

Abstract
Vitamin A deficiency (VAD) is a major public health problem and is associated with increased host susceptibility to infection; however, how VAD influences viral infection remains unclear. Using a persistent LCMV infection model, we showed in this study that while VAD did not alter innate type I interferon production, infected VAD mice had hyperactive, virus-specific T cell responses at both the acute and contraction stages, showing significantly decreased PD-1 but increased cytokine (IFN-γ, TNF-α and IL-2) expression by T cells. Compared with control mice, VAD mice displayed excessive inflammation and more severe liver pathology, with increased death during persistent infection. Of note, supplements of all-trans retinoic acid (RA), one of the important metabolites of vitamin A, downregulated hyperactive T cell responses and rescued the persistently infected-VAD mice. By using adoptive transfer of splenocytes, we found that the environmental vitamin A or its metabolites acted as rheostats modulating antiviral T cells. The analyses of T cell transcriptional factors and signaling pathways revealed the possible mechanisms of RA, as its supplements inhibited the abundance of NFATc1 (nuclear factor of activated T cells 1), a key regulator for T cell activation. Also, following CD3/CD28 cross-linking stimulation, RA negatively regulated the TCR-proximal signaling in T cells, via decreased phosphorylation of Zap70 and its downstream signals, including phosphorylated AKT, p38, ERK, and S6, respectively. Together, our data reveal VAD-mediated alterations in antiviral T cell responses and highlight the potential utility of RA for modulating excessive immune responses and tissue injury in infectious diseases.
1. Introduction
Vitamin A is one of the essential nutrients, playing an important role in physiological functions, including vision, growth, reproduction, immunity and cellular integrity (1). Vitamin A deficiency (VAD) is a significant public health problem worldwide, especially in low-income countries. Although the prevalence of VAD has declined in the past decade, which is attributed to vitamin A supplements, there is still a high prevalence in sub-Saharan Africa and south Asia (48% and 44% respectively) among children aged 6–59 months in 2013 (2). More than one hundred thousand deaths among under-5-year-olds from either diarrhea or measles are attributed to VAD in low-income and middle-income countries in 2013 (2). VAD is known to increase the risk of disease and death from severe and chronic viral infections. For instance, clinical studies have indicated that HIV-positive patients have lower serum retinol concentrations compared with HIV-negative individuals (3, 4). A high prevalence of VAD is also observed in hepatitis C patients throughout all stages of chronic liver disease, and the serum retinol concentration is related to the severity of disease development, complications and mortality (5, 6). Although the correlation of vitamin A and disease severity is observed in clinical studies, infectious diseases can precipitate VAD by decreasing intake and increasing excretion, raising a question as to whether VAD contributes to disease progress and eventual outcome (7). At present, the mechanism of vitamin A in determining anti-infectious immunity is not entirely understood.
The strategy to deliver vitamin A supplements to infants and children with or without HIV infection is performed in many countries, where VAD is a public health problem. Among HIV-positive children, vitamin A supplements reduced the mortality and morbidity; however, no beneficial effect was found for HIV-infected adults (1, 8, 9). These clinical trials suggest that vitamin A may not have direct anti-HIV effects. However, vitamin A and other retinoids have recently been demonstrated to inhibit measles virus and hepatitis C virus in vitro (10). The all-trans retinoic acid (RA), which is a predominate and natural metabolite of vitamin A, exhibited synergistic effects with PegIFN-α2a treatment in reducing viral load in HCV patients (11). These findings indicate that vitamin A and RA may regulate antiviral immune responses and benefit the host in viral infections. Recent studies highlight the role of RA in immunity and tolerance, especially in the intestinal tract. For instance, gut-associated lymphoid tissues (GALT) CD103+ dendritic cells (DCs) promote expression of homing markers α4β7 and CCR9 on effector T cells via RA (12, 13). The addition of RA to the spleen DC culture significantly enhanced regulatory T cell (Treg) induction in TGF-β- and retinoic acid receptor alpha (RARα)-dependent ways (14, 15). T helper 17 cells (Th17) were ablated in the GALT of VAD mice at steady state; however, RA treatment suppressed Th17 responses and reduced the pathology from bacterial infection (16). It has been reported that VAD mice have aberrant immunity, including increased Th1, but limited Th2 responses (17). Vitamin A and RA can downregulate IFN-γ production through modulating IFN-γ promoters and co-stimulating signals (18, 19). Thus, these findings indicate that RA orchestrates T cell activation and differentiation, contributing to T cell-mediated inflammation and tissue pathology. We previously reported that adenovirus infection increased the transcript level and enzyme activity of RALDH in hepatic stellate cells, indicating that endogenous RA may play a role in immune regulation during viral infection (20). Indeed, RA treatment attenuated liver injury and modulated T cell activation in acute viral infection, highlighting the therapeutic potential of RA in modulating immune responses and limiting tissue damage (20). However, the innate and adaptive immunity in VAD mice during different stages of viral infection remains not clear.
In this study, we established a systemic viral infection in VAD mice using a widely-used model pathogen, lymphocytic choriomeningitis virus (LCMV), and evaluated both innate and adaptive antiviral responses at acute, contraction and persistent stages. We also restored the appropriate T cell responses in VAD mice by RA treatment in persistent infection. To dissect the mechanism of RA on T cell activation, we treated mice with RA and measured the levels of cytokine production and transcriptional factor expression. Mechanistically, we analyzed the phosphorylation of TCR signaling and downstream molecules in T cells by RA treatment in vitro. Our study demonstrated that VAD contributes to a hyperactive T cell response to viral infection and that RA may have the potential to serve as a therapeutic agent for excessive immune responses and tissue injury caused by infectious diseases.
2. Materials and methods
2.1. Animals, infection and treatment
C57BL/6 (B6) mice from the Jackson Laboratory were bred and maintained under specific pathogen-free conditions in the UTMB animal care facility and used at 10–12 weeks of age for breeding. Vitamin A-deficient (TD.10991) and -control (20,000 IU vitamin A/kg, TD.10992) diets were purchased from Envigo (Huntingdon, UK). At day 14.5 of gestation, pregnant B6 females were fed with either vitamin A-deficient or a control diet and maintained on the relevant diet until weaning of the litter. Weanlings were maintained on a special diet throughout the study. Vitamin A-deficient and control mice (6–7 weeks of age) were i.v. injected with 2 × 106 and 1 × 105 focus forming units (FFU) of LCMV strain Clone 13 and Armstrong respectively. RA (Sigma, St. Louis, MO) was prepared in corn oil and DMSO was used as a control for RA. For long-term RA treatment in persistent infection, mice were i.p. injected with 25 μg RA daily. For RA treatment in acute infection, mice were i.p. injected with 200 μg RA at 1, 3, and 5 days post-infection (dpi). All procedures were approved by UTMB’s Institutional Animal Care and Use Committee and performed according to NIH Guidelines.
2.2. H&E and histological scores
Liver specimens were fixed in 10% buffered formalin. Paraffin-embedded sections were stained with H&E for histological evaluation using a modified Knodell scoring system (21).
2.3. Lymphocytes isolation and purification
Lymphocytes were isolated according to our previous method (22). Briefly, the liver and lungs were perfused and digested with 0.05% collagenase IV (Roche, Indianapolis, IN) at 37°C for 30 min. Cell suspensions were passed through a 70-μm nylon cell strainer to yield single-cell suspensions. Lymphocytes were enriched by centrifugation (400 g) at room temperature for 30 min over a 30/70% discontinuous Percoll gradient (Sigma, St. Louis, MO). Spleen and lymph nodes were removed from mice and gently meshed in the RPMI-1640 medium through a cell strainer. Red blood cells were removed by using Red Cell Lysis Buffer (Sigma, St. Louis, MO). Naïve CD4+ and CD8 T+ cells were isolated from spleens of naïve mice and purified using Naïve CD4+ and CD8+ T cell isolation kits respectively, based on the CD44− phenotype (Miltenyi Biotec, Auburn, CA). The purities of the target cells were higher than 95%.
