

Abstract
Diethylpyrocarbonate (DEPC)-based covalent labeling together with mass spectrometry is a promising tool for the higher-order structural analysis of antibody therapeutics. Reliable information about antibody higher-order structure can be obtained, though, only when the protein’s structural integrity is preserved during labeling. In this work, we have evaluated the applicability of DEPC reaction kinetics for ensuring the structural integrity of monoclonal antibodies (mAbs) during labeling. By monitoring the modification extent of selected proteolytic fragments as a function of DEPC concentration, we find that a common DEPC concentration can be used for different monoclonal antibodies in formulated samples without perturbing their higher-order structure. Under these labeling conditions, we find that the antibodies can accommodate up to four DEPC modifications without being structurally perturbed, indicating that multi-domain proteins can withstand more than one label, which contrasts to previously studied single-domain proteins. This more extensive labeling provides a more sensitive measure of structure, making DEPC-based covalent labeling-mass spectrometry suitable for the higher-order structural analyses of mAbs.
Keywords: Covalent Labeling, Diethylpyrocarbonate, Mass Spectrometry, Liquid Chromatography, Protein Higher-Order Structure, Protein Higher-Order Structural Analysis, Reaction Kinetics
GRAPHICAL ABSTRACT
Reaction kinetics can be used to ensure the structural integrity of multi-domain monoclonal antibodies (mAbs) during labeling, allowing reliable information about higher-order structure of mAbs to be obtained from covalent labeling.
INTRODUCTION
Mass spectrometry (MS) has emerged as one of the leading techniques used for the routine structural analysis of antibody therapeutics to ensure the quality, efficacy and safety throughout a product life cycle.1-4 Higher-order structure (HOS) is one of the quality attributes of therapeutic monoclonal antibody (mAb) that has to be characterized as changes in HOS can cause decrease in therapeutic efficacy, reduced stability, or possible immunogenicity.5-7 In addition, identifying specific residues involved in antigen-antibody interactions, i.e. epitope/paratope mapping, is particularly useful to evaluate epitope novelty, predict immunogenicity, and assess and optimize binding characteristics.8, 9 The wider use of MS-based methods to probe protein’s HOS results from (a) an ability to obtain residue-level structural information that optical spectroscopy cannot provide, and (b) a more rapid, sensitive, and sample-efficient analysis compared to X-ray crystallography and NMR spectroscopy.
The two most commonly used MS-based tools for studying mAb HOS are hydrogen-deuterium exchange (HDX) and covalent labeling (CL). HDX has been extensively used to investigate HOS changes and aggregation of mAbs during drug development.10-15 Epitope mapping and comparability studies are also examples of how HDX-MS has been used.14-22 CL of amino acid side chains is another technique that can be used to encode a protein’s structural properties into the mass of protein, allowing information about a protein’s structure in solution to be read-out through MS-based bottom-up approaches.23-27 CL-MS has more recently been applied to study mAb higher order structure. Compared to hydrogen-deuterium exchange (HDX), CL benefits from limited label loss and scrambling due to the irreversible nature of the labeling reaction. Non-specific reagents (e.g., hydroxyl radicals, carbenes, and diethylpyrocarbonate) can simultaneously probe a range of different side chains, allowing excellent structural resolution to be obtained. Hydroxyl radical footprinting (HRF) has been used to characterize the conformations and dimer interfaces of mAb therapeutics and to perform epitope mapping for various antigen-antibody interactions.28-33 Carboxyl group footprinting has also been used to characterize the HOS of mAbs and was found to provide complementary information to HRF.34
Our group has developed and used diethylpyrocarbonate (DEPC) as a labeling reagent because it is simple to use, commercially available, and labels a range of nucleophilic residues and the N-terminus of proteins once added into a solution. DEPC CL-MS has been used to reveal aggregation sites and HOS changes for heat-stressed murine IgG1 and rituximab, providing structural information for up to 30% of the residues in these mAbs.35, 36 An important principle of CL-MS experimental design is that a protein’s structure must not be perturbed during the labeling reaction, so that the probe can correctly report accurate structural information.23, 37 There are at least three general ways to assess a protein’s structural integrity upon CL. One way is to use a complementary measurement, such as CD spectroscopy, fluorescence spectroscopy, or an activity assay, to monitor any HOS change after labeling.36-38 These techniques are typically not sensitive enough to detect local structural perturbations. A second way is to ensure that the labeling reaction occurs faster than any label-induced HOS change can happen, guaranteeing a protein’s HOS is not affected during CL. This approach only works for fast labeling chemistries, e.g. HRF, that require laser or synchrotron sources.39-42
A third method, which is more reliable and effective, is to monitor labeling reaction kinetics. By plotting the unmodified fraction of a protein (or better, each of its peptide fragments) as function of reagent concentration, any labeling-induced structural perturbations can be revealed by deviations from the proper reaction order kinetics, e.g. pseudo first-order kinetics for HRF and second-order kinetics for DEPC CL.37, 43-45 Such dose-response plots of an intact protein are less useful than plots of peptide fragments as intact protein plots are ensemble averages, much like CD measurements, and do not provide site-specific information. Dose-response plots for proteolytic fragments obtained from bottom-up analysis of a labeled protein have been shown to be much more sensitive and reliable probes of possible protein structural perturbations as they provide information about any local structural perturbations caused by either nearby or distant modifications.37 Deviations in the kinetics allow the researcher to identify the highest reagent dose to use while still ensuring the structural integrity of the protein. Using the highest dose, while preventing structural perturbations, maximizes the labeling extent and thus the structural resolution of the method. Kinetic measurements via dose-response plots have been effective for ensuring the reliability of the structural information for proteins ranging from cytochrome c to lysozyme to gelsolin.43-45 These measurements, however, have only been used for, single-domain proteins, and so an interesting question is whether they are beneficial for multi-domain proteins like mAbs. Moreover, it would be valuable to know if CL on a residue in one domain of a protein (e.g. variable light chain in a mAb) can affect the structure and labeling of a distant domain (e.g. constant heavy chain in a mAb).
