

Abstract
Exposure of the brain to high levels of glucocorticoids during ischemia–reperfusion induces neuronal cell death. Oxidative stress alters blood–brain barrier (BBB) function during ischemia–reperfusion, and so we hypothesized that it might impair P-glycoprotein (P-gp)-mediated efflux transport of glucocorticoids at the BBB. Therefore, the purpose of this study was to clarify the molecular mechanism of this putative decrease of P-gp-mediated efflux function. First, we established that H2O2 treatment of a human in vitro BBB model (hCMEC/D3) reduced both P-gp efflux transport activity and protein expression on the plasma membrane within 20 min. These results suggested that the rapid decrease of efflux function might be due to internalization of P-gp. Furthermore, H2O2 treatment markedly increased tyrosine-14-phosphorylated caveolin-1, which is involved in P-gp internalization. A brain perfusion study in rats showed that cortisol efflux at the BBB was markedly decreased by H2O2 administration, and inhibitors of Abl kinase and Src kinase, which phosphorylate tyrosine-14 in caveolin-1, suppressed this decrease. Overall, these findings support the idea that oxidative stress-induced activation of Abl kinase and Src kinase induces internalization of P-gp via the phosphorylation of tyrosine-14 in caveolin-1, leading to a rapid decrease of P-gp-mediated cortisol efflux at the BBB.
Keywords: Blood–brain barrier, P-glycoprotein, oxidative stress, Abl kinase, Src kinase
Introduction
Despite improvements in the treatment of stroke, such as the introduction of thrombolytic therapy using tissue-plasminogen activator (t-PA), long-term care is required for between a third and a half of stroke survivors.1–3 Ischemia–reperfusion injury after t-PA therapy contributes to neuronal cell death in stroke patients, and there have been many attempts to develop neuroprotective agents. But, although over 100 neuroprotective compounds, such as NMDA antagonists,4 Ca2+ antagonists5 and Na+ channel inhibitors,6 have entered clinical trials, most have proved ineffective.7 Only edaravone, which is an active oxygen scavenger, has so far been approved for clinical use.8 However, the Inatomi historically controlled retrospective clinical study showed that although edaravone treatment for seven days could achieve a modest clinical improvement in patients with mild stroke (NIHSS ≤ 7), it was ineffective in patients with severe stroke (NIHSS > 7).9 Therefore, we need to identify new mechanism-based targets for the development of effective neuroprotective agents.
We focused on the facts that levels of glucocorticoids, which are stress-responsive hormones, are increased in the cerebrospinal fluid during the acute phase of stroke,10,11 and this increase is associated with neurotoxicity and decreased proliferation of nerve and hippocampus cells.12,13 Glucocorticoids were confirmed to be key mediators of nerve injury in a rat cerebral ischemia–reperfusion model.14 Nevertheless, nerve injury occurs the hippocampus of hypoxia/ischemic rats despite the fact that the blood glucocorticoid concentration does not change after recovery of blood flow.14 In addition, imaging studies have revealed almost no enhancement of blood–brain barrier (BBB) permeability for up to 2.1 h (median) after ischemia onset in patients with cerebral infarction,15 suggesting that the tight-junction integrity of the BBB was not impaired in the acute phase of blood flow recovery. Thus, there seem to be two possibilities to explain the increase of glucocorticoids in the brain in the early stage of human ischemia–reperfusion, i.e. enhanced influx transport function, or impaired efflux transport function. P-glycoprotein (P-gp/MDR1/mdr1a) is a key efflux transporter of the major primate glucocorticoid, cortisol,16 and oxidative stress influences the function of transporters at the BBB,17 therefore, decreased P-gp-mediated efflux of cortisol could play a role in ischemia–reperfusion injury in humans.
It was reported that long-term exposure (over 4 h) of rat brain microvessel endothelial cells to H2O2 increases the P-gp protein expression level and efflux transport activity in crude membrane fraction, whereas no change in P-gp expression was observed in the early phase of H2O2 exposure (up to 1 h).18 However, P-gp-mediated efflux transport activity is regulated not only by transcription, but also by post-translational mechanisms, such as changes of intracellular localization and binding proteins.19 In this context, we hypothesized that the elevated intracerebral concentration of cortisol in the acute phase of oxidative stress exposure might be caused by a rapid decrease of P-gp efflux transport activity. If this is the case, blocking the mechanism of the functional deterioration of P-gp should increase efflux of excess cortisol from the brain, reducing the degree of neuronal injury.
In the present study, we aimed to test the above hypothesis and to identify the mechanism involved. First, we established that H2O2 treatment of an in vitro BBB model (hCMEC/D3 cells) reduced P-gp efflux transport activity and P-gp content in the plasma membrane within 20 min. The rapidity of the response suggested that internalization of P-gp might be involved, so we next conducted a mechanistic investigation. Phosphoproteomic analysis and kinase inhibition studies suggested that Abl kinase- and Src kinase-induced phosphorylation of Tyr14 of caveolin-1 (Cav1), a scaffold protein that regulates P-gp function, was associated with the reduction of P-gp efflux function. Finally, we confirmed the in vitro results by means of a brain perfusion study in rats. Notably, cortisol concentrations in hippocampus and hypothalamus were markedly increased after H2O2 administration, and the administration of Abl kinase or Src kinase inhibitors attenuated the increase. Thus, our findings indicate that oxidative stress-induced activation of Abl kinase and Src kinase causes rapid phosphorylation of Tyr14 of Cav1, inducing internalization of P-gp, and thereby decreasing cortisol-efflux function at the BBB. We consider that inhibitors of Abl kinase and Src kinase may be promising candidate drugs for limiting the accumulation of excess cortisol in the brain after therapy to restore blood flow in patients with cerebral infarction.
Materials and methods
Reagents
[3H]Vinblastine sulfate was purchased from American Radioisotope Labeled Chemicals Inc. (MO, USA). PSC833 was purchased from Tocris Bioscience (Bristol, UK). Quinidine anhydrous, cortisol, dynasore hydrate, and SKF96365 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Raffinose, sucrose, cinchonine, dexamethasone and 30% hydrogen peroxide were purchased from Wako Pure Chemicals (Osaka, Japan). Imatinib mesylate was purchased from Biovision Inc. (CA, USA), Src kinase inhibitor PP2 from Cosmo Bio Co. Ltd (Tokyo, Japan), bis-ANS (4,4′-dianilino-1,1'-binaphthyl-5,5'-disulfonic acid) from Invitrogen (CA, USA), and KRIBB3 from Santa Cruz Biotechnology (CA, USA). Peptide probes were synthesized by Thermo Fisher Scientific (Waltham, MA, USA) or by Scrum Inc. (Tokyo, Japan) (>95% purity). Other reagents were commercial products of analytical grade.
Animals
Adult SD rats (male, eight weeks of age, 230–250 g) were purchased from CLEA Japan (Tokyo, Japan). Rats were maintained on a 12-h light/dark cycle in a temperature-controlled environment with free access to food and water. All experiments were approved by the Institutional Animal Care and Use Committee in Tohoku University, and were conducted and reported in accordance with the guidelines in Tohoku University and ARRIVE guidelines.
Uptake assay to estimate P-gp or MRP1-mediated [3H]vinblastine efflux activity
Human brain capillary endothelial model cell line (hCMEC/D3 cell)20 was kindly provided by Dr. Pierre-Olivier Couraud (Institut Cochin, Paris, France). Cell culture and uptake assay were performed as described previously.23 For details of uptake assay, see Supplementary Method 1.
siRNA transfection of hCMEC/D3 cells
siRNA transfection of hCMEC/D3 cells was performed as described previously.21 For details, see Supplementary Method 2.
Preparation of whole-cell lysate and plasma membrane fraction of hCMEC/D3 cells
The whole-cell lysate of hCMEC/D3 cells was obtained as described by Hoshi et al.21 The cells were washed twice with ice-cold phosphate-buffered saline (PBS) containing protease inhibitor cocktail (Sigma) at 4℃. The cell suspension was collected and centrifuged at 500 g for 5 min at 4℃, and the pellet was used for quantitative analysis. The plasma membrane fraction was isolated using a Minute Plasma Membrane Protein Isolation Kit (Invent Biotechnologies, Eden Prairie, MN, USA) according to the manufacturer's instructions. Total protein amount was measured by the Lowry method.
Quantitative targeted absolute proteomics in hCMEC/D3 cells
The absolute protein expression amounts of P-gp, MRP1 and Cav1 in hCMEC/D3 cells were determined using our established quantitative targeted absolute proteomics (QTAP) technique.22–26 For details, see Supplementary Table 5 and Supplementary Method 3.