2.4. Cell culture and stimulation
Splenocytes were cultured in an anti-CD3 Ab (BioLegend, 5 μg/mL)-coated plate with soluble anti-CD28 Ab (BioLegend, 4 μg/mL) for 3 or 4 days. RA was added into the culture at the beginning, and DMSO was used as a control. Brefeldin A (Thermo Fisher Scientific, Waltham, MA) was added into each well for the last 5 h of culture, followed by intracellular cytokine staining. For the T cell signaling assay, splenocytes were incubated with RA for 16 h in complete RPMI medium plus 10% FBS. Cells were collected and incubated on ice for 15 min with the anti-mouse CD3 (1 μg/mL) and anti-mouse CD28 (1 μg/mL). Cells were then stimulated by cross-linking the receptor-bound anti-CD3 and -CD28 for 10 mins in a 37°C water bath with 1 μg/ml of goat anti-hamster Ig (Thermo Fisher Scientific), followed by phosflow analysis.
2.5. Cell proliferation assay
Naïve CD4+ T cells and CD8+ T cells were labeled with CFSE (Sigma, St. Louis, MO), and washed three times. Cells (2 × 105/well) were seeded in an anti-CD3 Ab (5 μg/mL)-coated plate with soluble anti-CD28 Ab (4 μg/mL). After 4 days, cell proliferation was evaluated by flow cytometry.
2.6. Adoptive transfer
At 1 day prior to viral infection, splenocytes (1× 107 cells) from either naïve vitamin A control (VAC) or VAD mice were adoptively transferred into CD45.1 transgenic donor mice. In the parallel experiment, splenocytes (1× 107 cells) from naïve CD45.1 transgenic mice were adoptively transferred into control or VAD donor mice. Mice were i.v. infected with 2 × 106 FFU of LCMV Clone 13 and sacrificed at 6 dpi.
2.7. Flow cytometry
The following antibodies (Abs) were purchased from Thermo Fisher Scientific (San Diego, CA): Allophycocyanin-anti-IFN-γ (XMG1.2), PerCP-efluor 710-anti-TNF-α (MP6-XT22), FITC-anti-CD107a (eBio1D4B), PE-anti-Eomes (Dan11mag), Allophycocyanin-anti IL-10 (JES5-16E3), anti-Percp-Cy5.5 T-bet (ebio4B10), Allophycocyanin-anti-CXCR5 (SPRCL5), PE-anti-Foxp3 (FJK-16s) and Fixable Viability Dye eFluor 506. The following Abs were purchased from Biolegend: PE-Cy7-anti-CD3 (17A2), APC-Cy7-anti-CD8 (53-6.7), Pacific Blue-anti-CD4 (GK1.5), FITC-anti-CD19 (1D3), PE-anti-IL-2 (JES6-5H4), FITC-anti-PD-1 (J43), PE-anti-CD44 (IM3), Percp-Cy5.5-anti-CD45.1 (A20), FITC-anti-CD45.2 (104), PE-anti-NFATc1 (7A6), Allophycocyanin-anti-NK1.1 (PK136), Percp-Cy5.5-anti-CTLA4 (UC10–4B9) and purified anti-CD16/32 (2.4G2). The following phosflow Abs were purchased from Cell Signaling Technology (Danvers, MA): Phospho-Zap-70 (Tyr319)/Syk (Tyr352) (65E4), Phospho-p38 MAPK (Thr180/Tyr182) (D3F9), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E), Phospho-Akt (Ser473) (D9E) and Phospho-S6 Ribosomal Protein (Ser235/236) (D57.2.2E). The secondary antibody is anti-mouse IgG (H+L), F(ab’)2 Fragment (Alexa Fluor® 594 Conjugate). The PE-conjugated H-2Db/GP33 MHC tetramer was generated by the Advanced Technology Core of MHC Tetramer in Baylor College of Medicine. The goat anti–hamster Ig was from Southern Biotech (Birmingham, AL). The retinoic acid receptor (RAR) α-selective antagonist BMS195614 was from Cayman Chemical (Ann Arbor, Michigan). For surface staining, cells were first incubated with FcγR blocker (CD16/32), followed by fluorochrome-labeled Abs of surface markers. For intracellular cytokine staining, GP33 and GP61 peptides (5 μg/mL, Ana Spec, Fremont, CA) were used in the presence of Brefeldin A (BD Bioscience, San Jose, CA) to stimulate virus-specific CD8+ and CD4+ T cell responses, respectively. PMA (50 ng/ml) and ionomycin (750 ng/ml) from Sigma (St. Louis, MO) were also used in some experiments. After incubation, cells were stained for surface markers first, fixed by using an IC fixation buffer, and stained for intracellular cytokines (Thermo Fisher Scientific, Waltham, MA). For transcriptional factor staining, cells were fixed and permeabilized by eBioscience Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific, Waltham, MA). The phosflow experiments were performed according to the protocol from BD Bioscience (23). Briefly, cells were fixed immediately at the end of stimulation using a pre-warmed BD Phosflow Lyse/Fix Buffer at 37°C for 12 mins. Cells were permeabilized using chilled BD Phosflow Perm Buffer III for 1 h on ice, then washed and stained with Abs. Samples were processed on an LSRII FACSFortessa (Becton Dickinson, San Jose, CA) and analyzed by using FlowJo software (TreeStar, Ashland, OR).
2.8. ELISA and Bio-Plex assay
Tissue protein was extracted in RIPA buffer (Cell Signaling Technology, Danvers, MA) and quantified using a BCA kit (Thermo Fisher Scientific, Waltham, MA). Liver cytokine profiles and serum IFN-α/β were characterized respectively using Cytokine 17-Plex Mouse ProcartaPlex Panel and IFN alpha/IFN beta 2-Plex Mouse ProcartaPlex Panel (Thermo Fisher Scientific, Waltham, MA). The samples were read on a Bio-Rad Bio-Plex 200 System. Raw data were measured as the relative fluorescence intensity and then converted to the concentration according to the standard curve. For detecting IFN-γ and TNF-α in various organs, ELISA kits from BD Bioscience were also used.
2.9. Propagation and quantitation of virus
The LCMV stocks were prepared and titrated according to a modified method. Briefly, virus was incubated with baby hamster kidney (BHK) cells for 72 h. The culture fluid was centrifuged for 10 min at 350 g, 4°C, and stored at −70°C. For quantitation of the virus, Vero cells were cultured with a series of 10-fold virus dilutions for 90 min, followed by a methylcellulose overlay. After 4 days of culture, cells were first incubated with mouse anti-LCMV polyclonal antibody (Fitzgerald, Acton, MA), followed by incubation with Peroxidase (HRP)-conjugated anti-mouse IgG (Southern Biotech, Birmingham, AL). The AEC HRP Substrate Kit (Enzo life Sciences, Farmingdale, NY) was used for immunocytochemical procedures. Viral titers were calculated by counting the numbers of positive clusters.
2.10. Statistical analyses
Data were shown as mean ± SEM and analyzed by using the two-tailed Student’s t-test when compared between two groups. One-way ANOVA was used for statistical analysis of more than two groups. For analysis of the histological scores, the nonparametric Mann-Whitney test was used. *, ** or *** means P-value < 0.05, < 0.01 or < 0.001, respectively. Statistical analyses were operated by GraphPad Prism software 5.0 (GraphPad Software Inc., San Diego, CA).
