

Abstract
The dependency of prostate cancer (PCa) growth on androgen receptor (AR) signaling has been harnessed to develop first-line therapies for high-risk localized and metastatic PCa treatment. However, the occurrence of aberrant expression, mutated or splice variants of AR confers resistance to androgen ablation therapy (ADT), radiotherapy or chemotherapy in AR-positive PCa. Therapeutic strategies that effectively inhibit the expression and/or transcriptional activity of full-length AR, mutated AR and AR splice variants have remained elusive. In this study, we report that mithramycin (MTM), an antineoplastic antibiotic, suppresses cell proliferation and exhibits dual inhibitory effects on expression and transcriptional activity of AR and AR splice variants. MTM blocks AR recruitment to its genomic targets by occupying AR enhancers and causes downregulation of AR target genes, which includes key DNA repair factors in DNA damage repair (DDR). We show that MTM significantly impairs DDR and enhances the effectiveness of ionizing radiation or the radiomimetic agent Bleomycin in PCa. Thus, the combination of MTM treatment with RT or radiomimetic agents, such as bleomycin, may present a novel effective therapeutic strategy for patients with high-risk, clinically localized PCa.
Keywords: Mithramycin, Androgen receptor, DNA damage Repair, Ionizing radiation, Bleomycin
1. Introduction
Prostate cancer (PCa) is the most common malignancy in men and the second leading cause of male cancer-related death in the United States [1,2]. In the nucleus, AR binds to enhancers and promoters of its target genes, which are required for PCa progression and survival [3]. Following androgen deprivation therapy (ADT), prostate tumors in-variably recur and typically express high levels of nuclear AR and maintain expression of AR target genes. Multiple mechanisms of resistance to ADT have been described including intratumoral androgen synthesis, full-length AR (AR-FL) amplification/overexpression, expression of AR point mutations (ARmts) and AR splice variants (AR-Vs) that constitutively activate AR, such as AR-V7 [4]. Second generation agents including enzalutamide and abiraterone target AR signaling more effectively and have been proven to be effective against castration-resistant prostate cancer (CRPC), but these agents do not have a durable response and only provide an overall survival benefit of 4–5 months [5,6]. Importantly, in enzalutamide-resistant and abiraterone-resistant PCa, AR signaling remains active and drives the proliferation of CRPC through similar mechanisms of resistance [7]. While a more comprehensive strategy to target AR signaling may be effective in CRPC, the extensive mutation profile in CRPC, the heterogeneity and the emergence of AR-negative forms of CRPC may limit the utility of such approaches. However, in earlier stages of PCa, where PCa is still completely AR-driven, there is an opportunity for drugs that more effectively target AR signaling to enhance the effect of localized therapies and to prevent the progression to the incurable metastatic PCa [8].
Several studies have reported that AR regulates a transcriptional program of DNA repair genes to promote radiation resistance of prostate cancer [9–11]. Blockade of AR signaling with ADT or antiandrogens inhibits AR-driven transcription and DDR in PCa cells [12]. Recent studies have indicated that even short courses of ADT can induce the expression of both full-length AR and AR variants (AR-Vs) [13]. The induction of AR-Vs by ADT may represent a potential mechanism of radiation resistance during ADT, since AR-Vs can constitutively direct AR signaling and are not affected by ADT. We recently reported that AR-Vs alone could mediate DDR in PCa cells through direct interaction with the DNA protein kinase C (DNA-PKcs) [11]. Since most patients undergoing combined RT and ADT receive 2–3 months of ADT prior to initiating RT, these data indicate that AR-Vs may be induced within these prostate tissues and mediate radiation resistance.
Mithramycin (MTM) is an aureolic acid natural product from Streptomyces plicatus. As a DNA-binding antineoplastic antibiotic, MTM has been used to treat several malignant diseases, such as advanced testicular carcinoma [14], chronic myeloid leukemia [15], hy-percalcemia [16] and Ewing sarcoma [17]. MTM was also reported to decrease AR mRNA transcription by inhibiting Sp1 binding to the AR promoter [18] and enhance tumor sensitivity by targeting SP1 [19]. However, the biological effect of MTM on AR transcriptional activity and AR variants in PCa is unclear. Here we report that the antibiotic mithramycin (MTM) inhibits the genomic action of any form of AR, including AR-Vs, downregulates the expression of critical DNA damage repair genes, and sensitizes cancer cells and explants to ionizing radiation and DNA damaging agents in PCa.
2. Materials and methods
Cell culture and transfection.
22RV1 cells and VCaP cells were cultured in DMEM with 10% FBS. PC3 cells and LNCaP cells were cultured in RPMI with 10% FBS. R1-D567 and R1-AD1 were kind gifts from Dr. Scott Dehm (University of Minnesota) [20]. Plasmids were transfected with Effectene Transfection Reagent (Qiagen).
Antibodies and reagents.
The following antibodies were used for Western blot analysis: Phospho-ATM (Ser1981) Rabbit mAb (Cell Signaling, 13050), ATM antibody (Santa Cruz, sc-377293), Anti-DNA-PKcs (phospho S2056) antibody (Abcam, ab18192), DNA-PKcs Antibody (Santa Cruz, sc-9051), Phospho-ATR antibody (GeneTex, GTX128145), ATR Rabbit mAb (Cell Signaling, 13934), PARP-1 Antibody (Santa Cruz, sc-8007), Phospho-Chk2 (Thr68) Rabbit mAb (Cell Signaling, 2197), Chk2 Antibody (Santa cruz, sc-9064), Phospho-Chk1 (Ser317) Rabbit mAb (Cell Signaling, 12302), Chk1 Antibody (Santa cruz, sc-8408), anti-γ-H2A.X monoclonal antibody (Millipore, 05–636 or Santa cruz, sc-517348), H2AX Antibody (Santa Cruz, sc-517336), GAPDH (Cell Signaling Technology, 2118), Caspase 3 Antibody (Cell Signaling, 9665), Antigen Receptor (N-20) Antibody (Santa cruz, sc-816), Cox IV (Cell Signaling, 5247), ERG Antibody (Abcam, ab92513), PSA Antibody (Abcam, ab137330), XRCC4 Antibody (Santa cruz, sc-271087), RFC3 Antibody (Santa cruz, sc-390293), FANCC antibody (Epigentek, A62566), THE V5 Tag Antibody (Genscript, A01724); Immunohistochemistry: Ki67 Antibody (Abcam, ab15580). anti-γ-H2A.X monoclonal antibody (Millipore, 05–636).
The following reagents were used: Mithramycin (Cayman Chemical); Bleomycin (Cayman Chemical); Dihydrotestosterone (DHT) (Sigma-Aldrich); Enzalutamide (Selleck).
Plasmids.
cDNA fragments containing the reading frames of AR and ARv567es were amplified by PCR using primers appended with restriction site sequences and cloned into the pEGFP-N1 vector (Clontech) that EGFP gene had been replaced by a C terminal V5 tag. All primer sequences are listed in Table S1. The pGL4.24 and pGL4.73 vectors were used in luciferase reporter assay.
Cell viability assay.
Cells were plated onto 12-well plate with 20–30% confluence. After 24h, MTM was added in medium. Next cells were incubated in incubator until vehicle group got more than 95% confluence. Then, cells were stained by 0.5% crystal violet for 20min and rinsed twice by distilled water.
Cell proliferation assay.
1000 22RV1 cells, LNCaP cells or PC3 cells were seeded in 96 well culture plates (5000 VCaP cells). Cells were allowed to adhere overnight and were treated with Mithramycin or DMSO. LNCaP cells were transfected with AR or AR V567es plasmids for 48h then cells were trypsinized and seeded in 96 well plates. Relative cell proliferation was quantified over days after the treatments with the Cell Counting Kit-8 (Dojindo Molecular Technologies) on a spectrophotometer (BMG Labtech).
Clonogenic assay.
The clonogenic assay was modified from previous protocol [21]. Briefly, 300 22RV1 cells per well were seeded in 6-well plate for 24h. For MTM and IR, cells were pretreated by MTM for 24h before indicated dose of IR were employed. Then, plates were incubated at 37 °C with 5% CO2 for 14 days. For MTM and Bleomycin combination, cells were pretreated by MTM for 24h and then 10 μg/ml of Bleomycin for 1h. After PBS washed cells once, complete medium with MTM was added and plates were incubated at 37 °C with 5% CO2 for 14 days. Colonies are fixed and stained by 20% ethanol solution containing 0.5% crystal violet. Colonies are quantified by image J.
Purification of AR and ARv567es protein.
The plasmids containing AR-v5 or ARv567es-v5 were transfected into HEK293T cells for 48h. Cells were lysed in lysis buffer (50 mM HEPES [pH7.5], 400 mM NaCl, 0.2% Triton X-100, 1 mM DTT, 5% glycerol and protease inhibitor). AR-v5 and ARv567es-v5 were immunoprecipitated with V5-antibody (Genscript) for 3 h at 4 °C and GammaBind G Sepharose (GE healthcare) were added to harvest proteins for 1h. After washing beads, purified proteins were eluted by V5 peptide (Sigma) (50 mM HEPES [pH7.5], 400 mM NaCl, 0.05% Triton X-100, 1 mM DTT, 5% glycerol and 500 μg/ml V5 peptide) and dialyzed overnight.
Dose-response curves.
For dose-response curves, 1000 cells were plated into each well of a 96-well plate. After 24 h, the medium containing MTM or the combination of MTM and Belomycin was added into wells. After 4 days incubation, cell viability was determined using the Cell Counting Kit-8 (Dojindo Molecular Technologies) on a spectrophotometer (BMG Labtech). Curve fitting was performed in GraphPad Prism. Synergetic effects of drugs were analyzed in Combenefit [22] and the results shown are computed using the Bliss Model.
