

Abstract
Background & Aims:
Polycystic liver diseases (PLDs) are genetic disorders characterized by progressive development of multiple biliary cysts. Recently, novel PLD-causative genes, encoding for endoplasmic reticulum (ER)-resident proteins involved in protein biogenesis and transport, were identified. We hypothesized that aberrant proteostasis contributes to PLD pathogenesis, representing a potential therapeutic target.
Methods:
ER stress was analyzed at transcriptional (qPCR), proteomic (mass spectrometry), morphological (transmission electron microscopy, TEM) and functional (proteasome activity) levels in different PLD models. The effect of ER stress inhibitors [4-phenylbutyric acid (4-PBA)] and/or activators [tunicamycin (TM)] was tested in polycystic (PCK) rats and cystic cholangiocytes in vitro.
Results:
The expression levels of unfolded protein response (UPR) components were upregulated in liver tissue from PLD patients and PCK rats, as well as in primary cultures of human and rat cystic cholangiocytes, compared to normal controls. Cystic cholangiocytes showed altered proteomic profiles, mainly related to proteostasis (i.e., synthesis, folding, trafficking and degradation of proteins), marked enlargement of the ER lumen (by TEM), and hyperactivation of the proteasome. Notably, chronic treatment of PCK rats with 4-PBA decreased liver weight, as well as both liver and cystic volumes, of animals under baseline conditions or after TM administration compared to controls. In vitro, 4-PBA downregulated the expression (mRNA) of UPR effectors, normalized proteomic profiles related to protein synthesis, folding, trafficking and degradation, and reduced the proteasome hyperactivity in cystic cholangiocytes, reducing their hyperproliferation and apoptosis.
Conclusions:
Restoration of proteostasis in cystic cholangiocytes with 4-PBA halts hepatic cystogenesis, emerging as a novel therapeutic strategy.
Keywords: hepatic cystogenesis, cholangiocyte, endoplasmic reticulum stress, unfolded protein response (UPR), proteostasis, pathogenesis
Lay summary
Understanding the molecular mechanisms involved in the pathogenesis of PLDs is crucial in order to find new targets for therapy. This study provides evidence that cystic cholangiocytes are characterized by abnormal ER-related proteostasis that prompts adaptive pro-survival mechanisms, favouring PLD progression. Normalization of proteostasis in cystic cholangiocytes with the chemical chaperone 4-PBA halts hepatic cystogenesis in experimental models in vivo and in vitro, opening a new therapeutic avenue for PLDs.
Introduction
Polycystic liver disease (PLD) constitutes a group of genetic cholangiopathies characterized by bile duct dilatation and/or progressive development of multiple fluid-filled biliary cysts (>10), which are the main cause of morbidity.(1–3) Despite most patients with PLD remain clinically asymptomatic through the years, progressive hepatic cystogenesis may cause massive hepatomegaly in a proportion of them, potentially resulting in severe symptoms such as abdominal distention and bloating, dyspnea, back pain, gastroesophageal reflux, hypertension, as well as cyst hemorrhage, infection and rupture, among others.(1–3) To date, surgical and/or pharmacological strategies exert short-term and/or modest benefits, and liver transplantation emerges as the only curative option for patients with advanced PLD.(1–3)
According to the genetic traits, hepatic cystogenesis may solely affect the intrahepatic bile ducts [i.e., autosomal dominant polycystic liver disease (ADPLD)] or arise associated with renal cysts [i.e., autosomal dominant polycystic kidney disease (ADPKD), autosomal recessive polycystic kidney disease (ARPKD), Caroli’s disease and congenital hepatic fibrosis (CHF) in infants].(2, 3) Noteworthy, the majority of genes known to cause ADPLD (i.e., PRKCSH, SEC63, ALG8, GANAB and SEC61β)(2, 4) and ADPKD (i.e., PKD2 and GANAB)(2, 4) encode for endoplasmic reticulum (ER)− resident proteins.(1, 2, 4) These proteins play a relevant role in the ER protein biogenesis, governing the synthesis of functional membrane and secreted proteins.(1, 2, 4) In this regard, mutations (i.e., missense, nonsense, splice site and/or frameshift) in the aforementioned genes may result in partial or complete loss-of-function of the protein products, disrupting the folding, maturation and trafficking of nascent proteins and compromising ER proteostasis.(4, 5) Moreover, abnormalities in ER proteostasis may subsequently activate the unfolded protein response (UPR) signaling cascades, composed by sensor (i.e., IRE1α, PERK and ATF6) and effector (i.e., CHOP, GRP78 and XBP1) proteins, in order to promote protein folding, ER-associated protein degradation (ERAD), and the activation of pro-survival mechanisms.(6, 7) Nevertheless, when the UPR is unable to alleviate the burden of un/misfolded proteins, chronic/excessive ER stress might result in proteotoxicity-induced apoptosis.(6, 7)
In this study, we hypothesized that mutations in PLD-related genes could trigger proteostatic abnormalities in cystic cholangiocytes resulting in ER stress, which might contribute to hepatic cystogenesis, thus representing potential druggable targets.
Materials and methods
Human samples
Cystic wall tissue from patients with PLD (n=16), and healthy human liver (n=14) and gallbladder (n=14) biopsies were obtained from Radboud University Medical Center (Nijmegen, The Netherlands) and Donostia University Hospital (San Sebastian, Spain), respectively. Cystic tissues were collected from patients with ADPLD displaying either PRKCSH (protein kinase C substrate 80K-H: c.1341–2A>G or c.292+1G>C) or SEC63 (translocation protein SEC63 homolog: 1702delGAA) mutations, whereas healthy liver and gallbladder tissues were gathered from individuals after surgical resection of colon cancer metastasis or cholecystectomy, respectively. Supporting Table 1 summarizes the main demographic and clinical features of the patients included in the study. Research protocols were approved by the Clinical Research Ethics Committee of the supporting institutions (MSA-MMR-2017–01 and 2012/317), and an informed consent was obtained from all subjects.
PLD animal model
The polycystic (PCK) rat [PCK/CrljCrl-Pkhd1pck/Crl] constitutes a well-characterized animal model of ARPKD, presenting a mutation in the PKHD1 (polycystic kidney and hepatic disease 1) orthologous gene and developing hepato-renal cysts that recapitulate the course of the human disease.(8, 9) Hepatic cystogenesis, serum biochemical parameters and molecular markers were analyzed in wild-type (WT; n=12) and control PCK (n=12) rats, as well as in PCK rats chronically treated with 4-phenylbutyric acid sodium salt (4-PBA: 100 mM in drinking water; Scandinavian formulas, PA, USA; n=12) and/or Tunicamycin (TM: 0.02 mg/kg intraperitoneal injection; Sigma, MO, USA; n=12) for 5 months. All details are described in Supporting Information.
Human and rat cholangiocyte primary cultures
Normal human cholangiocytes (NHC) and polycystic human cholangiocytes were isolated and characterized by our group as previously described.(10) Two primary cultures of PLD cholangiocytes (i.e., ADPKD and ADPLD) were used. ADPKD cells have a missense mutation (c.2515C>T) in exon 22 of GANAB (glucosidase II alpha subunit) gene,(11) whereas ADPLD cells present a splice site mutation (c.292+1G>C) in intron 4 of the PRKCSH gene. In addition, PCK rat cholangiocytes, which hold a splicing mutation (IVS35–2A→T) in Pkhd1 gene,(8) as well as the corresponding normal rat cholangiocytes (NRC), were used.(12) All primary cultures were cultured in fully-supplemented DMEM/F-12 medium as previously described.(10)
RNA isolation, retrotranscription and quantitative polymerase chain reaction (qPCR)
RNA purification, retrotranscription and quantification of UPR factors (i.e., sensors and effectors), as well as, pro-fibrotic and pro-inflammatory genes were performed as detailed in Supporting Information. Primer sequences are included in Supporting Table 2.
Cell proliferation and death
The rates of cell proliferation and death were measured as described in Supporting Information.
Proteasome activity
Ubiquitinated protein degradation was determined measuring the proteasomal chymotrypsin-like activity with the Proteasome Activity Assay Kit (Abnova, Taiwan), according to the manufactureŕs instructions. Free 7-amino-4-methylcoumarin (AMC) fluorescence intensity was detected using the Infinite® 200 Pro plate reader (Tecan, Switzerland).
Transmission electron microscopy (TEM)
Ultrastructural examination of the ER morphology and size in human (i.e., NHC, ADPKD and ADPLD) and rat (i.e., NRC and PCK) cholangiocyte cultures were performed as previously described.(9) The total surface area occupied by the ER in each TEM image was determined with the ImageJ software version 1.50 (NIH, Bethesda, MA, USA) by calculating the sum of the area (μm2)/length (μm) ratio of each ER portion present in the captured images.
Mass spectrometry and proteomic analysis
Comparative proteomic analysis of whole cell extracts (WCEs) from normal and cystic cholangiocytes (both human and rat incubated with 4-PBA or vehicle) were performed by mass spectrometry as described in Supporting Information.
Statistical Analysis
Data are shown as mean ± standard error of mean (SEM). Once normality was assessed with Shapiro–Wilk test, parametric Student t test or non-parametric Mann-Whitney U test were used for statistical comparisons between two groups. For comparisons between more than two groups, the parametric one-way analysis of variance (ANOVA) test followed by Tukey’s post hoc test or the non-parametric Kruskal-Wallis test followed by a posteriori Dunn’s test were used. In the proteomics analysis, protein abundance differences and p-values were calculated and quantified using logarithmic values (log2 transformation and imputation). As samples did follow a Gaussian distribution, parametric Student t test was applied. Differences were considered significant when p<0.05 (*, ** and *** are p values <0.05, <0.01 and <0.001, respectively). Results were statistically analyzed using the GraphPad Prism version 6.01 software (La Jolla, CA, USA).
Results
UPR sensors and effectors are upregulated in human and rat PLD tissues and cystic cholangiocyte cultures
The levels (mRNA) of the main UPR components were measured in cystic wall tissue obtained from patients with PLD (i.e., ADPLD), as well as in healthy human gallbladder (mainly composed by cholangiocytes) and liver tissue. NHC were also included in this analysis, as they constitute a pure cellular population of healthy human cholangiocytes. Interestingly, human cysts exhibited an upregulation of the UPR components compared to normal controls (Fig. 1A). When compared with gallbladder tissue samples or NHCs, the expression levels of the three UPR sensors (i.e., ATF6, IRE1α and PERK) and the effectors GRP78 and CHOP were found upregulated in PLD tissue (Fig. 1A); likewise, increased levels of the effector XBP1 were observed in cystic tissue compared to NHC, while no differences were evident when compared to gallbladder tissue samples (Fig. 1A). In addition, PERK and CHOP were also found upregulated in cysts compared with healthy livers, whereas no significant differences in the expression of the other UPR components were noticed (Fig. 1A). Primary cultures of cystic human cholangiocytes (i.e., ADPKD and ADPLD) also exhibited upregulated levels of all the UPR components (i.e., sensors and effectors) when compared to NHC (Fig. 1B). Of note, ADPKD (GANAB-mutated) cholangiocytes displayed higher levels of the UPR effectors XBP1 and CHOP than ADPLD (PRKCSH-mutated) cholangiocytes (Fig. 1B).
Figure 1. Molecular and morphological ER features of human and rat cystic cholangiocytes in vivo and/or in vitro.
