

Abstract
Many natural products and medicinal drugs are heterocyclic amines possessing a chiral quaternary carbon atom in their heterocyclic ring. Herein, we report the first catalytic and asymmetric Staudinger–aza-Wittig reaction for the desymmetrization of ketones. This highly enantioselective transformation proceeds at room temperature to provide high yields—even on multigram scales—of nitrogen heterocycles featuring a chiral quaternary center. The products of this reaction are potential precursors for the synthesis of pharmaceuticals. A commercially available small P-chiral phosphine catalyst, HypPhos, induces the asymmetry and is recycled through in situ reduction of its oxide, mediated by phenylsilane in the presence of a carboxylic acid. The efficiency, selectivity, scalability, mild reaction conditions, and broad substrate scope portend that this process will expedite the syntheses of chiral heterocyclic amines of significance to chemistry, biology, and medicine.
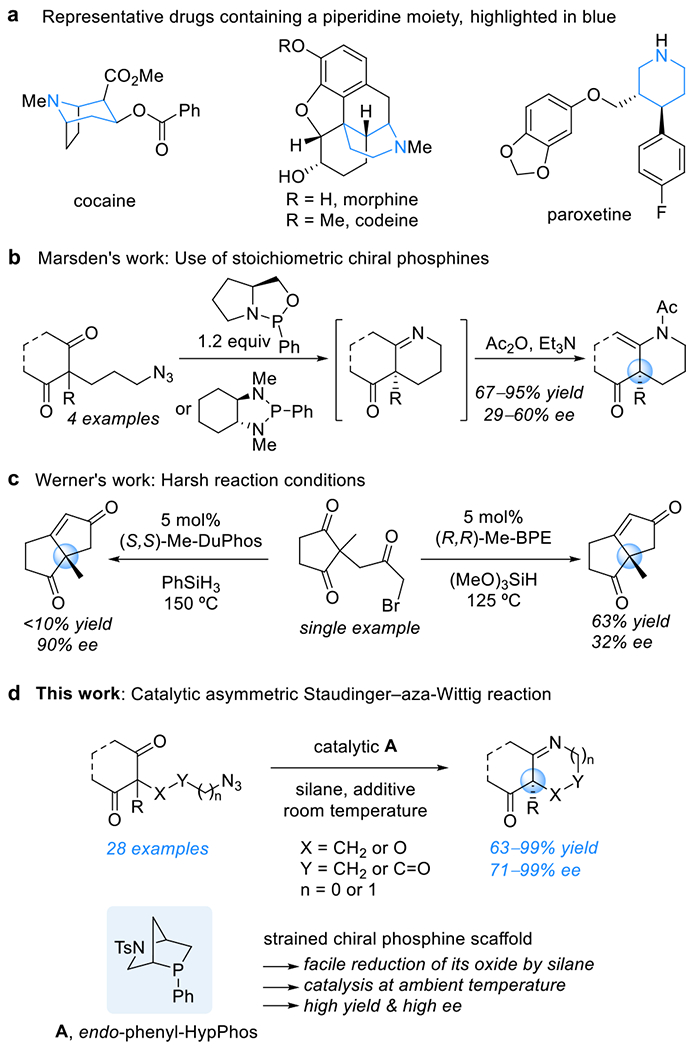
Chiral heterocyclic amines are very common structural elements within the plethora of biologically active natural products and medicinally relevant compounds. Indeed, more than 50% of the unique small-molecule drugs approved by the U.S. Food and Drug Administration through 2012 contained a nitrogen heterocycle.1,2 Six-membered heterocyclic amines, especially piperidines and piperazines, account for more than a quarter of the heterocyclic amine drugs; they appear, for example, in the structures of cocaine, morphine, codeine, and paroxetine (Scheme 1a). Among chiral heterocyclic amines, those possessing chiral quaternary carbon centers are particularly difficult to synthesize; more generally, the construction of quaternary centers in organic molecules remains one of the greatest problems in synthetic chemistry.3,4 This challenge increases in difficulty when all four of the substituents are unique, and the enantioselective preparation of such molecules is especially demanding.
Scheme 1.
Evolution of the Strategy toward the Catalytic Asymmetric Staudinger–aza-Wittig Reaction
Even today, pharmaceutical companies often rely on chemical resolution for the separation of racemic mixtures into pairs of enantiomers.5 Enantioselective synthesis is, in principle, more economical and more environmentally friendly because it avoids wasting the unneeded enantiomer.6 One method for preparing all-carbon quaternary stereocenters is the desymmetrization of prochiral substrates featuring a quaternary carbon center—ideally in a manner that is catalytic and asymmetric.4 We have discovered the first room-temperature catalytic asymmetric desymmetrization of ketones through a Staudinger–aza-Wittig reaction, and herein we demonstrate its use in the versatile preparation of five- and six-membered nitrogen heterocycles.
