

Abstract
Background
Alpha1‐antitrypsin deficiency (AATD) is an under‐diagnosed hereditary disorder characterized by reduced serum levels of alpha1‐antitrypsin (AAT) and increased risk to develop lung and liver diseases at an early age. AAT is encoded by the highly polymorphic SERPINA1 gene. The most common deficiency alleles are S and Z, but more than 150 rare variants lead to low levels of the protein. To identify these pathological allelic variants, sequencing is required. Since traditional sequencing is expensive and time‐consuming, we evaluated the accuracy of A1AT Genotyping Test, a new diagnostic genotyping kit which allows to simultaneously identify and genotype 14 deficiency variants of the SERPINA1 gene based on Luminex technology.
Methods
A total of 418 consecutive samples with AATD suspicion and submitted to the Italian Reference laboratory between January and April 2016 were analyzed both by applying the diagnostic algorithm currently in use, and by applying A1AT Genotyping Test.
Results
The assay gave the following results: 101 samples (24.2%) were positive for at least one of the 14 deficiency variants, 316 (75.6%) were negative for all the variants analyzed. The identified mutations showed a 100% correlation with the results obtained with our diagnostic algorithm. Seventeen samples (4%) resulted negative for the assay but sequencing identified other rare pathological variants in SERPINA1 gene.
Conclusion
The A1AT Genotyping Test assay was highly reliable and robust and allowed shorter diagnostic times. In few cases, it has been necessary to sequence the SERPINA1 gene to identify other rare mutations not included in the kit.
Keywords: alpha1‐antitrypsin, chemiluminescence, diagnosis, genotyping, SERPINA1
Abbreviations
- A1AT GT
Alpha1‐antitrypsin Genotyping Test
- AAT
alpha1‐antitrypsin
- AATD
alpha1‐antitrypsin deficiency
- BSA
bovine serum albumin
- COPD
chronic obstructive pulmonary disease
- CRP
C‐reactive protein
- DBS
dried blood spot
- DNA
deoxyribonucleic acid
- IEF
isoelectric focusing
- NE
neutrophil elastase
- PCR
polymerase chain reaction
- Pi
proteinase inhibitor
1. INTRODUCTION
Alpha1‐antitrypsin deficiency (AATD) is an under‐recognized and under‐diagnosed hereditary disorder characterized by reduced serum levels of alpha1‐antitrypsin (AAT) and increased risk to develop lung emphysema at an early age and chronic liver disease. AAT is a 52‐kD glycoprotein mainly produced by hepatocytes and secreted into the blood, where it acts as a circulating serine protease inhibitor.1 The principal role of AAT is to protect the lung tissues from proteolytic degradation by inactivating neutrophil elastase (NE). The circulating protein is responsible for more than 90% of the anti‐protease activity in the body.2
The protein is encoded by the protease inhibitor (Pi) locus on chromosome 14q32.1. The gene is called SERPINA1; it spans 12.2 kb and is organized into four coding (II, III, IV, and V) and three non‐coding (Ia, Ib, and Ic) exons.1 Normal AAT serum concentration in healthy individual is 1.00‐2.00 g/L.3 Reduced serum AAT concentrations can lead to lung and/or liver disease, depending on the associated genetic variant.
The World Health Organization and the medical societies of Europe and the US recommend AAT should be measured (quantitatively) at least once in the blood of all patients with COPD, regardless of whether they smoke. Testing is recommended for sibs of someone with AATD and should be considered for other relatives of someone with AATD or COPD. These same organizations recommend, at least once in a lifetime, measuring AAT in the blood of all patients with chronic liver disease of unknown etiology and especially if there are familial cases of liver disease and/or AATD. Severe adult asthma, antineutrophil cytoplasmic antibodies and systemic vasculitis, and relapsing panniculitis are other conditions for which diagnosis for AATD is recommended. If serum values of AAT are low (quantitatively measured by nephelometry), a qualitative confirmation of the patient's phenotype or genotype is recommended.4, 5, 6
Over 150 mutations have been described and have been named by a letter based on the migration speed of the resulting protein on isoelectric focusing.7 For clinical purposes, AAT variants have been classified into 3 major categories: normal (characterized by AAT plasma levels within general population reference ranges), deficient (characterized by decreased, but still detectable, AAT plasma levels, associated with an increased risk of developing lung and/or liver disease), and Null (currently designated Q0, with no detectable AAT plasma levels, associated with an increased risk of developing emphysema). Moreover, there are dysfunctional variants, that lead to abnormal function of AAT, with reduced binding to neutrophil elastase (as in the F variant) or, as with Pittsburgh, with structural abnormality that causes the protein to serve as a thrombin inhibitor rather than as an anti‐elastolytic protein, causing a bleeding diathesis.