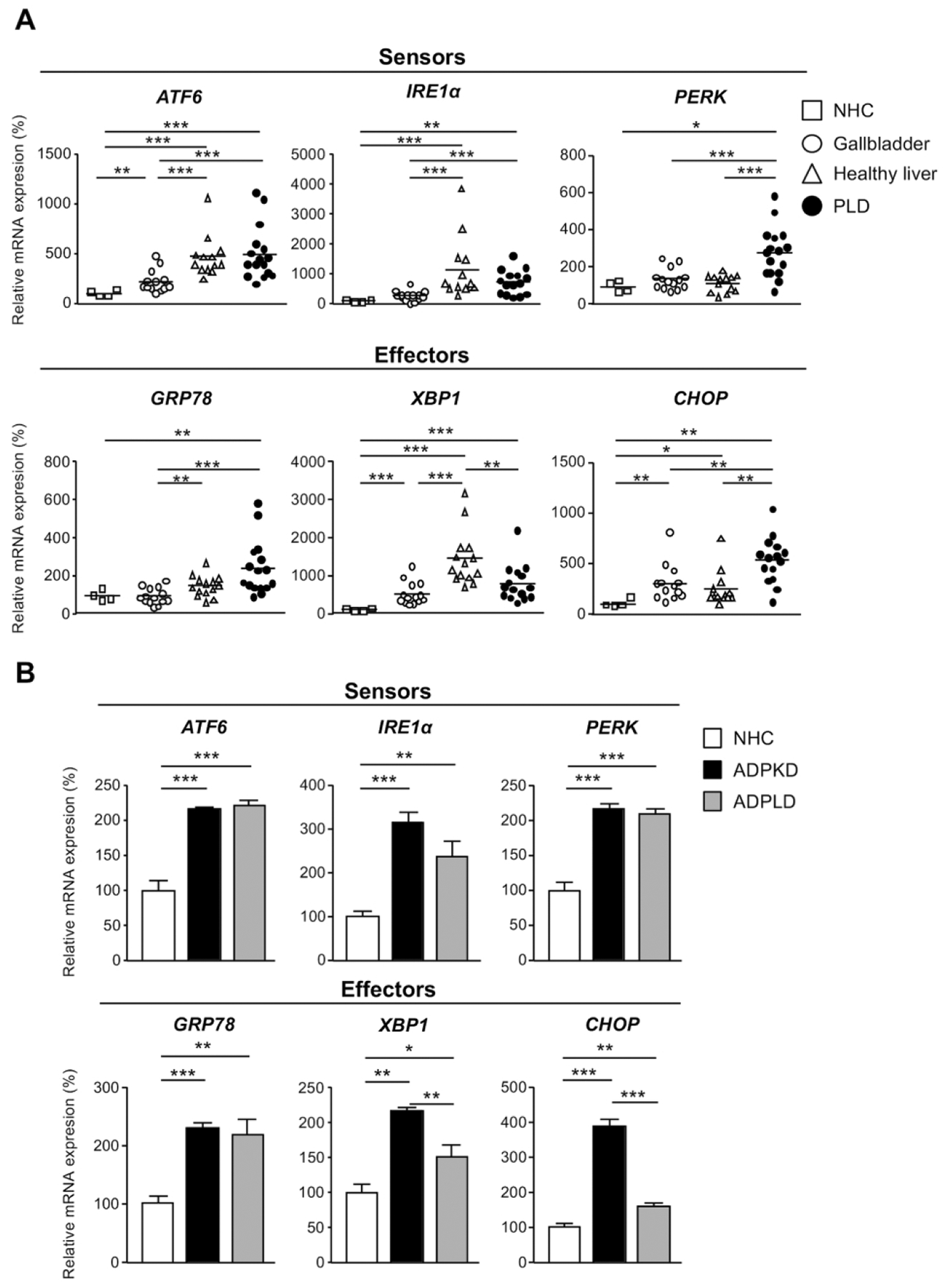
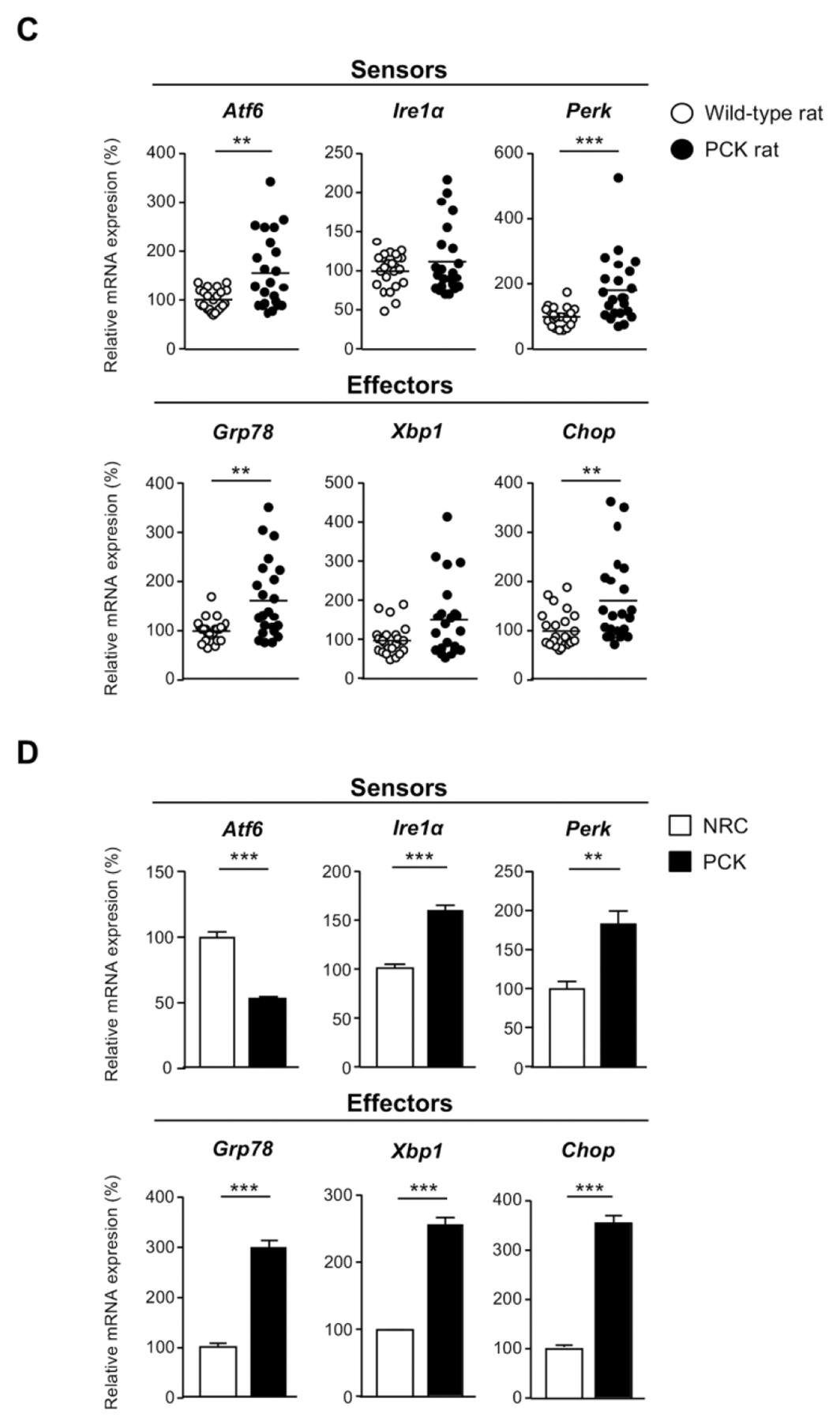
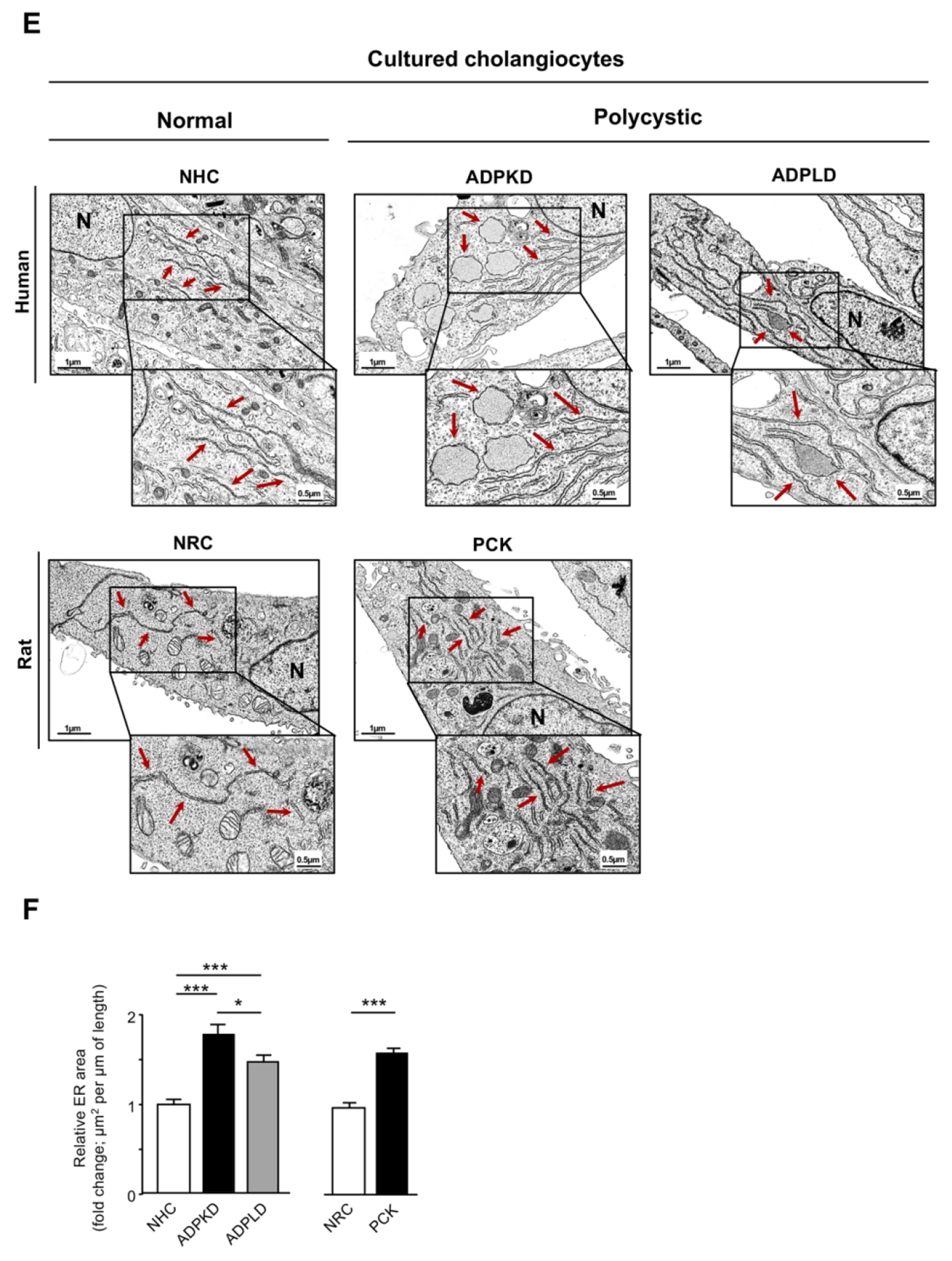
mRNA levels of UPR sensors and effectors in: (A) ADPLD cystic wall tissue (n=16) and healthy controls [liver (n=14), gallbladder (n=14), and NHC (n=4)], (B) normal (NHC; n=6) and cystic (ADPKD and ADPLD; n=6) human cholangiocytes in culture, (C) wild-type (n=20) and PCK rat (n=22) livers, and (D) normal (NRC; n=6) and cystic [PCK; n=6) rat cholangiocytes in culture. (E) Representative TEM images showing the ER ultrastructural morphology (red arrows) of normal and cystic cholangiocytes isolated from humans and rats. Scale bar= 0.5 μm. (F) Quantification of the ER lumen area in human [NHC (n=47), ADPKD (n=28), and ADPLD (n=28)] and rat [NRC (n=49) and PCK (n=52)] cholangiocytes.
The expression levels of the UPR components were also determined in both liver tissue samples and cultured cholangiocytes derived from WT and PCK rats. The UPR sensors Atf6 and Perk, as well as the effectors Grp78 and Chop, were all found upregulated in PCK rat livers compared to WT livers (Fig. 1C). Although Ire1α and Xbp1 expression levels displayed a tendency to increase in liver tissue from PCK rats, no significant differences were observed compared to matched liver tissue from healthy rats (Fig. 1C). Additionally, ER stress was also evidenced in PCK cholangiocytes in vitro, as shown by marked upregulation of most UPR components when compared to NRC (Fig. 1D).
Cystic cholangiocytes exhibit marked ER dilatation
Considering the overexpression of UPR components in human and rat cysts (Fig. 1A–D), which suggest aberrant ER proteostasis and potential accumulation of un/misfolded proteins within the ER lumen, the morphological and ultrastructural features of this organelle were evaluated. For this purpose, TEM images from normal and cystic cholangiocytes in culture were captured and analyzed. As expected, NHC and NRC exhibited a typical narrow ER lumen (Fig. 1E). However, pronounced dilatation of the ER lumen was found (Fig. 1E) in all primary cultures of cystic cholangiocytes (i.e., ADPKD, ADPLD, PCK) compared to normal controls, being this phenotype a well-documented feature linked to ER stress.(6, 7, 13) In particular, the ER area underwent a 1.79, 1.47 and 1.62-fold increase in ADPKD, ADPLD and PCK cholangiocytes respectively, compared to the respective normal controls (Fig. 1F). Of note, ADPKD cholangiocytes presented increased ER dilatation compared to ADPLD cholangiocytes.
The proteomic profile of cystic cholangiocytes is characteristic of aberrant proteostasis
The proteomic profiles of normal and cystic cholangiocytes were determined under baseline conditions using mass spectrometry. A total of 2878, 2775 and 2715 proteins were identified in NHC, ADPKD and ADPLD cholangiocytes, respectively (Fig. 2A). Among the significantly dysregulated proteins, 87 were found upregulated and 328 downregulated similarly in ADPKD and ADPLD (PLD) compared to normal cholangiocytes (Figs. 2B,C). Gene ontology (GO) analysis revealed that most of these dysregulated proteins are involved in ER proteostasis-related mechanisms (i.e., synthesis, folding, trafficking and degradation pathways) (Fig. 2D), while the STRING analysis revealed functional interactions between several protein clusters (Fig. 2E). Among these proteins, overexpression of ubiquitin-protein ligases (i.e., HERC3), flavoproteins that catalyze the formation of disulfide bonds (i.e., QSOX1) and enzymes that mediate lysosomal proteolysis (i.e., LGMN) stand out, whereas the levels of other proteins involved in the late stages of protein maturation [such as HSP90 family chaperones (i.e., HSP90AA1) and co-chaperones (i.e., UN45A)], ER to Golgi transport mediators (i.e., GDS1), and oxidoreductases (i.e., QOR) were found downregulated in cystic vs normal human cholangiocytes (Fig. 2F).
Figure 2. Proteomic profiles of normal and cystic human cholangiocyte primary cultures.