In this work, we have acquired DEPC labeling dose-response plots for proteolytic fragments of different mAbs to: (1) test the applicability of this method for ensuring the structural integrity of multi-domain mAbs during CL-MS-based structural analyses, (2) identify if a common DEPC molar excess might be suitable for DEPC-based CL-MS analyses of mAbs, and (3) evaluate the extent to which a multi-domain protein’s structure is sensitive to multiple DEPC modifications. Results suggest that multi-domain proteins can withstand more than one DEPC label without being perturbed structurally, which allows a more sensitive and higher resolution measure of structure because more modifications can be detected. In addition, based on the limited set of mAbs studied here, there seems to be a common DEPC molar ratio that can be used without worrying about structural perturbations.
EXPERIMENTAL
Materials
Rituximab (Rituxan® 100 mg/10 mL vial, lot#3209283, Genentech) and the NIST Monoclonal Antibody Reference Material 8671 (NISTmAb 10 mg/mL vial, lot#14HB-D-002) were ordered from Myoderm (Norristown, PA) and the National Institute of Standards and Technology (NIST, Gaithersburg, MD), respectively. Diethylpyrocarbonate (DEPC) (#D5758), imidazole (#I5513), iodoacetamide (#I6125), tris(2-carboxyethyl)phosphine (TCEP) (#C4706), and trypsin (#T1426) were all purchased from Sigma-Aldrich (St. Louis, MO). Sodium phosphate monobasic monohydrate (#S0710) was ordered from EM Science (Darmstadt, Hesse, Germany). Sodium phosphate dibasic anhydrous (#S374), LC/MS-grade formic acid (#A117), acetonitrile (#A998), and water (#W7) were obtained from Fisher Scientific (Fair Lawn, NJ). All reagents used in this study have no known potential hazards.
Sample Preparation and DEPC labeling reactions
Rituximab was used as is in its formulation, which contains 10 mg/mL rituximab, 0.7 mg/mL polysorbate 80, 7.35 mg/mL sodium citrate dihydrate and 9 mg/mL sodium chloride in water at pH 6.5. For the NISTmAb formulation, L-histidine was removed from the formulation using PD SpinTrap G-25 spin columns (#28918004, GE Healthcare Life Sciences, Marlborough, MA) to prevent this amino acid from interfering with the DEPC reaction. The NISTmAb was then buffer exchanged in 50 mM phosphate buffer at pH 6.0, which is the pH of the formulation before L-histidine removal. Aliquots of rituximab and the NISTmAb (10 mg/mL, 70 μM) with only minor dilution (to 60 μM) were reacted with DEPC. Stock solutions of DEPC (69 mM) were freshly prepared in acetonitrile, and the final solution of DEPC was then prepared in water. Labeling of each mAb was performed for 5 min at 37 °C and was initiated by adding various molar excesses of DEPC, from 2-fold up to 30-fold). The reaction was quenched by the addition of imidazole at a 1:50 DEPC to imidazole molar ratio. For experiments at each DEPC concentration, at least three replicates were performed on the rituximab and NISTmAb samples.
Proteolytic digestion
After quenching, the labeled mAb samples were diluted in 50 mM phosphate buffer at pH 7.4 and added into a urea-containing tube. The resulting mixture contained 8 M urea for protein denaturation at room temperature. TCEP was added at the final concentration of 25 mM to reduce the disulfide bonds, and iodoacetamide was simultaneously added at the same final concentration to alkylate the reduced Cys residues. The samples were then kept in the dark at room temperature for 20 min. For rituximab samples, DetergentOUT™ Tween® Micro spin columns (#786–214, G-Biosciences, St. Louis, MO) were used to remove Tween® 80 from the samples. Subsequently, overnight digestion at 37 °C was performed with trypsin at a 1:10 (w/w) enzyme to substrate ratio. Following digestion, trypsin was removed from the resulting rituximab or NISTmAb peptides through ultrafiltration using an Amicon® centrifugal filter with a 10 kDa molecular weight cutoff (#UFC501096, Millipore, Burlington, MA). The flow-through was collected, flash-frozen in liquid nitrogen, and stored at −20 °C until LC-MS/MS analysis.
HPLC separation
Online LC-MS/MS analyses were performed on all rituximab and NISTmAb digests. A sample containing approximately 5 μg of the digested protein was loaded on a Thermo Scientific Dionex Ultimate 3000 HPLC system (Waltham, MA). The separation of peptides was performed on a Thermo Scientific Acclaim™ PepMap™ RSLC C18 column (15 cm x 300 μm, 2 μm particle size, 100 Å pore size) with a flow rate of 4 μL/min. LC/MS-grade water (solvent A) and acetonitrile (solvent B), each containing 0.1% formic acid, were used as mobile phases. Desalting was performed at 5% B during the first 5 min after sample injection. A linear gradient of solvent B was increased from 5% B to 35% B over 65 min. The gradient was finally elevated to and held at 95% B to flush the column.
Mass Spectrometry
Mass spectra were acquired on a Thermo Scientific Orbitrap Fusion mass spectrometer (Waltham, MA). The electrospray needle voltage was kept at ~4 kV (positive mode), and the ion transfer tube temperature was set to 275 °C. Tandem mass spectrometry (MS/MS) was conducted on a set of selected mAb peptides. The precursor ions (unmodified or modified peptide ions) were selected using a quadrupole mass filter at an isolation width of 2.0. The AGC target and maximum injection time were set to 5 × 104 ions and 50 msec, respectively. Collision induced dissociation (CID) was performed in a linear quadrupole ion trap with a normalized collision energy of 35%. Mass spectra of product ions were acquired on an Orbitrap analyzer with a resolution of 30,000.
Dose-Response Plots
DEPC can modify His, Lys, Ser, Thr, Tyr, and N-termini, and the labeling results in a single type of modification product with mass addition of 72.02 Da (Scheme 1a and Figure S1). Under the conditions used in this study, the reaction of DEPC with a specific site in the protein follows second order kinetics37 (Equation 1),
| (1) |
where [P]0 is the initial concentration of unmodified mAb, [X]0 is the initial concentration of DEPC, [P] is the concentration of unmodified mAb at time t, [X] is the DEPC concentration at time t, and k is the second-order rate coefficient. Details about how this equation is derived can be found in the Supplementary Information.
Scheme 1.