3. Results
3.1. Vitamin A deficiency resulted in exaggerated responses of the T cell compartment and increased liver injury during LCMV infection
To investigate whether vitamin A deficiency affects type I interferon (IFN) responses, we measured serum levels of IFN-α and IFN-β of LCMV-infected mice at 12, 24 and 48 hours post-infection (hpi). Comparable levels of IFN-α and IFN-β, as well as viremia, were observed in VAD and control mice at these time-points (Fig. 1A and B), indicative of relatively normal innate antiviral responses in VAD mice during acute infection. Vitamin A is important for immune cell development and activation (24, 25). Naïve VAD and control mice had comparable T and B cell compartments in the spleen. However, VAD mice had lower percentages of B cells in the liver (Fig. S1A), which stores most of the body’s vitamin A. These aberrant T and B cell ratios were also observed in VAD mice in the liver, lung, mesenteric lymph nodes, but not in the spleen, inguinal or cervical lymph nodes at 2 dpi (Fig. 1C, D and S1B). In addition to the altered cell ratios, T cells in the liver, lung and mesenteric lymph nodes of VAD mice displayed higher activation status with upregulated IFN-γ expression (Fig. 1E). VAD mice exhibited increased liver injuries, as evidenced by higher serum ALT and AST levels, and pathologic changes at 7 dpi (Fig. 2A and B), which were associated with overzealous LCMV-specific T cell responses manifested by increased frequencies and numbers of IFN-γ+TNF-α+ T cells in the spleen and liver 7 dpi (Fig. 2C). The IFN-γ and TNF-α protein levels were also higher in the liver of VAD mice compared with that in control mice (Fig. 2D and S1C). We further analyzed virus-specific CD8+ T cells, and found increased GP33-tetramer+ CD8+ T cells in the liver of VAD mice. Importantly, vitamin A deficiency resulted in down-regulation of PD-1 on virus-specific CTLs (Fig. 2E). These exaggerated T cell responses in VAD animals were detrimental and did not lead to enhanced viral clearance at 7 and 15 dpi, as similar levels of viremia were observed in VAD and control mice (Fig. S1D). We also measured transcriptional factor T-bet and Eomes as well as CXCR5 on CD8+ T cells. The levels of T-bet were higher in the spleen of VAD mice, while T-bet was expressed on most of liver CD8+ T cells and its level was comparable between VAC and VAD samples (Fig. S2A). Expression of CXCR5 on CD8+ T cells were relative low and comparable in two groups. Although no difference was observed by Treg cell numbers, lower IL-10, but not CTLA-4 expression was found in VAD mice (Fig. S2B). We also infected mice with LCMV Armstrong and found the higher levels of ALT and AST in VAD mice at 6 dpi (Fig. S2C). Consistent with LCMV Clone 13 infection, the LCMV Armstrong-infected VAD mice displayed stronger immune responses, as evidenced by increased numbers of cytokine-producing T cells and higher expression of granzyme B and T-bet in spleen CD8+ T cells (Fig. S2D–E).
Figure 1. Vitamin A deficiency did not alter type I IFN responses, but increased T cell activation in early LCMV infection.

Vitamin A control (VAC) and vitamin A deficiency (VAD) mice were infected with LCMV Cl13 (2×106 FFU). A-B) Serum IFN-α and IFN-β, as well as viral loads were measured at 12, 24 and 48 hpi. C) Numbers of T cells, B cells, NK cells and NKT cells in spleen and livers at 48 hpi. D) Percentages of T cells and B cells in lymphoid nodes at 48 hpi. E) Lymphocytes were isolated from tissues and stimulated with PMA/Ionomycin in the presence of brefeldin A (BFA) for 4 h, followed by intracellular IFN-γ analysis using flow cytometry. The data are shown as mean ± SEM of three to six mice per group from a single representative experiment. The experiment was repeated three times independently. A two-tailed Student’s t-test was used to compare the two groups. * P<0.05, ** P<0.01.
Figure 2. VAD mice exhibited overzealous T cell responses and severe immunopathogenesis at the acute stage of viral infection.

VAC and VAD mice were infected with LCMV Cl13 (2×106 FFU) and sacrificed at 7 dpi. A) Serum ALT and AST. B) Liver histological scores. C) Lymphocytes were isolated from the spleen (S), liver (Lv) and lung (Lg), followed by stimulation with GP33 and GP61 peptides in the presence of BFA for 5 h. Intracellular IFN-γ and TNF-α were analyzed using flow cytometry. D) Liver IFN-γ and TNF-α levels were measured by ELISA kits. E) Virus-specific CD8+ T cells were detected by H-2Db/GP33 MHC tetramer. The PD-1 expression on GP33-tetramer+ CD8+ T cells was measured. The data are shown as mean ± SEM of three to six mice per group from a single representative experiment. The experiment was repeated three times independently. A two-tailed Student’s t-test was used to compare the two groups. A Mann-Whitney test was used to compare the histological scores. * P<0.05, ** P<0.01, *** P<0.001, NS, no significance.
The normal immune system enters the contraction phase following clonal expansion and viral reduction in the acute phase (26). Here, we examined the effect of vitamin A deficiency on immune regulation at 30 days post LCMV infection. We found that the levels of TNF-α and IL-2 in liver tissues were significantly higher in VAD mice compared with those in control mice (Fig. 3A). In addition, other pro-inflammatory cytokines, including IL-1β, IL-6, IL-18 and IL-23, as well as anti-inflammatory IL-10 and IL-13, were also elevated in VAD mice (Fig. 3A). PD-1 is an important check-point regulator (27) and plays a crucial role in immune contraction, T cell exhaustion and viral persistence during infection. Flow cytometry data showed that effector T cells of VAD mice displayed increased frequencies of IFN-γ+TNF-α+ and IFN-γ+IL-2+ T cells with significantly lower frequencies of PD-1+ T cells in the spleen, liver and lungs at 30 dpi (Fig. 3B–D). We further found the increased percentages and numbers of cytokine-expressing GP33 tetramer+ CD8+ T cells in spleen and livers of VAD mice (Fig. 3E). In line with the stronger T cell immune response in VAD mice at the contraction stage, their viremia was lower compared with that of control mice (Fig. S1D). Despite exaggerated responses of the T cell compartment in this phase, the percentages of B lymphocytes in the spleen, liver and lungs of VAD mice were significantly lower than those in control animals (data not shown). Together, these results demonstrated that vitamin A deficiency can cause profound dysregulation of adaptive immune responses to LCMV infection. Increased T cell activation and pro-inflammatory cytokine production can lead to tissue injury in viral infection.
Figure 3. RA treatment restored the hyperactive T cell functions of VAD mice at the contraction stage of viral infection.
