

Abstract
Genes with sex-biased expression in Drosophila are thought to underlie sexually dimorphic phenotypes and have been shown to possess unique evolutionary properties. However, the forces and constraints governing the evolution of sex-biased genes in the somatic tissues of Drosophila are largely unknown. By using population-scale RNA sequencing data, we show that sex-biased genes in the Drosophila brain are highly enriched on the X Chromosome and that most are biased in a species-specific manner. We show that X-linked male-biased genes, and to a lesser extent female-biased genes, are enriched for signatures of directional selection at the gene expression level. By examining the evolutionary properties of gene-flanking regions on the X Chromosome, we find evidence that adaptive cis-regulatory changes are more likely to drive the expression evolution of X-linked male-biased genes than other X-linked genes. Finally, we examine whether constraint owing to broad expression across multiple tissues and genetic constraint owing to the largely shared male and female genomes could be responsible for the observed patterns of gene expression evolution. We find that expression breadth does not constrain the directional evolution of gene expression in the brain. Additionally, we find that the shared genome between males and females imposes a substantial constraint on the expression evolution of sex-biased genes. Overall, these results significantly advance our understanding of the patterns and forces shaping the evolution of sexual dimorphism in the Drosophila brain.
Even though males and females share nearly identical genomes—with the exception of the Y Chromosome—they show clear phenotypic differences at physiological, morphological, and behavioral levels (Darwin 1871; Arnqvist and Rowe 2005). Thus, one of the most exciting challenges in evolutionary biology is understanding how males and females use shared genomes to produce sexually dimorphic phenotypes. A popular and tractable approach to understanding sexual dimorphism is to examine differences between the sexes at the transcriptome level (Jin et al. 2001; Ellegren and Parsch 2007; Grath and Parsch 2016). Historically, most work in Drosophila has focused on genes with sex-biased expression in either gonad or whole-body samples, where the differential expression signal is dominated by gonad expression (Parisi et al. 2003; Ranz et al. 2003; Whittle and Extavour 2019). Studies examining the forces influencing the evolution of these genes found that sex-biased genes—and male-biased genes in particular—are highly enriched for genes showing signatures of positive selection at the protein level, suggesting that the evolution of sex-biased genes is central to adaptive evolution in Drosophila (Pröschel et al. 2006; Grath and Parsch 2016). Relative to female-biased and unbiased genes, the expression levels of male-biased genes tend to diverge rapidly between species (Ranz et al. 2003; Zhang et al. 2007). However, genes frequently gain and lose sex bias over evolutionary time (Ranz et al. 2003; Zhang et al. 2007). It has also been shown that sex-biased genes within gonads and whole-body samples are distributed among chromosomes in a highly nonrandom manner: Male-biased genes are significantly depleted on the X Chromosome, whereas female-biased genes are significantly enriched on the X Chromosome, which may be attributable to the lack of X Chromosome dosage compensation in the male germline (Parisi et al. 2003; Meiklejohn et al. 2011; Meiklejohn and Presgraves 2012). Finally, several studies have examined sex-biased gene expression in the Drosophila melanogaster brain (Catalán et al. 2012; Huylmans and Parsch 2015; Pacifico et al. 2018). In contrast to findings in the gonads, male- and female-biased genes were both enriched on the X Chromosome (Catalán et al. 2012; Huylmans and Parsch 2015), suggesting that the chromosomal distributions of genes with sex-biased expression are highly tissue dependent. Despite these advances, the evolutionary properties of genes with sex-biased expression in sexually dimorphic somatic tissues such as the Drosophila brain remain largely unknown (Chang et al. 2011; Catalán et al. 2012).
Some possible forces, constraints, and phenomena associated with expression evolution include the faster-X effect, expression breadth across tissues, and genetic constraint. The faster-X effect is a phenomenon in which genes and regulatory regions tend to evolve more rapidly on the X Chromosome than on the autosomes (Charlesworth et al. 1987; Meisel et al. 2012; Meisel and Connallon 2013). This can be owing to the relatively rapid fixation of beneficial recessive mutations on the X Chromosome, and this effect has been observed in both sex-biased and unbiased genes but is usually most prominent in male-biased genes (Charlesworth et al. 1987; Orr and Betancourt 2001; Vicoso and Charlesworth 2006; Begun et al. 2007; Baines et al. 2008; Meisel et al. 2012; Meisel and Connallon 2013). Broad expression across multiple tissues may also act to constrain the expression evolution of genes in the brain. For instance, genes that are expressed in many tissues are likely to have an important role in at least several tissues, and hence, their expression levels may be subjected to greater selective constraint (Orr 2000; Chen and Dokholyan 2006; Meisel et al. 2012; Papakostas et al. 2014). Additionally, the genetic constraints imposed by the largely shared male and female genomes have been shown to constrain the evolution of sexually dimorphic phenotypes in various species, including Drosophila (Poissant et al. 2010; Griffin et al. 2013). By understanding the forces and constraints governing the evolution of sex-biased gene expression in the Drosophila brain, we may begin to uncover the molecular rules underpinning the evolution of sexually dimorphic behaviors.
To understand the evolutionary properties of genes showing sex-biased expression in the brain, we generated and analyzed gene expression data from female and male brains in multiple strains of D. melanogaster, Drosophila simulans, and Drosophila yakuba. To determine the types of selection affecting expression levels, we examined interspecific and intraspecific variability in gene expression using an analysis of variance (ANOVA) framework. We used this framework to study the selective forces affecting different classes of sex-biased and unbiased genes at the expression level. Finally, we examined how expression breadth across tissues and the shared genome between sexes constrains expression evolution.
Results
Large enrichment of sex-biased genes on the X Chromosome
By using strand-specific poly(A)+ RNA-seq data collected from the brains of D. melanogaster, D. simulans, and D. yakuba (Fig. 1A), we analyzed the expression patterns of each species, and identified 1246 female- and 989 male-biased genes in D. melanogaster, 767 female- and 725 male-biased genes in D. simulans, and 417 female- and 551 male-biased genes in D. yakuba at a false-discovery rate (FDR) of 0.05 (Fig. 1B; Supplemental Table S1). By looking at the chromosomal distribution of female- and male-biased genes, we found that both sets of genes were highly enriched on the X Chromosome in all three species (P < 1 × 10−5, by a factor of about two- to threefold in all species) and depleted on the autosomes, suggesting that the enrichment of X-linked genes with sex-biased expression in the brain is a conserved phenomenon (Fig. 2).
Figure 1.

Species tree and number of sex-biased genes. (A) A phylogenetic tree with the three species studied. (B) The fraction of the expressing genome showing sex-biased expression in each of the three species at an FDR of 0.05. The expressing genome was defined to be the set of all genes with finite P-values (from DESeq2) for a given species.
Figure 2.