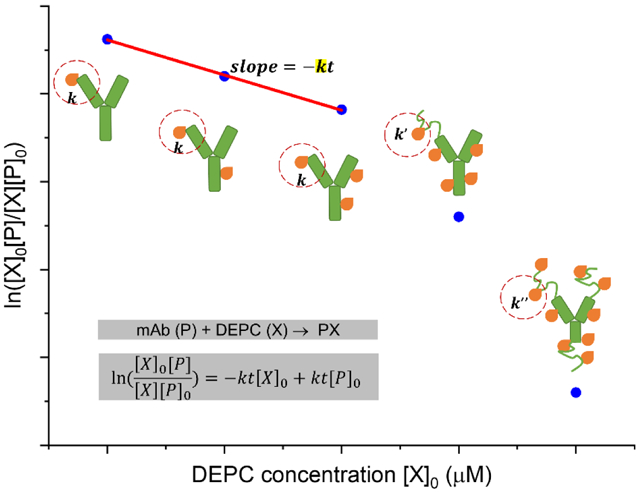
(a) DEPC labeling of nucleophilic side chains (His, Lys, Tyr, Ser, and Thr) in the antibodies. (b) A hypothetical dose-response plot for a given peptide fragment can be used to identify any labeling-induced structural perturbation to the protein in the region represented by the measured peptide. In this example, a break in linearity is indicative of a structural perturbation at a high DEPC concentration.
For each specific labeling site, a plot between and [X]0 was produced for a given peptide from LC-MS/MS data of that peptide. [P]/[P]0 is the ratio of the peak area for the unmodified peptide to the sum of the peak areas for the modified and unmodified peptide, and [X] is determined by the difference between the [P] and [P]0 values. A hypothetical dose-response plot for a second order reaction is shown in Scheme 1b.
A custom software pipeline specifically designed for protein CL-MS studies with DEPC35, 36 was used for the initial identification of the peptides that were chosen for MS/MS. Assignments of b and y ions from CID tandem mass spectra were performed with a mass tolerance of 0.5 Da. From the LC-MS/MS analyses on a set of selected mAb peptides, Thermo Scientific Xcalibur™ software was used to reconstruct product ion chromatograms of unmodified and modified peptides. Peptide identification and peak quantification were performed using tandem spectra and mass spectral peak areas, respectively.
RESULTS AND DISCUSSION
Dose-Response Plots as Indicators of Antibody Structural Changes upon DEPC Labeling
Reliable information about the HOS of mAbs can be obtained from CL-MS experiments only when the protein’s structural integrity is preserved during the labeling. Under the conditions that DEPC is normally used for labeling, its reaction with a specific site in a protein follows second order kinetics.37 Deviations from these kinetics at high reagent concentrations can indicate a structural perturbation, as the labeling kinetics are sensitive to any structural changes that affect the reactivity of a given residue. We have shown in previous work with DEPC that breaks in linearity in dose-response plots are indicators of labeling-induced structural perturbations in small proteins, such as cytochrome c and myoglobin.37 In addition, other investigators have used dose-response plots to measure structural changes during HRF and carboxyl group footprinting.34, 44 Because DEPC can hydrolyze in water over time, the DEPC reactions are conducted at a constant reaction time t, and a rate constant can be determined from a dose-response plot. Changes in local structural features like solvent accessibility and microenvironment23, 37, 46 that can occur at higher DEPC concentrations lead to breaks in the linearity of the plot, indicating these structural perturbations (Scheme 1b). Dose-response plots for individual proteolytic fragments of the labeled protein provide a sensitive measure of structural perturbations as they report on local changes. The experimental workflow to generate dose-response plots for proteolytic fragments is shown in Figure 1.
Figure 1.

The experimental workflow used to generate dose-response plots for selected proteolytic fragments of the mAbs studied here. The mAb reacts with DEPC at different reagent concentrations. The labeled mAb is subjected to proteolytic digestion, and the labeled peptides are analyzed using LC–MS/MS. Peak areas of unmodified and modified species are used to generate a dose-response plot.
The antibodies rituximab and NISTmAb were reacted with a range of DEPC concentrations, and dose-response plots were generated for a set of representative peptides with labeled sites spread throughout the antibody structure (Figure S2). One to two peptides per domain were selected, and these peptides (a) had no post-translational modifications and (b) were generated with no missed cleavages during the tryptic digestion. Several of the peptides that are reported here include residues in the complementarity-determining regions (CDRs), which makes them critically important for the antibody’s structure and function. Examples of tandem mass spectra and label site identifications of these representative peptides can be found in Figures S3 and S4.
Dose-response plots can be used to reveal labeling-induced structural perturbations for antibodies, which are multi-domain proteins.
Dose-response plots for selected proteolytic fragments of rituximab are shown in Figure 2 and S5. For all of the peptides, linear relationships are observed at low DEPC concentrations, suggesting that the protein’s HOS in different domains is maintained during the DEPC labeling at these concentrations. Breaks in linearity of the dose-response plots were determined from R2 values and standard errors of regression (sy/x).47, 48 Specifically, R2 values above 0.95 and sy/x below 30% were used as cutoffs for assessing linearity. The 30% value for sy/x was chosen because we found that the average %RSD for all the data for rituximab and NISTmAb is around 30%. One conclusion from the rituximab results is that the linearity of the dose-response plots is maintained for at least up to 6-fold DEPC in all cases, and for some residues it is maintained up to 10- or 15-fold DEPC [Table 1 and Figure S5 (j) to (p)]. This observation means that rituximab’s HOS can be reliably maintained at DEPC:protein molar ratios of 6-fold. An almost identical conclusion is made from the dose-response plots for proteolytic fragments of NISTmAb (Figure 3 and S6). The dose-response plots maintain linearity up to 6-fold for all peptides, while some peptides retain linearity at DEPC:protein ratios as high as 15-fold [Table 1 and Figure S6 (f) to (o)]. In the data for both mAbs, the error bars also tend to be larger at the higher DEPC concentrations, suggesting increased protein structural heterogeneity when label-induced structural changes occur. The fact that deviations in linearity occur at higher than 6-fold DEPC for both antibodies suggests that there might be a DEPC:protein ratio that could be commonly used for mAbs without worry of structural perturbations. Because therapeutic mAbs have similar overall folds and tend to have similar Fc domains, the results obtained here are probably transferrable to all mAbs. However, more studies would be needed to thoroughly evaluate this idea. Antibodies with different glycosylation profiles and antibody subclasses (e.g. IgG1, IgG2, IgG3 and IgG4) with different disulfide-bonding patterns have different stability profiles,49-53 and these differences might lead to different DEPC concentrations at which structural perturbations occur.
