

Abstract
Objective
To demonstrate the causal role in disease of the MT-TP m.15992A>T mutation observed in patients from 5 independent families.
Methods
Lactate measurement, muscle histology, and mitochondrial activities in patients; PCR-based analyses of the size, amount, and sequence of muscle mitochondrial DNA (mtDNA) and proportion of the mutation; respiration, mitochondrial activities, proteins, translation, transfer RNA (tRNA) levels, and base modification state in skin fibroblasts and cybrids; and reactive oxygen species production, proliferation in the absence of glucose, and plasma membrane potential in cybrids.
Results
All patients presented with severe exercise intolerance and hyperlactatemia. They were associated with prominent exercise-induced muscle swelling, conspicuous in masseter muscles (2 families), and/or with congenital cataract (2 families). MRI confirmed exercise-induced muscle edema. Muscle disclosed severe combined respiratory defect. Muscle mtDNA had normal size and amount. Its sequence was almost identical in all patients, defining the haplotype as J1c10, and sharing 31 variants, only 1 of which, MT-TP m.15992A>T, was likely pathogenic. The mutation was homoplasmic in all tissues and family members. Fibroblasts and cybrids with homoplasmic mutation had defective respiration, low complex III activity, and decreased tRNAPro amount. Their respiratory complexes amount and tRNAPro aminoacylation appeared normal. Low proliferation in the absence of glucose demonstrated the relevance of the defects on cybrid biology while abnormal loss of cell volume when faced to plasma membrane depolarization provided a link to the muscle edema observed in patients.
Conclusions
The homoplasmic MT-TP m.15992A>T mutation in the J1c10 haplotype causes exercise-induced muscle swelling and fatigue.
Mitochondrial diseases, due to oxidative phosphorylation defects, represent an extraordinary diagnostic challenge because of their diversity. The hundreds genes involved, located on either the mitochondrial or the nuclear DNA,1 or the frequent heteroplasmy of mitochondrial DNA (mtDNA) mutations, i.e., coexistence of wild-type and mutant mtDNA molecules,2 likely explain part of that diversity. In addition, deleterious homoplasmic mtDNA mutations, i.e., mutations affecting all mtDNA molecules, cause diseases with incomplete penetrance and tissue-specific expression that disclose the complexity of nuclear-mitochondrial interaction.3 This was the case with mutations causing Leber hereditary optic neuropathy4 or deafness.5 It was particularly striking for the homoplasmic MT-TI m.4300A>G mutation whose expression pattern ranged from infantile cardiac failure with severe cardiac mitochondrial defect to apparent health.6 Initial identification of these deleterious homoplasmic mutations thus required either their recurrence with a characteristic phenotype4 or the demonstration of the mitochondrial defect in the clinically targeted organ.6
Among deleterious mtDNA mutations, point mutations affecting transfer RNA (tRNA) genes are common (see Mitomap at mitomap.org). Their identification relies on a pathogenicity scoring system using criteria from clinical investigations, database analysis, and functional studies.7 Among the functional studies, cybrids, i.e., cytoplasmic hybrids with the patient's mtDNA and the nuclear genome from immortal cells8 bring the possibility to demonstrate the mtDNA origin of the mitochondrial defect and are thus considered a gold standard for the pathogenicity assessment of mtDNA mutations.7 However, as expected with mutations causing tissue-specific expression impairment, these cells, being a surrogate tissue for the disease, often disclosed only a mild mitochondrial defect.9,10
The impact of tRNA mutations depends on the minimal set of 22 mtDNA tRNAs using a nonuniversal genetic code and specific decoding rules for mitochondrial protein synthesis.11 In particular, all the codons with any base in the third codon position encoding the same amino acid are recognized by only 1 tRNA that carries an unmodified U in the wobble position of the anticodon.12
We here report the homoplasmic mtDNA m.15992A>T mutation of the wobble position of tRNAPro in 5 families with severe exercise intolerance and combined respiratory chain defect in muscle. We demonstrate the mutation pathogenicity in cybrids by showing its association with respiratory defect, decreased tRNAPro steady state, and altered response to depolarization reminiscent of the exercise-induced ionic disturbances observed in patients.
Patients and methods
Patients
Patient 1 (III-10 in family 1 in figure 1) presented with swelling of the temporal and masseter muscles during feeding since the neonatal period. Exercise intolerance occurred during early childhood, never associated with rhabdomyolysis. At age 22 years, she sought medical advice for myalgia and exhaustion after walking 500 m, followed with nausea, vomiting, and headache if effort was continued. Physical examination was normal at rest. Lactatemia was 2.1 mM in the fasting state and high after nonischemic forearm exercise test.13
Figure 1. Family trees of the patients with the m.15992A>T mutation.