Chromosomal distributions of genes with sex-biased expression in the Drosophila brain. Sex-biased genes are significantly enriched on the X Chromosome and depleted on the autosomes in the three Drosophila species. The red points indicate the observed number of sex-biased genes on a given chromosome arm (at an FDR of 0.05), and the box plots indicate the simulated numbers of sex-biased genes. (*****) P-value <1 × 10−5; (****) P-value <1 × 10−4; (***) P-value <1 × 10−3; (**) P-value <1 × 10−2; (*) P-value < 5 × 10−2. The P-values shown are two times the fraction of simulations, which resulted in a more extreme number of sex-biased genes on a given chromosome arm.
To determine if the significant enrichment of sex-biased genes on the X Chromosome was unique to the brain, we analyzed previously published expression data from D. melanogaster and D. yakuba in four other tissues: gonads, thorax, abdomen, and viscera (Yang et al. 2018). We found that the large enrichment of sex-biased genes on the X Chromosome was unusually large in the brain in both species (Supplemental Fig. S1).
To determine if certain biological processes were enriched in the sets of sex-biased genes, we used PANTHER Gene Ontology (Ashburner et al. 2000; Carbon et al. 2019) on lists of X chromosomal and autosomal male- and female-biased genes in all three species at an FDR of 0.05. In all three species, male-biased autosomal genes were enriched for Gene Ontology terms relating to metabolic processes (Supplemental Table S2). Other classes of genes were not consistently enriched for similar terms across the three species. In fact, with the exception of male-biased genes in D. yakuba, X-linked sex-biased genes were not enriched for any terms in any of the species.
We compared the expression levels of sex-biased and unbiased genes. The expression patterns were largely consistent between the three species, and male-biased autosomal genes had relatively high expression (Supplemental Fig. S2). We then examined the expression fold-changes and significance-levels of sex-biased genes and found that in all three species X Chromosome male-biased genes had a significantly higher fold-change than autosomal male-biased genes (Supplemental Fig. S3). Moreover, in all three species and in both sexes, sex-biased genes on the X Chromosome were more significantly sex-biased (smaller in q-value) than genes on the autosomes (Supplemental Fig. S3). Given that female-biased X Chromosome genes did not show higher fold-changes than autosomal female-biased genes (with the exception of D. simulans) but were more significantly sex-biased, our results suggest that female-biased autosomal genes may show a high degree of expression variability.
Rates of sex-bias turnover are high and chromosome specific
These observations prompted us to look at the evolution of sex-biased genes at the expression level. First, we looked at the turnover of sex-biased genes between the three Drosophila species. By only considering annotated 1:1:1 orthologs (Thurmond et al. 2019), we found that 57% of male-biased genes and 65% of female-biased genes within the three species are sex-biased in a species-specific manner (Fig. 3A), supporting that there is rapid sex-biased gene expression turnover in the brain. This result is consistent with findings in Heliconius butterflies (Catalán et al. 2018), suggesting that fast evolution and turnover of sex-biased genes in the brain may be a pervasive and conserved phenomenon in animals. In addition, we found the ratio of multispecies to single-species sex-biased genes to be substantially higher on the X Chromosome compared with the autosomes in both sexes (male-biased: 1.42 vs. 0.46; female-biased: 1.01 vs. 0.34; P < 2.2 × 10−16 for both; Fisher's exact test), suggesting that chromosome-dependent patterns of selection at the level of gene expression may be responsible for the accumulation of sex-biased genes on the X Chromosome. We also observed that a large majority (69 out of 97) (Supplemental Table S3) of sex-biased genes that have “switched” sex bias at some point in their recent evolutionary history (i.e., they are male-biased in at least one species and female-biased in at least one other Drosophila species) were located on the X Chromosome (Fig. 3B).
Figure 3.

Expression turnover of sex-biased genes in the brain. (A) The fraction of sex-biased genes that are sex-biased in one, two, or three species. Also shown is the fraction of sex-biased genes that have “switched” sex bias between species (i.e., genes that are male-biased in at least one species and female-biased in at least one other species). (B) The fraction of genes in each species number class (one, two, three, or switched) that are located on the X Chromosome. For any given gene, the chromosomal location was taken to be the location of the D. melanogaster ortholog.
Forces governing gene expression evolution in the brain are chromosome and sex-bias dependent
To infer the types of selection acting on gene expression levels in the Drosophila brain, we used an ANOVA framework, similar to the method described by Nuzhdin et al. (2004). In particular, we took the mean sum of squares for species (MSspecies) as a quantification of interspecific variability in expression and the mean sum of squares for strain (MSstrain) as a quantification of intraspecific variability in gene expression. We then calculated the ratio of MSspecies to MSstrain. Genes with higher ratios are presumably evolving under the influence of directional selection, whereas genes with low ratios may be evolving under balancing selection. Genes with intermediate ratios require a closer examination of their levels of between- and within-species variability. Relatively high levels of both between- and within-species variability is indicative of relaxed selective constraint, whereas genes with low interspecific and intraspecific variability are likely to be evolving under stabilizing selection (Nuzhdin et al. 2004).
Considering only 1:1:1 orthologs between the three species, we found that male-biased genes on the X Chromosome (defined as genes with male bias in at least one species) were enriched for signatures of directional selection in both sexes, and female-biased genes (defined as genes with female bias in at least one species) were enriched for signatures of directional selection in females (ratios and P-values see Fig. 4A; Supplemental Fig. S4; Supplemental Table S4). Female-biased autosomal genes showed lower than expected median values for MSspecies/MSstrain, which is indicative of balancing selection or adaptive variation. To determine if there is a faster-X effect for gene expression evolution in the brain, we compared MSspecies/MSstrain between genes on the X Chromosome and the autosomes. We found that sex-biased genes showed evidence of faster-X evolution in both males and females (Supplemental Table S5). No effect was found for unbiased X-linked genes.
Figure 4.

Ratio of interspecific to intraspecific variability in expression for genes in the Drosophila brain. (A) Male-biased genes on the X Chromosome show significantly elevated variability ratios, consistent with directional selection. Similarly, female-biased genes on the X Chromosome show signatures of directional selection in females. Female-biased autosomal genes show significantly reduced ratios, consistent with balancing selection on gene expression. Significance of medians relative to the genome-wide median is shown. P-values determined using 1 million simulations per group and Benjamini–Hochberg correction. (*****) P-value <1 × 10−5; (****) P-value <1 × 10−4; (***) P-value <1 × 10−3; (**) P-value <1 × 10−2; (*) P-value <5 × 10−2. A boxplot version of the data showing dispersion around the median is presented in Supplemental Figure S4. (B) Ratios of interspecific to intraspecific variability in males (on x-axis) show large correlations (Spearman's) with ratios in females (y-axis) irrespective of sex bias or chromosomal location. Only 1:1:1 orthologs were considered. Genes were considered male- or female-biased if they showed that bias in at least one species at an FDR of 0.05. For any given gene, the chromosomal location was taken to be the location of the D. melanogaster ortholog.
