

Abstract
Objective:
Coronavirus disease 2019 (COVID-19) is a global pandemic involving >5 500 000 cases worldwide as of May 26, 2020. The culprit is the severe acute respiratory syndrome coronavirus-2, which invades cells by binding to ACE2 (angiotensin-converting enzyme 2). While the majority of patients mount an appropriate antiviral response and recover at home, others progress to respiratory distress requiring hospital admission for supplemental oxygen. In severe cases, deterioration to acute respiratory distress syndrome necessitating mechanical ventilation, development of severe thrombotic events, or cardiac injury and dysfunction occurs. In this review, we highlight what is known to date about COVID-19 and cardiovascular risk, focusing in on the putative role of the endothelium in disease susceptibility and pathogenesis.
Approach and Results:
Cytokine-driven vascular leak in the lung alveolar-endothelial interface facilitates acute lung injury in the setting of viral infection. Given that the virus affects multiple organs, including the heart, it likely gains access into systemic circulation by infecting or passing from the respiratory epithelium to the endothelium for viral dissemination. Indeed, cardiovascular complications of COVID-19 are highly prevalent and include acute cardiac injury, myocarditis, and a hypercoagulable state, all of which may be influenced by altered endothelial function. Notably, the disease course is worse in individuals with preexisting comorbidities that involve endothelial dysfunction and may be linked to elevated ACE2 expression, such as diabetes mellitus, hypertension, and cardiovascular disease.
Conclusions:
Rapidly emerging data on COVID-19, together with results from studies on severe acute respiratory syndrome coronavirus-1, are providing insight into how endothelial dysfunction may contribute to the pandemic that is paralyzing the globe. This may, in turn, inform the design of biomarkers predictive of disease course, as well as therapeutics targeting pathogenic endothelial responses.
Keywords: biomarkers, cardiac injury, coronavirus, endothelium, pandemic
Highlights.
Preexisting chronic endothelial dysfunction coupled with virus-mediated endothelial activation, permeability, and hypercoagulable state is likely a driving factor in Coronavirus disease 2019 (COVID-19) severity.
Compensatory over-expression of ACE2 (angiotensin-converting enzyme 2) in the vasculature of patients with cardiovascular comorbidities may increase viral infection and dissemination.
The endothelium may be considered as a linchpin in COVID-19 pathogenesis; blocking viral binding to ACE2 receptors on the endothelium and preventing capillary barrier disruption and the hypercoagulable state should be explored as therapies for COVID-19.
Emerging and re-emerging zoonotic diseases represent a persistent and unpredictable threat to global health. In the last 2 decades, the world has witnessed 2 major epidemics from genetically distinct and highly infectious betacoronaviruses; severe acute respiratory syndrome coronavirus (SARS-CoV-1, 2002) and middle east respiratory coronavirus (MERS-CoV, 2012).1 Although causing respiratory, enteric, hepatic, and neurological diseases of varying severity, the cardiovascular complications associated with infection have been underappreciated until recently.
In early December 2019, China reported multiple cases of pneumonia of unknown etiology occurring in Wuhan, Hubei, China.2 By early January, viral sequencing identified the causative agent as a novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), with a 75% to 80% sequence identity to SARS-CoV-1. Between the index case in early December and May 26, 2020, >5 500 000 cases and >300 000 deaths due to coronavirus disease 2019 (COVID-19) were reported worldwide. Similar to SARS-CoV-1 and MERS-CoV, the spectrum of clinical presentation can range from asymptomatic infection to severe disease. Early clinical studies revealed that initial symptoms of COVID-19 are consistent with viral pneumonia, most commonly fever (≈88%–89%), cough (≈57%–68%), and myalgia or fatigue (≈36%).3 Although a majority of individuals seem to mount a controlled and appropriate viral response, others decompensate into respiratory distress requiring hospital admission for supplemental oxygen, or worse, develop acute respiratory distress syndrome (ARDS) necessitating mechanical ventilation and intensive care unit level care. Early meta-analyses of COVID-19 patient clinical characteristics revealed that ≈12% to 20% of all cases require intensive care, with the case fatality rate of admitted patients ranging from ≈4% to 26%.4,5 While the cause of death is largely related to ARDS, there is a strong correlation between markers of cardiac injury and mortality, with up to 7% dying as a result of substantial myocardial damage and 33% dying of combined cardiopulmonary and cardiovascular failure.6–9 Additionally, thrombotic events, both venous and arterial in nature, have been commonly observed in patients with COVID-19, further highlighting that cardiovascular complications are likely a strong predictor and contributor to the high mortality rate.8,10,11
Although rapidly evolving, our knowledge of COVID-19 pathophysiology remains incomplete. Consequently, our understanding of cardiovascular biology and the role of the endothelium is seen through the lens of lessons from SARS-CoV-1 and MERS-CoV, as well as emerging COVID-19 pathology reports.11,12 While elements of this review may be speculative regarding the role of the endothelium in COVID-19, a case series showing that SARS-CoV-2 infects endothelial cells causing endotheliitis,12 has begun to shed light on the unique pathogenesis of COVID-19 (Figure 1). Importantly, in this review, we endeavor to highlight research areas that will further our understanding and may lead to the development of therapies that address endothelial dysfunction.
Figure 1.
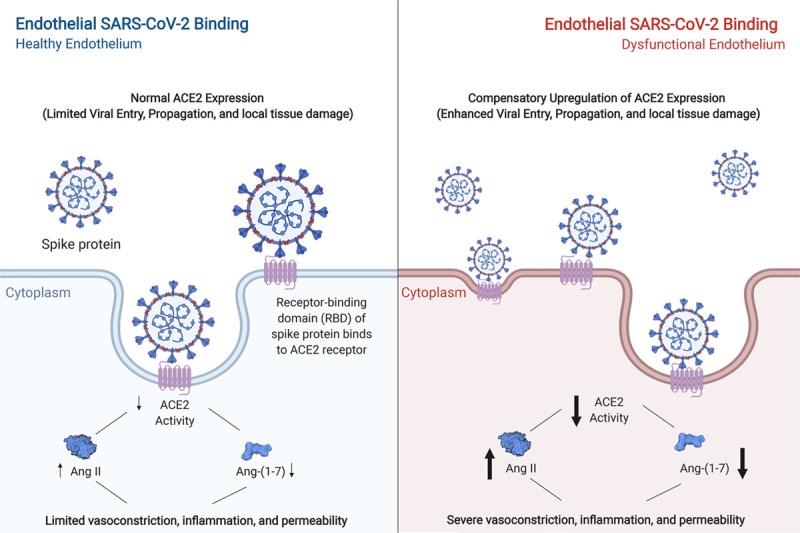
Proposed model of the pathological effect of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) on the endothelium. Left, under conditions of endothelial quiescence, binding of SARS-CoV-2 is mediated in part by ACE2 (angiotensin-converting enzyme 2). Internalization of ACE2, and subsequent receptor interference, results in an upregulation of AngII (angiotensin II) and downregulation of Ang-(1-7) (angiotensin-[1-7]). Although contributing to vasoconstriction, inflammation, and permeability, limited expression of ACE2 within a relatively quiescent endothelium may result in limited viral entry and local and systemic dysfunction. Right, paradoxically, although a dysfunctional endothelium may have higher baseline levels of ACE2, enhanced viral entry may increase the degradation of ACE2 in the lysosome, enhancing inflammation and tissue damage. The resulting downregulation of ACE2 activity, upregulation of Ang-II, and downregulation of Ang-(1-7) thereby may have a starker induction of vasoconstriction, inflammation, and permeability as compensatory mechanisms are abrogated. Arrow width corresponds to intensity. This figure was created with the assistance of www.BioRender.com.