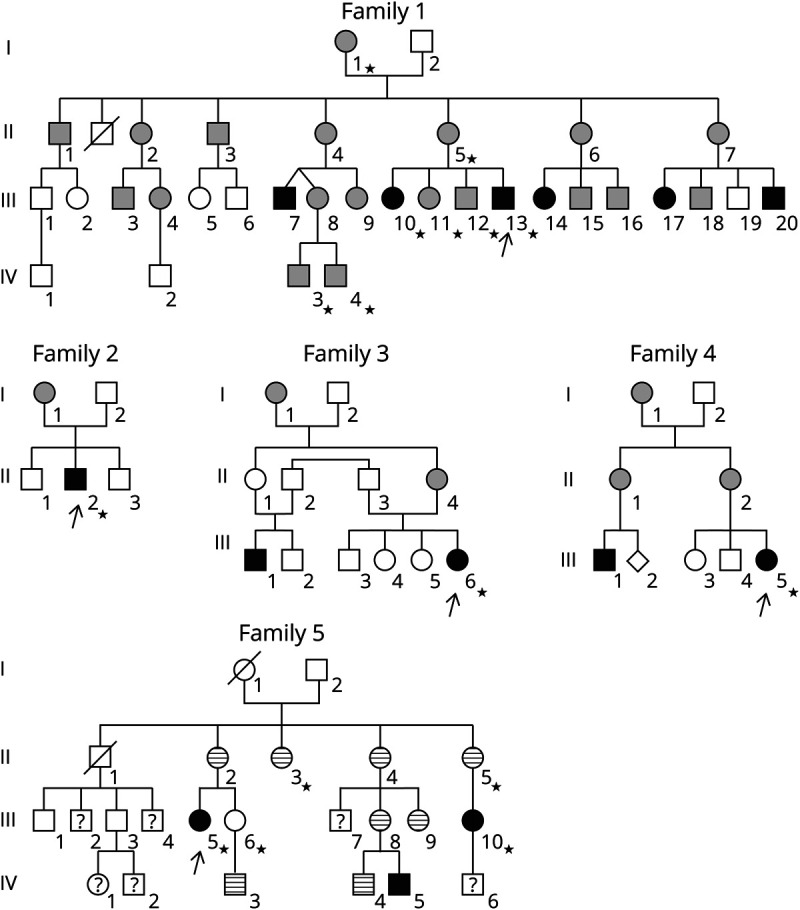
Arrows indicate the proband in each family; stars = subjects with samples for molecular studies; white forms = subjects asymptomatic or without sufficient information; black forms = patients with severe exercise intolerance, i.e., disabling daily life; gray forms = moderate exercise intolerance, i.e., not disabling daily life but clearly experienced; stripped forms = patients with isolated chewing-induced masseter swelling; question marks indicate unknown clinical status of the subject. In families 1 and 5, severe intolerance to exercise was constantly associated with chewing-induced masseters swelling, but the reverse was not true with several patients in family 5 presenting only chewing-induced masseters swelling.
Patient 2 (III-13 in family 1) sought medical advice at age 14 years for clinical and biological signs similar to patient 1.
Patient 3 (II-2 in family 2) presented at age 14 years with fatigability and muscle pain after moderate exercise (1 km cycling). He also presented with bilateral cataract, which was present in his mother. His physical examination was normal apart from short stature (−2.5 SD). Lactatemia was 6 mM in the postprandial state and high after moderate effort.
Patient 4 (III-6 in family 3) complained of myalgia and fatigability since early childhood, with vomiting if effort was continued. Physical examination only disclosed short stature (−3 SD). Lactatemia was 6.3 mM in the fasting state and high after effort.
Patient 5 (III-5 in family 4) complained of myalgia and muscle fatigue since age 3 years. Vomiting and malaise after prolonged efforts started after age 6 years. At age 13 years, she could only walk 200 m. Lactatemia was high in the fasting state (3.3 mM) and after nonischemic forearm exercise test.13
Patient 6 (III-5 in family 5 in figure 1) presented with congenital cataract. Severe exercise intolerance occurred in childhood and was associated with masseter swelling. At age 38 years, she could only walk 500 m because of myalgia and muscular exhaustion. Lactatemia was normal in the fasting state but high after exercise.
Methods
Patients' investigations
All patients gave their written informed consent for their analyses for diagnostic investigations and for their use in clinical research and publication, according to our institutional ethics board. The study received approval from our Institutional Review Board.
Brain imaging used a 1.5 T General Electric MRI with a head and neck coil, including coronal and axial T1 and short-tau inversion recovery (STIR) sequences.
Muscle fragments were immediately frozen and stored at −80°C until use. Standard procedures provided blood, buccal mucosa cells, urinary sediment, and cultured fibroblasts derived from a forearm skin biopsy (patients 1, 2, 3, and 4). Muscle histology followed standardized protocols.14,15
Molecular biology
Extraction of DNA from muscle and fibroblasts used standard methods based on proteinase K and SDS digestion; it used QIAamp DNA Mini Kit extraction (Qiagen) for blood, cells from buccal mucosa, or urinary sediment. Extraction of RNA used the miRNeasy Mini Kit (Qiagen) or TRIzol (Thermo Fisher Scientific).
Long-range PCR screened for large-scale deletions, while quantitative PCR evaluated the mtDNA copy number.16 The Sanger method provided mtDNA sequence. Mispairing PCR restriction quantified heteroplasmy using the DdeI site created by the mutation. Primers are in table e-1, links.lww.com/NXG/A282.
Quantification of mitochondrial tRNAs used Northern blotting of 8% polyacrylamide 8 M urea (Tris-borate) gels on Hybond N+ membranes and T4 polynucleotide kinase—5′-end 32P-labeled probes (table e-1, links.lww.com/NXG/A282). Radioactive signals were quantified using Typhoon Trio and ImageQuant software (GE Healthcare).