We then examined the marginal distributions of MSspecies and MSstrain and noticed that male-biased autosomal genes showed lower than expected values for both statistics in both sexes, which is suggestive of stabilizing selection (Supplemental Fig. S5A,B). To further investigate whether certain classes of genes were enriched for signatures of stabilizing selection, we examined the fold-enrichment of genes with lower than genome-wide median values for both MSspecies and MSstrain. We found an enrichment of signatures of stabilizing selection in two gene classes: (1) male-biased autosomal genes in both sexes and (2) unbiased genes on the X Chromosome in males (Supplemental Fig. S6). However, female-biased autosomal genes were depleted for signatures of stabilizing selection.
If the dosage compensation complex or other sex-specific trans-acting factors were the main drivers of adaptive gene expression evolution for X-linked sex-biased genes, one would expect correlations between variability ratios in males and females to be fairly low. However, we observed a high correlation (Spearman's r = 0.78–0.88) between the variability ratios of sex-biased genes in males and in females (Fig. 4B). This suggests that sex-specific trans-acting factors are unlikely to be the main drivers of adaptive gene expression evolution for X-linked sex-biased genes.
To determine if the observed patterns of gene expression evolution were associated with changes in sex-biased state, we compared genes with sex bias in only one species with genes with conserved sex bias in all three species (Supplemental Fig. S7). With the exception of female-biased autosomal genes, most sex-biased genes showed similar patterns regardless of conservation.
Sex-biased genes with large MSspecies/MSstrain ratios (i.e., whose expression is likely evolving under directional selection) include the X-linked gene cinnamon (FBgn0000316; ratio_male = 86.15, ratio_female = 302.26). Cinnamon, which is involved in the biosynthesis of the molybdenum cofactor and inter-male aggression (Kamdar et al. 1994; Ramin et al. 2019), has changed sex bias in its recent evolutionary history: It has female-biased expression in both D. melanogaster and D. simulans and male-biased expression in D. yakuba.
Patterns of gene expression evolution are associated with the evolution of flanking sequences
To determine if DNA sequence evolution in gene-flanking regions was associated with the observed patterns of selection on gene expression, we examined the levels of nucleotide diversity in regions 20 kb upstream of and downstream from sex-biased genes in D. melanogaster. We found, compared with unbiased genes and female-biased genes, a significant depression of nucleotide diversity (π) proximal to X-linked male-biased genes (Fig. 5A). This indicates that these genomic regions may have recently been under the influence of positive selection, which is consistent with our observation of directional selection acting on these genes at the expression level. We then used the Hudson–Kreitman–Aguade-like (HKAl) statistic (Hudson et al. 1987; Begun et al. 2007) to look for further evidence that the flanking regions of sex-biased X Chromosome genes may be evolving under directional selection. Lower values of HKAl are indicative of stronger deviation from neutral evolution. By considering the minimum HKAl value of genomic regions overlapping 20-kb flanking regions of genes on the X Chromosome (excluding the genes themselves), we found that male-biased genes had significantly lower minimum HKAl values than both unbiased and female-biased X Chromosome genes (P = 0.001 and P = 0.004, respectively; two-sided Mann–Whitney U test) (Supplemental Fig. S8A). Overall, these results suggest that the observed enrichment of directional selection on X-linked male-biased genes is at least partly owing to adaptive evolution in the flanking (putative regulatory) sequences of these genes.
Figure 5.

Nucleotide diversity in flanking regions of sex-biased genes. Nucleotide diversity (π) for various classes of genes on the X Chromosome and autosomes: (A) sex-biased versus unbiased genes, (B) diverged versus conserved genes, and (C) diverged sex-biased versus diverged unbiased genes. Diverged genes are defined as genes that are in the top 5% genome-wide in terms of MSspecies/MSstrain in either sex. Conserved genes are defined as genes that are not diverged in either sex. The 95% confidence intervals are shown in gray. Sex-biased genes are defined in D. melanogaster at an FDR of 0.05.
We noticed that female-biased X Chromosome genes showed no clear adaptive signatures in their flanking regions despite showing signatures of adaptation in their expression (Figs. 4A, 5). To explain this, we hypothesized that the expression evolution of male-biased X-linked genes is influenced by cis-regulatory changes to a greater extent than other classes of genes on the X Chromosome. To test this hypothesis, we first compared the nucleotide diversities in the flanking regions of diverged and conserved genes (at the gene expression level, we defined diverged genes as genes that have ratios of MSspecies to MSstrain in the top 5% genome wide; conserved genes are not diverged in either sex) and found significantly reduced nucleotide diversity in the flanking regions of diverged genes (Fig. 5B). We then compared the nucleotide diversities in the flanking regions of diverged sex-biased and unbiased genes. We observed that diverged male-biased genes on the X Chromosome had significantly reduced nucleotide diversity in their flanking regions compared with other X-linked genes (Fig. 5C). When comparing HKAl in the flanking regions of highly diverged sex-biased and unbiased genes, we noticed a similar trend; however, the differences were not significant (Supplemental Fig. S8C).
Broad expression across tissues does not play a major role in constraining the directional expression evolution of genes in the Drosophila brain
To examine whether broad expression played a role in constraining expression evolution in the brain, we calculated a tissue specificity statistic (Yanai et al. 2005) for genes expressed in males and females of both D. melanogaster and D. yakuba using published expression data from four tissues (abdomen, thorax, viscera, and gonads) (Yang et al. 2018), along with our brain data. A tissue specificity near zero indicates broad expression, whereas a tissue specificity near one indicates that expression is highly tissue specific. We found a very weak negative relationship between tissue specificity (in both sexes and species) and MSspecies/MSstrain in the brain, indicating that broad expression does not constrain directional gene expression evolution in the Drosophila brain (Fig. 6A–D).
Figure 6.

Expression breadth across tissues does not constrain gene expression evolution in the brain. There is a very weak but significant negative relationship between tissue specificity and MSspecies/MSstrain in the brain. The plots show the distribution of MSspecies/MSstrain for a range of tissue specificity in (A) D. melanogaster females, (B) D. melanogaster males, (C) D. yakuba females, and (D) D. yakuba males. A tissue specificity of one indicates expression in only one tissue, and a low tissue specificity indicates broad expression. Correlations shown are Spearman's correlations. Lines are generalized additive model smoothing curves. Sex-biased genes are defined in the same way as in Figure 4.
To determine if tissue specificity was associated with the observed patterns of directional expression evolution for sex-biased genes, we controlled for the effects of tissue specificity on MSspecies/MSstrain (Supplemental Fig. S9). We found that all of the patterns we observed earlier (Fig. 4A; Supplemental Fig. S4) still held. Hence, it appears as though broad expression imposes little constraint on the directional evolution of gene expression in the brain and it cannot explain the signatures of directional selection observed for sex-biased genes on the X Chromosome.
Despite the observation that expression breadth does not constrain adaptive gene expression evolution, we wondered if it could be constraining gene expression variability. Therefore, we examined the association between tissue specificity and interspecific and intraspecific gene expression variability (MSspecies and MSstrain, respectively) (Supplemental Fig. S10). We found that tissue specificity was positively correlated with both classes of gene expression variability in the brain. Broadly expressed genes tend to have low expression variability, whereas tissue-specific genes tend to have larger variability in expression.