The normal AAT protein is called M, and the most common mutations are Z (p.E366K c.1096G > A) and S (p.E288V c.863A > T).8 It has been estimated that combinations of M, S, and Z alleles encompass over 98% of the world population and therefore are the initial focus of AAT testing in the laboratory.5
The so‐called “rare variants” cover all that variants that differ from S and Z (including also the deficient M‐like variants and the Null variants).9
Little is known about genetic epidemiology of rare AAT variants. Analysis of 40 cohorts from Italy 10 showed that the frequencies of the S and Z alleles are highest in Northern Italy and decrease gradually from north to south. We have data showing that in the Italian Registry for AATD as many as 10% of the total AATD variants are rare variants. It seems that individuals carrying at least one rare AATD allele are characterized by a rather peculiar phenotypic profile, placing them in a precise position within the spectrum of genotype‐phenotype correlations in AATD. According to the specific molecular mechanism related to each rare variant, different degrees of risk for lung and/or liver diseases could be prognosticated. Moreover, the determination of the frequency and clinical impact of rare deficient phenotypes would help to establish the requirements for augmentation therapy in emphysema patients carrying these variants.11
Procedures for testing for AATD have been available since the 1960s, and new techniques have been introduced during the intervening years. These advances in methodology should facilitate the widespread application of more rapid, convenient, and cost‐effective tests for AATD and thus lead to an increase in the numbers of individuals diagnosed with the disorder.12
Although methods have been validated for testing for AATD, there is not one established algorithm for the detection and diagnosis of the disorder that is universally used by laboratories and countries around the world.13 The laboratory diagnosis of AATD requires a combination of different methods to perform both quantitative and qualitative tests, consisting in nephelometric serum AAT level determination, isoelectric focusing (IEF), and genotyping, as recently stated.6
The methodological difficulties associated with routine AATD assays are a common problem in the study of rare alleles.9, 14 To identify pathological allelic variants, different from S and Z, expanding genotyping, phenotyping, and sequencing of the gene coding regions are required.15
In reference laboratories, AATD diagnosis is usually lead on dried blood spot (DBS), drops of blood that have been dried onto a special filter paper, in order to enable the centralization of AATD diagnosis in some countries. This specimen represents an advantage of easier preservation and shipping of samples, thus allowing wide detection or screening programs for AATD.13 The applicability and reliability of the DBS matrix for AATD testing have already been described.15, 16
Sequencing demands time and resources since it entails the preparation of different reaction for each sample and running the sequencer to generate sequence reads. Moreover, once sequence reads are obtained, the operator must carry out sequence analysis comparing them to the reference one. Next‐generation sequencing could give some benefits on reducing costs than traditional sequencing but it is still far to be widely applied in the field of AATD laboratory diagnosis.
We evaluated the accuracy of A1AT Genotyping Test (A1AT GT) (Progenika, Grifols), a new diagnostic genotyping kit which allows identification of 14 deficiency variants of the SERPINA1 gene (including S and Z alleles) based on Luminex technology. The alleles detected by the assay are listed in Table 1. The 14 deficiency variants were chosen by the manufacturer; they include some of the more frequent rare variants (ie, Mmalton, Mprocida, I, F) and some other ultra‐rare variants, to offer a worldwide coverage.
Table 1.
Description of the allelic variants and associated alleles tested by A1AT Genotyping Test
| Allelic variants | Associated alleles |
|---|---|
| c.187C > T | PI*I |
| c.194C > T | PI*Mprocida |
| c.226_228delTTC | PI*Mmalton, PI*Mpalermo, PI*Mnichinan |
| c.230C > T | PI*Siyama |
| c.551_552delC | PI*Q0granite falls |
| c.647G > T | PI*Q0west |
| c.721A > T | PI*Q0bellingham |
| c.739C > T | PI*F |
| c.839A > T | PI*Plowell, PI*Pduarte, PI*Q0cardiff, PI*Ybarcelona |
| c.863A > T | PI*S |
| c.1096G > A | PI*Z |
| c.1130_1131insT | PI*Q0mattawa, PI*Q0ourem |
| c.1156_1157insC | PI*Q0clayton, PI*Q0Saarbruecken |
| c.1178C > T | PI*Mheerlen |
2. MATERIALS AND METHODS
2.1. Samples
A total of 418 consecutive DBS samples, submitted to the Italian Reference laboratory from January 2016 to April 2016 for AATD testing have been analyzed. Written consent has been included, according to the Institution's ethical recommendations.
