

Abstract
Newcastle disease virus (NDV) is an avian paramyxovirus that has been extensively studied as an oncolytic agent, in addition to being an economically important pathogen in the poultry industry. The establishment of a reverse genetics system for this virus has enabled the development of genetically modified recombinant NDV viruses with improved oncolytic and immunotherapeutic properties. In this chapter, we describe the materials and methods involved in the in vitro cloning and rescue of NDV expressing murine 4-1BBL as well as the in vivo evaluation of NDV expressing 4-1BBL in a B16-F10 murine melanoma model.
Keywords: Newcastle disease virus, Reverse genetics, Virus rescue, Oncolytic Vector, Immunotherapy
1. Introduction
Newcastle Disease Virus (NDV) is an avian paramyxovirus and an economically important pathogen for the poultry industry (1). NDV, or avian paramyxovirus type 1 (APMV-1), is classified as a member of the genus Avulavirus belonging to the family Paramyxoviridae (1)(2). It consists of a negative-sense, single-stranded RNA genome that is non-segmented and is approximately 15,186 nucleotides (nt) in length (3). Additionally, it encodes for six proteins including the nucleoprotein (NP), the phosphoprotein (P), the matrix protein (M), the fusion protein (F), the haemagglutinin-neuraminidase (HN) and the large polymerase protein (L) (4). NDV isolates can be grouped into velogenic (highly virulent), mesogenic (intermediate), or lentogenic (non-virulent) strains, depending on the virulence and pathogenicity in avian species and this is correlated with the cleavage site in the F protein (5). NDV LaSota is a naturally occurring lentogenic (non-virulent) strain commonly used as a live attenuated vaccine in the poultry industry, and has been demonstrated to be an effective and safe vaccine vector in multiple studies (6,7).
Besides being an important avian pathogen, NDV has been explored as a candidate for oncolytic immunotherapy since 1955 (8). Pioneering preclinical work by several labs including Sinkovics and Cassell from the 1960’s sought to establish NDV as an anti-neoplastic agent (9,10), with Cassell and Garrett observing in 1965 the development of anti-tumor immunity after viral oncolytic therapy with NDV (11,12). This precipitated the development of NDV oncolysates as an adjunctive immunotherapeutic vaccine for post-operative management of malignant melanoma patients, with two Phase II clinical studies involving 32 and 51 patients with Stage II malignant melanoma (13). A 10 year and a 15 year follow-up with these patients showed that over 60% and 55% were alive and free of recurrent disease, respectively, which was superior to historical controls (14,15). Another Phase II study involving 208 patients with locally advanced renal cell carcinoma was carried out with NDV oncolysates using the NDV 73T strain combined with low-dose recombinant interleukin-2 (IL-2) and interferon-α (IFNα), demonstrating improved disease-free survival (16). Later on, an attenuated veterinary vaccine strain NDV-MTH-68/N was tested out in a variety of previously chemo-refractory tumors by the Hungarian scientist Dr. Laszlo Csatary in a placebo-controlled Phase II clinical trial demonstrating an overall improvement in survival rates as compared to the control group (17,18). Furthermore, other strains such as the lentogenic NDV-HUJ strain (19), the lytic NDV PV701 (20,21) and ATV-NDV (22) have been studied in patients with glioblastoma multiforme (GBM) and a variety of other advanced cancers.
As evidenced, decades of research have demonstrated the natural and selective oncolytic capabilities of NDV in different mammalian cancer cell lines, animal tumor models, and clinical trials (23-25). The establishment of a reverse genetics systems for this virus has been particularly important as it has enabled the development of genetically modified recombinant NDV viruses with improved oncolytic and immunostimulatory properties (26,27). Several strategies have been attempted so far, including but not limited to the introduction of a polybasic cleavage site in the F protein of a lentogenic Hitchner B1 NDV strain, engineering of viruses to express IFNγ, GM-CSF, IL-2, or TNFα, as well as the generation of viruses which possess both modifications (28-33). Recent preclinical studies in a bilateral flank syngeneic melanoma model as well as in a bladder cancer model have shown that intratumoral administration of NDV in conjunction with systemic delivery of immune checkpoint blockade antibodies that target CTLA-4 and PD-1, could effectively activate both the innate and adaptive immune pathways to induce an abscopal effect. This abscopal effect was characterized by an enhanced immune infiltration into distant non-treated tumors (34,35). Additionally, Tcell effector function could be further enhanced within the tumor microenvironment with the stimulation of a relevant co-stimulatory pathway by the intratumoral delivery of a recombinant NDV expressing the ICOS ligand (ICOSL) (36). These studies demonstrate that stimulation of an adaptive immune response within the injected tumor has a potential to induce a stronger systemic anti-tumor immune response, which could result in superior anti-tumor efficacy not just against the injected, but also distant tumors.
Such strategies provide for a possibility of intratumoral delivery of immunostimulatory ligands that otherwise carry an increased potential for systemic toxicity. In this book chapter, we describe detailed methods for the generation of recombinant NDV with enhanced immunostimulatory effects. As a model, we use NDV expressing 4-1BB ligand (4-1BBL), a ligand that binds to the 4-1BB (CD137) receptor, signaling through which provides co-stimulatory function for cytotoxic T cells and NK cells, which has been explored for anti-cancer therapy using agonistic antibodies (37). An agonistic antibody to 4-1BB has been evaluated in a phase I study of 83 patients with advanced malignancies (38). Evidence of clinical activity was seen across a wide dose range, but the trial was suspended due to hepatotoxicity seen at higher doses. Intratumoral delivery of NDV expressing 4-1BBL has a potential to provide the initial co-stimulatory signal needed for T cell activation, while avoiding systemic toxicity. The chapter will be divided into five sections with section 1 and 2 describing the cloning and generation of NDV-LaSota full-length cDNA plasmid encoding 4-1BBL as well as the generation of the helper plasmids. Section 3 will detail the procedures for setting up rescue transfections, RT-PCR confirmation of positive rescues and tittering of NDV-LaSota viruses expressing 4-1BBL. Finally, sections 4 and 5 will demonstrate confirmation of surface expression of 4-1BBL in NDV-infected cells by flow cytometry and will demonstrate the ability of the virus to drive immune cell infiltration into both treated and non-treated tumors in vivo.
2. Materials
2.1. Construction of recombinant full-length NDV (rNDV) encoding 4-1BBL and helper plasmids
Codon-optimized synthesized gene ( sequence available upon request).