Viral Entry to Endothelial Cells and the Role of ACE2
At the cellular level, the pulmonary endothelium forms a key part of the alveolar-capillary unit, providing an interface for efficient gas exchange between the alveolar space and red blood cells within the capillaries of the lung. Under homeostatic conditions, the endothelial barrier is maintained through interactions between adjacent endothelial cells (ECs) via intercellular junctions—tight junctions and adherens junctions—that are tethered to the actin cytoskeleton.13 As a consequence of severe viral infection of the lower respiratory tract epithelium, there is attendant dysfunction and destruction of alveolar epithelia, with loss of junctional complexes between ECs leading to enhanced permeability of the capillary endothelium and edema in the lung. This is mediated in part by the impact of proinflammatory cytokines (referred to as cytokine storm) that are produced during the antiviral innate immune response.14 Subsequent loss of surfactant production, together with alveolar collapse and fluid accumulation, leads to acute lung injury, pneumonia, and ARDS: which are common complications of viral infection. Clinical studies have revealed that ≈29% of hospitalized patients diagnosed with COVID-19 develop ARDS, with up to 32% of these patients requiring intensive care.15,16 Concordant upregulation of EC inflammatory cascades promotes leukocyte recruitment, further driving EC activation, as well as activating coagulation pathways.14 Indeed, the endothelium has been shown to be a central orchestrator of cytokine storm and tissue damage during lung viral infections.17
Strategically located between the circulation and other vascular layers, the endothelium is responsible for dictating niche-specific vascular homeostasis through the balanced release of autocrine and paracrine molecules. In conditions of dysfunction, this balance is disrupted, making the endothelium susceptible to vasoconstriction, platelet activation, thrombosis, and vascular inflammation, among other detrimental effects. Fundamental features of EC dysfunction include reduced local production of the vasodilator NO (nitric oxide), decreased anticoagulant factors (eg, heparan and dipeptidyl peptidase-4), increased secretion of vWF (von Willebrand Factor) and tissue factor, upregulation of leukocyte adhesion molecules (eg, intercellular adhesion molecule 1, E-Selectin, P-Selectin), and increased generation of reactive oxygen species; all of which lead to compromised vascular homeostasis and inflammation.18–21
Among pathways responsible for the development of endothelial dysfunction, the renin-angiotensin-aldosterone system (RAAS) is a pivotal signalling axis. RAAS is a major regulator of blood pressure, fluid and electrolyte balance, cardiovascular homeostasis, and facilitates a complex series of enzymatic reactions that culminate in the generation of Ang II (angiotensin II); a potent vasoconstrictor and proinflammatory effector molecule.22,23 Although the initial conversion of Ang I to Ang II by angiotensin-converting enzyme 1 represents an important process in cardiovascular signalling, it is the negative regulator of this process, ACE2 (angiotensin-converting enzyme 2), which warrants in-depth examination in COVID-19. ACE2, a type I transmembrane zinc metallocarboxypeptidase, counterbalances angiotensin-converting enzyme 1 by converting Ang I and Ang II into Ang-(1-9) and Ang-(1-7), respectively, facilitating the attendant cardioprotective actions of NO release, vasodilation, and blunting of inflammation.24 The ability of ACE2 to provide negative regulation of Ang II and the receptor (angiotensin II receptor type 1) is of particular biological significance in pathological conditions where RAAS may be dysfunctional. In line with these observations, ACE2 broadly influences the cardiovascular function of multiple organs including the systemic vasculature, kidneys, liver, heart, and lungs through its broad expression in arterial and venous ECs, as well as vascular smooth muscle.25
Given its integral involvement in the regulation of cardiovascular function, it is unsurprising that ACE2 is dysregulated in numerous cardiovascular pathologies. Several clinical and experimental observations have suggested a beneficial role for ACE2 in cardiovascular function; yet, paradoxically, ACE2 expression is elevated at various stages of cardiovascular pathologies.25 Among comorbidities prevalent in the general population, diabetics have increased levels of circulating ACE2 when micro- and/or macrovascular disease is present, suggesting that the upregulation of ACE2 may act as a compensatory mechanism in maintaining homeostatic vascular function as a means to counteract RAAS activation.26,27 Increases in ACE2 have also been observed in cardiac tissue of patients with ischemic heart failure, idiopathic dilated cardiomyopathy, and pulmonary hypertension.28,29
In the setting of SARS-CoV-2 infection, enhanced levels of ACE2 may facilitate viral entry into multiple tissues (Figure 2).30 Recent cryo-electron microscopy studies have demonstrated that similar to SARS-CoV-1, the major spike glycoprotein (S1) of SARS-CoV-2 is capable of direct binding to ACE2, albeit with a 10- to 20-fold greater affinity.31–33 Although ACE2 has a seemingly prominent role in mediating viral entry as well as dictating cardiovascular phenotype, several other co-receptors/factors are likely involved in the process including transmembrane serine protease type II,34 which is expressed on ECs35,36 and plays a crucial role in viral entry by cleaving the viral spike protein into functional S1 and S2 subunits. While SARS-CoV-2 initially infects epithelial cells in the upper and lower respiratory tracts by binding to ACE2, viral dissemination to other organs requires the virus to cross the endothelium and enter the bloodstream. Indeed, SARS-CoV-2 RNA can be detected in the blood of patients that progress to severe disease and SARS-CoV-1 and SARS-CoV-2 have been shown to infect ECs, which are known to express ACE2 on their surface.37–40 Importantly, previous preclinical studies with the H5N1 virus revealed that blocking viral replication in the endothelium limits systemic viral spread, disease symptoms, and mortality, emphasizing the importance of infection of the endothelium in viral dissemination.41 Intriguingly, viral entry leads to internalization and degradation of ACE2, reducing its bioavailability, which may precariously reduce the much-needed activity of ACE2 in an already strained system.30,34,42 Indeed, in animal models, loss of ACE2 activity has been linked to decreased cardiac contractility (reduced systolic function), coronary vasoconstriction, microcirculatory dysfunction, and reductions in myocardial blood flow,25,28 while in the context of lung injury, loss of ACE2 results in worsened lung function, increased inflammatory cell infiltration, enhanced microvascular permeability, and decreased blood oxygenation.43
Figure 2.
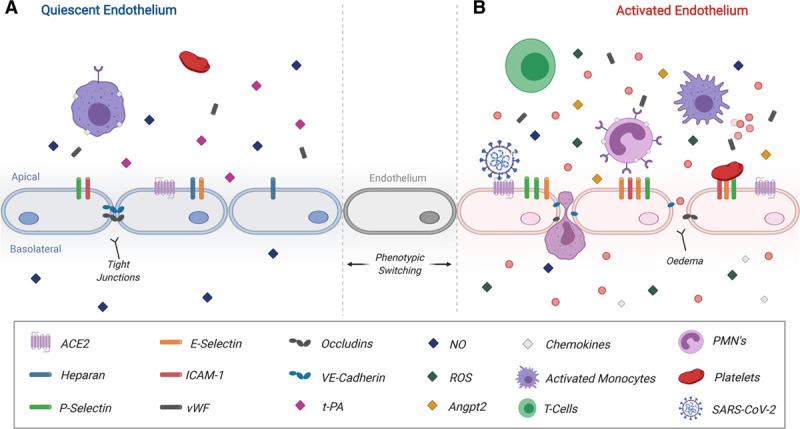
Proposed pathobiology of activated endothelium during severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2 infection). A, A quiescent endothelium is shown on the left. Endothelial quiescence facilitates normal responses to angiogenic signalling, facilitates controlled immune surveillance, and provides homeostatic cues for coagulation. B, Chronic activation of the endothelium is marked by stark upregulation of P-selectin, E-Selectin, ICAM-1, and ACE2 (angiotensin-converting enzyme 2), along with their respective soluble forms. The subsequent loss of tight junction expression expedites edema and facilitates enhanced recruitment, attachment, and extravasation of immune cells across the vascular endothelium. Disruption of coagulation cues through the loss of heparan, DDP-4, and t-PA (tissue-type plasminogen activator) and secretion of vWF (von Willebrand Factor) exacerbates endothelial injury and induces coagulopathies. Further disruption of endothelial phenotype results in local reductions of secreted NO, increases in secreted reactive oxygen species (ROS), and enhanced Angpt2 secretion. SARS-CoV-2 reduces ACE2 availability further propagating endothelial dysfunction. While intended to assist in infection control, innate and adaptive immune responses can instead induce a storm of chemokines and cytokines, which further propagates underlying inflammation and dysfunction. This figure was created with the assistance of www.BioRender.com. ICAM-1 indicates intercellular adhesion molecule; NO, nitric oxide; PMN, polymorphonuclear leukocyte; and VE-Cadherin, vascular endothelial cadherin.