Acid-denaturating gel separation and Northern blot analyzed the tRNA aminoacylation state17 while [35S]-methionine in vivo labeling in the presence of emetin, a specific inhibitor of cytosolic ribosomes, assessed mitochondrial translation18 and reverse transciption followed by PCR (RT-PCR) with extended oligonucleotides and sequencing with shorter primers (table e-1, links.lww.com/NXG/A282) searched for the presence of inosine at the wobble position of tRNA anticodon.19
Cell biology
Cytoplasmic hybrids (cybrids) were obtained from patients 1 and 4 fibroblasts.8
Cell respiration was analyzed in Oroboros high-resolution respirometer,20 whereas mitochondrial activities in muscle and fibroblasts used standardized spectrophotometric protocols.21 Mitochondrial production of superoxide ion was assessed by flow cytometry using 5 µM MitoSOX Red (Invitrogen).22
Mitochondrial pellet preparation and Western blot after blue native polyacrylamide gel electrophoresis were as described in Ref. 23. Primary antibodies were polyclonal antibodies against the F1 domain of bovine complex V, UQCRC2, MT-ND1, or MT-CO2 (respectively produced by Pr Joël Lunardi, Grenoble, by Dr Catherine Godinot, Lyon, or our group24) or monoclonal antibodies against the respiratory complex II SDHA subunit SDHA (Abcam), incubated overnight at 0°–4°C. Secondary antibodies were peroxidase conjugated (Sigma-Aldrich) and visualized with Pierce™ ECL Western Blotting Substrate (Life Technologies). Quantification used volumes and basal adjustment with rolling ball by Fusion FX (Vilber Company).
To analyze cell proliferation, 3,000 cells per well were cultured in four 24-well plates and Dulbecco Modified Eagle Medium with 1 mM pyruvate and either 1 g/L glucose or 200 µM glutamine without glucose.25 Cell numbering was performed, in triplicates for each condition, every 24 hours (T0, T24, T48, and T72 plates) using the neutral red method26 and expressed as fold increase relative to T0.
To evaluate plasma membrane, 100,000 cybrid cells were incubated in 96-well plate with 7.5 µM tetramethylrhodamine ethyl esther (TMRE) and 5 µM verapamil, an inhibitor of the multidrug resistance–associated proteins. Because resting membrane potential essentially depends on K diffusion gradient, progressive increase of external K concentration (from 10 to 80 mM) induced progressive depolarization. Osmolarity was kept at 300 mOsm/L. After 120-minute incubation, fluorescence was quantified by the Accuri™ C6 flow cytometer.
Statistics
Normality evaluation used the Shapiro-Wilk test. Depending on the distribution of data, comparison between 2 groups used the Mann-Whitney or t test. Differences were considered significant when p < 0.05.
Data availability
Any data not published within the article will be shared, as anonymized data, by request from any qualified investigator.
Results
The chewing-induced masticatory muscle swelling was due to edema
Nuclear MRI of facial muscles analyzed the exercise-induced muscle swelling in 2 members of family 5. Chewing a sandwich during 15 minutes induced a 20%–30% increase in thickness of the temporal and masseter muscles of patient 6 (figure 2, A and B) and her aunt (II-5 in family 5) (figure 2, C and D). Mild adipose infiltration of the masseter and lateral pterygoid muscles was present in patient 6's aunt. Prolonged edema was indicated by hypersignal in STIR, fat-suppression images, in all masticatory muscles of patient 6 (figure 2, E and F), and in the masseter, temporal, and lateral pterygoid muscles of her aunt (figure 2, G and H).
Figure 2. Chewing-induced edema of the facial muscles in 2 patients with the m.15992A>T mutation.

Coronal T1-weighted NMRI of the head of patient 6 (A and B) and her aunt (C and D); coronal STIR NMRI of patient 6 (E and F) and her aunt (G and H); images obtained at rest (A, C, E, and G) and 15 minutes after chewing a sandwich (B, D, F, and H). The measures show that after chewing, the diameter of temporal muscle increased by 22% in patient 6 (A vs B) and by 34% in her aunt who increased the diameter of her masseter muscle by 37% (C vs D). NMRI = nuclear magnetic resonance imaging; STIR = short-tau inversion recovery.
Mitochondrial myopathy underlay the muscle symptoms
Muscle histology disclosed mild alterations in patients 2, 3, 4, and 5, including moderate lipidosis and increased subsarcolemmal mitochondria (figure e-1A, links.lww.com/NXG/A279). It was considered normal for patient 6.
Spectrophotometric assays of the respiratory chain revealed severe combined defect of respiratory complexes I, III, and IV with increased citrate synthase in the muscle biopsies from all probands (table 1). Respiratory complex II, the only complex without mtDNA-encoded subunit, was either normal or elevated.
Table 1.
