

Abstract
Cerebral amyloid angiopathy (CAA) is present in over half of the elderly population and in 80–90% of Alzheimer’s disease (AD) patients. CAA is defined by the deposition of beta amyloid (Aβ) in small cerebral arteries and capillaries. Cardiovascular risk factors are associated with an increased incidence of CAA. We utilized 18-month-old endothelial nitric oxide synthase (eNOS) heterozygous knockout (+/−) mice, a clinically relevant model of endothelial dysfunction, to examine the role of endothelial nitric oxide (NO) in vascular Aβ accumulation. eNOS+/− mice had significantly higher vascular levels of Aβ40 (P < 0.05). Aβ42 was not detected. There was no difference in Aβ in brain tissue. Amyloid precursor protein and β-site APP cleavage enzyme 1 protein levels were unaltered, while levels of the α-secretase enzyme, a disintegrin and metalloproteinase 10, were significantly lower in eNOS + /− microvascular tissue (P < 0.05). Insulin degrading enzyme and low-density lipoprotein receptor-related protein 1 were significantly increased in eNOS+/− microvascular tissue, most likely an adaptive response to locally higher Aβ concentrations. Lastly, catalase and CuZn superoxide dismutase were significantly elevated in eNOS+/− microvascular tissue (P < 0.05). These data demonstrate decreased availability of endothelial NO leads to increased cerebrovascular concentration of Aβ along with compensatory mechanisms to protect the vasculature.
Keywords: ADAM10, beta amyloid, cerebral amyloid angiopathy, cerebrovascular, endothelial nitric oxide synthase
Introduction
Cerebral amyloid angiopathy (CAA) is present to some degree, asymptomatic to severe, in over half the elderly population.1 Furthermore, it is present in up to 80–90% of Alzheimer’s disease (AD) patients.2,3 CAA is characterized by the deposition of beta amyloid (Aβ) in small cerebral arteries, arterioles, and capillaries.1,4 The exact mechanism and source of the increased Aβ within the cerebral vessel wall are unknown. It has been suggested that several cardiovascular risk factors are associated with a higher incidence of CAA.5–7 A common feature of cardiovascular risk factors is endothelial dysfunction. Endothelial dysfunction is characterized by decreased bioavailability of endothelial nitric oxide (NO).8
Endothelial NO is produced and released by endothelial cells within the vascular wall. NO is highly diffusible and can act in an autocrine and paracrine fashion on the endothelium and smooth muscle cells (SMCs). Endothelial NO has many important functions, including: vasodilation and anti-inflammatory effects. Our previous studies also demonstrate an important role for endothelial NO in modulating amyloid precursor protein (APP) processing within human brain microvascular endothelial cells and mouse cerebral microvessels.9–12 Human brain microvascular endothelial cells treated with the nitric oxide synthase (NOS) inhibitor, N(G)-Nitro-L-Arginine Methyl Ester (L-NAME) led to increased APP and β-site APP cleaving enzyme (BACE)1 expression as well as increased Aβ production.12 Furthermore, increased APP and BACE1 expression were dependent on the NO second messenger, cyclic guanosine monophosphate (cGMP) as treatment with a soluble guanylyl cyclase inhibitor also led to increased APP and BACE1 expression in these cells. Lastly, phosphodiesterase 5 inhibition, resulting in increased cGMP levels, led to decreased APP and BACE1 expression.12
APP is highly expressed in the vessel wall in both vascular endothelial cells12–17 and SMCs.18–22 Both cell types express the secretase enzymes (α, β, and γ-secretase) required to generate APP cleavage products, including Aβ.12–22 Importantly, Aβ deposited within the vascular wall could be of vascular origin. Delineating the role of endothelial NO signaling in vascular Aβ deposition will provide mechanistic insights into the role of endothelial dysfunction in the pathogenesis of CAA and AD.
In our studies, we utilize aged (18 month old) endothelial (e)NOS heterozygous knockout (+/−) mice which represent a clinically relevant model of endothelial dysfunction, where there is a significant reduction in bioavailable NO but not a complete loss. We present novel findings demonstrating that decreased availability of endothelial NO leads to increased Aβ accumulation in the vascular tissue of aged eNOS+/− mice without any changes in the levels of Aβ in brain tissue.
Materials and methods
Animals
All experimental protocols were reviewed and approved by the Institutional Animal Care and Use Committee of Mayo Clinic. All protocols comply with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and with ARRIVE guidelines. Nos3™1Unc/J (eNOS−/−), stock #002684, and C57BL/6 (wild-type) mice, stock #000664 were purchased from Jackson Laboratory (Bar Harbor, ME). eNOS−/− mice were bred with eNOS+/+ (wild type) mice to generate eNOS+/− mice. eNOS+/− were bred to generate eNOS+/− and littermate wild type controls for use in experiments. Male mice were sacrificed at 17–19 months of age by a lethal dose of pentobarbital (200–250 mg/kg body weight, i.p.). Genotyping was performed using eNOS primers according to Jackson Laboratories. DNA was isolated from a 2–3 mm piece of mouse tail using the PureLink Genomic DNA mini kit (Invitrogen) following manufacturer’s instructions. The sequence for mutant (knock out) eNOS was 5′- AAT TCG CCA ATG ACA AGA CG -3′. The wild type eNOS sequence was 5′- AGG GGA ACA AGC CCA GTA GT -3′ and the common reverse primer for eNOS was 5′- CTT GTC CCC TAG GCA CCT CT -3′.
Glucose, cholesterol, and triglyceride measurements
Blood was collected via right ventricular puncture. Glucose was measured in whole blood using Accu Check (Roche Diagnostics, Indianapolis, IN). Blood was centrifuged (2000 r/min, 10 min, 4℃) and stored at −80° until all samples were collected. Total triglyceride and cholesterol levels were measured using the Hitachi 912 chemistry analyzer (Roche Diagnostics).
Tissue collection
Brains were carefully removed and immediately placed in ice cold modified Krebs-Ringer bicarbonate solution containing 118.6 mmol/L NaCl, 4.7 mmol/L KCl, 2.5 mmol/L CaCl2, 1.2 mmol/L KH2PO4, 25.1 mmol/L NaHCO3, 0.026 mmol/L EDTA, 10.1 mmol/L glucose as previously described (Austin et al 2010). Large cerebral arteries, including basilar and cerebral arteries were carefully removed prior to cerebral microvessel isolation or brain tissue homogenization.
