

Abstract
The following fictional case is intended as a learning tool within the Pathology Competencies for Medical Education (PCME), a set of national standards for teaching pathology. These are divided into three basic competencies: Disease Mechanisms and Processes, Organ System Pathology, and Diagnostic Medicine and Therapeutic Pathology. For additional information, and a full list of learning objectives for all three competencies, see http://journals.sagepub.com/doi/10.1177/2374289517715040.1
Keywords: pathology competencies, organ system pathology, kidney, congenital disorders, polycystic kidney disease, inheritance patterns, autosomal dominant
Primary Objective
Objective UTK4.1: Inherited Renal Disorders. Compare autosomal dominant and autosomal recessive polycystic kidney disease in terms of pathological anatomy, molecular pathogenesis, and clinical presentation.
Competency 2: Organ System Pathology; Topic UTK: Kidney; Learning Goal 4: Congenital Disorders of the Kidney.
Secondary Objective
Objective GM1.2: Inheritance Patterns. Compare and contrast the inheritance patterns of different types of Mendelian disorders and give examples of each type of pattern.
Competency 1: Disease Mechanisms and Processes; Topic GM: Genetic Mechanisms; Learning Goal 1: Genetic Mechanisms of Development and Functional Abnormalities.
Patient Presentation
A 40-year-old man with no significant past medical history presents as a restrained passenger in a motor vehicle collision with complaints of abdominal pain. Vital signs are remarkable for a blood pressure 150/92 mm Hg. He has normal bowel sounds and no localized abdominal pain, rebound tenderness, or guarding nor costovertebral angle tenderness on physical examination. However, bilateral flank fullness is noted, and contusions are visible across the chest and lower abdomen (seat belt sign).
Diagnostic Findings, Part I
Initial laboratory analysis is significant for an elevated serum creatinine of 1.8 mg/dL. To further rule out internal injury, computed tomography imaging with intravenous contrast of the abdomen and pelvis is performed. It shows no evidence of acute traumatic injury but does reveal both kidneys are enlarged and contain multiple, nonenhancing fluid-filled cysts without a solid component (Figure 1). No other pathologic process including liver cysts is identified.
Figure 1.
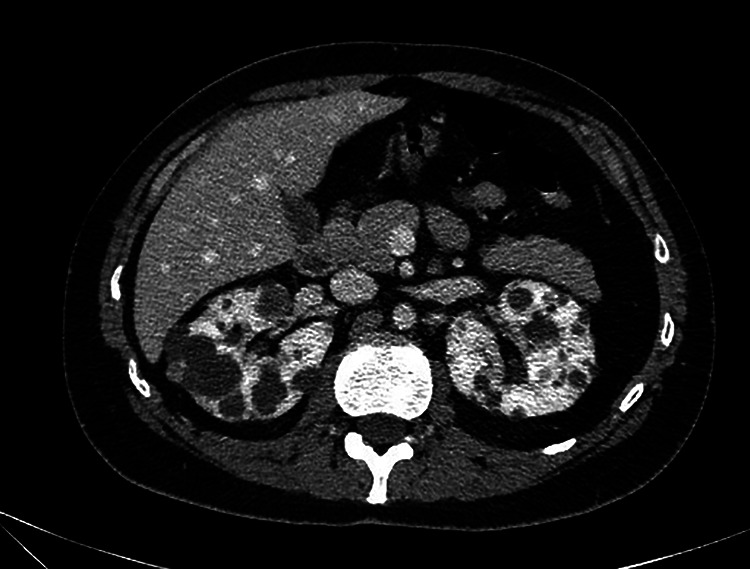
An axial computed tomography (CT) scan shows numerous cysts of variable sizes and locations in both kidneys.
Questions/Discussion Points, Part I
What Is the Differential Diagnosis for Cystic Kidneys in Adults?