Mitochondrial activities in the muscle of patients with homoplasmic m.15992A>T mutation

All patients shared the same homoplasmic MT-TP m.15992A>T mutation
Long-range PCR excluded the presence of large size rearrangement, whereas quantitative PCR ruled out depletion. Sequencing of the whole mtDNA sequence revealed that all patients had the same mtDNA, sharing 31 of 32 variants (table e-2, links.lww.com/NXG/A283). Thirty-one of these 32 variants were highly likely polymorphisms: 28 reported at least 1,462 times in GenBank database, a MT-RNR1 variant reported 37 times in different ethnic backgrounds, and 2 synonymous mutations (MTND2 c.5024C>T and MT-CO1 m.7028C>T) (mitomap.org). These polymorphisms defined the mtDNA haplotype as J1c10 for all the families. The last variant, common to all patients, was the m.15992A>T mutation in the MT-TP gene. Several criteria indicated its potential pathogenicity: (1) it was reported only once in more than 49,000 GenBank full-length mtDNA sequences; (2) it modified the strictly phylogenetically conserved wobble base in the tRNA anticodon (see MitotRNAdb/Mamit-tRNA website at mttrna.bioinf.uni-leipzig.de); and (3) alteration of the wobble position in mt-tRNA anticodon is a major deleterious factor in several mt-tRNA confirmed mutations.27
The mutation appeared homoplasmic (figure 3) with PCR restriction in the DNA samples from muscle of all the probands, from blood of all patients marked with a star in figure 1, and from urinary sediment and buccal cells of patients 1 and 2, their mother, and grandmother.
Figure 3. Homoplasmy of the m.15992A>T MT-TP mutation.
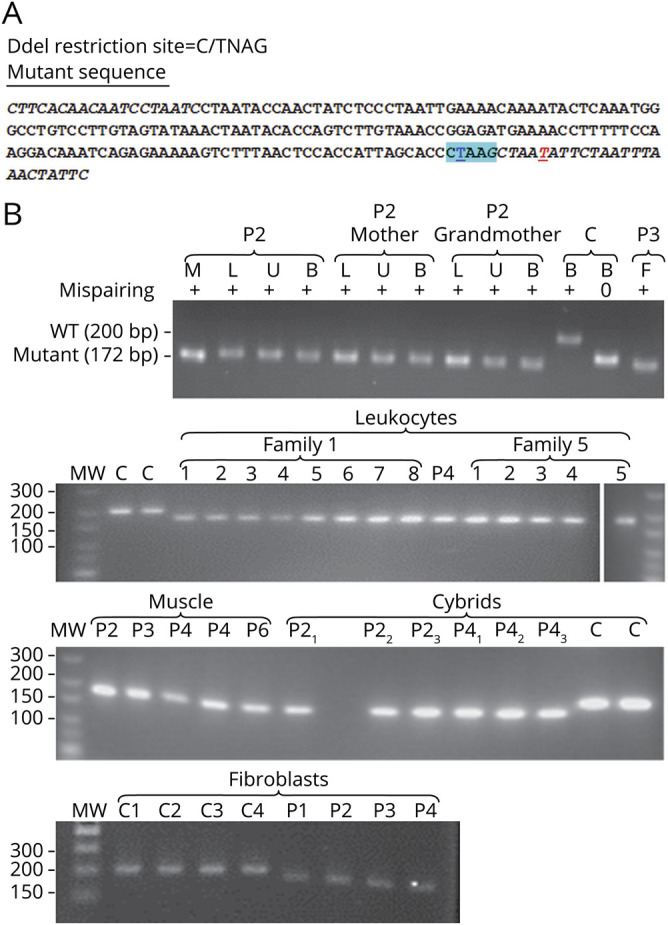
(A) Strategy of the mispairing PCR for restriction analysis of the m.15992A>T mutation proportion. Italic characters indicate the primers used to amplify the 200-bp-long mtDNA fragment from position 15820 to 16019; DdeI restriction site is highlighted in cyan blue; the nucleotide position 15992 in underlined bold blue character; the mispairing position in the backward primer is in underlined bold red. (B) Restriction fragments of mtDNA fragments cut with DdeI. P1, P2, P3, P4, P5, and P6 = patients 1, 2, 3, 4, 5, and 6 respectively; C = wild-type controls; M = DNA from muscle, L = DNA from blood leukocytes DNA, U = DNA from urinary sediment; B = DNA from buccal cells; F = DNA from cultured skin fibroblasts. The need for mispairing is shown in the upper panel with the control sample (C) amplified with and without mispairing and then cut with DdeI. Without mispairing, the WT restriction fragment run only slightly above the mutated fragments, in agreement with their length being only 5 bp longer than the mutant fragments. With mispairing abolishing the DdeI site at position 15,996, all samples amplified from patients' tissues run homogeneously at a longer distance than wild-type control mtDNA fragment in agreement with their 28 bp shorter length. The upper panel also shows that the mutation appeared homoplasmic in every tissue tested in several members of family 1. The rest of the panels show that the mutation appeared homoplasmic in every member analyzed in the 2 families where DNA from blood leukocytes was available (figure 1). It also appeared homoplasmic in cultured fibroblasts derived from a skin biopsy of patients 1, 2, 3, and 4 and in the cybrid clones obtained from either patient 2 or patient 4 fibroblasts. mtDNA = mitochondrial DNA; WT = wild type.
Transfer of the m.15992A>T mutation into cybrids demonstrated its deleterious potential on OXPHOS activities
Fibroblasts derived from a skin biopsy of patients 1, 2, 3, and 4 had 100% mutation (figure 3). They disclosed a mild but significant decrease of their basal respiration and respiration linked to adenosine triphosphate (ATP) production (figure 4A). Citrate synthase activity was significantly increased (figure 4B). Complexes I, III, IV, and V appeared decreased, reaching significance for complex III after normalization to citrate synthase (figure 4, B and C).