Tertiary phosphines are powerful reagents for transforming the C═O bonds of ketones and aldehydes into C═C and C═N bonds, through Wittig and aza-Wittig reactions, respectively.7–10 Enantioselective variants of these processes have been, however, only marginally successful to date, displaying poor substrate scope, providing products in low yields and with low enantioselectivities, requiring harsh conditions, and/or, most importantly, necessitating a stoichiometric amount of the chiral phosphine.11 In 2006, Marsden and co-workers reported the first enantioselective Staudinger–aza-Wittig reaction, in which a stoichiometric amount of a chiral oxazaphospholane or diazaphospholane was applied to form piperidines through desymmetrization of 2-(3-azidopropyl)-1,3-diones (Scheme 1b).11b More recently, the Werner group documented the first example of a catalytic asymmetric Wittig reaction, with either microwave or conventional heating used to drive the reduction of the phosphine oxide (Scheme 1c);11c a moderate yield or low enantioselectivity resulted in each case, presumably because of the harsh reaction conditions.
With the preceding observations in mind, we recognized that the key to achieving a catalytic enantioselective Staudinger–aza-Wittig reaction—ideally one that proceeds at room temperature—would be identifying a chiral phosphine that not only induces high enantioselectivity but also forms an oxide that is readily reduced by a silane. Silanes are the most functional group-tolerant reducing agents for the phosphine oxides produced at the end of each cycle of the Staudinger–aza-Wittig reaction.12 Recently, 2-phenyl-2-phosphabicyclo[2.2.1]heptane oxides have displayed efficiencies for silane-mediated reductions that are among the highest for all known phosphine oxides.13 Postulating that a chiral phosphine featuring a phosphabicyclo[2.2.1]heptane scaffold would undergo in situ reduction of its oxide under mild conditions, we tested the commercially available chiral phosphine “HypPhos,” derived from L-hydroxyproline (Hyp) (Scheme 1d).14 A stoichiometric amount of endo-phenyl-HypPhos A, containing a 2-aza-5-phosphabicyclo[2.2.1]-heptane scaffold, facilitated the asymmetric Staudinger–aza-Wittig reaction of the (3-azidopropyl)indanedione 1a in quantitative yield, with an ee of 79%, at room temperature (Figure 2a). For the silane-mediated reduction of endo-phenyl-HypPhos oxide A·[O] to proceed at an appreciable rate; however, heating at 80 °C was required. Indeed, employing 20 mol % of HypPhos A with 2 equiv of phenylsilane at 80 °C resulted in the formation of the same product, but with diminished enantioselectivity (71% ee).
Several research groups have demonstrated that the reductions of phosphine oxides are accelerated by acid additives, including phosphoric, benzoic, and triflic acids.15 To test whether our catalytic Staudinger–aza-Wittig reaction could be accelerated, such that it could proceed at appreciable rates (<2 days) at low temperatures (<80 °C), we tested the effects of various Brensted acids. 2-Nitrobenzoic acid provided the best results, facilitating the reaction at room temperature and giving the product in 95% yield and with 86% ee (Figure 1a). Increasing the loading of 2-nitrobenzoic acid from 20 mol % to 50 and 100 mol % decreased the enantioselectivity to 75 and 68% ee, respectively, presumably due to racemization induced through a combination of excess acid and adventitious water. By adding molecular sieves as a water-scavenger, the enantioselectivity improved further, to 90% ee, with the yield remaining near-quantitative at 99%.
Figure 1.