2.2. DBS preparation
To make precise blood spot punches, a Wallac 1296‐071 DBS Puncher (Perkin Elmer) is used. The DBS Puncher allows cutting 3.2 mm of diameter disks from the DBS samples.
2.3. Genotyping and sequence analysis
According to the standard testing flowchart15 (Figure 1A), all samples were subjected to AAT and C‐reactive protein level determinations, phenotyping by IEF and genotyping for the detection of S and Z variants by PCR with fluorescently labeled Taq‐Man probes (Vic or Fam labels) on a LigthCycler480 (Roche Diagnostics).3 Sequence analysis for coding exons (II‐V) of the SERPINA1 gene has been applied using the CEQ 8800 genetic analysis System (Beckman Coulter), in case of lack of correspondence between genotyping results, phenotyping, and concentration of AAT, as previously reported.15
Figure 1.
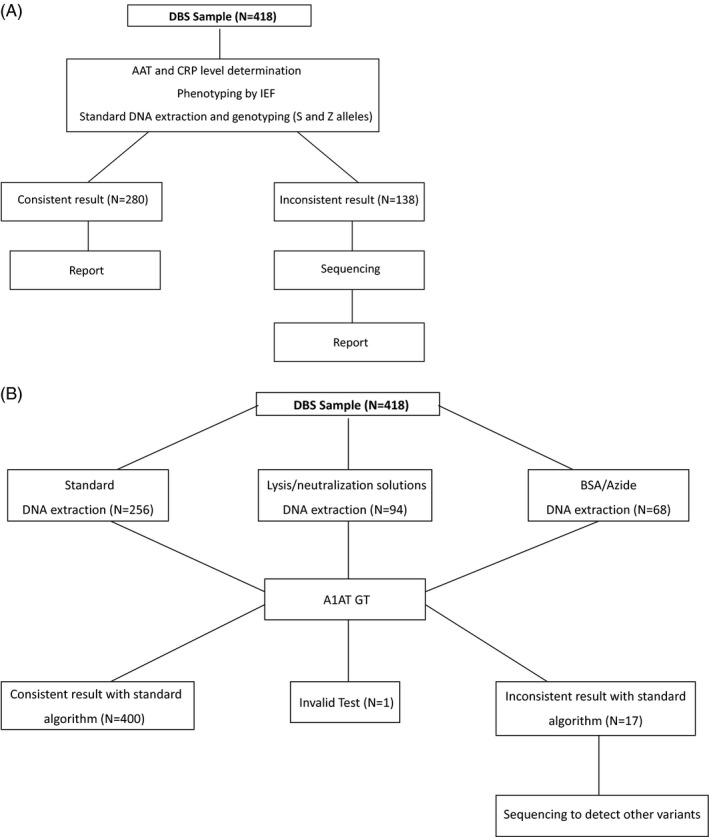
A, Standard testing flowchart. B, A1AT GT testing flowchart
2.4. DNA Extraction
For the analysis with the A1AT Genotyping Test, DNA was extracted from DBS with different methods using commercial kits and standard processing (QIAcube), Lysis/Neutralization Sigma Reagents Method or by an approved in‐house method and using a buffer containing BSA/Azide (Figure 1B).
For 256 of the 418 samples (61%), the extraction was performed using an automatic extractor (QIAcube) and the QIAamp DNA Mini Kit or Investigator Kit (QIAGEN). For each sample, three 3.2 mm disks with the Puncher were cut. Each sample was treated with the addition of 180 µL of ATL buffer—according to manufacturer's instructions and incubated at 85°C for 10 minutes with a shaker. Then, 20 µL of proteinase K was added to the samples that were incubated at 56°C for at least 1 hour with a shaker. The eluate was transferred to clean tubes and loaded into the QIAcube extractor, and then was processed according to the instrument's user manual. A final volume of 60 µL of extracted DNA was kept at 2‐8°C until the DNA amplification.