Figure 4. Mitochondrial activities in cells with the m.15992A>T mutation.

White bars = values obtained in control cells (C); black bars = values obtained in mutant cells (MT-TP) *p < 0.05, **p < 0.01 compared with the control group using the Mann-Whitney test or t test when data had a normal distribution according to the Shapiro-Wilk test. (A) Respiration rates measured in an Oroboros high-resolution respirometer and expressed as pmol·O2·sec−1·million cells−1. Data shown as mean and SEM of 12 independent measurements for mutant fibroblasts (4 mutant lines derived from patients 1, 2, 3, and 4, each assayed 3 times), 50 for control fibroblasts (14 different control lines), 8 for mutant cybrids (6 different mutant lines derived from 2 different fibroblasts lines), and 15 for control cybrids (derived from 6 different control fibroblasts lines); basal = basal respiration calculated by subtracting the nonrespiratory oxygen consumption (with 1 mM KCN) from the respiration in culture medium with 1g/L glucose; ATP = respiration linked to ATP production calculated by subtracting the respiration under complete inhibition of ATP synthase by 1 µM oligomycin from the respiration in culture medium with 1 g/L glucose; Max = maximal respiratory capacity calculated by subtracting the nonrespiratory oxygen consumption (with 1 mM KCN) from the maximal respiration observed after uncoupling with successive additions of the protonophore carbonyl cyanide m-chlorophenyl hydrazone. (B) Maximal velocities analyzed by spectrophotometric assays on frozen mitochondrial pellets and expressed as nanomoles·min−1·mg cellular proteins−1. Data shown as mean and SEM of 8 independent measurements for mutant fibroblasts (4 mutant lines, each assayed twice), 9 for control fibroblasts (6 control lines), 7 for mutant cybrids (5 mutant lines), and 7 for control cybrids (6 different control lines); CI = respiratory complex I (rotenone sensitive NADH ubiquinone oxidoreductase activity); CII = respiratory complex II (succinate ubiquinone oxidoreductase activity); CIII = respiratory complex III (antimycin sensitive ubiquinol cytochrome c oxidoreductase activity); CIV = respiratory complex IV (cytochrome c oxidase activity); CV = ATP synthase (oligomycin-sensitive ATP hydrolysis activity); CS = citrate synthase. (C) Oxidative phosphorylation pathway (OXPHOS) maximal velocities normalized to the citrate synthase activity of the sample.
Cybrid clones, derived from patient 2 and patient 4 fibroblasts, had homoplasmic m.15992A>T mutation (figure 3). They showed decreased respiration, either basal, linked to ATP production, or maximal (figure 4A). As in fibroblasts, cybrids had decreased activity for all the mtDNA-depending complexes, reaching significance for respiratory complex III (figure 4, B and C). The amount of the respiratory complexes appeared normal when analyzed by Western blot after blue native polyacrylamide gel electrophoresis (figure e-1B, links.lww.com/NXG/A279).
Mutant cells disclosed significantly slower proliferation than control cells in the absence of glucose, whereas their proliferation rate was identical in its presence (figure 5A). The mild oxidative phosphorylation pathway (OXPHOS) defect was therefore relevant, affecting the cell proliferation capacity in a medium without glucose.25,28,29 It had no apparent impact on the mitochondrial production of superoxide ion, which was similar in mutant and control cybrids in the basal state (figure 5B).30
Figure 5. Functional impact of the m.15992A>T mutation outside OXPHOS pathway.

WT = wild-type cybrids; MT-TP = cybrids with 100% m.15992A>T mutation; *p < 0.05, **p < 0.01, ***p < 0.001 compared with the control group using the Mann-Whitney test or t test when data had a normal distribution according to the Shapiro-Wilk test. (A) Proliferation assay in the presence of 1 g/L glucose (glu) or in its absence but in the presence of 200 µM glutamine (no glu). Number of experiments: 9 independent assays on 3 different control cybrid lines and 7 independent assays on 6 different mutant cybrid lines. (B) Evaluation of the superoxide ion production using the slope of MitoSOX fluorescence. Results shown as a box plot with the median, the quartile, and the centile as well as internal dots; number of experiments: 10 independent assays on 5 different control cybrid lines and 5 independent measurements on 5 different cybrid lines with 100% m.15992A>T mutation. (C) Cell size evaluation using the FSC given by the Accuri 6 flow cytometer. The FSC values in the basal state (i.e., in normal culture medium with TMRE and verapamil as explained in the methods section) are given in million for the sake of readability. The FSC values in the presence of 80 mM external K are given as % of the cells FSC in the basal state. (D) Evaluation of the plasma potential using the fluorescence signal of TMRE measured in channel A (FLA) of the Accuri 6 flow cytometer. The FLA values in the basal state are given in hundreds thousands for the sake of readability. The FLA values in the presence of 80 mM external K are given as % of the cells FLA in the basal state. FSC = forward scatter; TMRE = tetramethylrhodamine ethyl esther.
