- Winter PM, Miller JN (1976) Carbon monoxide poisoning. JAMA 236:1502–1504. 10.1001/jama.1976.03270140054029 [DOI] [PubMed] [Google Scholar]
- Wireko FC, Abraham DJ (1991) X-ray diffraction study of the binding of the antisickling agent 12C79 to human hemoglobin. Proc Natl Acad Sci USA 88:2209–2211. 10.1073/pnas.88.6.2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wireko FC, Kellogg GE, Abraham DJ (1991) Allosteric modifiers of hemoglobin. 2. Crystallographically determined binding sites and hydrophobic binding/interaction analysis of novel hemoglobin oxygen effectors. J Med Chem 34:758–767. 10.1021/jm00106a042 [DOI] [PubMed] [Google Scholar]
- Woods JA, Storey CJ, Babcock EE, Malloy CR (1998) Right-shifting the oxyhemoglobin dissociation curve with RSR13: effects on high-energy phosphates and myocardial recovery after low-flow ischemia. J Cardiovasc Pharmacol 31:359–363. 10.1097/00005344-199803000-00005 [DOI] [PubMed] [Google Scholar]
- Xu GG, Deshpande TM, Ghatge MS et al. (2015) Design, synthesis, and investigation of novel nitric oxide (NO)-releasing prodrugs as drug candidates for the treatment of ischemic disorders: insights into NO-releasing prodrug biotransformation and hemoglobin-NO biochemistry. Biochemistry 54:7178–7192. 10.1021/acs.biochem.5b01074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GG, Pagare PP, Ghatge MS et al. (2017) Design, synthesis, and biological evaluation of ester and ether derivatives of antisickling agent 5-HMF for the treatment of sickle cell disease. Mol Pharm 14:3499–3511. 10.1021/acs.molpharmaceut.7b00553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama T, Neya S, Tsuneshige A et al. (2006) R-state haemoglobin with low oxygen affinity: crystal structures of deoxy human and carbonmonoxy horse haemoglobin bound to the effector molecule L35. J Mol Biol 356:790–801. 10.1016/j.jmb.2005.12.018 [DOI] [PubMed] [Google Scholar]
- Yonetani T, Kanaori K (2013) How does hemoglobin generate such diverse functionality of physiological relevance? Biochim Biophys Acta 1834:1873–1884. 10.1016/j.bbapap.2013.04.026 [DOI] [PubMed] [Google Scholar]
- Yonetani T, Tsuneshige A (2003) The global allostery model of hemoglobin: an allosteric mechanism involving homotropic and heterotropic interactions. CR Biol 326:523–532. 10.1016/S1631-0691(03)00150-1 [DOI] [PubMed] [Google Scholar]
- Yonetani T, Park S-I, Tsuneshige A et al. (2002) Global allostery model of hemoglobin. Modulation of O(2) affinity, cooperativity, and Bohr effect by heterotropic allosteric effectors. J Biol Chem 277:34508–34520. 10.1074/jbc.M203135200 [DOI] [PubMed] [Google Scholar]
- Youssef AM, Safo MK, Danso-Danquah R et al. (2002) Synthesis and X-ray studies of chiral allosteric modifiers of hemoglobin. J Med Chem 45:1184–1195. 10.1021/jm010358l [DOI] [PubMed] [Google Scholar]
- Zago MA, Bottura C (1983) Splenic function in sickle-cell diseases. Clin Sci 65:297–302. 10.1042/cs0650297 [DOI] [PubMed] [Google Scholar]
- Zaugg RH, Walder JA, Klotz IM (1977) Schiff base adducts of hemoglobin. Modifications that inhibit erythrocyte sickling. J Biol Chem 252:8542–8548 [PubMed] [Google Scholar]
- Zhang Y, Berka V, Song A et al. (2014) Elevated sphingosine-1-phosphate promotes sickling and sickle cell disease progression. J Clin Invest 124:2750–2761. 10.1172/JCI74604 [DOI] [PMC free article] [PubMed] [Google Scholar]