COVID-19 and the Endothelium in Vascular and Cardiac Complications
Although COVID-19 is primarily considered a respiratory infection, it has important systemic effects on the cardiovascular system. Similar to other viral infections (eg, influenza, SARS-CoV-1, and MERS-CoV), SARS-CoV-2 infection seems to more readily predispose individuals with comorbidities to cardiopulmonary failure and death; perhaps as a result of existing dysregulation of the RAAS.44–46 In several large cohort studies, the clinical prevalence of these comorbidities among confirmed COVID-19 patients ranged between 8.3% and 10.5%; 7.3% for diabetes mellitus, 6.5% to 15.0% for hypertension, 2.5% for coronary artery diseases, and 1.4% for cerebrovascular disease.47 Recent reports have described a high prevalence of at least one comorbidity among intensive care unit-admitted COVID-19 patients, ranging from ≈68% (Italian cohort) to ≈72% (Chinese cohort).16,48 Hypertension and diabetes mellitus seem to be the most common, followed by other cardiovascular and pulmonary comorbidities.49 This has raised the question as to whether these comorbidities share feature(s) that put individuals at higher risk.50,51 As a result of these observations, collaborative efforts such as the COVID-19 host genetics initiative have been launched to better understand the role of host factors on COVID-19 susceptibility and severity.52 In line with epidemiological observations, there are noted changes in RAAS with these conditions,21 where the earliest stages of these disease conditions are often marked by structural and functional changes of the endothelium into a dysfunctional state (Figure 2).53
Similar to other viral outbreaks, worsening prognosis and increased adverse cardiovascular events associated with COVID-19 have been linked to the severity of inflammation and resulting cardiac dysfunction.54 Observed abnormalities from laboratory tests, including elevation of cardiac-centric molecules such as troponins and BNP, as well as increases in endothelial-centric factors such as vWF and D-dimers have been reported in patients with COVID-19, reflective of cardiac damage and increased thrombosis risk, respectively.3–5 Although the underlying mechanisms remain elusive, the importance of inflammation and their procoagulative effects highlight the need to focus on the role of the endothelium in virally induced cardiovascular pathology. While cardiac outcomes are prominent in older patients with COVID-19 with antecedent cardiovascular risk including hypertension, diabetes mellitus, and previous stroke, cardiac effects have also been observed in young and otherwise healthy patients with no known comorbidities.55 A key clinical question is whether the virus (1) directly infects cells within the heart, including the vascular endothelium, to cause pathology; (2) impairs cardiac function indirectly through pulmonary insufficiency and increased cardiac load; and/or (3) damages the heart via the host response (ie, cytokine storm), inducing systemic inflammation and disseminated intravascular coagulation (DIC). Indeed, it may be that all of these scenarios occur simultaneously.
Although ACE2 expressed on the pulmonary epithelium facilitates viral entry, the antiviral response—including contributions by the pulmonary endothelium—may subsequently drive systemic cytokine storm, whereby ACE2 expressed on cells of the cardiovascular system may facilitate viral propagation and subsequent dissemination to the heart.17 Notably, data from a small number of SARS-CoV-1 patients demonstrated that 35% of autopsied heart tissue had evidence of viral infection in the heart, with an average viral load of 4×106 copies/gram of tissue.56 Importantly, there was also evidence of significantly decreased ACE2 expression in these hearts, implying that viral infection impairs ACE2-dependent functions.56 Patients with COVID-19 and cardiac involvement had a significantly shorter duration of illness, suggestive of a more aggressive course.57 It is possible that ACE2 expression in either the endothelium or myocardium of the heart may correlate with the severity of COVID-19 infection. This will require detailed pathological analysis of postmortem tissues.56 Unfortunately, there are scant pathology reports from patients with COVID-19 at this time. Of the available evidence, results of biopsies from multiple tissues in 3 patients who died from COVID-19 reported that blood vessels in the lung were congested, edematous with immune cell infiltration, and had hyaline thrombi in a small number of microvessels.58 It is unclear whether the virus directly infects cells in the heart. Interestingly, single-cell sequencing datasets have revealed that 7.5% of myocardial cells express the mRNA that encodes for the ACE2 receptor.59 Additionally, another recent single-cell atlas of abandoned donor hearts demonstrated high ACE2 mRNA expression in pericytes with the dominant EC-pericyte crosstalk between EC receptors and pericyte ligands suggesting that SARS-CoV-2 may affect the cardiac microvasculature.60 Perhaps the most convincing experimental evidence to date of SARS-CoV-2 infection of the vasculature was shown by Monteil et al,39 who infected capillary organoids containing endothelial and pericyte cells with SARS-CoV-2 and showed that not only can viral RNA be recovered post-infection but also that progeny virus captured from supernatants are capable of infecting VeroE6 cells. Recent clinical evidence has also emerged with pathology reports demonstrating the presence of virions within ECs in conjunction with substantial endothelial cell inflammation and death.12,61 Endotheliitis as a direct consequence of viral infection may therefore explain the systemic impairment of microcirculatory function in different vascular beds, and the unique cardiovascular sequelae in patients with COVID-19.12,61
Although many reports of cardiovascular complications in the adult population appeared in the early weeks of the COVID-19 pandemic, reports of acute vasculitis and hyperinflammatory shock, suggestive of atypical Kawasaki disease, have been emerging in the pediatric population.62 Importantly, these pediatric cases reported elevated levels of C-reactive protein and D-dimers associated with COVID-19 exposure, despite initial negative tests for SARS-CoV-2, and alarmingly describe severe cardiovascular manifestations such as a fatal arrhythmia, and giant coronary aneurysms.63 Although the etiology of Kawasaki disease is largely unknown, existing evidence points toward an infectious trigger, with studies describing associations between viral respiratory infections and the presentation of the disease.64,65 Of relevance, during SARS-CoV-1, a case series of 3 patients described the findings of systemic vasculitis and infiltration of immune cells into vessel walls of numerous organs.66 Although the mechanism may be the same, with microangiopathy of vessels, there is no direct evidence to date uncovering the role of SARS-CoV-2 in Kawasaki disease.
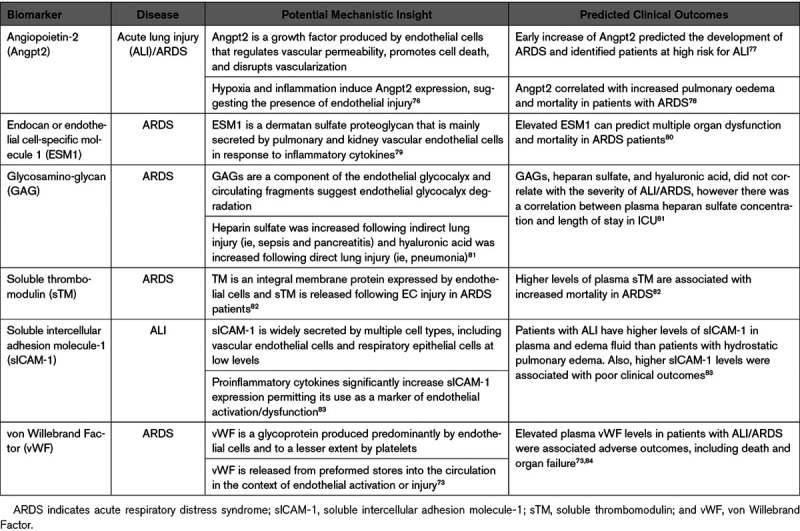
With the abundance of emerging evidence, it is likely that cardiac and vascular biomarkers could inform mechanisms of COVID-19 pathogenesis (Table 1). Several cardiac biomarkers have been found to be elevated in patients with COVID-19 and importantly, they portend substantially worse outcomes. Zhou et al10 observed an early increase in cardiac structural proteins including troponin I in nonsurvivors compared with survivors. In a cohort study from Wuhan on 416 hospitalized patients, 19.7% had elevated troponin I with a higher mortality rate (51.2%) compared with those with normal troponin levels (4.5%), illustrating the significantly worse outcomes associated with higher troponin I.6 In-hospital mortality for patients with elevated troponin T coupled with a history of cardiovascular disease was the highest at 69%.8 A recent case report described progressively elevated levels of vWF antigen and activity in a patient during clinical deterioration and onset of COVID-19-induced ARDS, suggestive of EC activation or damage.74 At this point, no endothelial-specific biomarkers have been routinely measured, apart from laboratory values that reflect generalized endothelial dysfunction such as D-dimers and DIC-related tests.75 It is however notable that markers of systemic inflammation (eg, CRP, IL-6) are elevated in those with poor outcomes, as this may imply the presence of EC activation.15,16,55 Together, the cardiac and generalized inflammatory biomarkers may provide useful indices across which endothelial-specific biomarkers could be compared. Assessment of endothelial-specific markers of dysfunction in animal models and clinical samples would provide insight into the contribution of EC dysfunction to disease course (Table 2). A starting point exists in the sepsis and infectious disease literature, for which there are several putative endothelial biomarkers, including Angpt2 (Angiopoietin-2), vWF, ADAMTS13 (a disintegrin-like and metallopeptidase with thrombospondin type 1 motif, 13), thrombomodulin, soluble E-selectin/ICAM-1 (intercellular adhesion molecule 1), VCAM-1 (vascular cell adhesion molecule 1), and VEGF (vascular endothelial growth factor), some of which have been linked to mortality in ARDS (eg, Angpt2; Table 3).85
Table 1.
Emerging Biomarkers of Poor Cardiovascular Outcomes in COVID-19
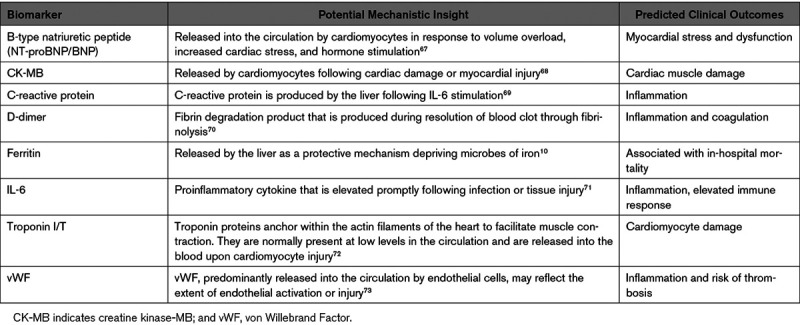
Table 2.