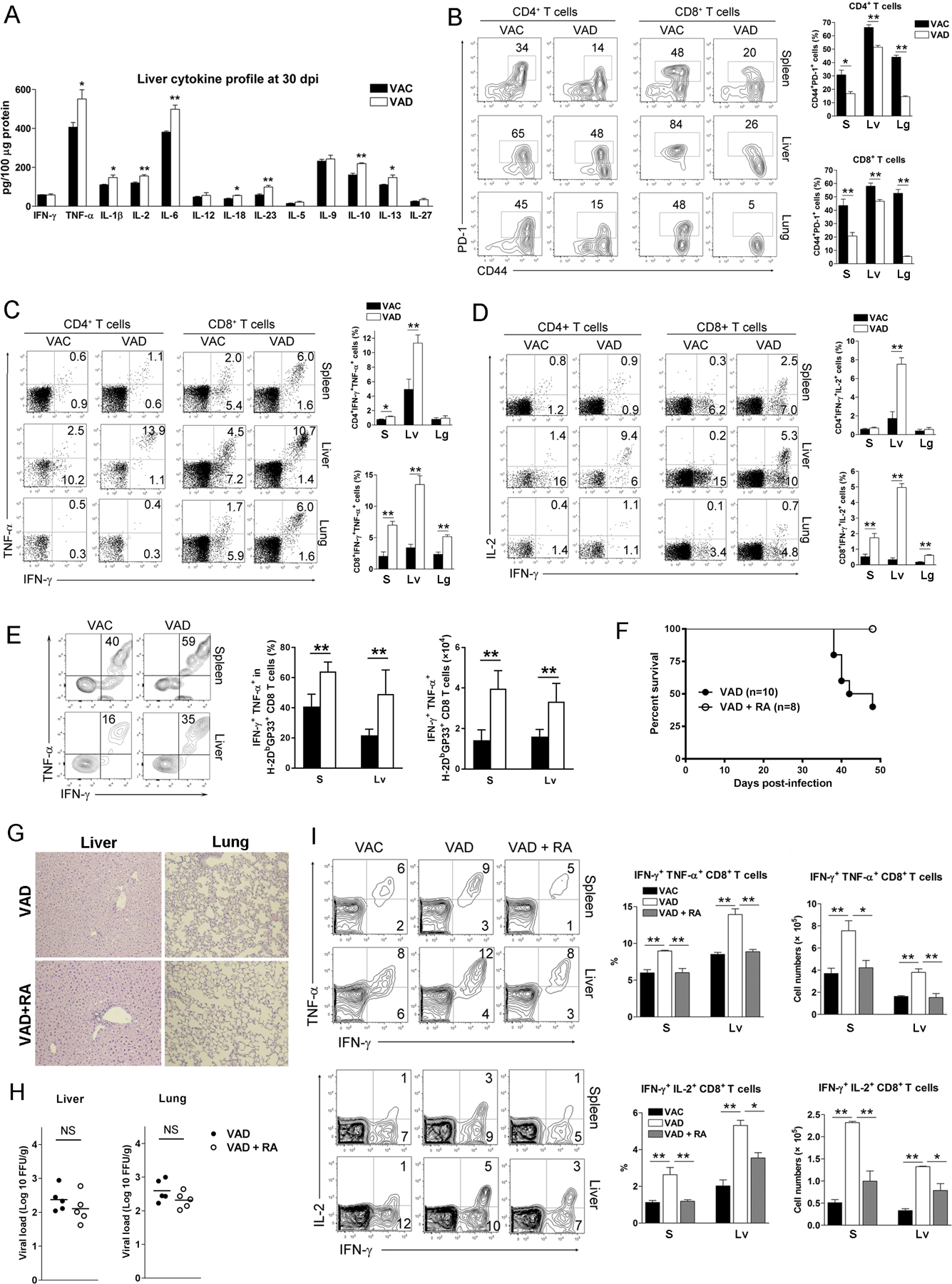
VAC and VAD mice were infected with LCMV Cl13 (2×106 FFU). A) Liver cytokine profile at 30 dpi. B-D) Lymphocytes were isolated from the spleen (S), liver (Lv) and lung (Lg), and stimulated with GP33 and GP61 peptides in the presence of BFA for 5 h. Percentages of CD44+PD-1+ T cells, IFN-γ+TNF-α+ T cells and IFN-γ+IL-2+ T cells were analyzed using flow cytometry. E) The H-2Db/GP33 MHC tetramer+ CD8+ T cells were gated first, followed by analysis of IFN-γ and TNF-α expression. F) Infected VAD mice treated with RA (25 μg/daily) starting from 12 dpi through 42 dpi. Animal survival rates were recorded. G) H&E staining and H) viral loads of liver and lungs at 30 dpi. I) Percentages of IFN-γ+TNF-α+ CD8+ T cells and IFN-γ+IL-2+ CD8+ T cells were measured in the spleen and livers at 30 dpi. The data are shown as mean ± SEM of at least six mice per group from a single representative experiment. The experiment was repeated three times independently. A two-tailed Student’s t-test was used to compare the two groups. * P<0.05, ** P<0.01, *** P<0.001, NS, no significance.
RA is one of the metabolites of vitamin A, and it plays an important role in immune function and regulation (28). We reported previously that RA treatment can attenuate liver inflammation and injury by restraining T cell functions in acute viral infection (20). However, whether RA can modulate exaggerated T cell responses during persistent infection is not known. Here, we treated the infected-VAD mice with RA daily starting from 12 dpi, and found that RA-treated VAD mice all survived, while 60% of VAD mice without RA treatment succumbed at 48 dpi (Fig. 3F). The histological results showed that the liver inflammation was mild and comparable in two groups; however, RA treated-VAD mice had lower pathogenic inflammation in the lungs at 30 dpi (Fig. 3G). Although RA treatment did not significantly inhibit viral loads in organs (Fig. 3H), it decreased the percentages and numbers of hyperactive effector cells, including IFN-γ+TNF-α+ and IFN-γ+IL-2+ CD8+ T cells at 30 dpi (Fig. 3I). These results demonstrated that RA treatment can partially restore hyperactive T cell functions and rescue VAD mice in persistent LCMV infection.
3.2. VAD mice failed to control viremia and succumbed to persistent infection
Normal mice reduce viral burden and survive persistent infection (29) (Fig. 4A). However, LCMV-infected VAD mice succumbed to the infection beginning at 32 dpi, and all died at 52 dpi (Fig. 4A). At 48 dpi, the moribund VAD mice exhibited a three-fold increase of PD-1 molecules on splenic CD8+ T cells compared with those of control mice (Fig. 4B). Additionally, around 80% of CD8+ T cells in the liver of VAD mice displayed a PD-1+ exhausted phenotype, compared to only half as much in control animals (Fig. 4B). The LCMV-specific multifunctional CTL responses were much lower in VAD mice, as evidenced by lower proportions of triple (IFN-γ+TNF-α+IL-2+ and IFN-γ+TNF-α+CD107a+), double (IFN-γ+CD107a+ and IFN-γ+TNF-α+) and single (IFN-γ+ and CD107a+) positive CD8+ T cells (Fig. 4C). Moreover, significant amounts of virus were detectable in the serum and lungs of VAD mice at 48 dpi (Fig. 4D). These data demonstrated that vitamin A deficiency resulted in T cell exhaustion, un-controlled viremia and animal death in persistent LCMV infection.
Figure 4. VAD mice exhibited T cell exhaustion and died in the persistent viral infection.

VAC and VAD mice were infected with LCMV Cl13 (2×106 FFU). A) Survival rates. B) Expression of PD-1 on CD8+ T cells. C) Multifunctional CTLs in the spleens. D) Viremia and lung viral loads at 48 dpi. The data are shown as mean ± SEM of four mice per group from a single representative experiment in B-D panels. The experiment was repeated twice independently. A two-tailed Student’s t-test was used to compare the two groups. * P<0.05, ** P<0.01, *** P<0.001.
3.3. Vitamin A was crucial for antiviral T cell functions in the periphery
To determine whether vitamin A deficiency causes anomalous thymic development of T cells or affects T cell functionality in the periphery, we evaluated the T cell activation and effector functions in vitro. We found that naïve T cells from both VAD and control mice performed equally well in proliferation and IFN-γ production in response to CD3/CD28 stimulation (Fig. 5A and B). To further determine the effect of VAD on T cell development in vivo, we isolated splenocytes from VAD and control mice on the CD45.2 background and adoptively transferred them into congenic CD45.1 mice, followed by LCMV infection. We found that donor cells from VAD or control mice displayed similar activation status in recipient animals, as evidenced by comparable cell percentages and cytokine production at 6 dpi (Fig. 5C). These results indicated that vitamin A deficiency did not grossly impair the thymic development of T cells.
Figure 5. VAD affected T cell functionality in the periphery in viral infection.