To test whether expression breadth could explain the enrichment of signatures of stabilizing selection on the expression levels of autosomal male-biased and X-linked unbiased genes, we examined the tissue specificities of each gene class (Supplemental Fig. S11). We observed that male-biased genes are generally broadly expressed and male-biased autosomal genes have particularly low tissue specificities. This suggests that expression breadth may partially explain the low inter- and intra-specific variability observed in male-biased autosomal genes. To examine this in greater depth, we controlled for the effects of tissue specificity on MSspecies and MSstrain and ran our test for stabilizing selection (Supplemental Fig. S12). The patterns we observed earlier (Supplemental Fig. S6) were similar, but significance for male-biased autosomal genes was lost. This suggests that broad expression may be responsible for our observation of stabilizing selection on the expression levels of male-biased autosomal genes.
The shared genome between the sexes constrains the expression evolution of sex-biased genes in the Drosophila brain
The shared genome has been shown to constrain the evolution of sexually dimorphic phenotypes in several species (Poissant et al. 2010; Griffin et al. 2013). An alternative to this is that sex-specific factors tend to govern the expression evolution of sex-biased genes. To distinguish between these two scenarios, we examined changes in the expression levels of sex-biased genes relative to their ancestral state in both sexes. The ancestral expression level for a given gene was estimated by maximizing the likelihood of a Brownian motion evolutionary model on the three-species phylogenetic tree. For both male-and female-biased genes in all three species, there was a strong correlation between the relative change in male expression and the relative change in female expression, indicating that male and female expression levels of sex-biased genes largely evolve in the same direction (Supplemental Fig. S13A,B).
We then sought to examine the role of genetic constraint in shaping the expression of sex-biased genes on a gene-by-gene basis. To do so, we looked at correlations between male and female expression levels for each sex-biased gene in D. melanogaster. Because males and females within a given strain possess nearly identical genomes, genes with positive correlations between male and female expression levels are genetically constrained. Regardless of sex bias and chromosomal location, the distribution of correlations was heavily positively skewed, indicating that the shared genome imposes a significant constraint on the expression evolution of sex-biased genes (Fig. 7). Given that we only have expression data from six D. melanogaster strains, it is likely that we consistently underestimated the correlations because it appears as though male–female correlations are generally strongly positive.
Figure 7.
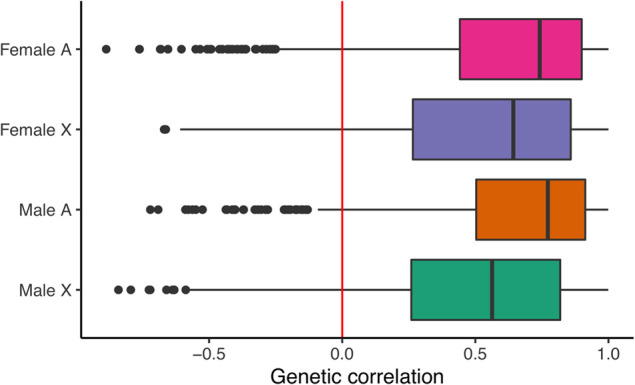
The shared genome constrains expression evolution of sex-biased genes in the Drosophila brain. Correlations between male and female expression levels for sex-biased genes in D. melanogaster. For each category (on the y-axis), a Pearson correlation coefficient (x-axis) was calculated for each gene using six male–female data points (one for each strain). The median correlations for each class of sex-biased gene are nonzero. One-sample Wilcoxon signed-rank test P-values <2.2 × 10−16 for each class (female autosomal, female X, male autosomal, and male X). Sex-biased genes are defined in D. melanogaster at an FDR of 0.05.
Evolution of sex-biased genes at the protein level in D. melanogaster
We then examined the forces governing the evolution of sex-biased genes at the protein level in D. melanogaster. First, we looked for evidence of selection on sex-biased genes by examining dN/dS values generated from published data comparing D. melanogaster to D. simulans (Zhao and Begun 2017). dN/dS is the ratio of nonsynonymous substitutions per nonsynonymous site to synonymous substitutions per synonymous site, with high ratios (>1) being indicative of positive selection and low ratios being indicative of purifying selection. We found that autosomal male-biased genes are significantly enriched for signatures of purifying selection (Supplemental Fig. S14). This result, along with the relatively high expression of autosomal male-biased genes, relatively low sex-biased fold-change in all three species, low gene expression variability within and between species, broad expression, and enrichment of metabolism-related GO terms, suggests that autosomal male-biased genes have important, conserved functions in Drosophila.
To further understand whether positive selection has influenced the evolution of coding sequences of sex-biased genes, we examined published McDonald–Kreitman (MK) test data of annotated protein-coding genes in D. melanogaster (Langley et al. 2012). The MK test infers the presence of positive selection by comparing the numbers of fixed and polymorphic, synonymous and nonsynonymous substitutions (McDonald and Kreitman 1991). We found that sex-biased genes on the X Chromosome were significantly enriched, relative to the set of all genes, for genes with significant MK tests (Supplemental Table S6A). However, when comparing the numbers of genes with positive MK tests between sex-biased genes on the X Chromosome and all genes on the X Chromosome, we found that sex-biased genes showed a slight but nonsignificant enrichment (Supplemental Table S6B).
Discussion
We have performed the first systematic study on the evolution of sex-biased gene expression in the Drosophila brain. The outcomes of our analysis in combination with past studies suggest that the evolutionary properties of sex-biased genes may be tissue specific (Naqvi et al. 2019). We found a significant enrichment of genes showing male- and female-biased expression on the X Chromosome in three species of Drosophila (Fig. 2). The X Chromosome harbored a significantly higher relative number of genes with conserved sex-biased expression in multiple species. In a possible paradox, however, we observed an enrichment of sex-biased genes with female bias in one species and male bias in at least one other species on the X Chromosome (Fig. 3).
When examining the evolutionary forces acting on gene expression, we noticed that sex-biased X-linked genes were enriched for signatures of directional selection (Fig. 4A). Moreover, when comparing X-linked genes with autosomal genes, we found evidence for a faster-X effect for the expression evolution of sex-biased genes (Supplemental Table S5). The assumptions underlying the most common explanation for faster-X evolution of gene expression are as follows: The mutations largely influencing gene expression on the X Chromosome are X-linked, and the mutations are at least partly recessive with respect to expression or fitness (Meisel et al. 2012; Meisel and Connallon 2013). The signatures of adaptive evolution we observed in the flanking regions of X-linked genes—especially in male-biased genes—are consistent with the first assumption (Fig. 5). Our observation of clearer signatures of adaptive evolution in genomic regions proximal to X-linked male-biased genes relative to other X-linked genes suggests that adaptive evolution in expression may be more likely to occur owing to adaptation in cis-regulatory regions for male-biased genes than unbiased and especially female-biased genes (Fig. 5C). This is consistent with previous findings suggesting that adaptive evolution of cis-regulatory regions for male-biased X-linked genes, in particular, could be partly responsible for a faster-X effect in expression for those genes (Coolon et al. 2015). Additionally, it has been observed that trans-factors responsible for the expression of X-linked genes tend to disproportionally reside on the X Chromosome (Coolon et al. 2015). Therefore, it is possible that evolution in trans-regulatory regions is partly responsible for adaptive expression evolution in sex-biased genes. Given that we observed a faster-X effect for expression in female-biased genes but not in unbiased genes may indicate that trans-factors regulating the expression of X-linked female-biased genes may be more likely to reside on the X Chromosome than trans-factors regulating unbiased X-linked genes.