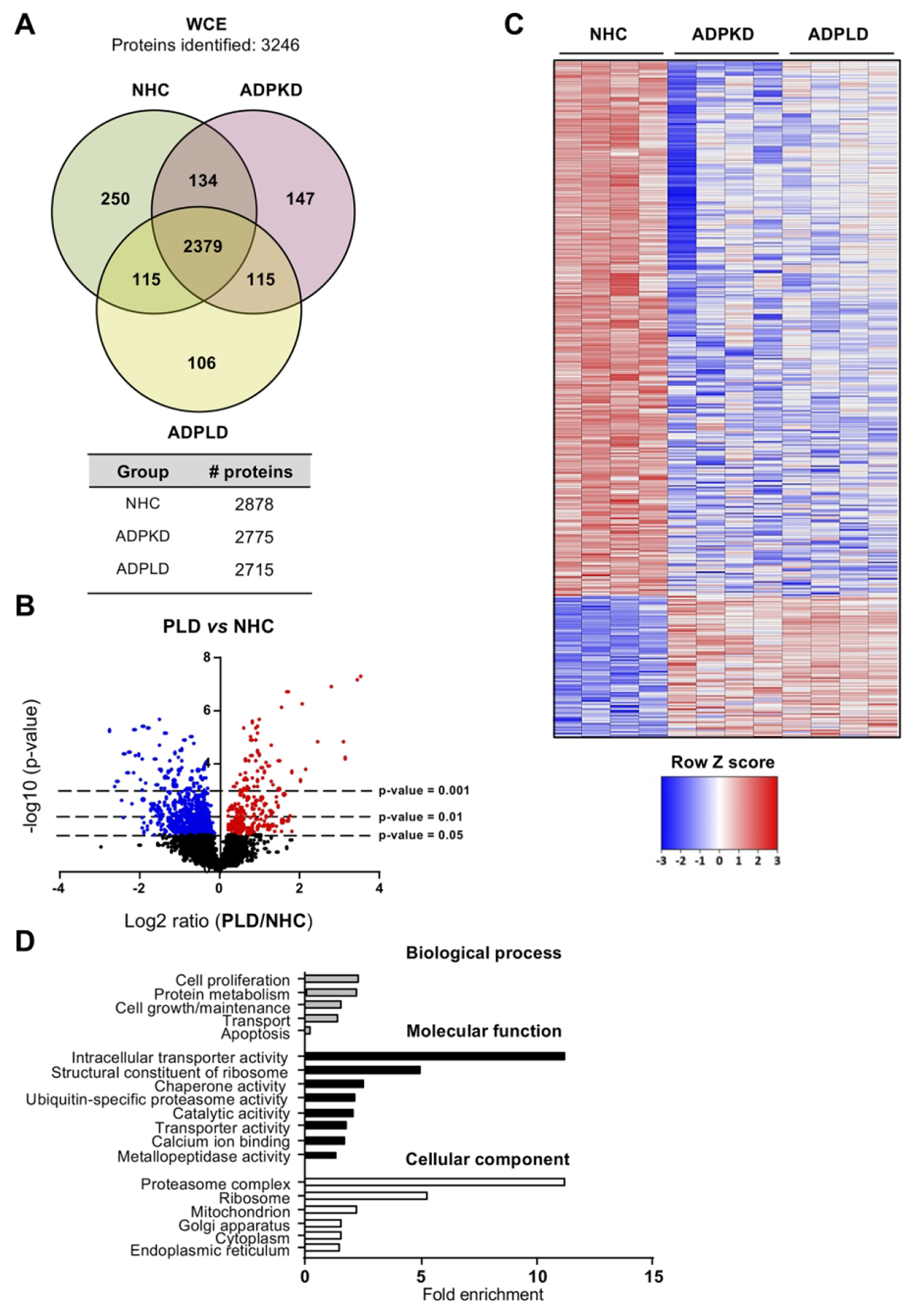
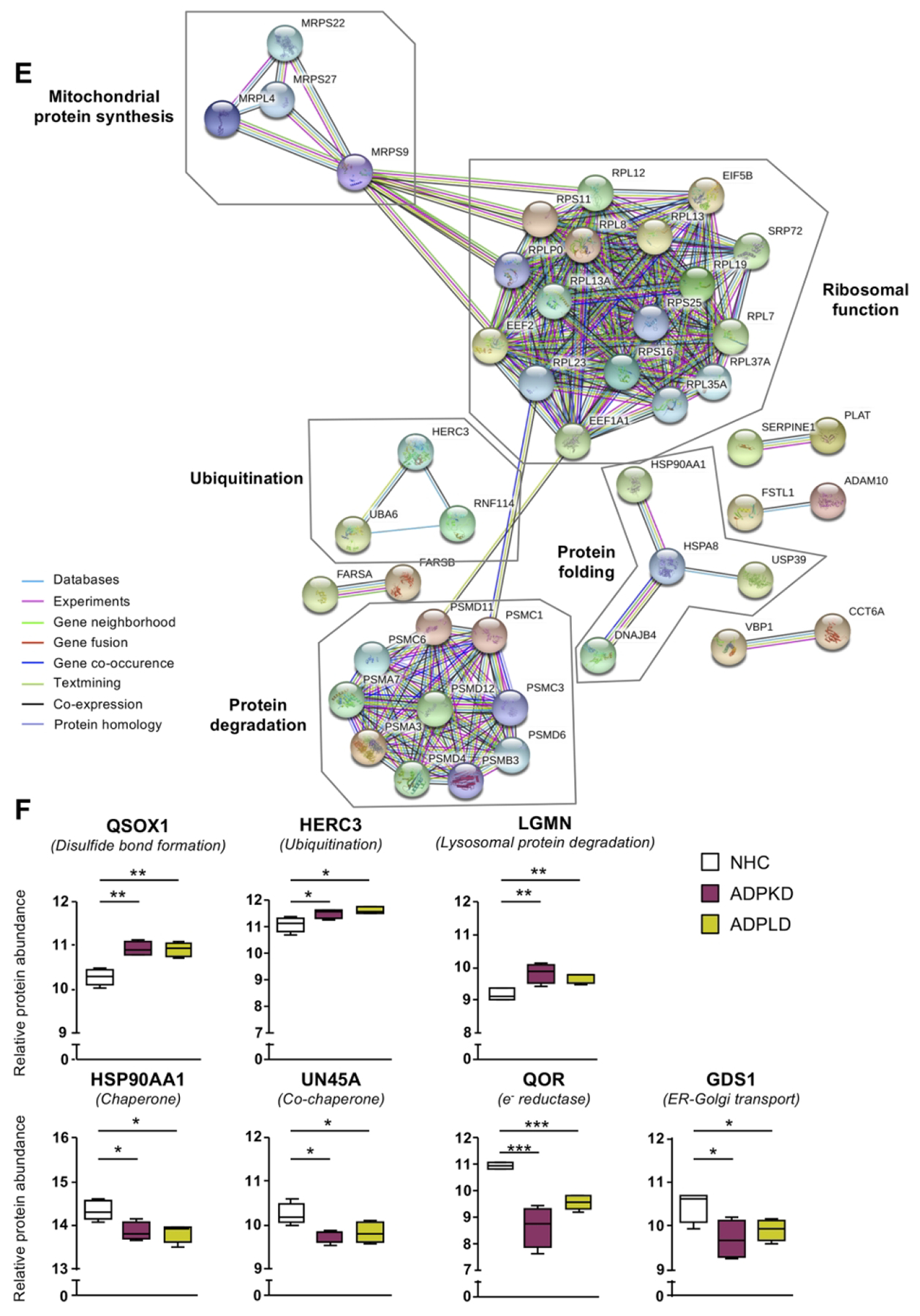
(A) Venn diagram with the proteins identified in each human cell type. (B) Volcano plot displaying the expression pattern and differences of all proteins identified in PLD vs NHC cholangiocytes. (C) Heatmap representing only the proteins differentially expressed in PLD (ADPKD and ADPLD) vs NHC. (D) GO classification of the proteins differentially expressed in PLD vs NHC. (E) STRING interaction analysis of the proteins differentially expressed in PLD vs NHC. (F) Box plot diagrams of the relative abundance of each selected protein in the three human cell types.
Similar to human cholangiocytes, the proteomic profiles of NRC and PCK rat cholangiocytes was determined. A total of 2575 and 2531 proteins were identified in NRC and PCK cholangiocytes, respectively (Fig. 3A). Among the significantly dysregulated proteins, 980 were found upregulated and 476 downregulated in PCK vs NRC cholangiocytes (Figs. 3B,C). Further functional analysis revealed a substantial enrichment of proteins involved in protein synthesis, folding, transport and degradation, as well as in other pivotal processes related to PLD pathogenesis such as ciliogenesis (Fig. 3D). The protein interaction network evidenced their roles along the different steps of protein maturation, from protein biogenesis to degradation (Fig. 3E). Among these proteins, overexpression of components related to protein synthesis (i.e., Eif3h) and folding (i.e., Calr, Pdia4), as well as of the 20S proteasome (i.e., Psmb3), Hsp70 family chaperones (i.e., Hspa4) and co-chaperones (i.e., Hspbp1) was evident, whereas the expression of other families of chaperones and mediators of nascent protein trafficking (i.e., Hspb1 and Bloc1s6, respectively) were downregulated in PCK vs NRC cholangiocytes (Fig. 3F).
Figure 3. Proteomic profiles of normal and cystic rat cholangiocyte primary cultures.
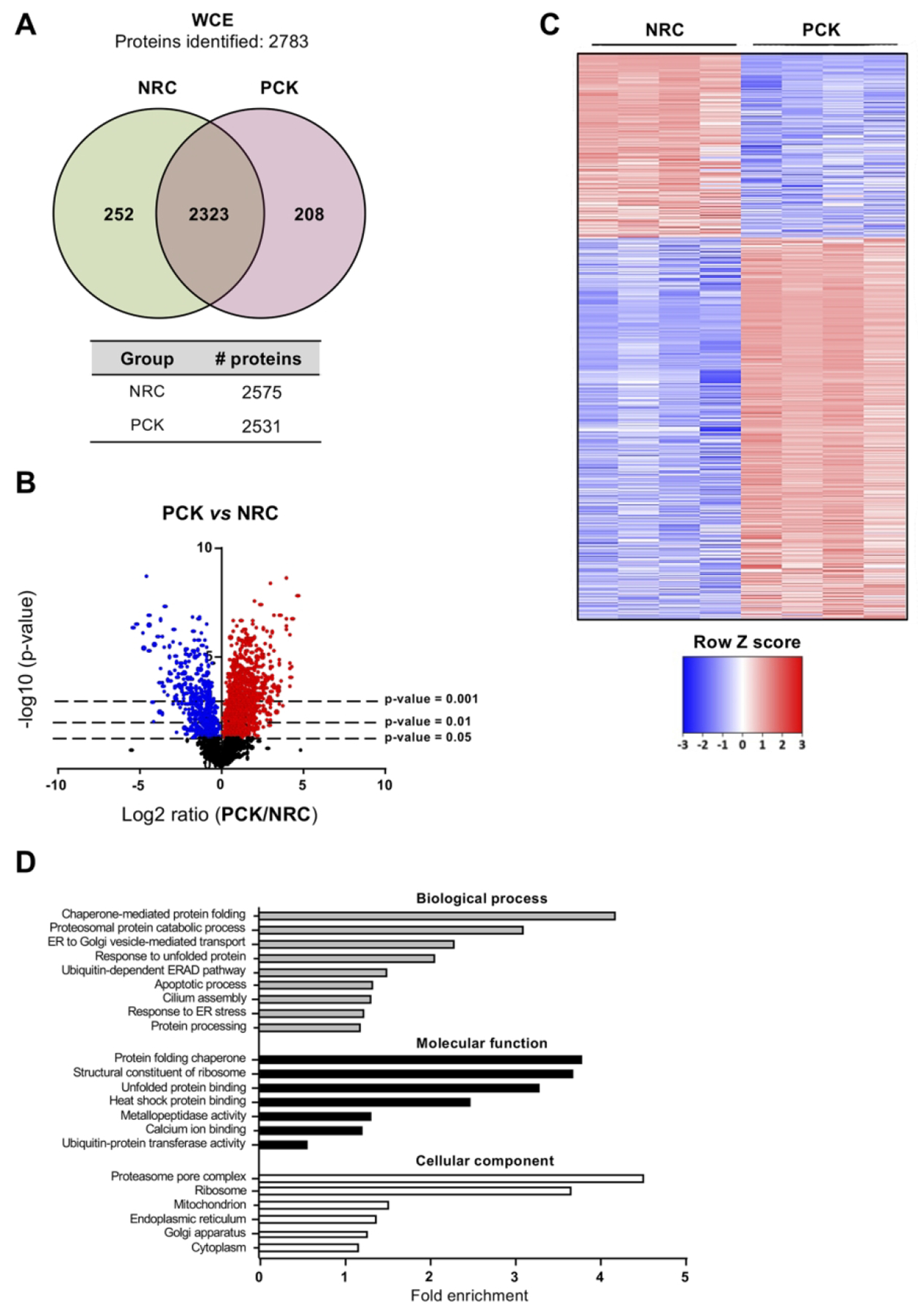
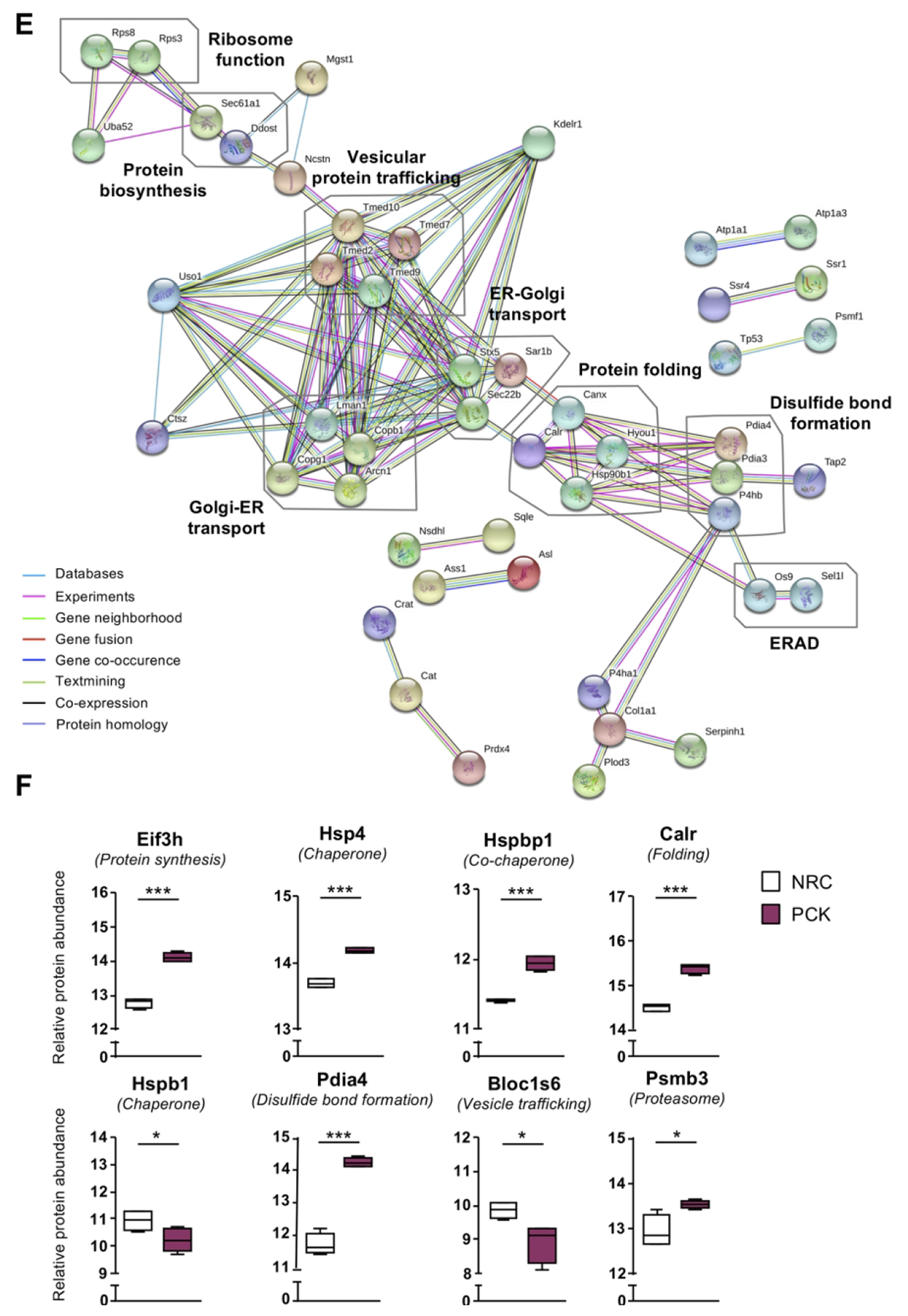
(A) Venn diagram with the proteins identified in normal and cystic rat cholangiocytes. (B) Volcano plot displaying the expression pattern and differences of all the identified proteins in PCK vs NRC. (C) Heatmap representing only the proteins differentially expressed in PCK vs NRC. (D) GO classification of the proteins differentially expressed in PCK vs NHC. (E) STRING interaction analysis of the proteins differentially expressed in PCK vs NRC. (F) Box plot diagrams of the relative abundance of each selected protein in normal and cystic rat cholangiocytes.