For 94 of the 418 samples, the commercial lysis and neutralization solution method (Sigma) was used for DNA extraction from DBS. Each 3.2 mm punch was transferred to a 96‐well reaction plate's well, and 20 µL of the lysis solution was added to each circle. The plate was spinned down, making sure that all punches and lysis buffer were in the bottom of the wells. The plate was then incubated at 55°C for 55 minutes in a thermal cycler block; 180 µL of the neutralization solution was added to each well of the extraction plate, mixing thoroughly by pipetting up‐down until a homogeneous mixture was obtained. Then, at least 50 µL of the mix was transferred to a new 96 well plate, that was kept at 2‐8°C until the DNA amplification.
For 68 of the 418 samples, a buffer containing 1 mg/mL of BSA and 1 mg/mL of azide in molecular biology water was used for DNA extraction from DBS. Each 3.2 mm punch was transferred to 96‐well reaction plate´s well, and 60 µL of BSA/azide buffer was added to each circle. The plate was spinned down, making sure that all punches and buffer were in the bottom of the wells and was then incubated at 2 steps: 56°C for 25 minutes and 100°C for 10 minutes in a thermal cycler block. The plate was then centrifuged for 5 minutes at 2880g. A volume of 40 µL of the mix was transferred to a new 96‐well reaction plate that was kept at 2‐8°C until the DNA amplification.
2.5. Genotyping with A1AT Genotyping Test
For the A1AT GT genotyping analysis, the A1AT Genotyping kit containing the ready‐to‐use reagents (A1AT polymerase chain reaction [PCR] Master Mix, A1AT Bead Master Mix, SAPE and SAPE Dilution) needed for the processing steps was used. First, targeted DNA regions were amplified through a multiplex PCR using biotinylated dCTP. The obtained PCR products were subsequently hybridized onto oligonucleotide probes, attached to microspheres and labeled with streptavidin‐conjugated phycoerythrin. The beads were finally analyzed with a Luminex® 200 system (Luminex Corporation). The raw data were processed in a simultaneous and automatic way for all samples with the A1AT Genotyping Test ANALYSIS SOFTWARE to obtain the genotypes for each of the allelic variants, as well as the Sample Result combining the results for all the individual allelic variants for a sample. The possible results of each sample by A1AT GT were as follows: valid test (genotype calls are displayed for all the allelic variants. Sample processing has been successful and the final report shows the corresponding Sample Result), invalid test (the sample analysis result is not acceptable), and unknown (a not known Sample Result due to an unknown allelic combination). When none of the 14 allelic variants included in the test have been detected, the A1AT Genotyping Test ANALYSIS SOFTWARE conventionally used the term M/M.
3. RESULTS
A total of 418 consecutive samples, submitted to the Italian Reference laboratory from January 2016 to April 2016 for AATD testing, were analyzed both by applying the diagnostic algorithm currently in use,15 and by applying the A1AT Genotyping Test, A1AT GT (Progenika, Grifols) based on Luminex 200 (Figure 1A and B).
Current genotyping analysis gave the following results: 55 samples were heterozygous for Z allele; 18 samples were heterozygous for S allele; 9 samples were homozygous for Z allele; 2 samples were heterozygous both for Z and S alleles; Z and S alleles have been excluded in 334 samples. A total of 138 samples (33%) (namely 16 heterozygous for Z allele, 4 heterozygous for S allele and 118 not S‐not Z) have been sequenced, because of inconsistency between quantitative data and phenotyping/genotyping.
The Progenika A1AT GT gave the following results: 101 samples (24.2%) were positive for at least one of the 14 deficiency variants, 316 (75.6%) were negative for all the variants analyzed, while one sample (0.2%), extracted with the BSA/azide buffer method, was invalid for the analysis, due to bad DNA quality. The identified mutations showed a 100% correlation with the results obtained with our diagnostic algorithm.
Sixty‐six samples resulted positive for the Z allele and 20 for the S allele.
Regarding the M‐like variants, 4 samples were positive for Mprocida, 2 for Mmalton, and 1 for Mheerlen. One sample resulted positive for the F allele, 8 for the I and 4 for the Plowell variants. Finally, in one sample, the Null variant Q0clayton was found.