Figure 2.

Dose-response plots for selected proteolytic fragments of rituximab after labeling with DEPC at concentrations varying from 2- to 30-fold DEPC:protein molar ratios. Plots of select reactive residues from each of the six antibody domains in heavy chain (HC) and light chain (LC), (a) VH, (b) CH1, (c) CH2, (d) CH3, (e) VL, and (f) CL, are shown here. The plots of other reactive sites in rituximab can be found in Figure S5. Linear relationships are observed between the unmodified fraction and the DEPC concentrations at low reagent concentrations. The rate coefficients (k) in M−1 s−1 are obtained by dividing the measured slopes by the reaction time (see Equation 1). Error bars for the rate coefficients are calculated from the standard error of the slope.
Table 1.
A summary of the dose-response data for residues in rituximab and the NISTmAb.
| Antibody Residuea | Secondary structure | Linearity up to |
Rate coefficient kb (M−1s−1) |
|---|---|---|---|
| Rituximab | |||
| LC S170/T171/Y172c | β-sheet & random coil | 6X | 7.4 ± 0.8 x 10−1 |
| HC H289/K292c | Random coil | 10X | 3.1 ± 0.2 x 10−1 |
| HC Y282 | β-sheet | 6X | 1.6 ± 0.1 x 10−1 |
| LC H33 | β-sheet | 6X | 1.0 ± 0.1 x 10−1 |
| HC S25/Y27c | Random coil | 10X | 7.2 ± 0.6 x 10−2 |
| LC Y48 | Random coil | 15X | 4.9 ± 0.3 x 10−2 |
| HC Y395/K396c | β-sheet | 10X | 2.2 ± 0.1 x 10−2 |
| HC T254 | β-sheet | 6X | 2.1 ± 0.4 x 10−2 |
| HC Y377 | Random coil | 6X | 1.34 ± 0.02 x 10−2 |
| HC S258 | Random coil | 6X | 1.31 ± 0.06 x 10−2 |
| HC Y153 | Random coil | 6X | 8.5 ± 0.1 x 10−3 |
| HC Y52 | β-sheet | 6X | 3.6 ± 0.2 x 10−3 |
| HC Y60/K63c | Random coil | 6X | 3.5 ± 0.2 x 10−3 |
| HC S185/S187/S188c | β-sheet | 15X | 2.5 ± 0.1 x 10−3 |
| HC S161 | β-sheet | 10X | 2.5 ± 0.2 x 10−3 |
| LC T108 | Random coil | 10X | 2.2 ± 0.1 x 10−3 |
| NISTmAb | |||
| HC K58/K59c | β-sheet & random coil | 6X | 8.0 ± 0.2 x 10−1 |
| HC Y281 | β-sheet | 10X | 3.6 ± 0.1 x 10−1 |
| HC H288/K291c | Random coil | 10X | 3.5 ± 0.1 x 10−1 |
| HC K13 | Random coil | 8X | 3.2 ± 0.2 x 10−1 |
| LC Y31 | Random coil | 15X | 4.3 ± 0.5 x 10−2 |
| LC H33 | β-sheet | 15X | 2.3 ± 0.2 x 10−2 |
| LC T50/S51/K52c | β-sheet & random coil | 6X | 1.22 ± 0.04 x 10−2 |
| LC S170/T171/Y172c | β-sheet & random coil | 10X | 1.22 ± 0.09 x 10−2 |
| HC Y376 | Random coil | 10X | 1.1 ± 0.1 x 10−2 |
| HC T253 | β-sheet | 6X | 4.7 ± 0.5 x 10−3 |
| HC Y152 | β-sheet | 10X | 4.5 ± 0.3 x 10−3 |
| HC S127 | β-sheet | 6X | 3.6 ± 0.1 x 10−3 |
| HC S257 | Random coil | 6X | 3.1 ± 0.2 x 10−3 |
| HC S134/S135/K136c | α-helix | 10X | 2.9 ± 0.2 x 10−3 |
| LC T108 | Random coil | 8X | 2.2 ± 0.1 x 10−3 |
LC refers to light chain, and HC refers to heavy chain.
The k values are obtained by dividing the measured slope by the reaction time (see Equation 1). Error bars are calculated from the standard error of the slope.
Tandem mass spectrometry does not enable the precise modification site to be definitively identified.
Figure 3.

Dose-response plots for selected proteolytic fragments of NISTmAb after labeling with DEPC at concentrations varying from 2- to 30-fold DEPC:protein molar ratios. Plots of select reactive residues from each of the six antibody domains in heavy chain (HC) and light chain (LC), (a) VH, (b) CH1, (c) CH2, (d) CH3, (e) VL, and (f) CL, are shown here. The plots of other reactive sites in NISTmAb can be found in Figure S6. Linear relationships are observed between the unmodified fraction and the DEPC concentrations at low reagent concentrations. The rate coefficients (k) in M−1 s−1 are obtained by dividing the measured slopes by the reaction time (see Equation 1). Error bars for the rate coefficients are calculated from the standard error of the slope.
A closer look at the labeling data reveals that some antibody domains are more susceptible to labeling-induced structural changes than others (Table 1 and Figure S7). Most notably, the Fab has a greater percentage of sites (60%) whose linearity goes beyond 6-fold DEPC. In contrast, the CH2 domain has a smaller percentage (38%) of sites that maintain linearity past 6-fold DEPC. These observations are consistent with the known stability differences of the Fab and CH2 domains. Previous studies have identified the melting temperatures (Tm) to be 71°C and 74°C for the CH2 and Fab domains of rituximab, respectively, and 69°C and 83°C for those of the NISTmAb.54, 55 The CH2 domains of both antibodies have lower Tm values than that of Fab domains, which may explain why the CH2 domain is more sensitive to labeling-induced structural perturbations. Upon comparing the Fab sites in each antibody, the NISTmAb has higher number of sites whose linearity goes beyond 6-fold DEPC (70%) than rituximab (50%). Perhaps the higher overall stability of NISTmAb’s Fab explains why it has higher number of sites whose linearity goes beyond 6-fold DEPC.