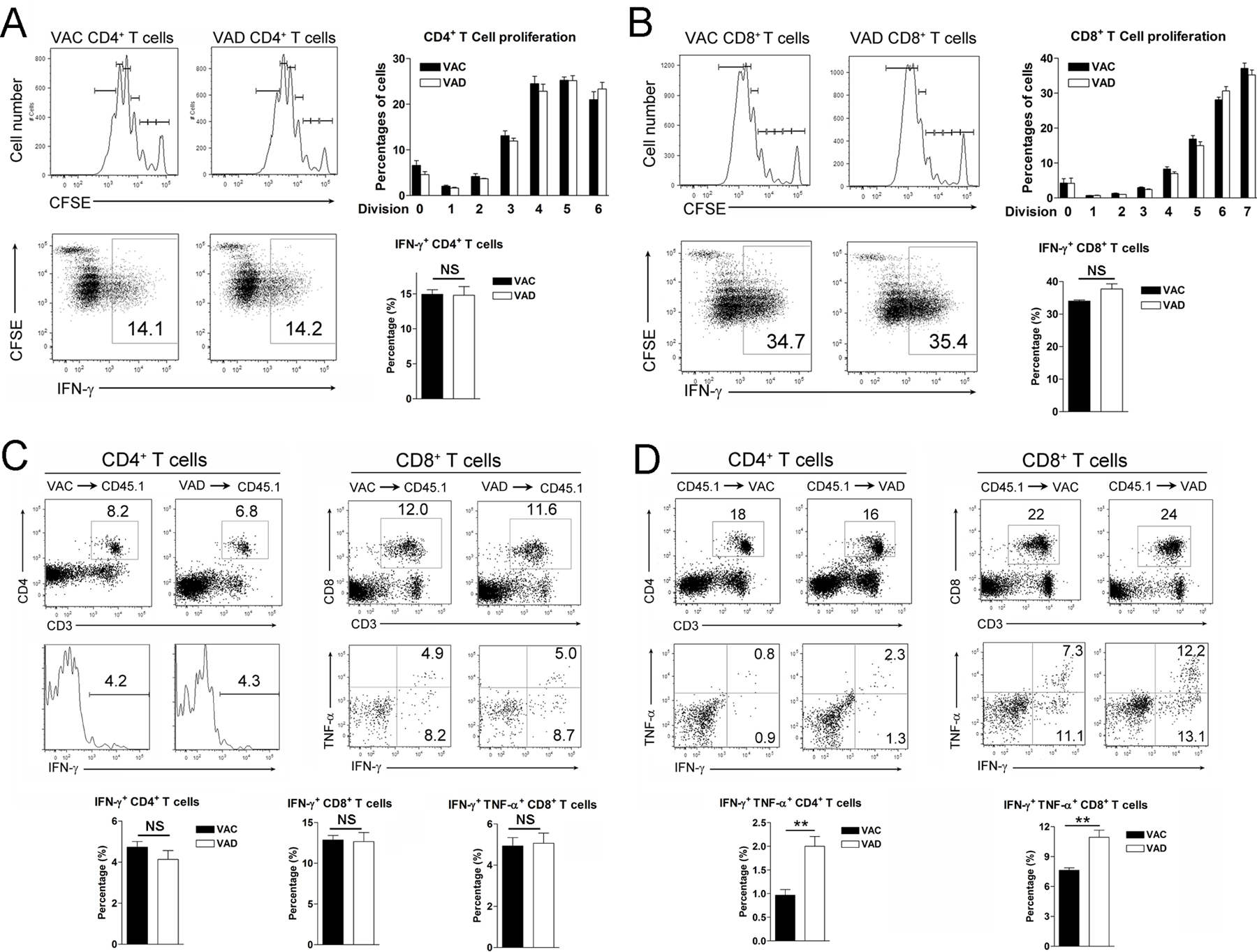
A) Naïve CD44−CD4+ and B) CD44−CD8+ T cells were purified from spleens of naïve mice. Cells were labeled with CFSE and cultured in vitro for 4 days with the anti-CD3/CD28 antibody stimulation. At last 5 h before harvest, PMA/Ionomycin and Brefeldin A were added into the culture system. Cell proliferation and intracellular IFN-γ expression were measured by flow cytometry. C) Splenocytes were isolated from VAC and VAD mice, followed by adoptively transferring into CD45.1 transgenic mice. D) Splenocytes were isolated from naïve CD45.1 transgenic mice, followed by adoptively transferring into VAC and VAD mice. These recipient mice were infected with LCMV and sacrificed at 6 dpi. The adoptive transferred cells were gated first, and the percentages of cytokine-producing T cells were analyzed by flow cytometry. The data are shown as mean ± SEM of three mice per group from a single representative experiment. The experiment was repeated three times independently. A two-tailed Student’s t-test was used to compare the two groups. ** P<0.01, NS, no significance.
To test if vitamin A deficiency affects T cell functionality in the periphery, we harvested naive splenocytes from CD45.1 congenic mice and transferred them into CD45.2 recipient mice either in VAD or control environment, followed by LCMV infection. We found that effector functions of donor T cells were elevated in VAD recipients compared with those in control mice, as evidenced by increased percentages of effector cytokine-producing CD45.1+ T cells in VAD mice (Fig. 5D). These data suggested that vitamin A in the peripheral organs played a critical role in regulating T cell functions and maintaining tissue homeostasis during viral infection.
3.4. RA inhibited T cell receptor signaling and NFAT proteins
The mechanism by which RA modulates T cell function in viral infection is not fully elucidated. T cell receptors (TCR) signaling plays key roles in T cell activation and differentiation of effector functions (30). To test the hypothesis that RA can regulate T cell responses through modulating TCR signaling, we cultured spleen cells of naïve mice in the presence of RA for 16 h. First, we found that RA of less than 10 μM concentrations was not cytotoxic. A higher dose of RA (20 μM) was toxic to T cells and reduced cell viability (Fig. S3A). By using Phosflow analysis, we found that RA treatment significantly attenuated TCR/CD28-mediated phosphorylation of several downstream signaling molecules of TCR, including Zap70, ERK, AKT and p38. Phosphorylated S6 (p-S6), a marker for mammalian target of rapamycin (mTOR) signaling pathway activation, was also reduced in T cells by RA treatment (Fig. 6A and S3B). Collectively, these data suggest a crucial role for RA in regulating TCR-proximal signaling events in both CD4+ and CD8+ T cells.
Figure 6. RA inhibited T cell receptor signaling and NFATc1 expression in T cells.

A) Naïve splenocytes were isolated and cultured with RA in vitro for 24 h, followed by the stimulation with anti-CD3 plus anti-CD28 using an antibody- cross-linking method. Cells were fixed immediately by BD Phosflow Lyse/Fix Buffer at 37 °C for 12 mins and permeabilized by BD Phosflow Perm Buffer III on ice for 30 mins. Cells were then incubated with surface antibodies and phosphorylated antibodies for 1 h, followed by flow cytometry analysis. Each group was in triplicates. B) LCMV-infected mice were treated with RA (200 μg/day) at 1, 3, and 5 dpi. The numbers of cytokine-producing T cells and C) the percentages and mean fluorescence intensity (MFI) of NFATc1 were examined at 7 dpi. D-E) Splenocytes of naïve mice were cultured in vitro by anti-CD3/CD28 antibody stimulation in the presence of various concentrations of RA. After 3-day culture, cytokine levels and NFATc1 expression were analyzed by flow cytometry. Each group was in triplicates and the RA treated groups were compared to the control group. All experiments were repeated two to three times independently. A two-tailed Student’s t-test was used to compare the two groups. One-way ANOVA was used to compare more than two groups. * P<0.05, ** P<0.01, *** P<0.001.
Nuclear factor of activated T-cells (NFAT) is a family of transcription factors important in T cell responses (31). As NFAT proteins are downstream of the TCR signaling pathway, we treated the LCMV-infected mice with RA and analyzed T cell activation at 7 dpi. Consistent with our previous finding (20), RA treatment significantly inhibited T cell activation in vivo as evidenced by decreased frequencies of IFN-γ- and IFN-γ TNF-α-producing T cells in both the spleen and liver (Fig. 6B). NFATc1 expression was also significantly reduced in CD8+ T cells of RA-treated mice (Fig. 6C). To confirm this finding, we cultured spleen cells with anti-CD3/CD28 antibodies in the presence of RA for 3 days. RA treatment significantly reduced the percentages of IFN-γ- and IFN-γ/TNF-α-expressing CD8+ and CD4+ T cells, as well as frequencies and levels of NFATc1 expression in these cells (Fig. 6D). A reduction of NFATc2 was also observed in RA-treated T cells (data not shown). Furthermore, the neutral retinoic acid receptor (RAR) α-selective antagonist BMS 195614 (1 μM) completely restored frequencies and levels of NFATc1 expression in RA-treated CD8+ and CD4+ T cells (Fig. S4). These data suggest that RA can restrain T cell effector functions by inhibiting a number of TCR downstream signaling molecules.