Cerebral microvessel isolation
Cerebral microvessels were isolated from the remaining brain tissue as previously described.11,12. Brain tissue, devoid of large vessels, was homogenized in ice cold PBS with Dounce homogenizer and centrifuged at 3000 r/min at 4℃. The supernatant, containing the parenchymal tissue, was discarded. The pellet was resuspended in PBS and centrifuged as described above. The resulting pellet was resuspended and layered over 15% Dextran (in PBS) (Sigma, St. Louis, MO) and centrifuged at 4000 r/min for 30 min at 4℃. The top layer was aspirated and discarded and the remaining pellet was resuspended in 1% bovine serum albumin (BSA) and the suspension was then passed through a 40 µm nylon mesh (BD Falcon). Microvessels retained on the mesh were washed with BSA/PBS and collected by centrifugation at 3000 r/min for 10 min at 4℃. Microvessels were rinsed with PBS, centrifuged at 4000 r/min for 5 min at 4℃ and then resuspended in appropriate buffer according to assay being performed.
Western blotting
To perform Western blot analyses, tissue homogenates were lysed in ice cold Triton lysis buffer (10 mmol/L Hepes, 50 mmol/L NaF, 50 mmol/L NaCl, 5 mmol/L EDTA, 5 mmol/L EGTA, 100 µmol/L Na3VO4, 50 mmol/L Na pyrophosphate and 1% Triton X-100) plus protease inhibitors as previously described.12 Equal protein amounts were resolved by SDS-PAGE and transferred to nitrocellulose membranes for Western blotting. Blots were probed with primary antibodies. β-site APP cleaving enzyme 1 (BACE1), presenilin (PS)1, PS2, low-density lipoprotein receptor-related protein 1 (LRP1), and neuronal (n) NOS were purchased from Cell Signaling (Danvers, MA). Endothelin-converting enzyme-1 (ECE1), insulin-degrading enzyme (IDE), and receptor for advanced glycation endproducts (RAGE) were obtained from Abcam (Cambridge, MA). APP was obtained from Upstate Cell Signaling (Temecula, CA) and cyclooxygenase (COX)-1 was purchased from Invitrogen (Camarillo, CA). COX-2, eNOS, and inducible (i) NOS were purchased from BD Transduction Laboratories (San Jose, CA). Manganese (Mn) superoxide dismutase (SOD), extracellular (EC) SOD, and Copper-Zinc (CuZn) SOD were purchased from Enso Life Sciences (Farmingdale, NY). Prostacyclin synthase (PGI2S) and thromboxane synthase (TXA2S) antibodies were obtained from Cayman Chemical (Ann Arbor, MI). Neprilysin and nicastrin antibodies were purchased from Millipore (Temecula, CA.). Anterior pharynx-defective 1 (Aph1) and a disintegrin and metalloproteinase (ADAM)10 antibodies were purchased from Thermoscientific and Chemicon International [of Thermoscientific] (Rockford, IL). Catalase and Actin were purchased from Sigma-Aldrich (St. Louis, MO). Presenilin enhancer 2 (Pen2) was purchased from ProSCi (Poway, CA.). For protein quantitation, the optical density of each protein of interest was normalized to its respective loading control’s optical density. Actin was used as the loading control. Data are presented as a ratio to wild type controls.
β-secretase activity assay
Brain and cerebral microvessels were homogenized with CelLytic MT lysis buffer (Sigma-Aldrich) and assayed for β-secretase activity via a commercially available fluorescent assay kit per manufacturer’s instructions (Sigma-Aldrich, St. Louis, MO).
Aβ ELISA
Aβ1-40 and Aβ1-42 levels from the circulating blood and from brain and microvascular tissue lysate were measured using commercially available ELISA kits (Invitrogen, Camarillo, CA).
sAPPα ELISA
Circulating and brain and microvascular tissue lysate levels of soluble (s)APPα were measured via a commercially available colorimetric ELISA per manufacturer’s instructions (IBL International, Japan).
Ex vivo measurement of secreted Aβ and sAPPα
Cerebral microvessels were isolated as described. Cerebral microvessels were then incubated overnight in endothelial basal medium 2 (EBM2; Lonza, Basel, Switzerland) at 37℃. Cultured medium was collected and assayed for secreted levels of Aβ and sAPPα.
Statistical analysis
Data are presented as mean ±SD. Graphs also display individual data points. Statistical analysis was performed using the unpaired Student’s t-test. For non-parametric data, statistical analysis was performed using Mann–Whitney Rank Sum. A value of P < 0.05 was considered statistically significant. When appropriate, power analysis tests were performed and significant data had a minimum power of 0.80 and an alpha of 0.05.
Results
Characteristics of aged (18 month old) eNOS+/− mice
Body weight of 18-month-old eNOS+/− mice was not significantly different than that of littermate wild type controls (Supplemental Table 1; P > 0.05). Prior studies established that arterial blood pressure was not different between 18-month-old eNOS+/− and wild type mice.23 Furthermore, circulating levels of total cholesterol, HDL cholesterol, and glucose were not different between wild type and eNOS+/− mice (Supplemental Table 1; P > 0.05). Circulating levels of triglycerides were significantly elevated in 18-month-old eNOS+/− mice as compared to littermate wild type controls (Supplemental Table 1; P < 0.05). Lastly, circulating plasma levels of APP cleavage products: sAPPα, Aβ40 and Aβ42 were also not different between wild type and eNOS+/− mice (Supplemental Table 1; P > 0.05).
Levels of APP cleavage products, Aβ and sAPPα in brain and cerebrovasculature
Aβ40 and Aβ42 levels were measured via ELISA from cerebral microvascular tissue lysate of 18-month-old male wild type and eNOS+/− mice. Levels of Aβ40 were significantly higher in cerebral microvascular tissue of eNOS+/− mice as compared to wild type (Figure 1(a); P < 0.05). Aβ42 was not detected in microvascular tissue lysate from either wild type or eNOS+/− mice (n = 4; data not shown). Interestingly, when we measured Aβ levels in brain tissue lysate there was no difference in either Aβ40 or Aβ42 between 18-month-old wild type and eNOS+/− mice (Figure 1(b) and (c), P > 0.05).
Figure 1.

Aβ40 levels were increased in eNOS+/− microvascular tissue. Microvascular tissue from 18-month-old eNOS+/− and wild type animals was analyzed for (a) Aβ40 via commercially available ELISA kit. Brain tissue from 18-month-old eNOS+/− and wild type animals was analyzed for (b) Aβ40 and (c) Aβ42 via commercially available ELISA kits. All data are presented as mean ±SD with individual data points shown (n = 4 animals; *P < 0.05).
To begin to examine the source of the increased microvascular Aβ, we next performed ex vivo experiments on isolated cerebral microvessels. First, we measured the secreted levels of Aβ from isolated cerebral microvessels of 18-month-old wild type and eNOS+/− mice. Aβ42 was not detected in the cultured medium of the microvessels of either wild type or eNOS+/− mice (n = 3; data not shown). Aβ40 levels were significantly higher in the conditioned medium of the cerebral microvessels isolated from 18-month-old eNOS+/− mice as compared to wild type (Figure 2(a); P < 0.05). We also measured the APP cleavage product, sAPPα, in the conditioned medium of isolated cerebral microvessels of 18-month-old wild type and eNOS+/− mice. sAPPα was significantly lower in the conditioned medium of isolated cerebral microvessels from eNOS+/− mice as compared to age-matched wild type controls (Figure 2(b); P < 0.05).