Taq DNA polymerase, High Fidelity.
Primer set (forward and reverse, 10 μM stock concentration).
dNTP mix.
10X Tris-acetate-EDTA (TAE) electrophoresis buffer: Mix 11.4 mL of glacial acetic acid, 3.72 g of EDTA, 48.4 g Tris base, 900mL deionized water. Adjust volume to 1 L with deionized water. Dilute 100 mL of 10X stock to 1L with deionized water to make 1X TAE buffer.
Ethidium Bromide.
1.5% Agarose-TAE gel: Measure 1.5 g agarose in 100 mL TAE. Microwave for 2-3 mins until the agarose is completely dissolved. Add 2-3 μL of ethidium bromide once the agarose solution has cooled down to about 60°C.
Gel loading dye, without SDS.
DNA Ladder (0.1-10.0 kb).
Gel and PCR Clean-up Kit.
SacII restriction enzyme.
Antarctic Phosphatase.
In-Fusion® HD Cloning Plus.
Stable Competent E. coli- High Efficiency.
Luria-Bertani (LB) -Ampicillin agar plates: Mix one pouch of pre-mixed, pre-sterilized LB growth medium including agar and ampicillin in 200 mL deionized water in an autoclaved flask. Gently heat the mixture in a microwave, without bringing it to a full boil so that it completely dissolves without degrading the ampicillin. Pour out the media into petridishes and let it cool at room temperature.
LB broth supplemented with ampicillin (100 μg/mL).
DNA Miniprep Kit.
Plasmid Midiprep Kit for purification of high purity, endotoxin-free DNA.
1.5 mL microcentrifuge tubes.
Micropipette tips, sterile.
PCR thermal cyclers.
Dry block heaters.
UV Transilluminator.
NanoDrop Spectrophotometer.
Water bath.
Super optimal broth with catabolic repressor (SOC) media
Microbiological incubators.
Sterile 1 L baffled flasks.
Shaking incubators.
Refrigerated tabletop centrifuge.
2.2. Rescue and amplification of recombinant NDV expressing the transgene
BSRT-7 cells.
Vero cells (ATCC, CCL-81).
Complete DMEM (cDMEM): Supplement Dulbecco’s Modified Eagle Medium (DMEM), high glucose, pyruvate. with 10% Fetal Bovine Serum (FBS) and 1% Penicillin-Streptomycin (P/S).
Trypsin-EDTA (0.05%), phenol red.
1X PBS, pH 7.4.
Opti-MEM™ Reduced Serum Medium, GlutaMAX™ Supplement.
High efficiency transfection reagent.
9 to 11-day-old specific pathogen-free (SPF) embryonated chicken eggs.
One Step RT-PCR with hot-start, thermostable Taq polymerase with enhanced specificity.
RNA Mini Kit.
Turkey red blood cells (RBC).
1% PBS-BSA: Mix 1mL of 10X BSA and 9mL of 1X PBS.
4% formaldehyde, methanol-free Fixative Solution.
Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488.
6-well tissue culture plates.
1.5 mL microcentrifuge tubes.
Micropipette tips, sterile.
Class II Biological Safety Cabinet.
37° C, 5% CO2 humidified incubator.
Cell scrapers.
Forceps.
Disposable spatula.
15 and 50mL conical centrifuge tubes.
Refrigerated tabletop centrifuge.
V-bottom 96-well plates.
96-well tissue culture plates.
PCR thermal cyclers.
Water bath.
UV Transilluminator.
Cryogenic vials.
2.3. Confirmation of ligand expression
Non-enzymatic EDTA-based cell dissociation solution.
FACS buffer: 1X PBS with 1% FBS.
6-well tissue culture plates.
Refrigerated tabletop centrifuge.
1.5 mL microcentrifuge tube.
96-well round bottom plate.
Flow Cytometer.
2.4. B16-F10 Tumor implantation and intratumoral treatment
B16-F10 melanoma cells (ATCC, CRL-6475).
Complete DMEM, Nutrient Mixture F-12 (cDMEM/F12). Supplement DMEM/F12 with 10% FBS and 1% P/S.
0.4% Trypan Blue Solution.
T175 flasks.
15 and 50mL conical centrifuge tubes.
Micropipette tips, sterile.
Refrigerated tabletop centrifuge.
Automated Cell Counter or Hemacytometer
70 μm strainers.
1 mL Sub-Q Syringe.
30G x 1/2 in. PrecisionGlide™ specialty use sterile hypodermic needle.
70% ethanol swabs.
Small animal trimmers.
Insulin syringes with 6mm x 31G needles.
2.5. Isolation of tumor-infiltrating lymphocytes
RPMI-0: RPMI-1640 Medium.
Complete RPMI (cRPMI): Supplement RPMI-0 with 10% FBS and 1% P/S.
2X Liberase/DNAse solution: Final concentration of Liberase in the sample will be 1.67 Wunsch U/mL. Prepare fresh. Add 7.5 mL of RPMI-0 to a 5 mg (26 U) vial of Liberase™ TL Research Grade and leave it on ice for 30 minutes. For each 7.5 mL of Liberase solution, add 150 μL of 100X DNAse (from a 20 mg/mL stock).
Pre-weighed 1.5 mL microcentrifuge tubes for tumors.
6-well plates for grinding tumors.
70 μm strainers.
15 mL centrifuge tubes.
Refrigerated tabletop centrifuge.
Flow cytometer.
3. Methods
3.1. Construction of recombinant full-length NDV (rNDV) encoding an immuno-modulatory transgene (4-1BBL)
pNDV-LaSota containing the full-length antigenomic cDNA of the NDV LaSota strain was constructed by reverse transcription-polymerase chain reaction (RT-PCR) of virion-derived genomic RNA with the addition of a unique restriction enzyme site SacII (nt 1747 to 1752), into a low-copy transcription vector as shown in Figure 1 (39). The antigenomic cDNA is under the control of the T7 promoter followed by a non-coding self-cleaving hepatitis delta virus (HDV) ribozyme site and a T7 terminator.
Fig. 1. Construction of a full-length antigenomic NDV plasmid containing the transgene for murine 4-1BBL (m4-1BBL).