Unanswered Questions Regarding the Role of the Endothelium in COVID-19 Pathogenesis
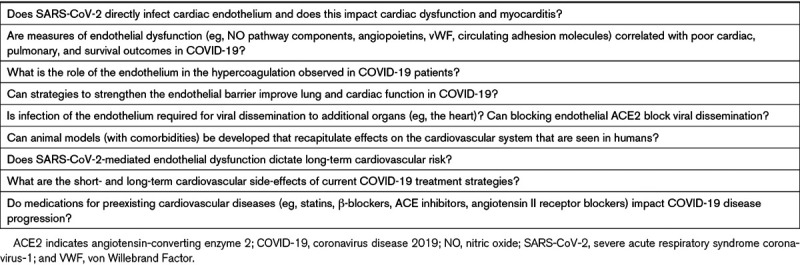
Table 3.
Circulating Endothelial-Derived Biomarkers That Predict the Development and/or Outcome of ARDS
COVID-19 and the Endothelium in Hemostasis and Thrombosis
There is a shared etiology of generalized endothelial dysfunction in driving COVID-19 related cardiac and vascular complications. Specifically, hypercoagulable events—both venous and arterial—have been observed in patients with COVID-19, and an elevation of circulating D-dimers has been correlated with the severity of disease and poor outcomes.86,87 While endothelial-specific biomarkers have not been broadly assessed in COVID-19, endothelial dysfunction in the setting of cytokine storm is likely a contributing factor to the hypercoagulable state and DIC reported in these patients (Table 3).88 Indeed, plasma markers of endothelial dysfunction are associated with mortality and severity of coagulopathy in patients with sepsis-associated DIC.89 Clinical observations have shown that mortality is higher for patients with COVID-19 with abnormal coagulation profiles, including elevated D-dimers and fibrin degradation products, with 71% of nonsurvivors meeting the criteria for DIC compared with 0.6% of survivors.90 Importantly, elevated levels of vWF activity, D-dimers, and fibrin degradation products point toward a severe state of hypercoagulability rather than consumptive coagulopathy, as seen in classical DIC.91 In support of this, analysis of critically ill patients from both Dutch and French cohorts has revealed a relatively high number of thrombotic events (adjusted cumulative incidence of up to 49% in the Dutch cohort), with the majority of events being pulmonary emboli.11,92 Notably, deep venous thrombosis (DVT) was found in 58% of patients (n-value of 12) in a recent prospective autopsy study in which it was not suspected before death—perhaps suggesting the need for routine DVT screening in this patient group.93 Although few cardiovascular imaging studies have been done to validate these findings, thoracic CT scans of patients with COVID-19 have described vascular thickening, vascular enlargement, or vascular congestion.94 Large vessel arterial occlusions causing strokes have also been reported in young, otherwise healthy adults.95 Importantly, Wang et al96 describe a prothrombotic DIC with high rates of venous thromboembolism, as well as micro- and macrovascular arterial thromboses (eg, stroke, acute limb ischemia) in critically ill patients with COVID-19 necessitating off-label use of tissue-type plasminogen activator treatment. Recent case reports and working hypotheses have additionally highlighted the role of inflammatory dysregulation in the development both micro- and macrovascular thrombosis.97–99 Although there is no direct evidence of a link between observed hypercoagulability and the endothelium, increased vWF alone points toward massive endothelial stimulation.74 It is additionally unclear whether this dysregulation involves the ACE2 receptor or co-receptors, direct effects of virus entry on the endothelium, an unknown vulnerability of the patient (eg, thrombophilia), or whether it merely reflects the collateral damage of an overwhelming host response. Regardless of how the system becomes dysregulated, it seems clear that endothelial dysfunction—both preexisting and acute—resulting from multiple damaging hits likely plays a role in the severity of COVID-19.
Modulating the Endothelium As a Potential Therapy for COVID-19
Despite a number of fast-tracked clinical trials testing existing antiviral and anti-inflammatory approaches for COVID-19, suitable preclinical models will be instrumental to understand cardiovascular complications and to develop novel treatments for COVID-19.100 Nonhuman primates are permissive to SARS-CoV-1 and SARS-CoV-2 infection, and while they develop mild lung pathology, the studies are typically short-term and have not assessed cardiovascular involvement.101,102 Interestingly, aged macaques develop an exaggerated innate immune response in response to SARS-CoV-1 infection, suggesting that comorbidities may be required to better model severe outcomes, and this may also be the case for cardiovascular pathology.102 Small animals, particularly transgenic murine models that express human ACE2 can be infected with SARS-CoV-1 and SARS-CoV-2, and mouse-adapted strains of SARS-CoV-1 can cause lethal infection, but pathological effects on the cardiovascular system have not been observed.38,103 Finally, while a hamster model is susceptible to SARS-CoV-1 and SARS-CoV-2 infection, they develop only mild cardiac disease and no mortality.38 Interestingly, an immunocompromised hamster model, which may better model the lymphocytopenia observed in human patients with severe symptoms, showed evidence of multiorgan pathology, including viral replication and myocardial inflammation as well as significant mortality.104 Considering existing evidence, the close association of underlying cardiovascular comorbidities with adverse outcomes of COVID-19—particularly cardiovascular outcomes—makes assessment of infection in rodent models of chronic comorbid disease a more likely source for valuable, clinically translatable information.
The implied necessity of SARS-CoV-2 to infect or transit the endothelium in an ACE2-dependent manner for dissemination outside the lungs to drive cardiovascular pathology represents a potential viral vulnerability. Proof of principle for the efficacy of ACE2 inhibition was provided with in vitro models of SARS-CoV-1, where small molecule inhibitors were able to inhibit SARS-CoV-1 entry by interfering with ACE2 binding and fusion.105 Recombinant human soluble ACE2 was also recently shown to suppress SARS-CoV-2 infection of engineered human blood vessel organoids.39 Additional strategies modulating the endothelium include preserving EC barrier function in the lung (and potentially other tissues) to preserve organ function, limiting systemic inflammatory responses, and impeding coagulation pathways, thereby limiting DIC. Importantly, therapies targeting cytokine storm have been largely unsuccessful in sepsis. This is in part due to the nuanced and biphasic role of inflammation in pathogenesis, making it difficult to determine the optimal timing of treatment.106 This will likely also be the case with COVID-19, despite emerging favorable reports.107–109 An alternative approach has been to utilize therapies that are intended to strengthen the endothelial barrier to withstand the detrimental effects of cytokine storm. Such approaches are effective in preclinical models of sepsis and influenza.110,111 Thus, therapies that preserve endothelial barrier function may improve the clinical features of ARDS and may limit systemic cardiovascular damage. Given that maintenance of barrier function in ECs is driven by cell surface and adaptor proteins, extracellular matrix, and the actin cytoskeleton, the opportunities for therapeutic design are plentiful. Candidate therapies include sphingosine-1-phosphate, Angiopoietin-1/Tie2, statins, interferon-β-1a, atrial natriuretic peptide, human recombinant soluble ACE2, and heparin, among others.112–116
Long-Term Effects of Coronavirus Infection on Cardiovascular Disease
Although significant resources and effort are being poured into addressing the immediate infection rate and reducing acute mortality, the long-term effects of COVID-19 on the cardiovascular system should be considered. Viral-mediated pneumonia is known to elicit long-term cardiovascular complications.117 Hospitalization with pneumonia has been associated with a subsequent increase in the risk of cardiovascular disease, with up to a 4-fold increase in the first 30 days, and 1.5-fold increase years later.118 Moreover, the magnitude of risk for cardiovascular disease events associated with pneumonia was similar or higher compared with the risk of cardiovascular disease associated with traditional risk factors, such as smoking, diabetes mellitus, and hypertension.118 Experimental models of infection have pointed toward the induction of strong proinflammatory signaling leading to changes in cellular composition of atherosclerotic lesions making individuals more susceptible to coronary and cerebrovascular events.119 Although studies of long-term follow-up in coronavirus patients indicate cardiovascular disease and altered metabolomics, the data are limited.120 Nonetheless, the connections between systemic viral infection, endothelial ACE2 dysregulation, and postinfection cardiovascular outcomes warrant investigation.