Labeling rate coefficients can be determined from dose-response plots
DEPC modification rate coefficients (k) range from 2.2 x 10−3 to 8.0 x 10−1 M−1s−1 (Table 1). These are ‘apparent’ rate coefficients because differences in ionization efficiencies between the labeled and unlabeled peptides make it difficult to obtain real rate coefficients from the experiments. The reaction rate coefficients tend to be higher for His and Lys residues than for Ser, Thr, and Tyr residues. Around 85% of His/Lys residues have k values greater than 1 x 10−2 M−1s−1, while about 50% of the Ser/Thr/Tyr residues have k values greater than 1 x 10−2 M−1s−1. These results are consistent with our previous studies that His and Lys side chains have higher intrinsic reactivities towards DEPC labeling than Ser, Thr, and Tyr side chains.23, 37, 46
Because the two mAbs have the same constant domain sequences, and presumably similar structures in these regions, we also can compare DEPC labeling rate coefficients for eight common residues in these constant regions (CL, CH1, and Fc) to understand how the different formulations affect the labeling rates. Upon comparing the common residues, we find that H289/K292 (H288/K291) and Y377 (Y376) in the heavy chain and T108 in the light chain have essentially the same rate coefficients for the two mAbs. Note that the NISTmAb’s residue numbers are shown in parentheses as its heavy chain has one less residue at the N-terminus than rituximab. In contrast, the rate coefficients for Y153 (Y152), Y282 (Y281), T254 (T253), and S258 (S257) in the heavy chain change by a factor of 2 to 4, and the rate coefficient for S170/T171/Y172 in the light chain changes by a factor of almost 60. The difference in labeling rate coefficients for these residues can be ascribed to (a) differences in the rituximab and NISTmAb formulations and/or (b) changes in the microenvironment around these residues due to the formulation differences or non-identical local structures in the constant regions of these mAbs.
The key differences of the rituximab and NISTmAb formulations are pH (6.5 and 6.0, respectively) and the presence of polysorbate 80 in the former. The residues Y153, Y282, and T254 have acidic residues within 4 Å. The difference in the pH of the two mAb formulations could affect the protonation states of these adjacent acidic residues, thereby influencing the pKa values and thus the DEPC reactivity of these Tyr and Thr residues.23 In contrast, there are no acidic residues adjacent to H289/K292, Y377, and T108, perhaps explaining why the rate coefficients for these residues are almost the same for the two mAbs. Interestingly, S170 has a nearby acidic residue in the NISTmAb, but in rituximab an Arg residue is close. It is reasonable that such a dramatic change in the electrostatic environment around S170 could explain why S170/T171/Y172 is 60-fold more reactive in rituximab. Moreover, the microenvironment around these residues in each mAb is quite different in terms of nearby hydrophobic residues and other polar residues.
Multi-domain antibodies can be labeled with more than one DEPC molecules before being structurally perturbed
The ‘single hit rule’ in CL indicates that the addition of subsequent covalent labels after the first will necessarily occur to a chemically perturbed protein and should thus be avoided. We were interested, though, to see how many DEPC labels a multi-domain protein could accommodate before structural changes actually occurred, as evidenced by changes in the reaction kinetics. Because the dose-response plots for both antibodies were still linear up to a 6-fold excess of DEPC, we separately reacted each antibody with this level of DEPC and then measured the extent of labeling on the intact light and heavy chains using LC-MS (see analysis details in the Supplementary Information). We find that the weighted average numbers of labels on the light chains of rituximab and NISTmAb are 0.6 and 0.7, respectively, whereas the weighted average number of labels on the two heavy chains of rituximab and NISTmAb are 2.3 and 2.7, respectively (Figure 4). Note that the heavy chain’s spectra are complicated by N-glycan heterogeneity. Enzymatic removal of the glycoform to reduce spectral complexity is possible, but we did not perform this because we wanted to limit any possible hydrolysis of the DEPC labeled sites during the relatively long enzymatic reaction time. Because each protein has two light chains, the total extent of labeling is 3.5 for rituximab and 4.1 for NISTmAb.
Figure 4.

Mass spectra obtained from LC-MS analyses of DEPC-labeled mAbs, showing the extent of modification for the light and heavy chains of (a) rituximab and (b) NISTmAb after labeling at a 6-fold DEPC:protein molar ratio. COEt refers to a carbethoxy group that is the product upon modification with DEPC. Because of glycan heterogeneity on the heavy chain (HC) and signal overlap with the light chain (LC), only the G1F/G1F glycoform of a non-reduced heavy chain (2HC) is shown.
The fact that both antibodies can accommodate 3 or 4 labels before any structural perturbation is observed suggests that the ‘single hit rule’ is not relevant to such large multi-domain proteins such as antibodies. Previous work in our group on relatively small (< 30 kDa) single-domain proteins showed that modification conditions that lead to 0.8 to 1.2 labels per protein on average can maintain the structural integrity of these proteins during the labeling.37, 56, 57 Being able to add more than one label per mAb is beneficial for CL-MS studies, as it allows a more sensitive measure of structure because more modifications are detected. It is perhaps not surprising that multi-domain proteins such as mAbs are capable of accommodating more than one label, as the multi-domain structure of these proteins likely make them less readily perturbed when modifications are occurring at distant sites (e.g. different domains). This explanation is supported by the success of therapeutic antibody-drug conjugates (ADCs) that have hydrophobic drugs attached. Conjugation of these drugs, often at multiple sites, do not significantly perturb the structure and function of the multi-domain antibody.58-61 While up to 4 DEPC modifications can be accommodated by the mAbs studied here, these proteins do have 12 domains, so modification-induced structural changes still do occur across domains. Overall, our findings indicate that multi-domain antibodies can be labeled with more than one DEPC molecule before being structurally perturbed, thus improving the CL-MS resolution substantially compared to the single-hit rule. In addition, these results could be helpful for understanding the extent to which ADCs could be conjugated with drug molecules before structural perturbations are observed.