We examined the role that broad expression and shared genomic constraints impose on the expression evolution of sex-biased genes and found that broad expression across tissues generally does not constrain the directional evolution of gene expression in the Drosophila brain (Fig. 6). However, we found that expression breadth does appear to constrain expression variability, both between and within species (Supplemental Fig. S10). The fact that expression breadth does not seem to inhibit directional gene expression evolution suggests that expression evolution in the brain might in part be attributable to selection acting on expression in other tissues which in turn “drag along” expression levels in the brain and vice versa.
In accordance with findings made in other systems, we found that the shared genome between the sexes significantly constrains the expression evolution of sex-biased genes (Fig. 7; Supplemental Fig. S13). This indicates that selection acting upon the expression level of a certain sex-biased gene in one sex will likely “drag along” the expression of that gene in the other sex. It has previously been shown that the chromatin structures of the X Chromosome in male and female Drosophila are similar, despite the fact that dosage compensation only occurs in males, and this might be because the need for dosage compensation in males imposes a constraint on the chromatin structure in females (Zhang and Oliver 2010). This suggests that dragging between sexes can even occur at the epigenetic level. Of course, dragging between sexes and tissues is only likely to occur if the effects of dragging are beneficial, neutral, or only slightly deleterious in the “dragged” tissue or sex. We speculate that gene expression dragging between sexes and tissues is a pervasive phenomenon that may be responsible for various observed instances of phenotypic evolution. If this is indeed the case, it will significantly alter our understanding of gene expression evolution, and the dragging effect on gene expression needs to be investigated in depth in the future.
The genes that we identified as rapidly evolving at the expression level may be thought of as candidate genes involved in the evolution of species-specific neural circuits and behaviors. Therefore, future research should systematically investigate the role that these genes play in rapidly evolving neural circuits and behaviors in Drosophila, both sexually dimorphic and otherwise (Seeholzer et al. 2018). Additionally, because mating alters gene expression patterns in Drosophila heads (Dalton et al. 2010; Ellis and Carney 2010), it would be interesting to see how the results of a study of sex-biased expression in unmated or young flies would compare to our results. Ultimately, the long-term goal is to reveal how sex-biased gene expression in somatic tissues contributes to reproduction and fitness in a species.
Methods
Fly lines and tissue collection
We collected data from inbred strains of three Drosophila species: The D. melanogaster strains come from the Drosophila melanogaster Genetic Reference Panel (DGRP) that are wild caught flies—RAL304, RAL307, RAL357, RAL360, RAL 399, RAL517—from Raleigh, North Carolina, USA (MacKay et al. 2012); D. simulans strains were FAIR3 (Zhao et al. 2015) and w501 (Begun et al. 2007); and D. yakuba strains were Tai18E2 (Drosophila 12 Genomes Consortium 2007) and CY07, which is a Cameroonian isofemale line (Bachtrog et al. 2006). All lines were maintained on standard media under a 12:12 light/dark cycle at 24°C.
Before the dissections, all fly lines were expanded and maintained under an undisturbed 12:12 light/dark cycle at 24°C in low density vials with fly food. All individuals were mated and precisely 4 d old. Flies were very briefly anesthetized with CO2, and heads were collected with a clean razor blade and then were transferred to a glass dissection plate with PBS for brain collection. Dissections were performed within a 3-h window always at the same circadian zeitgeber time (ZT1–ZT4; in this experiment the light was turned on at 8:00 a.m., and the dissections were performed between 9:00 a.m. and 12:00 p.m. within a 3-h interval). Male and female brains of the same line were dissected in parallel the same day at the same time. Dissected brains were immediately transferred into a low retention Eppendorf tube containing 100 µL TRIzol (Thermo Fisher Scientific) and kept frozen at −20°C until RNA extraction. We collected three biological replicates for each genotype by sex combination, and each replicate comprised a pool of exactly 15 brains.
RNA extractions and library preparation
All RNA extractions were performed according to TRIzol manufacturer protocol and immediately followed by a DNase treatment using the TURBO DNase from Thermo Fisher Scientific. RNA quality was assessed by a Bioanalyzer run of an Agilent eukaryote total RNA pico chip, whereas RNA quantity was measured with a NanoDrop One (ABI). About 50 ng total RNA was used for library preparation. Libraries were fragmented and enriched for mRNA using NEBNext poly(A) magnetic isolation module (NEB E7490) and prepared using NEBNext ultra II directional RNA library prep kit (NEB E7765) and dual indexing from the NEBNext multiplex oligos kit (NEB 7600) following manufacturer protocol including beads size selection for 200-bp inserts. Library quality was first assessed on Agilent D1000 ScreenTapes for TapeStation and then by Qubit and Agilent Bioanalyzer. Finally, 150-bp paired-ends libraries were sequenced on five lanes of an Illumina NextSeq 500 platform (Supplemental Table S7).
Read mapping and identification of sex-biased genes
Adaptors from RNA-seq reads were trimmed using Trimmomatic (Bolger et al. 2014). In addition, low-quality bases were trimmed using Trimmomatic settings LEADING:1 TRAILING: 1 SLIDINGWINDOW:20:25 and MINLEN:36. HISAT2 (Sirén et al. 2014) was then used to align reads to their respective reference genomes: dmel_r6.15 for D. melanogaster, dsim_r2.02 for D. simulans, and dyak_r1.05 for D. yakuba. Gene and transcript expression levels (transcripts per million [TPM]) were then quantified using StringTie, and read counts were extracted using a script, prepDE.py, provided by StringTie (Pertea et al. 2015). We performed quality control on the data obtained from the 60 libraries: Three of them (one replicate of male FAIR3, male R517, and female R360) were excluded from further analysis because of low correlation with other replicates or deviation from other samples in the PCA plot.
The read counts were then analyzed using DESeq2 (Love et al. 2014) to identify sex-biased genes in each species. Genes with fewer than 10 read counts (summed across samples within a species) were excluded from the analysis. The DESeq2 model used to determine differential expression between the sexes was counts ∼ strain + sex. Genes with statistically significant “sex” terms (FDR < 0.05) were considered to be sex-biased in the given species. In all future analyses in which we compared sex-biased genes to unbiased genes, we considered genes to be unbiased if DESeq2 assigned an insignificant P-value to the sex term for a gene. Genes for which a P-value could not be determined (“NA,” filtered out by DESeq2) were not considered for further analysis.