No samples were positive for the Siyama variant and for Q0granitefalls, Q0west, Q0bellingham e Q0mattawa variants.
In 400 samples, the A1AT GT analysis was in agreement with the AAT levels measured and with the phenotype; therefore, no further test was necessary to formulate the final genotype and allowed a complete report. Among them, 15 samples were homozygous (9 PI*ZZ) or compound heterozygous for 2 deficiency variants (2 PI*SZ, 1 PI*SPlowell, 1 PI*ZI, 1 PI*ZPlowell, and 1 PI*ZQ0clayton), and 83 were heterozygous for 1 deficiency variant. On the contrary, in 3 cases that resulted heterozygous after A1AT GT analysis displayed lower AAT levels than expected, therefore sequencing analysis was needed to determine the correct genotype, namely 1 PI*IMwurzburg, 1 PI*ZMwurzburg, and 1 PI*VZ (Table 2).
Table 2.
A1AT GT test results
| Results | Frequency (%) | Inconsistent* (N) | Final genotype | Sequencing Frequency (%) |
|---|---|---|---|---|
| No variants detected (MM) | 75.7 | 14 |
5 PI*MMwurzburg 3 PI*MV 1 PI*MQ0pordenone 1 PI*MQ0perugia 1PI*MSmunich 3 PI*M new mutation |
3.3 |
| MZ | 12.4 | 2 |
1 PI*ZMwurzburg 1 PI*VZ |
0.5 |
| MS | 4.1 | |||
| ZZ | 2.2 | |||
| MI | 1.7 | 1 | PI*Mwurzburg I | 0.2 |
| MMprocida | 1.0 | |||
| SZ | 0.5 | |||
| MMmalton | 0.5 | |||
| MPlowell | 0.5 | |||
| MMheerlen | 0.2 | |||
| MF | 0.2 | |||
| SPlowell | 0.2 | |||
| ZI | 0.2 | |||
| ZPlowell | 0.2 | |||
| ZQ0clayton | 0.2 | |||
| Invalid Test | 0.2 |
Inconsistent results with phenotyping and quantitative analysis of standard algorithm. These samples have been submitted to sequence analysis.
Moreover, in one case, the A1AT GT analysis identified a mutation (Mprocida) which was not recognized by standard algorithm, since the plasma concentration of AAT measured was normal (123.9 mg/dL) and, therefore, the sample was not sequenced.
Fourteen samples (3.3% of total) resulted negative for the A1AT GT; nevertheless, sequencing analysis identified other rare pathological variants. We found one sample positive for Q0pordenone variant, one for Q0perugia allele, and one for Smunich variant. Moreover, the Mwurzburg allele was found in 7 cases and the V allele in 4 samples (Table 2). The Mwurzburg mutation produces mutated AAT forming ordered polymers that are retained as inclusions within the endoplasmic reticulum of hepatocytes and is associated with a higher risk to develop both lung and liver diseases. The V allele, instead, is associated to a slightly decrease of AAT plasma level; consequently, the V variant is not pathological. However, in the presence of V variant, a particular IEF pattern and a double alpha1‐globulin peak in the serum protein electrophoresis are visible; for this reason, it is anyway important to identify also benign mutations, not to misdiagnose them. Finally, we found 3 new mutations, never identified in literature or databases at the time of diagnosis. The first mutation was found in exon 2 and consisted of a IleATC/AACAsn substitution in codon 50 (p.I74N); the mutation was found associated with the normal M1 allele in a patient with lower AAT plasma levels; it has been recently published as Tijarafe.17 The second mutation consisted in a GluGAA/LysAAA substitution at codon 205 (p.Q229K) in exon 3; the patient showed an IEF pattern similar to that obtained for the S allele but the mutation was associated with the M2 normal allele and the AAT plasma levels measured were almost normal. The third mutation was found in exon 5 at codon 362 (p.P386H), already reported in the NCBI database (rs143329723) but with unknown significance; the patient with this mutation had lower plasma levels of the protein.
4. DISCUSSION
It is well known that the genetic diagnosis of AATD based on genotyping of S and Z allele could be incomplete and non‐exhaustive. The recent European Respiratory Statement reported that “genotyping allows a rapid and precise identification/exclusion of S and Z alleles and other variants, where specific primers are available” and “gene sequencing remains necessary for those cases where a Null variant or a deficient variant other than Z and S is suspected”.6 Therefore, the examination of innovative diagnostic methods is advisable.