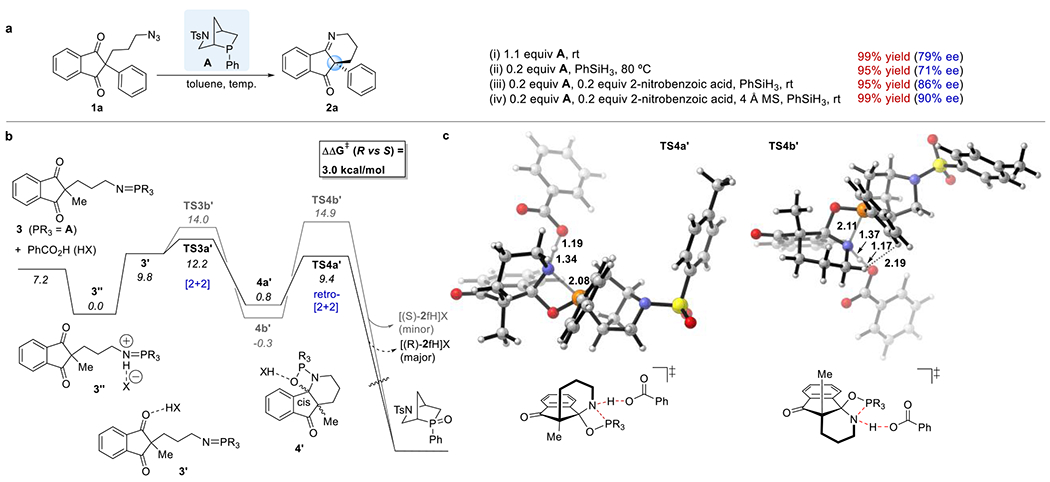
Rationalization of the observed enantioselectivity. (a) Optimization of the reaction conditions. (b) Free energy profile for the formation of the major (R, black lines) and minor (S, gray lines) enantiomers of the iminoketone in the aza-Wittig reaction of the iminophosphorane 3 in the presence of benzoic acid. (c) Structures of the transition states TS4a′ and TS4b′, highlighting the key stereodetermining interactions. Calculations performed with M06-2X/6-311++G(d,p)-SMD(toluene)//B3LYP/6-31G(d)-SMD(toluene). Numbers in italics are Gibbs free energies in kcal/mol.
To explore the origin of the enantioselectivity, we calculated the free energy profile for the aza-Wittig reaction of the iminophosphorane 3 derived from the chiral phosphine A and the azide 1f, with and without (see SI) benzoic acid as a model acid additive (Figure 1b,c). The calculations suggested that the reaction follows a [2+2]/retro-[2+2] cycloaddition pathway through the oxazaphosphetidine intermediate 4′.16 The enantioselectivity is induced by the chiral phosphine and is further enhanced by the acid.17 The acid protonates the imino phosphorane 3 to give the ion pair 3″.18 This ion pair can isomerize to the less stable hydrogen-bonded complex 3′, thereby activating the C═O group for the intramolecular [2+2] cycloaddition and, thereafter, accelerating the retro-[2+2] cycloaddition. The reactions leading to the major and minor enantiomers of the iminoketone have different rate-determining steps: namely, [2+2] cycloaddition for the major enantiomer (TS3a′) and retro-[2+2] cycloaddition for the minor enantiomer (TS4b′). The enantioselectivity is enhanced by the stabilization of TS4a′ being greater than that of TS4b′. The chiral phosphine strongly favors transition states where the apical positions of the P-trigonal bipyramid are occupied by the breaking bond (in this case, P—N) and the P—C bond to the bridgehead carbon. This arrangement, present in TS4a′ but not in TS4b′, allows the phenyl group of the HypPhos unit to stabilize the breaking P—N bond, it avoids repulsion between the HypPhos unit and the substrate C—H units, and it positions the piperidine N atom to be accessible for proton transfer from the acid cocatalyst. For TS4b′, in contrast, the HypPhos phenyl group interferes sterically with the N⋯acid interaction, such that proton transfer is less advanced in the transition state; steric repulsion occurs between the HypPhos phenyl group and the substrate C—H units (see dotted line in Figure 1c); and the HypPhos phenyl group is aligned less favorably for stabilizing the P—N breaking bond (dihedral angle, 63°; cf. −78° in TS4a′). The calculated difference in overall activation energies for the formation of the two enantiomeric products in this acid-assisted aza-Wittig reaction is 3.0 kcal/mol. This value matches the experimental enantioselectivity within computational error, predicting that product 2f would be formed with an ee of 99%. In addition to its accelerating influence on the rate of the aza-Wittig reaction, the acid is also likely to accelerate the reduction of the phosphine oxide back to the catalytically active phosphine and, thus, increase the catalytic turnover.19