4. Discussion
VAD is a public health problem, especially in sub-Saharan Africa and South Asia, leading to an increased risk of disease and death from severe infection (2). Vitamin A plays an essential role in immune processes, including immune cell development, proliferation, differentiation, migration and antibody production; however, its role and mechanism in viral infection are not entirely understood. VAD is linked with an abnormality of immune cell development. For instance, vitamin A is critical for B1 cell development through regulating the transcriptional factor NFATc1 (25). We observed decreased percentages of B cells in livers and mesenteric lymph nodes, but not in the spleen, inguinal or cervical lymph nodes at early stages during viral infection (Fig. 1C–D). Notably, aged VAD mice also displayed decreased numbers of B cells in the spleen (25). Since vitamin A is mainly stored in the liver, our results indicate that the impaired immune cell development by VAD may occur first in the gut-liver axis (32). Moreover, we provided both in vitro and in vivo evidence that T cells from VAD mice displayed comparable capabilities for proliferation and activation as those from control mice (Fig. 5). Other researchers also reported that VAD results in more myeloid cells in the periphery (33). These findings suggest that vitamin A may differently regulate the development of immune cell subpopulations.
Malnutrition may lead to abnormal type I interferon responses (34), which is essential for viral control and following adaptive virus-specific immunity (35). Vitamin A and RA have been demonstrated to inhibit viral replication through induction of interferon-stimulated genes (ISGS) and upregulation of IFN-αR in vitro (10, 36); however, we found comparable type I IFN production and viremia in VAD mice at 12, 24 and 48 hpi (Fig. 1A and B), indicative of the dispensable role of endogenous vitamin A and retinols for the type I interferon response and early viral control. In addition to innate immunity, vitamin A and its metabolite RA are essential for T cell activation and differentiation (Figs. 2–3), which determine the outcome of viral infection. Several reports demonstrated recently that RA inhibits Th17 polarization via inhibiting IL-6 signaling (16, 37, 38), but increases TGF-β -mediated Treg cell expansion and Foxp3 expression in naïve T cells through TGF-β-enhanced RAR (15, 39). Overproduction of IFN-γ was reported in VAD mice, while dietary vitamin A supplements down-regulated IFN-γ expression in Th1 cells (17, 18, 40). These studies suggest vitamin A and RA act as potent immunomodulators for T cell responses. In this study, we found significantly increased numbers and cytokine production of T cells in the liver, lung and mesenteric lymph nodes of VAD mice at 2 dpi (Fig. 1C–E). This overzealous T cell response was further observed in both acute and contraction stages, resulting in increased inflammatory cytokines and severe tissue damage (Figs. 2–3). Vitamin D is reported to upregulate PD-1/PD-L1 signaling in Crohn’s disease patients (41), indicating that vitamin might be involved in checkpoint regulation. In our study, the lower levels of PD-1 expression in various organs of VAD mice may suggest the essential role of vitamin A in the regulation of immune check-point molecules. However, the underlying mechanism is still not well known and need further investigation. Interestingly, naïve T cells isolated from either VAD or control mice displayed a similar capacity for proliferation and IFN-γ production in vitro and in vivo. However, donor T cells in adoptive transfer experiments exhibited a more activation phenotype in VAD recipient mice (Fig. 5), suggesting that the upregulated T cell response in VAD mice was attributed to the lack of environmental vitamin A. In addition, we also found decreased B cell percentages in VAD mice at 30 dpi (data not shown), suggesting that VAD mice may have impaired antibody production in persistent infections (17). Our results highlight the role of vitamin A in regulating antiviral T cell responses and subsequent tissue pathogenesis following viral infection. However, the increased T cell responses in VAD mice may also contribute to viral clearance in the contraction stage (Fig. S1D). Interestingly, VAD led to impaired multifunctional T cell responses, an inability to clear the virus, and animal death in the persistent infection (Fig. 4). Moreover, vitamin A deficiency may also lead to impaired T regulatory cell function (Fig. S2B). These findings indicate that the overzealous T cell activity and immune-mediated tissue damage may ultimately cause T cell exhaustion and viral relapse in chronic infection. Importantly, RA treatment delayed VAD mice death in the persistent infection (Fig. 3F). It is reported that LCMV-infected mice may die due to the leakage of exudate into the lung that compromised respiration (29). We observed that LCMV-infected VAD mice did not have any obvious signs of illness (Data not shown). Since vitamin A plays a key role in maintaining lung epithelial integrity, it is highly possible that VAD mice may succumb to infection due to lung pathology. Our histological staining results showed that RA treatment may rescue mice through decreasing lung inflammation (Fig. 3G). Further analysis showed that RA treatment decreased the numbers of cytokine-producing T cells during the T cell contraction stage (Fig. 3I). Therefore, our results suggest to us that dietary RA supplements may inhibit the overactivation of T cells and protect against T cell exhaustion in chronic infections.
Our previous and current studies have demonstrated that RA treatment inhibited T cell activation both in vitro and in vivo (Fig. 6) (20). RA contributes to T cell responses through several ways, including modulating dendritic cell migration, inducing immunosuppressive molecule arginase-I, regulating cytokine production, and determining T cell-homing markers (20, 28). The strength of the TCR signal is pivotal for T cell activation and differentiation. TCR signaling is initiated by the activation of the protein tyrosine kinase Lck and the phosphorylation of the TCR-signaling chain CD3ζ, followed by recruitment and phosphorylation of the tyrosine kinase Zap70 (42, 43). Activated Zap70 then phosphorylates several other signaling molecules, including p38, ERK, NF- κB, AKT and mTOR, transducing the TCR signals to downstream signaling pathways. Here, we found that RA can target TCR signaling and associated-downstream molecules. By using the CD3/CD28 cross-linking stimulation, we found that RA negatively regulated the TCR-proximal signaling, as evidenced by decreased phosphorylation of Zap70. Consistently, the phosphorylation of several downstream signaling molecules, including p38, ERK, AKT and S6, were all down-regulated by RA in a dose-dependent manner (Fig. 6). It has been demonstrated that the strength of the TCR signal controls Th1/Th2 polarization (44–46). A weak TCR signal primes IL-4-producing Th2 cells, while a strong TCR signal drives Th1 differentiation. Accordingly, upon TCR engagement, the phosphorylation profiles were weaker in Th2 than that in Th1 clones. Interestingly, RA treatment induces a Th2 polarization and impairs Th1 responses in both human and mouse purified T cells under TCR stimulation (47, 48). Thus, RA may contribute to T cell differentiation by regulating TCR signals. Notably, it is reported that RARα-deficient CD4+ T cells have poor effector responses and impaired signaling activation by anti-CD3 stimulation, indicating that RA is necessary for T cell activation (49). Although the reason for the discrepancies from different models is not known at present, we demonstrated here that exogenous RA played a critical role in regulating TCR signals.
The transcriptional factor NFATs are downstream of the TCR, playing a critical role in the development and differentiation of immune cells (31). T cells can express NFATc1 (NFAT2), NFATc2 (NFAT1) and NFATc3 (NFAT4). The expression of NFTAc1 is inducible and can be upregulated by TCR stimulation, resulting in the activation of T cells (50). We demonstrated here that RA treatment can decrease the expression of NFATc1 in CD8+ T cells in vivo (Fig. 6C). In vitro study showed that RA inhibited NFATc1 expression in both CD4+ and CD8+ T cells, while the selective RARα antagonist restored NFATc1 expression in the presence of RA (Fig. 6D–E and S4). Our results may imply that RA modulates T cell function through regulating NFATs (51, 52). However, it is reported that VAD mice have decreased NFATc1 expression in B1 cells, resulting in an inability to mount an antibody response against bacterial infection (25). The discrepancies in these studies indicate that RA may diversely regulate NFAT functions in different immune cells.