Quantification of phosphorylated Cav1 and Src kinase in whole-cell lysate of hCMEC/D3 cells
To quantify the absolute protein expression amounts of phosphorylated Cav1 (Tyr6, Tyr14, both Tyr6 and Tyr14, Tyr25 and Tyr42) and Src kinase (Tyr419), we enriched phosphorylated peptides with Titansphere® Phos-TiO (GL Science) according to the manufacturer's instructions. Internal standard peptides were added to a tryptic digest of whole-cell lysate equivalent to 1800 µg protein, and applied to a Titansphere®Phos-TiO column. The phosphorylated peptides-enriched fraction was acidified with trifluoroacetic acid and desalted using SDB and GC tips (GL Science) for subsequent LC-MS/MS analysis.
LC-MS/MS conditions for QTAP analysis
LC-MS/MS analysis for the quantification of protein expression amount was performed as described by Hoshi et al.21 For details, see Supplementary Method 4.
LC-MS/MS conditions for phosphoproteomic analysis
Comprehensive comparative phosphoproteomic analysis was performed as described by Hoshi et al.21 For details, see Supplementary Method 5.
Immunoblotting assay for Abl kinase
Immunoblotting assay was performed as described by Kamiie et al.25 Protein samples (10 µg/lane) were resolved by 10% SDS-polyacrylamide gel electrophoresis and electrotransferred to polyvinylidene difluoride (PVDF) membranes. The PVDF membranes were rotated in blocking buffer (4% skimmed milk in 25 mM Tris–HCl (pH 8.0), 125 mM NaCl, 0.1% Tween 20) for 1 h at room temperature and incubated with anti-Abl kinase antibody (K-12, Santa Cruz) or anti-Na+/K+ ATPase antibody (clone C464.6, Upstate Biotechnology, Inc., NY, USA) at 4℃ overnight. The membranes were washed three times with washing buffer (25 mM Tris–HCl (pH 8.0), 125 mM NaCl, 0.1% Tween 20) and incubated with horseradish peroxidase-conjugated antibody. Signals were visualized with an enhanced chemiluminescence kit (ECL prime, Millipore, Bedford, MA, USA).
In situ brain perfusion
In situ brain perfusion was performed as described in Hosoya et al.27 with some modifications. For details, see Supplementary Method 6.
Statistical analysis
All statistical analyses were performed under the null hypothesis, assuming that the means of the compared groups were equal. Comparison between two groups was performed by an unpaired two-tailed Student's t-test (equal variance) or Welch's test (unequal variance) according to the result of the F test. For more than two groups, a one-way analysis of variance (ANOVA) followed by Bonferroni-corrected Student's t-tests were performed to determine the statistical significance of differences between datasets. If the p-value was less than 0.05, the difference was considered as statistically significant and the null hypothesis was rejected. No formal power calculation was performed to estimate the required sample size, as a sample size of 4–6 is typically used in rat studies to examine changes in Rbr of P-gp substrates,28 while a sample size of 3–4 is used in uptake assay and QTAP analysis using hCMEC/D3 cells.21 No randomization or blinding was performed in this study.
Results
Rapid and reversible reduction of P-gp efflux transport activity by H2O2 in hCMEC/D3 cells
We first examined the change of P-gp efflux transport activity at the BBB in the acute phase of oxidative stress by using H2O2-exposed hCMEC/D3 cells as an in vitro model. The P-gp efflux transport activity was calculated from the inhibitory effect of PSC833, a P-gp inhibitor, on vinblastine excretion, as described in Materials and Methods. In the presence of H2O2, the efflux transport activity of P-gp decreased within 20 min in a concentration-dependent manner (Figure 1(a) and (b)). Specifically, 0.5 mM, 1 mM, 2.5 mM and 5 mM H2O2 significantly decreased P-gp efflux transport activity to 67.2%, 52.7%, 34.0% and 28.8% of that at 0 mM H2O2. We also examined the effect of MK571, an inhibitor of multidrug resistance-associated protein (MRP) transporters. The MK571-sensitive vinblastine efflux transport activity was not affected by treatment with 5 mM H2O2 for 20 min (Figure 1(c) and (d)). Therefore, the reduction of vinblastine efflux transport activity by H2O2 does not involve MRP transporters.
Figure 1.

Effect of H2O2 on P-gp- and MRP1-mediated [3H]vinblastine efflux activity in hCMEC/D3 cells. (a, c) hCMEC/D3 cells were pre-incubated in extracellular fluid (ECF) buffer containing DMSO, 10 µM PSC833 (P-gp inhibitor) or 50 µM MK571 (MRP1 inhibitor) for 30 min at 37℃, and then the cellular uptake of the P-gp substrate vinblastine was measured for 20 min in the presence of H2O2 (0.05 mM to 5 mM) or ECF buffer (0 mM H2O2). To effectively inhibit P-gp or MRP1 efflux activity, PSC833 or MK571 was present during both the 30 min pre-incubation and the 20-min uptake period. The cellular uptake was expressed as the cell-to-medium ratio, as described in Materials and methods. Each column represents mean ± SD (n = 3). (b, d) Based on the data of Figure 1(a) and (c), P-gp or MRP1 efflux transport activity was calculated from the cell-to-medium ratio in the absence and presence of PSC833 or MK571 according to Materials and methods. (e) Cellular uptake of vinblastine was measured at 5 min, 20 min and 60 min after addition of 5 mM H2O2 (closed circles). To determine the effect of 5, 20 or 60 min treatment with H2O2 on the P-gp efflux transport activity, hCMEC/D3 cells were pre-incubated in ECF buffer containing DMSO or 10 µM PSC833 for 30 min at 37℃, and then the cellular uptake of vinblastine was measured for 5, 20 or 60 min with or without 5 mM H2O2. To examine reversibility, the medium was replaced with fresh medium after pre-incubation with 5 mM H2O2 for 20 min (open circles), and the cellular uptake of vinblastine was measured for 20 min in the absence of 5 mM H2O2. Both pre-incubation and vinblastine uptake were conducted in the presence of DMSO or PSC833. Each value represents the mean ± SD (n = 3). Statistical significance was determined by one-way ANOVA followed by post hoc Bonferroni-corrected Student's t-test. *p < 0.05, **p < 0.01, ***p < 0.005.
Next, we examined the time dependence of the reduction of P-gp efflux transport activity by H2O2. 5 mM H2O2 reduced P-gp efflux transport activity in the range of 5 min to 60 min (Figure 1(e)). Upon removal of H2O2, the transport activity of P-gp recovered to the control level within 20 min (Figure 1(e)). These data demonstrate that the decrease of P-gp efflux transport activity by H2O2 is both rapid and reversible.
Dynamin-dependent internalization of P-gp by H2O2 treatment in hCMEC/D3 cells
In order to clarify the effect of H2O2 on the P-gp protein expression amount, we quantified P-gp by means of LC-MS/MS-based proteomics. The P-gp protein expression amount in whole-cell lysate of hCMEC/D3 cells did not change during treatment with H2O2 in the concentration range of 0.25 mM to 5 mM (Figure 2(a)). But, in contrast, P-gp in the plasma membrane fraction was decreased in a H2O2 concentration-dependent manner to 83.4% (0.5 mM), 77.8% (1 mM), 66.0% (2.5 mM), and 55.3% (5 mM), as shown in Figure 2(b). There was no change in MRP1 expression either in whole-cell lysate or in plasma membrane fraction (Figure 2(c) and (d)). Therefore, it appeared that H2O2 induces a change of P-gp localization from the cell membrane to an intracellular site. To confirm that P-gp was internalized, we used dynasore, a dynamin inhibitor, because dynamin is a key molecule in endocytosis.29 In the presence of dynasore, the decrease of P-gp protein in the plasma membrane fraction induced by 0.5 mM H2O2 was significantly suppressed (Figure 2(e)). Furthermore, dynasore significantly suppressed the decrease of P-gp efflux transport activity by 0.5 mM H2O2 (Figure 2(f)). These results suggest that the reduction of P-gp efflux transport activity by H2O2 is due to dynamin-dependent internalization of P-gp.
Figure 2.