ChIP-seq.
We performed ChIP in VCaP cells as previously described (Kittler et al., 2013). Barcoded libraries of ChIP and input DNA were generated with the TruSeq® ChIP Sample Preparation Kit (Illumina®), and were sequenced under 75-nt single-end reads in the Illumina HiSeq 2500 system. AR-bound regions were identified as genomic regions with a significant read enrichment and binding peak profile in the AR reads over the input reads by using the HOMER software tool (v.4.7) with 1% FDR. The findMotifsGenome module in HOMER was employed to analyze de novo motif for AR-bound regions.
ChIP-qPCR.
The LNCaP/GFP, LNCaP/AR-V7 and LNCaP/ARv567es cells were induced by 1 μg/ml doxycycline for 24h. Then 50 nM MTM was added into medium for another 24h treatment. ChIP was performed as ChIP-seq protocol. Next, purified DNA was utilized for qPCR using specific primers.
RNA-seq.
VCaP cells were treated by MTM for 24h and then RNA was extracted from MTM or Vehicle-treated cells using the Aurum Total RNA Mini Kit (Bio-rad). RNA quality was determined in an Agilent TapeStation 4200. Five micrograms of total RNA from each sample was used to generate libraries using Illumina’s TruSeq Stranded Total RNA Ribo-Zero kit (Illumina). Libraries were sequenced on the Illumina HiSeq 2500 and 75-nucleotide single-end reads. RNA-seq reads were mapped to hg19 genome build using tophat (v 2.0.12), and reads per kilobase per million mapped reads (RPKM) calculation and differential expression analysis was performed using cufflinks (v 2.2.1).
Immunofluorescence analysis of γ-H2AX foci.
Cells were seeded on BD Biocoat™ 12 mm round coverslips in a 24-well plate or Lab TekTM 8-well chamber slide for 24h. For MTM and IR, cells were pretreated by MTM for 24h before 2Gy ionizing radiation were employed. Then, cells were incubated at 37 °C with 5% CO2 for indicated time points (1h, 6h and 24h). For MTM and Bleomycin combination, cells were pretreated by MTM for 24h and then 10 μg/ml of Bleomycin for 1h. After PBS washed cells once, complete medium with MTM was added and cells were incubated for indicated time points (1h and 24h). Then cells were fixed for 15 min in 4% paraformaldehyde/PBS, washed three times in PBS, permeabilized for 10 min in PBS containing 0.2% Triton-X, washed three times in PBS and blocked for 30 min in PBST (0.1% Tween-20 in PBS) with 2% BSA. Next, cells were incubated with mouse anti-γ-H2AX monoclonal antibody (Millipore Cat #05–636) in blocking buffer for 1 h at room temperature, washed three times with PBST, incubated for 1 h with Alexa Fluor 488-anti-mouse IgG (Invitrogen) in blocking buffer, and washed three times with PBST. Cells were then incubated with DAPI for 5 min, and the slides were finally mounted with Shandon™ Immu-Mount™ (Thermo Scientific). Images acquisition was performed with a Leica DM5500B microscope. For quantification of γH2AX foci a minimum of ten fields each containing at least 20 cells were counted in Image J.
Surviving fraction analysis.
22RV1 cells were trypsinized and 200 cells per well (counted in Beckman Coulter Z2 Particle Counter) were plated in 6-well plate. After 24h, Cells were pretreated by MTM for another 24h. Then plates were irradiated at various doses (2Gy, 4Gy and 6Gy) using a 137Cs irradiator (Mark 1–68 irradiator, J.L. Shepherd and associates). Irradiated plates were incubated for 14 days. Colonies are fixed and stained by 20% ethanol solution containing 0.5% crystal violet. Colonies are quantified by image J. The surviving cell fraction was calculated and visualized in GraphPad.
Explant experiments and Immunohistochemistry.
Fresh prostate cancer tissues (n = 4) were obtained with informed consent from men undergoing radical prostatectomy at the Hospitals of the University of Texas Southwestern Medical Center (Dallas, TX). The tissues were dis-sected into 1 mm3 pieces and randomly placed in triplicates on a pre-soaked 1 cm3 veterinary dental sponge (Novartis, East Hanover, NJ) inside the wells of a 12-well plate containing 600 ml RPMI 1640 with 5% heat inactivated FBS, 100 units/mL penicillin-streptomycin and vehicle (DMSO) alone or 250 nM MTM. Tissues were cultured at 37 °C for 24 h and then formalin-fixed and paraffin embedded for immunohistochemistical analyses. Number of positive tumor cells in relation to the total number of cells encountered and the intensity of nuclear staining (weak or strong) for each of the markers (Ki-67) were quantified manually per tissue core by a staff pathologist, who was blinded from the clinical data. The percentage of positively staining tumor cells was assessed.
Statistical Analysis.
Statistical significance was determined by a two-tailed unpaired Student’s t-test and p values < 0.05 were considered statistically significant.
3. Results
3.1. Mithramycin inhibits growth of PCa cells by targeting AR and AR variants
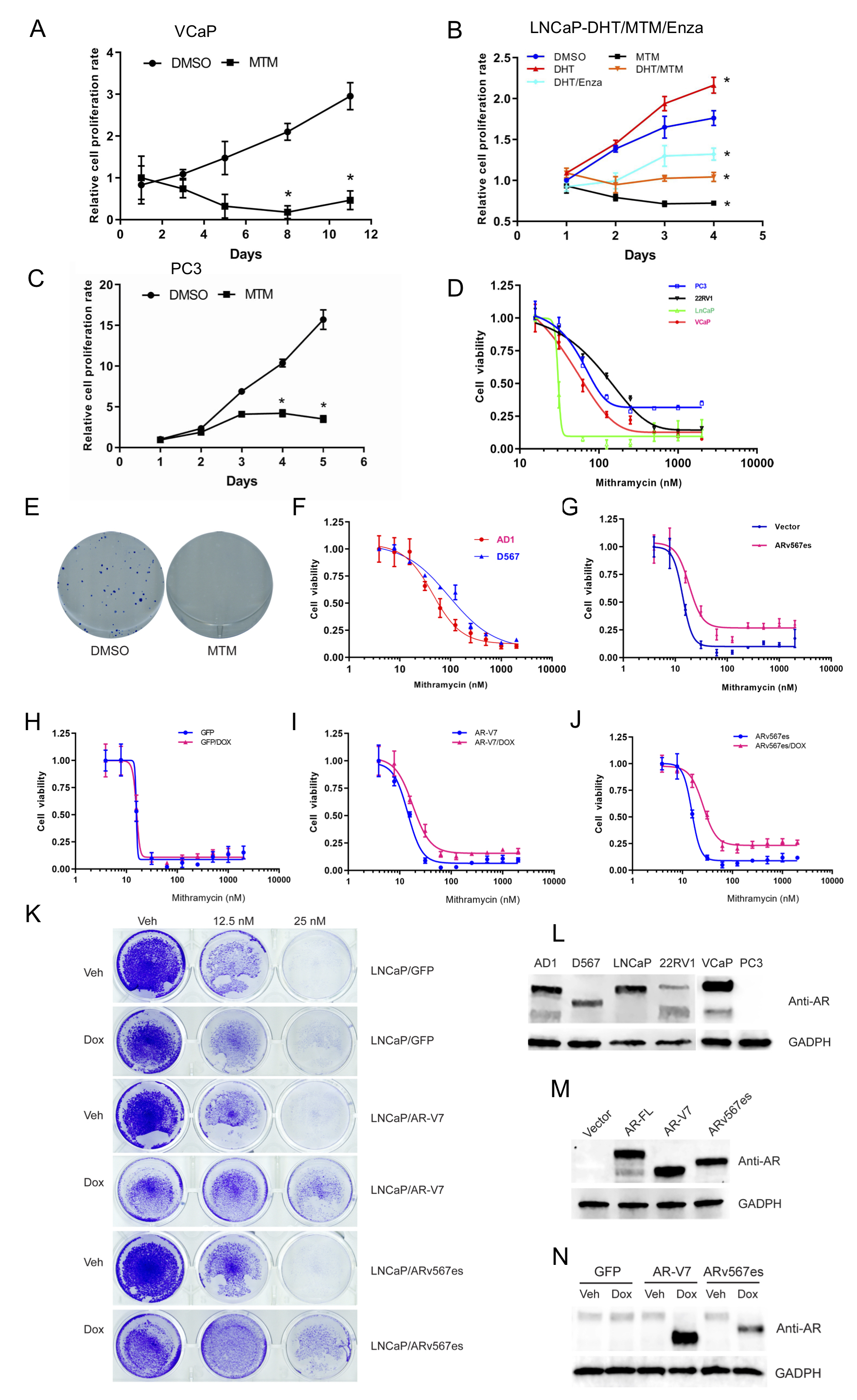
To test the biological function of MTM in PCa, we first analyzed the antiproliferative effect of MTM on prostate cancer cells. As shown in Fig. 1A, even low concentrations of MTM, such as 25 nmol/L, can nearly completely inhibit cell proliferation in AR-positive LNCaP cells. However, in AR-negative PC3 cells, cell growth is only partially inhibited upon 25 nmol/L MTM treatment. Also, two PCa cell lines engineered via TALEN (transcription activator-like effector nuclease) to express AR, R1-AD1 cells (which only express AR-FL protein) and R1-D567 cells (which only express ARv567es protein) (Supplementary Figure 1L) [20], displayed obvious cytotoxicity to MTM (Fig. 1A). Next, cell growth was measured by colorimetric assays in PCa cells upon MTM treatment. To the AR-positive LNCaP, R1-AD1, R1-D567, VCaP and 22RV1 cells, MTM almost completely abolished cell growth at 100 nmol/L (Fig. 1B–E, Supplementary Fig. 1A). Also, both the co-treatment with DHT and Enzalutamide or DHT and MTM, demonstrate that both MTM and Enzalutamide exhibit an inhibitory effect on cell growth in LNCaP cells, which suggests that Enzalutamide and MTM have similar antiandrogenic effects (Supplementary Fig. 1B). However, some recovery of cell growth was observed at 3–5 days in AR-negative PC3 cells (Supplementary Fig. 1C). Similarly, cell viability assays of MTM in PC3 cells showed that 35% of PC3 cells remain viability even up to 2 μmol/L MTM treatment, which is consistent with the higher IC50 of MTM in PC3 cells than LNCaP cells (Supplementary Fig. 1D). These findings indicate that the inhibition of AR actions may be critical for the anticancer effects of MTM in AR-positive prostate cancer in addition to its inhibitory effects on SP1, which may be the main target of MTM in AR-negative prostate cancer. Furthermore, clonogenic growth assays showed that 50 nmol/L of MTM could inhibit cell growth of AR-positive 22RV1 (Supplementary Fig. 1E).