ER proteostasis disturbance and stress contributes to hepatic cystogenesis in vivo
The contribution of ER proteostasis disturbance and proteotoxic stress to hepatic cystogenesis, as well as the potential therapeutic value of its modulation, was evaluated in PCK rats. 4-PBA is a chemical chaperone that inhibits ER stress,(14) while tunicamycin (TM) prevents the glycosylation and proper folding of proteins within the ER, being used as a “bona fide” ER stressor.(15) Therefore, 4-PBA and/or TM were administered to 8-weeks-old PCK rats for 5 months. In comparison to WT animals, control PCK rats developed hepatomegaly as shown by a significant increase in liver weight and volume (Fig. 4A), without displaying any differences in total body weight, thus resulting in an increased liver/body weight ratio (Supporting Table 3). Remarkably, chronic administration of 4-PBA to PCK rats reduced their total liver weight, as well as the liver and cystic volumes, when compared with control PCK rats (Fig. 4A,B). Administration of TM to PCK rats did not further increase their liver weight, volume and cystogenesis (Figs. 4A,B), but it induced a significant increase of the total liver/body weight ratio and in the levels of serum markers of cholestasis (ALP) and liver injury (AST), when compared to control PCK rats (Supporting Table 3). Noteworthy, 4-PBA was also able to decrease the liver weight and volume, as well as the hepatic cystic volume, of TM-induced PCK rats, when compared with rats exposed to TM alone (Fig. 4A and 4B, respectively).
Figure 4. Effect of 4-PBA and/or TM on the liver of PCK rats.
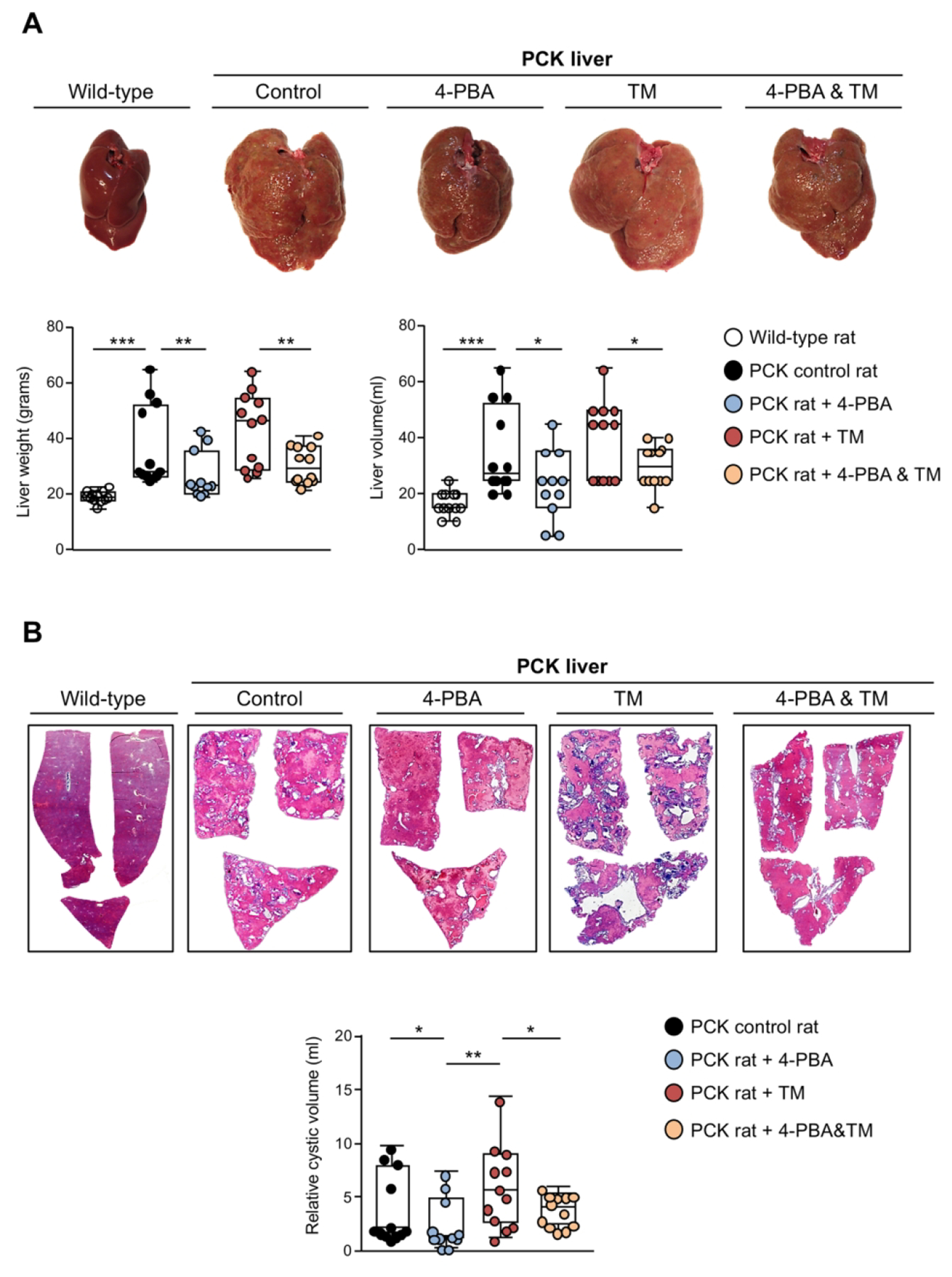
(A) Representative macroscopic images of liver tissue and corresponding quantification of its total weight and volume in wild-type and PCK rats [control, 4-PBA (100 mM), TM (0.02 mg/kg) and 4-PBA&TM; n=12 per group]. (B) Representative histological images of liver sections stained with hematoxylin/eosin and corresponding measurement of the total cystic volume in the aforementioned in vivo experimental groups (n=12 per group).