Given the limited number of substrates in the previous reports of asymmetric Wittig processes,11b,c we were gratified to find that the scope of our catalytic asymmetric Staudinger–aza-Wittig reaction is indeed extensive and that the reaction is readily scalable (Scheme 2). When run using 2.5 g of the starting material 1a, the reaction produced the expected product 2a in 97% yield and with 89.4% ee. Both aryl and alkyl substituents at the quaternary center rendered products with high enantioselectivities and in excellent yields. Azidoindanediones presenting m-benzyloxyphenyl, p-tolyl, and o-anisyl substituents produced their iminoketones 2b, 2c, and 2d, respectively, with 84–90% ee’s and in almost quantitative yields. A 2-thienyl group was compatible with the reaction, furnishing its indanopiperidine 2e with 85% ee and in 91% yield. Methyl-, ethyl-, isopropyl-, allyl-, and benzyl-substituted indanediones formed their desired products 2f–2j in 89–97% yields and with 84–99% ee’s. Cyclopentyl-, cyclohexyl-, and cycloheptyl-substituted azidoindanediones gave the iminoketone products 2k–2m in 92–99% yields and with 90–96% ee’s. Subsequently, we varied the substitution pattern of the indane motif. Switching the benzene ring to a naphthalene unit had little effect on the reactivity, resulting in 2n being formed in 92% yield and with 89% ee. In contrast, changing the electronic properties of the indane moiety influenced the facility of the Staudinger–aza-Wittig reaction. The presence of electron-donating dimethoxy groups necessitated a slightly elevated reaction temperature (35 °C) to ensure an appreciable reaction rate, but led to the product 2o being obtained with lower enantioselectivity. Electron-withdrawing dichloro groups accelerated the reaction, furnishing the products 2p–2v with greater than 90% ee’s and in high yields. The azide chain can incorporate ether and ester linkages, allowing efficient syntheses of the dihydrooxazinone 2w (90% yield, 80% ee) and the dihydrooxazine 2x (92% yield, 87% ee). Furthermore, we constructed both tetrahydropyridine and dihydropyrrole scaffolds having a phenalendione backbone. For the tetrahydropyridine 2y, the exo-phenyl-HypPhos B produced the iminoketone in 63% yield and with 71% ee. For dihydropyrrole formation, both the HypPhos catalysts A and B displayed similar reactivities. Phenyl-, p-anisyl-, and o-tolyl-substituted azidophenalendiones delivered their dihydropyrrole products 2z–2ab in 85–95% yields and with 84–87% ee’s. X-ray crystallographic analysis of compound 2q established the (S)-configurations of these products.
Scheme 2. Scope of the Catalytic Asymmetric Staudinger–aza-Wittig Reaction.
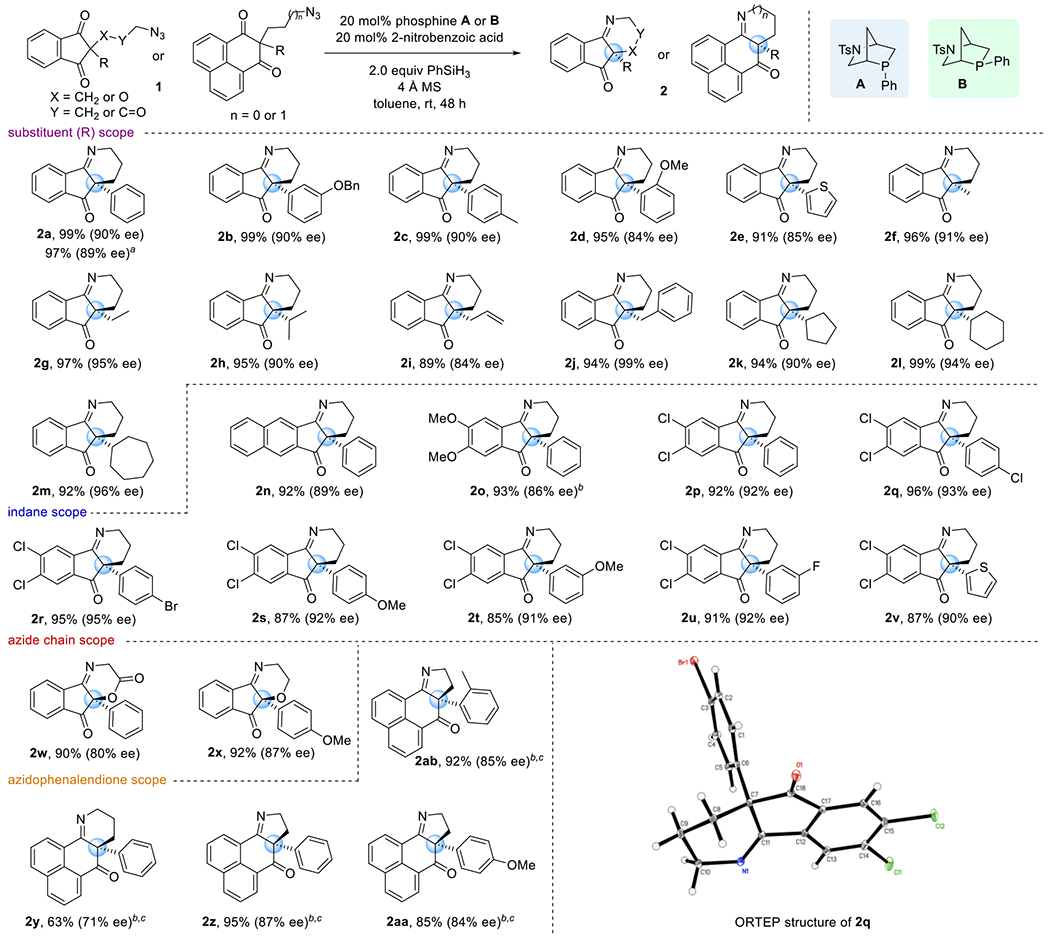
aReaction performed using 2.5 g of the substrate. bReaction performed at 35 °C. cHypPhos B used as the catalyst.