Fig. 1. MTM inhibits proliferation of prostate cancer cells and blocks DNA-binding ability of AR-FL and ARvs.
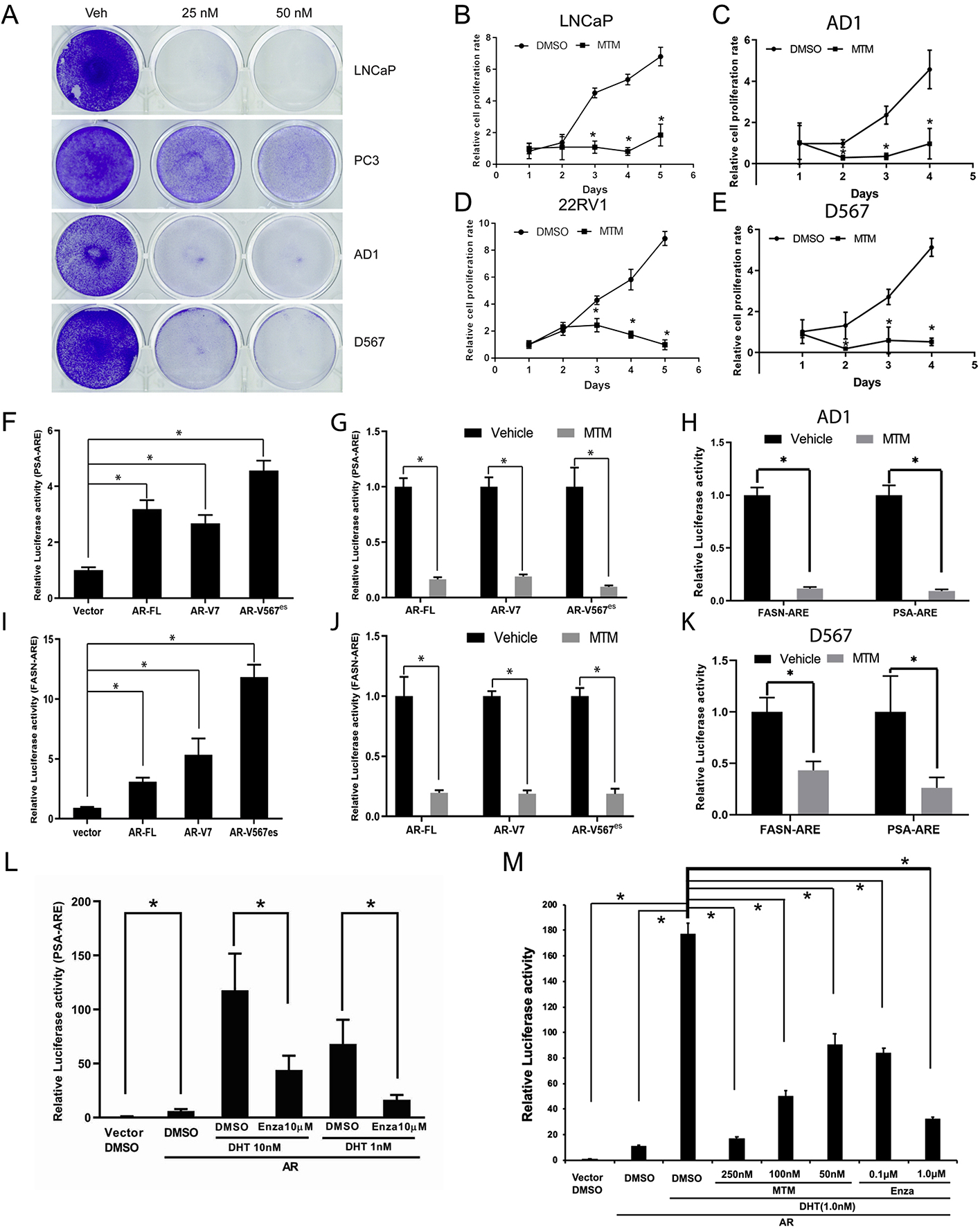
A, Crystal violet staining of PCa cells with MTM treatment. B, MTM inhibits cell growth of LNCaP cells. C, MTM inhibits cell growth of AD1 cells D, MTM inhibits cell growth of 22RV1 cells. E, MTM inhibits cell growth of D567 cells. F, Luciferase activity of PSA-ARE in HEK293T cells with overexpressing AR-FL, AR-V7 and ARv567es. G, The effect of MTM on luciferase activity of PSA-ARE in HEK293T cells with overexpressing AR-FL, AR-V7 and ARv567es. H, Luciferase activity of PSA-ARE and FASN-ARE in AD1 cells with MTM treatment. I, Luciferase activity of FASN-ARE in HEK293T cells with overexpressing AR-FL, AR-V7 and ARv567es. J, The effect of MTM on luciferase activity of FASN-ARE in HEK293T cells with overexpressing AR-FL, AR-V7 and ARv567es. K, Luciferase activity of PSA-ARE and FASN-ARE in D567 cells with MTM treatment. L, Effects of DHT and Enzalutamide on the luciferase activity of the PSA-ARE reporter vector in HEK293T cells overexpressing AR-FL. M, Effects of DHT and Enzalutamide and MTM on the luciferase activity of the PSA-ARE reported vector in HEK293T cells overexpressing AR-FL. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Next, we determined the IC50 of MTM in LNCaP cells, 22RV1 cells, VCaP cells and PC3 cells (Supplementary Fig. 1D). Interestingly, IC50 values of MTM in 22RV1 cells (150 nmol/L) and VCaP cells (80 nmol/L) were higher than the value in LNCaP cells (30 nmol/L). This may be due to higher expression of AR variants in these cells, as expression of AR variants, such as ARv567es and AR-V7, has been reported to correlate to resistance to ADT [23] and HSP90 inhibitors [24]. To validate this hypothesis, we performed transient overexpression and inducible expression of AR variants in LNCaP cells. As shown in Supplementary Figs. 1F–1K, overexpression of AR-V7 and ARv567es moderately enhanced resistance to MTM in LNCaP cells. Also, R1-D567 cells that only express ARv567es had a higher IC50 for MTM than the isogenic R1-AD1 cells (Supplementary Fig. 1F).
To further test if MTM inhibits AR activity, we analyzed transactivation by AR with reporter gene constructs that contain androgen-response elements (AREs) by ectopically expressing AR-Vs in AR-negative H293T cells (Supplementary Figure 1M). Using two different AREs, PSA-ARE and FASN-ARE, we found the transcriptional activity of AR-V7 and ARv567es could be inhibited by MTM (Fig. 1F–G, 1I–1J). Furthermore, similar effects were observed in R1-AD1 and R1-D567 cells (Fig. 1H–K). Finally, we tested the effect of the combination of DHT and Enzalutamide or DHT and MTM on AR’s ability to bind to the PSA-ARE and transactivate reporter gene expression (Fig. 1L–M). These results suggest that the activating effect of DHT and inhibitory effect of Enzalutamide or MTM occur in a dose-dependent manner. Collectively, these results suggest that MTM may reduce the transcription activity of AR and AR-Vs by directly blocking their DNA binding.
3.2. Effects of Mithramycin on AR targeted genes in PCa cells
To test the effect of MTM on expression of AR mRNA and protein in AR positive cells, we treated VCaP cells with 100 nmol/L MTM for 24h. RNA-seq analysis showed that MTM only caused a modest reduction (by ~15%) of AR mRNA expression. This was validated by qRT-PCR and western blot, which showed that at a MTM concentration of 50 nmol/L, AR-FL mRNA were only reduced by 20% in comparison to vehicle and AR protein levels were not significantly change. Despite this lack of change of AR-FL expression at the protein level, the expression of the AR target genes KLK3 (PSA) and TMPRSS2-ERG were significantly reduced at both the protein and mRNA level (Supplementary Figs. 2A and 2B). These findings suggest that AR is one of major targets of MTM by inhibiting its transcriptional activity.
To characterize the impact of MTM treatment on the genetic program regulated by AR, we analyzed the AR cistrome and transcriptome in VCaP cells. For the cistrome analysis, we used chromatin immunoprecipitation followed by deep sequencing (ChIP-seq), which express AR-FL and AR variants at high levels. We found significantly decreased occupancy for AR in MTM-treated cells (with 8758 lost peaks, Fig. 2A–B). De novo motif analysis found that AR binding sites with reduced AR occupancy following MTM treatment were highly enriched for the presence of the canonical androgen-response element (p = 1e-118). Additional motifs significantly lost at AR binding sites following MTM treatment included the motifs for HOXD13, GATA1 and ERG as well as the NF1-half site motif (Fig. 2C). To further examine whether MTM directly blocked the binding of AR-variants to chromatin, we performed ChIP-qPCR for AR in LNCaP cells that ectopically express GFP(LNCaP/GFP), AR-V7(LNCaP/AR-V7) and ARv567es(LNCaP/ARv567es). Consistent with the data in VCaP cells, MTM treatment reduced occupancy of AR-V7, ARv567es at the PSA enhancer in these LNCaP cells (Fig. 2D, Supplementary Figure 1M). Similar results were obtained for the enhancer of another AR target gene, FKBP5 (Fig. 2E), indicating that MTM directly inhibits the binding of AR to chromatin.
