

Abstract
Background
Mammalian cells must constantly reprogram the distribution of mitochondria in order to meet the local demands for energy, calcium, redox balance, and other mitochondrial functions. Mitochondrial localization inside the cell is a result of a combination of movement along the microtubule tracks plus anchoring to actin filaments.
Recent findings
Recent advances show that subcellular distribution of mitochondria can regulate tumor cell growth, proliferation/motility plasticity, metastatic competence, and therapy responses in tumors. In this review, we discuss our current understanding of the mechanisms by which mitochondrial subcellular distribution is regulated in tumor cells.
Conclusions
Mitochondrial trafficking is dysregulated in tumors. Accumulation of mitochondria at the leading edge of the cell supports energy expensive processes of focal adhesion dynamics, cell membrane dynamics, migration, and invasion.
Keywords: cancer, dynein, kinesin, mitochondria, myosin, trafficking
1. INTRODUCTION
Mitochondria are a keystone organelle in the cell responsible for metabolism, maintaining calcium homeostasis, buffering reactive oxygen species (ROS), surveying cell wellness, and initiating apoptosis. Not surprisingly, mitochondria have been linked to many hallmarks of cancer.1 To meet the ever‐changing demands of cancer cells, the mitochondrial pool is surveyed by quality control mechanisms and dynamically reprogrammed to maximize function. One such mechanism that allows for adaptation to systemic, extracellular, and intracellular conditions is mitochondrial movement. Mitochondrial subcellular localization is key to maintaining cell polarity, morphology, and cell homeostasis. A growing body of evidence links alterations of mitochondrial movement to mitochondrial dysfunction and metabolic alterations associated with motor neuron diseases,2 lethal encephalopathy,3 Charcot‐Marie‐Tooth hereditary neuropathy type 2A (CMT2A),4 Alzheimer's disease,5 diabetes, and cancer. In this review, we discuss recent progress in our understanding of the underpinnings by which mitochondrial subcellular distribution influences the metastatic ability of tumor cells.
2. REGULATION OF MITOCHONDRIAL MOVEMENT
Any mammalian cell must constantly reprogram the distribution of mitochondria in order to meet the local demands for energy, calcium, redox balance, and other mitochondrial functions. Mitochondrial localization inside the cell is a result of a combination of movement along the microtubule tracks and anchoring to actin filaments6 (Table 1). The molecular machinery responsible for mitochondrial movements along the cytoskeleton has been largely characterized in neurons. These studies showed that long‐range intracellular mitochondrial transport occurs primarily via the microtubule cytoskeleton using a trafficking machinery that involves mitochondrial Rho GTPases (MIRO1/2), trafficking adapter proteins that bind to kinesin (TRAK1/2), and Kinesin‐1/3 and Dynein motors (extensively reviewed in Lin and Sheng7). Anterograde movement of mitochondria (towards the + end of microtubules) is dependent on Kinesin‐1/3 motors (Figure 1A) and transport mitochondria in the retrograde fashion (towards the − end of microtubules) is achieved by Dynein motors (Figure 1A). More recently, additional Kinesin‐1 binding partners have been identified, including fasciculation and elongation protein zeta 1 (FEZ1), syntabulin (SYBU), and Ran‐binding protein 2 (RanBP2) (Figure 1B). Thus, neurons rely on several Kinesin‐1 containing complexes to move mitochondria. In non‐neuronal cells, it has been shown that MIRO1/Kinesin‐1 complexes regulate mitochondrial movements. However, further studies are needed to address whether Kinesin‐1 complexes with FEZ1, SYBU, or RANBP2 are conserved in non‐neuronal cells.
Table 1.
Composition of the motor complexes discussed in this review
| Motor | Filament Type | Mitochondrial Adaptor | Implication in Cancer | Reference |
|---|---|---|---|---|
| Kinesin | Microtuble | FEZ1/LZTS1 | Proliferation, chemotherapy resistance | Zhou et al92 Al Nakouzi et al93 and Lovat et al94 |
| MIRO1/2 | Cell migration and invasion | Desai et al22 and Caino et al24 | ||
| RanBP2 | Segregation errors | Vecchione et al95 and Navarro et al96 | ||
| SYBU | ? | — | ||
| TRAK1/2/Milton | Cell invasion | Onodera et al21 | ||
| Myosin | Actin | ? | Cell invasion, chemotherapy resistance, EMT | Ouderkirk and Krendel56 Galland et al57 and Yi et al55 Fernández‐Pérez et al58 and Lan et al61 |
Figure 1.
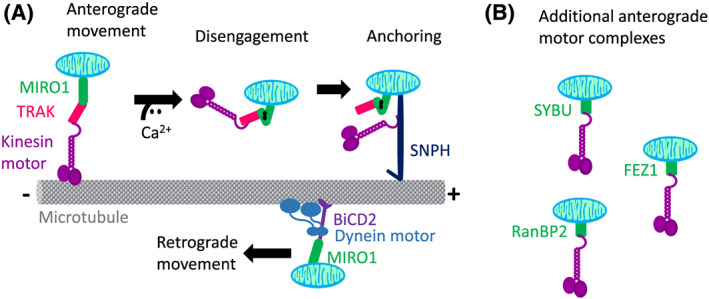
Microtubule‐mediated mitochondrial trafficking. A, Kinesin motor complexes containing TRAK and MIRO1 are responsible for moving mitochondria towards the + end of microtubules. Calcium negatively regulates movement by inducing a conformational change on MIRO1, which in turn disengages mitochondria from the microtubules. SNPH is an outer membrane mitochondrial protein that anchors mitochondria via direct binding to microtubules and Kinesin. Dynein motor complexes containing bicaudal 2 (BiCD2) adaptor and MIRO1 move mitochondria towards the − end of microtubules. B, Additional kinesin‐containing complexes have been described in neurons, containing SYBU, FEZ1, or RanBP2
Short‐range mitochondrial movements rely on actin filaments and unconventional myosin motors (MYO19, MYO6, and MYO5) (Table 1 and Figure 2). MYOs move along actin filaments in either anterograde (+ end) or retrograde (− end) directions.8, 9 The mechanisms by which MYOs regulate mitochondrial movements are poorly studied, and it is currently unknown how the MYOs bind to mitochondria (e.g., the adapters have not been identified yet).
Figure 2.
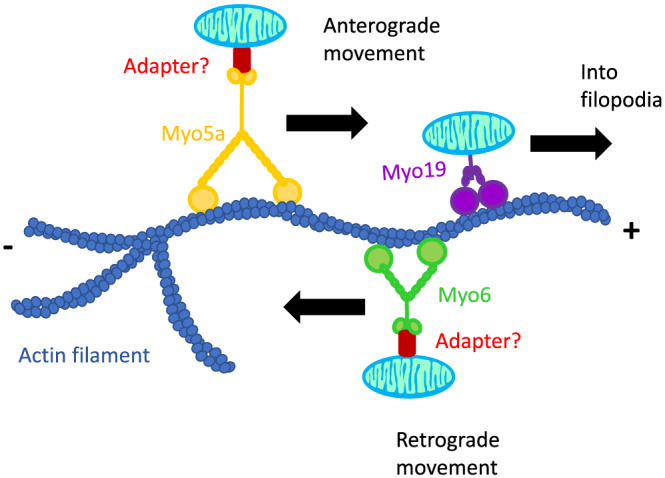
Actin‐mediated short‐range mitochondrial movement. Both MYO5A and MYO19 motors move mitochondria towards the + end of the actin filaments. MYO19 has been shown to traffick mitochondria into filopodia as well. MYO6 motors move mitochondria towards the − end of the actin filaments. While MYO19 is an outer membrane mitochondrial protein, the other Myosins would require a yet to be identified adapter protein to link the cargo to the motors
Finally, mitochondrial movement is negatively regulated by immobilization of organelles to the cytoskeleton. To date, four alternative ways to immobilize mitochondria have been described: (1) MYO‐dependent binding to actin,10, 11 (2) anchoring to microtubules via syntaphilin (SNPH),12 (3) calcium‐induced disengagement from microtubules,6 and (4) irreversible removal of the Kinesin‐1/TRAK complex by proteasomal degradation.6
Although many fundamental questions on the field of mitochondrial movement remain unanswered, it has become clear that subcellular distribution of mitochondria can regulate tumor cell growth, proliferation/motility plasticity, metastatic competence, and therapy responses in tumors (reviewed in Caino and Altieri13).