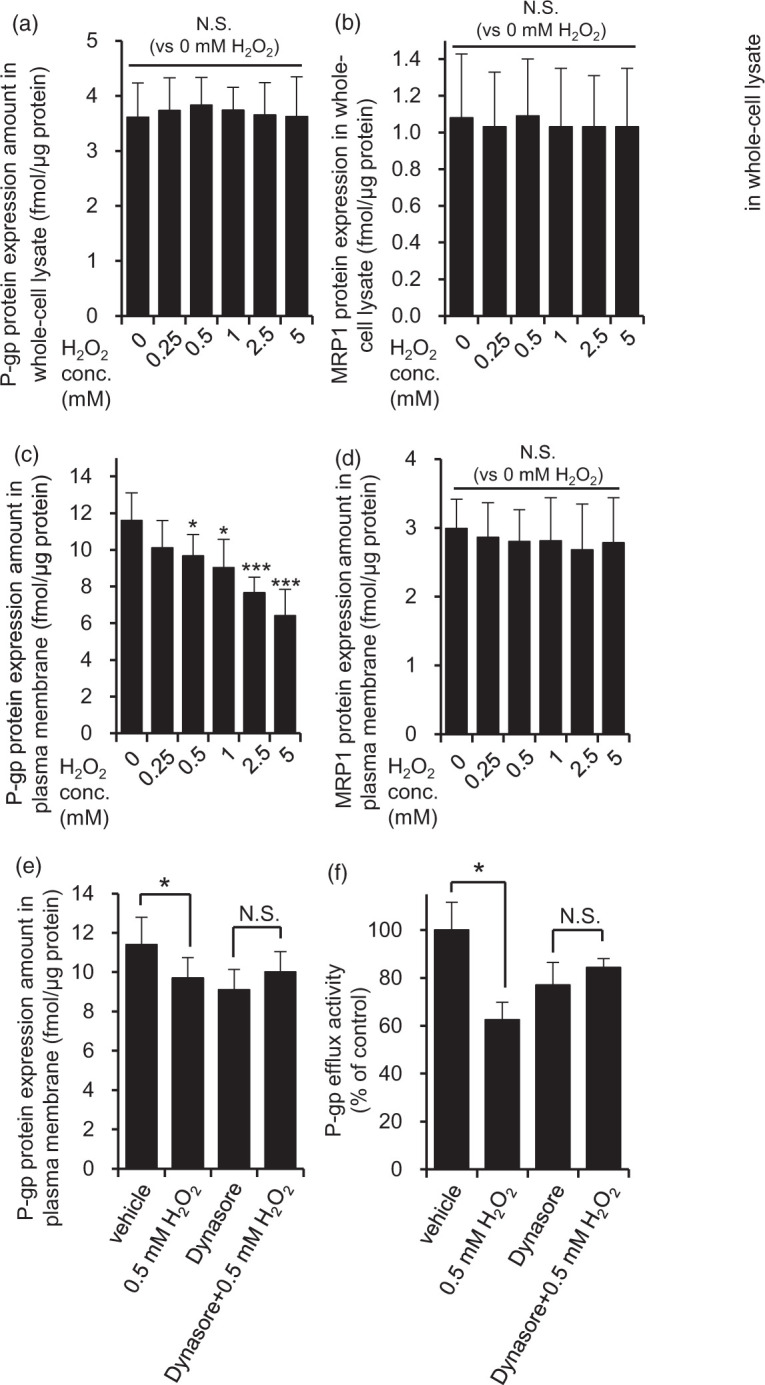
H2O2 facilitated dynamin-dependent P-gp internalization in hCMEC/D3 cells. The effect of H2O2 on P-gp and MRP1 protein expression amounts in whole-cell lysate (a, b) and plasma membrane fraction (c, d) was examined. hCMEC/D3 cells were exposed to 0.25 mM to 5 mM H2O2 for 20 min in ECF buffer. Whole-cell lysate and plasma membrane fraction of hCMEC/D3 cells were digested with lysyl endopeptidase and trypsin. The digests were subjected to LC-MS/MS together with internal standard peptides. Each value represents the mean ± SD of 8–16 SRM/MRM transitions in three to four independent samples. One-way ANOVA with Bonferroni-corrected Student's t-test was performed to determine the statistical significance of differences between protein expression levels under control and H2O2-treated conditions. (e) Dynasore attenuated the decrease of P-gp protein expression amount in the plasma membrane fraction caused by 0.5 mM H2O2 exposure. hCMEC/D3 cells were pre-incubated with dynasore (80 µM) or DMSO (vehicle) for 30 min, and then co-incubated with 0.5 mM H2O2 and dyansore or DMSO in ECF buffer for 20 min at 37℃. P-gp protein expression amounts in plasma membrane fraction were determined by LC-MS/MS analysis as described above. Each value represents the mean ± SD of 9-12 SRM/MRM transitions in three independent samples. (f) Dynasore attenuated the decrease of P-gp efflux transport activity caused by 0.5 mM H2O2 treatment. hCMEC/D3 cells were pre-incubated with dynasore (80 µM) for 30 min, and then the cellular uptake of vinblastine was measured with or without 0.5 mM H2O2, PSC833 and dynasore for 20 min. Each value represents the mean ± SD (n = 3). *p < 0.05, ***p < 0.005 (Student’s t-test).
Identification of phosphorylated proteins as candidate regulators of P-gp efflux transport activity in H2O2-treated hCMEC/D3 cells
Since phosphorylation is a rapid and transient regulatory mechanism, we hypothesized that it could play a role in the change of P-gp efflux function in response to oxidative stress. To find candidate substrates, we conducted comprehensive comparative phosphoproteomics. Since the P-gp efflux transport activity was significantly decreased by H2O2 in the range of 0.5 mM to 5 mM, we focused on the nine phosphorylated proteins that commonly changed their phosphorylation levels in cells treated with 0.5 mM and with 5 mM H2O2 (Supplementary Figure 1 and Supplemental Table 1). Among them, we further examined four for which phosphorylation inhibitors are available: Cav1, stromal interaction molecule 1 (STIM1), microtubule-associated protein 4 (MAP4), and heat shock factor binding protein 1 (HSBP1). Cav1 is reported to be phosphorylated by two kinases, Abl kinase and Src kinase, in fibroblasts in the presence of H2O2.30,31 Therefore, to block Cav1 signaling, we employed imatinib and PP2 as Abl kinase and Src kinase inhibitors, respectively. STIMl is involved in Ca2+ uptake, and we used the Ca2+ uptake inhibitor SKF96365 as a STIM1 inhibitor.32 MAP4 is a microtubule-binding protein, so we used Bis-ANS as an inhibitor of microtubule interaction.33 KRIBB3, an inhibitor of blocking protein kinase C-dependent phosphorylation of HSPB1,34 was used to suppress HSBP1 signaling. The concentrations of inhibitors were set as equal to or higher than those reported in the literature as significantly suppressing the activity or expression levels of the target proteins, Abl kinase,35 Src kinase,36 STIM1,37 MAP433 and HSPB1.34 In the presence of imatinib and PP2, the decrease of P-gp efflux transport activity by 5 mM H2O2 was significantly attenuated (Figure 3(a) and (b)). However, SKF96365, Bis-ANS and KRIBB3 had no effect (Figure 3(c) to (e)). These results suggest that phosphorylation of Cav1 by Abl kinase and Src kinase is involved, at least in part, in the decrease of P-gp function at the BBB under acute oxidative stress.
Figure 3.

Effect of inhibitors of candidate molecules potentially involved in the reduction of P-gp efflux transport activity by H2O2. hCMEC/D3 cells were pre-incubated with inhibitors of candidate molecules for the indicated period of time at 37℃, and the P-gp efflux activity was measured by vinblastine uptake assay with or without PSC833, candidate molecule inhibitors and 5 mM H2O2. (a) Imatinib (10 µM), an Abl kinase inhibitor, was present during pre-incubation for 30 min and incubation for 20 min for vinblastine uptake assay in ECF buffer. (b) PP2 (10 µM), a Src kinase inhibitor, was present during pre-incubation for 30 min and incubation for 20 min for vinblastine uptake in ECF buffer. (c) SKF-96365 (20 µM), a Ca2 + influx inhibitor, was present during pre-incubation for 10 min and incubation for 20 min for vinblastine uptake in ECF buffer. (d) Bis-ANS (10 µM), an inhibitor of microtubule interaction with MAP4, was present during pre-incubation for 30 min and incubation for 20 min for vinblastine uptake in ECF buffer. (e) KRIBB3 (10 µM), an inhibitor of protein kinase C-dependent phosphorylation of HSPB1, was present during pre-incubation for 4 h in culture medium and incubation for 20 min for vinblastine uptake in ECF buffer. Each value represents the mean ± SD (n = 3). *p < 0.05 (Student's t-test).
Abl kinase- and Src kinase-mediated Tyr14 phosphorylation of Cav1 is required for the internalization of P-gp and Cav1 in hCMEC/D3 cells
In order to examine how the changes described above are linked, we investigated the mechanism of internalization of P-gp under oxidative stress. First, in order to confirm that imatinib and PP2 inhibit Cav1 phosphorylation, tyrosine-14 (Tyr14)-phosphorylated Cav1 was quantitated by means of LC-MS/MS. Although 0.5 mM H2O2 did not change the total expression amount of Cav1 (Figure 4(a)), Tyr14-phosphorylated Cav1 was significantly increased (Figure 4(b)). Imatinib suppressed the increase of Tyr14-phosphorylated Cav1 by 0.5 mM H2O2 from 2.58 fmol/µg protein to 1.12 fmol/µg protein (Figure 4(b)), while PP2 suppressed it to 0.968 fmol/µg protein (Figure 4(b)).
Figure 4.