Indano[1,2-b]piperidine motifs have been excellent sources of new medicinal leads.21 Recently, scientists at Astellas Pharma established them as potent antagonists of the N-methyl-d-aspartate (NMDA) receptor, which is associated with several neurological disorders, including Alzheimer’s and Parkinson’s diseases (Scheme 3a).22 Substituents at the quaternary carbon center of the indanopiperidines influence their activities, as does the stereochemistry of the ring juncture. Notably, the enantiomers of the most potent (ethyl-substituted) indanopiperidine were chemically resolved at Astellas Pharma through three rounds of selective salt formation using enantiopure dibenzoyltartaric acid. Given the high bioactivity of indanopiperidine derivatives, manipulation of the Staudinger–aza-Wittig reaction products 2 could produce potentially useful compounds for drug discovery. Lithium aluminum hydride (LAH)-mediated reduction of 2a and subsequent mesylation rendered the methanesulfonate 6 as a single diastereoisomer (92% yield over two steps) (Scheme 3b). Global reduction of 6 with magnesium in methanol removed the sulfonate group and reduced the imino group to form the desired indanopiperidine 7 with a cis ring juncture. Reductive amination with formaldehyde then formed the tertiary amine 8. When we performed the reduction of 6 with sodium borohydride (NaBH4) in methanol, selective reduction of the imino group delivered the diastereoisomerically pure indanopiperidine 9, having a trans ring juncture, in 78% yield. Selective removal of the sulfonate group of 6 was achieved using sodium naphthalenide, providing the imine 10 in 90% yield. The indanopiperidine 11, having a trans ring juncture, was obtained in two ways: from the mesylate 9 (through removal of the sulfonate) in 78% yield and from the imine 10 (through reduction of the imino group) in 85% yield.
Scheme 3.
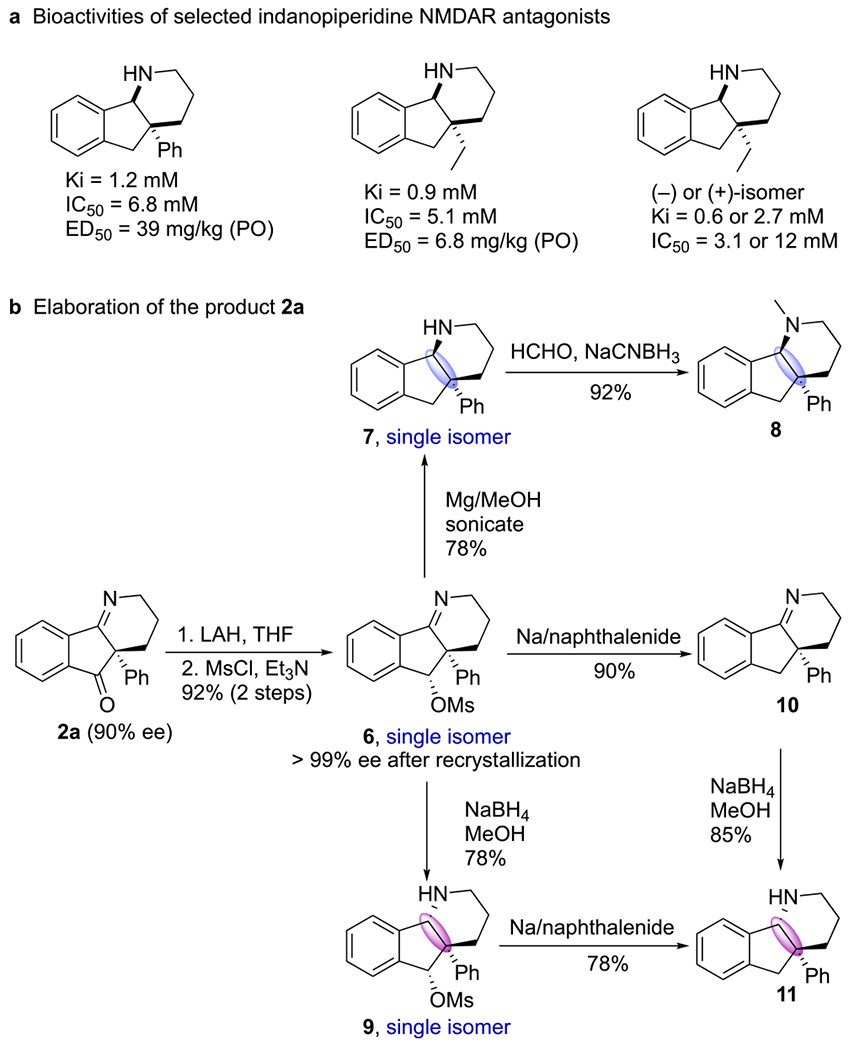
Transformations of a Staudinger–aza-Wittig Product: Syntheses of NMDAR Antagonists
Many contemporary methods for the construction of chiral heterocyclic amines are based on transition metal catalysis.23 The methodological advance presented herein involves main group-based catalysis. Fundamentally, the combination of a P-chiral phosphine and a Brønsted acid enables catalysis at room temperature, providing a strategic blueprint for taking a canonically stoichiometric phosphine-mediated reaction and rendering it catalytic and enantioselective. The ready availability of the HypPhos catalysts and the high efficiency, selectivity, and practicality of this catalytic enantioselective Staudinger–aza-Wittig process foretell its lasting impact on the preparation of chiral nitrogen heterocycles having relevant biological and medicinal applications.
Supplementary Material
ACKNOWLEDGMENTS
We thank the NIH (GM071779 to O.K.), Boehringer Ingelheim (Green Chemistry Award to O.K.), the NSF (CHE-1764328 to K.N.H.), the ARC (DP150103131 and DP180103047 to E.H.K.), and the University of Queensland (to E.H.K.) for financial support. We also thank Andrew J. Smaligo and Xiangyang Chen for assistance with the preparation of the figures. This study was supported by shared instrumentation grants from the NSF (CHE-1048804) and the NIH NCRR (S10RR025631). Calculations were performed on the Hoffman2 cluster at UCLA, supported by the NSF (Grant OCI-1053575), and using the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the Australian National Computational Infrastructure and the UQ Research Computing Centre.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.9b04803.