Conclusions and Perspectives for Future Research
The endothelium plays a key role in the pathogenesis of infectious diseases, contributing to dysfunction of inflammation, vascular permeability, coagulation, and immune activation. Inferences from influenza, SARS-CoV-1, and MERS-CoV, although limited in their examinations of the endothelium, provide valuable pathophysiological insights into SARS-CoV-2. The abundance of ACE2 in the cardiovascular system and the inherent role of the endothelium as a gatekeeper to other organs highlights its potentially critical role as a mediator of disease severity, but many questions remain (Table 2). Emerging literature continues to suggest a key role for dysregulated endothelial function in SARS-CoV-2 infection. Detailed investigations are needed to unravel the complex interplay between SARS-CoV-2, the RAAS, and endothelial cell responses. Whether the expression of ACE2, as well as potential co-receptors and their modulation as a result of comorbidities, serve as a metric of disease susceptibility and severity warrants further investigation. Comprehensive disease-course assessments of endothelial activation and RAAS status may be relevant in guiding the appropriate implementation of treatment, as well as to uncover the potential long-term consequences of COVID-19.
Acknowledgments
The authors would like to thank all of the front-line and essential workers who continue to perform their duties in the face of COVID-19.
Sources of Funding
Preparation of this paper used the resources of the Toronto General Hospital Research Institute, University Health Network, Toronto, Canada. Research in the laboratory of J.E.F. is supported by a Project Grant from the Canadian Institutes of Health Research (Grant: PJT148487); a Pilot Grant from the Banting & Best Diabetes Centre and the Heart & Stroke Richard Lewar Centre of Excellence in Cardiovascular Research; a Seed Grant from the Ted Rogers Centre for Heart Research; a Medicine by Design Team Grant; and an infrastructure funding from the Canada Foundation for Innovation, the John R. Evans Leaders Fund, and the Ontario Research Fund. J.E.F. is supported by a Tier 2 Canada Research Chair in Vascular Cell and Molecular Biology from the Canadian Institutes of Health. K.L.H. is supported by funding from the University Health Network, Peter Munk Cardiac Centre (PMCC), Division of Vascular Surgery at the University of Toronto, and an innovation grant from the PMCC. K.L.H. and J.E.F. are jointly supported by COVID-19 innovation research grants provided by the Peter Munk Cardiac Center and the Ted Rogers Centre for Heart Research.
Disclosures
None.
Footnotes
Nonstandard Abbreviations and Acronyms
- ACE2
- angiotensin-converting enzyme 2
- Ang II
- angiotensin II
- Angpt2
- angiopoietin-2
- ARDS
- acute respiratory distress syndrome
- COVID-19
- coronavirus disease 2019
- DIC
- disseminated intravascular coagulation
- ECs
- endothelial cells
- MERS-CoV
- Middle east respiratory coronavirus
- NO
- nitric oxide
- RAAS
- renin-angiotensin-aldosterone system
- SARS-CoV-1
- severe acute respiratory syndrome coronavirus-1
- vWF
- von Willebrand Factor
For Sources of Funding and Disclosures, see page 1826.
References
- 1.Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 2020395565–574doi: 10.1016/S0140-6736(20)30251-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. Novel Coronavirus (2019-nCoV)-SITUATION REPORT-1. 2020 https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200121-sitrep-1-2019-ncov.pdf?sfvrsn=20a99c10_4. Accessed April 6, 2020.
- 3.Li LQ, Huang T, Wang YQ, Wang Z-P, Liang Y, Huang T-B, Zhang H-Y, Sun W, Wang Y. COVID-19 patients’ clinical characteristics, discharge rate, and fatality rate of meta-analysis [published online March 12, 2020 ]. J Med Virol. doi: 10.1002/jmv.25757. doi: 10.1002/jmv.25757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodriguez-Morales AJ, Cardona-Ospina JA, Gutiérrez-Ocampo E, Villamizar-Peña R, Holguin-Rivera Y, Escalera-Antezana JP, Alvarado-Arnez LE, Bonilla-Aldana DK, Franco-Paredes C, Henao-Martinez AF, et al. ; Latin American Network of Coronavirus Disease 2019-COVID-19 Research (LANCOVID-19) Electronic address: https://www.lancovid.org. Clinical, laboratory and imaging features of COVID-19: A systematic review and meta-analysis. Travel Med Infect Dis 202034101623.doi: 10.1016/j.tmaid.2020.101623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grasselli G, Zangrillo A, Zanella A, Antonelli M, Cabrini L, Castelli A, Cereda D, Coluccello A, Foti G, Fumagalli R, et al. Baseline characteristics and outcomes of 1591 patients infected with SARS-CoV-2 admitted to ICUs of the Lombardy Region, Italy. JAMA 20203231574–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, Gong W, Liu X, Liang J, Zhao Q, et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China [published online March 25, 2020]. JAMA Cardiol. :e200950. doi: 10.1001/jamacardio.2020.0950. doi: 10.1001/jamacardio.2020.0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med 202046846–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, Wang H, Wan J, Wang X, Lu Z. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19) [published online March 27, 2020]. JAMA Cardiol. :e201017. doi: 10.1001/jamacardio.2020.1017. doi: 10.1001/jamacardio.2020.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cdc Covid-19 Response Team Severe outcomes among patients with coronavirus disease 2019 (COVID-19) - United States, February 12-March 16, 2020. MMWR Morb Mortal Wkly Rep 202069343–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, Xiang J, Wang Y, Song B, Gu X, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 20203951054–1062doi: 10.1016/S0140-6736(20)30566-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Helms J, Tacquard C, Severac F, Leonard-Lorant I, Ohana M, Delabranche X, Merdji H, Clere-Jehl R, Schenck M, Gandet FF, et al. High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study [published online May 4, 2020]. Intensive Care Med 1–10doi: 10.1007/s00134-020-06062-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, Mehra MR, Schuepbach RA, Ruschitzka F, Moch H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 20203951417–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sukriti S, Tauseef M, Yazbeck P, Mehta D. Mechanisms regulating endothelial permeability. Pulm Circ 20144535–551doi: 10.1086/677356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Armstrong SM, Darwish I, Lee WL. Endothelial activation and dysfunction in the pathogenesis of influenza A virus infection. Virulence 20134537–542doi: 10.4161/viru.25779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020395497–506doi: 10.1016/S0140-6736(20)30183-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, Wang B, Xiang H, Cheng Z, Xiong Y, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA 20203231061–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 2011146980–991doi: 10.1016/j.cell.2011.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krijnen PA, Hahn NE, Kholová I, Baylan U, Sipkens JA, van Alphen FP, Vonk AB, Simsek S, Meischl C, Schalkwijk CG, et al. Loss of DPP4 activity is related to a prothrombogenic status of endothelial cells: implications for the coronary microvasculature of myocardial infarction patients. Basic Res Cardiol. 2012;107:233. doi: 10.1007/s00395-011-0233-5. doi: 10.1007/s00395-011-0233-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McEver RP. Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc Res 2015107331–339doi: 10.1093/cvr/cvv154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vita JA, Keaney JF., Jr. Endothelial function: a barometer for cardiovascular risk? Circulation 2002106640–642doi: 10.1161/01.cir.0000028581.07992.56 [DOI] [PubMed] [Google Scholar]
- 21.Rabelo LA, Alenina N, Bader M. ACE2-angiotensin-(1-7)-Mas axis and oxidative stress in cardiovascular disease. Hypertens Res 201134154–160doi: 10.1038/hr.2010.235 [DOI] [PubMed] [Google Scholar]
- 22.Thomas MC, Pickering RJ, Tsorotes D, Koitka A, Sheehy K, Bernardi S, Toffoli B, Nguyen-Huu TP, Head GA, Fu Y, et al. Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ Res 2010107888–897doi: 10.1161/CIRCRESAHA.110.219279 [DOI] [PubMed] [Google Scholar]
- 23.Boehm M, Nabel EG. Angiotensin-converting enzyme 2–a new cardiac regulator. N Engl J Med 20023471795–1797doi: 10.1056/NEJMcibr022472 [DOI] [PubMed] [Google Scholar]