In conclusion, our study revealed that VAD caused an aberrant T cell response to viral infection, resulting in increased tissue damage and host mortality; while, RA can down-regulate T cell activation and rescue mice from death in the persistent infection. VAD did not cause an intrinsic deficiency of T cell function, but the micro-environmental vitamin A or its metabolites played an essential role in T cell function during viral infection. Moreover, RA may modulate T cell activation by regulating TCR signals and NFAT transcriptional factors, indicating that RA may represent a potential immunomodulator in infectious diseases.
Supplementary Material
Key points:
Vitamin A deficiency causes overzealous T cell responses and tissue damage.
Retinoic acid protects vitamin A deficient mice in viral infection.
Retinoic acid downregulates TCR-proximal signals.
Acknowledgements
We thank Dr. Sherry Haller for assistance with manuscript preparation. The authors wish to express gratitude to other members of the UTMB Joint Immunology Working Group (Drs. Cong, Stephens, Rajsbaum, Hu, and their trainees) for many helpful discussions.
This work was supported in part by grants from the NIH (AI126371 to JS; AI126343 and AI132674 to LS). Panpan Yi and Xiaofang Wang were visiting scientists partially supported by the Department of Infectious Diseases, Xiangya Hospital, China and the National Natural Science Foundation of China (#81800506 to PY). Biao Zhang was a visiting scientist partially supported by National Natural Science Foundation of China (#81873885), and China Scholarship Council in 2018 (File#201808440556).
Abbreviations:
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CLN
cervical lymph mode
- DCs
dendritic cells
- ILN
inguinal lymph node
- LCMV
lymphocytic choriomeningitis virus
- MLN
mesenteric lymph node
- RA
all-trans retinoic acid
- VAC
vitamin A control
- VAD
vitamin A deficiency
Footnotes
Disclosures: The authors declare no financial or commercial conflict of interest.
References
- 1.Villamor E, and Fawzi WW 2005. Effects of vitamin a supplementation on immune responses and correlation with clinical outcomes. Clin Microbiol Rev 18:446–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stevens GA, Bennett JE, Hennocq Q, Lu Y, De-Regil LM, Rogers L, Danaei G, Li G, White RA, Flaxman SR, Oehrle SP, Finucane MM, Guerrero R, Bhutta ZA, Then-Paulino A, Fawzi W, Black RE, and Ezzati M 2015. Trends and mortality effects of vitamin A deficiency in children in 138 low-income and middle-income countries between 1991 and 2013: a pooled analysis of population-based surveys. Lancet Glob Health 3:e528–536. [DOI] [PubMed] [Google Scholar]
- 3.Baum M, Cassetti L, Bonvehi P, Shor-Posner G, Lu Y, and Sauberlich H 1994. Inadequate dietary intake and altered nutrition status in early HIV-1 infection. Nutrition 10:16–20. [PubMed] [Google Scholar]
- 4.Semba RD, Caiaffa WT, Graham NM, Cohn S, and Vlahov D 1995. Vitamin A deficiency and wasting as predictors of mortality in human immunodeficiency virus-infected injection drug users. J Infect Dis 171:1196–1202. [DOI] [PubMed] [Google Scholar]
- 5.Peres WA, Chaves GV, Goncalves JC, Ramalho A, and Coelho HS 2011. Vitamin A deficiency in patients with hepatitis C virus-related chronic liver disease. Br J Nutr 106:1724–1731. [DOI] [PubMed] [Google Scholar]
- 6.Bitetto D, Bortolotti N, Falleti E, Vescovo S, Fabris C, Fattovich G, Cussigh A, Cmet S, Fornasiere E, Ceriani E, Pirisi M, and Toniutto P 2013. Vitamin A deficiency is associated with hepatitis C virus chronic infection and with unresponsiveness to interferon-based antiviral therapy. Hepatology 57:925–933. [DOI] [PubMed] [Google Scholar]
- 7.Stephensen CB 2001. Vitamin A, infection, and immune function. Annu Rev Nutr 21:167–192. [DOI] [PubMed] [Google Scholar]
- 8.Fawzi WW, Msamanga GI, Spiegelman D, Urassa EJ, McGrath N, Mwakagile D, Antelman G, Mbise R, Herrera G, Kapiga S, Willett W, and Hunter DJ 1998. Randomised trial of effects of vitamin supplements on pregnancy outcomes and T cell counts in HIV-1-infected women in Tanzania. Lancet 351:1477–1482. [DOI] [PubMed] [Google Scholar]
- 9.Fawzi WW, Msamanga GI, Spiegelman D, Wei R, Kapiga S, Villamor E, Mwakagile D, Mugusi F, Hertzmark E, Essex M, and Hunter DJ 2004. A randomized trial of multivitamin supplements and HIV disease progression and mortality. N Engl J Med 351:23–32. [DOI] [PubMed] [Google Scholar]
- 10.Trottier C, Colombo M, Mann KK, Miller WH Jr., and Ward BJ 2009. Retinoids inhibit measles virus through a type I IFN-dependent bystander effect. Faseb J 23:3203–3212. [DOI] [PubMed] [Google Scholar]
- 11.Bocher WO, Wallasch C, Hohler T, and Galle PR 2008. All-trans retinoic acid for treatment of chronic hepatitis C. Liver Int 28:347–354. [DOI] [PubMed] [Google Scholar]
- 12.Johansson-Lindbom B, Svensson M, Pabst O, Palmqvist C, Marquez G, Forster R, and Agace WW 2005. Functional specialization of gut CD103+ dendritic cells in the regulation of tissue-selective T cell homing. J Exp Med 202:1063–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, and Song SY 2004. Retinoic acid imprints gut-homing specificity on T cells. Immunity 21:527–538. [DOI] [PubMed] [Google Scholar]
- 14.Benson MJ, Pino-Lagos K, Rosemblatt M, and Noelle RJ 2007. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J Exp Med 204:1765–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hill JA, Hall JA, Sun CM, Cai Q, Ghyselinck N, Chambon P, Belkaid Y, Mathis D, and Benoist C 2008. Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi Cells. Immunity 29:758–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, and Cheroutre H 2007. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317:256–260. [DOI] [PubMed] [Google Scholar]
- 17.Cantorna MT, Nashold FE, and Hayes CE 1995. Vitamin A deficiency results in a priming environment conducive for Th1 cell development. Eur J Immunol 25:1673–1679. [DOI] [PubMed] [Google Scholar]
- 18.Cantorna MT, Nashold FE, Chun TY, and Hayes CE 1996. Vitamin A down-regulation of IFN-gamma synthesis in cloned mouse Th1 lymphocytes depends on the CD28 costimulatory pathway. J Immunol 156:2674–2679. [PubMed] [Google Scholar]
- 19.Cippitelli M, Ye J, Viggiano V, Sica A, Ghosh P, Gulino A, Santoni A, and Young HA 1996. Retinoic acid-induced transcriptional modulation of the human interferon-gamma promoter. J Biol Chem 271:26783–26793. [DOI] [PubMed] [Google Scholar]
- 20.Jie Z, Liang Y, Yi P, Tang H, Soong L, Cong Y, Zhang K, and Sun J 2017. Retinoic acid regulates immune responses by promoting IL-22 and modulating S100 proteins in viral hepatitis. J Immunol 198:3448–3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knodell RG, Ishak KG, Black WC, Chen TS, Craig R, Kaplowitz N, Kiernan TW, and Wollman J 1981. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology 1:431–435. [DOI] [PubMed] [Google Scholar]