Modulation of ER stress impacts on cholangiocyte proliferation and cell death in vitro
The effect of ER stress regulation was also evaluated in normal and cystic (ADPKD and ADPLD) cholangiocytes in vitro. The mRNA levels of the UPR sensors ATF6 and PERK remained mostly constant in normal and cystic cholangiocytes after 4-PBA and/or TM incubation, whereas IRE1α was upregulated in both cell types under the presence of 4-PBA and/or TM (Supporting Fig. 1). Of note, 4-PBA reduced the expression of CHOP, GRP78 and XBP1 in ADPKD, but not in ADPLD cholangiocytes and also reduced the levels of GRP78 and XBP1 in NHC (Fig. 5A). On the other hand, TM induced the expression of the UPR effectors CHOP, GRP78 and XBP1 in both normal and cystic human cholangiocytes compared to corresponding control conditions, with 4-PBA almost reverting these alterations to basal levels (Fig. 5A).
Figure 5. Effect of 4-PBA and/or TM on normal and cystic human cholangiocytes in culture.
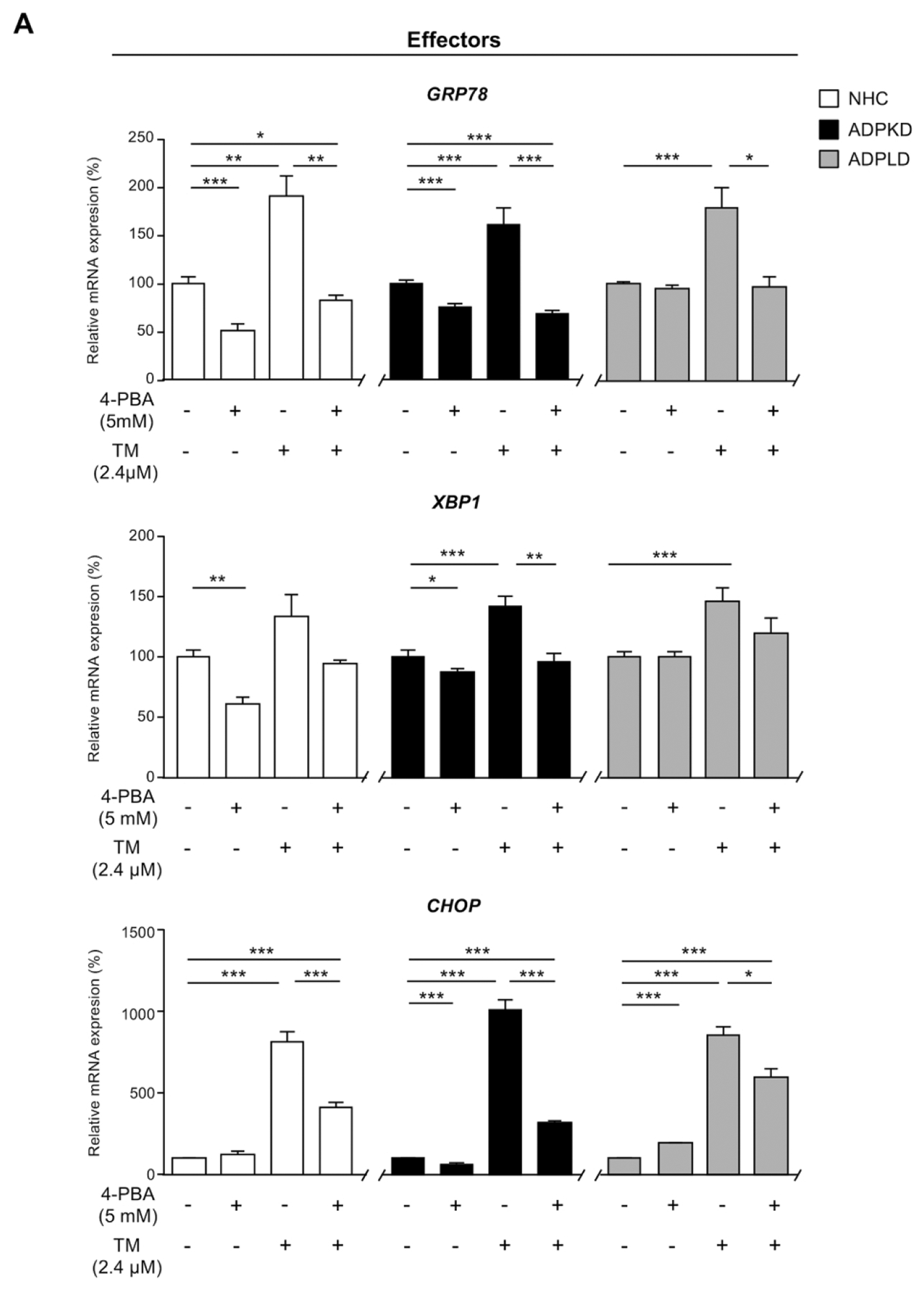
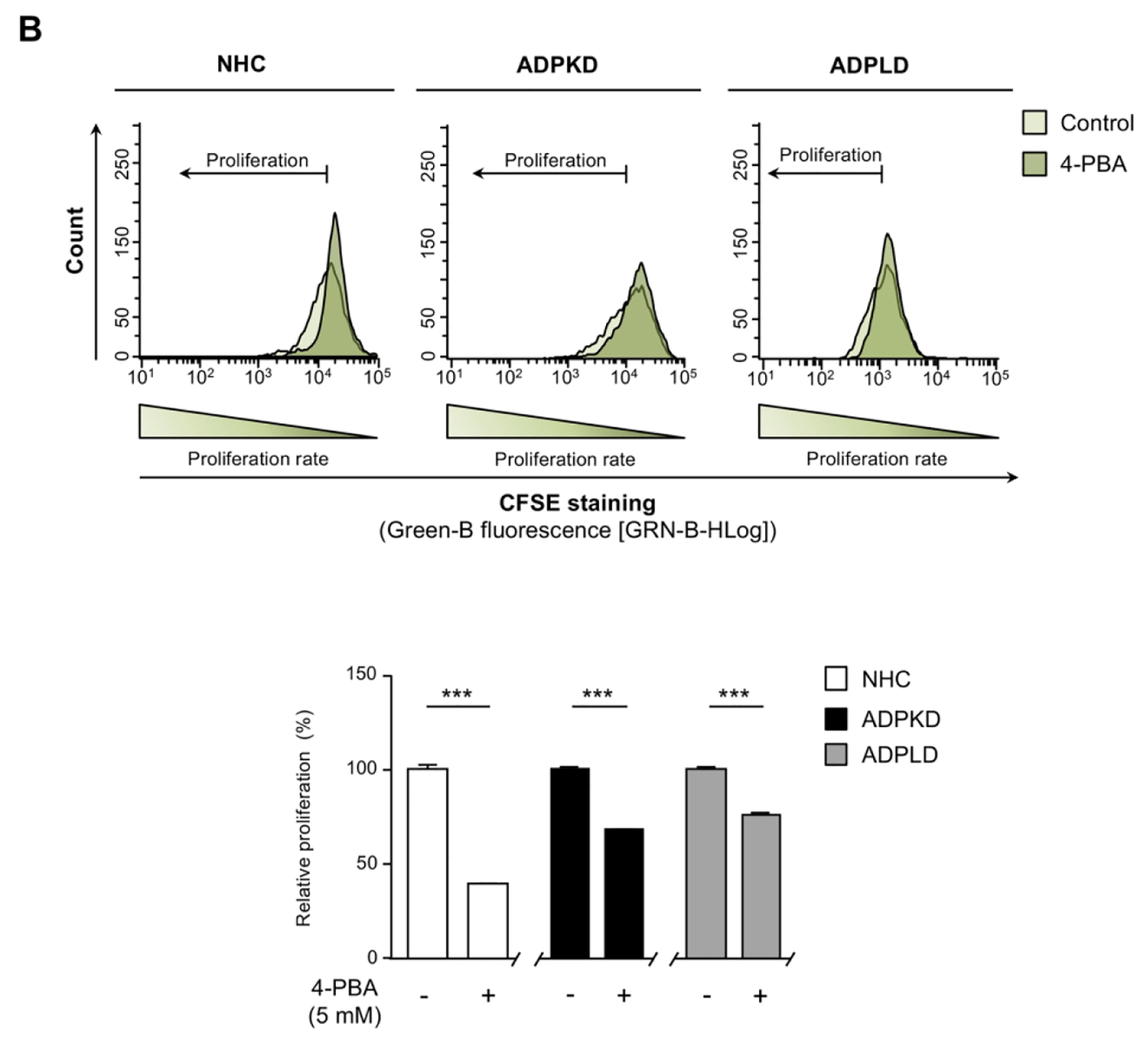
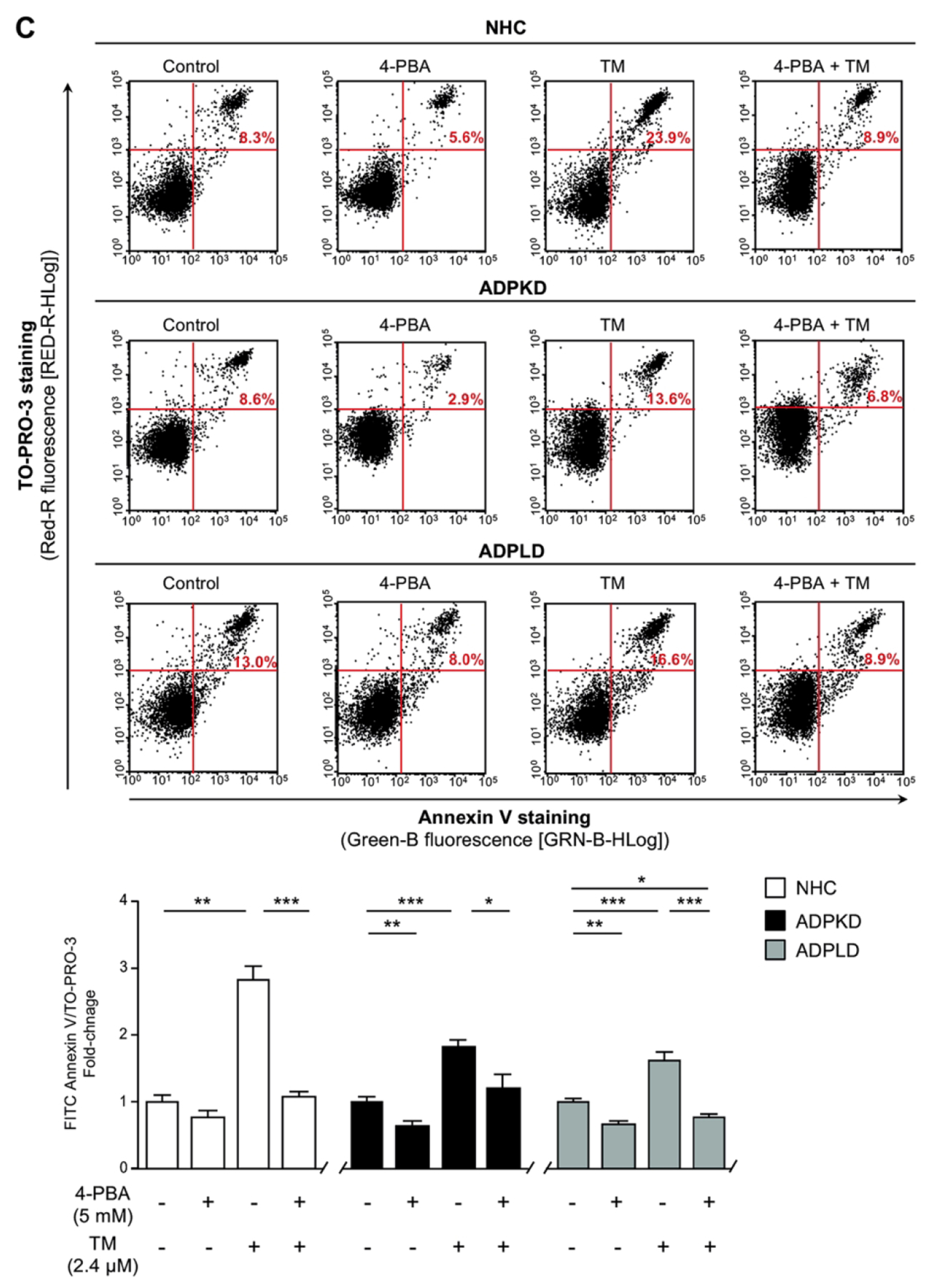
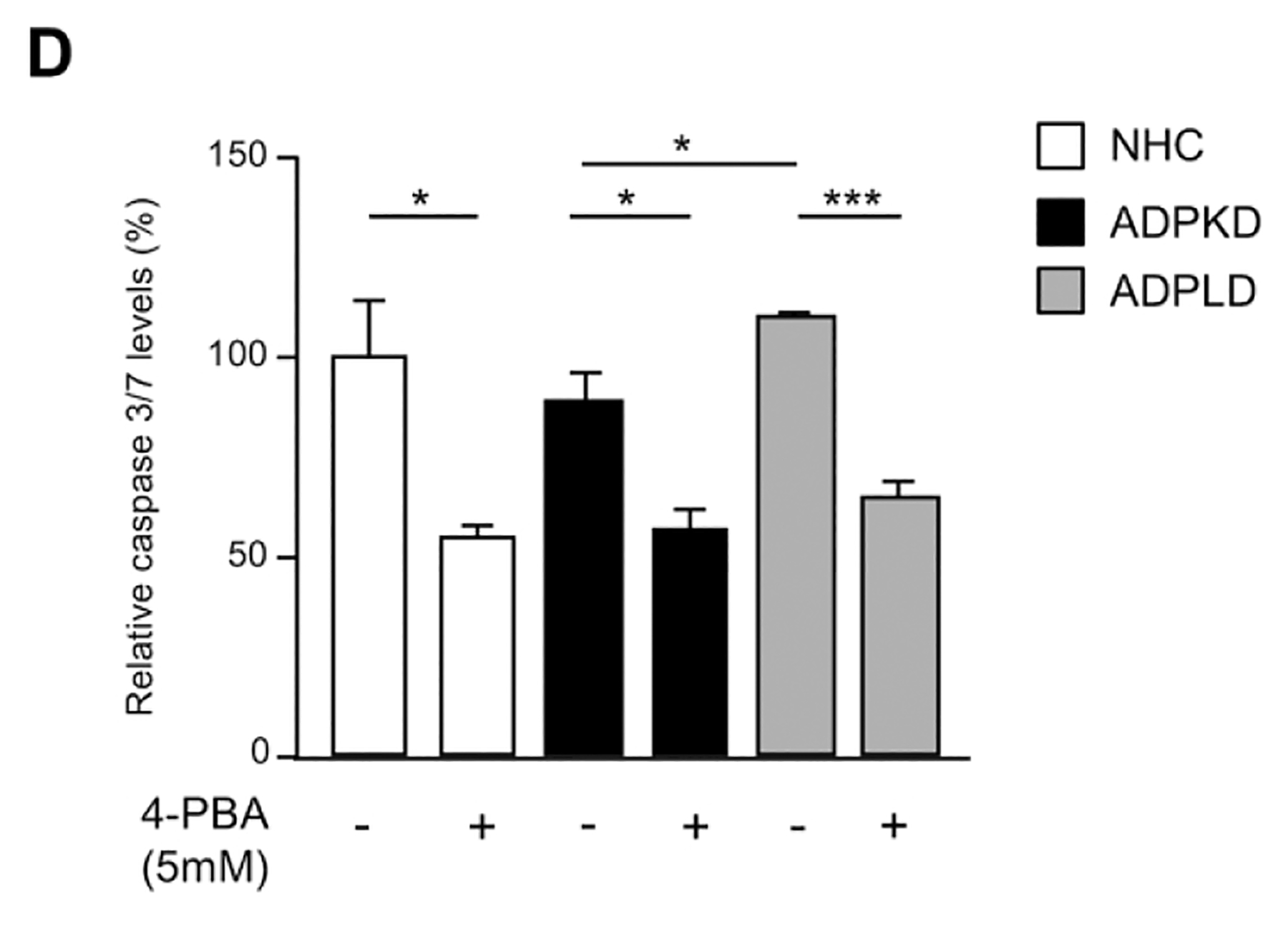
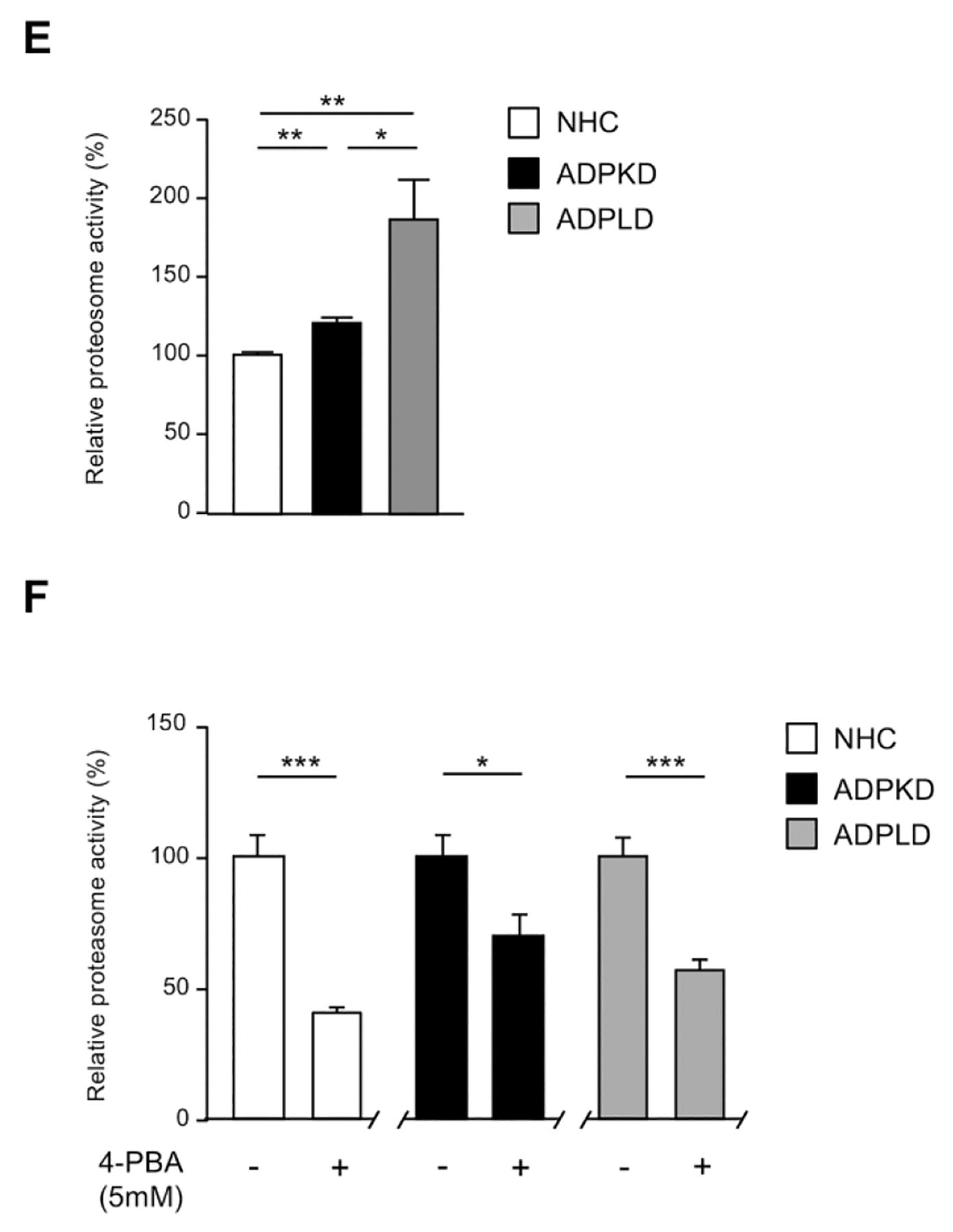
(A) Transcriptional analysis of the UPR effectors in normal (NHC; n=5) and cystic [ADPKD and ADPLD; n=4–5] cholangiocytes after incubation with 4-PBA and/or TM. (B) Representative proliferation histograms and corresponding quantification in NHC, ADPKD and ADPLD (n=4) cholangiocytes incubated with 4-PBA or vehicle. (C) Representative scatterplots indicating the apoptotic rate of normal and cystic cholangiocytes (n=6) incubated with 4-PBA and/or TM. (D) Caspase 3/7 activity in NHC, ADPKD and ADPLD (n=4) cholangiocytes incubated with 4-PBA or vehicle. Relative 20S proteasome activity of NHC, ADPKD and ADPLD cholangiocytes (n=4–5) under (E) baseline and (F) 4-PBA incubation (n=6).