The differential diagnosis for renal cysts in adults includes inherited diseases such as multicystic renal dysplasia, medullary sponge kidney, medullary cystic disease, and autosomal dominant polycystic kidney disease (ADPKD); acquired cystic disease; simple cortical cysts; and neoplasms, including renal cell carcinoma and mesenchymal tumors (eg, renal sarcomas and angiomyolipoma). Some of these neoplasms are also associated with familial syndromes, including von Hippel-Lindau syndrome, tuberous sclerosis, Birt-Hogg-Dubé syndrome (benign skin lesions, lung cysts, and renal cysts and tumors), and hereditary papillary renal cell carcinoma.2-4 Cysts arising from structures other than renal parenchyma (cortex and medulla) may also be identified. These include lesions arising from a calyx or the pelvis (also referred to as a calyceal diverticulum, pyelogenic cyst, or pyelocalyceal cyst) or those that arise within the hilum, such as lymphangitic cysts.3,4
Diagnostic Findings, Part II
A more detailed history reveals that the patient’s father had a kidney transplant in his 60s for renal cysts, and his paternal grandmother died in her 50s from “bleeding in her head.” Additional laboratory studies disclose microscopic hematuria.
Questions/Discussion Points, Part II
What Is the Most Likely Diagnosis in This Patient?
Given the patient’s family history, hypertension, laboratory results, and the finding of numerous bilateral renal cysts without other radiologic abnormalities or past medical history, the most likely diagnosis in this patient is ADPKD. In a patient with a family history of ADPKD, the finding of at least 2 cysts in each kidney (patients ages 30-59 years) is both 100% sensitive and specific for this disease.2
What Is the Typical Clinical Presentation of Autosomal Dominant Polycystic Kidney Disease?
Autosomal dominant polycystic kidney disease is initially a clinically silent genetic disorder and is often discovered incidentally on radiology studies or following workup of abnormal laboratory tests performed for other reasons. With an estimated prevalence as high as 1 in 400, patients are often not diagnosed until their fifth decade of life.2 Autosomal dominant polycystic kidney disease is a clinically significant entity, as it is a common cause of renal failure.2
Flank pain is the most common presenting complaint.3 This pain can be due to infection within or bleeding of cysts or due to kidney stones. Patients with ADPKD also commonly present with flank fullness, gross or microscopic hematuria, and hypertension.3
Describe the Pathogenesis of Autosomal Dominant Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease is the result of an inherited mutation that most commonly occurs in either the PKD1 (85% of cases) or PKD2 genes.4 All cells in the patient’s body have the mutation. Cyst formation is thought to occur in discrete areas of the kidney when cells lose their second copy of their respective gene.5 This makes ADPKD an excellent example of the Knudson “two-hit” hypothesis, which was first described in retinoblastoma patients.6 The protein product of the PKD1 gene is named polycystin-1, which is expressed on tubular epithelial cells. Notably, this large, membrane protein is highly expressed in the distal nephron. Currently, its precise function is unknown; however, it contains domains that are putatively involved in cell–cell and cell–matrix interactions. The protein product of the PKD2 gene is polycystin-2, a Ca2+-permeable cation channel within the cell membrane of renal tubules as well as tissues outside the kidneys.4 Notably, time to renal failure is closely correlated with the associated causative genetic mutation, as patients with mutated PKD1 often develop end-stage renal disease (ESRD) by their 50s and those with PKD2 mutations not until their 70s.3
The dominant hypothesis regarding the pathogenesis of ADPKD focuses on the role of the cilia–centrosome complex of tubular epithelial cells.4 Disorders that result in defects in this complex are termed “ciliopathies,” and many of the associated disorders have renal cysts as a component of their pathology. The cilia act as mechanosensors, and abnormalities in this function, along with changes in calcium flux, contribute to the process of cyst formation. This process occurs through the combination of increased proliferation of cells, secretion of fluid, and alterations of extracellular matrix. The 3 main cellular defects that lead to cyst formation are increased cell proliferation and fluid secretion, decreased cell differentiation, and abnormal extracellular matrix.2 Due to the change in membrane permeability attributed to the altered polycystin-1 or polycystin-2, subsequent cell signaling pathways related to growth, apoptosis, and secretion are purportedly altered.4 These changes in cell growth and secretory function drive cyst formation and development. Similarly, dysregulation of cell membranes leads to the presence of inflammatory mediators. These inflammatory mediators can be found in cyst fluid and are thought to play a role in the fibrosis of the cellular matrix and thickening of the epithelial basement membrane.7
What Are the Expected Gross Features of ADPKD as Seen in Figure 2? Described the Early and Late Histologic Findings in This Disease as Seen in Figures 3 and 4
Figure 2.