Figure 2.
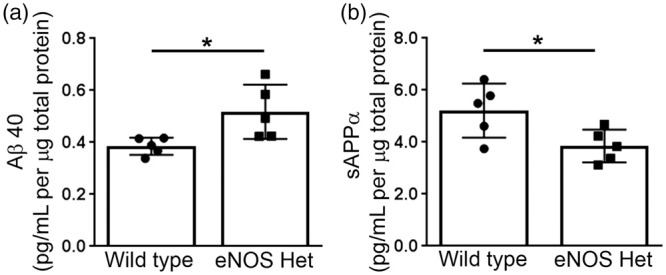
Ex vivo secretion of Aβ40 is increased, while secretion of sAPPα is decreased from microvessels isolated from eNOS+/− mice. Cerebral microvessels were isolated from 18-month-old eNOS+/− and wild type mice and incubated overnight in fresh endothelial basal media. Supernatant was collected and analyzed for Aβ40 and sAPPα. Ex vivo secreted levels of (a) Aβ40 and (b) sAPPα from cerebral microvessels of 18-month-old eNOS+/− and wild type mice were measured via commercially available ELISA kits. All data are presented as mean ±SD with individual data points shown (n = 5 animals; *P < 0.05).
Levels of APP and its cleavage enzymes
Next, we examined if protein expression of the major α and β-secretase enzymes was altered in microvascular or brain tissue of 18-month-old wild type or eNOS+/− mice. APP, ADAM10, the major α-secretase,24,25 and BACE1 protein levels were measured by Western blot analyses. Cerebral microvessel protein levels of APP and BACE1 were not different between 18-month-old wild type and eNOS+/− mice (Figure 3(a) to (c)), P > 0.05). However, cerebral vascular ADAM10 protein levels were significantly lower in 18-month-old eNOS+/−mice as compared to littermate wild type controls (Figure 3(a) and (d); P < 0.05). Notably, levels of APP, BACE1, and ADAM10 were not altered in the brain tissue of 18-month-old eNOS+/− mice as compared to wild type control (Supplemental Figure 1(a) to (d)); P > 0.05). Importantly, this demonstrates that alterations in APP processing and Aβ are restricted to microvascular tissue.
Figure 3.

Mature ADAM10 protein levels were significantly reduced in eNOS+/− microvascular tissue. (a) Microvascular tissue from 18-month-old eNOS+/− and wild type animals was Western blotted using anti-APP, anti-BACE1, anti-ADAM10, and anti-Actin (loading control) antibodies. Representative image is shown. Densitometric analysis was performed for (b) APP (n = 6 animals), (c) BACE1 (n = 7 animals), and (d) ADAM10 (n = 5 animals; *P < 0.05). BACE enzymatic activity was measured from 18-month-old eNOS+/− and wild type (e) microvascular tissue via a commercially available BACE activity assay kit (n = 3–4). All data are presented as mean ±SD with individual data points shown.
We also measured enzyme activity of BACE from brain and cerebral microvascular tissue of 18-month-old wild type and eNOS+/− mice. BACE enzyme activity in microvascular tissue was not different between littermate wild type and eNOS+/− mice (Figure 3(e); P > 0.05). BACE enzymatic activity was decreased in the brain tissue of 18-month-old eNOS+/− mice as compared to wild type (Supplemental Figure 1(e)). The difference was very small but it did reach statistical significance (wild type: 0.454 ± 0.0152, eNOS+/−: 0.415 ± 0.0058; n = 4, P < 0.05).
We examined protein levels of the γ-secretase complex: PS1, PS2, nicastrin, Aph1, and Pen2 in microvascular tissue. PS1, but not PS2, protein expression was significantly increased in the cerebral microvascular tissue of 18-month-old eNOS+/− mice as compared to littermate wild type mice (Figure 4(a) and (b)); P < 0.05) while the other γ-secretase protein levels were unchanged (Figure 4(a) and (c) to (f)); P > 0.05).
Figure 4.
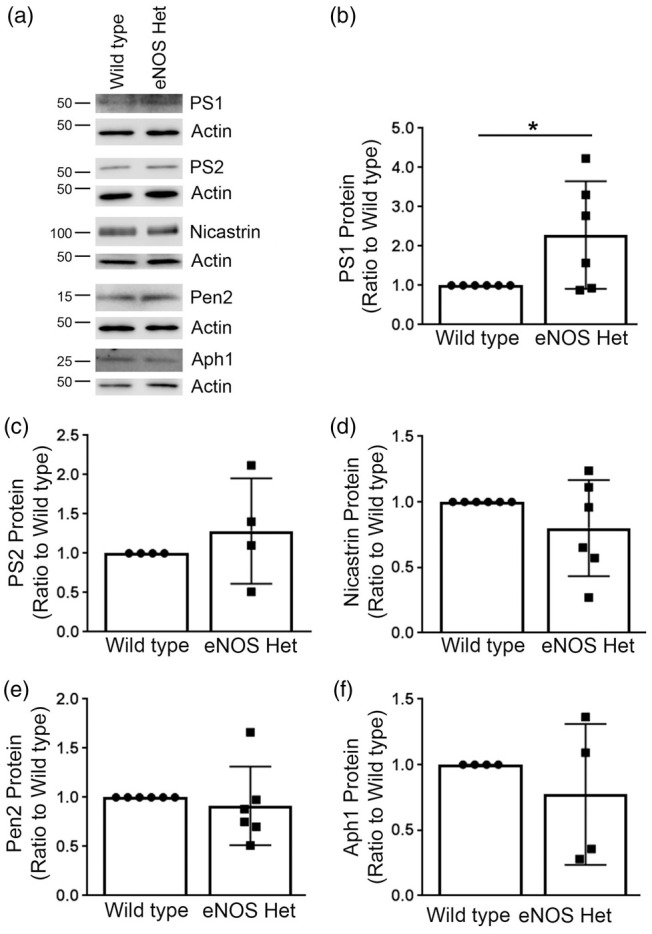
PS1, a major γ-secretase component, was significantly increased in microvascular tissue of eNOS+/− mice. (a) Microvascular tissue from 18-month-old eNOS+/− and wild type animals was analyzed by Western blot analyses for γ-secretase components: PS1, PS2, nicastrin, Pen2, and Aph1. Representative image is shown. Densitometric analysis was performed for (b) PS1 (n = 6 animals; *P < 0.05), (c) PS2 (n = 4 animals), (d) nicastrin (n = 6 animals), (e) Pen2 (n = 6 animals), and (f) Aph1 (n = 4 animals). All data are presented as mean±SD with individual data points shown.