Fig. 2.
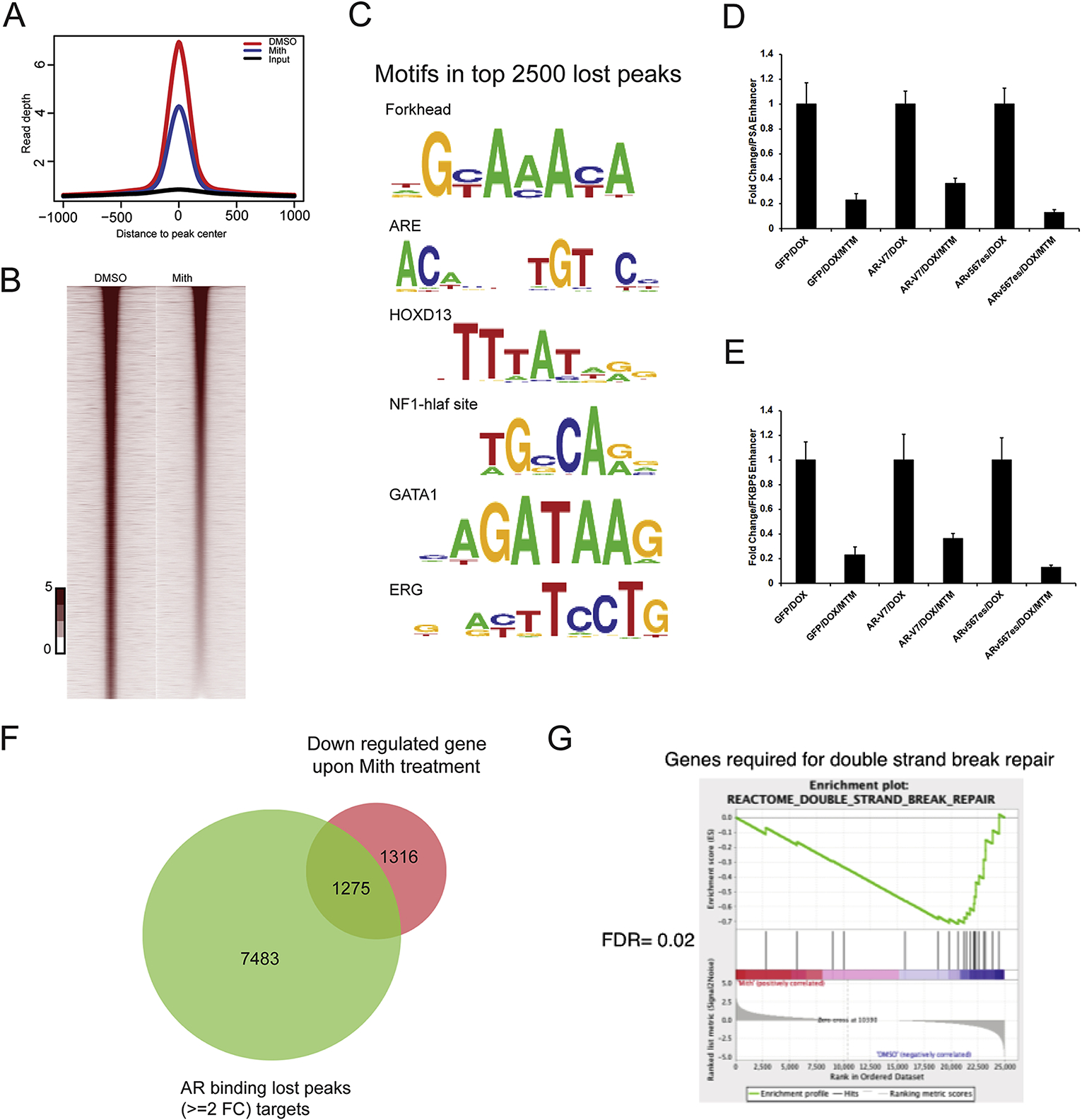
MTM weakens the transcription activity of AR in PCa. A, Density plots of ChIP-seq reads for AR occupied regions in VCaP cells with MTM treatment. The x-axis shows 2 kb 5′ and 3′ to the predicted peak summits. The y-axis depicts read depth in ChIP-seq. B, Heat map of ChIP-seq signals around AR peak in VCaP cells with MTM treatment. C, A list of the lost gene motifs in AR bound regions under MTM treatment. D, Fold change of ChIP-qPCR of PSA enhancer in LNCaP/GFP, LNCaP/AR-V7 and LNCaP/ARv567es cells with MTM treatment. E, Fold change of ChIP-qPCR of FKBP5 enhancer in LNCaP/GFP, LNCaP/AR-V7 and LNCaP/ARv567es cells with MTM treatment. F, Venn diagram representing AR binding lost peaks (≥2 Folds change) and downregulated genes upon MTM treatment in VCaP cells. G, GSEA of double strand break repair in VCaP cells with MTM treatment.
To test if reduced AR occupancy in putative AR-bound enhancers following MTM treatment also translated into reduced expression of AR target genes, we profiled the transcriptome of VCaP cells following MTM treatment by RNA-seq. Treatment with 100 nmol/L MTM for 24 h caused downregulation of bona fide AR target genes consistent with the loss of AR-binding peaks in enhancer regions of these genes (Supplementary Fig. 2C). In total, 1509 up-regulated genes and 2581 down-regulated genes were found in the transcriptome of VCaP cells treated with MTM (Supplementary Fig. 2D). Integration of our ChIP-seq and RNA-seq datasets after MTM treatment showed that nearly half of the genes downregulated by MTM treatment were direct AR target genes where MTM decreased AR binding (p = 1.87e-57, hypergeo-metric test, Fig. 2F), suggesting that transcriptional changes caused by MTM were to a large extent caused by disruption of AR binding. Importantly, genes that were differentially expressed following MTM treatment were enriched for gene sets related to AR-upregulated steady state and nascent transcripts in PCa. Through gene set enrichment analysis (GSEA), we found that genes relevant to DNA damage response were among the gene sets that were both targets of AR with reduced occupancy and transcriptionally repressed following MTM treatment (FDR = 0.02, Fig. 2G). AR was previously reported to upregulate a set of DNA damage repair genes that promotes prostate cancer radioresistance [9]. Additionally, our previous work has shown that the AR-Vs alone can mediate DDR in PCa cells through direct interaction with the DNA-dependent protein kinase catalytic subunit (DNA-PKcs/PRKDC) [11]. Considering that MTM effectively inhibits the AR-FL and AR-Vs, we hypothesize that MTM may sensitize cells to ionizing radiation or DNA damage agents.
3.3. MTM enhances the effectiveness of ionizing radiation in PCa
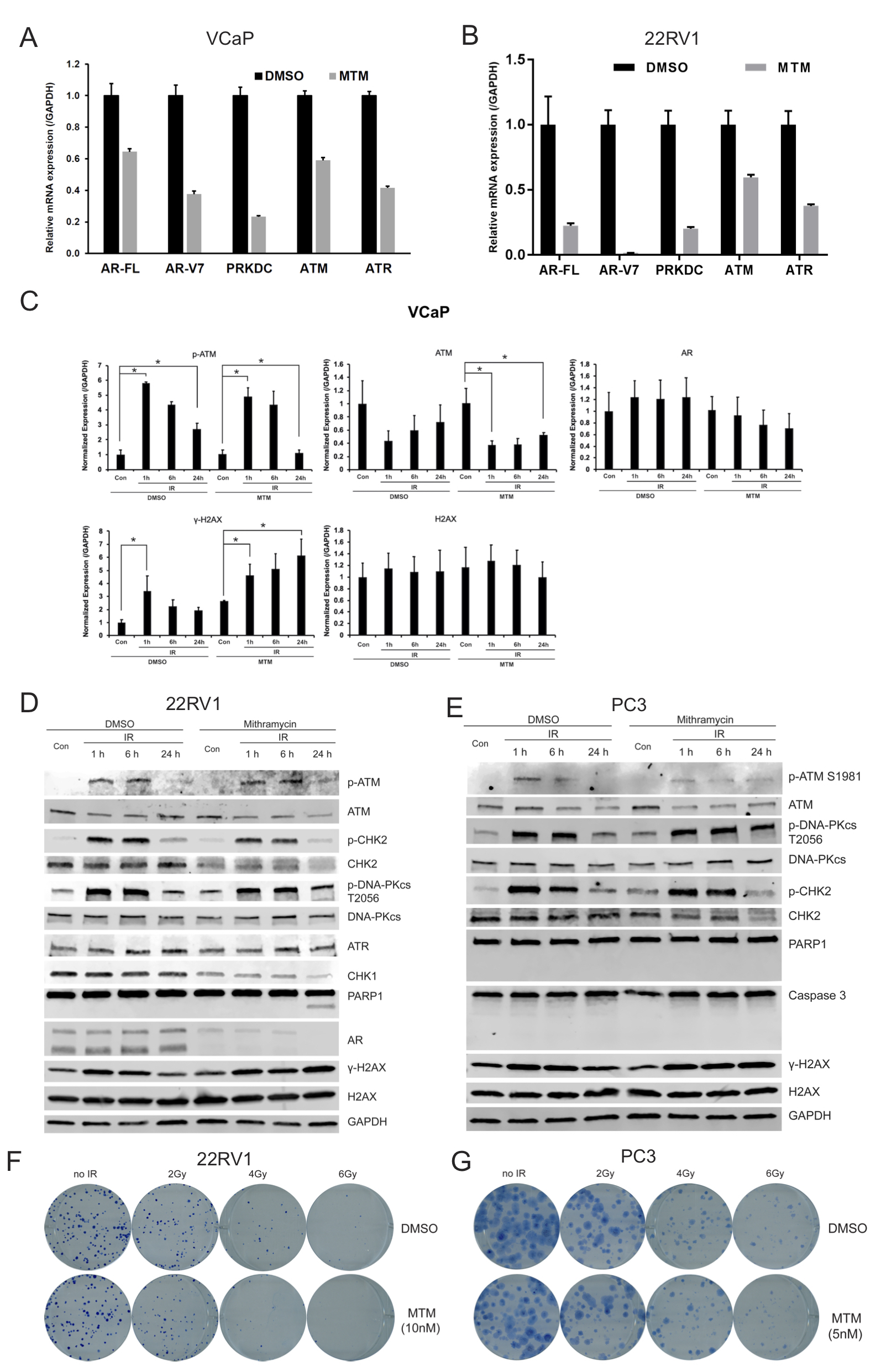
To access the effect of MTM on the DNA damage response following ionizing radiation, we exposed AR-positive and AR-negative prostate cancer cells that were pretreated for 24h with MTM (50 nmol/L or 100 nmol/L) or vehicle to 4 Gy of ionizing radiation. Subsequently the levels of DNA damage repair-related proteins were assayed by immunoblotting at various timepoints. As shown in Fig. 3A–B, γ-H2AX (Ser139) levels increased after radiation but returned to baseline levels at 24 h post-radiation in the vehicle control. However, in the MTM-treated samples, γ-H2AX (Ser139) levels remained at the same or a higher level 24h post-radiation as compared to 1h post-radiation, indicating that MTM inhibited DNA damage repair (Fig. 3C and Supplementary Fig. 3C). Additionally, we noticed that the expression of AR-Vs was dramatically ablated at both the mRNA and protein level in MTM-treated AR-Vs-positive cells, which suggests that MTM may inhibit the ability of AR-Vs to drive DNA damage repair.