To directly compare gene expression levels between samples from different species we used trimmed mean M (TMM) values normalized TPM values. TMM normalization was implemented with the “R” package edgeR (Robinson et al. 2010).
Chromosomal distributions of sex-biased genes
To determine significance for the enrichment of sex-biased genes on the X Chromosome we used random sampling. For each simulation, the total number of male- and female-biased genes was taken to be the same as the number observed at an FDR of 0.05. That number of genes was then randomly sampled from a list of all genes with finite P-values (from DESeq2), and the number of sampled genes on each chromosome arm was recorded. Simulations were repeated 100,000 times for each bias-species combination. The P-values were calculated as two times the fraction of simulations, which resulted in a more extreme number of sex-biased genes on a given chromosome arm.
Expression turnover of sex-biased genes
To get a conservative estimate for the expression turnover of sex-biased genes, equal numbers of genes were taken to be sex-biased in each of the three species. This was performed by increasing the FDR in D. simulans and D. yakuba so that the numbers of sex-biased genes in each species was the same as the number found in D. melanogaster at an FDR of 0.05. Within these three sets, we subsequently only considered 1:1:1 orthologs identified using a list obtained from FlyBase (Thurmond et al. 2019). Genes that are sex-biased in one, two, or all three species were identified, as well as genes that “switched” sex bias in their recent evolutionary history. “Switched” genes are genes that are male-biased in at least one species and female-biased in at least one other species.
Gene expression evolution
One-to-one-to-one orthologs between the three species were identified using a list obtained from FlyBase (Thurmond et al. 2019). TMM normalization of TPM values was then applied across all samples and replicates simultaneously for orthologous genes. Only genes with expression higher than 1 TPM in at least one of the three species were considered for further analysis. The resulting expression values were then log2 transformed and Pareto scaled. For every single gene, we fit a model expression ∼ species + strain. Strain was taken to be a random effect and was nested within species. The model was fit using the MIXED procedure in SAS. The model was fit using the type III sum of squares. We then took the ratio of mean sum of squares (MS) for the species term as a quantification of interspecific variability and MS for the strain term as a quantification of intraspecific variability. A high value for the ratio MSspecies/MSstrain was taken as indicative of directional selection on expression, whereas low values of the ratio were taken as indicative of balancing selection. To compare the ratios, interspecific variabilities, and intraspecific variabilities of various gene classes to genome-wide medians, we used simulations. We randomly sampled gene sets of identical size to the gene set of interest and computed medians for the statistic of interest. Simulations were run 1 million times, for each set of interest, and P-values were defined as twice the proportion of simulations where the median value for the statistic of interested was more extreme than the observed median. Genes were considered “male-biased” or “female-biased” if they were male-biased or female-biased in at least one of the species. Genes that were male-biased in at least one species and female-biased in at least one other species were removed from the analysis.
Calculation of tissue specificity
RNA-seq data from the thorax, abdomen, gonads, and viscera of D. melanogaster and D. yakuba were obtained from Yang et al. (2018). Reads were aligned and expression was quantified in the same manner as for our brain data (except adaptors were not trimmed before alignment). Within each species, sex, and tissue (including our brain data), we computed median TPM values, which were then log2 transformed. Tissue specificity (τ) was then calculated in each sex and species using the formula
where xi is the expression of gene x in tissue i (Yanai et al. 2005). The sum and maximum are taken over all tissues, with n being the total number of tissues.
Controlling for tissue specificity
To control for the effects of tissue specificity on MSspecies/MSstrain, MSspecies, and MSstrain, we used a generalized additive model fit to the relationship between log(“statistic”) and tissue specificity (τ), where “statistic” is the statistic of interest. To do so, we used the function “gam,” from the “R” library mgcv, with formula y ∼ s(x, bs = “cs”) (R Core Team 2019). The residuals were then calculated and plotted.
Direction of gene expression evolution and the shared genome constraint
To infer ancestral expression levels for a given gene, we maximized the likelihood of a Brownian motion model on the three species phylogenetic tree. Because our results provide evidence that sex-biased genes are enriched for signatures of selection at the expression level, we only used a Brownian model as an approximation. In particular, we used TMM normalized and log2 transformed expression levels (across all samples in all species) in combination with the anc.ML function from the “R” library phytools to infer the ancestral expression levels (R Core Team 2019). This was performed for sex-biased genes with 1:1:1 orthologs in all three species. Additionally, we required that all genes considered had a median expression TPM > 1 in at least one species in each sex. The ancestral state was estimated independently for males and females. For a given gene, the change in expression relative to its ancestral state was defined as (current – ancestral)/abs(ancestral), where “current” is the log2(TPM + 1) of the extant expression and “ancestral” is the log2(TPM + 1) of the inferred ancestral expression. Pearson correlations between male and female changes relative to ancestral state were then calculated.
To investigate the role of genetic constraint on a gene-by-gene basis, we first TMM-normalized the D. melanogaster TPM data. Then for each sex, strain, and gene, we calculated median expression, leading to six male–female pairs for each gene. By using these six data points (per gene), we calculated Pearson correlations for each sex-biased gene in D. melanogaster.
Analysis of nucleotide diversity in gene-flanking regions
SNP data for D. melanogaster were obtained from DGRP freeze2 (MacKay et al. 2012). Nucleotide diversity (π) within all DGRP strains was then calculated genome-wide in 500-bp windows using VCFtools (Danecek et al. 2011). Genes were considered “diverged” at the expression level if they had a ratio of MSspecies/MSstrain in the highest 5% genome wide in either sex. For each gene in a given class (i.e., male-biased X Chromosome, unbiased autosomes, etc.), π windows overlapping ±20-kb flanking regions were considered. The π of the gene is represented as a single point at zero. All of the data (distance from gene for each π window and the value of π within each window) from each gene in a given class were aggregated, and generalized additive model smoothing was performed. To smooth the data, we used the function “gam” from the “R” library mgcv, with formula y ∼s(x, bs = “cs”), distribution family “Gamma,” and link function “log” (R Core Team 2019). The resulting curves were plotted with the function “geom_smooth” from the “R” library “ggplot2” (Wickham 2016).
Protein evolution analysis
Protein coding gene sequences and orthologous information were downloaded from FlyBase (Thurmond et al. 2019). One-to-one orthologous genes in D. melanogaster and D. simulans were aligned using GeneWise to remove insertions and deletions and then realigned using PRANK with the –codon option (Zhao and Begun 2017). We then used a KaKs calculator (Zhang et al. 2006) to calculate dN and dS for aligned gene pairs between each of the two species using YN model (Yang and Nielsen 2000). The D. melanogaster MK test and HKAl results, including the Raleigh and Malawi samples, were reported by Langley et al. (2012). We estimated the proportion of adaptive amino acid fixations (α) of each gene according to the method of Smith and Eyre-Walker (2002) and the direction of selection (DoS) index according to Stoletzki and Eyre-Walker (2011).