Measurement of proteins involved in Aβ clearance mechanisms
Next, we measured protein levels of the three major Aβ-degradation enzymes: ECE1, IDE, and neprilysin. ECE1 and neprilysin protein levels were not significantly altered in eNOS+/− mice as compared to littermate wild type control (Figure 5). IDE protein levels were significantly higher in 18-month-old cerebral microvessels of eNOS+/− mice as compared to littermate wild type controls (Figure 5(a) and (c); P < 0.05). We also measured the protein levels of the two major Aβ receptors responsible for Aβ efflux (LRP1) and influx (RAGE) into the brain. Protein levels of LRP1 were significantly higher in the cerebral microvascular tissue of 18-month-old eNOS+/− mice as compared to littermate wild type controls (Figure 6(a) and (b); P < 0.05). Protein levels of RAGE were not altered between cerebral microvascular tissue of 18-month-old wild type and eNOS+/− mice (Figure 6(a) and (c); P > 0.05).
Figure 5.
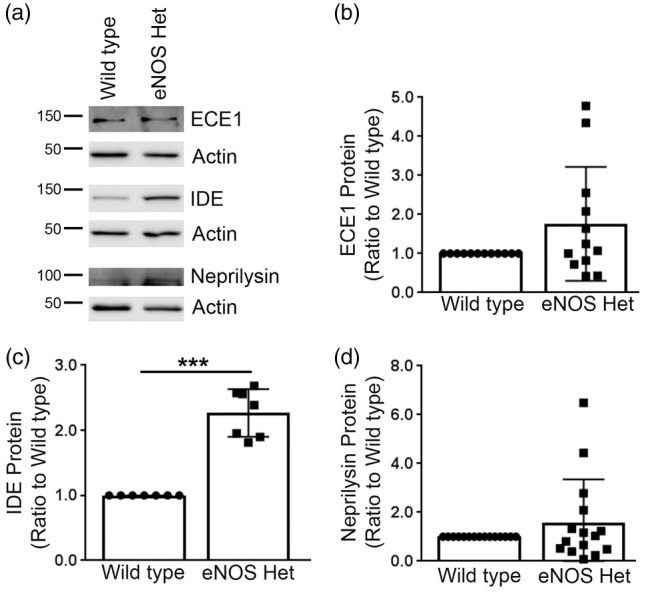
IDE was significantly increased in eNOS+/− microvascular tissue. (a) Microvascular tissue from 18-month-old eNOS+/− and wild type animals was Western blotted using anti-ECE1, anti-IDE, anti-neprilysin, and anti-Actin (loading control) antibodies. Representative image is shown. Densitometric analysis was performed for (b) ECE1 (n = 12 animals), (c) IDE (n = 7 animals; ***P < 0.0001), and (d) neprilysin (n = 15 animals). All data are presented as mean ±SD with individual data points shown.
Figure 6.
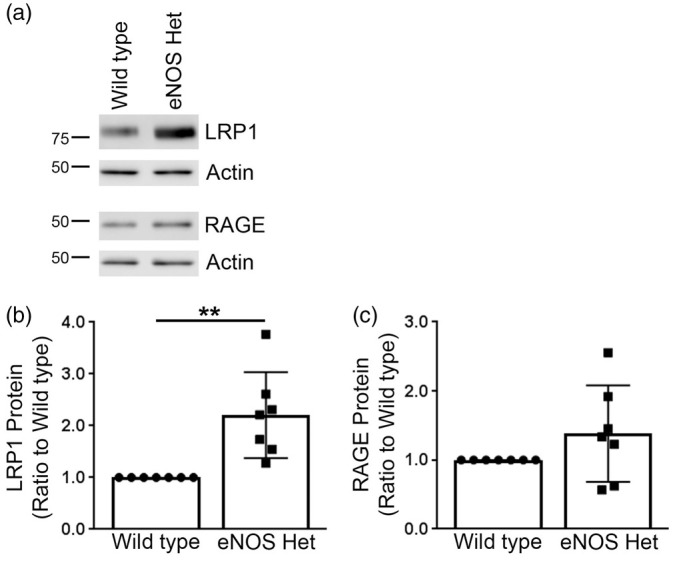
Aβ receptor, LRP1, was significantly increased in microvascular tissue of eNOS+/− mice. (a) Microvascular tissue from 18-month-old eNOS+/− and wild type animals was analyzed by Western blot analyses for Aβ receptors: LRP1 and RAGE. Representative image is shown. Densitometric analysis was performed for (b) LRP1 (n = 7 animals; **P < 0.01) and (c) RAGE (n = 7 animals). All data are presented as mean ±SD with individual data points shown.
Characterization of proteins important for endothelial signaling pathways
We measured levels of all NOS isoforms in the cerebral microvessels of 18-month-old wild type and eNOS+/− mice. As expected, eNOS was significantly reduced in the cerebral microvessels of 18-month-old eNOS+/− mice as compared to littermate wild type controls (Supplemental Figure 2(a) and (b); P < 0.05). iNOS protein levels were unchanged and nNOS was not detected in cerebral microvessels of wild type and eNOS+/− mice (Supplemental Figure 2(a) and (c) and (d)).
Next, we measured levels of critical enzymes involved in the antioxidant system, including: catalase, CuZn SOD, EC SOD, and Mn SOD. Levels of catalase and CuZn SOD were significantly increased in cerebral microvessels of 18-month-old eNOS+/− mice as compared to littermate wild type controls (Figure 7(a) to (c)); P < 0.05), while levels of EC SOD and Mn SOD were not altered in 18-month-old eNOS+/− mice (Figure 7(a) and (d) and (e)); P > 0.05).
Figure 7.

Protein levels of catalase and CuZn SOD were significantly increased in eNOS+/− microvascular tissue. (a) Microvascular tissue from 18-month-old eNOS+/− and wild type animals was Western blotted using anti-catalase, anti-CuZn SOD, anti-EC SOD, anti-Mn SOD, and anti-Actin (loading control) antibodies. Representative image is shown. Densitometric analysis was performed for (b) catalase (n = 9 animals; **P < 0.01), (c) CuZn SOD (n = 7 animals; **P < 0.01), (d) EC SOD (n = 5 animals), (e) Mn SOD (n = 6 animals). All data are presented as mean ±SD with individual data points shown.
Lastly, we measured levels of COX-1, COX-2, PGI2S, and TXA2S protein. COX-2 protein levels were significantly higher in the cerebral microvessels of 18-month-old eNOS+/− mice as compared to littermate wild type controls (Supplemental Figure 3(a) and (c); P<0.05). TXA2S protein levels tended to be increased but did not quite reach significance (Supplemental Figure 3(a) and (e); P = 0.0699, n = 12).