Fig. 3. MTM enhance effectiveness of Ionizing Radiation and Bleomycin in PCa.
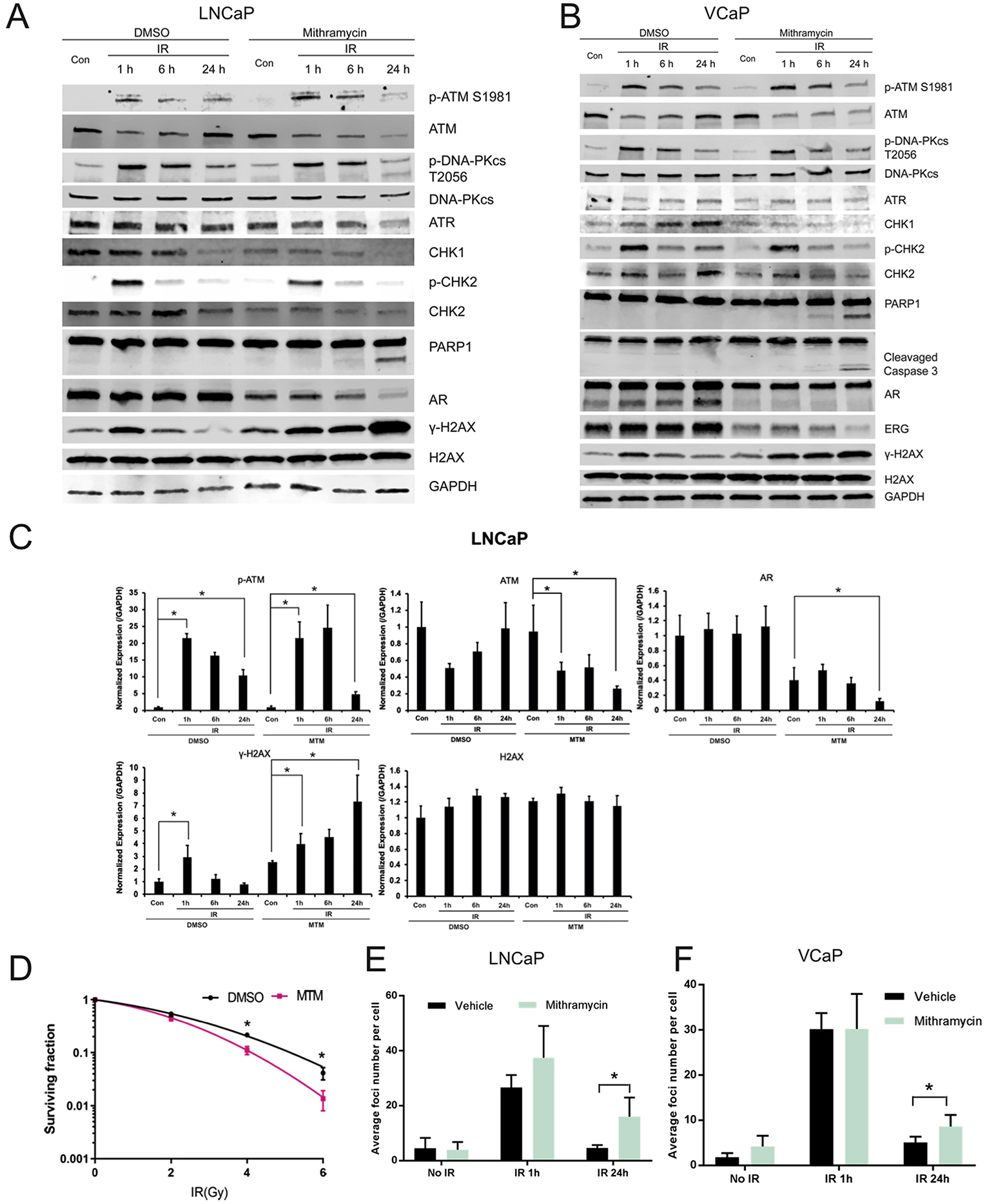
A, Western blot of DNA repair-related genes in MTM and IR co-treated LNCaP cells. B, Western blot of DNA repair-related genes in MTM and IR co-treated VCaP cells. C, Quantification of Western data in LNCaP cells with Image J using GAPDH intensity for normalization. D, Decreased survival when IR in the presence of MTM versus mock in 22RV1 cells (*, P ≤ 0.05). E, Average number of γ-H2AX Foci in MTM and 2Gy IR co-treated LNCaP cells. F, Average number of γ-H2AX Foci in MTM and 2Gy IR co-treated VCaP cells.
Next, we investigated the molecular mechanisms underlying the enhanced effectiveness of ionizing radiation by MTM. First, DNA damage response kinases, including the phosphatidylinositol 3-kinase-related kinases (PIKK) family members DNA-PKcs, ATR and ATM, were evaluated by qPCR and western blot. The mRNA levels of DNA-PKcs, ATR and ATM were downregulated upon MTM treatment (Supplementary Figs. 3A and 3B), which are consistent with prior data which shows DNA-PKcs, ATR and ATM are key targets of AR [9,25]. Although the levels of phosphorylated DNA-PKcs (S2056) and ATM (S1981) and total DNA-PKcs in both DMSO and MTM groups was similar 24h post-radiation (Fig. 3A–B), it is possible that the decreased protein expression of ATM or ATR results in the inability to effectively repair DNA damage. To this impaired DNA repair the observed reduction in the levels of CHK1, which is downstream of ATR, and CHK2, which is downstream of ATM, in MTM-treated cells may also contribute. Also, reduced phosphorylation of CHK2 upon MTM treatment suggest that the ATM-CHK2 pathway might play a dominant role in the DDR of IR-induced damage that is affected by MTM. We found that treatment with MTM and IR resulted in increased cleavage of poly (ADP-ribose) polymerase 1 (PARP-1) and Caspase 3, markers for apoptosis, in AR-positive cell lines LNCaP, VCaP and 22RV1 (Fig. 3A and B and Supplementary Fig. 3D). Interestingly, however, the combination of MTM and radiation did not result in increased apoptosis in AR-negative PC3 cells (Supplementary Fig. 3E). This finding suggests that the radiosensitizing effect of MTM may be due to the inhibition of the AR’s role in DDR [9].
The ability of MTM to sensitize AR-positive PCa to radiation was further corroborated by clonogenic assays using 22RV1 cells. Compared to the vehicle control, the AR-positive 22RV1 cells pretreated with MTM (10 nmol/L) showed a decreased clonogenic potential following IR (Fig. 3D, Supplementary Fig. 3F). In contrast, the clonogenic potential of AR-negative PC3 cells following radiation was not decreased by MTM (Supplementary Fig. 3G).
To determine if MTM increased DNA damage (with or without IR) or prevented DNA damage repair induced by IR, the number of γ-H2AX foci were determined immediately following and at 24 h after radiation (the 2Gy dose was used to avoid oversaturation of the γ-H2AX staining, which we observed for doses > 2Gy). We found that in both AR-positive LNCaP and VCaP cells the foci numbers between MTM and DMSO pretreated cells peaked at 1h post-radiation had no significant difference, whereas the foci number was significantly increased in MTM-pretreated cells 24h after radiation (Fig. 3E–F). This is consistent with immunoblotting data showing that MTM at this concentration does not cause significant DNA damage, but greatly reduces the ability of cells to effectively repair DNA.