The A1AT GT assay was found to be highly reliable because it gave results that are overlapping 100% with those previously obtained by using our diagnostic algorithm. Moreover, we proved that the assay is robust, since the test gave an inconclusive result only for the 0.2% of the samples. For analyzing with A1A Genotyping Test, DNA was extracted from DBS with different methods (2 standard, and 1 in‐house) and only in one case the result was invalid. DNA for this sample had been extracted with the BSA/Azide method though having given a worse outcome than the rest of the samples for which DNA had been extracted with standard methods. Finally, the examined test allows faster analysis and its results are easy for interpretation. Nevertheless, the conventionally used term “M/M,” if none of the 14 allelic variants included in the test have been detected, is confounding for the reader. We therefore suggest to clearly explain it in the final report to eliminate any doubt.
Allelic variants included in the A1AT GT (Progenika, Grifols) are chosen to offer worldwide coverage. Therefore, among the 14 variants detectable by the test, some (Siyama18 and Q0west,19 in particular) are completely absent in the European population. On the other hand, mutations that are frequently seen in Caucasian‐descent population (Mwurzburg20 and V21 that can be detected by IEF, in particular) are not included in the kit panel. Accordingly, in certain cases (4%), it has been necessary to sequence the SERPINA1 gene to identify other rare mutations found in the samples analyzed in the Italian reference Center and not included in the assay.
Although some allelic variants detected by sequencing were not included in the test, we calculated that the A1AT GT assay could allow us to decrease the rate of samples to be sequenced in the Italian reference center and as a result, it reduces the diagnostic time frame. Indeed, for those samples in which the results agree, and, consequently, they do not need sequencing, the diagnostic time is reduced by 66%: the reason is that sequencing is a quite long method, as it takes at least 3 days between sample preparation, loading in the instrument and reading of each sequence. On the contrary, the A1AT GT test can be performed in a single day. Regarding the comparison of costs, it was not the matter of the present paper; due to the importance of this theme, it could be the object for future investigations.
In conclusion, the A1AT GT assay is a robust and efficient method to quickly diagnose most of deficient variants of SERPINA1 gene. The customization of the variant panel would be welcome to improve diagnostic efficiency on a country‐based view.
Ottaviani S, Barzon V, Buxens A, et al. Molecular diagnosis of alpha1‐antitrypsin deficiency: A new method based on Luminex technology. J Clin Lab Anal. 2020;34:e23279 10.1002/jcla.23279
Funding information
The Center for Diagnosis of Inherited Alpha1‐antitrypsin Deficiency, Laboratory of Biochemistry and Genetics, Institute for Respiratory Disease, Department of Internal Medicine and Therapeutics, University of Pavia, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy is supported by Grifols and CSL Behring. This study has been also supported by grant of Fondazione Cariplo to AGC (#2013‐0967) and by Fondazione IRCCS Policlinico San Matteo—Ricerca Corrente (RC721). All authors have read the journal's policy on disclosure of potential conflicts of interest. None of the authors has personal or financial relationship with organizations that could potentially be perceived as influencing the described research. A. Buxens, A. Larruskain and R. El Hamss are employees of Progenika Biopharma‐Grifols.