The T7 RNA Polymerase transcribes the antigenome starting at the T7 promoter (T7p). The HDV ribozyme site ensures the cleavage of the transcript. The ORF for 4-1BBL is flanked at the beginning with the gene end (GE) and gene start (GS) signals followed by the Kozak sequence and with the SacII site at the end after the stop codon.
As the viral RNA polymerase synthesizes and transcribes the genome in a polar and sequential manner by a start and stop mechanism, starting from the 3’ entry site, there is a gradient of expression with respect to the position of the transgene relative to the 3’ start end (40). Furthermore, studies have shown that the optimal insertion site for a transgene for efficient expression is in between the P and M genes (41). Foreign genes are inserted into the SacII cloning site as a transcriptional unit in between the P and M gene and are flanked by short conserved sequence motifs that guide transcription, namely the gene start and gene end sequences (42).
Digest and linearize pNDV-LaSota plasmid (2-5 μg) with 2-5 μL of SacII restriction enzyme for 1 hour at 37°C in a total reaction volume of 50 μL. Heat inactivate the enzyme by incubating at 65°C for 20 mins (see Note 1).
To prevent re-circularization of the linearized pNDV-LaSota plasmid, incubate the above reaction mixture with Antarctic Phosphatase for 1 hour at 37°C. Purify with the Gel and PCR Clean-up Kit following the manufacturer’s instruction. Determine the DNA concentration using a NanoDrop Spectrophotometer (see Note 1).
Generate a codon-optimized insert using gene synthesis. Make sure to add a Kozak consensus sequence before the start codon to enhance translation efficiency of the inserted transgene. In addition, take care to ensure that the cloned transgene follows the “rule of six” to ensure optimal viral replication. This can be achieved with careful primer design, with adding additional stop codons to the transgene (see Note 2, 3 and 4).
Amplify the insert by PCR with primers specifically designed to include 15 nucleotides of 5’ overhangs that are complementary to the 5’ and 3’ cloning ends of the linearized pNDV-LaSota plasmid (Table 1). These overhangs are necessary for the In-Fusion® HD system-based cloning.
Mix the PCR reactions with the gel loading dye and load into individual wells on a 1.5% Agarose-TAE gel in a gel box filled with 1X TAE buffer, including one well with the DNA ladder. Run the gel at 80-150 V for 45-60 mins or until the dye front is 80% of the way down the gel.
After visualization on a UV transilluminator, cut the gene band of the appropriate size from the gel and place it into a microcentrifuge tube.
Purify the DNA using the Gel and PCR Clean-up Kit following the manufacturer’s instruction. Determine the DNA concentration using the NanoDrop Spectrophotometer.
Clone the insert into the linearized pNDV-LaSota plasmid using the In-Fusion® HD Cloning Plus kit following the manufacturer’s instructions.
Transform 50 μL of Stable Competent E. coli cells with 2.5 μL of the In-Fusion reaction mixture for 30 mins on ice (see Note 5).
Heat-shock the transformation reaction for 30 secs at 42°C and incubate it for 2 mins on ice.
Add 1000 μL of SOC medium to the transformation reaction and incubate for 1 hour at 37°C in a shaker.
Centrifuge the transformation reaction at 6000 rpm for 5 mins. Discard the supernatant and resuspend the pellet in 100 μL of SOC medium. Plate all of the transformation onto a 10 cm LB-Ampicillin agar plate and incubate overnight at 30°C.
The next day, pick individual colonies and grow them overnight in a shaker with LB broth supplemented with ampicillin at 30°C.
Isolate plasmid DNA using the Miniprep Kit following the manufacturer’s instructions and sequence the plasmids with the screening primers listed in Table 1 to confirm presence of the transgene and absence of any mutations introduced during the PCR or cloning procedure.
The correct clone is then grown overnight in 200 mL of LB broth supplemented with ampicillin at 30°C.
Isolate high purity, endotoxin-free plasmid DNA using the plasmid Midiprep Kit following the manufacturer’s instructions. Measure DNA concentration using the NanoDrop Spectrophotometer.
Table 1:
List of primers
| Primer | Sequence |
|---|---|
| 4-1BBL Cloning for | GCACCGAGTTCCCCCCCGCGGTTAGAAAAAATACGGGTAGAACCGCCACCATGGACCGCGCCGTTAGC |
| 4-1BBL Cloning Rev | AGTTGGACCTTGGGTCCGCGGATTATCATTCCCATGGGTTGTC |
| Sequencing/RT-PCR primer For | GATATCTAGATTAGAAAAAATACGGGTAGAACCGCCACC |
| Sequencing/RT-PCR primer Rev | CAAAGTACAGCCCAATTGTCC |
3.2. Construction of the NDV NP, P and L and expression plasmids
The rescue of rNDV viruses requires the co-transfection of cDNA encoding the ribonucleoprotein complex or RNP, which consists of the viral polymerase (P and L proteins), the nucleoprotein (NP) and the full-length antigenomic RNA of the virus.
Genes encoding each of the L, P and NP proteins are cloned into a pTM1 expression vector under the transcriptional control of a T7 promoter. The expression vector also includes T7 terminator, a pUC19 origin of replication and an ampicillin resistance gene. The full-length DNAs of each gene for construction of the plasmids are obtained by RT-PCR amplification from viral RNA. Additionally a codon-optimized T7 RNA polymerase is cloned into a pCAGGs expression vector (pCAGGs-T7opt) (43).
3.3. Rescue and amplification of recombinant NDV expressing the transgene
The plasmid-based rescue of recombinant NDV viruses as shown in Figure 2 entails the transfection of cells that constitutively express T7 polymerase such as BSRT7 cells, with 1) plasmid DNA encoding the L, NP and P proteins of NDV under the transcriptional control of a T7 promoter (pTM1-L, pTM1-NP and pTM1-P, respectively), 2) pCAGGs expression vector encoding T7 RNA polymerase and 3) a plasmid DNA encoding the NDV antigenome with the transgene under the transcriptional control of a T7 promoter. Subsequently, the rescue of infectious NDV viruses is facilitated by the amplification and transcription of the NDV antigenome in embryonated chicken eggs.
Figure 2: Rescue of recombinant NDVs expressing m4-1BBL.

BSRT7 cells are co-transfected with plasmids encoding L, NP and P proteins under the transcriptional control of a T7 promoter and the plasmid encoding the NDV antigenome with the cloned transgene (4-1BBL) under the transcriptional control of a T7 promoter. The supernatants of transfected cells are injected into the allantoic cavities of 9 to 11-day-old embryonated chicken eggs.