CONCLUSION
Measuring the kinetics of DEPC CL reactions can reveal the DEPC concentrations at which labeling-induced structural perturbations occur for antibodies. For two separate antibodies that have different formulations, we find that a 6-fold molar excess of DEPC can be used without perturbing the protein’s structure, suggesting that this is a reliable DEPC concentration for CL studies of antibodies. This practical finding should avoid the need for time-consuming optimization of DEPC CL conditions for antibodies. Moreover, at these DEPC:mAb labeling ratios, up to four DEPC modifications can be added, indicating that these multi-domain proteins can accommodate more than one label without being structurally perturbed. Interestingly, most of the domains in the NISTmAb maintain their structural integrity at higher DEPC concentrations than rituximab, which is consistent with the greater stability of this mAb. This observation may suggest that more thermally stable proteins could accommodate even higher levels of labeling, allowing a more sensitive measure of HOS by DEPC CL. Overall, these studies improve our ability to use DEPC CL to study the HOS of protein therapeutics.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by a National Institutes of Health (NIH) Small Business Innovation Research (SBIR) grant (R43 GM116211) and supported by NIH grant R01 GM075092. We thank QuarryBio for permission to use their customized software pipeline for some of the DEPC labeling experiments described here. The authors acknowledge Dr. Cedric E. Bobst for his help with the Thermo Scientific Orbitrap Fusion mass spectrometer. The Thermo Scientific Orbitrap Fusion mass spectrometer was funded by a National Institutes of Health grant S10OD010645. P.K.L. also acknowledges his doctoral fellowship from the Faculty of Pharmaceutical Sciences, Chulalongkorn University (Bangkok, Thailand).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: https://doi.org/10.1021/jasms.0c00067
The authors declare no competing financial interest.
REFERENCES
- 1.Berkowitz SA; Engen JR; Mazzeo JR; Jones GB, Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat. Rev. Drug Discovery 2012, 11 (7), 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang H; Cui W; Gross ML, Mass spectrometry for the biophysical characterization of therapeutic monoclonal antibodies. FEBS Lett. 2014, 588 (2), 308–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rogstad S; Faustino A; Ruth A; Keire D; Boyne M; Park J, A Retrospective Evaluation of the Use of Mass Spectrometry in FDA Biologics License Applications. J. Am. Soc. Mass Spectrom 2017, 28 (5), 786–794. [DOI] [PubMed] [Google Scholar]
- 4.Rathore D; Faustino A; Schiel J; Pang E; Boyne M; Rogstad S, The role of mass spectrometry in the characterization of biologic protein products. Expert Rev. Proteomics 2018, 15 (5), 431–449. [DOI] [PubMed] [Google Scholar]
- 5.Wang W; Singh S; Zeng DL; King K; Nema S, Antibody Structure, Instability, and Formulation. J. Pharm. Sci 2007, 96 (1), 1–26. [DOI] [PubMed] [Google Scholar]
- 6.Dingman R; Balu-Iyer SV, Immunogenicity of Protein Pharmaceuticals. J. Pharm. Sci 2019, 108 (5), 1637–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frokjaer S; Otzen DE, Protein drug stability: a formulation challenge. Nat. Rev. Drug Discovery 2005, 4 (4), 298–306. [DOI] [PubMed] [Google Scholar]
- 8.Abbott WM; Damschroder MM; Lowe DC, Current approaches to fine mapping of antigen-antibody interactions. Immunology 2014, 142 (4), 526–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Opuni KFM; Al-Majdoub M; Yefremova Y; El-Kased RF; Koy C; Glocker MO, Mass spectrometric epitope mapping. Mass Spectrom Rev 2018, 37 (2), 229–241. [DOI] [PubMed] [Google Scholar]
- 10.Majumdar R; Middaugh CR; Weis DD; Volkin DB, Hydrogen-Deuterium Exchange Mass Spectrometry as an Emerging Analytical Tool for Stabilization and Formulation Development of Therapeutic Monoclonal Antibodies. Journal of Pharmaceutical Sciences 2015, 104 (2), 327–345. [DOI] [PubMed] [Google Scholar]
- 11.Iacob RE; Bou-Assaf GM; Makowski L; Engen JR; Berkowitz SA; Houde D, Investigating Monoclonal Antibody Aggregation Using a Combination of H/DX-MS and Other Biophysical Measurements. Journal of Pharmaceutical Sciences 2013, 102 (12), 4315–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bommana R; Chai Q; Schöneich C; Weiss WF; Majumdar R, Understanding the Increased Aggregation Propensity of a Light-Exposed IgG1 Monoclonal Antibody Using Hydrogen Exchange Mass Spectrometry, Biophysical Characterization, and Structural Analysis. Journal of Pharmaceutical Sciences 2018, 107 (6), 1498–1511. [DOI] [PubMed] [Google Scholar]
- 13.Zhang A; Singh SK; Shirts MR; Kumar S; Fernandez EJ, Distinct Aggregation Mechanisms of Monoclonal Antibody Under Thermal and Freeze-Thaw Stresses Revealed by Hydrogen Exchange. Pharmaceutical Research 2012, 29 (1), 236–250. [DOI] [PubMed] [Google Scholar]
- 14.Wei H; Mo J; Tao L; Russell RJ; Tymiak AA; Chen G; Iacob RE; Engen JR, Hydrogen/Deuterium Exchange Mass Spectrometry for Probing Higher Order Structure of Protein Therapeutics: Methodology and Applications. Drug discovery today 2014, 19 (1), 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weis DD, ed. Hydrogen Exchange mass Spectrometry of Proteins: Fundamentals, Methods, and Applications. John Wiley & Sons; West Sussex, England, United Kingdom: 2016. [Google Scholar]
- 16.Houde D; Berkowitz SA; Engen JR, The Utility of Hydrogen/Deuterium Exchange Mass Spectrometry in Biopharmaceutical Comparability Studies. J. Pharm. Sci 2011, 100 (6), 2071–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coales SJ; Tuske SJ; Tomasso JC; Hamuro Y, Epitope mapping by amide hydrogen/deuterium exchange coupled with immobilization of antibody, on-line proteolysis, liquid chromatography and mass spectrometry. Rapid Commun. Mass Spectrom 2009, 23 (5), 639–647. [DOI] [PubMed] [Google Scholar]