Cystic cholangiocytes exhibit higher proliferative rates than normal cholangiocytes.(16, 17) Noteworthy, the proliferation of both ADPKD and ADPLD cholangiocytes was significantly reduced upon 4-PBA treatment, an effect that was also evident in NHC (Fig. 5B). On the other hand, no significant differences in the baseline apoptotic rate were observed between normal and cystic cholangiocytes in vitro (Fig. 5C). 4-PBA also decreased ADPKD and ADPLD cholangiocytes baseline cell death, while no differences were evident in NHC (Fig. 5C). Moreover, 4-PBA protected against TM-induced apoptosis in both normal and cystic human cholangiocytes (Fig. 5C). In line with this, baseline caspase 3/7 activity was significantly diminished after 4-PBA exposure in normal and cystic cholangiocytes (Fig. 5D), further substantiating the anti-apoptotic effect of 4-PBA.
The ER stress and ubiquitin-proteasome system are interconnected in cystic cholangiocytes
An integrated part of the ERAD pathway is the ubiquitin-proteasome system (UPS), which ultimately leads to the degradation of the terminally un/misfolded proteins retrotranslocated from the ER,(18) in order to alleviate the burden of accumulated structurally aberrant proteins, in an attempt to decrease the ER stress.(19) Data showed that the 20S proteasome activity is increased in cystic human cholangiocytes (i.e., ADPKD and ADPLD) under baseline conditions, compared to the respective controls (Fig. 5E). In addition, ADPLD cholangiocytes exhibited increased 20S proteasome activity compared to ADPKD cholangiocytes (Fig. 5E). Interestingly, a decline of 30–60% of the 20S proteasome activity was observed in NHC, ADPKD and ADPLD cholangiocytes under the presence of 4-PBA (Fig. 5F).
4-PBA improves ER proteostasis in cystic cholangiocytes
The impact of 4-PBA administration on the proteomic profile and protein biogenesis pathways of cystic cholangiocytes were further analyzed. A total of 2250 proteins were commonly expressed in cystic cholangiocytes (i.e., ADPKD and ADPLD) under baseline and 4-PBA conditions (Fig. 6A). Importantly, the expression levels of 266 dysregulated proteins in ADPKD and ADPLD cholangiocytes became normalized after 4-PBA administration (Fig. 6B). Among them, the expression of 8 proteins commonly dysregulated in ADPKD and ADPLD cholangiocytes and involved in protein folding, trafficking and degradation were reverted to baseline levels after 4-PBA administration (Fig. 6C). As shown in Fig. 6D, the levels of most of the aforementioned key proteins found dysregulated under baseline conditions in cystic cholangiocytes (Fig. 2F) were reverted by 4-PBA. Similarly, the levels of 225 from a total of 2092 proteins differentially expressed between PCK and NRC cholangiocytes were subsequently normalized, at least in part, by 4-PBA (Figs. 7A–C).
Figure 6. Effect of 4-PBA on the proteomic profiles of normal and cystic human cholangiocyte.
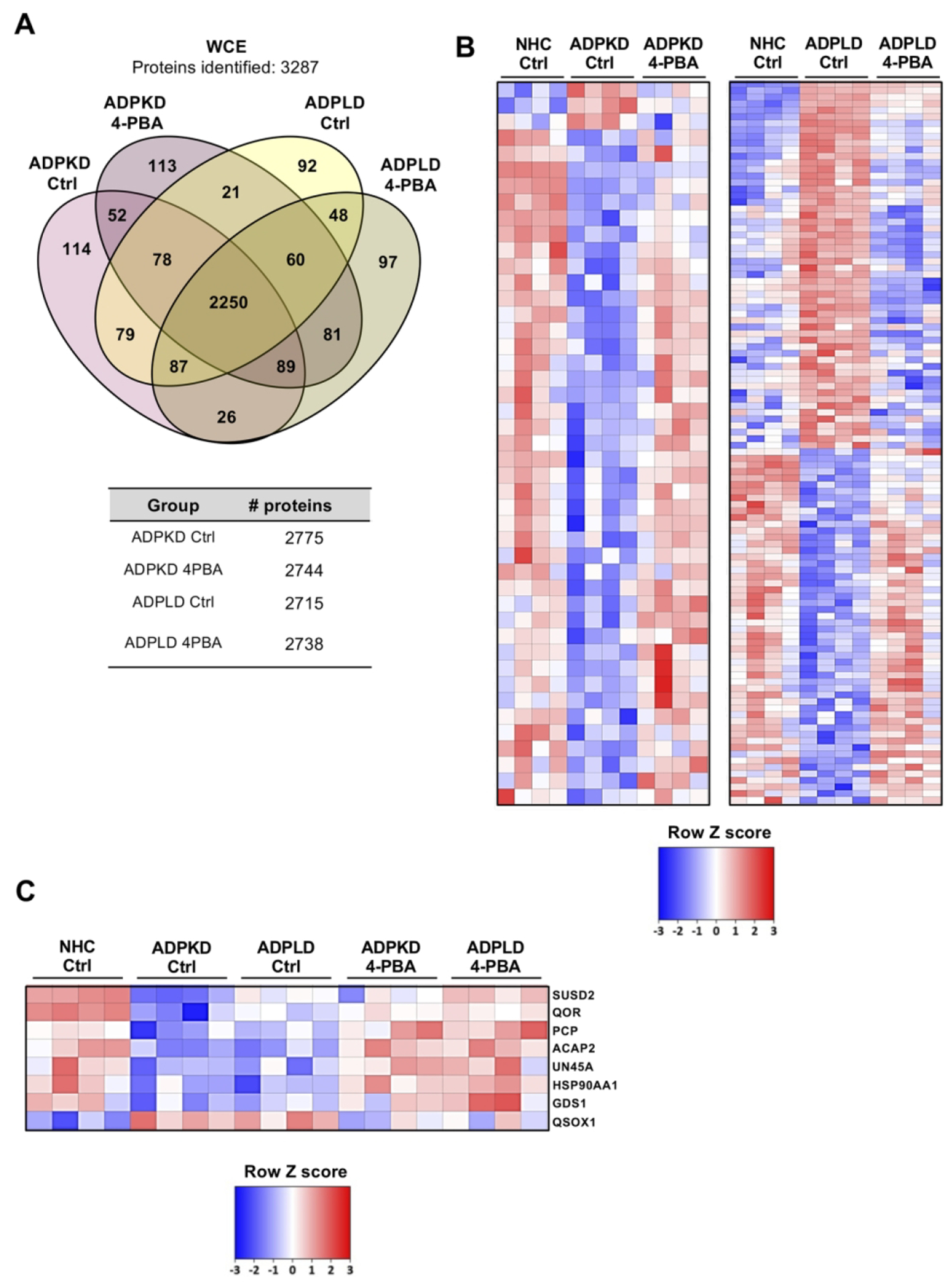
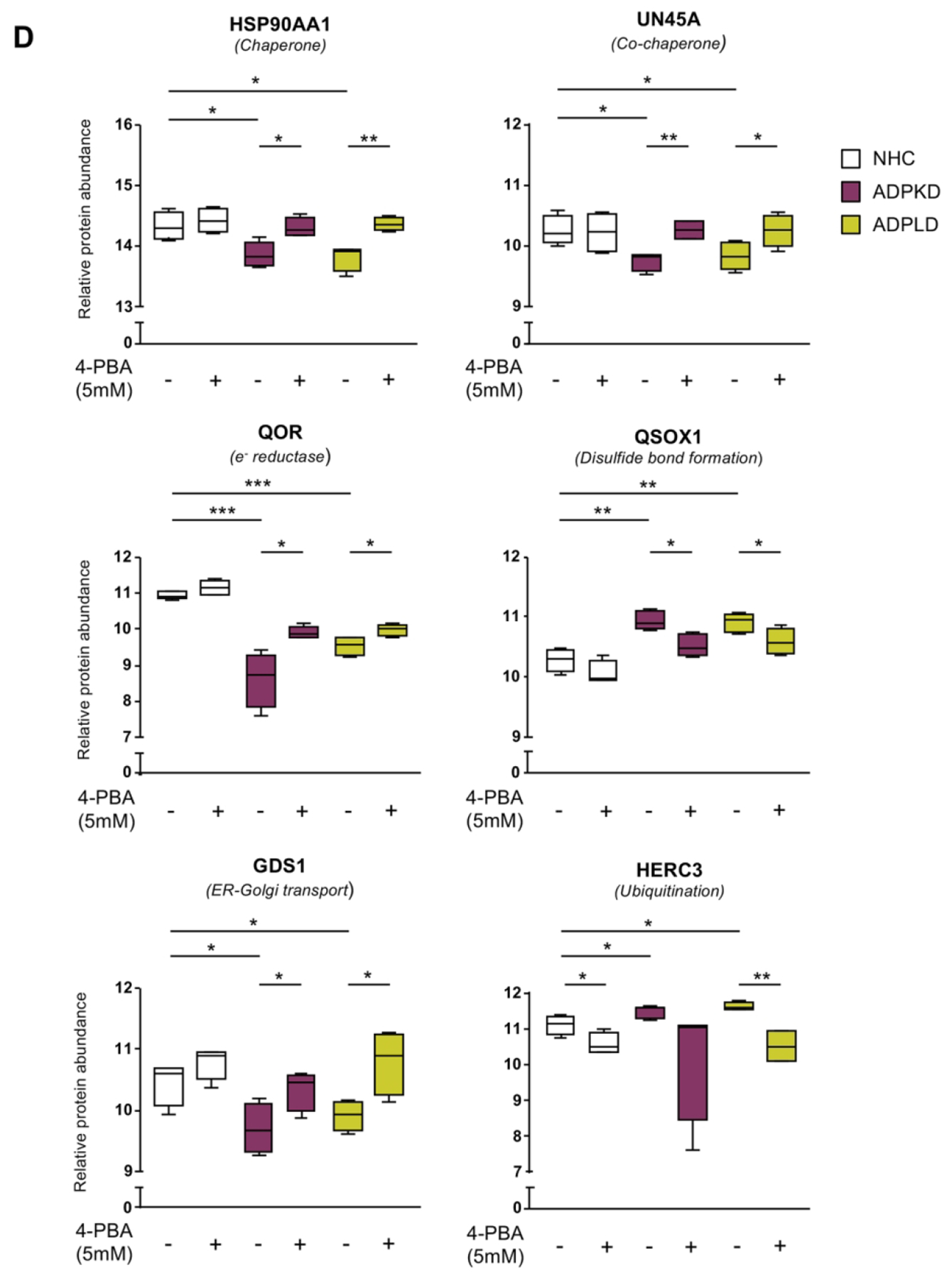
(A) Venn diagram depicting the number of proteins identified in cystic human cholangiocytes (ADPKD and ADPLD) under baseline and 4-PBA incubated conditions. (B) Heatmaps showing the effect of 4-PBA on the expression pattern of proteins dysregulated in ADPKD and ADPLD cholangiocytes compared to NHC. (C) Heatmap showing those proteins that were similarly dysregulated in ADPKD and ADPLD cholangiocytes and equally modulated by 4-PBA. (D) Box plot diagrams showing the modulatory effects of 4-PBA on the relative abundance of each selected protein in normal and cystic human cholangiocytes.
Figure 7. Effect of 4-PBA on the proteomic profiles of normal and cystic rat cholangiocyte.