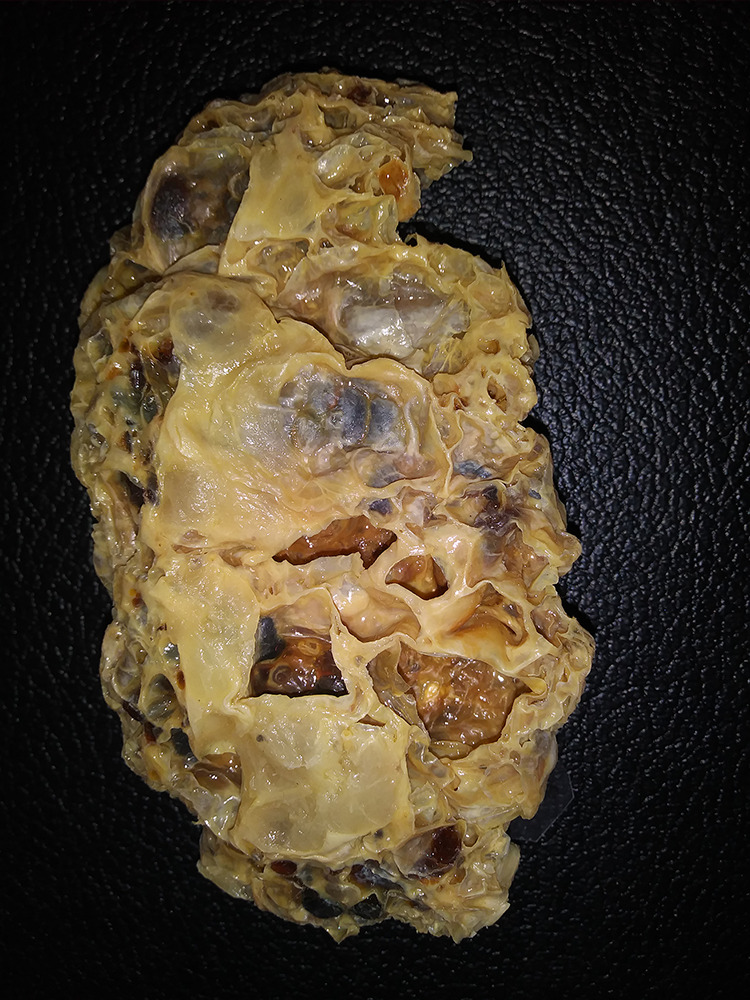
Grossly, the kidney is enlarged and distorted by numerous cysts of varying sizes in a random distribution. Normal parenchyma is not grossly apparent.
Figure 3.
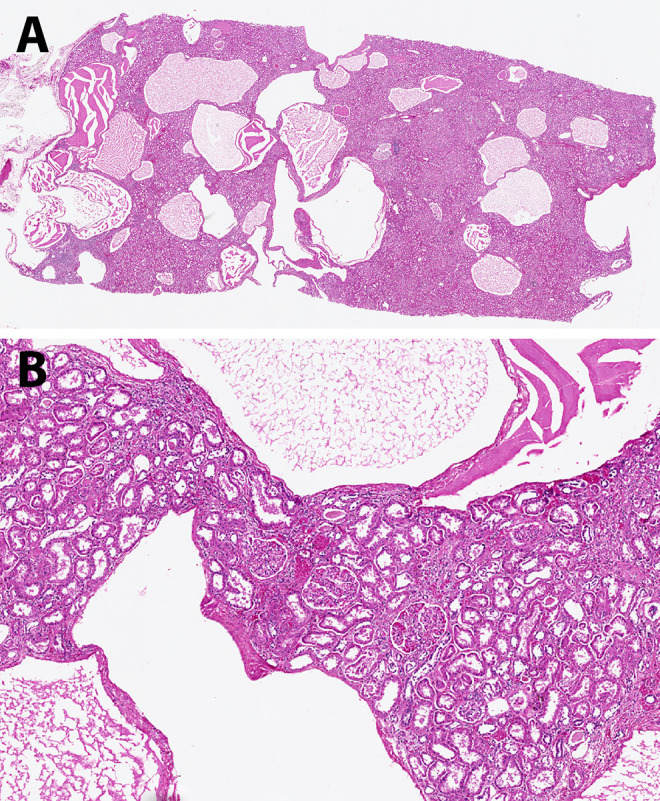
A, Multiple cysts, here mostly containing proteinaceous material, are present throughout the cortex in this earlier stage of autosomal dominant polycystic kidney disease (ADPKD). B, The renal parenchyma between the cysts is still fairly unremarkable (hematoxylin and eosin, scanning magnification [A] and low power [B]).