3.3.1. Setting up rescue transfections:
Maintain BSRT7 cells in cDMEM. One day before rescue transfections, split BSRT7 cells into 6-well tissue culture plates at a concentration of 3 × 105 cells per well to achieve 75 to 80% confluency on the day of transfection (see Note 6).
The next day, perform transfections in the tissue culture hood. Warm the transfection reagent TransIT-LT1 to room temperature and gently vortex before using (see Note 7).
Prepare the transfection plasmid cocktail in 250 μL of Opti-MEM Reduced-Serum Medium in a sterile 1.5 mL microcentrifuge tube. For a single transfection, add 1 μg of pTM1-NP, 0.5 μg of pTM1-P, 0.5 μg of pTM1-L, 1 μg of pCAGGs-T7opt and 2 μg of pNDV-LaSota into the microcentrifuge tube with 250 μL of Opti-MEM and mix gently by pipetting (see Note 7).
Add dropwise 15 μL of the TransIT-LT1 reagent to the plasmid-Opti-MEM mixture and mix gently by pipetting. Incubate the transfection plasmid cocktail mix at room temperature for 30 mins (see Note 8).
Add the transfection plasmid cocktail mix dropwise to the wells and gently rock the plate back and forth to evenly distribute the transfection plasmid cocktail mix. Incubate at 37°C, 5% CO2 for 48 hours.
Scrape the cells along with the tissue culture supernatant into a sterile 1.5 mL microcentrifuge tube and inoculate 500 μL directly into the allantoic cavity of 9 to 11-day-old SPF embryonated chicken eggs. Incubate the eggs at 37°C for 2-3 days.
After incubation, place the infected eggs at 4°C overnight.
Harvest the allantoic fluid in a biosafety cabinet by carefully taping the apical section of the egg, over the air sac with a spoon after sterilizing the eggs with 70% ethanol.
Remove the cracked eggshells with forceps and expose the allantoic fluid by carefully peeling back the allantoic membrane without damaging the blood vessels and the yolk.
Press down the embryo with a disposable spatula. Collect the allantoic fluid and transfer into a 15 mL centrifuge tube on ice (see Note 9).
Centrifuge the allantoic fluid at 1500 rpm for 5 mins at 4°C to remove any debris and transfer the supernatant into a new sterile 15 mL centrifuge tube.
3.3.2. Hemagglutination (HA) assay confirming positive rNDV rescue:
A positive rescue is confirmed by a hemagglutination (HA) assay using 0.5% turkey RBC in 1X PBS, with approximately 106 plaque forming units (pfu) per mL giving a positive signal.
HA assays are carried out in V-bottom 96-well plates with negative controls (either 1X PBS or uninfected allantoic fluid) and/or positive controls (NDV virus grown in allantoic fluid) included to validate the assay.
Pipette 50 μL of 1X PBS per well into all wells, except for the first row.
Pipette 100 μL of the allantoic fluid from individual eggs into the wells in the first row of the plate and perform 2-fold serial dilutions across the plate.
Pipette 50 μL of 0.5% turkey RBC into all the wells and gently tap the plate to mix.
Incubate the plate at 4°C (or on ice) for 30-45 mins or until a clear pellet is observed in the negative control wells. Absence of a red pellet in rows 2, 3 and 4 as shown in Figure 3 demonstrates positive viral rescue of rNDV-41BBL.
Figure 3: Hemagglutination assay (HA) confirming positive rNDV rescue.

Postive rescues with the presence of viral particles induce hemagglutination, whereas negative rescues or the abscence of viruses induce a red pellet in the bottom of the well. The first row is the negative control (1X PBS) and rows 2, 3 and 4 show positive rescue of rNDVs expressing 4-1BBL.
3.3.3. RT-PCR confirmation of viral transgene:
Confirm presence of the transgene by extracting the viral RNA using the RNA Mini Kit following manufacturer’s instructions.
Set up an RT-PCR for the transgene with the screening primers listed in Table 1 using wild-type NDV (NDV-WT) as control. Run the products of the RT-PCR reaction on a 1.5% Agarose-TAE gel in a gel box filled with 1X TAE buffer, with one well containing the DNA ladder.
Once the bands have been confirmed to be of the right size as shown in Figure 4, gel extract the appropriate bands using the Gel and PCR Clean-up Kit following the manufacturer’s instructions.
Confirm sequence fidelity of the extracted DNA by Sanger sequencing using the primers listed in Table 1.
Once the sequence has been confirmed, grow stocks of the newly rescued virus by dilution purification. Inoculate 9 to 10-day-old SPF embryonated chicken eggs with 100 μL each of 10−7 serially diluted virus in 1X PBS.
Repeat steps 7 to 11 in Section 3.3.1 and steps in Section 3.3.2 to collect the allantoic fluid and confirm positive viral growth by an HA assay.
Aliquot 0.5 mL of the confirmed virus in cryogenic tubes and store up to 10 years at −80°C.
Figure 4: Confirmation of presence of transgene by RT-PCR.
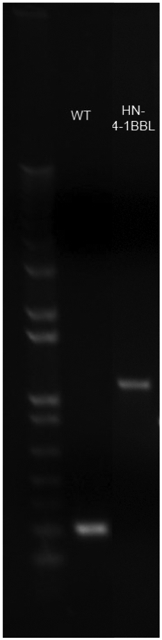
Viral RNA was extracted from positive rNDV rescues and using primers mentioned in Table 1, RT-PCR was set up to confirm transgene presence. Viral RNA from NDV-WT was used as a positive control.
3.3.4. Titration of rNDV stocks by immunofluorescence
One day before infection set up a 96-well plate of Vero cells at a concentration of 2 × 104 cells/mL (see Note 10).
On the day of the infection, thaw out one vial each of the rescued virus and wild-type NDV (NDV-WT) as control. Perform a five-fold serial dilution of the viruses in a separate sterile 96-well plate with 1X PBS.
Aspirate the medium from the Vero cells in the 96-well plate.
Pipette 50 μL of the virus dilutions from the dilution plate into the 96-well plate with the Vero cells using a multi-channel pipette.
Incubate at 37°C, 5% CO2 for 1 hour. Gently rock the plates every 15 mins to ensure the cells are evenly infected with the virus dilutions.
Aspirate the virus dilutions off the Vero cells and add 100 μL of cDMEM .