Tyr14 Cav1 phosphorylation by both Abl kinase and Src kinase is correlated with the reduction of P-gp efflux activity by H2O2. (a) The effects of imatinib and PP2, Abl kinase and Src kinase inhibitors, respectively, on the protein expression amount of Cav-1. hCMEC/D3 cells were incubated with 0.5 mM H2O2 for 20 min at 37℃ after pre-incubation with 10 µM imatinib or 10 µM PP2 for 30 min in ECF buffer. The whole-cell lysate of hCMEC/D3 cells was digested with lysyl endopeptidase and trypsin. Cav1 protein expression amounts were determined by LC-MS/MS analysis. (b) The effects of imatinib and PP2 on the Tyr14 phosphorylation amount of Cav1. Tyr14 phosphorylation levels of Cav1 in the whole-cell lysate of hCMEC/D3 cells were determined by LC-MS/MS analysis as described above. (c, d) Effect of Abl kinase knockdown with siRNA on the protein expression amount or Tyr14 phosphorylation amount of Cav1. hCMEC/D3 cells were transfected with negative control siRNA or Abl kinase siRNA. At 72 h post transfection, LC-MS/MS analysis was performed to determine the protein expression amount or Tyr14 phosphorylation amount of Cav1 in the whole-cell lysate of hCMEC/D3 cells. (e) Confirmation of Abl kinase knockdown by Western blotting. Whole-cell lysate of hCMEC/D3 cells (10 µg protein) was loaded on each lane. Na+/K+ ATPase was detected as a loading control. Three independent experiments were performed and representative data are shown. (f) Confirmation of the PP2 effect on Src kinase activity. Tyr419 phosphorylation amounts of Src kinase in the whole-cell lysate of hCMEC/D3 cells were determined by LC-MS/MS analysis. (g) Relationship between P-gp efflux transport activity and Tyr6, Tyr14, both Tyr6 and Tyr14, Tyr25 and Tyr42 phosphorylation amounts of Cav1 in 0.25 mM to 5 mM H2O2-treated hCMEC/D3 cells. P-gp efflux transport activity after H2O2 treatment was taken from Figure 1(b). After 20 min incubation of H2O2 in ECF buffer, the whole-cell lysate of hCMEC/D3 cells was digested with lysyl endopeptidase and trypsin. The underlined amino acids in the Cav1 sequence are potential phosphorylation sites. The YVDSEGHLYTVPIR peptide contains two phosphorylation sites, so Tyr6-phosphorylated, Tyr14-phosphorylated and both Tyr6/Tyr14-phosphorylated peptides were separately quantified in this study. The phosphorylation levels of Tyr25 and Tyr42 were under the detection limit in 0.25 mM to 5 mM H2O2-treated hCMEC/D3 cells. The limit of quantification values for Tyr25 and Tyr42 was 0.0100 fmol/µg protein and 0.0244 fmol/µg protein, respectively. Quantitative value determined by LC-MS/MS represents the mean ± SD of 9-12 SRM/MRM transitions in three independent analyses. *p < 0.05, ***p < 0.005. NC siRNA: negative control siRNA-transfected cells; Abl siRNA: Abl kinase siRNA-transfected cells; U.L.Q: under the limit of quantification; R: Pearson’s correlation coefficient.
Next, we set out to clarify the roles of Abl kinase and Src kinase in the phosphorylation of Tyr14 in Cav1 at the BBB. To do this, we firstly conducted Abl kinase knockdown experiments using siRNA. We confirmed that knockdown of Abl kinase with siRNA was effective (Figure 4(e)), and did not affect Cav1 expression (Figure 4(c)). The knockdown attenuated the H2O2-induced increase of Tyr14-phosphorylated Cav1 from 3.38 fmol/µg protein to 1.78 fmol/µg protein (Figure 4(d)). Secondly, we examined the effect of PP2 on Tyr419 phosphorylation. Since the phosphorylation of Src kinase at Tyr419 activates the kinase activity,38 the expression amount of Tyr419-phosphorylated Src kinase is a good measure of Src kinase activation state. PP2 blocked the H2O2-induced increase of Tyr419-phosphorylated Src kinase, reducing the amount from 1.73 fmol/µg protein to below the quantitation limit (LQ; 0.371 fmol/µg protein) (Figure 4(f)). Under the same conditions, PP2 attenuated the increase of Tyr14-phosphorylated Cav1 induced by 0.5 mM H2O2 (Figure 4(b)). These results indicate that the expression amount of Tyr14-phosphorylated Cav1 is regulated through both Abl kinase and Src kinase at the BBB in the presence of oxidative stress.
We then directly examined the phosphorylation sites in Cav1. Among peptides containing Tyr residues in human Cav1, those containing Tyr6, Tyr14, Tyr25 and Tyr42 fulfil the in silico peptide selection criteria described by Uchida et al.,26 so phosphorylation at these sites is expected to be detectable by LC-MS/MS analysis. In a previous study, Tyr6 and Tyr14 were identified as major Tyr phosphorylation sites of Cav1 by Src kinase.39 As shown in Supplementary Table 4, we found that phosphorylated peptides containing Tyr6, Tyr14, Tyr25 and Tyr42 were below the quantitation limit in the H2O2-untreated condition; the limits of quantification were 0.00654 fmol/µg protein (Tyr6), 0.000500 fmol/µg protein (Tyr14), 0.0100 fmol/µg protein (Tyr25) and 0.0244 fmol/µg protein (Tyr42). On the other hand, after treatment with 0.25 mM to 5 mM H2O2, phosphorylation of Tyr6 and Tyr14 of Cav1 became detectable (Figure 4(g), Supplementary Table 4). These were the same phosphorylation sites detected in the phosphoproteomic analysis (Supplementary Table 1). In the case of 5 mM H2O2 treatment, the expression amount of Tyr14-phosphorylated Cav1 was the greatest, 4.79 fmol/µg protein, which was about 200-fold or more larger than the limit-of-quantification values of Tyr25 and Tyr42. Small amounts of phosphorylation at Tyr6, and at both Tyr 6/Tyr14, were also detected. The phosphorylation ratio (%) at Tyr14 (calculated by dividing the expression amount of Tyr14-phosphorylated Cav1 by the protein expression amount of total Cav1) was 16.6%. Importantly, the quantitative value of Tyr14-phosphorylated peptide was significantly correlated with the decrease in efflux transport activity of P-gp, whereas the quantitative values of Tyr6- or both Tyr6 and Tyr14-phosphorylated peptides were not (Figure 4(g)). This result suggests that Tyr14 of Cav1 is a major phosphorylation site involved in the decrease of efflux transport activity of P-gp.
Next, we investigated the relationship between Tyr14-phosphorylation of Cav1 and the internalization of P-gp. In pulmonary capillary endothelial cells, H2O2 treatment for 30 min accelerates Tyr14 phosphorylation of Cav1 and albumin uptake via caveolae endocytosis.40 Since 80% of P-gp expressed in human brain capillaries is co-localized with Cav1 in caveolae fractions,41 P-gp could be internalized together with Cav1 in response to oxidative stress. Thus, we examined the changes in the expression amounts of P-gp and Cav1 in the plasma membrane fraction of hCMEC/D3 cells caused by H2O2 treatment, and found that Cav1 in the plasma membrane fraction was significantly decreased to 57.9% (0.25 mM H2O2), 46.2% (0.5 mM), 54.3% (1 mM), 37.0% (2.5 mM), and 21.7% (5 mM) (Figure 5(a) and (b)). Under the same conditions, there was no change in the expression amount of Cav1 in whole-cell lysate (Figure 4(a) and (c); concentrations other than 0.5 mM H2O2 are not shown). These results indicate that Cav1 is internalized at the BBB under conditions of oxidative stress. Further, there was a statistically significant correlation between the amounts of P-gp and Cav1 in the plasma membrane fraction (Figure 5(a)). The efflux transport activity of P-gp was also well correlated with the expression level of Cav1 in the plasma membrane fraction (Figure 5(b)). These findings support the idea that that P-gp is internalized together with Cav1 at the BBB under conditions of oxidative stress.
Figure 5.