3.4. MTM increases the effectiveness of Bleomycin in PCa
Considering that MTM appears to impair DNA damage repair following radiation, we surmised that MTM could also increase the therapeutic efficacy of DNA damaging agents in PCa. To substantiate this hypothesis, we treated PCa for 1h with bleomycin (10 mg/L), a radiomimetic agent, which induces DNA single-strand breaks (SSB) and double-strand breaks (DSB). We then determined γ-H2AX levels 24h post-treatment to assess DNA damage repair efficiency. Like for the combination of ionizing radiation and MTM, γ-H2AX levels peaked after 1h bleomycin treatment and decreased to baseline after 24h in the control. However, for the MTM treated cells, while γ-H2AX levels were similar to DMSO at 1h post treatment, significantly increased γ-H2AX levels were found in the MTM treated group 24h after bleomycin treatment in comparison to DMSO in AR-positive VCaP, 22RV1 and LNCaP cells (Fig. 4A–B, Supplemental Fig. 4A). This defect in DNA damage repair following MTM treatment did not occur in PC3 cells (Supplemental Fig. 4B). Also, similar to the combination of ionizing radiation and MTM, MTM pretreatment moderately decreased the protein levels of DNA-PKcs, ATM and ATR, in AR-positive PCa (Fig. 4A–B, Supplemental Figs. 4A) and a reduction of phospho-CHK2 levels further suggest that the AR-ATM-CHK2 pathway might be the main target of MTM in PCa. These results suggest that MTM attenuates repair ability of bleomycin-induced DNA damage in AR-positive PCa.
Fig. 4.
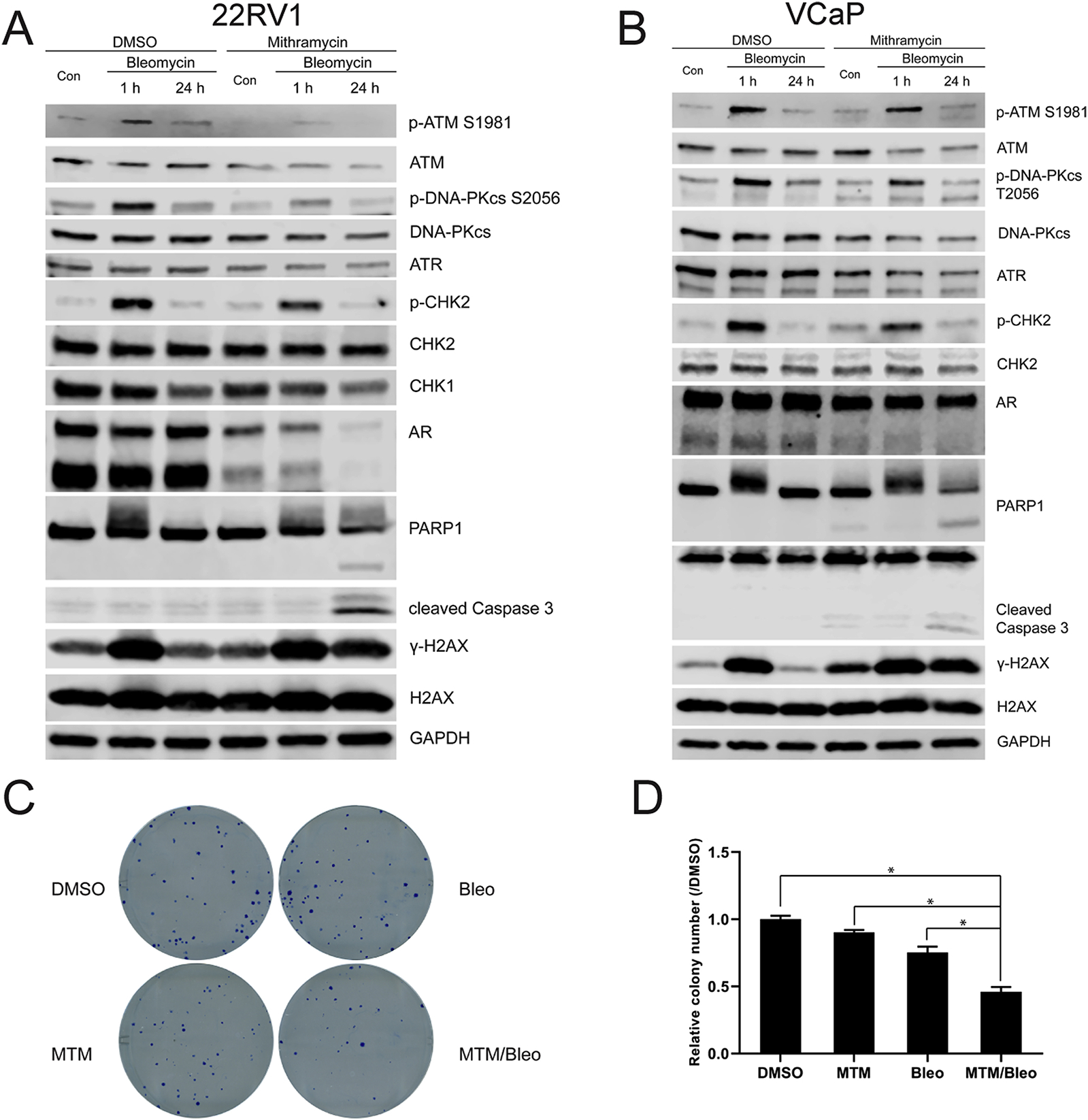
MTM intensifies effectiveness of DNA damaging agent in PCa. A, Western blot of DNA repair-related genes in MTM and Bleomycin co-treated 22RV1 cells. B, Western blot of DNA repair-related genes in MTM and Bleomycin co-treated VCaP cells. C, Representative images of clonogenic assay of 22RV1 cells upon MTM and Bleomycin co-treatment. D, Quantification of colonies in clonogenic assay of 22RV1 cells in C.
The apparent AR-dependent defect in DNA damage repair ultimately resulted in cell death (as demonstrated by cleaved PARP1 and caspase 3) in AR-positive LNCaP, VCaP and 22RV1 cells cotreated with MTM and bleomycin, but not in AR-negative PC3 cells (Fig. 4A–B and Supplementary Figs. 4A and 4B). We further validated this finding using clonogenic assays and found synergistic effects in AR-positive 22RV1 cells with MTM and bleomycin combination treatment (Fig. 4C–D; Supplementary Fig. 4D). In contrast, there was no synergistic effect for combination of MTM and bleomycin on cell viability in AR-negative PC3 cells (Supplementary Fig. 4E).
3.5. MTM inhibits cell proliferation and recovery from DNA damage in primary PCa explants
The study of radioresistance in PCa is especially challenging because few models exist where PCa tissues are in their normal physiological context. Cell line models differ greatly from actual human PCa in their hormone and nutrient levels, and lack any stromal interactions, all of which may affect the response to radiation. Furthermore, current PCa cell lines are generated from metastatic tumors and may not be clinically relevant to study DDR of clinically localized PCa. To this end, we have adapted our ex vivo explant culture system that enables short-term maintenance of primary human PCa tumors in their native microenvironment on a gelatin sponge for evaluation of therapeutic responsiveness [26](Fig. 5A).
Fig. 5. MTM inhibits cell growth and strengthens efficacy of IR in prostate cancer explants.
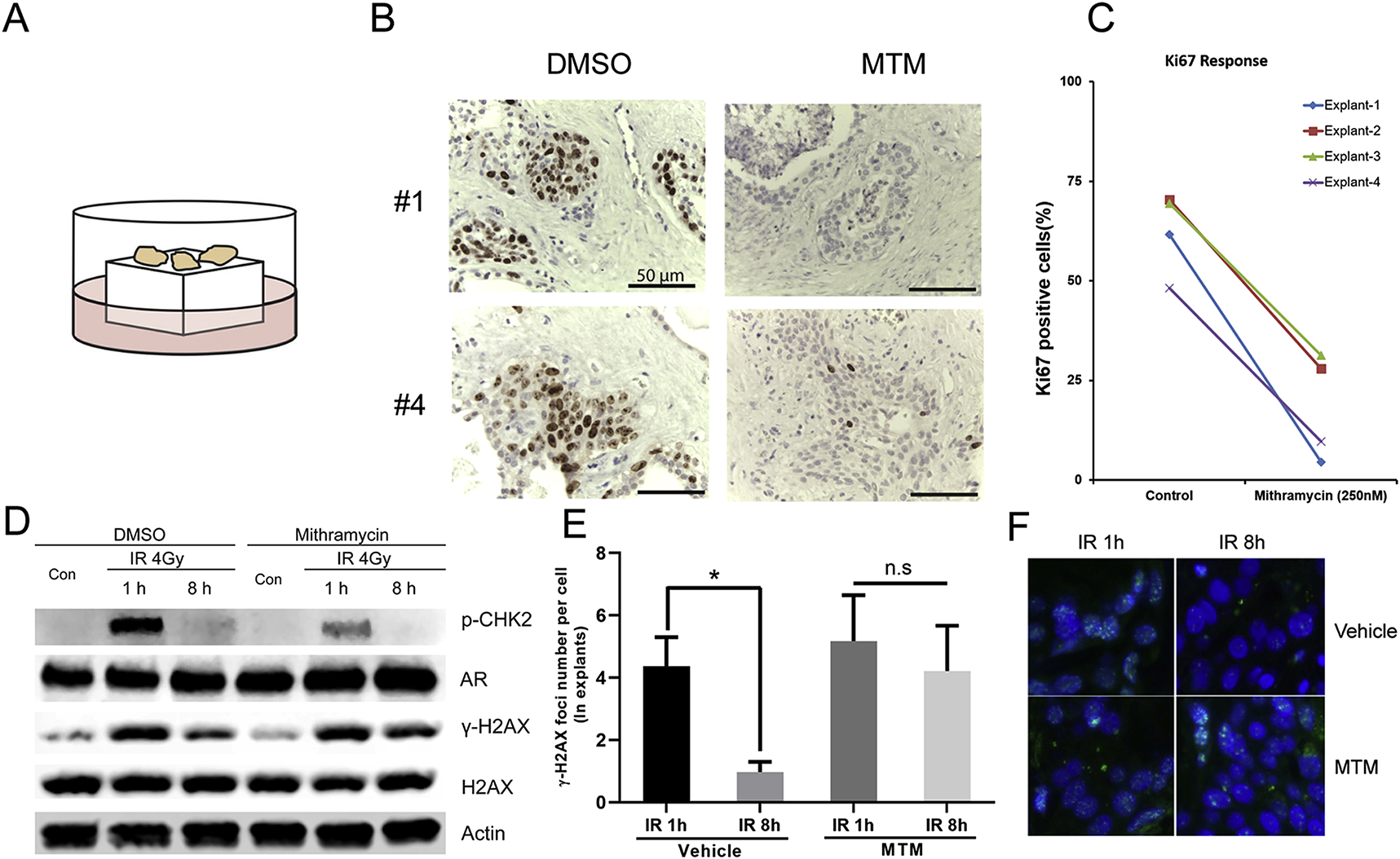
A, Culture model of prostate cancer explants. B, Representative images of Ki-67 staining in PCa explants with MTM treatment. C, Quantification of Ki67 positive cells in PCa explants with MTM treatment. D, Western blot of γ-H2AX, p-Chk2 and AR in MTM and IR co-treated PCa explants. E, Average number of γ-H2AX Foci in MTM and IR co-treated PCa explants. F, Representative images of γ-H2AX Foci in MTM and IR co-treated PCa explants.