Aspirate the medium from the infected Vero cells and wash the cells once with 100 μL of 1X PBS.
Fix cells with 100 μL of Fixative Solution at 4°C for 10 mins protected from light.
Aspirate the 4% formaldehyde and wash once with 100 μL of 1X PBS.
Block cells with 1% PBS-BSA for 2 hours at 4°C.
Incubate cells with the primary antibody either overnight at 4°C or at room temperature for 2 hours (Primary antibody: anti-NDV polyclonal rabbit serum diluted in 1X PBS at 1:500).
Aspirate the primary antibody and wash the cells four times with 100 μL of 1X PBS.
Incubate the cells with the secondary antibody for one hour at room temperature protected from light (Secondary antibody: anti-rabbit-Alexa Fluor 488 diluted in 1X PBS at 1:1000).
Aspirate the secondary antibody and wash the cells four times with 100 μL of 1X PBS.
Count the infected green cells, starting from the undiluted wells until you find a well down the dilution series with a countable number of cells. Count the cells in the entire well.
The titer of the virus can be determined by the following formula:
3.4. Confirmation of ligand expression
For confirmation of expression of the transgene in the infected cells, one can utilize any human or murine cancer cell line known to be susceptible to NDV infection. For the purposes of subsequent testing in syngeneic tumor models, B16-F10 melanoma is commonly used. For confirmation of surface expression, flow cytometry is a commonly used method.
Split B16-F10 cells into 6-well tissue culture plates to achieve approximately 80% confluency the following day. Ensure that a sufficient number of wells are available to allow for treatment with 1X PBS, wild-type NDV (NDV-WT), and rNDV-4-1BBL.
On the day of infection, dilute NDV to 8×106 pfu/mL in cDMEM..
Aspirate the medium, rinse with 2 mL PBS, and infect the cells at an MOI of 2 with 200 μL of diluted control or rNDV-4-1BBL. Incubate the plates at room temperature for 1 hour. Gently rock the plates every 15 mins to ensure even distribution of the virus over the cells and to prevent the cells from drying.
After 1 hour, add 2 mL of cDMEM to each plate. Incubate for 24 hours at 37°C.
Aspirate the medium and wash the cells with 2 mL of 1X PBS twice. Add 1 mL of Cellstripper™ solution to each well and incubate at 37°C for up to 5 mins. Cells may require pipetting or a mechanical stripper to detach from the plate.
Collect the cells in microcentrifuge tubes and spin down at 4°C at 5000 rpm for 5 mins.
Meanwhile, prepare the antibody staining solution in FACS buffer according to the recommended staining concentration.
Resuspend the cells in 100 μL of antibody staining solution and transfer to a 96-well round bottom plate. Cover and incubate on ice for 30 mins.
Spin down the plate at 2500 rpm in a refrigerated tabletop centrifuge for 2 mins. Discard the staining solution by inverting the plate.
Resuspend the cells in 200 μL of FACS buffer, spin down, and discard. Repeat the wash step 2 more times.
Resuspend the cells in 200 μL FACS buffer and analyze on a flow cytometer.
3.5. Model assay of efficacy using intratumoral administration
As a model system, the murine B16-F10 melanoma tumor derived from C57BL/6 mice is the most widely used (44). To assess the therapeutic efficacy of the virus both in the treated tumor and the non-treated tumor, a bilateral flank B16-F10 melanoma model is established to characterize the local and abscopal effect. Once tumors have been established and treated with intratumoral injections of rNDV-41BBL, both the treated and the non-treated tumors will be analyzed for immune infiltration, as a read-out for anti-tumor efficacy.
3.5.1. B16-F10 Tumor implantation:
B16-F10 cells are maintained in cDMEM/F12 (see Note 11).
Two days prior to implantation, split B16-F10 cells and expand into several T175cm2 flasks to achieve 60 to 75% confluency on the day of implantation. The number of T175cm2 flasks depends on the number of mice that need to be implanted.
On the day of implantation, rinse B16-F10 cells with 1X PBS and detach using 3mL of trypsin-EDTA (0.05%).
Once the cells have lifted off, add 7 mL of cDMEM/F12 medium and pipette up and down to obtain a single-cell suspension.
Transfer the cells from all of the flasks into a 50 mL conical centrifuge tube and pellet the cells by spinning it at 1500 rpm for 5 mins at 4°C.
Decant the supernatant and resuspend the pellet in 10 mL of ice-cold 1X PBS and pipette it through a 70 μm cell strainer into a new sterile 50 mL conical centrifuge tube.
Count the viable cells on a hemacytometer or an automated cell counter using Trypan Blue (see Note 12).
Resuspend the tumor cells at a concentration of 2 × 106 cells/mL in ice-cold 1X PBS. This final cell suspension is kept on ice throughout the implantation. It is critical to implant the mice as quickly as possible, as viability of the cells decreases over time.
4-6 week old female C57BL/6 mice are used for implantation. After the mice have been anesthetized appropriately, shave the fur off the right flank and wipe the area with a 70% ethanol swab.
Resuspend the cells by inverting the tube kept on ice 2-3 times and using a 1 mL Sub-Q syringe without the needle attached to it to draw up the cells. Attach a new sterile 30G X 1/2 needle to the syringe for implantation.
Gently insert the needle very superficially under the skin and inject 100 μL of cell suspension intradermally. Avoid injecting the underlying muscle and gently withdraw the needle. Place the mice back in the cage and allow them to recover.
For tumor implantation on the left flank on Day 4, repeat the above steps and dilute the cells to 1 × 106 cells/mL in ice-cold 1X PBS before implantation. Repeat steps 9 to 11 for implantation.
On Day 6 randomize the mice among the different treatment cohorts prior to the start of the experiment.
3.5.2. Intratumoral injection of tumors with rNDV-4-1BBL
On Day 7, thaw out the virus on ice and depending on the titer of the virus, dilute it down so that 1 × 107 pfu is delivered into the tumor in a volume of 100 μL.
Anesthetize the tumor-implanted mice appropriately.
Draw up 100 μL of the virus in an insulin syringe or a syringe with a 31G needle. Slowly glide the needle into the tumor, towards the middle and inject.
Place the mice back in the cage and allow them to recover.
Repeat steps 1-4 on days 9, 11 and 13 with freshly thawed out virus.
3.5.3. Isolation of tumor-infiltrating lymphocytes
Euthanize animals and remove tumors. Be careful not to remove too much connective tissue with the tumors. Work with one animal cage at a time.