- 24.Sampaio WO, Souza dos Santos RA, Faria-Silva R, da Mata Machado LT, Schiffrin EL, Touyz RM. Angiotensin-(1-7) through receptor Mas mediates endothelial nitric oxide synthase activation via Akt-dependent pathways. Hypertension 200749185–192doi: 10.1161/01.HYP.0000251865.35728.2f [DOI] [PubMed] [Google Scholar]
- 25.Oudit GY, Crackower MA, Backx PH, Penninger JM. The role of ACE2 in cardiovascular physiology. Trends Cardiovasc Med 20031393–101doi: 10.1016/s1050-1738(02)00233-5 [DOI] [PubMed] [Google Scholar]
- 26.Soro-Paavonen A, Gordin D, Forsblom C, Rosengard-Barlund M, Waden J, Thorn L, Sandholm N, Thomas MC, Groop PH; FinnDiane Study Group Circulating ACE2 activity is increased in patients with type 1 diabetes and vascular complications. J Hypertens 201230375–383doi: 10.1097/HJH.0b013e32834f04b6 [DOI] [PubMed] [Google Scholar]
- 27.Wu YH, Li JY, Wang C, Zhang L-M, Qiao H. The ACE2 G8790A polymorphism: involvement in type 2 diabetes mellitus combined with cerebral stroke. J Clin Lab Anal. 2017;31:e22033. doi: 10.1002/jcla.22033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patel VB, Mori J, McLean BA, Basu R, Das SK, Ramprasath T, Parajuli N, Penninger JM, Grant MB, Lopaschuk GD, et al. ACE2 deficiency worsens epicardial adipose tissue inflammation and cardiac dysfunction in response to diet-induced obesity. Diabetes 20166585–95doi: 10.2337/db15-0399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goulter AB, Goddard MJ, Allen JC, Clark KL. ACE2 gene expression is up-regulated in the human failing heart. BMC Med. 2004;2:19. doi: 10.1186/1741-7015-2-19. doi: 10.1186/1741-7015-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, Raizada MK, Grant MB, Oudit GY. Angiotensin-converting enzyme 2: SARS-CoV-2 receptor and regulator of the renin-angiotensin system: celebrating the 20th anniversary of the discovery of ACE2. Circ Res 20201261456–1474doi: 10.1161/CIRCRESAHA.120.317015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020181281–292.e6doi: 10.1016/j.cell.2020.02.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 2020;94:e00127-20. doi: 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 20203671260–1263doi: 10.1126/science.abb2507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020181271.e8–280.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aimes RT, Zijlstra A, Hooper JD, Ogbourne SM, Sit ML, Fuchs S, Gotley DC, Quigley JP, Antalis TM. Endothelial cell serine proteases expressed during vascular morphogenesis and angiogenesis. Thromb Haemost 200389561–572 [PubMed] [Google Scholar]
- 36.Bertram S, Heurich A, Lavender H, Gierer S, Danisch S, Perin P, Lucas JM, Nelson PS, Pöhlmann S, Soilleux EJ. Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts. PLoS One. 2012;7:e35876. doi: 10.1371/journal.pone.0035876. doi: 10.1371/journal.pone.0035876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 2004203631–637doi: 10.1002/path.1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roberts A, Vogel L, Guarner J, Hayes N, Murphy B, Zaki S, Subbarao K. Severe acute respiratory syndrome coronavirus infection of golden Syrian hamsters. J Virol 200579503–511doi: 10.1128/JVI.79.1.503-511.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, Leopoldi A, Garreta E, Hurtado Del Pozo C, Prosper F, et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 2020181905–913.e7doi: 10.1016/j.cell.2020.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen W, Lan Y, Yuan X, Deng X, Li Y, Cai X, Li L, He R, Tan Y, Deng X, et al. Detectable 2019-nCoV viral RNA in blood is a strong indicator for the further clinical severity. Emerg Microbes Infect 20209469–473doi: 10.1080/22221751.2020.1732837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tundup S, Kandasamy M, Perez JT, Mena N, Steel J, Nagy T, Albrecht RA, Manicassamy B. Endothelial cell tropism is a determinant of H5N1 pathogenesis in mammalian species. PLoS Pathog. 2017;13:e1006270. doi: 10.1371/journal.ppat.1006270. doi: 10.1371/journal.ppat.1006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang K, Gheblawi M, Oudit GY. Angiotensin converting enzyme 2: a double-edged sword [published online March 26, 2020]. Circulation. doi: 10.1161/CIRCULATIONAHA.120.047049. doi: 10.1161/CIRCULATIONAHA.120.047049. [DOI] [PubMed] [Google Scholar]
- 43.Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005436112–116doi: 10.1038/nature03712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Booth CM, Matukas LM, Tomlinson GA, Rachlis AR, Rose DB, Dwosh HA, Walmsley SL, Mazzulli T, Avendano M, Derkach P, et al. Clinical features and short-term outcomes of 144 patients with SARS in the greater Toronto area. JAMA 20032892801–2809doi: 10.1001/jama.289.21.JOC30885 [DOI] [PubMed] [Google Scholar]
- 45.Badawi A, Ryoo SG. Prevalence of comorbidities in the Middle East respiratory syndrome coronavirus (MERS-CoV): a systematic review and meta-analysis. Int J Infect Dis 201649129–133doi: 10.1016/j.ijid.2016.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li B, Yang J, Zhao F, Zhi L, Wang X, Liu L, Bi Z, Zhao Y. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin Res Cardiol 2020109531–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in china: summary of a report of 72 314 cases from the Chinese center for disease control and prevention [published online February 24, 2020]. JAMA. doi: 10.1001/jama.2020.2648. doi: 10.1001/jama.2020.2648. [DOI] [PubMed] [Google Scholar]
- 48.Yang X, Yu Y, Xu J, Shu H, Xia J, Liu H, Wu Y, Zhang L, Yu Z, Fang M, et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: a single-centered, retrospective, observational study. Lancet Respir Med 20208475–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang J, Zheng Y, Gou X, Pu K, Chen Z, Guo Q, Ji R, Wang H, Wang Y, Zhou Y. Prevalence of comorbidities in the novel Wuhan coronavirus (COVID-19) infection: a systematic review and meta-analysis. Int J Infect Dis 20209491–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Balakumar P, Maung-U K, Jagadeesh G. Prevalence and prevention of cardiovascular disease and diabetes mellitus. Pharmacol Res 2016113Pt A600–609doi: 10.1016/j.phrs.2016.09.040 [DOI] [PubMed] [Google Scholar]
- 51.Bloch MJ. Worldwide prevalence of hypertension exceeds 1.3 billion. J Am Soc Hypertens 201610753–754doi: 10.1016/j.jash.2016.08.006 [DOI] [PubMed] [Google Scholar]
- 52.COVID-19 Host Genetics Initiative The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur J Hum Genet 202028715–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Widlansky ME, Gokce N, Keaney JF, Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol 2003421149–1160doi: 10.1016/s0735-1097(03)00994-x [DOI] [PubMed] [Google Scholar]
- 54.Bonow RO, Fonarow GC, O’Gara PT, Yancy CW. Association of coronavirus disease 2019 (COVID-19) with myocardial injury and mortality [published online March 27, 2020]. JAMA Cardiol. doi: 10.1001/jamacardio.2020.1105. doi: 10.1001/jamacardio.2020.1105. [DOI] [PubMed] [Google Scholar]
- 55.Du RH, Liu LM, Yin W, Wang W, Guan L-L, Yuan M-L, Li Y-L, Hu Y, Li X-Y, Sun B, et al. Hospitalization and critical care of 109 decedents with COVID-19 pneumonia in Wuhan, China [published online April 7, 2020]. Ann Am Thorac Soc. doi: 10.1513/AnnalsATS.202003-225OC. doi: 10.1513/AnnalsATS.202003-225OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oudit GY, Kassiri Z, Jiang C, Liu PP, Poutanen SM, Penninger JM, Butany J. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest 200939618–625doi: 10.1111/j.1365-2362.2009.02153.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Han H, Xie L, Liu R, Yang J, Liu F, Wu K, Chen L, Hou W, Feng Y, Zhu C. Analysis of heart injury laboratory parameters in 273 COVID-19 patients in one hospital in Wuhan, China [published online March 31, 2020]. J Med Virol. doi: 10.1002/jmv.25809. doi: 10.1002/jmv.25809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yao XH, Li TY, He ZC, Ping YF, Liu HW, Yu SC, Mou HM, Wang LH, Zhang HR, Fu WJ, et al. [A pathological report of three COVID-19 cases by minimal invasive autopsies]. Zhonghua Bing Li Xue Za Zhi 202049411–417doi: 10.3760/cma.j.cn112151-20200312-00193 [DOI] [PubMed] [Google Scholar]