We first tested the effect of MTM on the cell proliferation of orga-notypically cultured prostate tumors from clinical prostate cancer specimens. Ki67 IHC analysis indicated that MTM significantly decreased cell proliferation of primary PCa explants (Fig. 5B and C). We then irradiated several explants that were pretreated with MTM to determine how MTM affected DNA repair in these primary human tumors. Both immunoblotting and IHC demonstrated that MTM pretreatment significantly impaired DNA damage response, as evidenced by a reduced decline of γ-H2AX levels/foci in MTM pretreated explants as compared to vehicle controls at 8 h post radiation (Fig. 4D–F). In addition, phospho-CHK2 levels were reduced upon MTM treatment, which is consistent with our findings in PCa cell lines, which supports our model that MTM may suppress the ATM-CHK2 pathway in AR-driven PCa.
4. Discussion
Because of the loss of the ligand-binding domain, AR splice variants may confer resistance to hormone therapy in PCa. Among several AR-Vs, AR-V7 has been predominantly studied in mCRPC. Although the understanding of the precise functional role of AR-Vs in the progression of castration-resistant PCa is incomplete, the frequently observed induction of AR-Vs by ADT has catapulted the identification of small molecules that inhibit AR-Vs into the forefront of therapeutics development for CRPC. Currently, several small molecules have been suggested to target AR-Vs, including EPI-001 analogs [27], methylselenol prodrug [28], PKC inhibitor Ro31–8220 [29] and Niclosamide [30]. In this study, we identified Mithramycin as a novel inhibitor of AR and AR-Vs. MTM can inhibit both the expression and transcriptional activity of AR and AR-Vs. Prior work has shown that MTM decreases mRNA expression of AR by restraining SP1 activity [18]. Our work shows that at a low concentration, MTM treatment has a variable effect on AR mRNA expression (e.g. with only a modest effect in VCaP cells), but appears to effectively inhibit the transcriptional activity of AR (Supplementary Fig. 2B). Also, our work revealed that MTM inhibits AR-V7 and ARv567es transcriptional activity and reduces the recruitment of AR-V7 to canonical AREs. These findings provide another mechanism for MTM on inhibiting AR signaling in PCa.
Our findings indicate that the DNA-binding small molecule MTM reduces AR recruitment to its genomic targets thereby downregulating the expression of AR target genes, which includes key DNA damage repair factors required for DDR/radioresistance. The dual activity of MTM on PCa proliferation and DDR through the inhibition of AR and AR-Vs DNA binding provides a mechanistic rationale for the combination of MTM and RT. Thus, the combination of MTM treatment with RT or radiomimetic agents, such as bleomycin, may present a novel effective therapeutic strategy for patients with high-risk clinically localized PCa (Fig. 6). Bleomycin is a chemotherapeutic agent from Streptomyces verticillus and could induce DNA double-strand breaks (DSBs) and single-strand breaks (SSBs) via generating free radicals in cells [31,32]. The mechanism of bleomycin on DSBs is similar to the indirect effect of ionizing radiation on DNA damage [32,33]. Therefore, bleomycin is considered as a radiomimetic agent and is widely used in the treatment of different types of cancer, such as lung cancer and cervical cancer [32]. Our data showed that through its radiomimetic characteristics, bleomycin demonstrates the synergetic effect in prostate cancer with the combination with MTM.
Fig. 6.
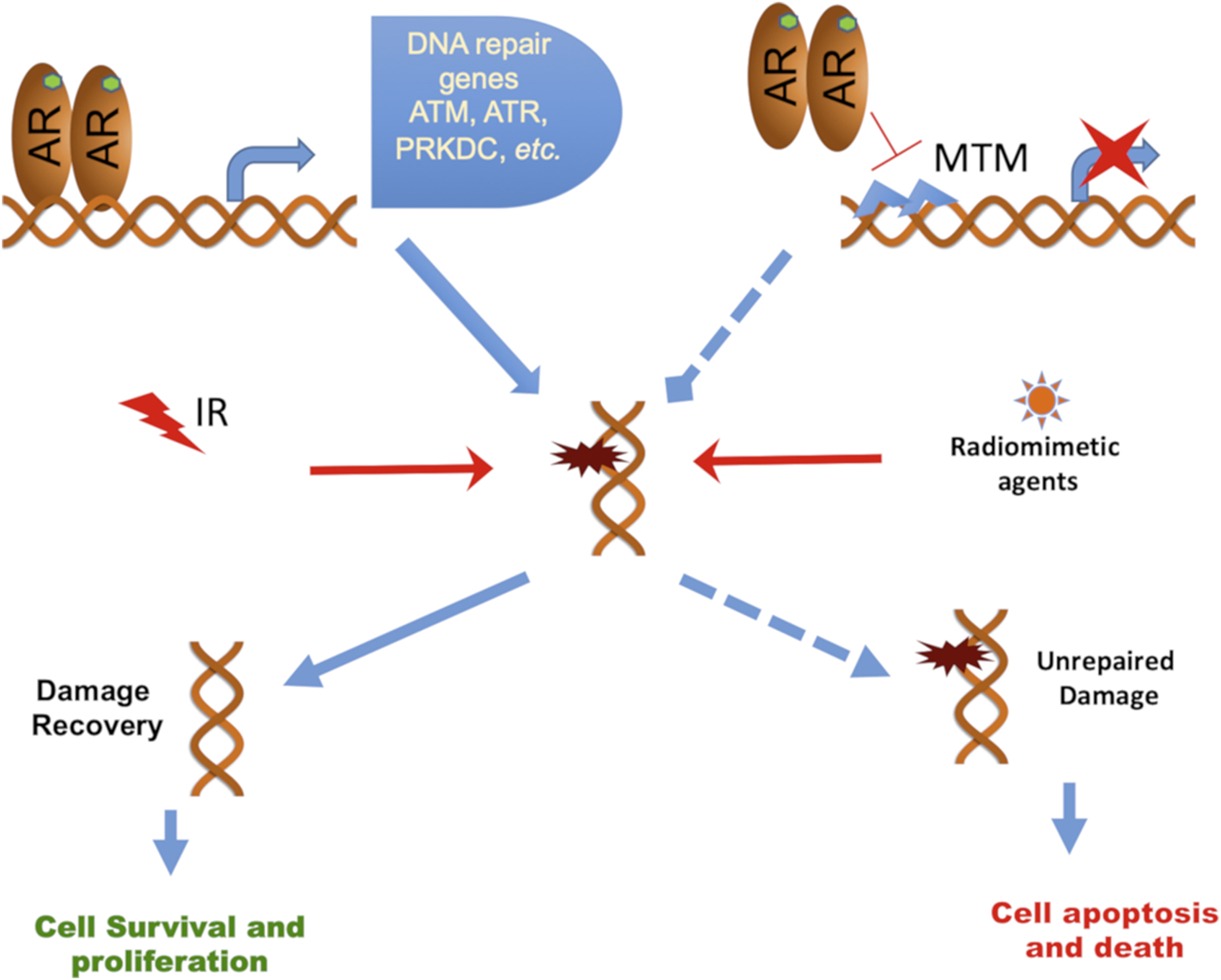
Schematic model of MTM in prostate cancer cells.
While many strategies for direct inhibition of DDR were proposed to enhance RT, our strategy is novel as we indirectly target DDR by interfering with the transcriptional activity of all forms of AR, which may benefit patients with high-risk clinically localized PCa.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
This study was supported by a grant from the National Cancer Institute of the National Institutes of Health (R01CA200787), the CPRIT Scholar in Cancer Research Award (R1002) of the Cancer Prevention and Research Institute of Texas and the John L. Roach Endowment in Biomedical Research to R.K.
Footnotes
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.canlet.2020.05.027.