Place the tumors into the pre-weighed microcentrifuge tubes.
After each cage, weigh and record the tube weight and add 500 μL of RPMI-0 (without serum) to each tube and place on ice.
When all tumors have been collected in tubes, prepare 2X Liberase/DNAse mix and put on ice.
Mince the tumors with scissors into small pieces.
Add 0.5 mL RPMI-0/DNAse/Liberase solution to tumors and incubate at 37°C on a shaker for 30 mins.
At the end of the incubation, transfer the tumors into 70 μm strainers in 6-well plates.
Pass the tumors through the strainers using the back end of a 1 or 3 mL syringe plunger.
Rinse the strainers with 3 mL of cRPMI.
Transfer to pre-labeled 15 mL centrifuge tubes.
Spin down the cells at 2000 rpm for 5 min.
Resuspend the cells in cRPMI.
Proceed with labeling for flow cytometry.
Following dissociation, the treated and non-treated tumors were analyzed for tumor-infiltrating lymphocyte (TIL) infiltration by flow cytometry. As shown in Figure 7, introduction of 4-1BBL into NDV resulted in enhanced T cell infiltration in both virus-injected and distant tumors, when compared to the parental NDV.
Figure 7: NDV-HN-4-1BBL induces increased T cell infiltration in the injected and distant tumors.

Animals bearing bilateral flank B16 melanoma tumors were treated intratumorally with the indicated virus as previously. After 4 treatments, animals were euthanized and tumor-infiltrating lymphocytes from bilateral tumors were analyzed by flow cytometry.
3.6. Conclusions:
The ability to genetically engineer NDV to express immunostimulatory agents has a potential to enhance its immune-potentiating properties. We demonstrated that introduction of immunomodulatory genes into NDV has the potential to potentiate T cell infiltration into both treated and distant tumors. Such properties have the potential to obviate the need for systemic administration of NDV, as they rely primarily on NDV-induced anti-tumor immune responses. Administration of such agents within the context of systemic immune checkpoint blockade carries a potential to have a strong systemic anti-tumor effect as we demonstrated previously.
The agents for intratumoral modulation may span a range of genes targeting both innate and adaptive arms of the immune system. Optimal targets for intratumoral modulation using NDV are currently unknown and may be influenced by the differences in tumor types, levels of virus replication, or even differences in the composition in tumor microenvironment in the same tumor type. Selection of optimal agents for intratumoral modulation may require evaluation of recombinant NDV in several syngeneic tumor models in order to determine the generalizability of the treatment to different tumor types and to define potential biomarkers that may guide tumor selection.
Figure 5: Confirmation of surface expression of the transgene (4-1BBL) in B16-F10 infected cells by flow cytometry.

B16-F10 cells were infected with NDV-WT or rNDV-4-1BBL at an MOI of 2. Twenty-four hours later, the cells were collected using Cell Stripper, processed for staining of surface 4-1BBL and analyzed by flow cytometry. Blue: NDV-WT, red: rNDV-m4-1BBL.
Figure 6: Treatment schema for intratumoral administration of rNDV-4-1BBL in a B16-F10 murine melanoma tumor model.
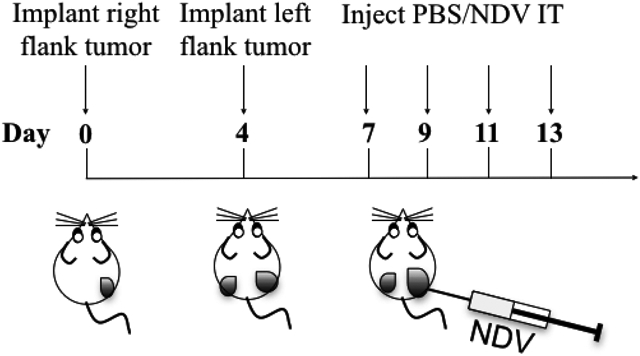
Bilateral flank tumor models were established by injecting C57BL/6 mice with 2 × 105 B16-F10 cells in the right flank intradermally (i.d.) on day 0 and 1 × 105 cells in the left flank on day 4. On days 7, 9, 11 and 13, mice were treated with four 100 μL intratumoral injections of rNDV-4-1BBL in 1X PBS (1 × 107 pfu). On day 15 the animals were euthanized and the tumors were processed for analysis of tumor-infiltrating lymphocytes.
Acknowledgements:
D.Z. received funding from the Ovarian Cancer Research Foundation, and the Department of Defense Ovarian Cancer Research Academy (OC150111). D.Z. is a member of the Parker Institute for Cancer Immunotherapy, which supports the MSKCC Cancer Immunotherapy Program. MSKCC is supported by the NCI Core grant P30 CA008748.
Footnotes
Competing financial interests:
D.Z. is an inventor on a patent concerning the uses of recombinant Newcastle Disease Virus for cancer therapy.
When digesting pNDV-LaSota plasmid with SacII restriction enzyme, overnight incubation at 37°C prior to Antarctic Phosphatase treatment can enhance linearization efficiency with reduction in background. Run the linearized vector on a 1.5% Agarose-TAE gel to ensure optimal digestion. After visualization on a UV transilluminator, excise the appropriate band using a razor blade or a scalpel and place it into a 1.5 mL microcentrifuge tube. Purify the linearized vector using the Gel and PCR Clean-up Kit following the manufacturer’s instructions.
When designing primers to PCR-amplify the insert for InFusion cloning, the optimal length of homologous overlap between the terminus of the linearized plasmid and insert can vary between 15 nts for a single insert and 20 nts for multiple inserts. Desalted, high-quality PCR primers are preferred.
Codon optimization of the transgene can enhance gene expression.
Insertion of large transgenes (>3 kb) can attenuate the virus.
When selecting a competent E. coli strain for cloning applications, select one that is compatible with cloning large unstable DNA.
BSRT7 cells that are used for transfection should be maintained at a low passage number and under mycoplasma-free conditions to enhance transfection efficiency.
While any high efficiency transfection reagent could be used, for this particular protocol we use TransIT®-LT1 Transfection Reagent from Mirus. Transfection protocol can be modified based on reagent of choice.
When adding the TransIT-LT1 Reagent to the plasmid-Opti-MEM mix, avoid touching the sides of the plastic tube.