Figure 4.
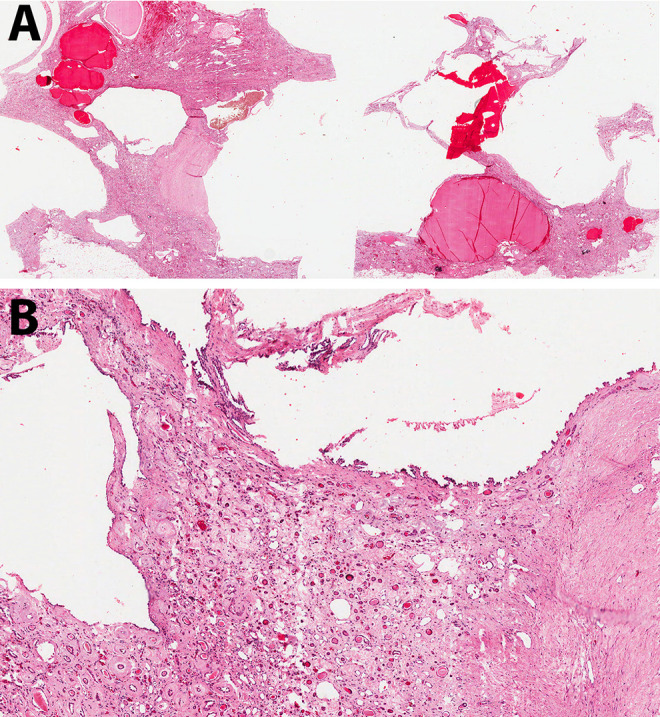
A, This kidney from a patient later in the course of autosomal dominant polycystic kidney disease (ADPKD) contains cysts of variable size filled with blood or serous fluid. The intervening tissue appears more eosinophilic than that in Figure 3. B, Abundant fibrosis surrounds the few residual normal structures (hematoxylin and eosin, scanning magnification [A] and low power [B]).
Over time, both kidneys become enlarged and appear predominantly composed of discrete cysts that develop randomly throughout the kidney (Figure 2). The cysts are filled with fluid that can be clear (serous), cloudy, or red-brown (hemorrhagic).4 Histologically, ADPKD also shows progression over time as seen in Figures 3 and 4. Earlier in the process, the cysts can be seen interspersed between renal parenchymal elements (Figure 3). Later, the normal renal elements are damaged or absent, and fibrosis develops between more numerous cysts (Figure 4). Since cysts develop from any portion of the nephron, variable-appearing epithelium lines the cysts, ranging from flattened to cuboidal (Figure 5). A small fraction (1%-2%) of the cysts even form within the glomeruli.3
Figure 5.
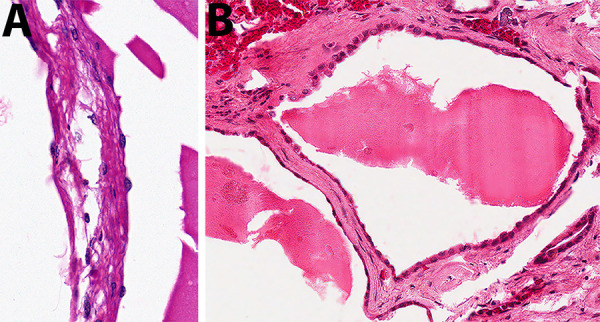
The cysts in autosomal dominant polycystic kidney disease (ADPKD) can be lined by variable-appearing epithelium, including flattened (A) and cuboidal (B), (hematoxylin and eosin, high power).
Describe the Inheritance Pattern of Autosomal Dominant Polycystic Kidney Disease
Autosomal dominant polycystic kidney disease is an example of a Mendelian disorder, that is, a condition resulting from a mutation in a single gene. Single-gene disorders are usually inherited in an autosomal dominant, autosomal recessive, or X-linked pattern.8 Examples of disorders inherited through each transmission pattern are provided in Table 1.