Characterization of 18-month-old eNOS+/− brain tissue
As stated earlier, no increases in APP expression, processing, or Aβ generation were observed in the brains of 18-month-old eNOS+/− mice; however, we also examined NOS isoforms, Aβ degradation enzymes, and receptors in brain tissue.
As anticipated, eNOS protein was significantly decreased in the brain tissue of eNOS+/− mice as compared to littermate wild type mice (Supplemental Figure 4(a) to (d); P > 0.05). This reflects decreased microvascular expression of eNOS protein as eNOS expression is restricted to endothelial cells.26,27 No compensatory changes in other NOS isoforms were detected (Supplemental Figure 4(a) and (c) and (d); P > 0.05).
Protein levels of IDE were significantly higher in 18-month-old eNOS+/− mice as compared to littermate wild type controls (Supplemental Figure 5(a) and (c); P < 0.05). All other proteins involved in degradation or proteins acting as Aβ receptors were unchanged in eNOS+/− mice (Supplemental Figures 5 and 6; P > 0.05).
Discussion
Our data demonstrate that partial loss of endothelial NO, a clinically relevant model of endothelial dysfunction, leads to increased Aβ40 production and accumulation within microvascular tissue. We were unable to detect Aβ42 in vascular lysates. CAA is present to some degree in over half the elderly population and is present in up to 80–90% of AD patients.1–3 Aβ40 is the predominant Aβ species present in vascular tissue, while Aβ42 is the prominent species in parenchymal plaques.28,29 In line with this, we report increased Aβ40 within the vasculature. Importantly, microvascular changes are independent of alterations in brain tissue Aβ production as brain tissue from 18-month-old eNOS+/− mice lacked any changes in APP expression or processing. Our previous results as well as those from others have demonstrated the ability of endothelial NO to inhibit amyloidogenic processing of APP in both human cells and mouse cerebrovascular tissue.9,11,12,23,30
Vascular dysfunction is a significant feature of CAA and AD.31,32 Endothelial dysfunction is an early event in a mouse model of AD.33,34 Faraco et al.35 demonstrated profound endothelial dysfunction in Tg2576 mice that was exacerbated with angiotensin II induced hypertension. It is possible that in these mice, loss of endothelial NO played a role in the angiotensin II-induced exacerbation of amyloid pathologies.
Partial loss of endothelial NO is a clinically relevant model of endothelial dysfunction as complete loss of endothelial NO does not occur in human disease. Indeed, cardiovascular risk factors including aging, hypertension, hypercholesterolemia, smoking, and sedentary lifestyle cause partial loss of expression and function of eNOS. Several human eNOS genetic polymorphisms have been identified.36,37 Polymorphisms that are associated with enhanced eNOS signaling are associated with decreased risk of peripheral artery disease and stroke, while polymorphisms that result in decreased eNOS signaling correlate with higher risks.37 The effects of eNOS polymorphisms on CAA, AD, and cognitive function remain to be determined. In mice, heterozygous eNOS gene deficiency does not result in any overt phenotypic characteristics. This is likely a result of adaptive mechanisms activated in response to impairment of eNOS signaling (reviewed36). In order to unmask the vulnerability of the vasculature when only one copy of eNOS is present, the animals must be stressed. In our studies, we are using aging to stress eNOS+/− mice.
It is important to note that phenotypically 18-month-old eNOS+/− mice have metabolic parameters similar to their littermate wild type controls. Importantly, Tan et al.23 demonstrated that mean arterial blood pressure does not differ between 6- and 18-month-old eNOS+/− and wild type mice.23 We report here that body weight, glucose, total and HDL cholesterol are not different between 18-month-old eNOS+/− and wild type mice. Triglycerides were significantly higher in 18-month-old eNOS+/− mice. The effect of elevated triglyceride levels, if any, on vascular Aβ production remains to be determined. Taken together, these data demonstrate that alterations observed in the microvascular and brain tissue of 18-month-old eNOS+/− mice are likely not caused by an increase in arterial blood pressure or by systemic alterations of lipid or glucose metabolism.
Most notably, we observed increased microvascular Aβ levels; however, we did not observe changes in APP or BACE1 expression or activity in microvascular tissue. Interestingly, we did observe decreased expression of mature ADAM10, the primary α-secretase enzyme.24,25 Endothelial NO may affect transcription or post translational modification of ADAM10 protein. The promoter region for ADAM10 has several functional binding sites, including stimulating protein (Sp)-1, nuclear factor (NF)κB, and peroxisome proliferator-activated receptors (PPARs) response elements which can be modulated by NO.38–43 The mechanism by which loss of endothelial NO leads to decreased ADAM10 expression remains to be determined. Regardless of the mechanism by which endothelial NO modulates ADAM10 protein levels, a reduction in mature ADAM10 could result in less full length APP being processed via the α-secretase pathway thereby leading to a larger pool of full length APP available to be processed by BACE1. α and β secretase pathways appear to compete for the same pool of APP.44,45 In BACE−/− mice and in wild type mice treated with BACE inhibitior, sAPPβ and Aβ levels were significantly reduced, while sAPPα levels were increased.44 Furthermore, overexpression of BACE in HEK293 cells resulted in increased Aβ production and decreased sAPPα levels.45 Taken together, these data demonstrate that a shift in enzymatic activity or expression in one pathway likely shifts processing to the other pathways as they are competing for the same pool of APP. Indeed, we do see reduced sAPPα production accompanied by increased Aβ production in cerebral microvessels of 18-month-old eNOS+/− mice. Furthermore, we observed an increase in PS1 protein levels, a key component of the γ-secretase complex. Taken together, these data may explain why we have observed a decrease in sAPPα production and an increase in Aβ levels without changes in APP or BACE1 protein expression or activity.
sAPPα is largely accepted as a vascular and neuroprotective molecule. Numerous studies have demonstrated the neuroprotective effects of sAPPα.46–49 sAPPα derived from vascular tissue may play a role in protecting cells within the vasculature and nearby neuronal tissue. d’Uscio et al.50 demonstrated endothelial dysfunction in endothelial specific APP knockout mice. Lastly, sAPPα may have a role in memory learning in animal models.51,52 Tan et al.23 described impaired spatial learning and memory in 18-month-old eNOS+/− mice.23 The loss of sAPPα, in addition to the detrimental effects of increased vascular Aβ seen in this model of eNOS deficiency has serious implications for cerebrovascular and cognitive health in conditions where endothelial dysfunction is present.