- 59.Zou X, Chen K, Zou J, Han P, Hao J, Han Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med 202014185–192doi: 10.1007/s11684-020-0754-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res 20201161097–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bradley BT, Maioli H, Johnston R, Chaudhry I, Fink SL, Xu H, Najafian B, Marshall D, Lacy JM, Williams T, et al. Histopathology and ultrastructural findings of fatal COVID-19 infections [published online April 21, 2020]. medRxiv. doi: 10.1101/2020.04.17.20058545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jones VG, Mills M, Suarez D, Hogan CA, Yeh D, Segal JB, Nguyen EL, Barsh GR, Maskatia S, Mathew R. COVID-19 and Kawasaki disease: novel virus and novel case. Hosp Pediatr 202010537–540doi: 10.1542/hpeds.2020-0123 [DOI] [PubMed] [Google Scholar]
- 63.Riphagen S, Gomez X, Gonzalez-Martinez C, Wilkinson N, Theocharis P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 20203951607–1608doi: 10.1016/S0140-6736(20)31094-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Turnier JL, Anderson MS, Heizer HR, Jone PN, Glodé MP, Dominguez SR. Concurrent respiratory viruses and kawasaki disease. Pediatrics 2015136e609–e614doi: 10.1542/peds.2015-0950 [DOI] [PubMed] [Google Scholar]
- 65.Shirato K, Imada Y, Kawase M, Nakagaki K, Matsuyama S, Taguchi F. Possible involvement of infection with human coronavirus 229E, but not NL63, in Kawasaki disease. J Med Virol 2014862146–2153doi: 10.1002/jmv.23950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ding Y, Wang H, Shen H, Li Z, Geng J, Han H, Cai J, Li X, Kang W, Weng D, et al. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol 2003200282–289doi: 10.1002/path.1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cao Z, Jia Y, Zhu B. BNP and NT-proBNP as diagnostic biomarkers for cardiac dysfunction in both clinical and forensic medicine. Int J Mol Sci. 2019;20:1820. doi: 10.3390/ijms20081820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vassiliadis E, Barascuk N, Didangelos A, Karsdal MA. Novel cardiac-specific biomarkers and the cardiovascular continuum. Biomark Insights 2012745–57doi: 10.4137/BMI.S9536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sproston NR, Ashworth JJ. Role of C-reactive protein at sites of inflammation and infection. Front Immunol. 2018;9:754. doi: 10.3389/fimmu.2018.00754. doi: 10.3389/fimmu.2018.00754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cerinic MM, Valentini G, Sorano GG, D’Angelo S, Cuomo G, Fenu L, Generini S, Cinotti S, Morfini M, Pignone A, et al. Blood coagulation, fibrinolysis, and markers of endothelial dysfunction in systemic sclerosis. Semin Arthritis Rheum 200332285–295doi: 10.1053/sarh.2002.50011 [DOI] [PubMed] [Google Scholar]
- 71.Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6:a016295. doi: 10.1101/cshperspect.a016295. doi: 10.1101/cshperspect.a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Park KC, Gaze DC, Collinson PO, Marber MS. Cardiac troponins: from myocardial infarction to chronic disease. Cardiovasc Res 20171131708–1718doi: 10.1093/cvr/cvx183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ware LB, Eisner MD, Thompson BT, Parsons PE, Matthay MA. Significance of von Willebrand factor in septic and nonseptic patients with acute lung injury. Am J Respir Crit Care Med 2004170766–772doi: 10.1164/rccm.200310-1434OC [DOI] [PubMed] [Google Scholar]
- 74.Escher R, Breakey N, Lämmle B. Severe COVID-19 infection associated with endothelial activation. Thromb Res. 2020;190:62. doi: 10.1016/j.thromres.2020.04.014. doi: 10.1016/j.thromres.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Han H, Yang L, Liu R, Liu F, Wu K-L, Li J, Liu X-H, Zhu C-L. Prominent changes in blood coagulation of patients with SARS-CoV-2 infection [published online March 16, 2020]. Clin Chem Lab Med. doi: 10.1515/cclm-2020-0188. doi: 10.1515/cclm-2020-0188. [DOI] [PubMed] [Google Scholar]
- 76.Lee SJ, Lee CK, Kang S, Park I, Kim YH, Kim SK, Hong SP, Bae H, He Y, Kubota Y, et al. Angiopoietin-2 exacerbates cardiac hypoxia and inflammation after myocardial infarction. J Clin Invest 20181285018–5033doi: 10.1172/JCI99659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Agrawal A, Matthay MA, Kangelaris KN, Stein J, Chu JC, Imp BM, Cortez A, Abbott J, Liu KD, Calfee CS. Plasma angiopoietin-2 predicts the onset of acute lung injury in critically ill patients. Am J Respir Crit Care Med 2013187736–742doi: 10.1164/rccm.201208-1460OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Calfee CS, Gallagher D, Abbott J, Thompson BT, Matthay MA; NHLBI ARDS Network Plasma angiopoietin-2 in clinical acute lung injury: prognostic and pathogenetic significance. Crit Care Med 2012401731–1737doi: 10.1097/CCM.0b013e3182451c87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cox LA, van Eijk LT, Ramakers BP, Dorresteijn MJ, Gerretsen J, Kox M, Pickkers P. Inflammation-induced increases in plasma endocan levels are associated with endothelial dysfunction in humans in vivo. Shock 201543322–326doi: 10.1097/SHK.0000000000000320 [DOI] [PubMed] [Google Scholar]
- 80.Tang L, Zhao Y, Wang D, Deng W, Li C, Li Q, Huang S, Shu C. Endocan levels in peripheral blood predict outcomes of acute respiratory distress syndrome. Mediators Inflamm. 2014;2014:625180. doi: 10.1155/2014/625180. doi: 10.1155/2014/625180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schmidt EP, Li G, Li L, Fu L, Yang Y, Overdier KH, Douglas IS, Linhardt RJ. The circulating glycosaminoglycan signature of respiratory failure in critically ill adults. J Biol Chem 20142898194–8202doi: 10.1074/jbc.M113.539452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sapru A, Calfee CS, Liu KD, Kangelaris K, Hansen H, Pawlikowska L, Ware LB, Alkhouli MF, Abbott J, Abbot J, et al. ; NHLBI ARDS Network Plasma soluble thrombomodulin levels are associated with mortality in the acute respiratory distress syndrome. Intensive Care Med 201541470–478doi: 10.1007/s00134-015-3648-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Calfee CS, Eisner MD, Parsons PE, Thompson BT, Conner ER, Jr, Matthay MA, Ware LB; NHLBI Acute Respiratory Distress Syndrome Clinical Trials Network Soluble intercellular adhesion molecule-1 and clinical outcomes in patients with acute lung injury. Intensive Care Med 200935248–257doi: 10.1007/s00134-008-1235-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rubin DB, Wiener-Kronish JP, Murray JF, Green DR, Turner J, Luce JM, Montgomery AB, Marks JD, Matthay MA. Elevated von Willebrand factor antigen is an early plasma predictor of acute lung injury in nonpulmonary sepsis syndrome. J Clin Invest 199086474–480doi: 10.1172/JCI114733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Page AV, Liles WC. Biomarkers of endothelial activation/dysfunction in infectious diseases. Virulence 20134507–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gao Y, Li T, Han M, Li X, Wu D, Xu Y, Zhu Y, Liu Y, Wang X, Wang L. Diagnostic utility of clinical laboratory data determinations for patients with the severe COVID-19 [published online March 17, 2020]. J Med Virol. doi: 10.1002/jmv.25770. doi: 10.1002/jmv.25770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang H, Luo S, Shen Y, Li M, Zhang Z, Dong Y, Zhou H, Lin L, Guo W, Kang Z, et al. Multiple enzyme release, inflammation storm and hypercoagulability are prominent indicators for disease progression in COVID-19: a multi-centered, correlation study with CT imaging score [published online March 2, 2020]. SSRN. doi: 10.2139/ssrn.3544837. [DOI] [Google Scholar]
- 88.Lillicrap D. Disseminated intravascular coagulation in patients with 2019-nCoV pneumonia. J Thromb Haemost 202018786–787doi: 10.1111/jth.14781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Walborn A, Rondina M, Mosier M, Fareed J, Hoppensteadt D. Endothelial dysfunction is associated with mortality and severity of coagulopathy in patients with sepsis and disseminated intravascular coagulation. Clin Appl Thromb Hemost. 2019;25:1076029619852163. doi: 10.1177/1076029619852163. doi: 10.1177/1076029619852163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost 202018844–847doi: 10.1111/jth.14768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Spiezia L, Boscolo A, Poletto F, Cerruti L, Tiberio I, Campello E, Navalesi P, Simioni P. COVID-19-related severe hypercoagulability in patients admitted to intensive care unit for acute respiratory failure. Thromb Haemost 2020120998–1000doi: 10.1055/s-0040-1710018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers D, Kant KM, Kaptein FHJ, van Paassen J, Stals MAM, Huisman MV, et al. Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: an updated analysis. Thromb Res 2020191148–150doi: 10.1016/j.thromres.2020.04.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wichmann D, Sperhake JP, Lütgehetmann M, Steurer S, Edler C, Heinemann A, Heinrich F, Mushumba H, Kniep I, Sophie Schröder A, et al. Autopsy findings and venous thromboembolism in patients with COVID-19: a prospective cohort study [published online May 6, 2020]. Ann Intern Med 2020M20–2003doi: 10.7326/M20-2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Qanadli SD, Beigelman-Aubry C, Rotzinger DC. Vascular changes detected with thoracic CT in coronavirus disease (COVID-19) might be significant determinants for accurate diagnosis and optimal patient management [published online April 7, 2020]. AJR Am J Roentgenol. :W1. doi: 10.2214/AJR.20.23185. doi: 10.2214/AJR.20.23185. [DOI] [PubMed] [Google Scholar]