Table 1.
Examples of Mendelian Disorders by Inheritance Pattern.
| Inheritance pattern | Disease |
|---|---|
| Autosomal dominant | Autosomal dominant polycystic kidney disease Neurofibromatosis (1 and 2) Familial polyposis coli Marfan syndrome Huntington disease Tuberous sclerosis von Hippel-Lindau syndrome Myotonic dystrophy Familial hypercholesterolemia Achondroplasia |
| Autosomal recessive | Autosomal recessive polycystic kidney disease Cystic fibrosis Sickle cell anemia Thalassemias Hemochromatosis Xeroderma pigmentosum Tay-Sachs disease Phenylketonuria Galactosemia Gaucher disease |
| X-linked | Hemophilia (A and B) Duchenne muscular dystrophy Red-green color blindness Glucose-6-phosphate dehydrogenase (G6PD) deficiency Adrenoleukodystrophy Lesch-Nyhan syndrome Wiskott-Aldrich syndrome X-linked agammaglobulinemia Fabry disease Vitamin D-resistant ricketsa |
a This is an X-linked dominant condition; all others listed are recessive.
Autosomal dominant polycystic kidney disease is inherited in an autosomal dominant manner as the name implies. This means that if one parent is heterozygous for the trait, then there is a 50% likelihood that their offspring will receive the trait and have the disease. However, not all patients will have the same level of disease, as ADPKD has 100% penetrance but variable expressivity.4 An autosomal recessive disorder such as autosomal recessive polycystic kidney disease (ARPKD) requires inheritance of 2 mutated gene alleles, one from each parent. The parents are usually unaffected, although each sibling also has a 25% chance of disease.8 X-linked disorders are associated with the X sex chromosome. Most are recessive and so usually affect only males, who then have the potential to pass the trait to their daughters. However, females can have some phenotypic changes due to random X inactivation. X-linked dominant conditions are rare.8
What Is the Difference Between Penetrance and Expressivity, Especially as It Relates to This Condition?
“Penetrance” is the degree to which a person expresses features of their genes. Complete penetrance occurs when the person expresses their genotype, whereas incomplete penetrance occurs when the inherited trait does not cause a phenotypic change.9 In the case of ADPKD, a disorder with complete penetrance, a mutation in PKD1 or PKD2 results in expression of a mutated phenotype: cyst development in the kidneys. “Expressivity” is a term also used to describe phenotypic results of an altered genotype. This word describes differences in the degree of phenotypic anomalies.9 In a sense, penetrance is like a light switch: is the gene on in this patient or off in this patient? Conversely, expressivity is like installing a dimmer switch for the light—the result is variable. The gene is expressed (the light is on), but to what degree: mild, moderate, severe (how bright is the light)? In a patient, any mutation of either PKD1 or PKD2 results in phenotypic ADPKD. This means that ADPKD has 100% penetrance. However, not all patients with ADPKD, or even all patients with PKD1 mutations, present the same way. This, in turn, speaks to the condition’s variable expressivity. The reasons for these differences in disease phenotype are not entirely known at this point, but other genes or environment may play a role. Persons with ADPKD may have mild manifestations or death before diagnosis, and thus, a family history may not be known despite autosomal dominant inheritance. New mutations can also occur within gametes. In contrast, autosomal recessive disorders tend to have complete penetrance and more similar expression of phenotype.8
What Complications (Renal and Extrarenal) and Other Conditions Are Associated With Autosomal Dominant Polycystic Kidney Disease?