Expression of IDE and LRP1 was increased in the cerebral microvessels of 18-month-old eNOS+/− mice. We think that this is a compensatory adaptation made by microvascular tissue in attempt to help lower rising Aβ levels. It is also possible that the decreased levels of mature ADAM10 led to a reduction in cleavage of LRP1. ADAM10 is one of the enzymes responsible for shedding LRP153 to a smaller soluble LRP1. We also observed increased IDE expression in brain tissue in the absence of increased brain Aβ levels. Further studies will need to be done to examine other important Aβ related proteins, such as Aβ scavenger receptors, including: CD36, CD47, and scavenger receptor-A which have been suggested to modulate Aβ accumulation.54–56
It appears that these compensatory increases in IDE and LRP1 expression are not sufficient to lower Aβ levels. Endothelial dysfunction (lower bioavailability of endothelial NO) as in eNOS+/− microvascular tissue is accompanied by an increase oxidative stress.57 Cordes et al.58 report that oxidative stress can alter enzymatic activity of IDE so even though expression levels are increased, enzymatic activity of IDE may be lower. Oxidized LRP1 does not bind Aβ as efficiently and therefore clearance of Aβ may also be lower even though LRP1 protein levels are increased.59 We speculate that LRP1 and IDE may be oxidized in the eNOS+/− model and thus net degradation and clearance of Aβ are reduced.
We report here that partial loss of endothelial NO leads to increased expression of catalase and CuZn SOD. Increased expression of these two major antioxidant enzymes may be a compensatory response to increased oxidative stress. Indeed, increased Aβ leads to oxidative stress and increased production of superoxide anion in endothelial and SMCs of the vasculature.60–68 Increased superoxide anion is detrimental in many ways, but specific to endothelial cells, it inactivates endothelial NO leading to the formation of peroxynitrite which could further damage the endothelium and surrounding cells.69–71
COX-2 expression is increased in microvascular tissue of eNOS+/− mice. TXA2S protein tended to be increased. COX-2 and TXA2S are typical proinflammatory mediators which are induced by a variety of stimuli, including Aβ oxidative stress.72–80 Importantly, TXA2 is a powerful vasoconstrictor and stimulator of platelet aggregation.81,82 Loss of endothelial NO and the resulting increased production of TXA2 puts the vasculature at greater risk of thrombotic events. Indeed, Tan et al.23 reported increased thrombotic microinfarcts in aged eNOS+/− mice.23
It is important to note that eNOS-deficiency first leads to Aβ changes within the vasculature and not the brain parenchyma. This suggests that cerebrovascular alterations occur independent of changes within the brain. Defining the role of endothelial NO signaling in vascular Aβ deposition provides mechanistic insights into the role of endothelial dysfunction in the pathogenesis of CAA as well as AD. In AD with CAA present, these data suggest that vascular alterations may initiate or exacerbate changes within the brain. Our findings have important clinical implications. Indeed, activation of NO/cGMP signaling in the cerebrovascular endothelium and SMCs may offer a new therapeutic approach in the prevention and treatment of CAA.
Supplemental Material
Supplemental Material for Partial loss of endothelial nitric oxide leads to increased cerebrovascular beta amyloid by Susan A Austin and Zvonimir S Katusic in Journal of Cerebral Blood Flow & Metabolism
Authors’ contributions
SAA contributed to experimental design and performance, data collection and analysis, and manuscript writing. ZSK participated in experimental design and analysis, and manuscript writing.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institutes of Health grant HL-131515 (Z.S.K.), AHA Scientist Development Award AHA# 14SDG20410063 (S.A.A), and the Mayo Foundation.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr. Katusic served as a consultant to Ironwood Pharmaceuticals (Massachusetts).
Supplemental material
Supplementary material for this paper can be found at the journal website: http://journals.sagepub.com/home/jcb
References
- 1.Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke 1987; 18: 311–324. [DOI] [PubMed] [Google Scholar]
- 2.Castellani RJ, Smith MA, Perry G, et al. Cerebral amyloid angiopathy: major contributor or decorative response to Alzheimer’s disease pathogenesis. Neurobiol Aging 2004; 25: 599–602. discussion 3–4. [DOI] [PubMed] [Google Scholar]
- 3.Ellis RJ, Olichney JM, Thal LJ, et al. Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, part XV. Neurology 1996; 46: 1592–1596. [DOI] [PubMed] [Google Scholar]
- 4.Herzig MC, Van Nostrand WE, Jucker M. Mechanism of cerebral beta-amyloid angiopathy: murine and cellular models. Brain Pathol 2006; 16: 40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the american heart association/american stroke association. Stroke 2011; 42: 2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Izumihara A, Suzuki M, Ishihara T. Recurrence and extension of lobar hemorrhage related to cerebral amyloid angiopathy: multivariate analysis of clinical risk factors. Surg Neurol 2005; 64: 160–164. discussion 4. [DOI] [PubMed] [Google Scholar]
- 7.Purnell C, Gao S, Callahan CM, et al. Cardiovascular risk factors and incident Alzheimer disease: a systematic review of the literature. Alzheimer Dis Assoc Disord 2009; 23: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vanhoutte PM, Zhao Y, Xu A, et al. Thirty years of saying no: sources, fate, actions, and misfortunes of the endothelium-derived vasodilator mediator. Circul Res 2016; 119: 375–396. [DOI] [PubMed] [Google Scholar]
- 9.Austin SA, d’Uscio LV, Katusic ZS. Supplementation of nitric oxide attenuates AbetaPP and BACE1 protein in cerebral microcirculation of eNOS-deficient mice. J Alzheimers Dis 2013; 33: 29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Austin SA, Katusic ZS. Loss of endothelial nitric oxide synthase promotes p25 generation and tau phosphorylation in a murine model of Alzheimer’s disease. Circul Res 2016; 119: 1128–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Austin SA, Santhanam AV, Hinton DJ, et al. Endothelial nitric oxide deficiency promotes Alzheimer’s disease pathology. J Neurochem 2013; 127: 691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Austin SA, Santhanam AV, Katusic ZS. Endothelial nitric oxide modulates expression and processing of amyloid precursor protein. Circul Res 2010; 107: 1498–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Austin SA, Combs CK. Amyloid precursor protein mediates monocyte adhesion in AD tissue and apoE(-)/(-) mice. Neurobiol Aging 2010; 31: 1854–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Austin SA, Sens MA, Combs CK. Amyloid precursor protein mediates a tyrosine kinase-dependent activation response in endothelial cells. J Neurosci 2009; 29: 14451–14462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.d’Uscio LV, He T, Katusic ZS. Expression and processing of amyloid precursor protein in vascular endothelium. Physiology 2017; 32: 20–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forloni G, Demicheli F, Giorgi S, et al. Expression of amyloid precursor protein mRNAs in endothelial, neuronal and glial cells: modulation by interleukin-1. Brain Res Mol Brain Res 1992; 16: 128–134. [DOI] [PubMed] [Google Scholar]