The mutant tRNAPro had a lower steady-state but normal aminoacylation
Northern blot analysis in several cell lines revealed significant decrease of the amount of the mitochondrial tRNAPro in all the mutant cell lines, fibroblasts, or cybrids (figure e-2A, links.lww.com/NXG/A280). Cybrids also disclosed an increase of tRNAVal, which could suggest an increase in the mitochondrial ribosomes because tRNAVal is one of their integral components.31
In accordance, mitochondrial translation, studied in 2 patients' fibroblast lines by [35S]-methionine pulse-chase incorporation, showed a moderately decreased translation of most mtDNA-encoded proteins (figure e-2B, links.lww.com/NXG/A280).
As expected, because the mutated base was not known as involved in the prolyl-tRNA synthetases recognition of the tRNAPro, the in vivo aminoacylation state of the mutant tRNA appeared as normal17 (figure e-3A, links.lww.com/NXG/A281).
Because tRNAPro is encoded in the light mtDNA strand, its sequence is in the reverse sense. Mutation m.159992A>T thus replaces the wild-type uridine in the tRNA sequence by an adenine at the wobble position of the anticodon. A nonmodified adenine in the wobble position of tRNA is very rare and is almost always deaminated into inosine to improve the decoding process.32 We investigated whether that was the case for the mutant mitochondrial tRNAPro, using the fact that inosine is read as guanosine during the RT-PCR reaction.19 In both fibroblasts and cybrids, we could not detect any trace of inosine in the mutant mitochondrial tRNAPro (figure e-3B, links.lww.com/NXG/A281). This result fitted with the absence of the needed enzymatic apparatus in mitochondria.33
Abnormal decrease of the cell volume in mutant cybrids on depolarization by external potassium was reminiscent of the exercise-induced muscle edema observed in patients
Ionic disturbances were a striking observation in 2 families with the m.15992A>T mutation. They were reminiscent of ionic disturbances observed in patients with homoplasmic MT-ATP6 mutations and recurrent paralysis episodes associated with significant plasma membrane depolarization in fibroblasts.22 Core facilities providing patch-clamp electrophysiologic analyses are scarce. Therefore, to analyze the plasma membrane potential in cybrids, we used an indirect determination based on TMRE, a fluorescent probe that follows Nernst equation and thus may be used to analyze membrane potentials.34 Because 7.5 µM TMRE significantly decreases respiration,34 the mitochondrial membrane potential does not influence the TMRE fluorescence signal, which thus essentially represents both the cell size and plasma membrane potential. As expected, increasing in isosmotic conditions the external K concentration from 5 to 10, 20, 30, 40, and 80 mM led to progressive decrease of TMRE fluorescent signal to 91 ± 9, 78 ± 12, 73 ± 7, 61 ± 9, and 38 ± 10% of its initial value, respectively (analysis of 33 independent cell populations, mutant and wild type grouped). At the basal state, the size (evaluated by the forward scatter) and the TMRE fluorescence (evaluated by the fluorescence area FLA) did not differ between wild-type and mutant cybrids with either the m.15992A>T mutation or the m.9185T>C mutation previously associated with permanent plasma membrane depolarization22 (figure 5, C and D). In contrast, at 80 mM external K concentration, both types of mutant cybrids had significantly decreased their size, whereas wild-type cybrids had maintained a size similar to the basal state (figure 5C). All 3 cybrid types had decreased their TMRE signal, in accordance with the induced decrease of plasma membrane potential, but mutant cybrids to a level significantly lower than wild-type cybrids.
Discussion
In this article, we demonstrate the pathogenicity of an original homoplasmic MT-TP mutation targeting skeletal muscle. In muscle, the severe combined respiratory chain defect demonstrated the mtDNA origin of the disease. However, we could not exclude a nuclear DNA alteration solely affecting muscle mtDNA expression because the absence of paternal transmission by the few affected fathers was insufficient to demonstrate maternal inheritance. Therefore, we had to provide demonstration of the mutation deleterious potential.
As observed with other deleterious homoplasmic mtDNA mutations with restricted clinical expression, fibroblasts and cybrids presented with very mild enzymatic defect.6,35 However, that defect was statistically significant with unbiased nonparametric statistical tests. In addition, it induced significant reduction of the mutant cells proliferation in the absence of glucose showing its relevance to cell physiology.36 Because most organs essentially comprise postmitotic cells, cell proliferation is not relevant for clinical symptoms. Therefore, we addressed the ionic disturbances that were a striking aspect of the disease, prominent in 2 families. When faced to an isosmotic high potassium concentration, inducing plasma membrane depolarization, cybrids with the MT-TP mutation differed from wild-type cybrids by losing part of their cell volume, most probably through loss of internal fluid. That behavior fitted with the edema observed in patients using muscle imaging after 15-minute chewing effort.
At the molecular level, the mutant mitochondrial tRNAPro had a reduced steady-state level in cells. It bore an unmodified adenosine at the wobble position of its anticodon, which thus should optimally recognize only codon CCU that represents less than 25% of the total mtDNA proline codons. Special mitochondrial decoding rules probably allowed the mutant tRNA to recognize all the 4 proline codons, but less efficiently. These 2 anomalies (reduced tRNA steady-state and decoding problems) might have a cumulative effect on mitochondrial translation. Why the impact on OXPHOS was mild in fibroblasts and cybrids but drastic in muscle remained hypothetical in the absence of appropriate muscle fragments.