Experimental procedures and analytical data (PDF)
Crystallographic data for 2q (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).Vitaku E; Smith DT; Njardarson JT Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem 2014, 57, 10257. [DOI] [PubMed] [Google Scholar]
- (2).(a) McGrath NA; Brichacek M; Njardarson JT A graphical journey of innovative organic architectures that have improved our lives. J. Chem. Educ 2010, 87, 1348. [Google Scholar]; (b) Ilardi EA; Vitaku E; Njardarson JT An in-pharm-ative educational poster anthology highlighting the therapeutic agents that chronicle our medicinal history. J. Chem. Educ 2013, 90, 1403. [Google Scholar]
- (3).Quasdorf KW; Overman LE Catalytic enantioselective synthesis of quaternary carbon stereocenters. Nature 2014, 516, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Zeng XP; Cao ZY; Wang YH; Zhou F; Zhou J Catalytic enantioselective desymmetrization reactions to all-carbon quaternary stereocenters. Chem. Rev 2016, 116, 7330. [DOI] [PubMed] [Google Scholar]
- (5).Mane S Racemic drug resolution: A comprehensive guide. Anal. Methods 2016, 8, 7567. [Google Scholar]
- (6).Catalytic asymmetric synthesis, 3rd ed.; Ojima I, Ed.; John Wiley & Sons: Hoboken, NJ, 2010. [Google Scholar]
- (7).(a) Pommer H The Wittig reaction in industrial practice. Angew. Chem., Int. Ed. Engl 1977, 16, 423. [Google Scholar]; (b) Maryanoff BE; Reitz AB The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions. Stereochemistry, mechanism, and selected synthetic aspects. Chem. Rev 1989, 89, 863. [Google Scholar]; (c) Hoffmann RW Wittig and his accomplishments: Still relevant beyond his 100th birthday. Angew. Chem., Int. Ed 2001, 40, 1411. [DOI] [PubMed] [Google Scholar]
- (8).Palacios F; Alonso C; Aparicio D; Rubiales G; de los Santos J The aza-Wittig reaction: An efficient tool for the construction of carbon–nitrogen double bonds. Tetrahedron 2007, 63, 523. [Google Scholar]
- (9).Lao Z; Toy P Catalytic Wittig and aza-Wittig reactions. Beilstein J. Org. Chem 2016, 12, 2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).For examples of catalytic aza-Wittig reactions, see:; (a) Marsden SP; McGonagle AE; McKeever-Abbas B Catalytic aza-Wittig cyclizations for heteroaromatic synthesis. Org. Lett 2008, 10, 2589. [DOI] [PubMed] [Google Scholar]; (b) van Kalkeren HA; te Grotenhuis C; Haasjes FS; Hommersom CA; Rutjes FPJT; van Delft FL Catalytic Staudinger/aza-Wittig sequence by in situ phosphane oxide reduction. Eur. J. Org. Chem 2013, 2013, 7059. [Google Scholar]; (c) Wang L; Wang Y; Chen M; Ding M Reversible P(III)/P(V) redox: Catalytic reaction for the synthesis of 4(3H)-quinazolinones and the natural product vasicinone. Adv. Synth. Catal 2014, 356, 1098. [Google Scholar]; (d) Wang L; Qin R; Yan H; Ding M New efficient synthesis of 1,4-benzodiazepin-5-ones by catalytic aza-Wittig reaction. Synthesis 2015, 47, 3522. [Google Scholar]; (e) Wang L; Xie Y; Huang N; Yan J; Hu W; Liu M; Ding M . Catalytic aza-Wittig reaction of acid anhydride for the synthesis of 4H-benzo[d][1,3]oxazin-4-ones and 4-benzylidene-2-aryloxazol-5(4H)-ones. ACS Catal. 