3. MITOCHONDRIAL LOCALIZATION IMPACTS CELLULAR FUNCTION
How exactly does the altered mitochondrial trafficking regulate metastatic properties of tumor cells? Our current understanding is that accumulation of mitochondria to the leading edge supports tumor cell invasion by providing a local source of energy and facilitating signal transduction at the leading edge of the cell (Figure 3).
Figure 3.
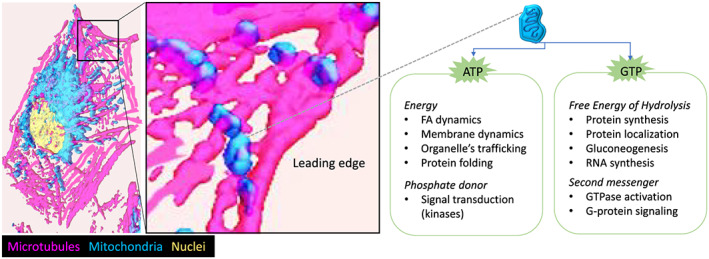
Cortical mitochondria provide a regional source of energy. Possible mechanisms by which mitochondria accumulation on the cortical cytoskeleton enhance tumor cell motility and invasion. When mitochondria reach the leading edge of the cell, local production of ATP and GTP by mitochondrial bioenergetics enable energy‐expensive cellular processes. Examples of the consumption of ATP and GTP for signal transduction and energy‐dependent molecular, cellular, and structural homeostasis in cells are provided
The importance of regional adenosine triphosphate (ATP) production for mitochondrial localization at the leading edge was demonstrated by studying tumor cells that bear oxidative phosphorylation (OxPhos)–deficient mitochondria (named ρ0 cells).14 Interestingly, ρ0 cells failed to reposition mitochondria proximal to focal adhesion (FA) complexes. An independent approach that relied on misfolding of the OxPhos complex II subunit SDHB by treatment with the mitochondrial‐Hsp90 inhibitor Gamitrinib also prevented accumulation of mitochondria to FAs.14 Finally, pharmacological inhibitors of OxPhos complexes I, III, and V (rotenone, antimycin A, and oligomycin) or the mitochondrial uncoupler (carbonyl cyanide m‐chlorophenyl hydrazine, CCCP) inhibited mitochondrial repositioning to the cortical cytoskeleton.14 In all cases, this reduction of mitochondria to the vicinity of FAs was associated with impaired focal adhesion dynamics and tumor cell invasion when compared with OxPhos‐proficient (wild type [WT]) cells or vehicle‐treated cells. Overall, these studies suggest that only bioenergetically active mitochondria travel to the cortical cytoskeleton and localize to FAs.15 These cortical mitochondria might support enhanced cell motility and invasion by numerous mechanisms (Figure 3), including providing a local source of energy (ATP and guanosine triphosphate [GTP]) to fuel signal transduction and cytoskeleton dynamics at the leading edge. For instance, it was shown that positioning of mitochondria influences the shape of energy gradients in living mouse embryonic fibroblasts (MEFs).16 Preventing accumulation of cortical mitochondria in Miro1−/− MEFs led to reduced ATP/ADP ratios at the cell periphery and impaired actin dynamics, lamellipodia protrusion, and membrane ruffling. Furthermore, Miro1−/− MEFs showed slower migration than WT MEFs. Local energy production was also important in ovarian cancer cells, where mitochondria led to increased ATP concentration at the leading edge of lamellipodia.17 As energy in the form of ATP and GTP is responsible for fueling a myriad of molecular and cellular processes, mitochondrial localization provides a means to concentrate energy where is most needed. Thus, a cancer cell can reprogram mitochondrial localization to respond to extracellular and intracellular cues and switch between highly proliferative (when mitochondria are perinuclear) and a highly invasive (when mitochondria are at the leading edge) phenotype. Indeed, it has been shown that key regulators of mitochondrial trafficking via microtubules control the balance between proliferation and motility18 (see below).
4. MICROTUBULE‐BASED MITOCHONDRIAL MOVEMENT
The earliest studies suggesting a role for mitochondrial trafficking in cancer involve TRAK1. TRAK1 is upregulated in gastric19 and colorectal cancer,20 and higher TRAK1 levels correlated with poorer prognosis of patients. In a recent study, TRAK1 was shown to crosstalk to the Arf6–AMAP1 pathway to control of mitochondrial positioning in highly invasive breast cancer cells. TRAK1 was key to buffer oxidative stress and sustain tumor cell invasion.21
More recently, MIRO1 has emerged as a regulator of cancer cell motility.22 In these studies, anterior localization of mitochondria to the migrating front of the cells led to faster migration velocities and increased directional persistence. Depletion of MIRO1 efficiently reduced cell motility in breast cancer cells.22 Since then, MIRO1 has emerged as a common modulator of tumor cell migration in breast,22 pancreatic,23 glioblastoma,24 and colon cancer.25 Furthermore, pancreatic cancer is associated with upregulated expression of MIRO1,23 and higher levels of MIRO1 correlate with lymph node metastasis and reduced overall survival.26 MIRO2 was consistently upregulated in tumors of disparate origin, including breast, brain, prostate, lung, and melanoma, among others.24 The role of MIRO2 in tumor cell invasion was context dependent, while MIRO2 was dispensable for cell motility defects on SNPH‐deficient cells (see below), it was shown to mediate therapy‐adaptive responses.24 While all evidence points to a general role of MIRO1 as a positive regulator of tumor cell motility, conclusive evidence that links MIRO1's role in mitochondrial trafficking to its effect on tumor cell invasion is missing.
Kinesins have also been implicated in cancer. For instance, KIF5B prevents epithelial to mesenchymal transition.27 Thus, knockdown of KIF5B resulted in diminished cell proliferation, cell shape changes and increased cell migration. KIF5B also participates in exocytosis of membrane‐tethered membrane type 1–matrix metalloproteinase (MT1‐MMP).28 The binding of phospholipase D2–generated signaling lipid phosphatidic acid and KIF5B was required for the vesicular association of KIF5B and surface localization of MT1‐MMP.29 Of note, both functions are independent from kinesin's role as a mitochondrial motor. KIF5B also participates in common lung cancer–driving fusions with ALK,30 MET,31 and RET.32 Notably, KIF5B‐RET–driven tumors show enhanced sensitivity to molecular therapies.33, 34 Another member of the KIF5 family, KIF5A, is upregulated in breast cancer.35 Functional studies concluded that KIF5A overexpression in basal breast cancer confers resistance to taxane‐based chemotherapy.35, 36 Interestingly, the bromodomain protein ANCCA/ATAD2, previously shown to be an estrogen‐induced chromatin regulator, plays a crucial role in the upregulating kinesins by estrogen.37 Thus, in ER+ breast cancer, KIF5A and B are highly expressed. Another study suggests a potential role for KIF5A in the pathogenesis of prostate cancer, since KIF5A was among commonly mutated genes.38 To validate this possibility, functional studies will be required. However, the numerous kinesin‐bound cargoes (vesicles, organelles, protein complexes, and mRNAs) preclude from drawing conclusions as to what exactly is the contribution of mitochondria transport to these phenotypes.