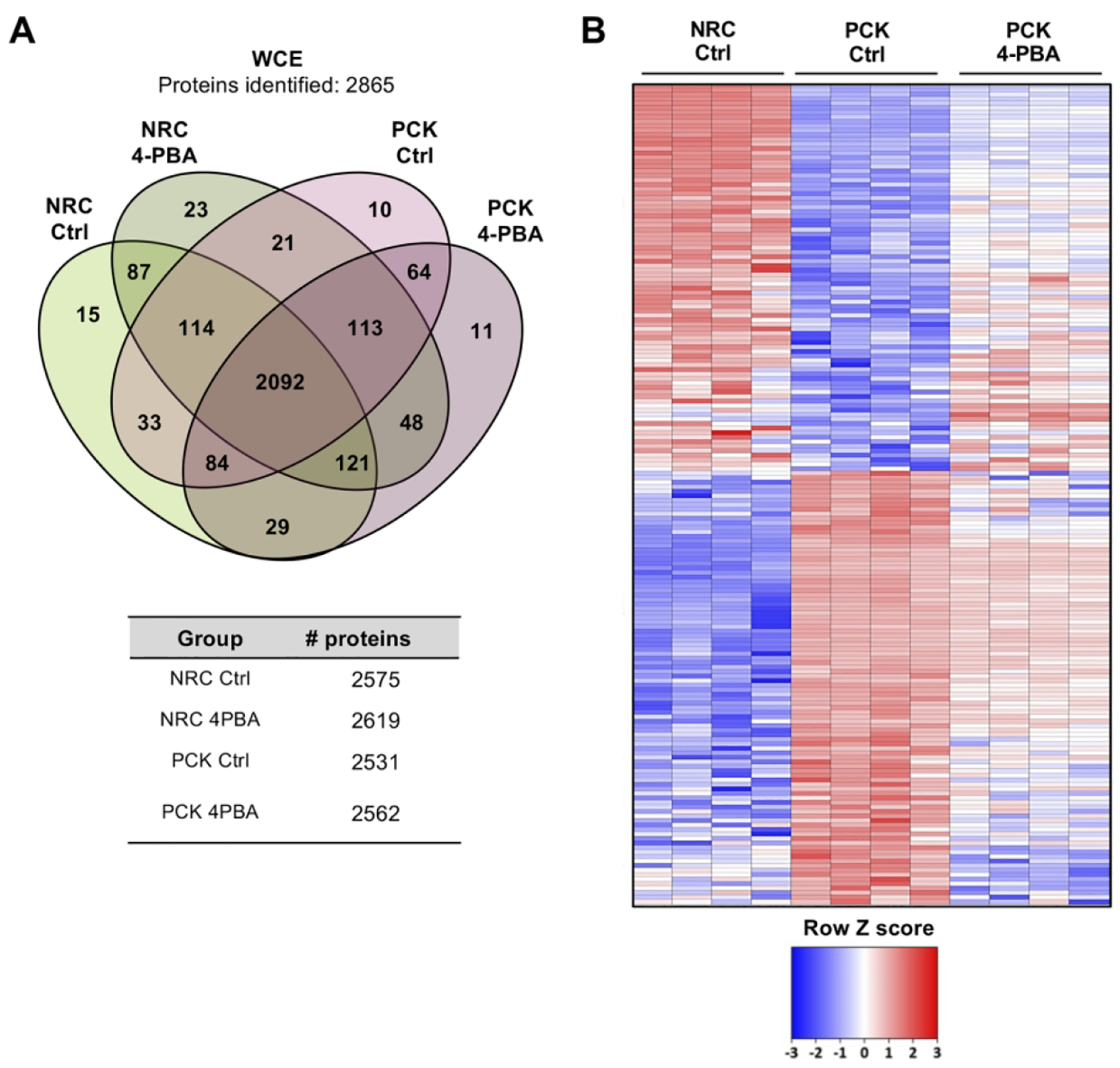
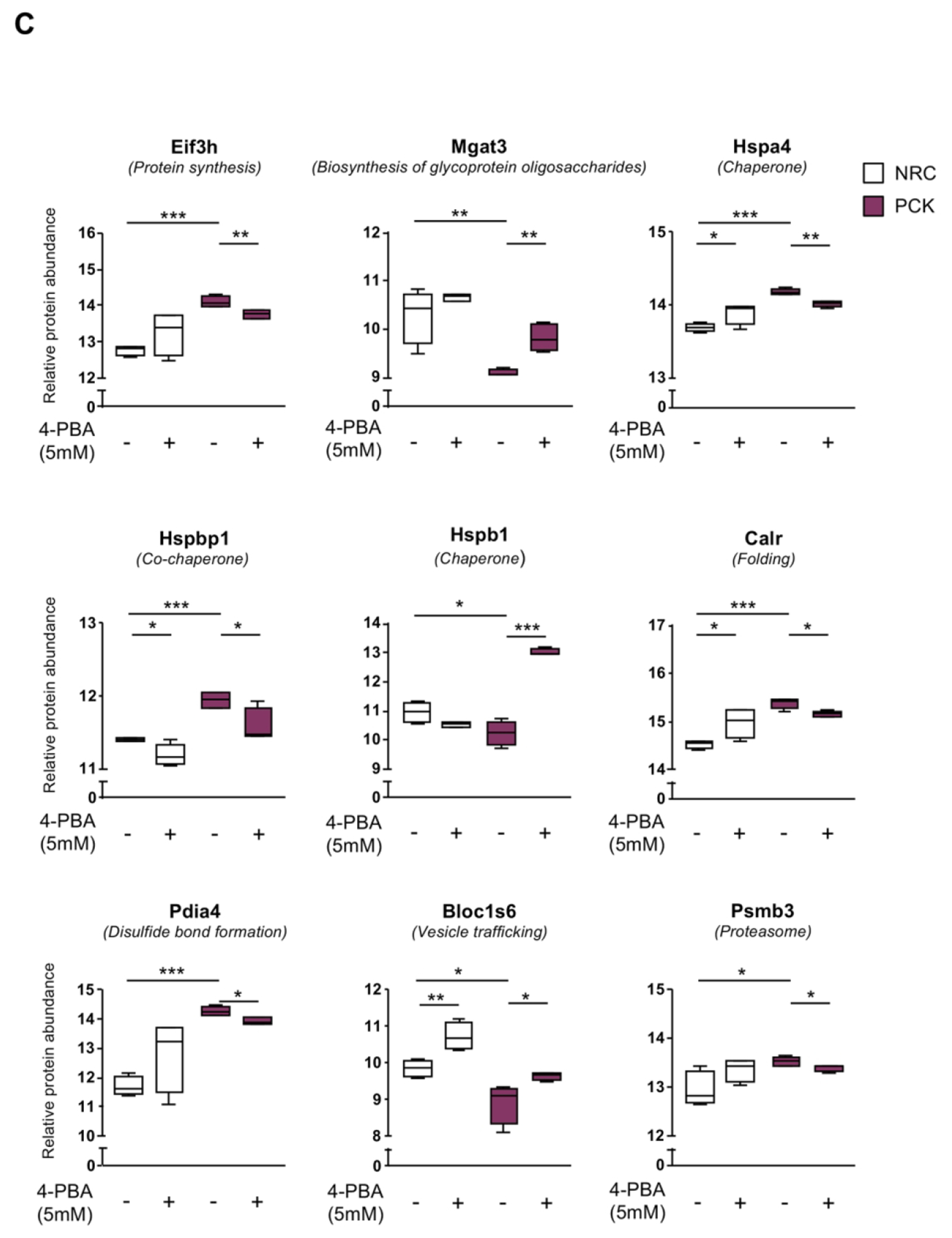
(A) Venn diagram depicting the number of proteins identified in both normal and cystic rat cholangiocytes under baseline and 4-PBA incubated conditions. (B) Heatmap showing the effect of 4-PBA on the expression pattern of proteins significantly dysregulated in PCK compared to NRC cholangiocytes. (C) Box plot diagrams showing the modulatory effects of 4-PBA on the relative abundance of each selected protein in normal and cystic rat cholangiocytes.
Discussion
The findings reported in this study indicate that: (I) the UPR signaling pathways are induced in PLD, which is revealed by increased expression of ER stress sensors and effectors in cystic tissue from patients with PLD and in cholangiocytes in culture; (II) morphologically, the ER lumen of cystic cholangiocytes undergoes a marked dilatation under baseline conditions, which is a characteristic of ER stress; (III) the proteomic profile of cystic cholangiocytes is consistent with alterations in protein synthesis, folding, trafficking and degradation; (IV) chronic administration of the 4-PBA chaperone reduces liver weight and volume, as well as the hepatic cystic volume in PCK rats, both in the presence or absence of the ER stress inducer TM, thus delaying disease progression; (V) 4-PBA exerts anti-proliferative and anti-apoptotic effects, and re-establishes ER proteostatic mechanisms in cystic cholangiocytes in vitro; (VI) TM increases the expression of the UPR effectors and promotes cell death, two events that were efficiently attenuated by 4-PBA; (VII) cystic cholangiocytes exhibits 20S proteasome hyperactivity, which is reduced after 4-PBA incubation in vitro. All these data are consistent with our hypothesis that abnormal proteostasis and ER stress play a pivotal role in the pathogenesis of PLD, while its attenuation may halt hepatic cystogenesis, ameliorating the disease pathogenesis and thus representing a novel and promising therapeutic strategy.
Different molecular mechanisms are behind the pathogenesis of PLD, including primary cilium and centrosome dysfunctions, matrix metalloproteinase hyperactivity, aberrant fluid secretion, epigenetic alterations, and dysregulated intracellular levels of cAMP and Ca2+, leading to the characteristic hyperproliferative phenotype of cystic cholangiocytes that govern the onset and progression of hepatic cystogenesis.(3, 17) However, the fact that the majority of PLD-causative genes encode for ER-resident proteins involved in the biogenesis and trafficking of nascent proteins,(2) suggests that these processes may be primarily or secondary altered in PLD, contributing to cyst growth and disease severity. In this regard, it was proposed that impaired biogenesis and/or transport of polycystin-1 to the primary cilia reduce its functional dosage, which is the rate-limiting determinant of cystogenesis and consequently of PLD severity.(4, 5) Taking these observations into consideration, we aimed to study the morphological and molecular features of the ER in cystic cholangiocytes, ascertain their role in the development and progression of PLD, and evaluate the therapeutic value of its regulation. Our data indicate that mutations in the PLD-related genes PRKCSH, SEC63, GANAB and Pkhd1 are associated with upregulation of the UPR signaling pathways (i.e., sensors and effectors). Further proteomic studies confirmed an abnormal ER-related proteostasis in cystic cholangiocytes associated with overexpression of the Hsp70 family, as a potential compensatory mechanism, but without changes in the expression other chaperone families involved in late stages of protein folding (such as Hsp90, among others). In line with this, pronounced enlargement of the ER lumen, 20S proteasome hyperactivity and overexpression of several ubiquitin ligases were evident in cystic cholangiocytes. PRKCSH, SEC63 and GANAB encode for the ER-resident proteins glucosidase II beta subunit, SEC63, and glucosidase II alpha subunit respectively, which play key roles in co- and post-translational modifications of nascent proteins.(4) Hence, partial or total abolishment of the function of these proteins may impair the folding, maturation and/or trafficking of nascent ER proteins, promoting their accumulation inside the ER and triggering ER stress. Of note, ADPKD (GANAB mutant) cholangiocytes exhibited increased levels of the UPR effectors XBP1 and CHOP, as well as enlarged ER lumen, compared to ADPLD (PRKCSH mutant) cholangiocytes at baseline conditions. These effects were in line with the 20S proteasome activity in both types of diseased cholangiocytes, supporting the notion that the 20S proteasome activity aims to relieve the levels of un/misfolded proteins and the subsequent ER stress in cystic cholangiocytes. Based on all these data, the levels of ER stress and the interplay between the ER proteostasis and the proteasome activity seem to be dependent on the type of gene found mutated in PLD. On the other hand, Pkhd1 encodes for fibrocystin/polyductin, a protein localized in the primary cilium that seems to participate in tubulogenesis and the maintenance of ductal lumen epithelium architecture.(3, 17) The splicing mutation of the Pkhd1 orthologous gene in PCK rats compromise the correct maturation of fibrocystin/polyductin, which is retained within the ER and rapidly degraded, reducing the amount of protein located in the primary cilia, where it usually interacts with polycystin-2 (PC2) and modulate the activity of this calcium channel.(20) Furthermore, fibrocystin/polyductin also interacts with the calcium modulating cyclophilin ligand, a primary cilia and ER-associated protein involved in the regulation of cytosolic Ca2+ levels.(21) Interestingly, ER stress is also induced when the ER-calcium storage is depleted, as some ER chaperones require an optimal Ca2+ concentration for the correct folding and maturation of nascent proteins,(14, 22) supporting our data. Under all of these pathological circumstances, and in order to restore the protein homeostasis within the ER lumen and resolve ER stress, UPR signaling cascades become activated, leading to the upregulation of genes involved in both protein folding and ERAD.(6, 7) It is well documented that the transcription factors XBP1 and ATF6 (i.e., N-terminal cytosolic domain) translocate to the nucleus,(23) where they induce the expression of GRP78 (a member of the Hsp70 family of chaperones), XBP1 and components of the ERAD machinery to promote the folding and UPS-mediated degradation of structurally aberrant proteins, respectively.(23) Interestingly, both mechanisms counteract the mild pro-apoptotic signals motivated by the transcriptional activation of CHOP via ATF4.(23) However, when the UPR is unable to relieve the burden of un/misfolded proteins, excessive/chronic ER stress might result in proteotoxicity-induced cell death.(6, 7) In this scenario, it has been reported that ATF4 and ATF6 markedly upregulate the expression of CHOP,(7, 24) reaching levels that are unmanageable by the adaptive responses inducing apoptosis. Of note, sustained UPR activation was previously identified as a pivotal pathogenic factor in several human diseases, such as diabetes mellitus, neurodegenerative disorders, viral infections, cancer, heart failure and a wide range of liver diseases.(25, 26)
Several reports have demonstrated the chaperone-like activity of the small molecule 4-PBA, which promotes protein folding and trafficking, thus preventing the aggregation of un/misfolded proteins within the ER and alleviating the ER stress.(14, 27) Indeed, the therapeutic effects of 4-PBA were assessed in a variety of experimental models of genetic,(28) inflammatory,(29) metabolic,(30) and liver(31) diseases, exerting promising benefits. The results obtained in our in vitro and in vivo experimental models of PLD are consistent with these findings, as 4-PBA halted the disease progression in PCK rats. On the other hand, the ER stressor TM did not exacerbate the hepatomegaly and liver cystogenesis of PCK rats, probably as a consequence of proteotoxic-induced cell death processes that could be linked to the elevation in AST and ALP levels observed in TM-administered PCK animals. Notably, 4-PBA exerted similar therapeutic benefits when it was administered in combination with TM. Furthermore, 4-PBA markedly attenuated the UPS adaptive response (i.e., proteasome activity and expression of ubiquitin-ligases) in cystic cholangiocytes due to its capacity to assist both folding and trafficking of newly synthesized proteins, contributing to restore ER proteostasis and resolve ER stress. Accordingly, 4-PBA inhibited the hyperproliferation, as well as the baseline and TM-induced apoptosis in cystic cholangiocytes, maintaining a suitable balance that delays the progression of PLD.(32)
Our data are also in line with a previous report indicating that cystic cholangiocytes are characterized by increased autophagy,(12) as overexpression of lysosomal proteolysis components was also evident here in cystic cholangiocytes. In this regard, emerging evidences indicate that activation of UPR signaling can also promote the clearance of un/misfolded proteins via autophagy, specially, of those proteins that are not efficiently degraded by the ERAD-proteasome system.(33–35)
In summary (Fig. 8), this study provides strong evidence that cystic cholangiocytes are characterized by abnormal ER proteostasis, leading to the accumulation of a burden of un/misfolded proteins within the ER lumen that causes ER stress and the subsequent activation of pro-survival mechanisms that promote hepatic cystogenesis, thus representing a novel and promising therapeutic target for patients with PLD.
Figure 8. Working model.