Both Abl kinase and Src kinase are involved in the internalization of P-gp and Cav1 in H2O2-treated hCMEC/D3 cells. (a) Relationship between P-gp efflux transport activity and Cav1 expression amounts in the plasma membrane fraction of hCMEC/D3 cells. P-gp efflux transport activity was taken from Figure 1(b). (b) Comparison between P-gp and Cav1 in the plasma membrane fraction of H2O2-treated hCMEC/D3 cells. hCMEC/D3 cells were incubated with 0.25 mM, 0.5 mM, 1 mM, 2.5 mM or 5 mM H2O2 for 20 min at 37℃ in ECF buffer. The plasma membrane fraction was isolated with an IVB plasma membrane isolation kit. The plasma membrane fraction of hCMEC/D3 cells was digested with lysyl endopeptidase and trypsin. The protein expression amounts were determined by LC-MS/MS analysis. P-gp and Cav1 expression amounts in the plasma membrane fraction under the H2O2-treated condition were taken from Figures 2(c) and 5(a), respectively. (c, e, g) Effect of Abl kinase knockdown on the protein expression amounts of P-gp, Cav1 and MRP1 in the plasma membrane fraction of H2O2-treated hCMEC/D3 cells. hCMEC/D3 cells were transfected with negative control siRNA or siRNA directed against Abl kinase. At 72 h post-transfection, hCMEC/D3 cells were incubated with 0.5 mM H2O2 for 20 min, and then the plasma membrane fraction was isolated and used for LC-MS/MS analysis. To compare the effect of H2O2 on the internalization of P-gp, Cav1 and MRP1 in the negative control siRNA-transfected cell and Abl kinase siRNA-transfected cells, the H2O2-dependent decrease was calculated by subtracting the ratio of the protein expression amounts between H2O2-treated cells per control cells (percent) from 100%. MRP1 was also quantified as a H2O2 non-sensitive membrane marker, because no internalization or reduction of its efflux transport activity was observed in H2O2-treated hCMEC/D3 cells (Figures 1(d) and 2(d)). (d, f, h) Effect of PP2, a Src kinase inhibitor, on the protein expression amounts of P-gp, Cav1 and MRP1 in plasma membrane fraction of H2O2-treated hCMEC/D3 cells. hCMEC/D3 cells were pre-incubated with 10 µM PP2 or DMSO (vehicle) for 30 min, and then 0.5 mM H2O2 was treated with hCMEC/D3 cells for 20 min. The protein expression amounts of P-gp, Cav1 and MRP1 in the plasma membrane fraction were determined by LC-MS/MS analysis. Each value represents the mean ± SD of 10-12 SRM/MRM transitions in three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.005 (Student's t-test). N.S: not significant.
Finally, in order to confirm that internalization of P-gp and Cav1 is mediated by activation of Abl kinase and Src kinase, the amounts of P-gp and Cav1 in the plasma membrane fraction were measured under conditions of Abl kinase knockdown and PP2 (Src kinase inhibitor) treatment. Abl kinase knockdown significantly attenuated the H2O2-induced decrease in the plasma membrane expression of P-gp, as compared with negative control siRNA-transfected cells (Figure 5(c)). The amount of internalized Cav1 was also significantly suppressed (Figure 5(e)). In contrast, the plasma membrane expression of MRP1, which did not show H2O2-induced decrease of efflux transport activity (Figure 1(c) and (d)), did not change (Figure 5(g)). PP2 also significantly attenuated the H2O2-induced decrease of P-gp and Cav1 (Figure 5(d) and (f)). These results support the idea that activation of both Abl kinase and Src kinase mediates the internalization of P-gp and Cav1 at the BBB under conditions of oxidative stress.
Imatinib and PP2 attenuate the reduction of P-gp efflux transport activity by H2O2 in rat BBB
To see whether the same mechanism operates in vivo, we conducted a brain perfusion study to measure P-gp efflux function at the rat BBB using a typical P-gp substrate, quinidine. The ratio of brain concentration per perfusate concentration of quinidine, Rbr, was increased significantly in rats treated with 5 mM H2O2 (Figure 6(a), left panel). This increase was significantly suppressed by imatinib or PP2 (Figure 6(a), left panel). The Rbr of raffinose, a non-permeable marker, was statistically significantly increased in PP2-administered rats, while no marked change was observed among vehicle-treated and 5 mM H2O2-treated or imatinib-treated rats (Figure 6(a), right panel), indicating that tight-junction integrity was well maintained. Thus, H2O2 decreases the efflux transport activity of P-gp through the activation of Abl kinase and Src kinase in vivo.
Figure 6.

Effect of H2O2, imatinib and PP2 on the in vivo brain distributions of quinidine, cortisol and raffinose. Brain distributions of quinidine and raffinose in the whole cerebrum (a), cortisol and raffinose in the hippocampus (b), hypothalamus (c) and other cerebrum regions (d). Rats were pre-infused with modified Ringer solution containing 328 µM imatinib, 328 µM PP2 or DMSO (vehicle) for 30 min at a flow rate of 50 µL/min from the left side of the external carotid artery. In this pre-treatment, imatinib or PP2 concentration in the blood at the luminal side of the brain microvessels was estimated to be 5 µM. Details of the estimation of the concentration at the luminal side of the blood microvessels are given in Materials and methods. After pre-treatment, rats were perfused with modified Ringer solution containing 1 µM quinidine, 500 µM raffinose and 5 mM H2O2 for 10 min at a flow rate of 2 mL/min from the external carotid artery. In the case of imatinib or PP2 treatment, rats were perfused with modified Ringer solution containing 275 nM imatinib or 275 nM PP2. The concentration of 275 nM imatinib in perfusate is similar to the free concentration of 5 µM imatinib in blood, because the free fraction of imatinib is 5.5% in rat plasma. To determine quinidine distribution to the brain, the left cerebral hemisphere was collected for LC-MS/MS analysis. For examining cortisol distribution to specific regions of the brain, the hippocampus and hypothalamus were collected from the left cerebrum. The remaining cerebrum was used as other cerebrum regions. The brain distributions of quinidine, raffinose and cortisol were represented as Rbr, which is the ratio between perfusate and tissue concentration. Each column represents the mean ± SD of four to six rats. *p < 0.05, **p < 0.01, ***p < 0.005 (Bonferroni corrected Student’s t-test). N.S: not significant.
Next, we examined the uptake of cortisol. The Rbr values of cortisol in hippocampus, hypothalamus and other cerebrum regions were significantly elevated after the administration of 5 mM H2O2 (Figure 6(b) to (d), left panel). These elevations were significantly attenuated by imatinib or PP2 (Figure 6(b) to (d), left panel), while there was no significant alteration in the Rbr of raffinose (Figure 6(b) to (d), right panel). These results suggest that an oxidative stress-induced, Abl kinase- and Src kinase-mediated decrease of P-gp efflux transport activity contributes to the elevation of cortisol concentration in the brain.
Discussion
Abl kinase and Src kinase inhibitors are reported to reduce neurotoxicity in ischemia–reperfusion injury. A phase II clinical trial of imatinib, aiming to protect the brain after t-PA treatment for thrombolysis in patients with acute cerebral infarction, resulted in improved functional autonomy.42 Also, Src kinase inhibitors SKI-606 and SKS-927 improved the neurological score of rats after middle cerebral artery occlusion (MCAO).43 These inhibitors are reported to work by suppressing the decrease in expression of tight junction molecules at the BBB and by inhibiting release of inflammatory cytokines.43,44 Our results here suggest an additional mechanism of action. Specifically, we found that oxidative stress (H2O2 treatment) induced the dynamin-dependent internalization of P-gp, resulting in a rapid decrease of P-gp-mediated efflux transport activity. This response is mediated by Abl kinase and Src kinase, which phosphorylate Tyr14 in the P-gp-binding molecule Cav1, inducing concomitant internalization of both molecules. We also confirmed that the resulting decrease of P-gp function is associated with an increase of cortisol concentration in the brain. Since exposure to high levels of glucocorticoids in the brain induces hippocampal neuronal cell death,13,14 the suppression of elevated cortisol concentration in the brain may have contributed to the improvement of cognitive function by Abl kinase and Src kinase inhibitors observed in the previous studies.
It is well established that a rapid increase of reactive oxygen species after ischemia–reperfusion alters BBB function. Previous studies have documented increased expression of P-gp45 and transcriptional activation of P-gp46 in H2O2-treated rat brain capillary primary-cultured cells. In contrast, we found that the expression amount of P-gp in the plasma membrane fraction of hCMEC/D3 cells was significantly decreased within 20 min in response to H2O2 treatment, without any change of P-gp protein amount in the whole-cell lysate. The decreased expression of P-gp was attenuated in the presence of dynasore, which is an inhibitor of dynamin-dependent endocytosis. These results indicated that intracellular trafficking of P-gp is involved in the post-translational regulation of P-gp during the acute phase of oxidative stress exposure at the BBB.