- 17.Goldgaber D, Harris HW, Hla T, et al. Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc Natl Acad Sci U S A 1989; 86: 7606–7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coma M, Guix FX, Ill-Raga G, et al. Oxidative stress triggers the amyloidogenic pathway in human vascular smooth muscle cells. Neurobiol Aging 2008; 29: 969–980. [DOI] [PubMed] [Google Scholar]
- 19.Frackowiak J, Potempska A, LeVine H, et al. Extracellular deposits of A beta produced in cultures of Alzheimer disease brain vascular smooth muscle cells. J Neuropathol Exp Neurol 2005; 64: 82–90. [DOI] [PubMed] [Google Scholar]
- 20.Kalaria RN, Premkumar DR, Pax AB, et al. Production and increased detection of amyloid beta protein and amyloidogenic fragments in brain microvessels, meningeal vessels and choroid plexus in Alzheimer’s disease. Brain Res Mol Brain Res 1996; 35: 58–68. [DOI] [PubMed] [Google Scholar]
- 21.Natte R, de Boer WI, Maat-Schieman ML, et al. Amyloid beta precursor protein-mRNA is expressed throughout cerebral vessel walls. Brain Res 1999; 828: 179–183. [DOI] [PubMed] [Google Scholar]
- 22.Wisniewski HM, Frackowiak J, Mazur-Kolecka B. In vitro production of beta-amyloid in smooth muscle cells isolated from amyloid angiopathy-affected vessels. Neurosci Lett 1995; 183: 120–123. [DOI] [PubMed] [Google Scholar]
- 23.Tan XL, Xue YQ, Ma T, et al. Partial eNOS deficiency causes spontaneous thrombotic cerebral infarction, amyloid angiopathy and cognitive impairment. Mol Neurodegenerat 2015; 10: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lammich S, Kojro E, Postina R, et al. Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A 1999; 96: 3922–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vingtdeux V, Marambaud P. Identification and biology of alpha-secretase. J Neurochem 2012; 120(Suppl 1): 34–45. [DOI] [PubMed] [Google Scholar]
- 26.Demas GE, Kriegsfeld LJ, Blackshaw S, et al. Elimination of aggressive behavior in male mice lacking endothelial nitric oxide synthase. J Neurosci 1999; 19: RC30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Chen K, Sloan SA, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 2014; 34: 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gravina SA, Ho L, Eckman CB, et al. Amyloid beta protein (A beta) in Alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43). J Biol Chem 1995; 270: 7013–7016. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki N, Iwatsubo T, Odaka A, et al. High tissue content of soluble beta 1-40 is linked to cerebral amyloid angiopathy. Am J Pathol 1994; 145: 452–460. [PMC free article] [PubMed] [Google Scholar]
- 30.Kwak YD, Wang R, Li JJ, et al. Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol Neurodegenerat 2011; 6: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de la Torre JC. Cerebromicrovascular pathology in Alzheimer’s disease compared to normal aging. Gerontology 1997; 43: 26–43. [DOI] [PubMed] [Google Scholar]
- 32.Faraci FM. Protecting against vascular disease in brain. Am J Physiol Heart Circ Physiol 2011; 300: H1566–H1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iadecola C, Zhang F, Niwa K, et al. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci 1999; 2: 157–161. [DOI] [PubMed] [Google Scholar]
- 34.Niwa K, Kazama K, Younkin L, et al. Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol Heart Circ Physiol 2002; 283: H315–H323. [DOI] [PubMed] [Google Scholar]
- 35.Faraco G, Park L, Zhou P, et al. Hypertension enhances Abeta-induced neurovascular dysfunction, promotes beta-secretase activity, and leads to amyloidogenic processing of APP. J Cereb Blood Flow Metab 2016; 36: 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Didion SP. Heterozygous eNOS deficient mice as a model to examine the effects of eNOS haploinsufficiency on the cerebral circulation. J Neurol Neuromed 2017; 2: 6–9. [PMC free article] [PubMed] [Google Scholar]
- 37.Emdin CA, Khera AV, Klarin D, et al. Phenotypic consequences of a genetic predisposition to enhanced nitric oxide signaling. Circulation 2018; 137: 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berendji-Grun D, Kolb-Bachofen V, Kroncke KD. Nitric oxide inhibits endothelial IL-1[beta]-induced ICAM-1 gene expression at the transcriptional level decreasing Sp1 and AP-1 activity. Mol Med 2001; 7: 748–754. [PMC free article] [PubMed] [Google Scholar]
- 39.Peng HB, Libby P, Liao JK. Induction and stabilization of I kappa B alpha by nitric oxide mediates inhibition of NF-kappa B. J Biol Chem 1995; 270: 14214–14219. [DOI] [PubMed] [Google Scholar]
- 40.Prinzen C, Muller U, Endres K, et al. Genomic structure and functional characterization of the human ADAM10 promoter. FASEB J 2005; 19: 1522–1524. [DOI] [PubMed] [Google Scholar]
- 41.Ptasinska A, Wang S, Zhang J, et al. Nitric oxide activation of peroxisome proliferator-activated receptor gamma through a p38 MAPK signaling pathway. FASEB J 2007; 21: 950–961. [DOI] [PubMed] [Google Scholar]
- 42.Tippmann F, Hundt J, Schneider A, et al. Up-regulation of the alpha-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J 2009; 23: 1643–1654. [DOI] [PubMed] [Google Scholar]
- 43.Von Knethen A, Brune B. Activation of peroxisome proliferator-activated receptor gamma by nitric oxide in monocytes/macrophages down-regulates p47phox and attenuates the respiratory burst. J Immunol 2002; 169: 2619–2626. [DOI] [PubMed] [Google Scholar]
- 44.Nishitomi K, Sakaguchi G, Horikoshi Y, et al. BACE1 inhibition reduces endogenous Abeta and alters APP processing in wild-type mice. J Neurochem 2006; 99: 1555–1563. [DOI] [PubMed] [Google Scholar]
- 45.Vassar R, Bennett BD, Babu-Khan S, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999; 286: 735–741. [DOI] [PubMed] [Google Scholar]
- 46.Furukawa K, Sopher BL, Rydel RE, et al. Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J Neurochem 1996; 67: 1882–1896. [DOI] [PubMed] [Google Scholar]
- 47.Han P, Dou F, Li F, et al. Suppression of cyclin-dependent kinase 5 activation by amyloid precursor protein: a novel excitoprotective mechanism involving modulation of tau phosphorylation. J Neurosci 2005; 25: 11542–11552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma T, Zhao Y, Kwak YD, et al. Statin’s excitoprotection is mediated by sAPP and the subsequent attenuation of calpain-induced truncation events, likely via rho-ROCK signaling. J Neurosci 2009; 29: 11226–11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mattson MP, Cheng B, Culwell AR, et al. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron 1993; 10: 243–254. [DOI] [PubMed] [Google Scholar]