The mitochondria‐anchoring protein SNPH is a negative regulator of mitochondrial trafficking in neurons. In a recent study, we showed that SNPH antagonizes cancer cell invasion in glioblastoma, breast, lung, and prostate models.24 SNPH was downregulated across disparate tumor types, and lower levels of SNPH correlated with tumor progression, metastatic dissemination, and poorer survival of patients with lung, colon, prostate, and breast cancer (Figure 4).24 At the cellular level, SNPH inhibited the speed and distance travelled by individual mitochondria and suppressed organelle shape changes. In turn, repositioning of mitochondria away from focal adhesions blocked chemotaxis and metastasis, in vivo.24 Mechanistically, both MIRO1 and Kinesin‐1 were required for SNPH‐dependent inhibition of cell motility. Surprisingly, MIRO2 was dispensable for SNPH's functions. In follow‐up studies, we uncovered a novel isoform of SNPH that localizes to the mitochondrial matrix and maximizes the activity of the OxPhos complex II.18 Thus, SNPH knockdown increased mitochondrial superoxide production and oxidative stress‐dependent tumor cell motility. Importantly, endogenous metastatic tumors in mice displayed lower SNPH levels and enhanced markers of oxidative stress and epithelial to meshenchymal transition (EMT).18 Furthermore, this novel intramitochondrial SNPH isoform was able to block metastatic dissemination in a syngeneic melanoma mice model.18 Given this role of SNPH in modulating cell metabolism and proliferation versus motility of cancer cells, it was not surprising that hypoxic or oxidative stress quickly downregulated SNPH expression.18 SNPH was also downregulated in tumors with mutations or deletions of von Hippel‐Lindau (VHL),18 the negative regulator of the hypoxia inducible transcription factors.
Figure 4.
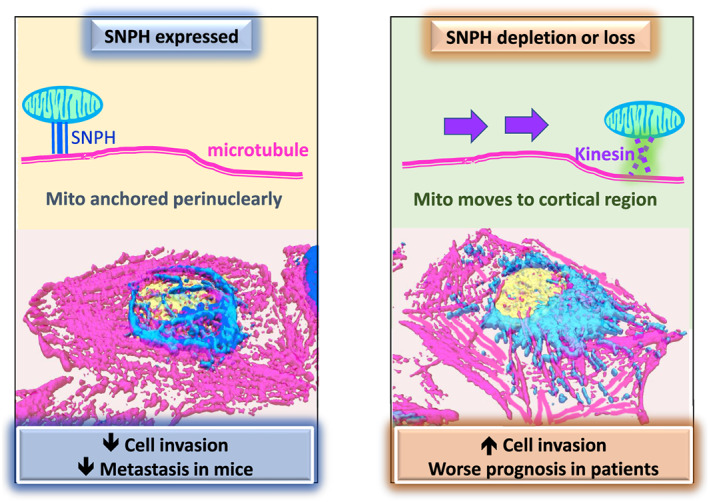
SNPH prevents metastasis. In tumors with high expression of SNPH, mitochondria are anchored perinuclearly, leading to lower cell invasion and preventing metastatic dissemination in mice. In tumors with loss of SNPH expression, mitochondria are free to move to the cortical cytoskeleton via Kinesin/MIRO1 complexes. These cortical mitochondria fuel enhanced tumor cell invasion and correlate with poorer prognosis in patients
In summary, we know that mitochondrial trafficking in cancer cells follows the neuronal model, relying on microtubules and Kinesin‐1 complexes with MIRO1 and TRAK. One important difference is that SNPH anchors mitochondria perinuclearly (− end of microtubules) on cancer cells, but it accumulates mitochondria in axons (+ end of microtubules) on neurons. Since higher levels of SNPH favor tumor cell proliferation, it might be that SNPH's anchoring of mitochondria is essential for cell division. Mechanisms by which SNPH levels and function are regulated on cancer are less clear, raising a number of questions. For example, how are SNPH's levels downregulated in tumors? Which signaling pathways converge on SNPH to activate mitochondrial anchoring? Are the levels or activity of SNPH regulated by stimuli other than hypoxia and oxidative stress?
We also know that tipping the balance towards increased mitochondrial trafficking (e.g., by downregulation of SNPH) leads to enhanced metastatic competence of tumor cells (Figure 4). Given that SNPH is commonly downregulated in tumors and further reduced in metastatic tissues, further investigation into the molecular underpinnings that control mitochondrial trafficking in SNPH‐low tumor cells is warranted. While MIRO1 and Kinesin‐1 were essential to restrain enhanced tumor cell invasion on SNPH‐low cells, it remains to be tested whether ablating MIRO1/Kinesin‐1 in tumors can block metastatic dissemination in vivo. On the long term, these studies will shed light into potential actionable targets to reduce metastatic dissemination in SNPH‐low tumors.
5. ACTIN‐BASED MITOCHONDRIAL MOVEMENT
Currently, MYOs are the only known actin‐based motor. There are over 20 classes of MYOs in humans. MYOs type I and II are conventional and make up basic contractile units of skeletal muscle and heart tissue.39, 40 Traditionally, MYOs were thought to only participate in or makeup muscle fibers, thus all other MYOs discovered thereafter are considered unconventional. MYO19, MYO5, and MYO6 are unconventional MYOs that have provided the most evidence of MYO‐guided mitochondrial movement. Many studies highlight their dysregulation in cancer and are strong factors in invasion and avoidance of apoptosis. Despite these advances, the adapter proteins and regulators have yet to be explored.
Within the last decade, the unconventional MYO19 gained attention as the canonical mitochondrial transporter that moves towards the + end of actin filaments.9, 41 The earliest studies on MYO19 established that its overexpression led to increased mitochondrial movement in lung adenocarcinoma.11 Since then, studies on MYO19 have followed suit to elucidate its function. There is evidence that MYO19 is partially responsible in ensuring mitochondrial distribution during cell division. MYO19 siRNA knockdowns in HeLa cells caused daughter cells to asymmetrically inherit mitochondria, but this was not lethal.42 Another study reported MYO19‐dependent induction of filopodia in glucose‐starved conditions in U2OS cells. Mitochondrial localization was in part attributed to the buildup of ROS, and the addition of H2O2 could also elicit mitochondrial polarization.43, 44, 45 Interestingly, the MYO19‐specific mitochondria outer membrane association domain (MyMOMA) is sufficient to localize with mitochondria in vitro. However, acidic conditions liberated mitochondria from the MyMOMA domain. The authors described amino acids 882 and 883 essential for mitochondrial interaction; however, the mechanism by which MYO19 binds to mitochondria in neutral pH conditions remains unknown.11, 46 Future studies are needed to identify an adapter protein (e.g., an outer mitochondrial membrane protein) that anchors MYO19 to mitochondria.