McCaffrey et al.47 reported that P-gp, Cav1 and Tyr14-phosphorylated Cav1 formed a high-molecular-weight complex in isolated rat brain capillaries 3 h after administration of λ-carrageenan in a hyperalgesic rat model. Subcellular fractionation of isolated rat brain capillaries showed that the complex of P-gp and Cav1 was redistributed from the plasma membrane protein-rich fractions to nuclear membrane protein-rich fractions. This indicates that P-gp and Cav1 trafficking to the intracellular compartment was facilitated in λ-carrageenan-induced hyperalgesic rats. Also, the P-gp and Cav1 complex was dissociated by reducing agents, suggesting that the complex was produced by disulfide-bond formation between P-gp and Cav1 via oxidative reaction. Based on these findings, Davis’s group proposed that redox-dependent rearrangements of P-gp complexes could maintain P-gp in an inactive form in the intracellular compartment.19 Here, we show that oxidative stress can indeed be a trigger for P-gp internalization. In addition, we revealed the involvement of dynamin in the H2O2-mediated P-gp internalization. Dynamin is a key player in caveolae-dependent, clathrin-dependent and interleukin 2 receptor-dependent endocytosis.29 Our finding of a significant correlation between P-gp function and Cav1 expression amount in the plasma membrane indicates that P-gp forms a complex with Cav1 and this complex is internalized via caveolae-dependent endocytosis in H2O2-treated hCMEC/D3 cells. This is consistent with a previous report showing that immortalized rat brain capillary endothelial cells (RBE4) expressing mutant Cav1 with point mutation of Tyr14 formed a smaller amount of P-gp and Cav1 complex as compared with cells expressing normal Cav1.48 It is known that Tyr14 of Cav1 is phosphorylated by Abl kinase and Src kinase in H2O2-treated fibroblasts,30,31 and the present work indicates that Tyr14 in Cav1 is the predominant target of these kinases in an H2O2-treated human BBB model, at least among Tyr6, Tyr14, Tyr25 and Tyr42. Notably, the amount of Tyr14-phosphorylated Cav1 was well correlated with the decrease of P-gp efflux transport activity. We also showed that Abl kinase and Src kinase inhibitors attenuated the H2O2-mediated reduction of P-gp efflux transport activity in a rat model in vivo. Overall, our study has provided experimental evidence for the redox-dependent trafficking of P-gp that was previously proposed by Davis’s group, and we also newly identified dynamin, Abl kinase and Src kinase as key molecules involved in signaling for P-gp translocation (Figure 7).
Figure 7.

A schematic overview of the proposed molecular mechanism of the rapid reduction in P-gp efflux activity at the BBB under oxidative stress. During oxidative stress, both Abl and Src kinases increase the phosphorylation level of Tyr14 in Cav1. This induces internalization of both P-gp and Cav1 through dynamin-dependent endocytosis, with loss of P-gp efflux activity. It is not clear whether intracellular Cav1 remains phosphorylated, but Abl and Src kinases could be continuously activated during oxidative stress, so we show intracellular Cav1 as phosphorylated. When the oxidative stress is removed, P-gp efflux activity recovers. This should be due to trafficking of intracellular P-gp to the plasma membrane, because the recovery of P-gp efflux activity was seen within 20 min after removal of oxidative stress (Figure 1(e)).
Dan et al.49 have reported that the permeability coefficient-surface area product (PS value) of the P-gp substrate domperidone in ischemia–reperfusion treated rats was up to 3.6 times higher than that in a sham-treated group, and the BBB penetration rate constant (Kin) of domperidone in mdr1a (–/–) knock-out mice was 3.1 times higher than that of wild-type mice,50 suggesting that P-gp efflux function is largely suppressed in ischemia–reperfusion-treated rats. However, when the rats were also given the P-gp inhibitor verapamil, the PS value of domperidone increased to 43.3 times that in the sham-treated group. This result indicated that domperidone efflux at the BBB might be mediated not only by P-gp, but also by other efflux transporters, and verapamil inhibited all of them. Therefore, it could not be concluded from their findings whether or not P-gp efflux transport activity is decreased during cerebral ischemia-reperfusion. In our case, the decrease of P-gp efflux transport activity caused by H2O2 treatment in hCMEC/D3 cells was measured in terms of PSC833-sensitive vinblastine accumulation. It is reported that a high concentration of PSC833 can inhibit MRP1-mediated vinblastine efflux transport.51 Also, MRP1 and MRP4 are expressed in hCMEC/D3 cells.52 However, in the uptake analysis using MK571, an MRP family inhibitor, vinblastine efflux transport activity was unaffected by 5 mM H2O2 treatment. Furthermore, the expression amounts of MRP1 in plasma membrane fraction and whole-cell lysate did not change significantly. Thus, we can conclude that the H2O2-dependent decrease in vinblastine efflux transport in hCMEC/D3 cells was specifically due to P-gp. Furthermore, Uchida et al.53 successfully reconstructed the Kp brain value of quinidine in mice by using the free fraction of quinidine in plasma and the contribution rate of P-gp to the brain migration of quinidine, which were calculated from data obtained in mdr1a (−/−) knock-out mice and wild-type mice. Their findings suggested that the major transporter contributing to brain migration of quinidine in mice is P-gp, and other transporters have a negligible effect. Thus, we have shown for the first time that the P-gp efflux transport activity is decreased by oxidative stress. This decrease of P-gp efflux transport activity would have contributed to the increased brain migration of domperidone observed by Dan et al.
We also established that the intracerebral concentrations of quinidine and cortisol were significantly elevated in 5 mM H2O2-administered rats. However, it is important to consider whether P-gp efflux function might actually be decreased during human cerebral ischemia–reperfusion. In this connection, Mathru et al.54 investigated the concentration changes of H2O2 in whole blood during lower extremity surgery; the value before hemostasis was 133 µM, while it reached 796 µM (about 6-fold increase) in local blood downstream of the hemostasis site at 5 min after reperfusion following 126 min of hemostasis. Furthermore, a microdialysis study found that the H2O2 concentration in the intercellular fluid in rat striatum was about 25 µM in normal rats, whereas it reached 150–175 µM (5 - to 7-fold increase) after ischemia–reperfusion.55 The H2O2 concentration in human whole blood was reported to be 114–577 µM,56 so if we assume that there is a 5-fold increase during cerebral ischemia-reperfusion, it is plausible that blood concentration of H2O2 in the lumen of the brain capillaries reaches 0.57 mM to 2.9 mM H2O2. Our results in hCMEC/D3 cells suggested that 0.5 mM and 2.5 mM H2O2 would decrease P-gp efflux transport activity by about 33% and 66%, respectively. Overall, these findings support the idea that P-gp efflux transport activity at the BBB would be substantially decreased in the H2O2 concentration range that could be reached during human cerebral ischemia–reperfusion.
Takasato et al.57 first developed the brain perfusion technique, which is now considered a well-established method for measuring net transport across the BBB under near-physiological conditions. There is a good correlation between values of the permeability coefficient of nonelectrolytes measured by the brain perfusion and intravenous injection methods,57 suggesting that the integrity of BBB is well maintained during brain perfusion. Furthermore, a brain perfusion study by Hawkins et al. showed that the inhibition of P-gp by 8 µM cyclosporine A increased the brain distribution of verapamil 4.3-fold relative to vehicle controls.37 In an intravenous injection study, 5.8 µM and 9.3 µM cyclosporine A in blood increased the brain concentration of verapamil 2.6 - and 7.9-fold, respectively.58 These results suggest that the brain perfusion technique well reflects the physiological function of P-gp in the BBB in vivo.
Although the hCMEC/D3 cell line is commonly used in the field, there are some important differences between the hCMEC/D3 cell model and the in vivo human BBB. For example, P-gp expression level in hCMEC/D3 cells is extremely low compared to that in freshly isolated human cerebral capillaries.59 Thus, our study may have underestimated the in vivo P-gp function. There may also be differences in the protein expression levels of Abl kinase, Src kinase and Cav1 between hCMEC/D3 cells and human cerebral capillaries. Further studies will be needed in order to evaluate whether or not our findings in hCMEC/D3 cells can be extrapolated to humans.
It is well known that BBB integrity is impaired in the early phase of ischemia–reperfusion.15 This loss of integrity might enable increased entry of cortisol into the brain. However, there is evidence that P-gp can limit migration of its substrates into the brain even under conditions where the tight junctions are disrupted. For example, in mice subjected to cerebral ischemia–reperfusion, the concentrations of the P-gp substrates FK506 and rifampicin were increased by co-administration of a P-gp inhibitor, tariquidar.60 Similarly, in λ-carrageenan-induced hyperalgesic rats in which the tight junctions are impaired,61 the brain migration of morphine was increased by co-administration of the P-gp inhibitor cyclosporin A.62 We found that the Rbr of raffinose was significantly increased in PP2-administered rats, but the H2O2-induced increase of Rbr of quinidine was markedly suppressed in PP2-co-administered rats. Thus, cortisol efflux transport mediated by P-gp could play an important role even under conditions where the integrity of the BBB is compromised. This in turn suggests that Abl kinase and Src kinase inhibitors could be effective for maintaining P-gp-mediated efflux of glucocorticoids even during ischemia–reperfusion in humans.
Supplemental Material
Supplemental Material for Oxidative stress-induced activation of Abl and Src kinases rapidly induces P-glycoprotein internalization via phosphorylation of caveolin-1 on tyrosine-14, decreasing cortisol efflux at the blood–brain barrier by Yutaro Hoshi, Yasuo Uchida, Masanori Tachikawa, Sumio Ohtsuki, Pierre-Olivier Couraud, Takashi Suzuki and Tetsuya Terasaki in Journal of Cerebral Blood Flow & Metabolism
Acknowledgements
We thank A. Niitomi and N. Handa for their secretarial assistance.