Avoid damaging the yolk sac membrane and minimize the amount of blood in the allantoic fluid when extracting it from the egg.
When plating Vero cells for virus titration, use poly D-lysine coated plates to prevent the cells from sloughing off during the wash steps.
Maintain B16-F10 cells at a low passage number to enhance implantation efficiency.
Make sure the viability of the B16-F10 cells to be implanted is >90%.
Bibliography
- 1.Lancaster JE (2007) A History of Newcastle Disease with Comments on its Economic Effects. World’s Poultry Science Journal 32 (2):167–175. doi: 10.1079/WPS19760001 [DOI] [Google Scholar]
- 2.Fields BN (2007) Paramyxoviridae: the viruses and their replication. [Google Scholar]
- 3.Nagai Y, Hamaguchi M, Toyoda T (1989) Molecular biology of Newcastle disease virus. Progress in veterinary microbiology and immunology 5:16–64 [PubMed] [Google Scholar]
- 4.Krishnamurthy S, Samal SK (1998) Nucleotide sequences of the trailer, nucleocapsid protein gene and intergenic regions of Newcastle disease virus strain Beaudette C and completion of the entire genome sequence. J Gen Virol 79 ( Pt 10):2419–2424. doi: 10.1099/0022-1317-79-10-2419 [DOI] [PubMed] [Google Scholar]
- 5.Panda A, Huang Z, Elankumaran S, Rockemann DD, Samal SK (2004) Role of fusion protein cleavage site in the virulence of Newcastle disease virus. Microbial pathogenesis 36 (1):1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bukreyev A, Huang Z, Yang L, Elankumaran S, Claire M. St., Murphy BR, Samal SK, Collins PL (2005) Recombinant Newcastle Disease Virus Expressing a Foreign Viral Antigen Is Attenuated and Highly Immunogenic in Primates. Journal of Virology 79 (21):13275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim SH, Samal SK (2016) Newcastle Disease Virus as a Vaccine Vector for Development of Human and Veterinary Vaccines. Viruses 8 (7). doi: 10.3390/v8070183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flanagan AD, Love R, Tesar W (1955) Propagation of Newcastle disease virus in Ehrlich ascites cells in vitro and in vivo. Proc Soc Exp Biol Med 90 (1):82–86 [DOI] [PubMed] [Google Scholar]
- 9.Sinkovics J (1957) Studies on the biological characteristics of the Newcastle disease virus (NDV) adapted to the brain of newborne mice. Arch Gesamte Virusforsch 7 (4):403–411 [DOI] [PubMed] [Google Scholar]
- 10.Eaton MD, Levinthal JD, Scala AR (1967) Contribution of antiviral immunity to oncolysis by Newcastle disease virus in a murine lymphoma. Journal of the National Cancer Institute 39 (6):1089–1097 [PubMed] [Google Scholar]
- 11.Cassel WA, Garrett RE (1965) Newcastle disease virus as an antineoplastic agent. Cancer 18 (7):863–868. doi: [DOI] [PubMed] [Google Scholar]
- 12.Cassel WA, Garrett RE (1966) Tumor immunity after viral oncolysis. J Bacteriol 92 (3):792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murray DR, Cassel WA, Torbin AH, Olkowski ZL, Moore ME (1977) Viral oncolysate in the management of malignant melanoma. II. Clinical studies. Cancer 40 (2):680–686 [DOI] [PubMed] [Google Scholar]
- 14.Cassel WA, Murray DR (1992) A ten-year follow-up on stage II malignant melanoma patients treated postsurgically with Newcastle disease virus oncolysate. Medical oncology and tumor pharmacotherapy 9 (4):169–171 [DOI] [PubMed] [Google Scholar]
- 15.Batliwalla FM, Bateman BA, Serrano D, Murray D, Macphail S, Maino VC, Ansel JC, Gregersen PK, Armstrong CA (1998) A 15-year follow-up of AJCC stage III malignant melanoma patients treated postsurgically with Newcastle disease virus (NDV) oncolysate and determination of alterations in the CD8 T cell repertoire. Mol Med 4 (12):783–794 [PMC free article] [PubMed] [Google Scholar]
- 16.Kirchner HH, Anton P, Atzpodien J (1995) Adjuvant treatment of locally advanced renal cancer with autologous virus-modified tumor vaccines. World journal of urology 13 (3):171–173 [DOI] [PubMed] [Google Scholar]
- 17.Csatary LK, Eckhardt S, Bukosza I, Czegledi F, Fenyvesi C, Gergely P, Bodey B, Csatary CM (1993) Attenuated veterinary virus vaccine for the treatment of cancer. Cancer detection and prevention 17 (6):619–627 [PubMed] [Google Scholar]
- 18.Csatary LK, Gosztonyi G, Szeberenyi J, Fabian Z, Liszka V, Bodey B, Csatary CM (2004) MTH-68/H oncolytic viral treatment in human high-grade gliomas. Journal of neuro-oncology 67 (1–2):83–93 [DOI] [PubMed] [Google Scholar]
- 19.Freeman AI, Zakay-Rones Z, Gomori JM, Linetsky E, Rasooly L, Greenbaum E, Rozenman-Yair S, Panet A, Libson E, Irving CS, Galun E, Siegal T (2006) Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther 13 (1):221–228. doi: 10.1016/j.ymthe.2005.08.016 [DOI] [PubMed] [Google Scholar]
- 20.Pecora AL, Rizvi N, Cohen GI, Meropol NJ, Sterman D, Marshall JL, Goldberg S, Gross P, O’Neil JD, Groene WS, Roberts MS, Rabin H, Bamat MK, Lorence RM (2002) Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced solid cancers. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 20 (9):2251–2266. doi: 10.1200/jco.2002.08.042 [DOI] [PubMed] [Google Scholar]
- 21.Lorence RM, Pecora AL, Major PP, Hotte SJ, Laurie SA, Roberts MS, Groene WS, Bamat MK (2003) Overview of phase I studies of intravenous administration of PV701, an oncolytic virus. Current opinion in molecular therapeutics 5 (6):618–624 [PubMed] [Google Scholar]
- 22.Schirrmacher V (2016) Fifty Years of Clinical Application of Newcastle Disease Virus: Time to Celebrate! Biomedicines 4 (3). doi: 10.3390/biomedicines4030016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reichard KW, Lorence RM, Cascino CJ, Peeples ME, Walter RJ, Fernando MB, Reyes HM, Greager JA (1992) Newcastle disease virus selectively kills human tumor cells. The Journal of surgical research 52 (5):448–453 [DOI] [PubMed] [Google Scholar]