Human MYO5 family members include MYO5A, MYO5B, and MYO5C. Yeasts harbor a homolog of the human class V MYO, Myo2. There appears to be a consensus that Myo2 ensures symmetrical mitochondrial inheritance.47, 48 Myo2 is directly related to mitochondrial localization,49, 50 since its overexpression results in inefficient polarization of mitochondria.51 These findings are recapitulated in Drosophila neurons, diminishing MyoV liberates mitochondria, suggesting a docking role.10 More recently, human MYO5s have been linked to actin‐mediated mitochondrial transport.
MYO5A is largely expressed in neuronal and skin tissues and is the best understood of the human class V MYOs.52 Like yeast Myo2, human MYO5A was shown to directly associate with mitochondria in melanoma and mouse pancreatic cells.53, 54 In addition to associating with mitochondria, there is mounting evidence that class V MYOs are highly expressed in cancer.55, 56 Namely, microarray analysis of pituitary adenomas pointed to stronger invasive phenotypes when MYO5A was overexpressed compared to noninvasive samples.57 In melanoma, MYO5A was shown to export methotrexate and knockdowns subsequently sensitized cells to treatment.58 Another group verified that diminishing MYO5A in melanoma reduced migration and invasion in vitro.59 Bcl‐xL, a negative regulator of the apoptotic pathway, was shown to physically associate with MYO5A in immunoprecipitations in a mouse model of pancreatic islet tumorigenesis. In this context, MYO5A is thought to synergize with the antiapoptotic and protumorigenic effects of Bcl‐xL.60 This work has underscored the importance of the MYO5A on migratory phenotypes and possible activation and crosstalk with pathways other than apoptosis. Although MYO5A was associated with cancer, we still do not know the mechanism of how it becomes pathogenic or the inciting stimuli that may alter normal function.
Modern research has emphasized the link between MYO5A and EMT. Snail, a master regulator of EMT, was shown to bind the promoter of MYO5A in both lung and colon cancer models. MYO5A RNAi resulted in a decreased invasion in vitro and reduced metastases in vivo.61 Congruent with this idea, MYO5A was associated with worse survival in previously untreated, patient‐derived esophageal squamous cell carcinoma (ESCC). In these cases, MYO5A also correlated with the increase in Vimentin mRNA and the loss of E‐cadherin mRNA. Furthermore, MYO5A ablation resulted in decreased migratory behavior.62 Taken together, there is a growing evidence that class V MYOs are instrumental in the metastatic cascade and driving cancer development. Snail‐mediated activation of MYO5A is probably one of many possible explanations for the changes in migratory behavior; however, MYO5A was not shown to localize to mitochondria in that context. While these studies suggest that mitochondria might be required for EMT, further studies should unequivocally address this idea. Additionally, there should be a focus on MYO5A as an integrator of signaling cascades downstream of EMT or apoptosis to help explain divergent functions of MYO5A depending on cancer context.
The other two human MYOs have not been yet associated with mitochondrial movements; however, they have been shown to correlate with patient prognosis. In a meta‐analysis, MYO5B was identified as a biomarker in colorectal cancers for poorer patient outcome.63 MYO5B is present in some neural tissues, colon, and testes. MYO5B participates in recycling endosome trafficking and membrane recycling through the Rab family members.64, 65 MYO5C is the least characterized class V MYO and is associated with epithelial, glandular, and secretory tissues. MYO5C retains only half of the sequence similarity compared with the other human MYOs but is still structurally similar.52 It is shown to interact with secretory granules and localizes distinctly from other MYO cargo.66 Although MYO5C has not yet been shown to interact with mitochondria, it has been proposed as a putative biomarker for prostate cancer. Further studies are needed to delineate MYO5C's role in prostate cancer, yet alone whether MYO5C plays a role in mitochondrial trafficking at all.
MYO6 is an emerging player in cytoskeletal dynamics and cancer progression. In contrast to MYO5, MYO6 is a − end directed motor.67 MYO6 has been used as a marker for invasive border cells in ovarian cancer. Microarray analyses revealed that increased expression of MYO6 is associated with an aggressive phenotype compared with benign ovarian tumors. In this study, they revealed that silencing MYO6 reduced migration in vitro and metastases in nude mice but did not affect proliferation rates.68 Contrarily, microarray analysis of medium‐grade prostate cancer was associated with an increase of MYO6 expression. Interestingly, higher grade, androgen‐independent cell lines showed a reduction in expression of MYO6. Depleting MYO6 also caused a marked increase in a tumor suppressor, vitamin D3 upregulated protein 1 (VDUP1), suggesting a protumorigenic role for MYO6.69 Overall, MYO6 is a contributor to some aspects of pathogenesis but dispensable in other contexts. Its presence in cancer still stresses MYO6 as a potential modulator of aberrant behavior.
Although MYO6 has not been shown to directly drive the typical cancer hallmarks, it was shown to interact with mitophagy mediators.70 Shortly after that initial finding, MYO6 was verified to interact with the E3 ubiquitin ligase Parkin and ubiquitin‐marked mitochondria upon induced mitochondrial damage in HEK293 cells. In these studies, MYO6 knockdown in MEFs resulted in inefficient mitophagy. Other evidence points out an involvement of MYO6 in mitophagy. MYO6 was shown promote F‐actin encapsulating waves to sequester mitochondria destined for degradation. Consistent with this role, MYO6 was required for full mitochondrial operating capacity.71, 72 From these studies, we see a divergent role for MYO6 in the mitophagy cascade. Interestingly, the authors showed that MYO6's association with mitochondria was Parkin‐dependent. To our knowledge, the other instance in which MYO6 was shown to interact with mitochondria was in Drosophila oocytes.73 As mitochondria are needed to support several of the hallmarks of cancer,1 we propose that MYO6‐dependent mitophagy could serve as initiators or promoters of precancerous phenotypes. Further work should be conducted to determine if MYO6 associates with mitochondria exclusively during mitophagy or if it has other functions pertaining to mitochondria.
In summary, MYOs drive some of the hallmarks of cancer progression. Specifically, inhibiting apoptotic signaling and altering distinctive epithelial and mesenchymal markers to induce invasion. From this body of evidence, we are left with many questions pertaining to the specificity and regulation of MYOs. Many of the MYOs affect migration, invasion, and metastases in vivo. An additional layer of complexity might be contributed by the fact that multiple MYOs might be coexpressed in the same cell types and thus compete for mitochondria binding. What converging feature allows MYOs to have distinct functions in their respective tissues, yet all alter cell mobility? One alternative is that different MYO‐cargoes, including mitochondria, would mediate divergent functions of MYOs. Yet another possibility is that mitochondria are the common denominator that explains the cell motility phenotypes for all the MYOs, seeing as all of them (except MYO5B and MYO5C) have evidence of interaction with mitochondria. This idea is supported by the fact that invasion and migration require copious amounts of energy, thus requiring and adaptation and the need for mitochondria in select locations. Another open question is how are these mitochondria recruited to the MYOs? Aside from the MYO19 outer membrane association domain, there are no known MYO5 or MYO6 adapters to mitochondria. The mechanisms of regulation of these MYOs are largely unknown. Realistically, the involvement of MYO/mitochondria in metastatic pathology is probably a multifaceted process that requires integration and transduction of many signals; all of which must be identified.