- 95.Oxley TJ, Mocco J, Majidi S, Kellner CP, Shoirah H, Singh IP, De Leacy RA, Shigematsu T, Ladner TR, Yaeger KA, et al. Large-Vessel Stroke as a Presenting Feature of Covid-19 in the Young. N Engl J Med. 2020;382:e60. doi: 10.1056/NEJMc2009787. doi: 10.1056/NEJMc2009787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang J, Hajizadeh N, Moore EE, Moore EE, McIntyre RC, Moore PK, Veress LA, Yaffe MB, Moore HB, Barrett CD. Tissue Plasminogen Activator (tPA) treatment for COVID-19 associated Acute Respiratory Distress Syndrome (ARDS): a case series [published online April 8, 2020]. J Thromb Haemost. doi: 10.1111/jth.14828. doi: 10.1111/jth.14828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ciceri F, Beretta L, Scandroglio AM, Colombo S, Landoni G, Ruggeri A, Peccatori J, D'Angelo A, De Cobelli F, Rovere-Querini P, et al. Microvascular COVID-19 lung vessels obstructive thromboinflammatory syndrome (MicroCLOTS): an atypical acute respiratory distress syndrome working hypothesis [published online April 14, 2020]. Crit Care Resusc. doi: 10.51893/2020.2.pov2. https://europepmc.org/article/med/32294809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhou B, She J, Wang Y, Ma X. Venous thrombosis and arteriosclerosis obliterans of lower extremities in a very severe patient with 2019 novel coronavirus disease: a case report [published online April 18, 2020]. J Thromb Thrombolysis. doi: 10.1007/s11239-020-02084-w. doi: 10.1007/s11239-020-02084-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, Baxter-Stoltzfus A, Laurencef J. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases [published online April 15, 2020]. Transl Res. doi: 10.1016/j.trsl.2020.04.007. S1931-5244(20)30070-0. doi: 10.1016/j.trsl.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang W, Zhao Y, Zhang F, Wang Q, Li T, Liu Z, Wang J, Qin Y, Zhang X, Yan X, et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin Immunol. 2020;214:108393. doi: 10.1016/j.clim.2020.108393. doi: 10.1016/j.clim.2020.108393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rockx B, Kuiken T, Herfst S, Bestebroer T, Lamers MM, Oude Munnink BB, de Meulder D, van Amerongen G, van den Brand J, Okba NMA, et al. Comparative pathogenesis Of COVID-19, MERS And SARS in a non-human primate model. Science 20203681012–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Smits SL, de Lang A, van den Brand JM, Leijten LM, van IJcken WF, Eijkemans MJ, van Amerongen G, Kuiken T, Andeweg AC, Osterhaus AD, et al. Exacerbated innate host response to SARS-CoV in aged non-human primates. PLoS Pathog. 2010;6:e1000756. doi: 10.1371/journal.ppat.1000756. doi: 10.1371/journal.ppat.1000756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Day CW, Baric R, Cai SX, Frieman M, Kumaki Y, Morrey JD, Smee DF, Barnard DL. A new mouse-adapted strain of SARS-CoV as a lethal model for evaluating antiviral agents in vitro and in vivo. Virology 2009395210–222doi: 10.1016/j.virol.2009.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Schaecher SR, Stabenow J, Oberle C, Schriewer J, Buller RM, Sagartz JE, Pekosz A. An immunosuppressed Syrian golden hamster model for SARS-CoV infection. Virology 2008380312–321doi: 10.1016/j.virol.2008.07.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Adedeji AO, Severson W, Jonsson C, Singh K, Weiss SR, Sarafianos SG. Novel inhibitors of severe acute respiratory syndrome coronavirus entry that act by three distinct mechanisms. J Virol 2013878017–8028doi: 10.1128/JVI.00998-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cavaillon JM, Singer M, Skirecki T. Sepsis therapies: learning from 30 years of failure of translational research to propose new leads. EMBO Mol Med. 2020;12:e10128. doi: 10.15252/emmm.201810128. doi: 10.15252/emmm.201810128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Alattar R, Ibrahim TBH, Shaar SH, Abdalla S, Shukri K, Daghfal JN, Khatib MY, Aboukamar M, Abukhattab M, Alsoub HA, et al. Tocilizumab for the treatment of severe coronavirus disease 2019 [published online May 5, 2020]. J Med Virol. doi: 10.1002/jmv.25964. doi: 10.1002/jmv.25964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Toniati P, Piva S, Cattalini M, Garrafa E, Regola F, Castelli F, Franceschini F, Airò P, Bazzani C, Beindorf E-A, et al. Tocilizumab for the treatment of severe COVID-19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: A single center study of 100 patients in Brescia, Italy. Autoimmun Rev. 2020;19:102568. doi: 10.1016/j.autrev.2020.102568. doi: 10.1016/j.autrev.2020.102568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cunningham L, Kimber I, Basketter DA, McFadden JP. Why judiciously timed anti-IL 6 therapy may be of benefit in severe COVID-19 infection. Autoimmun Rev. 2020;19:102563. doi: 10.1016/j.autrev.2020.102563. doi: 10.1016/j.autrev.2020.102563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.London NR, Zhu W, Bozza FA, Smith MC, Greif DM, Sorensen LK, Chen L, Kaminoh Y, Chan AC, Passi SF, et al. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Sci Transl Med. 2010;2:23ra19. doi: 10.1126/scitranslmed.3000678. doi: 10.1126/scitranslmed.3000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sugiyama MG, Armstrong SM, Wang C, Hwang D, Leong-Poi H, Advani A, Advani S, Zhang H, Szaszi K, Tabuchi A, et al. The Tie2-agonist Vasculotide rescues mice from influenza virus infection. Sci Rep. 2015;5:11030. doi: 10.1038/srep11030. doi: 10.1038/srep11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Millar FR, Summers C, Griffiths MJ, Toshner MR, Proudfoot AG. The pulmonary endothelium in acute respiratory distress syndrome: insights and therapeutic opportunities. Thorax 201671462–473doi: 10.1136/thoraxjnl-2015-207461 [DOI] [PubMed] [Google Scholar]
- 113.Müller HC, Hellwig K, Rosseau S, Tschernig T, Schmiedl A, Gutbier B, Schmeck B, Hippenstiel S, Peters H, Morawietz L, et al. Simvastatin attenuates ventilator-induced lung injury in mice. Crit Care. 2010;14:R143. doi: 10.1186/cc9209. doi: 10.1186/cc9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jacobson JR, Barnard JW, Grigoryev DN, Ma SF, Tuder RM, Garcia JG. Simvastatin attenuates vascular leak and inflammation in murine inflammatory lung injury. Am J Physiol Lung Cell Mol Physiol 2005288L1026–L1032doi: 10.1152/ajplung.00354.2004 [DOI] [PubMed] [Google Scholar]
- 115.Birukova AA, Xing J, Fu P, Yakubov B, Dubrovskyi O, Fortune JA, Klibanov AM, Birukov KG. Atrial natriuretic peptide attenuates LPS-induced lung vascular leak: role of PAK1. Am J Physiol Lung Cell Mol Physiol 2010299L652–L663doi: 10.1152/ajplung.00202.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost 2020181094–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brack MC, Lienau J, Kuebler WM, Witzenrath M. Cardiovascular sequelae of pneumonia. Curr Opin Pulm Med 201925257–262doi: 10.1097/MCP.0000000000000584 [DOI] [PubMed] [Google Scholar]
- 118.Corrales-Medina VF, Alvarez KN, Weissfeld LA, Angus DC, Chirinos JA, Chang CC, Newman A, Loehr L, Folsom AR, Elkind MS, et al. Association between hospitalization for pneumonia and subsequent risk of cardiovascular disease. JAMA 2015313264–274doi: 10.1001/jama.2014.18229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kaptoge S, Seshasai SR, Gao P, Freitag DF, Butterworth AS, Borglykke A, Di Angelantonio E, Gudnason V, Rumley A, Lowe GD, et al. Inflammatory cytokines and risk of coronary heart disease: new prospective study and updated meta-analysis. Eur Heart J 201435578–589doi: 10.1093/eurheartj/eht367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wu Q, Zhou L, Sun X, Yan Z, Hu C, Wu J, Xu L, Li X, Liu H, Yin P, et al. Altered Lipid Metabolism in Recovered SARS Patients Twelve Years after Infection. Sci Rep. 2017;7:9110. doi: 10.1038/s41598-017-09536-z. doi: 10.1038/s41598-017-09536-z. [DOI] [PMC free article] [PubMed] [Google Scholar]