The slowly enlarging cysts in ADPKD lead to a decline in kidney function, often resulting in ESRD and the development of a dialysis requirement or necessitating renal transplant.10 When the cysts become large, they can push upon the pelvocalyceal system and thus lead to defects in this manner.4 As mentioned above, flank pain, renal infections, and nephrolithiasis are additional complications. Of note, the frequency of renal cell carcinoma does not appear to be increased in patients with ADPKD, though there is increased incidence of bilateral renal cell carcinoma, notably in a younger population.3 Patients with ADPKD often have elevated erythropoietin levels, which can mask the anemia of ESRD, should the disease process progress to that point.10 Patients may also experience an impact on their gastrointestinal function due to mass effect from the markedly enlarged kidneys.3
Patients with ADPKD are also at increased risk for comorbidities in other organs related to the mutated PKD1 and PKD2 gene products, as the protein is found in several other locations in the body. The most clinically significant comorbid conditions are berry aneurysms, often found in the circle of Willis, and liver cysts derived from biliary epithelium. The former is of particular interest, as berry aneurysms are at increased risk of rupture due to the renin-driven hypertension found in patients with ADPKD. This makes blood pressure control a high priority in these patients.2 The combination of these ruptured aneurysms along with intracerebral bleeding due to hypertension account for 15% of deaths in patients with ADPKD.4 Left ventricular hypertrophy is also associated with hypertension and can interestingly develop even in patients who are normotensive.2 In addition to the liver, cyst formation has also been reported in the seminal vesicles, arachnoid membrane, and pancreas.3 Other significant abnormalities include colonic diverticulosis and mitral valve prolapse.2
How Does Autosomal Dominant Polycystic Kidney Disease Differ From Autosomal Recessive Polycystic Kidney Disease? Consider the Genetic, Histopathologic, and Clinical Differences
While there is some overlap between ADPKD and ARPKD, these disorders have several distinct differences which are outlined in Table 2. As denoted by the names of each condition, they have different genetic inheritance patterns. Similarly, they have different genetic loci: PKD1 and PKD2 are located on chromosomes 16 and 4, respectively, while PKHD1 is located on chromosome 6. However, both disorders are examples of ciliopathies. Notably, each condition presents in different times in life: ADPKD presents during late adolescence (in severe cases) or may be clinically silent throughout an individual’s life, whereas ARPKD is detectable in utero.3 The prognoses differ significantly: the earlier ARPKD is diagnosed, the more severe it usually is, with 50% of affected newborns diagnosed with ARPKD dying in the first few days of life.11 Those patients who survive later into childhood have more prominent liver disease, congenital hepatic fibrosis, that may lead to portal hypertension. This hepatic disease is histologically characterized by bile ductular proliferation and portal fibrosis.4
Table 2.
Comparative Features of Autosomal Dominant Polycystic Kidney Disease (ADPKD) Versus Autosomal Recessive Polycystic Kidney Disease (ARPKD).
| ADPKD | ARPKD | |
|---|---|---|
| Inheritance pattern | Autosomal dominant | Autosomal recessive |
| Chromosome/gene/protein | 16p13.3/PKD1/polycystin-1; 4q21/PKD2/polycystin-2 | 6p21-p23/PKHD1/fibrocystin |
| Typical age at presentation | Adult | Infant/child |
| Symptoms at presentation | Renal insufficiency, hematuria, pain, polyuria, hypertension | Oligohydramnios, abnormalities on fetal ultrasound |
| Pathology of kidneys | Large, multicystic (up to 3-4 cm), haphazard cysts with variable lining. Cysts can originate from any portion of the nephron | Large, smooth kidneys; elongated channels at right angles to cortical surface; uniform lining of cuboidal cells (collecting duct origin) |
| Extrarenal abnormalities | Hepatic cysts (40%); cysts in pancreas, spleen, lungs; intracranial berry aneurysms; mitral valve prolapse (20%-25%) | Congenital hepatic fibrosis (ductal plate malformation) |
| Usual outcome | Chronic renal failure starting in adulthood (age 40-60) | Death in infancy or childhood without kidney transplant |
Gross and microscopic differences are present in both disorders as well. The kidneys in ARPKD are often described as “sponge-like” with a smooth exterior. Gross section reveals “channels” running from medullary and cortical cysts, perpendicular to the cortical surface.4
The major difference between ADPKD and ARPKD on histological inspection is the location of the cysts and, subsequently, the lining of the cysts.3 This is a result of differences in origins of the cysts. Autosomal recessive polycystic kidney disease originates from the epithelial cells in the collecting ducts, as opposed to ADPKD which arises from any portion of the nephron.4 Hafer and Conran previously published a case study of ARPKD with a more complete discussion and representative images.12
Teaching Points
Autosomal dominant polycystic kidney disease originates from mutations in the PKD1 or PKD2 gene loci. The inheritance pattern is autosomal dominant with complete penetrance but variable expressivity.