One interesting feature of microtubule‐mediated transport is that SNPH can sense the OxPhos capacity of transported mitochondria. Thus, mitochondria that are deficient in OxPhos are not engaged in long‐term movements. It can be postulated that a similar mechanism of quality control might be at place on actin‐mediated transport. In this line of thought, a yet to be identified sensor might select active versus inactive mitochondria for engagement with MYOs and active transport.
6. SIGNALING PATHWAYS THAT REGULATE MITOCHONDRIAL LOCALIZATION
Within the past decade, it has become evident that signaling components regulate mitochondrial movement via calcium‐induced conformational changes and posttranslational modifications mediated by kinases, GTPases, and E3 ubiquitin ligases.
High concentrations of calcium inhibit mitochondrial trafficking, by regulating MIRO1/2. When MIRO1's EF‐hands bind Ca2+, binding to KIF5 is reduced. Therefore, Ca2+ inhibits mitochondrial movement by disengaging MIRO from its respective motor protein.74 The prominent role of calcium as a second messenger suggests that mitochondrial trafficking might be regulated by a myriad of signaling pathways that culminate in calcium release into the cytosol. Furthermore, mitochondria survey and buffer intracellular levels of calcium; thus, one might hypothesize that regulation of MIRO coupling to kinesins might provide a way to locally sense calcium spikes in the mitochondrial surface. In this manner, mitochondrial trafficking can be switched on and off according to fluctuations in calcium signaling.
A second signaling mechanism to control mitochondrial trafficking is ubiquitination of SNPH. In a recent study, it was demonstrated that ubiquitination of several residues in SNPH is required for proper association of SNPH with Tubulin, thus tethering the mitochondria to the microtubule.75 Mechanistic studies showed that SNPH is modified by the ubiquitin ligase, CHIP/STUB1, and deubiquitinated in an USP7‐dependent manner. These studies pointed to ubiquitination of SNPH as a key regulator of mitochondrial trafficking and tumor cell motility and invasion.75
Environmental cues such as ROS, nutrients, or oxygen concentration have all been shown to regulate mitochondrial trafficking. For instance, ROS drive cancer motility through the actions of polarized mitochondria.43, 76, 77 Mechanistically, the ROS‐sensitive MAPK, p38, was shown to target the MIRO/TRAK complexes to decrease mitochondrial trafficking.78
Glucose fluctuations can also elicit post‐translational modifications that directly affect mitochondrial movement. For instance, high extracellular glucose induced O‐GlcNAcylation on the mitochondrial adapter TRAK1 in hippocampal neurons. As a result, mitochondrial movement is negatively regulated by glucose starvation in an O‐GlcNac transferase (OGT)–dependent manner.79 It is conceivable that a similar mechanism is at play in tumor cells; however, future studies will need to look into this possibility.
Lastly, hypoxia‐upregulated mitochondrial movement receptor (HUMMR) was induced to associate with the MIRO/TRAK complex and inhibited mitochondrial movement under a hypoxic environment. While this is a mechanism that was described in neuronal cells,80 one might postulate that it is shared by cancer cells. In this context, recent evidence supports alternate mechanisms by which hypoxia controls mitochondria localization in cancer cells. Tumor cells exposed to hypoxia quickly downregulated SNPH protein and mRNA levels, which led to enhanced cortical mitochondrial localization and invasiveness of glioblastoma cells.18 Interestingly, tumors where HIF1α was stabilized due to mutations or deletions in its negative regulator VHL correlated with lower expression of SNPH, arguing that this mechanism is relevant in vivo.18
Another mechanism that controls mitochondria's movement is physical removal of MIRO by degradation or mitophagy. This is mediated primarily by the actions of the E3 ubiquitin ligase, Parkin. The PTEN‐induced Kinase 1 (PINK1)/Parkin pathway was discovered as the most common hereditary driver of Parkinson's disease when mutated. Under basal conditions, PINK1 is degraded by the inner membrane serine protease, PARL.81 When mitochondria are damaged, PINK1 accumulates on the surface of the mitochondria and directly phosphorylates Parkin.82, 83 Once Parkin is activated by phosphorylation, it can initiate mitophagy by ubiquitination of outer mitochondrial membrane proteins.84, 85, 86 In addition, PINK1 targets MIRO. PINK1‐induced phosphorylation on MIRO1 at S156 induces mitochondrial arrest and induces MIRO1 degradation. However, this phosphorylation site is not sufficient to induce mitophagy.87, 88
In summary, an increasing number of signaling pathways are being linked to mitochondrial trafficking. Since this is an emerging field of investigation, it is highly probable that there are other nodes of regulation yet to be identified. Molecules such as MIRO, SNPH, TRAK, and FEZ appear to be pivotal regulators of mitochondrial trafficking that have repercussions intracellularly (subcellular mitochondrial localization) and possibly extracellularly (mitochondrial‐mediated metastasis). Further studies are needed to fully characterize posttranslational modifications and signaling pathways that regulate the activation, availability, and degradation of the core trafficking components.
7. CONCLUSIONS
In order for cancer cells to grow and evolve, they must carefully balance the many roles of mitochondria as metabolic and signaling hubs, apoptosis, and calcium/ROS homeostasis. Over the past few years, we have learned that mitochondria are not static, solitary organelles, but they rather undergo constant changes in morphology and subcellular distribution to meet the metabolic and homeostatic demands of the cell.
Up to date, most of the studies have shown that dysregulated mitochondrial trafficking fuels high cell motility and invasion, leading to metastasis of cancer cells. While these mechanisms of mitochondrial trafficking were characterized and mostly studied in neurons, only recently it has been appreciated that tumor cells exploit mitochondrial trafficking to fuel metastatic traits. This is an emerging field of study, where many open questions remain unanswered. Some of the most basic questions include what are the types and compositions of trafficking complexes present in cancer cells? As it has been shown that kinesin complexes might use different alternate adapters in neurons (SYBU, MIRO1, FEZ1, and RanBP2), it might be possible that several complexes cooperate to move mitochondria along microtubules in cancer cells as well. Likewise, the three MYOs (MYO19, MYO5A, and MYO6) might compete or cooperate for short‐term movement of mitochondria along actin filaments. Knowledge of the different complexes might open opportunities to characterize the spatio‐temporal regulation of trafficking. One possibility is that cells prefer a motor complex for trafficking between certain subcellular regions (e.g., MYO19 from the cortical cytoskeleton into the filopodia, but not into invadopodia). Alternatively, migrating cells might utilize a preferred motor for moving mitochondria into the migratory front versus the retracting tail of the cells.
Another question that warrants further investigation is whether there is crosstalk between the tubulin and actin transport systems. How is the sharing and transfer of cargoes between kinesin, dynein, and MYOs coordinated? One study bridged this gap where it modeled MYO5A gliding over microtubules, suggesting a mechanism that allows the switching of microtubule and actin cargoes.89 Similarly, the actin‐binding domain of the adapter protein Melanophilin could be phosphorylated and change MYO5A's preference from actin to microtubules.90 This evidence also adds a layer of complexity that not only the motor domain but also the adapters could dictate crosstalk between these transport systems. Despite these exciting findings, the subject of filament switching still requires further investigation and validation in other cell systems. We also need to know whether there is competitive binding of the different complexes to mitochondria and how is this regulated and also whether mitochondria competes with other cargoes for binding to molecular motors. Another related issue to examine is whether mitochondria recruitment to a particular motor complex is specific on the context?