The role of the J1c10 haplotype in the mutation pathophysiology was disputable. That haplotype was constant in the 5 families here reported and in the sole GenBank report of the mutation. It is not pathogenic in itself, being observed in 2.5%–5% of the people living in the western half of France, but it is a significant modifier in Leber hereditary optic neuropathy37,38 and in the cybrid resistance to rotenone toxicity.39
According to the pathogenicity scoring system for tRNA mutations updated in 2011,7 mutation m.159992A>T has a score of 13, which indicated a mutation definitely pathogenic (2 for independent pedigrees, 2 for phylogenetic conservation, 2 for muscle histology, 2 for biochemical defects, and 5 for cybrid analysis).
The homoplasmic m.15992A>T MT-TP mutation in the J1c10 haplotype causes severe intolerance to exercise, often associated with exercise-induced muscle edema.
Acknowledgment
The authors thank Frédéric Bouillaud, INSERM U1016, for his insightful discussion of the data; Dr. Armelle Magot, Centre de Référence Maladies Neuromusculaires AOC Hôtel-Dieu, CHU Nantes, for providing the results of patient 6 muscle biopsy; and Rafael Boucher, Clément Vaz, and Caroline L'Hermitte-Stead, INSERM U1016, for their efficient technical help.
Glossary
- mtDNA
mitochondrial DNA
- mt-tRNA
mitochondrial transfer RNA
- STIR
short-tau inversion recovery
- TMRE
tetramethylrhodamine ethyl esther
Appendix 1. Authors
Study funding
No targeted funding reported.
Disclosure
A. Lombès and I. Tarassov are financially supported by INSERM and CNRS. A. Lombès received grants from the Fondation pour la Recherche Médicale (FRM) (grant DPM20121125550), the AFM Telethon, and the AMMi (Association contre les Maladies Mitochondriales). I. Tarassov received financial support from the University of Strasbourg, the LabEx MitoCross, and the Graduate School IMCBio (National Program PIA, Programme Investissement d'Avenir). Go to Neurology.org/NG for full disclosures.
References
- 1.Craven L, Alston CL, Taylor RW, Turnbull DM. Recent advances in mitochondrial disease. Annu Rev Genomics Hum Genet 2017;18:257–275. [DOI] [PubMed] [Google Scholar]
- 2.Shanske S, Moraes CT, Lombès A, et al. Widespread tissue distribution of mitochondrial DNA deletions in Kearns-Sayre syndrome. Neurology 1990;40:24–28. [DOI] [PubMed] [Google Scholar]
- 3.Carelli V, Giordano C, d'Amati G. Pathogenic expression of homoplasmic mtDNA mutations needs a complex nuclear-mitochondrial interaction. Trends Genet 2003;19:257–262. [DOI] [PubMed] [Google Scholar]
- 4.Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science 1988;242:1427–1430. [DOI] [PubMed] [Google Scholar]
- 5.Prezant TR, Agapian JV, Bohlman MC, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet 1993;4:289–294. [DOI] [PubMed] [Google Scholar]
- 6.Taylor RW, Giordano C, Davidson MM, et al. A homoplasmic mitochondrial transfer ribonucleic acid mutation as a cause of maternally inherited hypertrophic cardiomyopathy. J Am Coll Cardiol 2003;41:1786–1796. [DOI] [PubMed] [Google Scholar]
- 7.Yarham JW, Al-Dosary M, Blakely EL, et al. A comparative analysis approach to determining the pathogenicity of mitochondrial tRNA mutations. Hum Mutat 2011;32:1319–1325. [DOI] [PubMed] [Google Scholar]
- 8.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science 1989;246:500–503. [DOI] [PubMed] [Google Scholar]
- 9.Jun AS, Trounce IA, Brown MD, Shoffner JM, Wallace DC. Use of transmitochondrial cybrids to assign a complex I defect to the mitochondrial DNA-encoded NADH dehydrogenase subunit 6 gene mutation at nucleotide pair 14459 that causes leber hereditary optic neuropathy and dystonia. Mol Cell Biol 1996;16:771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toompuu M, Tiranti V, Zeviani M, Jacobs HT. Molecular phenotype of the np 7472 deafness-associated mitochondrial mutation in osteosarcoma cell cybrids. Hum Mol Genet 1999;8:2275–2283. [DOI] [PubMed] [Google Scholar]
- 11.Fender A, Sissler M, Florentz C, Giege R. Functional idiosyncrasies of tRNA isoacceptors in cognate and noncognate aminoacylation systems. Biochimie 2004;86:21–29. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe K. Unique features of animal mitochondrial translation systems. The non-universal genetic code, unusual features of the translational apparatus and their relevance to human mitochondrial diseases. Proc Jpn Acad Ser B Phys Biol Sci 2010;86:11–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hogrel JY, Laforêt P, Ben Yaou R, Chevrot M, Eymard B, Lombès A. A non-ischemic forearm exercise test for the screening of patients with exercise intolerance. Neurology 2001;56:1733–1738. [DOI] [PubMed] [Google Scholar]
- 14.Dubowitz V, Sewry CA, Oldfors A. Muscle Biopsy: A Practical Approach. Philadelphia, PA: Saunders Elsevier; 2013. [Google Scholar]