2016, 6, 4010. [Google Scholar]; (f) Yan Y; Rao Y; Ding M One-pot synthesis of multisubstituted benzimidazoles via sequential Ugi and catalytic aza-Wittig reaction starting from 2-aminobenzoyl azides. J. Org. Chem 2016, 81, 1263. [DOI] [PubMed] [Google Scholar]; (g) Ren Z; Liu J; Ding M A Facile synthesis of 4-tetrazolyl-substituted 4H-3,1-benzoxazines through sequential Passerini-azide/acylation/catalytic aza-Wittig reaction. Synthesis 2017, 49, 745. [Google Scholar]; (h) Yan Y; Li H; Ren J; Wang S; Ding M One-pot selective synthesis of multisubstituted quinoxalin-2(1H)-ones by a Ugi 4CR/catalytic aza-Wittig sequence. Synlett 2018, 29, 1447. [Google Scholar]
- (11).(a) Trost BM; Curran DP An enantiodirected cyclopentenone annulation. Synthesis of a useful building block for condensed cyclopentanoid natural products. J. Am. Chem. Soc 1980, 102, 5699. [Google Scholar]; (b) Lertpibulpanya D; Marsden SP; Rodriguez-Garcia I; Kilner CA Asymmetric aza-Wittig reaction: Enantioselective synthesis of β-quaternary azacycles. Angew. Chem., Int. Ed 2006, 45, 5000. [DOI] [PubMed] [Google Scholar]; (c) Werner T; Hoffmann M; Deshmukh S First enantioselective catalytic Wittig reaction. Eur. J. Org. Chem 2014, 2014, 6630. [Google Scholar]; (d) Schneider LM; Schmiedel VM; Pecchioli T; Lentz D; Merten C; Christmann M Asymmetric synthesis of carbocyclic propellanes. Org. Lett 2017, 19, 2310. [DOI] [PubMed] [Google Scholar]; (e) Schmiedel VM; Hong YJ; Lentz D; Tantillo DJ; Christmann M Synthesis and structure revision of dichrocephones A and B. Angew. Chem., Int. Ed 2018, 57, 2419. [DOI] [PubMed] [Google Scholar]
- (12).O’Brien CJ; Tellez J; Nixon ZS; Kang LJ; Carter AL; Kunkel SR; Przeworski KC; Chass GA Recycling the waste: The development of a catalytic Wittig reaction. Angew. Chem., Int. Ed 2009, 48, 6836. [DOI] [PubMed] [Google Scholar]
- (13).Zhang K; Cai L; Yang Z; Houk KN; Kwon O Bridged [2.2.1] bicyclic phosphine oxide facilitates catalytic γ-umpolung addition–Wittig olefination. Chem. Sci 2018, 9, 1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Henry CE; Xu Q; Fan YC; Martin TJ; Belding L; Dudding R; Kwon O Hydroxyproline-derived pseudoenantiomeric [2.2.1] bicyclic phosphines: Asymmetric synthesis of (+)- and (−)-pyrrolines. J. Am. Chem. Soc 2014, 136, 11890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Li Y; Lu LQ; Das S; Pisiewicz S; Junge K; Beller M Highly chemoselective metal-free reduction of phosphine oxides to phosphines. J. Am. Chem. Soc 2012, 134, 18325. [DOI] [PubMed] [Google Scholar]; (b) O’Brien CJ; Lavigne F; Coyle EE; Holohan AJ; Doonan BJ Breaking the ring through a room temperature catalytic Wittig reaction. Chem. - Eur. J 2013, 19, 5854. [DOI] [PubMed] [Google Scholar]; (c) Hérault D; Nguyen DH; Nuel D; Buono G Reduction of secondary and tertiary phosphine oxides to phosphines. Chem. Soc. Rev 2015, 44, 2508. [DOI] [PubMed] [Google Scholar]; (d) Schirmer ML; Jopp S; Holz J; Spannenberg A; Werner T Organocatalyzed reduction of tertiary phosphine oxide. Adv. Synth. Catal 2016, 358, 26. [Google Scholar]