For the actin‐MYO trafficking, more studies should focus on the regulation of the recruitment of MYO5a and MYO6 to the mitochondria. MYO6 was shown to switch from a motile to anchoring position in adenosine diphosphate (ADP)–rich conditions, which allowed a competing MYO5 “win” the cargo,91 but this still requires examination. In addition, the adaptor proteins that link MYO5A/6 to mitochondria have not been identified yet. Is there an actin‐anchoring protein similar to SNPH? This putative actin‐anchoring protein might contribute to negative regulation of mitochondrial movement, by immobilizing organelles at sites of intensive energy requirements or where other mitochondrial functions are locally needed.
From a cancer‐related standpoint, we have limited knowledge on potential differences between normal and cancer tissues regarding expression and genomic/epigenetic alterations on the molecular machinery that regulates trafficking. Another point in need of studies is the role of each of the individual components of the trafficking complexes during tumor progression. While the evidence shows that MIRO1, SNPH, MYO19, and MYO5A are common regulators of tumor cell motility, invasion and EMT in cell culture and experimental models of metastasis, we lack studies that address the requirements of these genes in endogenous models of tumor progression. Thus, future studies should focus on understanding the spatio‐temporal regulation of mitochondrial trafficking, emphasizing on the molecular complexes, during the natural progression of cancer. Likewise, there is limited information on how the genetic, epigenetic, and microenvironmental factors influence mitochondrial trafficking or which signaling pathways integrate extracellular stimuli with mitochondrial localization in cancer.
Finally, an area in need of further investment is the development of selective small molecule inhibitors that target these pathways. The fact that several tumors show increased expression of kinesin or MYO complexes might warrant further effort in this area.
CONFLICT OF INTEREST
The authors declare that no conflicts of interest exist.
AUTHORS' CONTRIBUTION
All authors had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization, M.C.C.; Methodology, M.C.C. and M.F.; Investigation, M.C.C. and M.F.; Writing ‐ Original Draft, M.C.C. and M.F.; Writing ‐ Review & Editing, M.C.C. and M.F.; Supervision, M.C.C.; Funding Acquisition, M.C.C.
ACKNOWLEDGMENTS
The authors would like to apologize to those colleagues whose work could not be cited or discussed in sufficient detail due to space limitation. M.F. is supported by a training grant NCI T32‐GM007635‐40 from the National Cancer Institute (NCI). M.C.C. is supported by ACS IRG #16‐184‐56 from the American Cancer Society. The content is solely the responsibility of the author and does not necessarily represent official views of the ACS. The funders had no role in decision to publish or preparation of the manuscript.
Furnish M, Caino MC. Altered mitochondrial trafficking as a novel mechanism of cancer metastasis. Cancer Reports. 2020;3:e1157. 10.1002/cnr2.1157
REFERENCES
- 1. Zong WX, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell. 2016;61(5):667‐676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chevalier‐Larsen E, Holzbaur EL. Axonal transport and neurodegenerative disease. Biochim Biophys Acta. 2006;1762(11–12):1094‐1108. [DOI] [PubMed] [Google Scholar]
- 3. Barel O, Malicdan MCV, Ben‐Zeev B, et al. Deleterious variants in TRAK1 disrupt mitochondrial movement and cause fatal encephalopathy. Brain. 2017;140(3):568‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhao C, Takita J, Tanaka Y, et al. Charcot‐Marie‐Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105(5):587‐597. [DOI] [PubMed] [Google Scholar]
- 5. Kamal A, Almenar‐Queralt A, LeBlanc JF, Roberts EA, Goldstein LSB. Kinesin‐mediated axonal transport of a membrane compartment containing beta‐secretase and presenilin‐1 requires APP. Nature. 2001;414(6864):643‐648. [DOI] [PubMed] [Google Scholar]
- 6. Schwarz TL. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol. 2013;5(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin MY, Sheng ZH. Regulation of mitochondrial transport in neurons. Exp Cell Res. 2015;334(1):35‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xiao Q, Hu X, Wei Z, Tam KY. Cytoskeleton molecular motors: structures and their functions in neuron. Int J Biol Sci. 2016;12(9):1083‐1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Frederick RL, Shaw JM. Moving mitochondria: establishing distribution of an essential organelle. Traffic. 2007;8(12):1668‐1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pathak D, Sepp KJ, Hollenbeck PJ. Evidence that myosin activity opposes microtubule‐based axonal transport of mitochondria. J Neurosci. 2010;30(26):8984‐8992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Quintero OA, DiVito MM, Adikes RC, et al. Human Myo19 is a novel myosin that associates with mitochondria. Curr Biol. 2009;19(23):2008‐2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kang JS, Tian JH, Pan PY, et al. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short‐term facilitation. Cell. 2008;132(1):137‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Caino MC, Altieri DC. Molecular pathways: mitochondrial reprogramming in tumor progression and therapy. Clin Cancer Res. 2016;22(3):540‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Caino MC, Ghosh JC, Chae YC, et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc Natl Acad Sci U S A. 2015;112(28):8638‐8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Caino MC, Altieri DC. Cancer cells exploit adaptive mitochondrial dynamics to increase tumor cell invasion. Cell Cycle. 2015;14(20):3242‐3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schuler MH, Lewandowska A, Caprio GD, et al. Miro1‐mediated mitochondrial positioning shapes intracellular energy gradients required for cell migration. Mol Biol Cell. 2017;28(16):2159‐2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cunniff B, McKenzie AJ, Heintz NH, Howe AK. AMPK activity regulates trafficking of mitochondria to the leading edge during cell migration and matrix invasion. Mol Biol Cell. 2016;27(17):2662‐2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Caino MC, Seo JH, Wang Y, et al. Syntaphilin controls a mitochondrial rheostat for proliferation‐motility decisions in cancer. J Clin Invest. 2017;127(10):3755‐3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang F, Ren G, Lu Y, et al. Identification of TRAK1 (trafficking protein, kinesin‐binding 1) as MGb2‐Ag: a novel cancer biomarker. Cancer Lett. 2009;274(2):250‐258. [DOI] [PubMed] [Google Scholar]
- 20. An Y, Zhou Y, Ren G, et al. Elevated expression of MGb2‐Ag/TRAK1 is correlated with poor prognosis in patients with colorectal cancer. Int J Colorectal Dis. 2011;26(11):1397‐1404. [DOI] [PubMed] [Google Scholar]
- 21. Onodera Y, Nam JM, Horikawa M, Shirato H, Sabe H. Arf6‐driven cell invasion is intrinsically linked to TRAK1‐mediated mitochondrial anterograde trafficking to avoid oxidative catastrophe. Nat Commun. 2018;9(1):2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Desai SP, Bhatia SN, Toner M, Irimia D. Mitochondrial localization and the persistent migration of epithelial cancer cells. Biophys J. 2013;104(9):2077‐2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li Q, Yao L, Wei Y, Geng S, He C, Jiang H. Role of RHOT1 on migration and proliferation of pancreatic cancer. Am J Cancer Res. 2015;5(4):1460‐1470. [PMC free article] [PubMed] [Google Scholar]