- 22.Liang Y, Jie Z, Hou L, Aguilar-Valenzuela R, Vu D, Soong L, and Sun J 2013. IL-33 induces nuocytes and modulates liver injury in viral hepatitis. J Immunol 190:5666–5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang Y, Yi P, Yuan DMK, Jie Z, Kwota Z, Soong L, Cong Y, and Sun J 2019. IL-33 induces immunosuppressive neutrophils via a type 2 innate lymphoid cell/IL-13/STAT6 axis and protects the liver against injury in LCMV infection-induced viral hepatitis. Cell Mol Immunol 16:126–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ross AC, Chen Q, and Ma Y 2011. Vitamin A and retinoic acid in the regulation of B-cell development and antibody production. Vitam Horm 86:103–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maruya M, Suzuki K, Fujimoto H, Miyajima M, Kanagawa O, Wakayama T, and Fagarasan S 2011. Vitamin A-dependent transcriptional activation of the nuclear factor of activated T cells c1 (NFATc1) is critical for the development and survival of B1 cells. Proc Natl Acad Sci U S A 108:722–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bocharov G, Argilaguet J, and Meyerhans A 2015. Understanding Experimental LCMV Infection of Mice: The Role of Mathematical Models. J Immunol Res 2015:739706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun C, Mezzadra R, and Schumacher TN 2018. Regulation and function of the PD-L1 checkpoint. Immunity 48:434–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bono MR, Tejon G, Flores-Santibanez F, Fernandez D, Rosemblatt M, and Sauma D 2016. Retinoic acid as a modulator of T cell immunity. Nutrients 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oldstone MBA, Ware BC, Horton LE, Welch MJ, Aiolfi R, Zarpellon A, Ruggeri ZM, and Sullivan BM 2018. Lymphocytic choriomeningitis virus Clone 13 infection causes either persistence or acute death dependent on IFN-1, cytotoxic T lymphocytes (CTLs), and host genetics. Proc Natl Acad Sci U S A 115:E7814–E7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Courtney AH, Lo WL, and Weiss A 2018. TCR Signaling: Mechanisms of Initiation and Propagation. Trends Biochem Sci 43:108–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macian F 2005. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 5:472–484. [DOI] [PubMed] [Google Scholar]
- 32.Kane MA, Folias AE, Wang C, and Napoli JL 2008. Quantitative profiling of endogenous retinoic acid in vivo and in vitro by tandem mass spectrometry. Anal Chem 80:1702–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuwata T, Wang IM, Tamura T, Ponnamperuma RM, Levine R, Holmes KL, Morse HC, De Luca LM, and Ozato K 2000. Vitamin A deficiency in mice causes a systemic expansion of myeloid cells. Blood 95:3349–3356. [PubMed] [Google Scholar]
- 34.Reynolds JA, Rosenberg AZ, Smith CK, Sergeant JC, Rice GI, Briggs TA, Bruce IN, and Kaplan MJ 2016. Vitamin D deficiency is associated with endothelial dysfunction and increases type I interferon gene expression in a murine model of systemic lupus erythematosus. Arthritis Rheumatol 68:2929–2935. [DOI] [PubMed] [Google Scholar]
- 35.Snell LM, McGaha TL, and Brooks DG 2017. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol 38:542–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hamamoto S, Fukuda R, Ishimura N, Rumi MA, Kazumori H, Uchida Y, Kadowaki Y, Ishihara S, and Kinoshita Y 2003. 9-cis retinoic acid enhances the antiviral effect of interferon on hepatitis C virus replication through increased expression of type I interferon receptor. J Lab Clin Med 141:58–66. [DOI] [PubMed] [Google Scholar]
- 37.Brown CC, Esterhazy D, Sarde A, London M, Pullabhatla V, Osma-Garcia I, Al-Bader R, Ortiz C, Elgueta R, Arno M, de Rinaldis E, Mucida D, Lord GM, and Noelle RJ 2015. Retinoic acid is essential for Th1 cell lineage stability and prevents transition to a Th17 cell program. Immunity 42:499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elias KM, Laurence A, Davidson TS, Stephens G, Kanno Y, Shevach EM, and O’Shea JJ 2008. Retinoic acid inhibits Th17 polarization and enhances FoxP3 expression through a Stat-3/Stat-5 independent signaling pathway. Blood 111:1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takahashi H, Kanno T, Nakayamada S, Hirahara K, Sciume G, Muljo SA, Kuchen S, Casellas R, Wei L, Kanno Y, and O’Shea JJ 2012. TGF-beta and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat Immunol 13:587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carman JA, and Hayes CE 1991. Abnormal regulation of IFN-gamma secretion in vitamin A deficiency. J Immunol 147:1247–1252. [PubMed] [Google Scholar]
- 41.Bendix M, Greisen S, Dige A, Hvas CL, Bak N, Jorgensen SP, Dahlerup JF, Deleuran B, and Agnholt J 2017. Vitamin D increases programmed death receptor-1 expression in Crohn’s disease. Oncotarget 8:24177–24186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, Kadlecek TA, Au-Yeung BB, Goodfellow HE, Hsu LY, Freedman TS, and Weiss A 2010. ZAP-70: an essential kinase in T-cell signaling. Cold Spring Harb Perspect Biol 2:a002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chan AC, Iwashima M, Turck CW, and Weiss A 1992. ZAP-70: a 70 kd protein-tyrosine kinase that associates with the TCR zeta chain. Cell 71:649–662. [DOI] [PubMed] [Google Scholar]
- 44.Tao X, Constant S, Jorritsma P, and Bottomly K 1997. Strength of TCR signal determines the costimulatory requirements for Th1 and Th2 CD4+ T cell differentiation. J Immunol 159:5956–5963. [PubMed] [Google Scholar]
- 45.Hannier S, Bitegye C, and Demotz S 2002. Early events of TCR signaling are distinct in human Th1 and Th2 cells. J Immunol 169:1904–1911. [DOI] [PubMed] [Google Scholar]
- 46.van Panhuys N, Klauschen F, and Germain RN 2014. T-cell-receptor-dependent signal intensity dominantly controls CD4(+) T cell polarization In Vivo. Immunity 41:63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dawson HD, Collins G, Pyle R, Key M, Weeraratna A, Deep-Dixit V, Nadal CN, and Taub DD 2006. Direct and indirect effects of retinoic acid on human Th2 cytokine and chemokine expression by human T lymphocytes. BMC Immunol 7:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iwata M, Eshima Y, and Kagechika H 2003. Retinoic acids exert direct effects on T cells to suppress Th1 development and enhance Th2 development via retinoic acid receptors. Int Immunol 15:1017–1025. [DOI] [PubMed] [Google Scholar]
- 49.Hall JA, Cannons JL, Grainger JR, Dos Santos LM, Hand TW, Naik S, Wohlfert EA, Chou DB, Oldenhove G, Robinson M, Grigg ME, Kastenmayer R, Schwartzberg PL, and Belkaid Y 2011. Essential role for retinoic acid in the promotion of CD4(+) T cell effector responses via retinoic acid receptor alpha. Immunity 34:435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu CC, Hsu SC, Shih HM, and Lai MZ 2003. Nuclear factor of activated T cells c is a target of p38 mitogen-activated protein kinase in T cells. Mol Cell Biol 23:6442–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Balkan W, Rodriguez-Gonzalez M, Pang M, Fernandez I, and Troen BR 2011. Retinoic acid inhibits NFATc1 expression and osteoclast differentiation. J Bone Miner Metab 29:652–661. [DOI] [PubMed] [Google Scholar]
- 52.Lee MO, Kang HJ, Kim YM, Oum JH, and Park J 2002. Repression of FasL expression by retinoic acid involves a novel mechanism of inhibition of transactivation function of the nuclear factors of activated T-cells. Eur J Biochem 269:1162–1170. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.