Authors’ contributions
Yutaro Hoshi and Yasuo Uchida: Study design/conception, analysis/acquisition of data, drafting of manuscript. Masanori Tachikawa, Sumio Ohtsuki, Tetsuya Terasaki: Study design/conception, revision of manuscript. Pierre-Olivier Couraud: Providing the hCMEC/D3 cells. Takashi Suzuki: Providing the human brain tissues.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by Grants-in-Aids from the Japanese Society for the Promotion of Science (JSPS) for Young Scientists (A) [KAKENHI: 16H06218], Young Scientists (B) [KAKENHI: 23790170], Scientific Research (A) [KAKENHI: 24249011], Bilateral Open Partnership Joint Research Program (between Finland and Japan) and JSPS Research Fellow [KAKENHI: 266383]. This study was also supported in part by Grants-in-Aids from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) for Scientific Research on Innovative Areas [KAKENHI: 18H04534], and from Nakatomi Foundation.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Tetsuya Terasaki and Sumio Ohtsuki are full professors at Tohoku University and Kumamoto University, and are also directors of Proteomedix Frontiers Co., Ltd. This study was not supported by Proteomedix Frontiers Co., Ltd, and their positions at Proteomedix Frontiers Co., Ltd did not influence the design of the study, the collection of the data, the analysis or interpretation of the data, the decision to submit the manuscript for publication, or the writing of the manuscript and did not present any financial conflicts. The other authors declare no competing interests. All experiments were conducted in compliance with the ARRIVE guidelines.
Supplementary material
Supplementary material for this paper can be found at the journal website: http://journals.sagepub.com/home/jcb
References
- 1.Luengo-Fernandez R, Paul NL, Gray AM, et al. Population-based study of disability and institutionalization after transient ischemic attack and stroke: 10-year results of the Oxford Vascular Study. Stroke 2013; 44: 2854–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kelly-Hayes M, Beiser A, Kase CS, et al. The influence of gender and age on disability following ischemic stroke: the Framingham study. J Stroke Cerebrovasc Dis 2003; 12: 119–126. [DOI] [PubMed] [Google Scholar]
- 3.National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group. Tissue plasminogen activator for acute ischemic stroke. N Engl J Med 1995; 333: 1581–1587. [DOI] [PubMed] [Google Scholar]
- 4.Davis SM, Lees KR, Albers GW, et al. Selfotel in acute ischemic stroke: possible neurotoxic effects of an NMDA antagonist. Stroke 2000; 31: 347–354. [DOI] [PubMed] [Google Scholar]
- 5.Horn J, Limburg M. Calcium antagonists for ischemic stroke: a systematic review. Stroke 2001; 32: 570–576. [DOI] [PubMed] [Google Scholar]
- 6.Diener HC, Cortens M, Ford G, et al. Lubeluzole in acute ischemic stroke treatment: a double-blind study with an 8-hour inclusion window comparing a 10-mg daily dose of lubeluzole with placebo. Stroke 2000; 31: 2543–2551. [DOI] [PubMed] [Google Scholar]
- 7.Xu SY, Pan SY. The failure of animal models of neuroprotection in acute ischemic stroke to translate to clinical efficacy. Med Sci Monit Basic Res 2013; 19: 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edaravone Acute Infarction Study Group. Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction. Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc Dis 2003; 15: 222–229. [DOI] [PubMed] [Google Scholar]
- 9.Inatomi Y, Takita T, Yonehara T, et al. Efficacy of edaravone in cardioembolic stroke. Intern Med 2006; 45: 253–257. [DOI] [PubMed] [Google Scholar]
- 10.Selakovic VM, Jovanovic MD, Jovicic A. Changes of cortisol levels and index of lipid peroxidation in cerebrospinal fluid of patients in the acute phase of completed stroke. Vojnosanit Pregl 2002; 59: 485–491. [DOI] [PubMed] [Google Scholar]
- 11.Selakovic V. Cortisol in plasma and cerebrospinal fluid of patients with brain ischemia. Med Pregl 2004; 57: 354–358. [DOI] [PubMed] [Google Scholar]
- 12.Abraham IM, Harkany T, Horvath KM, et al. Action of glucocorticoids on survival of nerve cells: promoting neurodegeneration or neuroprotection? J Neuroendocrinol 2001; 13: 749–760. [DOI] [PubMed] [Google Scholar]
- 13.Anacker C, Cattaneo A, Luoni A, et al. Glucocorticoid-related molecular signaling pathways regulating hippocampal neurogenesis. Neuropsychopharmacology 2013; 38: 872–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krugers HJ, Maslam S, Korf J, et al. The corticosterone synthesis inhibitor metyrapone prevents hypoxia/ischemia-induced loss of synaptic function in the rat hippocampus. Stroke 2000; 31: 1162–1172. [DOI] [PubMed] [Google Scholar]
- 15.Giraud M, Cho TH, Nighoghossian N, et al. Early blood brain barrier changes in acute ischemic stroke: a sequential MRI study. J Neuroimaging 2015; 25: 959–963. [DOI] [PubMed] [Google Scholar]
- 16.Uhr M, Holsboer F, Muller MB. Penetration of endogenous steroid hormones corticosterone, cortisol, aldosterone and progesterone into the brain is enhanced in mice deficient for both mdr1a and mdr1b P-glycoproteins. J Neuroendocrinol 2002; 14: 753–759. [DOI] [PubMed] [Google Scholar]
- 17.Terasaki Y, Liu Y, Hayakawa K, et al. Mechanisms of neurovascular dysfunction in acute ischemic brain. Curr Med Chem 2014; 21: 2035–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Felix RA, Barrand MA. P-glycoprotein expression in rat brain endothelial cells: evidence for regulation by transient oxidative stress. J Neurochem 2002; 80: 64–72. [DOI] [PubMed] [Google Scholar]
- 19.Davis TP, Sanchez-Covarubias L, Tome ME. P-glycoprotein trafficking as a therapeutic target to optimize CNS drug delivery. Adv Pharmacol 2014; 71: 25–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weksler BB, Subileau EA, Perriere N, et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J 2005; 19: 1872–1874. [DOI] [PubMed] [Google Scholar]
- 21.Hoshi Y, Uchida Y, Tachikawa M, et al. Actin filament-associated protein 1 (AFAP-1) is a key mediator in inflammatory signaling-induced rapid attenuation of intrinsic P-gp function in human brain capillary endothelial cells. J Neurochem 2017; 141: 247–262. [DOI] [PubMed] [Google Scholar]
- 22.Uchida Y, Ohtsuki S, Katsukura Y, et al. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J Neurochem 2011; 117: 333–345. [DOI] [PubMed] [Google Scholar]
- 23.Ito K, Uchida Y, Ohtsuki S, et al. Quantitative membrane protein expression at the blood-brain barrier of adult and younger cynomolgus monkeys. J Pharm Sci 2011; 100: 3939–3950. [DOI] [PubMed] [Google Scholar]
- 24.Hoshi Y, Uchida Y, Tachikawa M, et al. Quantitative atlas of blood-brain barrier transporters, receptors, and tight junction proteins in rats and common marmoset. J Pharm Sci 2013; 102: 3343–3355. [DOI] [PubMed] [Google Scholar]
- 25.Kamiie J, Ohtsuki S, Iwase R, et al. Quantitative atlas of membrane transporter proteins: development and application of a highly sensitive simultaneous LC/MS/MS method combined with novel in-silico peptide selection criteria. Pharm Res 2008; 25: 1469–1483. [DOI] [PubMed] [Google Scholar]
- 26.Uchida Y, Tachikawa M, Obuchi W, et al. A study protocol for quantitative targeted absolute proteomics (QTAP) by LC-MS/MS: application for inter-strain differences in protein expression levels of transporters, receptors, claudin-5, and marker proteins at the blood-brain barrier in ddY, FVB, and C57BL/6J mice. Fluids Barriers CNS 2013; 10: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hosoya K, Saeki S, Terasaki T. Activation of carrier-mediated transport of L-cystine at the blood-brain and blood-retinal barriers in vivo. Microvasc Res 2001; 62: 136–142. [DOI] [PubMed] [Google Scholar]
- 28.Cannon RE, Peart JC, Hawkins BT, et al. Targeting blood-brain barrier sphingolipid signaling reduces basal P-glycoprotein activity and improves drug delivery to the brain. Proc Natl Acad Sci U S A 2012; 109: 15930–15935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blouin CM, Lamaze C. Interferon gamma receptor: the beginning of the journey. Front Immunol 2013; 4: 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanguinetti AR, Mastick CC. c-Abl is required for oxidative stress-induced phosphorylation of caveolin-1 on tyrosine 14. Cell Signal 2003; 15: 289–298. [DOI] [PubMed] [Google Scholar]