Data access
The RNA-seq data generated in this study have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject/) under accession number PRJNA553234.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
We thank Roger Vaughan and Caroline Jiang for their help with the ANOVA analysis. We thank David Begun for sharing the Drosophila strains. We thank Grace Y.C. Lee and Jen Perry for critical reading of the manuscript and members of the Zhao laboratory for helpful discussions during the work. L.Z. was supported by the Robertson Foundation, a Monique Weill-Caulier Career Scientist Award, an Alfred P. Sloan Research Fellowship (FG-2018-10627), and a Rita Allen Foundation Scholar Program. This work was supported by a National Institute of General Medical Sciences/National Institutes of Health (NIGMS/NIH) Maximizing Investigators’ Research Award under the award number R35GM133780.
Author contributions: L.Z., N.S., and S.K. conceived the project; L.Z. and N.S. designed the experiments; N.S. and S.M.D. generated the data; S.K. performed data analysis with the input from L.Z. and N.S.; S.K. and L.Z. wrote the manuscript with the input from all authors.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.259069.119.
References
- Arnqvist G, Rowe L. 2005. Sexual conflict. Princeton University Press, Princeton. [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. 2000. Gene Ontology: tool for the unification of biology. Nat Genet 25: 25–29. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtrog D, Thornton K, Clark A, Andolfatto P. 2006. Extensive introgression of mitochondrial DNA relative to nuclear genes in the Drosophila yakuba species group. Evolution (N Y) 60: 292–302. [PubMed] [Google Scholar]
- Baines JF, Sawyer SA, Hartl DL, Parsch J. 2008. Effects of X-linkage and sex-biased gene expression on the rate of adaptive protein evolution in Drosophila. Mol Biol Evol 25: 1639–1650. 10.1093/molbev/msn111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun DJ, Holloway AK, Stevens K, Hillier LW, Poh Y-P, Hahn MW, Nista PM, Jones CD, Kern AD, Dewey CN, et al. 2007. Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol 5: e310 10.1371/journal.pbio.0050310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S, Douglass E, Dunn N, Good B, Harris NL, Lewis SE, Mungall CJ, Basu S, Chisholm RL, Dodson RJ, et al. 2019. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 47: D330–D338. 10.1093/nar/gky1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalán A, Hutter S, Parsch J. 2012. Population and sex differences in Drosophila melanogaster brain gene expression. BMC Genomics 13: 654 10.1186/1471-2164-13-654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalán A, Macias-Muñoz A, Briscoe AD. 2018. Evolution of sex-biased gene expression and dosage compensation in the eye and brain of Heliconius butterflies. Mol Biol Evol 35: 2120–2134. 10.1093/molbev/msy111 [DOI] [PubMed] [Google Scholar]
- Chang PL, Dunham JP, Nuzhdin SV, Arbeitman MN. 2011. Somatic sex-specific transcriptome differences in Drosophila revealed by whole transcriptome sequencing. BMC Genomics 12: 364 10.1186/1471-2164-12-364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Coyne JA, Barton NH. 1987. The relative rates of evolution of sex chromosomes and autosomes. Am Nat 130: 113–146. 10.1086/284701 [DOI] [Google Scholar]
- Chen Y, Dokholyan NV. 2006. The coordinated evolution of yeast proteins is constrained by functional modularity. Trends Genet 22: 416–419. 10.1016/j.tig.2006.06.008 [DOI] [PubMed] [Google Scholar]
- Coolon JD, Stevenson KR, McManus CJ, Yang B, Graveley BR, Wittkopp PJ. 2015. Molecular mechanisms and evolutionary processes contributing to accelerated divergence of gene expression on the Drosophila X chromosome. Mol Biol Evol 32: 2605–2615. 10.1093/molbev/msv135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton JE, Kacheria TS, Knott SRV, Lebo MS, Nishitani A, Sanders LE, Stirling EJ, Winbush A, Arbeitman MN. 2010. Dynamic, mating-induced gene expression changes in female head and brain tissues of Drosophila melanogaster. BMC Genomics 11: 541 10.1186/1471-2164-11-541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darwin C. 1871. The descent of man, and selection in relation to sex, Vol. 1 John Murray, London. [Google Scholar]
- Drosophila 12 Genomes Consortium. 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450: 203–218. 10.1038/nature06341 [DOI] [PubMed] [Google Scholar]
- Ellegren H, Parsch J. 2007. The evolution of sex-biased genes and sex-biased gene expression. Nat Rev Genet 8: 689–698. 10.1038/nrg2167 [DOI] [PubMed] [Google Scholar]
- Ellis LL, Carney GE. 2010. Mating alters gene expression patterns in Drosophila melanogaster male heads. BMC Genomics 11: 558 10.1186/1471-2164-11-558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grath S, Parsch J. 2016. Sex-biased gene expression. Annu Rev Genet 50: 29–44. 10.1146/annurev-genet-120215-035429 [DOI] [PubMed] [Google Scholar]
- Griffin RM, Dean R, Grace JL, Ryden P, Friberg U. 2013. The shared genome is a pervasive constraint on the evolution of sex-biased gene expression. Mol Biol Evol 30: 2168–2176. 10.1093/molbev/mst121 [DOI] [PubMed] [Google Scholar]
- Hudson RR, Kreitman M, Aguadé M. 1987. A test of neutral molecular evolution based on nucleotide data. Genetics 116: 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huylmans AK, Parsch J. 2015. Variation in the X:autosome distribution of male-biased genes among Drosophila melanogaster tissues and its relationship with dosage compensation. Genome Biol Evol 7: 1960–1971. 10.1093/gbe/evv117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Riley RM, Wolfinger RD, White KP, Passador-Gurgel G, Gibson G. 2001. The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat Genet 29: 389–395. 10.1038/ng766 [DOI] [PubMed] [Google Scholar]
- Kamdar KP, Shelton ME, Finnerty V. 1994. The Drosophila molybdenum cofactor gene cinnamon is homologous to three Escherichia coli cofactor proteins and to the rat protein gephyrin. Genetics 137: 791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley CH, Stevens K, Cardeno C, Lee YCG, Schrider DR, Pool JE, Langley SA, Suarez C, Corbett-Detig RB, Kolaczkowski B, et al. 2012. Genomic variation in natural populations of Drosophila melanogaster. Genetics 192: 533–598. 10.1534/genetics.112.142018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKay TFC, Richards S, Stone EA, Barbadilla A, Ayroles JF, Zhu D, Casillas S, Han Y, Magwire MM, Cridland JM, et al. 2012. The Drosophila melanogaster genetic reference panel. Nature 482: 173–178. 10.1038/nature10811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JH, Kreitman M. 1991. Adaptive protein evolution at the Adh locus in Drosophila. Nature 351: 652–654. 10.1038/351652a0 [DOI] [PubMed] [Google Scholar]