REFERENCES
- 1. Greene CM, Marciniak SJ, Teckman J, et al. α1‐Antitrypsin deficiency. Nat Rev Dis Primers. 2016;2:16051. [DOI] [PubMed] [Google Scholar]
- 2. Janciauskiene SM, Bals R, Koczulla R, Vogelmeier C, Köhnlein T, Welte T. The discovery of alpha 1 antitrypsin and its role in health and disease. Respir Med. 2011;105(8):1129‐1139. [DOI] [PubMed] [Google Scholar]
- 3. Ferrarotti I, Thun GA, Zorzetto M, et al. Serum levels and genotype distribution of α1‐antitrypsin in the general population. Thorax. 2012;67(8):669‐674. [DOI] [PubMed] [Google Scholar]
- 4. Alpha1‐antitrypsina deficiency: memorandum from WHO meeting. Bull World Health Organ. 1997;75(5):397‐415. [PMC free article] [PubMed] [Google Scholar]
- 5. American Thoracic Society/European Respiratory Society . American Thoracic Society/European Respiratory Society statement: standards for the diagnosis and management of individuals with alpha‐1antitrypsin deficiency. Am J Respir Crit Care Med. 2003;168(7):818‐900. [DOI] [PubMed] [Google Scholar]
- 6. Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in a1‐antitrypsin deficiency. Eur Respir J. 2017; 50 (5): pii:1700610. [DOI] [PubMed] [Google Scholar]
- 7. Giacopuzzi E, Laffranchi M, Berardelli R, et al. Real‐world clinical applicability of pathogenicity predictors assessed on SERPINA1 mutations in alpha‐1‐antitrypsin deficiency. Hum Mutat. 2018;39(9):1203‐1213. [DOI] [PubMed] [Google Scholar]
- 8. Lomas DA, Parfrey H. Alfa1‐Antitrypsin deficiency * 4: molecular pathophysiology. Thorax. 2004;59(6):529‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rodriguez‐Frias F, Miravitlles M, Vidal R, Camos S, Jardi R. Rare alpha‐1‐antitrypsin variants: are they really so rare? Ther Adv Respir Dis. 2012;6(2):79‐85. [DOI] [PubMed] [Google Scholar]
- 10. De Serres FJ, Luisetti M, Ferrarotti I, Blanco I, Fernández‐Bustillo E. Alpha‐1 antitrypsin deficiency in Italy: regional differences of the PIS and PIZ deficiency alleles. Monaldi Arch Chest Dis. 2005;63(3):133‐141. [DOI] [PubMed] [Google Scholar]
- 11. Ferrarotti I, Baccheschi J, Zorzetto M, et al. Prevalence and phenotype of subjects carrying rare variants in the Italian Registry for Alpha1‐antitrypsin deficiency. J Med Genet. 2005;42(3):282‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Campbell EJ. Alpha1‐antitrypsin deficiency: incidence and detection program. Respir Med. 2000;94(Suppl C):S18‐S21. [DOI] [PubMed] [Google Scholar]
- 13. Miravitlles M, Herr C, Ferrarotti I, et al. Laboratory testing of individuals with severe alpha1‐antitrypsin deficiency in three European centres. Eur Respir J. 2010;35(5):960‐968. [DOI] [PubMed] [Google Scholar]
- 14. Ferrarotti I, Carroll TP, Ottaviani S, et al. Identification and characterization of eight novel SERPINA1 Null mutations. Orph J Rare Dis. 2014;9:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferrarotti I, Scabini R, Campo I, et al. Laboratory diagnosis of alpha1‐antitrypsin deficiency. Transl Res. 2007;150(5):267‐274. [DOI] [PubMed] [Google Scholar]
- 16. Gorrini M, Ferrarotti I, Lupi A, et al. Validation of a rapid and simple method to measure a1‐antitrypsin. Clin Chem. 2006;52(5):899‐901. [DOI] [PubMed] [Google Scholar]
- 17. Matamala N, Lara B, Gomez‐Mariano G, et al. Characterization of novel missense variants of SERPINA1 causing alpha1‐antitrypsin deficiency. Am J Respir Cell Mol Biol. 2018;58(6):706‐716. [DOI] [PubMed] [Google Scholar]
- 18. Takabe K, Seyama K, Shinada H, et al. A new variant of alpha‐1‐antitrypsin deficiency (Siiyama) associated with pulmonary emphysema. Intern Med. 1992;31(5):702‐707. [DOI] [PubMed] [Google Scholar]
- 19. Laubach VE, Ryan WJ, Brantly M. Characterization of a human alpha 1‐antitrypsin null allele involving aberrant mRNA splicing. Hum Mol Genet. 1993;2(7):1001‐1005. [DOI] [PubMed] [Google Scholar]
- 20. Poller W, Merklein F, Schneider‐Rasp S, et al. Molecular characterisation of the defective a1‐antitrypsin alleles PI Mwurzburg (Pro369Ser), Mheerlen (Pro369Leu), and Q0lisbon (Thr68lle). Eur J Hum Genet. 1999;7(3):321‐331. [DOI] [PubMed] [Google Scholar]
- 21. Faber JP, Poller W, Weidinger S, et al. Identification and DNA sequence analysis of 15 new a1‐antitryspin variants, including two PI*Q0 alleles and one deficient PI*M allele. Am J Hum Genet. 1994;55(6):1113‐1121. [PMC free article] [PubMed] [Google Scholar]