- 24. Caino MC, Seo JH, Aguinaldo A, et al. A neuronal network of mitochondrial dynamics regulates metastasis. Nat Commun. 2016;7(1):13730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mills KM, Brocardo MG, Henderson BR. APC binds the Miro/Milton motor complex to stimulate transport of mitochondria to the plasma membrane. Mol Biol Cell. 2016;27(3):466‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiang H, He C, Geng S, et al. RhoT1 and Smad4 are correlated with lymph node metastasis and overall survival in pancreatic cancer. PLoS One. 2012;7(7):e42234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cui J, Jin G, Yu B, Wang Z, Lin R, Huang JD. Stable knockdown of Kif5b in MDCK cells leads to epithelial‐mesenchymal transition. Biochem Biophys Res Commun. 2015;463(1–2):123‐129. [DOI] [PubMed] [Google Scholar]
- 28. Marchesin V, Castro‐Castro A, Lodillinsky C, et al. ARF6‐JIP3/4 regulate endosomal tubules for MT1‐MMP exocytosis in cancer invasion. J Cell Biol. 2015;211(2):339‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Z, Zhang F, He J, et al. Binding of PLD2‐generated phosphatidic acid to KIF5B promotes MT1‐MMP surface trafficking and lung metastasis of mouse breast cancer cells. Dev Cell. 2017;43(2):186‐197. e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rosenbaum JN, Bloom R, Forys JT, et al. Genomic heterogeneity of ALK fusion breakpoints in non‐small‐cell lung cancer. Mod Pathol. 2018;31(5):791‐808. [DOI] [PubMed] [Google Scholar]
- 31. Plenker D et al. Structural alterations of MET trigger response to MET kinase inhibition in lung adenocarcinoma patients. Clin Cancer Res. 2017. [DOI] [PubMed] [Google Scholar]
- 32. Roskoski, R., Jr. And Sadeghi‐Nejad A., Role of RET protein‐tyrosine kinase inhibitors in the treatment RET‐driven thyroid and lung cancers. Pharmacol Res, 2017;128:1–17. [DOI] [PubMed] [Google Scholar]
- 33. Lin C, Wang S, Xie W, Zheng R, Gan Y, Chang J. Apatinib inhibits cellular invasion and migration by fusion kinase KIF5B‐RET via suppressing RET/Src signaling pathway. Oncotarget. 2016;7(37):59236‐59244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li GG, Somwar R, Joseph J, et al. Antitumor activity of RXDX‐105 in multiple cancer types with RET rearrangements or mutations. Clin Cancer Res. 2017;23(12):2981‐2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. De S et al. Overexpression of kinesins mediates docetaxel resistance in breast cancer cells. Cancer Res. 2009;69(20):8035‐8042. [DOI] [PubMed] [Google Scholar]
- 36. Tan MH, de S, Bebek G, et al. Specific kinesin expression profiles associated with taxane resistance in basal‐like breast cancer. Breast Cancer Res Treat. 2012;131(3):849‐858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zou JX, Duan Z, Wang J, et al. Kinesin family deregulation coordinated by bromodomain protein ANCCA and histone methyltransferase MLL for breast cancer cell growth, survival, and tamoxifen resistance. Mol Cancer Res. 2014;12(4):539‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lindberg J, Mills IG, Klevebring D, et al. The mitochondrial and autosomal mutation landscapes of prostate cancer. Eur Urol. 2013;63(4):702‐708. [DOI] [PubMed] [Google Scholar]
- 39. Chantler PD, Wylie SR, Wheeler‐Jones CP, McGonnell IM. Conventional myosins—unconventional functions. Biophysical Reviews. 2010;2(2):67‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sellers JR. Myosins: a diverse superfamily. Biochimica et Biophysica Acta (BBA) ‐ Molecular Cell Research. 2000;1496(1):3‐22. [DOI] [PubMed] [Google Scholar]
- 41. Lu Z, Ma XN, Zhang HM, et al. Mouse Myosin‐19 is a plus‐end‐directed, high‐duty ratio molecular motor. J Biol Chem. 2014;289(26):18535‐18548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rohn JL, Patel JV, Neumann B, et al. Myo19 ensures symmetric partitioning of mitochondria and coupling of mitochondrial segregation to cell division. Curr Biol. 2014;24(21):2598‐2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shneyer BI et al. ROS induced distribution of mitochondria to filopodia by Myo19 depends on a class specific tryptophan in the motor domain. Sci Rep. 2017;7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ušaj M, Henn A. Kinetic adaptation of human Myo19 for active mitochondrial transport to growing filopodia tips. Sci Rep. 2017;7(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shneyer BI, Ušaj M, Henn A. Myo19 is an outer mitochondrial membrane motor and effector of starvation‐induced filopodia. J Cell Sci. 2016;129(3):543‐556. [DOI] [PubMed] [Google Scholar]
- 46. Hawthorne JL, Mehta PR, Singh PP, Wong NQ, Quintero OA. Positively charged residues within the MYO19 MyMOMA domain are essential for proper localization of MYO19 to the mitochondrial outer membrane. Cytoskeleton. 2016;73(6):286‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Altmann K, Frank M, Neumann D, Jakobs S, Westermann B. The class V myosin motor protein, Myo2, plays a major role in mitochondrial motility in Saccharomyces cerevisiae. J Cell Biol. 2008;181(1):119‐130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Böckler S, Chelius X, Hock N, et al. Fusion, fission, and transport control asymmetric inheritance of mitochondria and protein aggregates. J Cell Biol. 2017;216(8):2481‐2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boldogh IR, Ramcharan SL, Yang HC, Pon LA. A type V myosin (Myo2p) and a Rab‐like G‐protein (Ypt11p) are required for retention of newly inherited mitochondria in yeast cells during cell division. Mol Biol Cell. 2004;15(9):3994‐4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Eves PT, Jin Y, Brunner M, Weisman LS. Overlap of cargo binding sites on myosin V coordinates the inheritance of diverse cargoes. J Cell Biol. 2012;198(1):69‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Donovan KW, Bretscher A. Head‐to‐tail regulation is critical for the in vivo function of myosin V. J Cell Biol. 2015;209(3):359‐365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rodriguez OC, Cheney RE. Human myosin‐Vc is a novel class V myosin expressed in epithelial cells. J Cell Sci. 2002;115(5):991‐1004. [DOI] [PubMed] [Google Scholar]
- 53. Nascimento AAC, Amaral RG, Bizario JCS, Larson RE, Espreafico EM. Subcellular localization of myosin‐V in the B16 melanoma cells, a wild‐type cell line for the dilute gene. Mol Biol Cell. 1997;8(10):1971‐1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Varadi A, Tsuboi T, Rutter GA. Myosin Va transports dense Core secretory vesicles in pancreatic MIN6 β‐cells. Mol Biol Cell. 2005;16(6):2670‐2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Li Y‐R, Yang W‐X. Myosins as fundamental components during tumorigenesis: diverse and indispensable. Oncotarget. 2016;7(29):46785‐46812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ouderkirk JL, Krendel M. Non‐muscle myosins in tumor progression, cancer cell invasion, and metastasis. Cytoskeleton. 2014;71(8):447‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Galland F, Lacroix L, Saulnier P, et al. Differential gene expression profiles of invasive and non‐invasive non‐functioning pituitary adenomas based on microarray analysis. Endocr Relat Cancer. 2010;17(2):361‐371. [DOI] [PubMed] [Google Scholar]
- 58. Fernández‐Pérez MP, Montenegro MF, Sáez‐Ayala M, et al. Suppression of antifolate resistance by targeting the myosin Va trafficking pathway in melanoma. Neoplasia (New York, NY). 2013;15(7):826‐839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Alves CP, Moraes MH, Sousa JF, et al. Myosin‐Va contributes to manifestation of malignant‐related properties in melanoma cells. Journal of Investigative Dermatology. 2013;133(12):2809‐2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yi M, Weaver D, Hajnóczky G. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol. 2004;167(4):661‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lan L, Han H, Zuo H, et al. Upregulation of myosin Va by snail is involved in cancer cell migration and metastasis. Int J Cancer. 2010;126(1):53‐64. [DOI] [PubMed] [Google Scholar]