- 50.d’Uscio LV, He T, Santhanam AV, et al. Endothelium-specific amyloid precursor protein deficiency causes endothelial dysfunction in cerebral arteries. J Cereb Blood Flow Metab 2018; 38: 1715–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meziane H, Dodart JC, Mathis C, et al. Memory-enhancing effects of secreted forms of the beta-amyloid precursor protein in normal and amnestic mice. Proc Natl Acad Sci U S A 1998; 95: 12683–12688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Taylor CJ, Ireland DR, Ballagh I, et al. Endogenous secreted amyloid precursor protein-alpha regulates hippocampal NMDA receptor function, long-term potentiation and spatial memory. Neurobiol Dis 2008; 31: 250–260. [DOI] [PubMed] [Google Scholar]
- 53.Liu H, Shim AH, He X. Structural characterization of the ectodomain of a disintegrin and metalloproteinase-22 (ADAM22), a neural adhesion receptor instead of metalloproteinase: insights on ADAM function. J Biol Chem 2009; 284: 29077–29086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bamberger ME, Harris ME, McDonald DR, et al. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci 2003; 23: 2665–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron 1996; 17: 553–565. [DOI] [PubMed] [Google Scholar]
- 56.Park L, Zhou J, Zhou P, et al. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc Natl Acad Sci U S A 2013; 110: 3089–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Faraci FM, Sigmund CD, Shesely EG, et al. Responses of carotid artery in mice deficient in expression of the gene for endothelial NO synthase. Am J Physiol 1998; 274: H564–H570. [DOI] [PubMed] [Google Scholar]
- 58.Cordes CM, Bennett RG, Siford GL, et al. Redox regulation of insulin degradation by insulin-degrading enzyme. PLoS One 2011; 6: e18138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Drake J, Link CD, Butterfield DA. Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid beta-peptide (1-42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging 2003; 24: 415–420. [DOI] [PubMed] [Google Scholar]
- 60.Fonseca AC, Moreira PI, Oliveira CR, et al. Amyloid-beta disrupts calcium and redox homeostasis in brain endothelial cells. Mol Neurobiol 2015; 51: 610–622. [DOI] [PubMed] [Google Scholar]
- 61.Koizumi K, Wang G, Park L. Endothelial dysfunction and amyloid-beta-induced neurovascular alterations. Cell Mol Neurobiol 2016; 36: 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Niwa K, Carlson GA, Iadecola C. Exogenous A beta1-40 reproduces cerebrovascular alterations resulting from amyloid precursor protein overexpression in mice. J Cereb Blood Flow Metab 2000; 20: 1659–1668. [DOI] [PubMed] [Google Scholar]
- 63.Park L, Anrather J, Forster C, et al. Abeta-induced vascular oxidative stress and attenuation of functional hyperemia in mouse somatosensory cortex. J Cereb Blood Flow Metab 2004; 24: 334–342. [DOI] [PubMed] [Google Scholar]
- 64.Park L, Anrather J, Zhou P, et al. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci 2005; 25: 1769–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park L, Zhou P, Pitstick R, et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A 2008; 105: 1347–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reynolds MR, Singh I, Azad TD, et al. Heparan sulfate proteoglycans mediate Abeta-induced oxidative stress and hypercontractility in cultured vascular smooth muscle cells. Mol Neurodegenerat 2016; 11: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thomas T, McLendon C, Sutton ET, et al. beta-Amyloid-induced cerebrovascular endothelial dysfunction. Ann N Y Acad Sci 1997; 826: 447–451. [DOI] [PubMed] [Google Scholar]
- 68.Thomas T, McLendon C, Sutton ET, et al. Cerebrovascular endothelial dysfunction mediated by beta-amyloid. Neuroreport 1997; 8: 1387–1391. [DOI] [PubMed] [Google Scholar]
- 69.Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxidants Redox Signal 2011; 15: 1583–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 1986; 320: 454–456. [DOI] [PubMed] [Google Scholar]
- 71.Rubanyi GM, Vanhoutte PM. Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor. Am J Physiol 1986; 250: H822–H827. [DOI] [PubMed] [Google Scholar]
- 72.Ricciotti E, FitzGerald GA. Prostaglandins and inflammation. Arterioscleros Thromb Vasc Biol 2011; 31: 986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wong WT, Tian XY, Chen Y, et al. Bone morphogenic protein-4 impairs endothelial function through oxidative stress-dependent cyclooxygenase-2 upregulation: implications on hypertension. Circul Res 2010; 107: 984–991. [DOI] [PubMed] [Google Scholar]
- 74.Wong WT, Wong SL, Tian XY, et al. Endothelial dysfunction: the common consequence in diabetes and hypertension. J Cardiovasc Pharmacol 2010; 55: 300–307. [DOI] [PubMed] [Google Scholar]
- 75.Choi JY, Yeo IJ, Kim KC, et al. K284-6111 prevents the amyloid beta-induced neuroinflammation and impairment of recognition memory through inhibition of NF-kappaB-mediated CHI3L1 expression. J Neuroinflamm 2018; 15: 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li J, Ma X, Wang Y, et al. Methyl salicylate lactoside protects neurons ameliorating cognitive disorder through inhibiting amyloid beta-induced neuroinflammatory response in Alzheimer’s disease. Front Aging Neurosci 2018; 10: 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu N, Zhuang Y, Zhou Z, et al. NF-kappaB dependent up-regulation of TRPC6 by Abeta in BV-2 microglia cells increases COX-2 expression and contributes to hippocampus neuron damage. Neurosci Lett 2017; 651: 1–8. [DOI] [PubMed] [Google Scholar]
- 78.Qi Y, Ji XF, Chi TY, et al. Xanthoceraside attenuates amyloid beta peptide1-42-induced memory impairments by reducing neuroinflammatory responses in mice. Eur J Pharmacol 2018; 820: 18–30. [DOI] [PubMed] [Google Scholar]
- 79.Shen MY, Hsiao G, Fong TH, et al. Amyloid beta peptide-activated signal pathways in human platelets. Eur J Pharmacol 2008; 588: 259–266. [DOI] [PubMed] [Google Scholar]
- 80.Yagami T, Takahara Y, Ishibashi C, et al. Amyloid beta protein impairs motor function via thromboxane A2 in the rat striatum. Neurobiol Dis 2004; 16: 481–489. [DOI] [PubMed] [Google Scholar]
- 81.Chamorro A. TP receptor antagonism: a new concept in atherothrombosis and stroke prevention. Cerebrovasc Dis 2009; 27(Suppl 3): 20–27. [DOI] [PubMed] [Google Scholar]
- 82.Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med 2007; 357: 2482–2494. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material for Partial loss of endothelial nitric oxide leads to increased cerebrovascular beta amyloid by Susan A Austin and Zvonimir S Katusic in Journal of Cerebral Blood Flow & Metabolism