- 31.Cao H, Sanguinetti AR, Mastick CC. Oxidative stress activates both Src-kinases and their negative regulator Csk and induces phosphorylation of two targeting proteins for Csk: caveolin-1 and paxillin. Exp Cell Res 2004; 294: 159–171. [DOI] [PubMed] [Google Scholar]
- 32.Zou JJ, Gao YD, Geng S, et al. Role of STIM1/Orai1-mediated store-operated Ca(2)(+) entry in airway smooth muscle cell proliferation. J Appl Physiol 2011; 110: 1256–1263. [DOI] [PubMed] [Google Scholar]
- 33.Mazumdar M, Parrack PK, Mukhopadhyay K, et al. Bis-ANS as a specific inhibitor for microtubule-associated protein induced assembly of tubulin. Biochemistry 1992; 31: 6470–6474. [DOI] [PubMed] [Google Scholar]
- 34.Shin KD, Lee MY, Shin DS, et al. Blocking tumor cell migration and invasion with biphenyl isoxazole derivative KRIBB3, a synthetic molecule that inhibits Hsp27 phosphorylation. J Biol Chem 2005; 280: 41439–41448. [DOI] [PubMed] [Google Scholar]
- 35.Yang J, Campobasso N, Biju MP, et al. Discovery and characterization of a cell-permeable, small-molecule c-Abl kinase activator that binds to the myristoyl binding site. Chem Biol 2011; 18: 177–186. [DOI] [PubMed] [Google Scholar]
- 36.Hawkins BT, Sykes DB, Miller DS. Rapid, reversible modulation of blood-brain barrier P-glycoprotein transport activity by vascular endothelial growth factor. J Neurosci 2010; 30: 1417–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liou J, Kim ML, Heo WD, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol 2005; 15: 1235–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brown MT, Cooper JA. Regulation, substrates and functions of src. Biochim Biophys Acta 1996; 1287: 121–149. [DOI] [PubMed] [Google Scholar]
- 39.Li S, Seitz R, Lisanti MP. Phosphorylation of caveolin by src tyrosine kinases. The alpha-isoform of caveolin is selectively phosphorylated by v-Src in vivo. J Biol Chem 1996; 271: 3863–3868. [PubMed] [Google Scholar]
- 40.Sun Y, Hu G, Zhang X, et al. Phosphorylation of caveolin-1 regulates oxidant-induced pulmonary vascular permeability via paracellular and transcellular pathways. Circ Res 2009; 105: 676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jodoin J, Demeule M, Fenart L, et al. P-glycoprotein in blood-brain barrier endothelial cells: interaction and oligomerization with caveolins. J Neurochem 2003; 87: 1010–1023. [DOI] [PubMed] [Google Scholar]
- 42.Wahlgren N, Thoren M, Hojeberg B, et al. Randomized assessment of imatinib in patients with acute ischaemic stroke treated with intravenous thrombolysis. J Intern Med 2017; 281: 273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang S, Pong K, Gonzales C, et al. Neuroprotective profile of novel SRC kinase inhibitors in rodent models of cerebral ischemia. J Pharmacol Exp Ther 2009; 331: 827–835. [DOI] [PubMed] [Google Scholar]
- 44.Merali Z, Leung J, Mikulis D, et al. Longitudinal assessment of imatinib's effect on the blood-brain barrier after ischemia/reperfusion injury with permeability MRI. Transl Stroke Res 2015; 6: 39–49. [DOI] [PubMed] [Google Scholar]
- 45.Nwaozuzu OM, Sellers LA, Barrand MA. Signalling pathways influencing basal and H(2)O(2)-induced P-glycoprotein expression in endothelial cells derived from the blood-brain barrier. J Neurochem 2003; 87: 1043–1051. [DOI] [PubMed] [Google Scholar]
- 46.Robertson SJ, Kania KD, Hladky SB, et al. P-glycoprotein expression in immortalised rat brain endothelial cells: comparisons following exogenously applied hydrogen peroxide and after hypoxia-reoxygenation. J Neurochem 2009; 111: 132–141. [DOI] [PubMed] [Google Scholar]
- 47.McCaffrey G, Staatz WD, Sanchez-Covarrubias L, et al. P-glycoprotein trafficking at the blood-brain barrier altered by peripheral inflammatory hyperalgesia. J Neurochem 2012; 122: 962–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barakat S, Demeule M, Pilorget A, et al. Modulation of p-glycoprotein function by caveolin-1 phosphorylation. J Neurochem 2007; 101: 1–8. [DOI] [PubMed] [Google Scholar]
- 49.Dan Y, Murakami H, Koyabu N, et al. Distribution of domperidone into the rat brain is increased by brain ischaemia or treatment with the P-glycoprotein inhibitor verapamil. J Pharm Pharmacol 2002; 54: 729–733. [DOI] [PubMed] [Google Scholar]
- 50.Dagenais C, Avdeef A, Tsinman O, et al. P-glycoprotein deficient mouse in situ blood-brain barrier permeability and its prediction using an in combo PAMPA model. Eur J Pharm Sci 2009; 38: 121–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leier I, Jedlitschky G, Buchholz U, et al. The MRP gene encodes an ATP-dependent export pump for leukotriene C4 and structurally related conjugates. J Biol Chem 1994; 269: 27807–27810. [PubMed] [Google Scholar]
- 52.Ohtsuki S, Ikeda C, Uchida Y, et al. Quantitative targeted absolute proteomic analysis of transporters, receptors and junction proteins for validation of human cerebral microvascular endothelial cell line hCMEC/D3 as a human blood-brain barrier model. Mol Pharm 2013; 10: 289–296. [DOI] [PubMed] [Google Scholar]
- 53.Uchida Y, Ohtsuki S, Kamiie J, et al. Blood-brain barrier (BBB) pharmacoproteomics: reconstruction of in vivo brain distribution of 11 P-glycoprotein substrates based on the BBB transporter protein concentration, in vitro intrinsic transport activity, and unbound fraction in plasma and brain in mice. J Pharmacol Exp Ther 2011; 339: 579–588. [DOI] [PubMed] [Google Scholar]
- 54.Mathru M, Dries DJ, Barnes L, et al. Tourniquet-induced exsanguination in patients requiring lower limb surgery. An ischemia-reperfusion model of oxidant and antioxidant metabolism. Anesthesiology 1996; 84: 14–22. [DOI] [PubMed] [Google Scholar]
- 55.Hyslop PA, Zhang Z, Pearson DV, et al. Measurement of striatal H2O2 by microdialysis following global forebrain ischemia and reperfusion in the rat: correlation with the cytotoxic potential of H2O2 in vitro. Brain Res 1995; 671: 181–186. [DOI] [PubMed] [Google Scholar]
- 56.Varma SD, Devamanoharan PS. Hydrogen peroxide in human blood. Free Radic Res Commun 1991; 14: 125–131. [DOI] [PubMed] [Google Scholar]
- 57.Takasato Y, Rapoport SI, Smith QR. An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am J Physiol 1984; 247: H484–H493. [DOI] [PubMed] [Google Scholar]
- 58.Hsiao P, Sasongko L, Link JM, et al. Verapamil P-glycoprotein transport across the rat blood-brain barrier: cyclosporine, a concentration inhibition analysis, and comparison with human data. J Pharmacol Exp Ther 2006; 317: 704–710. [DOI] [PubMed] [Google Scholar]
- 59.Hartz AM, Schulz JA, Sokola BS, et al. Isolation of cerebral capillaries from fresh human brain tissue. J Vis Exp 2018; 139: e57346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spudich A, Kilic E, Xing H, et al. Inhibition of multidrug resistance transporter-1 facilitates neuroprotective therapies after focal cerebral ischemia. Nat Neurosci 2006; 9: 487–488. [DOI] [PubMed] [Google Scholar]
- 61.Huber JD, Hau VS, Borg L, et al. Blood-brain barrier tight junctions are altered during a 72-h exposure to lambda-carrageenan-induced inflammatory pain. Am J Physiol Heart Circ Physiol 2002; 283: H1531–H1537. [DOI] [PubMed] [Google Scholar]
- 62.Seelbach MJ, Brooks TA, Egleton RD, et al. Peripheral inflammatory hyperalgesia modulates morphine delivery to the brain: a role for P-glycoprotein. J Neurochem 2007; 102: 1677–1690. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material for Oxidative stress-induced activation of Abl and Src kinases rapidly induces P-glycoprotein internalization via phosphorylation of caveolin-1 on tyrosine-14, decreasing cortisol efflux at the blood–brain barrier by Yutaro Hoshi, Yasuo Uchida, Masanori Tachikawa, Sumio Ohtsuki, Pierre-Olivier Couraud, Takashi Suzuki and Tetsuya Terasaki in Journal of Cerebral Blood Flow & Metabolism