- Meiklejohn CD, Presgraves DC. 2012. Little evidence for demasculinization of the Drosophila X chromosome among genes expressed in the male germline. Genome Biol Evol 4: 1007–1016. 10.1093/gbe/evs077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiklejohn CD, Landeen EL, Cook JM, Kingan SB, Presgraves DC. 2011. Sex chromosome-specific regulation in the Drosophila male germline but little evidence for chromosomal dosage compensation or meiotic inactivation. PLoS Biol 9: e1001126 10.1371/journal.pbio.1001126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP, Connallon T. 2013. The faster-X effect: integrating theory and data. Trends Genet 29: 537–544. 10.1016/j.tig.2013.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisel RP, Malone JH, Clark AG. 2012. Faster-X evolution of gene expression in Drosophila. PLoS Genet 8: e1003013 10.1371/journal.pgen.1003013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi S, Godfrey AK, Hughes JF, Goodheart ML, Mitchell RN, Page DC. 2019. Conservation, acquisition, and functional impact of sex-biased gene expression in mammals. Science 365: eaaw7317 10.1126/science.aaw7317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuzhdin SV, Wayne ML, Harmon KL, McIntyre LM. 2004. Common pattern of evolution of gene expression level and protein sequence in Drosophila. Mol Biol Evol 21: 1308–1317. 10.1093/molbev/msh128 [DOI] [PubMed] [Google Scholar]
- Orr HA. 2000. Adaptation and the cost of complexity. Evolution (N Y) 54: 13–20. 10.1111/j.0014-3820.2000.tb00002.x [DOI] [PubMed] [Google Scholar]
- Orr HA, Betancourt AJ. 2001. Haldane's sieve and adaptation from the standing genetic variation. Genetics 157: 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacifico R, MacMullen CM, Walkinshaw E, Zhang X, Davis RL. 2018. Brain transcriptome changes in the aging Drosophila melanogaster accompany olfactory memory performance deficits. PLoS One 13: e0209405 10.1371/journal.pone.0209405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papakostas S, Vøllestad LA, Bruneaux M, Aykanat T, Vanoverbeke J, Ning M, Primmer CR, Leder EH. 2014. Gene pleiotropy constrains gene expression changes in fish adapted to different thermal conditions. Nat Commun 5: 4071 10.1038/ncomms5071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi M, Nuttall R, Naiman D, Bouffard G, Malley J, Andrews J, Eastman S, Oliver B. 2003. Paucity of genes on the Drosophila X chromosome showing male-biased expression. Science 299: 697–700. 10.1126/science.1079190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertea M, Pertea GM, Antonescu CM, Chang T-C, Mendell JT, Salzberg SL. 2015. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol 33: 290–295. 10.1038/nbt.3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poissant J, Wilson AJ, Coltman DW. 2010. Sex-specific genetic variance and the evolution of sexual dimorphism: a systematic review of cross-sex genetic correlations. Evolution (N Y) 64: 97–107. 10.1111/j.1558-5646.2009.00793.x [DOI] [PubMed] [Google Scholar]
- Pröschel M, Zhang Z, Parsch J. 2006. Widespread adaptive evolution of Drosophila genes with sex-biased expression. Genetics 174: 893–900. 10.1534/genetics.106.058008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramin M, Li Y, Chang W-T, Shaw H, Rao Y. 2019. The peacefulness gene promotes aggression in Drosophila. Mol Brain 12: 1 10.1186/s13041-018-0417-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranz JM, Castillo-Davis CI, Meiklejohn CD, Hartl DL. 2003. Sex-dependent gene expression and evolution of the Drosophila transcriptome. Science 300: 1742–1745. 10.1126/science.1085881 [DOI] [PubMed] [Google Scholar]
- R Core Team. 2019. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: http://www.R-project.org/. [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140. 10.1093/bioinformatics/btp616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeholzer LF, Seppo M, Stern DL, Ruta V. 2018. Evolution of a central neural circuit underlies Drosophila mate preferences. Nature 559: 564–569. 10.1038/s41586-018-0322-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirén J, Välimäki N, Mäkinen V. 2014. HISAT2: fast and sensitive alignment against general human population. IEEE/ACM Trans Comput Biol Bioinforma 11: 375–388. [DOI] [PubMed] [Google Scholar]
- Smith NGC, Eyre-Walker A. 2002. Adaptive protein evolution in Drosophila. Nature 415: 1022–1024. 10.1038/4151022a [DOI] [PubMed] [Google Scholar]
- Stoletzki N, Eyre-Walker A. 2011. Estimation of the neutrality index. Mol Biol Evol 28: 63–70. 10.1093/molbev/msq249 [DOI] [PubMed] [Google Scholar]
- Thurmond J, Goodman JL, Strelets VB, Attrill H, Gramates LS, Marygold SJ, Matthews BB, Millburn G, Antonazzo G, Trovisco V, et al. 2019. FlyBase 2.0: the next generation. Nucleic Acids Res 47: D759–D765. 10.1093/nar/gky1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso B, Charlesworth B. 2006. Evolution on the X chromosome: unusual patterns and processes. Nat Rev Genet 7: 645–653. 10.1038/nrg1914 [DOI] [PubMed] [Google Scholar]
- Whittle CA, Extavour CG. 2019. Selection shapes turnover and magnitude of sex-biased expression in Drosophila gonads. BMC Evol Biol 19: 60 10.1186/s12862-019-1377-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. 2016. ggplot2: elegant graphics for data analysis. Springer-Verlag, New York. [Google Scholar]
- Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, et al. 2005. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21: 650–659. 10.1093/bioinformatics/bti042 [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol Biol Evol 17: 32–43. 10.1093/oxfordjournals.molbev.a026236 [DOI] [PubMed] [Google Scholar]
- Yang H, Jaime M, Polihronakis M, Kanegawa K, Markow T, Kaneshiro K, Oliver B. 2018. Re-annotation of eight Drosophila genomes. Life Sci Alliance 1: e201800156 10.26508/lsa.201800156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Oliver B. 2010. An evolutionary consequence of dosage compensation on Drosophila melanogaster female X-chromatin structure? BMC Genomics 11: 6 10.1186/1471-2164-11-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Li J, Zhao X-Q, Wang J, Wong GK-S, Yu J. 2006. KaKs_Calculator: calculating Ka and Ks through model selection and model averaging. Genomics Proteomics Bioinformatics 4: 259–263. 10.1016/S1672-0229(07)60007-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Sturgill D, Parisi M, Kumar S, Oliver B. 2007. Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature 450: 233–237. 10.1038/nature06323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Begun DJ. 2017. Genomics of parallel adaptation at two timescales in Drosophila. PLoS Genet 13: e1007016 10.1371/journal.pgen.1007016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Wit J, Svetec N, Begun DJ. 2015. Parallel gene expression differences between low and high latitude populations of Drosophila melanogaster and D. simulans. PLoS Genet 11: e1005184 10.1371/journal.pgen.1005184 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.