References
- [1].Siegel RL, Miller KD, Jemal A, Cancer statistics CA A Cancer J. Clin 68 (2018) 7–30 2018. [DOI] [PubMed] [Google Scholar]
- [2].Siegel RL, Miller KD, Jemal A, Cancer statistics CA A Cancer J. Clin 69 (2019) 7–34 2019. [DOI] [PubMed] [Google Scholar]
- [3].Matsumoto T, Sakari M, Okada M, Yokoyama A, Takahashi S, Kouzmenko A, Kato S, The androgen receptor in health and disease, Annu. Rev. Physiol 75 (2013) 201–224. [DOI] [PubMed] [Google Scholar]
- [4].Karantanos T, Evans CP, Tombal B, Thompson TC, Montironi R, Isaacs WB, Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level, Eur. Urol 67 (2015) 470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Flechon A, Mainwaring P, Fleming M, Hainsworth JD, Hirmand M, Selby B, Seely L, de Bono JS, Investigators A, Increased survival with enzalutamide in prostate cancer after chemotherapy, N. Engl. J. Med 367 (2012) 1187–1197. [DOI] [PubMed] [Google Scholar]
- [6].Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, Fizazi K, Mainwaring P, Piulats JM, Ng S, Carles J, Mulders PF, Basch E, Small EJ, Saad F, Schrijvers D, Van Poppel H, Mukherjee SD, Suttmann H, Gerritsen WR, Flaig TW, George DJ, Yu EY, Efstathiou E, Pantuck A, Winquist E, Higano CS, Taplin ME, Park Y, Kheoh T, Griffin T, Scher HI, Rathkopf DE, Investigators CA, Abiraterone in metastatic prostate cancer without previous chemotherapy, N. Engl. J. Med 368 (2013) 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zadra G, Ribeiro CF, Chetta P, Ho Y, Cacciatore S, Gao X, Syamala S, Bango C, Photopoulos C, Huang Y, Tyekucheva S, Bastos DC, Tchaicha J, Lawney B, Uo T, D’Anello L, Csibi A, Kalekar R, Larimer B, Ellis L, Butler LM, Morrissey C, McGovern K, Palombella VJ, Kutok JL, Mahmood U, Bosari S, Adams J, Peluso S, Dehm SM, Plymate SR, Loda M, Inhibition of de novo lipogenesis targets androgen receptor signaling in castration-resistant prostate cancer, Proc. Natl. Acad. Sci. U. S. A 116 (2019) 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Copeland BT, Pal SK, Bolton EC, Jones JO, The androgen receptor malignancy shift in prostate cancer, Prostate 78 (2018) 521–531. [DOI] [PubMed] [Google Scholar]
- [9].Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, Arora VK, Yen WF, Cai L, Zheng D, Carver BS, Chen Y, Watson PA, Shah NP, Fujisawa S, Goglia AG, Gopalan A, Hieronymus H, Wongvipat J, Scardino PT, Zelefsky MJ, Jasin M, Chaudhuri J, Powell SN, Sawyers CL, Androgen receptor signaling regulates DNA repair in prostate cancers, Canc. Discov 3 (2013) 1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Spratt DE, Evans MJ, Davis BJ, Doran MG, Lee MX, Shah N, Wongvipat J, Carnazza KE, Klee GG, Polkinghorn W, Tindall DJ, Lewis JS, Sawyers CL, Androgen receptor upregulation mediates radioresistance after ionizing radiation, Canc. Res 75 (2015) 4688–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yin Y, Li R, Xu K, Ding S, Li J, Baek G, Ramanand SG, Ding S, Liu Z, Gao Y, Kanchwala MS, Li X, Hutchinson R, Liu X, Woldu SL, Xing C, Desai NB, Feng FY, Burma S, de Bono JS, Dehm SM, Mani RS, Chen BPC, Raj GV, Androgen receptor variants mediate DNA repair after prostate cancer irradiation, Canc. Res 77 (2017) 4745–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zelefsky MJ, Reuter VE, Fuks Z, Scardino P, Shippy A, Influence of local tumor control on distant metastases and cancer related mortality after external beam radiotherapy for prostate cancer, J. Urol 179 (2008) 1368–1373 discussion 1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Yu Z, Chen S, Sowalsky AG, Voznesensky OS, Mostaghel EA, Nelson PS, Cai C, Balk SP, Rapid induction of androgen receptor splice variants by androgen deprivation in prostate cancer, Clin. Canc. Res 20 (2014) 1590–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kennedy BJ, Torkelson JL, Long-term follow-up of stage III testicular carcinoma treated with mithramycin (plicamycin), Med. Pediatr. Oncol 24 (1995) 327–328. [DOI] [PubMed] [Google Scholar]
- [15].Dutcher JP, Coletti D, Paietta E, Wiernik PH, A pilot study of alpha-interferon and plicamycin for accelerated phase of chronic myeloid leukemia, Leuk. Res 21 (1997) 375–380. [DOI] [PubMed] [Google Scholar]
- [16].Parsons V, Scott G, Baum M, Molland E, Makin J, Mithramycin treatment of hy-percalcaemia and renal failure in a patient with paratesticular embryonic sarcoma, Br. J. Canc 25 (1971) 306–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Grohar PJ, Glod J, Peer CJ, Sissung TM, Arnaldez FI, Long L, Figg WD, Whitcomb P, Helman LJ, Widemann BC, A phase I/II trial and pharmacokinetic study of mithramycin in children and adults with refractory Ewing sarcoma and EWS–FLI1 fusion transcript, Canc. Chemother. Pharmacol 80 (2017) 645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wang LG, Ferrari AC, Mithramycin targets sp1 and the androgen receptor transcription level-potential therapeutic role in advanced prostate cancer, Transl. Oncogenomics 1 (2006) 19–31. [PMC free article] [PubMed] [Google Scholar]
- [19].Carles J, Gallardo E, Doménech M, Font A, Bellmunt J, Figols M, Mellado B, Sáez MI, Suárez C, Méndez MJ, Maroto P, Luque R, Portugal T, Aldabo R, Bonfill T, Morales-Barrera R, García J, Maciá S, Maldonado X, Foro P, Phase 2 randomized study of radiation therapy and 3-year androgen deprivation with or without concurrent weekly docetaxel in high-risk localized prostate cancer patients, Int. J. Radiat. Oncol. Biol. Phys 103 (2019) 344–352. [DOI] [PubMed] [Google Scholar]
- [20].Nyquist MD, Li Y, Hwang TH, Manlove LS, Vessella RL, Silverstein KA, Voytas DF, Dehm SM, TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer, Proc. Natl. Acad. Sci. U. S. A 110 (2013) 17492–17497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C, Clonogenic assay of cells in vitro, Nat. Protoc 1 (2006) 2315–2319. [DOI] [PubMed] [Google Scholar]
- [22].Di Veroli GY, Fornari C, Wang D, Mollard S, Bramhall JL, Richards FM, Jodrell DI, Combenefit: an interactive platform for the analysis and visualization of drug combinations, Bioinformatics 32 (2016) 2866–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, Page ST, Coleman IM, Nguyen HM, Sun H, Nelson PS, Plymate SR, Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant, J. Clin. Invest 120 (2010) 2715–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Shafi AA, Cox MB, Weigel NL, Androgen receptor splice variants are resistant to inhibitors of Hsp90 and FKBP52, which alter androgen receptor activity and expression, Steroids 78 (2013) 548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, Ma T, Den RB, Dicker AP, Feng FY, Knudsen KE, A hormone-DNA repair circuit governs the response to genotoxic insult, Canc. Discov 3 (2013) 1254–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang S, Kollipara RK, Srivastava N, Li R, Ravindranathan P, Hernandez E, Freeman E, Humphries CG, Kapur P, Lotan Y, Fazli L, Gleave ME, Plymate SR, Raj GV, Hsieh JT, Kittler R, Ablation of the oncogenic transcription factor ERG by deubiquitinase inhibition in prostate cancer, Proc. Natl. Acad. Sci. U. S. A 111 (2014) 4251–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Myung JK, Banuelos CA, Fernandez JG, Mawji NR, Wang J, Tien AH, Yang YC, Tavakoli I, Haile S, Watt K, McEwan IJ, Plymate S, Andersen RJ, Sadar MD, An androgen receptor N-terminal domain antagonist for treating prostate cancer, J. Clin. Invest 123 (2013) 2948–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhan Y, Cao B, Qi Y, Liu S, Zhang Q, Zhou W, Xu D, Lu H, Sartor O, Kong W, Zhang H, Dong Y, Methylselenol prodrug enhances MDV3100 efficacy for treatment of castration-resistant prostate cancer, Int. J. Canc 133 (2013) 2225–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Shiota M, Yokomizo A, Takeuchi A, Imada K, Kashiwagi E, Song Y, Inokuchi J, Tatsugami K, Uchiumi T, Naito S, Inhibition of protein kinase C/Twist1 signaling augments anticancer effects of androgen deprivation and enzalutamide in prostate cancer, Clin. Canc. Res 20 (2014) 951–961. [DOI] [PubMed] [Google Scholar]
- [30].Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP, Gao AC, Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer, Clin. Canc. Res 20 (2014) 3198–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Regulus P, Duroux B, Bayle PA, Favier A, Cadet J, Ravanat JL, Oxidation of the sugar moiety of DNA by ionizing radiation or bleomycin could induce the formation of a cluster DNA lesion, Proc. Natl. Acad. Sci. U. S. A 104 (2007) 14032–14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bolzan AD, Bianchi MS, DNA and chromosome damage induced by bleomycin in mammalian cells: an update, Mutat. Res 775 (2018) 51–62. [DOI] [PubMed] [Google Scholar]
- [33].Lomax ME, Folkes LK, O’Neill P, Biological consequences of radiation-induced DNA damage: relevance to radiotherapy, Clin. Oncol 25 (2013) 578–585. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.