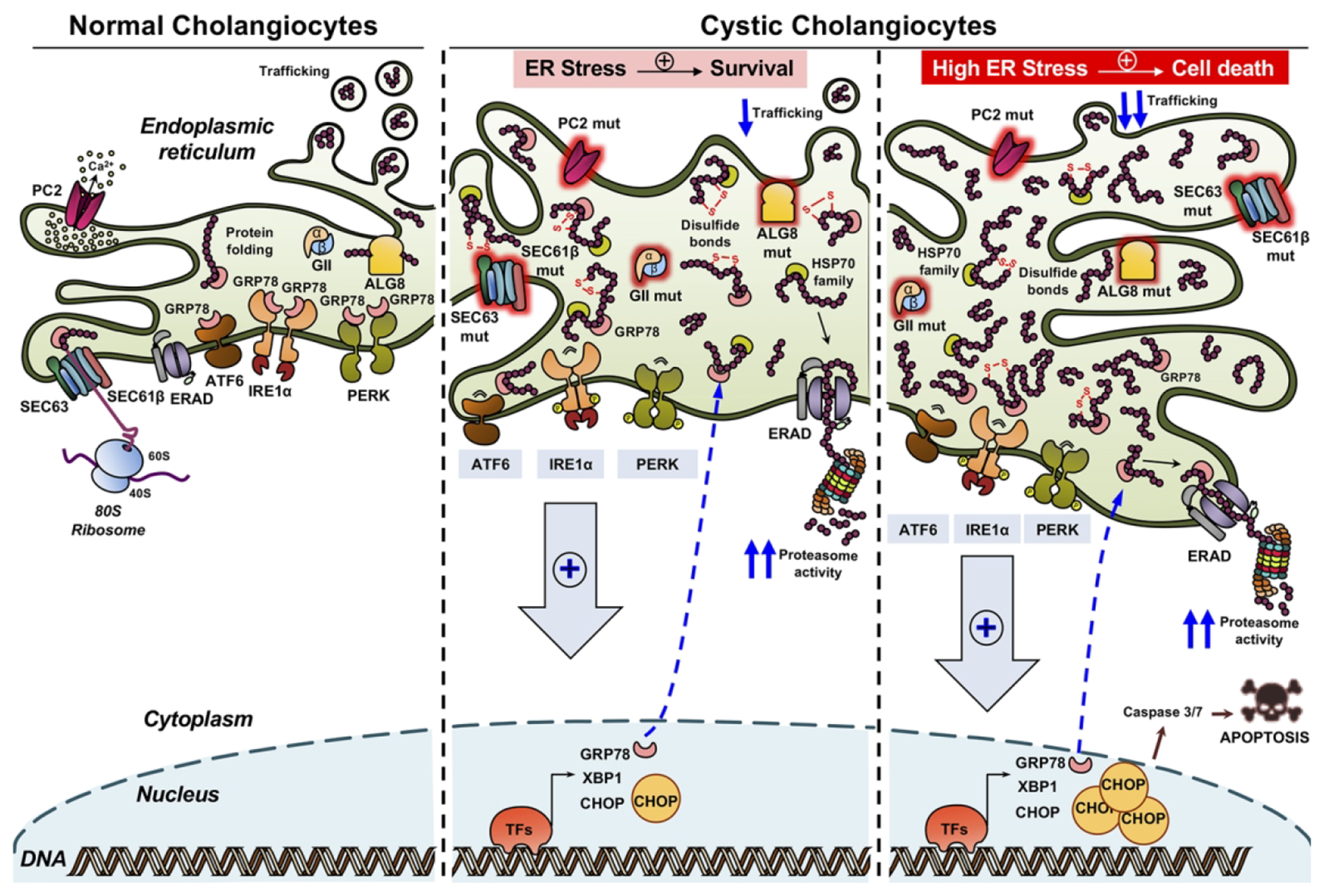
Mutations in PLD-related genes that encode for ER-resident proteins (i.e., PC2, ALG8, SEC63, SEC61B, GIIa and GIIb) compromise the ER proteostasis and promote the accumulation of aberrant structurally nascent proteins within the ER lumen, leading to the induction of the UPR signaling cascades, enlargement of the ER lumen and hyperactivation of the cellular degradation mechanisms mediated by the UPS. When ER stress levels are low the three adaptive mechanisms are able to restore the proteostasis and resolve the ER stress reinforcing the survival and proliferation of cystic cholangiocytes. Nevertheless, if the ER stress becomes chronic and excessive the adaptive mechanism fails to restore ER proteostasis, inducing cholangiocyte apoptosis by CHOP and caspase 3/7 activation.
Supplementary Material
Acknowledgements:
Dr. Aura D. Urribarri (CIMA of the University of Navarra, Spain) and Charlotte de Mooij (Erasmus student, The Netherlands) for preliminary data assistance.
Fianancial Support: Spanish Carlos III Health Institute (ISCIII) [J.M. Banales (FIS PI12/00380, PI15/01132, PI18/01075 and Miguel Servet Program CON14/00129); M.J. Perugorria (FIS PI14/00399, PI17/00022)] cofinanced by “Fondo Europeo de Desarrollo Regional” (FEDER); CIBERehd (M.J. Perugorria, J.M. Banales and L. Bujanda), Spain; “Diputación Foral de Gipuzkoa” (J.M. Banales: DFG15/010, DFG16/004; BIOEF (Basque Foundation for Innovation and Health Research: EiTB Maratoia BIO15/CA/016/BD to J.M. Banales); Department of Health of the Basque Country (M.J. Perugorria: 2015111100 and J.M. Banales: 2017111010) and Euskadi RIS3 (J.M. Banales: 2016222001, 2017222014, 2018222029). La Caixa Scientific Foundation (J.M. Banales: HR17-00601). “Fundación Científica de la Asociación Española Contra el Cáncer” (AECC Scientific Foundation, to J.M. Banales). National Institutes of Health (NIH) of United States of America (DK24031 to N.F. LaRusso). MJ Perugorria was funded by the Spanish Ministry of Economy and Competitiveness (Ramón y Cajal Program RYC-2015-17755), FJ Caballero by the Spanish Ministry of Economy and Business (MINECO: BES-2014-069148) and A Santos-Laso by the Basque Government (PRE_2018_2_0195) and European Molecular Biology Organization (EMBO, Short-Term Fellowship Award 2019; #8223).
Abbreviations:
- PLDs
polycystic liver diseases
- ER
endoplasmic reticulum
- qPCR
quantitative polymerase chain reaction
- TEM
transmission electron microscopy
- 4-PBA
4-phenylbutyric acid sodium salt
- TM
tunicamycin
- PCK
polycystic
- UPR
unfolded protein response
- mRNA
messenger ribonucleic acid
- ADPLD
autosomal dominant polycystic liver disease
- ADPKD
autosomal dominant polycystic kidney disease
- ARPKD
autosomal recessive polycystic kidney disease
- CHF
congenital hepatic fibrosis
- PRKCSH
protein kinase C substrate 80K-H
- SEC63
translocation protein SEC63 homolog
- ALG8
asparagine-linked glycosylation 8
- GANAB
glucosidase II alpha subunit
- SEC61β
SEC61 translocon beta subunit
- PKD2
polycystic kidney disease 2
- IRE1α
inositol-requiring enzyme 1 alpha
- PERK
PRKR-like endoplasmic reticulum kinase
- ATF6
activating transcription factor 6
- CHOP
C/EBP-homologous protein
- GRP78
glucose-regulated protein 78kDa
- XBP1
x-box binding protein 1
- ERAD
endoplasmic reticulum-associated protein degradation
- WT
wild-type
- NHC
normal human cholangiocytes
- NRC
normal rat cholangiocytes
- DMEM/F-12
dulbecco’s modified eagle medium/nutrient mixture F-12
- PKHD1
polycystic kidney and hepatic disease 1
- AMC
7-amino-4-methylcoumarin
- NIH
national institutes of health
- WCEs
whole cell extracts
- SEM
standard error of the mean
- ANOVA
analysis of variance
- GO
gene ontology
- ALP
alkaline phosphatase
- AST
aspartate aminotransferase
- UPS
ubiquitin proteasome system
- cAMP
cyclic adenosine monophosphate
- Ca2+
calcium
- PC2
polycystin-2
- ATF4
activating transcription factor 4
Footnotes
Conflict of interest: authors disclose no conflicts.
References
- 1.van Aerts RMM, van de Laarschot LFM, Banales JM, Drenth JPH. Clinical management of polycystic liver disease. J Hepatol. 2018;68(4):827–37. [DOI] [PubMed] [Google Scholar]
- 2.Perugorria MJ, Banales JM. Genetics: Novel causative genes for polycystic liver disease. Nat Rev Gastroenterol Hepatol. 2017;14(7):391–2. [DOI] [PubMed] [Google Scholar]
- 3.Santos-Laso A, Izquierdo-Sanchez L, Lee-Law PY, Perugorria MJ, Marzioni M, Marin JJ, et al. New Advances in Polycystic Liver Diseases. Semin Liver Dis. 2017;37(1):45–55. [DOI] [PubMed] [Google Scholar]
- 4.Besse W, Dong K, Choi J, Punia S, Fedeles SV, Choi M, et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J Clin Invest. 2017;127(5):1772–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fedeles SV, So JS, Shrikhande A, Lee SH, Gallagher AR, Barkauskas CE, et al. Sec63 and Xbp1 regulate IRE1alpha activity and polycystic disease severity. J Clin Invest. 2015;125(5):1955–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hetz C, Chevet E, Oakes SA. Proteostasis control by the unfolded protein response. Nat Cell Biol. 2015;17(7):829–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hetz C, Papa FR. The Unfolded Protein Response and Cell Fate Control. Mol Cell. 2018;69(2):169–81. [DOI] [PubMed] [Google Scholar]
- 8.Mason SB, Liang Y, Sinders RM, Miller CA, Eggleston-Gulyas T, Crisler-Roberts R, et al. Disease stage characterization of hepatorenal fibrocystic pathology in the PCK rat model of ARPKD. Anat Rec. 2010;293(8):1279–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masyuk TV, Huang BQ, Masyuk AI, Ritman EL, Torres VE, Wang X, et al. Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. Am J Pathol. 2004;165(5):1719–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urribarri AD, Munoz-Garrido P, Perugorria MJ, Erice O, Merino-Azpitarte M, Arbelaiz A, et al. Inhibition of metalloprotease hyperactivity in cystic cholangiocytes halts the development of polycystic liver diseases. Gut. 2014;63(10):1658–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porath B, Gainullin VG, Cornec-Le Gall E, Dillinger EK, Heyer CM, Hopp K, et al. Mutations in GANAB, Encoding the Glucosidase IIalpha Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am J Hum Genet. 2016;98(6):1193–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Masyuk AI, Masyuk TV, Lorenzo Pisarello MJ, Ding JF, Loarca L, Huang BQ, et al. Cholangiocyte autophagy contributes to hepatic cystogenesis in polycystic liver disease and represents a potential therapeutic target. Hepatology. 2018;67(3):1088–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schonthal AH. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica. 2012;2012:857516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lebeaupin C, Vallee D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol. 2018;69(4):927–47. [DOI] [PubMed] [Google Scholar]
- 15.Guha P, Kaptan E, Gade P, Kalvakolanu DV, Ahmed H. Tunicamycin induced endoplasmic reticulum stress promotes apoptosis of prostate cancer cells by activating mTORC1. Oncotarget. 2017;8(40):68191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Banales JM, Masyuk TV, Gradilone SA, Masyuk AI, Medina JF, LaRusso NF. The cAMP effectors Epac and protein kinase a (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD). Hepatology. 2009;49(1):160–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perugorria MJ, Masyuk TV, Marin JJ, Marzioni M, Bujanda L, LaRusso NF, et al. Polycystic liver diseases: advanced insights into the molecular mechanisms. Nat Rev Gastroenterol Hepatol. 2014;11(12):750–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christianson JC, Ye Y. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol. 2014;21(4):325–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menendez-Benito V, Verhoef LG, Masucci MG, Dantuma NP. Endoplasmic reticulum stress compromises the ubiquitin-proteasome system. Hum Mol Genet. 2005;14(19):2787–99. [DOI] [PubMed] [Google Scholar]
- 20.Kim I, Fu Y, Hui K, Moeckel G, Mai W, Li C, et al. Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J Am Soc Nephrol. 2008;19(3):455–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagano J, Kitamura K, Hujer KM, Ward CJ, Bram RJ, Hopfer U, et al. Fibrocystin interacts with CAML, a protein involved in Ca2+ signaling. Biochem Biophys Res Commun. 2005;338(2):880–9. [DOI] [PubMed] [Google Scholar]
- 22.Torres M, Encina G, Soto C, Hetz C. Abnormal calcium homeostasis and protein folding stress at the ER: A common factor in familial and infectious prion disorders. Commun Integr Biol. 2011;4(3):258–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corazzari M, Gagliardi M, Fimia GM, Piacentini M. Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front Oncol. 2017;7:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu H, Tian M, Ding C, Yu S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front Immunol. 2018;9:3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dara L, Ji C, Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology. 2011;53(5):1752–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kolb PS, Ayaub EA, Zhou W, Yum V, Dickhout JG, Ask K. The therapeutic effects of 4-phenylbutyric acid in maintaining proteostasis. Int J Biochem Cell Biol. 2015;61:45–52. [DOI] [PubMed] [Google Scholar]
- 28.Besio R, Iula G, Garibaldi N, Cipolla L, Sabbioneda S, Biggiogera M, et al. 4-PBA ameliorates cellular homeostasis in fibroblasts from osteogenesis imperfecta patients by enhancing autophagy and stimulating protein secretion. Biochim Biophys Acta Mol Basis Dis. 2018;1864(5 Pt A):1642–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim HJ, Jeong JS, Kim SR, Park SY, Chae HJ, Lee YC. Inhibition of endoplasmic reticulum stress alleviates lipopolysaccharide-induced lung inflammation through modulation of NF-kappaB/HIF-1alpha signaling pathway. Sci Rep. 2013;3:1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Montane J, de Pablo S, Castano C, Rodriguez-Comas J, Cadavez L, Obach M, et al. Amyloid-induced beta-cell dysfunction and islet inflammation are ameliorated by 4-phenylbutyrate (PBA) treatment. FASEB J. 2017;31(12):5296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ren LP, Song GY, Hu ZJ, Zhang M, Peng L, Chen SC, et al. The chemical chaperon 4-phenylbutyric acid ameliorates hepatic steatosis through inhibition of de novo lipogenesis in high-fructose-fed rats. Int J Mol Med. 2013;32(5):1029–36. [DOI] [PubMed] [Google Scholar]
- 32.Lee EJ. Cell Proliferation and Apoptosis in ADPKD. Adv Exp Med Biol. 2016;933:25–34. [DOI] [PubMed] [Google Scholar]
- 33.Rashid HO, Yadav RK, Kim HR, Chae HJ. ER stress: Autophagy induction, inhibition and selection. Autophagy. 2015;11(11):1956–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol. 2017;13(11):681–96. [DOI] [PubMed] [Google Scholar]
- 35.Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci. 2015;40(3):141–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.