Patients with ADPKD may be clinically silent or present in adulthood with flank pain, hematuria, hypertension, or flank fullness. While some will have clinically insignificant consequences, others will progressively lose renal function and require dialysis or transplant.
Extrarenal conditions associated with ADPKD include berry aneurysms, hepatic cysts, and mitral valve prolapse.
The kidneys in ADPKD are enlarged and contain numerous cysts of variable size and location and differences in the appearance of lining cells. This reflects the possibility that cysts can arise from any part of the nephron.
Autosomal recessive polycystic kidney disease is the result of a mutation of the PKHD1 gene inherited in an autosomal recessive pattern. It presents in utero or early in life and is often fatal. The cysts are elongated, more uniform (all arise from collecting ducts), and arranged perpendicular to the renal capsule.
Both ADPKD and ARPKD are examples of ciliopathies, disorders resulting from mutations affecting primary cilia. This group of disorders typically causes renal cysts.
In addition to autosomal dominant and autosomal recessive, the third Mendelian inheritance pattern is X-linked.
Footnotes
Author’s Note: The opinions expressed herein are those of the author and are not necessarily representative of those of the Uniformed Services University of the Health Sciences (USUHS), the Department of Defense (DOD), or the United States Army, Navy, or Air Force.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD: Alison R. Huppmann  https://orcid.org/0000-0003-0739-1721
https://orcid.org/0000-0003-0739-1721
References
- 1. Knollmann-Ritschel BEC, Regula DP, Borowitz MJ, Conran R, Prystowsky MB. Pathology competencies for medical education and educational cases. Acad Pathol. 2017;4 doi:10.1177/2374289517715040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou J, Pollak MR. Polycystic kidney disease and other inherited disorders of tubule growth and development In: Jameson J, Fauci AS, Kasper DL, et al. eds. Harrison’s Principles of Internal Medicine. 20th ed McGraw-Hill; 2018. Accessed March 31, 2020 https://accessmedicine.mhmedical.com/content.aspx?sectionid=192281464&bookid=2129#192281475 [Google Scholar]
- 3. Pope JC. Renal dysgenesis and cystic disease of the kidney In: Partin AW, Peters CA, Kavoussi LR, et al. eds. Campbell-Walsh Urology. 12th ed Elsevier; 2020:741–775. [Google Scholar]
- 4. Alpers CE, Chang A. The kidney In: Kumar V, Abbas A, Aster A, eds. Robbins and Cotran Pathologic Basis of Disease. 9th ed Elsevier Saunders; 2015:897–955. [Google Scholar]
- 5. Pei Y. A ‘two-hit’ model of cystogenesis in autosomal dominant polycystic kidney disease? Trends Mol Med. 2001;7:151–156. [DOI] [PubMed] [Google Scholar]
- 6. Knudson AG. Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci. 1971;68:820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bremmer M. Polycystic kidney disease: inheritance, pathophysiology, prognosis, and treatment. Int J Nephrol Renovasc Dis. 2010:69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kumar V, Abbas A, Aster A. Genetic disorders In: Kumar V, Abbas A, Aster A, eds. Robbins and Cotran Pathologic Basis of Disease. 9th ed Elsevier Saunders; 2015:137–183. [Google Scholar]
- 9. Schaefer GB, Thompson JN. Mendelian genetics: patterns of gene transmission In: Schaefer GB, Thompson JN, eds. Medical Genetics: An Integrated Approach. McGraw-Hill; 2014. Accessed March 31, 2020 http://accessmedicine.mhmedical.com/content.aspx?bookid=2247§ionid=173744374 [Google Scholar]
- 10. Gabow PA. Autosomal dominant polycystic kidney disease. N Engl J Med. 1993;329:332–342. [DOI] [PubMed] [Google Scholar]
- 11. Kaplan BS, Kaplan P, Rosenberg HK, Lamothe E, Rosenblatt DS. Polycystic kidney diseases in childhood. J Pediatr. 1989;115:867–880. [DOI] [PubMed] [Google Scholar]
- 12. Hafer AS, Conran RM. Autosomal recessive polycystic kidney disease. Acad Pathol. 2017;4 doi:10.1177/2374289517718560 [DOI] [PMC free article] [PubMed] [Google Scholar]