- 24.Zorn U, Dallmann I, Grosse J, Kirchner H, Poliwoda H, Atzpodien J (1994) Induction of cytokines and cytotoxicity against tumor cells by Newcastle disease virus. Cancer biotherapy 9 (3):225–235 [DOI] [PubMed] [Google Scholar]
- 25.Sinkovics JG, Horvath JC (2000) Newcastle disease virus (NDV): brief history of its oncolytic strains. J Clin Virol 16 (1):1–15 [DOI] [PubMed] [Google Scholar]
- 26.Peeters BP, de Leeuw OS, Koch G, Gielkens AL (1999) Rescue of Newcastle disease virus from cloned cDNA: evidence that cleavability of the fusion protein is a major determinant for virulence. J Virol 73 (6):5001–5009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molouki A, Peeters B (2017) Rescue of recombinant Newcastle disease virus: a short history of how it all started. Arch Virol 162 (7):1845–1854. doi: 10.1007/s00705-017-3308-2 [DOI] [PubMed] [Google Scholar]
- 28.Vigil A, Park MS, Martinez O, Chua MA, Xiao S, Cros JF, Martinez-Sobrido L, Woo SL, Garcia-Sastre A (2007) Use of reverse genetics to enhance the oncolytic properties of Newcastle disease virus. Cancer Res 67 (17):8285–8292. doi: 10.1158/0008-5472.can-07-1025 [DOI] [PubMed] [Google Scholar]
- 29.Vigil A, Martinez O, Chua MA, Garcia-Sastre A (2008) Recombinant Newcastle disease virus as a vaccine vector for cancer therapy. Mol Ther 16 (11):1883–1890. doi: 10.1038/mt.2008.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zamarin D, Martinez-Sobrido L, Kelly K, Mansour M, Sheng G, Vigil A, Garcia-Sastre A, Palese P, Fong Y (2009) Enhancement of oncolytic properties of recombinant newcastle disease virus through antagonism of cellular innate immune responses. Mol Ther 17 (4):697–706. doi: 10.1038/mt.2008.286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Song KY, Wong J, Gonzalez L, Sheng G, Zamarin D, Fong Y (2010) Antitumor efficacy of viral therapy using genetically engineered Newcastle disease virus [NDV(F3aa)-GFP] for peritoneally disseminated gastric cancer. J Mol Med (Berl) 88 (6):589–596. doi: 10.1007/s00109-010-0605-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li P, Chen CH, Li S, Givi B, Yu Z, Zamarin D, Palese P, Fong Y, Wong RJ (2011) Therapeutic effects of a fusogenic newcastle disease virus in treating head and neck cancer. Head & neck 33 (10):1394–1399. doi: 10.1002/hed.21609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zamarin D, Palese P (2012) Oncolytic Newcastle disease virus for cancer therapy: old challenges and new directions. Future Microbiol 7 (3):347–367. doi: 10.2217/fmb.12.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zamarin D, Holmgaard RB, Subudhi SK, Park JS, Mansour M, Palese P, Merghoub T, Wolchok JD, Allison JP (2014) Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Science translational medicine 6 (226):226ra232. doi: 10.1126/scitranslmed.3008095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oseledchyk A, Ricca JM, Gigoux M, Ko B, Redelman-Sidi G, Walther T, Liu C, Iyer G, Merghoub T, Wolchok JD, Zamarin D (2018) Lysis-independent potentiation of immune checkpoint blockade by oncolytic virus. Oncotarget 9 (47):28702–28716. doi: 10.18632/oncotarget.25614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zamarin D, Holmgaard RB, Ricca J, Plitt T, Palese P, Sharma P, Merghoub T, Wolchok JD, Allison JP (2017) Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nature communications 8:14340. doi: 10.1038/ncomms14340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watts TH (2005) TNF/TNFR family members in costimulation of T cell responses. Annual review of immunology 23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839 [DOI] [PubMed] [Google Scholar]
- 38.Sznol M, Hodi FS, Margolin DF, McDermott D, Ernstoff JM, Kirkwood JM, Wojtaszek C, Feltquate D, Logan T (2008) Phase I study of BMS-663513, a fully human anti-CD137 agonist monoclonal antibody, in patients (pts) with advanced cancer (CA). Paper presented at the 2008 ASCO Annual Meeting, Chicago, IL, [Google Scholar]
- 39.Nakaya T, Cros J, Park MS, Nakaya Y, Zheng H, Sagrera A, Villar E, Garcia-Sastre A, Palese P (2001) Recombinant Newcastle disease virus as a vaccine vector. J Virol 75 (23):11868–11873. doi: 10.1128/jvi.75.23.11868-11873.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao H, Peeters BP (2003) Recombinant Newcastle disease virus as a viral vector: effect of genomic location of foreign gene on gene expression and virus replication. J Gen Virol 84 (Pt 4):781–788. doi: 10.1099/vir.0.18884-0 [DOI] [PubMed] [Google Scholar]
- 41.Zhao W, Zhang Z, Zsak L, Yu Q (2015) P and M gene junction is the optimal insertion site in Newcastle disease virus vaccine vector for foreign gene expression. J Gen Virol 96 (Pt 1):40–45. doi: 10.1099/vir.0.068437-0 [DOI] [PubMed] [Google Scholar]
- 42.Marcos F, Ferreira L, Cros J, Park MS, Nakaya T, Garcia-Sastre A, Villar E (2005) Mapping of the RNA promoter of Newcastle disease virus. Virology 331 (2):396–406. doi: 10.1016/j.virol.2004.10.040 [DOI] [PubMed] [Google Scholar]
- 43.Beaty SM, Park A, Won ST, Hong P, Lyons M, Vigant F, Freiberg AN, tenOever BR, Duprex WP, Lee B (2017) Efficient and Robust Paramyxoviridae Reverse Genetics Systems. mSphere 2 (2). doi: 10.1128/mSphere.00376-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Overwijk WW, Restifo NP (2001) B16 as a Mouse Model for Human Melanoma. Current protocols in immunology / edited by John E Coligan [et al. ] CHAPTER:Unit-20.21. doi: 10.1002/0471142735.im2001s39 [DOI] [PMC free article] [PubMed] [Google Scholar]