- 18.Malito E; Faleri A; Lo Surdo P; Veggi D; Maruggi G; Grassi E; Cartocci E; Bertoldi I; Genovese A; Santini L; Romagnoli G; Borgogni E; Brier S; Lo Passo C; Domina M; Castellino F; Felici F; van der Veen S; Johnson S; Lea SM; Tang CM; Pizza M; Savino S; Norais N; Rappuoli R; Bottomley MJ; Masignani V, Defining a protective epitope on factor H binding protein, a key meningococcal virulence factor and vaccine antigen. Proc. Natl. Acad. Sci. U.S.A 2013, 110 (9), 3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Q; Willison LN; Tripathi P; Sathe SK; Roux KH; Emmett MR; Blakney GT; Zhang H-M; Marshall AG, Epitope Mapping of a 95 kDa Antigen in Complex with Antibody by Solution-Phase Amide Backbone Hydrogen/Deuterium Exchange Monitored by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem 2011, 83 (18), 7129–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang RYC; Krystek SR Jr.; Felix N; Graziano RF; Srinivasan M; Pashine A; Chen G, Hydrogen/deuterium exchange mass spectrometry and computational modeling reveal a discontinuous epitope of an antibody/TL1A Interaction. mAbs 2018, 10 (1), 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hudgens JW; Gallagher ES; Karageorgos I; Anderson KW; Filliben JJ; Huang RYC; Chen G; Bou-Assaf GM; Espada A; Chalmers MJ; Harguindey E; Zhang H-M; Walters BT; Zhang J; Venable J; Steckler C; Park I; Brock A; Lu X; Pandey R; Chandramohan A; Anand GS; Nirudodhi SN; Sperry JB; Rouse JC; Carroll JA; Rand KD; Leurs U; Weis DD; Al-Naqshabandi MA; Hageman TS; Deredge D; Wintrode PL; Papanastasiou M; Lambris JD; Li S; Urata S, Interlaboratory Comparison of Hydrogen–Deuterium Exchange Mass Spectrometry Measurements of the Fab Fragment of NISTmAb. Anal. Chem 2019, 91 (11), 7336–7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang RYC; Kuhne M; Deshpande S; Rangan V; Srinivasan M; Wang Y; Chen G, Mapping binding epitopes of monoclonal antibodies targeting major histocompatibility complex class I chain-related A (MICA) with hydrogen/deuterium exchange and electron-transfer dissociation mass spectrometry. Anal. Bioanal. Chem 2020, 412 (7), 1693–1700. [DOI] [PubMed] [Google Scholar]
- 23.Limpikirati P; Liu T; Vachet RW, Covalent labeling-mass spectrometry with non-specific reagents for studying protein structure and interactions. Methods 2018, 144, 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mendoza VL; Vachet RW, Probing protein structure by amino acid-specific covalent labeling and mass spectrometry. Mass Spectrom Rev 2009, 28 (5), 785–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu G; Chance MR, Hydroxyl Radical-Mediated Modification of Proteins as Probes for Structural Proteomics. Chemical Reviews 2007, 107 (8), 3514–3543. [DOI] [PubMed] [Google Scholar]
- 26.Zhang B; Cheng M; Rempel D; Gross ML, Implementing fast photochemical oxidation of proteins (FPOP) as a footprinting approach to solve diverse problems in structural biology. Methods 2018, 144, 94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J; Chen G, The use of fast photochemical oxidation of proteins coupled with mass spectrometry in protein therapeutics discovery and development. Drug Discovery Today 2019, 24 (3), 829–834. [DOI] [PubMed] [Google Scholar]
- 28.Deperalta G; Alvarez M; Bechtel C; Dong K; McDonald R; Ling V, Structural analysis of a therapeutic monoclonal antibody dimer by hydroxyl radical footprinting. mAbs 2013, 5 (1), 86–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li J; Wei H; Krystek SR; Bond D; Brender TM; Cohen D; Feiner J; Hamacher N; Harshman J; Huang RYC; Julien SH; Lin Z; Moore K; Mueller L; Noriega C; Sejwal P; Sheppard P; Stevens B; Chen G; Tymiak AA; Gross ML; Schneeweis LA, Mapping the Energetic Epitope of an Antibody/Interleukin-23 Interaction with Hydrogen/Deuterium Exchange, Fast Photochemical Oxidation of Proteins Mass Spectrometry, and Alanine Shave Mutagenesis. Anal. Chem 2017, 89 (4), 2250–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y; Wecksler AT; Molina P; Deperalta G; Gross ML, Mapping the Binding Interface of VEGF and a Monoclonal Antibody Fab-1 Fragment with Fast Photochemical Oxidation of Proteins (FPOP) and Mass Spectrometry. J. Am. Soc. Mass Spectrom 2017, 28 (5), 850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones LM; Sperry JB; Carroll JA; Gross ML, Fast Photochemical Oxidation of Proteins for Epitope Mapping. Anal. Chem 2011, 83 (20), 7657–7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watson C; Sharp JS, Conformational Analysis of Therapeutic Proteins by Hydroxyl Radical Protein Footprinting. The AAPS Journal 2012, 14 (2), 206–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li KS; Chen G; Mo J; Huang RYC; Deyanova EG; Beno BR; O’Neil SR; Tymiak AA; Gross ML, Orthogonal Mass Spectrometry-Based Footprinting for Epitope Mapping and Structural Characterization: The IL-6 Receptor upon Binding of Protein Therapeutics. Anal. Chem 2017, 89 (14), 7742–7749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaur P; Tomechko SE; Kiselar J; Shi W; Deperalta G; Wecksler AT; Gokulrangan G; Ling V; Chance MR, Characterizing monoclonal antibody structure by carboxyl group footprinting. mAbs 2015, 7 (3), 540–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Limpikirati P; Hale JE; Hazelbaker M; Huang Y; Jia Z; Yazdani M; Graban EM; Vaughan RC; Vachet RW, Covalent labeling and mass spectrometry reveal subtle higher order structural changes for antibody therapeutics. mAbs 2019, 11 (3), 463–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Borotto NB; Zhou Y; Hollingsworth SR; Hale JE; Graban EM; Vaughan