- (16).Cossío FP; Alonso C; Lecea B; Ayerbe M; Rubiales G; Palacios F Mechanism and stereoselectivity of the aza-Wittig reaction between phosphazenes and aldehydes. J. Org. Chem 2006, 71 , 2839. [DOI] [PubMed] [Google Scholar]
- (17).Computations on the acid-free aza-Wittig reaction of 3 are reported in the Supporting Information. The difference in barriers (ΔΔG‡) between the major and minor enantiodiscriminating transition states under acid-free conditions is calculated to be 1.3 kcal/mol, corresponding to an ee of 80% at 25 °C.
- (18).For earlier examples of acid additives used in nucleophilic phosphine catalysis, see:; (a) Trost BM; Li CJ Phosphine-catalyzed isomerization-addition of oxygen nucleophiles to 2-alkynoates. J. Am. Chem. Soc 1994, 116, 10819. [Google Scholar]; (b) Rychnovsky SD; Kim J Triphenylphosphine-catalyzed isomerizations of enynes to (E,E,E)-trienes: Phenol as a cocatalyst. J. Org. Chem 1994, 59, 2659. [Google Scholar]; For selected examples of enantioselectivity enhancement in asymmetric phosphine catalysis, see:; (c) Smith SW; Fu GC Asymmetric carbon–carbon bond formation γ to a carbonyl group: Phosphine-catalyzed addition of nitromethane to allenes. J. Am. Chem. Soc 2009, 131, 14231. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Yao W; Yu Z; Wen S; Ni H; Ullah N; Lan Y; Lu Y Chiral phosphine-mediated intramolecular [3 + 2] annulation: Enhanced enantioselectivity by achiral Brønsted acid. Chem. Sci 2017, 8, 5196. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Xu Q; Dupper NJ; Smaligo AJ; Fan YC; Cai L; Wang Z; Langenbacher AD; Chen J; Kwon O Catalytic enantioselective synthesis of guvacine derivatives through [4 + 2] annulations of imines with α-methylallenoates. Org. Lett 2018, 20, 6089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Computational studies of the phosphine recycling reaction will be reported in due course:; Krenske EH; Kirk AM; Coyle EE; O’Brien CJ Unpublished results.
- (20).The alkyl-substituted products slowly racemized over several days at room temperature when exposed to air. No racemization occurred when the products were kept at −20 °C for 2 days. We surmise that the Indane scaffold, which features extended conjugation to both the imino and keto functionalities, stabilizes the iminoketone product both electronically and sterically, thereby inhibiting its racemization.
- (21).(a) Froimowitz M Conformational analysis of cocaine, the potent analog 2β-carbomethoxy-3β-(4-fluorophenyl)tropane (CFT), and other dopamine reuptake blockers. J. Comput. Chem 1993, 14, 934. [Google Scholar]; (b) De Wit T; van Emelen K; Maertens F; Hoornaert GJ; Compernolle F cis-Fused hexahydro-4aH-indeno[1,2-b]pyridines: Transformation of bridgehead ester group and conversion to tricyclic analogues of NK-1 and dopamine receptor ligands. Tetrahedron Lett. 2001, 42, 4919. [Google Scholar]
- (22).(a) Hayashibe S; Yamasaki S; Shiraishi N; Hoshii H; Tobe T Fused Indane compound. Patent WO 2009/069610, 2009.; (b) Finsinger DD; Bruge DD; Buchstaller HD; Emde UD Indane. Patent WO 2006/094602, 2006.
- (23).Advances in transition-metal mediated heterocyclic synthesis; Sole D, Fernandez I, Eds.; Elsevier: Amsterdam, 2018. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.