- 62. Sato N, Fujishima F, Nakamura Y, et al. Myosin 5a regulates tumor migration and epithelial‐mesenchymal transition in esophageal squamous cell carcinoma: utility as a prognostic factor. Hum Pathol. 2018;80:113‐122. [DOI] [PubMed] [Google Scholar]
- 63. Letellier E, Schmitz M, Ginolhac A, et al. Loss of myosin Vb in colorectal cancer is a strong prognostic factor for disease recurrence. Br J Cancer. 2017;117(11):1689‐1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schafer JC, Baetz NW, Lapierre LA, McRae RE, Roland JT, Goldenring JR. Rab11‐FIP2 interaction with MYO5B regulates movement of Rab11a‐containing recycling vesicles. Traffic. 2014;15(3):292‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lapierre, L.A. and Goldenring J.R., Interactions of Myosin Vb with Rab11 Family Members and Cargoes Traversing the Plasma Membrane Recycling System, in GTPases Regulating Membrane Targeting and Fusion. 2005:715–723. [DOI] [PubMed]
- 66. Jacobs DT, Weigert R, Grode KD, Donaldson JG, Cheney RE. Myosin Vc is a molecular motor that functions in secretory granule trafficking. Mol Biol Cell. 2009;20(21):4471‐4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. De La Cruz EM, Wells AL, Rosenfeld SS, Ostap EM, Sweeney HL. The kinetic mechanism of myosin V. Proc Natl Acad Sci. 1999;96(24):13726‐13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yoshida H, Cheng W, Hung J, et al. Lessons from border cell migration in the Drosophila ovary: a role for myosin VI in dissemination of human ovarian cancer. Proc Natl Acad Sci U S A. 2004;101(21):8144‐8149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dunn TA, Chen S, Faith DA, et al. A novel role of myosin VI in human prostate cancer. Am J Pathol. 2006;169(5):1843‐1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sarraf SA, Raman M, Guarani‐Pereira V, et al. Landscape of the PARKIN‐dependent ubiquitylome in response to mitochondrial depolarization. Nature. 2013;496(7445):372‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kruppa AJ, Buss F. Actin cages isolate damaged mitochondria during mitophagy. Autophagy. 2018;14(9):1644‐1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kruppa AJ, Kishi‐Itakura C, Masters TA, et al. Myosin VI‐dependent actin cages encapsulate Parkin‐positive damaged mitochondria. Dev Cell. 2018;44(4):484‐499. e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bohrmann J. Drosophila unconventional myosin VI is involved in intra‐ and intercellular transport during oogenesis. Cell Mol Life Sci. 1997;53(8):652‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Macaskill AF et al. Miro1 is a calcium sensor for glutamate receptor‐dependent localization of mitochondria at synapses. Neuron. 2009;61(4):541‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Seo JH, Agarwal E, Bryant KG, et al. Syntaphilin ubiquitination regulates mitochondrial dynamics and tumor cell movements. Cancer Res. 2018;78(15):4215‐4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hemachandra LP et al. Mitochondrial superoxide dismutase has a protumorigenic role in ovarian clear cell carcinoma. Cancer Res. 2015;75(22):4973‐4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Comito G, Calvani M, Giannoni E, et al. HIF‐1alpha stabilization by mitochondrial ROS promotes met‐dependent invasive growth and vasculogenic mimicry in melanoma cells. Free Radic Biol Med. 2011;51(4):893‐904. [DOI] [PubMed] [Google Scholar]
- 78. Debattisti V, Gerencser AA, Saotome M, Das S, Hajnóczky G. ROS control mitochondrial motility through p38 and the motor adaptor Miro/Trak. Cell Rep. 2017;21(6):1667‐1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pekkurnaz G, Trinidad JC, Wang X, Kong D, Schwarz TL. Glucose regulates mitochondrial motility via Milton modification by O‐GlcNAc transferase. Cell. 2014;158(1):54‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li Y, Lim S, Hoffman D, Aspenstrom P, Federoff HJ, Rempe DA. HUMMR, a hypoxia‐ and HIF‐1alpha‐inducible protein, alters mitochondrial distribution and transport. J Cell Biol. 2009;185(6):1065‐1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Narendra DP, Jin SM, Tanaka A, et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8(1):e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shiba‐Fukushima K, Imai Y, Yoshida S, et al. PINK1‐mediated phosphorylation of the Parkin ubiquitin‐like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep. 2012;2(1):1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kazlauskaite A, Kondapalli C, Gourlay R, et al. Parkin is activated by PINK1‐dependent phosphorylation of ubiquitin at Ser65. Biochem J. 2014;460(1):127‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin‐mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119‐131. [DOI] [PubMed] [Google Scholar]
- 85. Chan NC, Salazar AM, Pham AH, et al. Broad activation of the ubiquitin‐proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20(9):1726‐1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AHV, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin‐dependent manner upon induction of mitophagy. Hum Mol Genet. 2010;19(24):4861‐4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wang X, Winter D, Ashrafi G, et al. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell. 2011;147(4):893‐906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Shlevkov E, Kramer T, Schapansky J, LaVoie MJ, Schwarz TL. Miro phosphorylation sites regulate Parkin recruitment and mitochondrial motility. Proc Natl Acad Sci U S A. 2016;113(41):E6097‐E6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ali MY, Krementsova EB, Kennedy GG, et al. Myosin Va maneuvers through actin intersections and diffuses along microtubules. Proc Natl Acad Sci. 2007;104(11):4332‐4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Oberhofer A, Spieler P, Rosenfeld Y, et al. Myosin Va's adaptor protein melanophilin enforces track selection on the microtubule and actin networks in vitro. Proc Natl Acad Sci. 2017;114(24):E4714‐E4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ali MY, Kennedy GG, Safer D, Trybus KM, Sweeney HL, Warshaw DM. Myosin Va and myosin VI coordinate their steps while engaged in an in vitro tug of war during cargo transport. Proc Natl Acad Sci. 2011;108(34):E535‐E541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Zhou W, He MR, Jiao HL, et al. The tumor‐suppressor gene LZTS1 suppresses colorectal cancer proliferation through inhibition of the AKT‐mTOR signaling pathway. Cancer Lett. 2015;360(1):68‐75. [DOI] [PubMed] [Google Scholar]
- 93. Al Nakouzi N, Cotteret S, Commo F, et al. Targeting CDC25C, PLK1 and CHEK1 to overcome docetaxel resistance induced by loss of LZTS1 in prostate cancer. Oncotarget. 2014;5(3):667‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Lovat F, Ishii H, Schiappacassi M, et al. LZTS1 downregulation confers paclitaxel resistance and is associated with worse prognosis in breast cancer. Oncotarget. 2014;5(4):970‐977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Vecchione L, Gambino V, Raaijmakers J, et al. A vulnerability of a subset of colon cancers with potential clinical utility. Cell. 2016;165(2):317‐330. [DOI] [PubMed] [Google Scholar]
- 96. Navarro MS, Bachant J. RanBP2: a tumor suppressor with a new twist on TopoII, SUMO, and centromeres. Cancer Cell. 2008;13(4):293‐295. [DOI] [PubMed] [Google Scholar]