RC; Vachet RW, Investigating Therapeutic Protein Structure with Diethylpyrocarbonate Labeling and Mass Spectrometry. Anal. Chem 2015, 87 (20), 10627–10634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mendoza VL; Vachet RW, Protein Surface Mapping Using Diethylpyrocarbonate with Mass Spectrometric Detection. Anal. Chem 2008, 80 (8), 2895–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manzi L; Barrow AS; Scott D; Layfield R; Wright TG; Moses JE; Oldham NJ, Carbene footprinting accurately maps binding sites in protein–ligand and protein–protein interactions. Nat. Commun 2016, 7 (1), 13288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gau BC; Sharp JS; Rempel DL; Gross ML, Fast Photochemical Oxidation of Protein Footprints Faster than Protein Unfolding. Anal. Chem 2009, 81 (16), 6563–6571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jumper CC; Schriemer DC, Mass Spectrometry of Laser-Initiated Carbene Reactions for Protein Topographic Analysis. Anal. Chem 2011, 83 (8), 2913–2920. [DOI] [PubMed] [Google Scholar]
- 41.Hambly DM; Gross ML, Laser Flash Photolysis of Hydrogen Peroxide to Oxidize Protein Solvent-Accessible Residues on the Microsecond Timescale. J. Am. Soc. Mass Spectrom 2005, 16 (12), 2057–2063. [DOI] [PubMed] [Google Scholar]
- 42.Jumper CC; Bomgarden R; Rogers J; Etienne C; Schriemer DC, High-Resolution Mapping of Carbene-Based Protein Footprints. Anal. Chem 2012, 84 (10), 4411–4418. [DOI] [PubMed] [Google Scholar]
- 43.Gupta S; Celestre R; Petzold CJ; Chance MR; Ralston C, Development of a microsecond X-ray protein footprinting facility at the Advanced Light Source. J. Synchrotron Radiat 2014, 21 (4), 690–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kiselar JG; Janmey PA; Almo SC; Chance MR, Structural Analysis of Gelsolin Using Synchrotron Protein Footprinting. Mol. Cell. Proteomics 2003, 2 (10), 1120. [DOI] [PubMed] [Google Scholar]
- 45.Kiselar JG; Maleknia SD; Sullivan M; Downard KM; Chance MR, Hydroxyl radical probe of protein surfaces using synchrotron X-ray radiolysis and mass spectrometry. Int. J. Radiat. Biol 2002, 78 (2), 101–114. [DOI] [PubMed] [Google Scholar]
- 46.Limpikirati P; Pan X; Vachet RW, Covalent Labeling with Diethylpyrocarbonate: Sensitive to the Residue Microenvironment, Providing Improved Analysis of Protein Higher Order Structure by Mass Spectrometry. Anal. Chem 2019, 91 (13), 8516–8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asuero AG; Sayago A; González AG, The Correlation Coefficient: An Overview. Crit. Rev. Anal. Chem 2006, 36 (1), 41–59. [Google Scholar]
- 48.Miller JN; Miller JC, Statistics and chemometrics for analytical chemistry. 6th ed.; Pearson/Prentice Hall: Harlow, England, United Kingdom, 2005. [Google Scholar]
- 49.Zhang A; Fang J; Chou RYT; Bondarenko PV; Zhang Z, Conformational Difference in Human IgG2 Disulfide Isoforms Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry. Biochemistry 2015, 54 (10), 1956–1962. [DOI] [PubMed] [Google Scholar]
- 50.Tian Y; Han L; Buckner AC; Ruotolo BT, Collision Induced Unfolding of Intact Antibodies: Rapid Characterization of Disulfide Bonding Patterns, Glycosylation, and Structures. Anal. Chem 2015, 87 (22), 11509–11515. [DOI] [PubMed] [Google Scholar]
- 51.Ferguson CN; Gucinski-Ruth AC, Evaluation of Ion Mobility-Mass Spectrometry for Comparative Analysis of Monoclonal Antibodies. J. Am. Soc. Mass Spectrom 2016, 27 (5), 822–833. [DOI] [PubMed] [Google Scholar]
- 52.Le Basle Y; Chennell P; Tokhadze N; Astier A; Sautou V, Physicochemical Stability of Monoclonal Antibodies: A Review. J. Pharm. Sci 2020, 109 (1), 169–190. [DOI] [PubMed] [Google Scholar]
- 53.Wada R; Matsui M; Kawasaki N, Influence of N-glycosylation on effector functions and thermal stability of glycoengineered IgG1 monoclonal antibody with homogeneous glycoforms. mAbs 2019, 11 (2), 350–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andersen CB; Manno M; Rischel C; Thórólfsson M; Martorana V, Aggregation of a multidomain protein: A coagulation mechanism governs aggregation of a model IgG1 antibody under weak thermal stress. Protein Sci. 2010, 19 (2), 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.NISTmAb Common Technical Document Case Study. National Institute of Standards and Technology. [Google Scholar]
- 56.Liu T; Marcinko TM; Kiefer PA; Vachet RW, Using Covalent Labeling and Mass Spectrometry To Study Protein Binding Sites of Amyloid Inhibiting Molecules. Anal. Chem 2017, 89 (21), 11583–11591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu T; Limpikirati P; Vachet RW, Synergistic Structural Information from Covalent Labeling and Hydrogen–Deuterium Exchange Mass Spectrometry for Protein–Ligand Interactions. Anal. Chem 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pan LY; Salas-Solano O; Valliere-Douglass JF, Antibody Structural Integrity of Site-Specific Antibody-Drug Conjugates Investigated by Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem 2015, 87 (11), 5669–5676. [DOI] [PubMed] [Google Scholar]
- 59.Huang RYC; O’Neil SR; Lipovšek D; Chen G, Conformational Assessment of Adnectin and Adnectin-Drug Conjugate by Hydrogen/Deuterium Exchange Mass Spectrometry. J. Am. Soc. Mass Spectrom 2018, 29 (7), 1524–1531. [DOI] [PubMed] [Google Scholar]
- 60.Pan LY; Salas-Solano O; Valliere-Douglass JF, Localized conformational interrogation of antibody and antibody-drug conjugates by site-specific carboxyl group footprinting. mAbs 2017, 9 (2), 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pan LY; Salas-Solano O; Valliere-Douglass JF, Conformation and Dynamics of Interchain Cysteine-Linked Antibody-Drug Conjugates as Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem 2014, 86 (5), 2657–2664. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.