- 15.Possekel S, Lombès A, Ogier de Baulny H, et al. Immunohistochemical analysis of muscle cytochrome c oxidase deficiency in children. Histochem 1995;103:59–68. [DOI] [PubMed] [Google Scholar]
- 16.Agier V, Oliviéro P, Lainé J, et al. Defective mitochondrial fusion, altered respiratory function, and distorted cristae structure in skin fibroblasts with heterozygous OPA1 mutations. Biochim Biophys Acta 2012;1822:1570–1580. [DOI] [PubMed] [Google Scholar]
- 17.Varshney U, Lee CP, RajBhandary UL. Direct analysis of aminoacylation levels of tRNAs in vivo. Application to studying recognition of Escherichia coli initiator tRNA mutants by glutaminyl-tRNA synthetase. J Biol Chem 1991;266:24712–24718. [PubMed] [Google Scholar]
- 18.Karicheva OZ, Kolesnikova OA, Schirtz T, et al. Correction of the consequences of mitochondrial 3243A>G mutation in the MT-TL1 gene causing the MELAS syndrome by tRNA import into mitochondria. Nucleic Acids Res 2011;39:8173–8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawahara Y. Quantification of adenosine-to-inosine editing of microRNAs using a conventional method. Nat Protoc 2012;7:1426–1437. [DOI] [PubMed] [Google Scholar]
- 20.Pesta D, Gnaiger E. High-resolution respirometry: OXPHOS protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol Biol 2012;810:25–58. [DOI] [PubMed] [Google Scholar]
- 21.Medja F, Allouche S, Frachon P, et al. Development and implementation of standardized respiratory chain spectrophotometric assays for clinical diagnosis. Mitochondrion 2009;9:331–339. [DOI] [PubMed] [Google Scholar]
- 22.Auré K, Dubourg O, Jardel C, et al. Episodic weakness due to mitochondrial DNA MT-ATP6/8 mutations. Neurology 2013;81:1810–1818. [DOI] [PubMed] [Google Scholar]
- 23.Haraux F, Lombes A. Kinetic analysis of ATP hydrolysis by complex V in four murine tissues: towards an assay suitable for clinical diagnosis. PLoS One 2019;14:e0221886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Auré K, Fayet G, Leroy JP, Lacène E, Romero NB, Lombès A. Apoptosis in mitochondrial myopathies is linked to mitochondrial proliferation. Brain 2006;129:1249–1259. [DOI] [PubMed] [Google Scholar]
- 25.Auré K, Mamchaoui K, Frachon P, Butler-Browne GS, Lombès A, Mouly V. Impact on oxidative phosphorylation of immortalization with the telomerase gene. Neuromuscul Disord 2007;17:368–375. [DOI] [PubMed] [Google Scholar]
- 26.Repetto G, del Peso A, Zurita JL. Neutral red uptake assay for the estimation of cell viability/cytotoxicity. Nat Protoc 2008;3:1125–1131. [DOI] [PubMed] [Google Scholar]
- 27.Yasukawa T, Suzuki T, Ishii N, Ohta S, Watanabe K. Wobble modification defect in tRNA disturbs codon-anticodon interaction in a mitochondrial disease. EMBO J 2001;20:4794–4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem 1979;254:2669–2676. [PubMed] [Google Scholar]
- 29.Will Y, Dykens J. Mitochondrial toxicity assessment in industry—a decade of technology development and insight. Expert Opin Drug Metab Toxicol 2014;10:1061–1067. [DOI] [PubMed] [Google Scholar]
- 30.Quinlan CL, Gerencser AA, Treberg JR, Brand MD. The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J Biol Chem 2011;286:31361–31372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rorbach J, Gao F, Powell CA, et al. Human mitochondrial ribosomes can switch their structural RNA composition. Proc Natl Acad Sci U S A 2016;113:12198–12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grosjean H, de Crecy-Lagard V, Marck C. Deciphering synonymous codons in the three domains of life: co-evolution with specific tRNA modification enzymes. FEBS Lett 2010;584:252–264. [DOI] [PubMed] [Google Scholar]
- 33.de Crecy-Lagard V, Boccaletto P, Mangleburg CG, et al. Matching tRNA modifications in humans to their known and predicted enzymes. Nucleic Acids Res 2019;47:2143–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scaduto RC Jr, Grotyohann LW. Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys J 1999;76:469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Howell N. Leber hereditary optic neuropathy: how do mitochondrial DNA mutations cause degeneration of the optic nerve? J Bioenerg Biomembr 1997;29:165–173. [DOI] [PubMed] [Google Scholar]
- 36.Rossignol R, Letellier T, Malgat M, Rocher C, Mazat JP. Tissue variation in the control of oxidative phosphorylation: implication for mitochondrial diseases. Biochem J 2000;347(pt 1):45–53. [PMC free article] [PubMed] [Google Scholar]
- 37.Carelli V, Achilli A, Valentino ML, et al. Haplogroup effects and recombination of mitochondrial DNA: novel clues from the analysis of Leber hereditary optic neuropathy pedigrees. Am J Hum Genet 2006;78:564–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hudson G, Carelli V, Spruijt L, et al. Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am J Hum Genet 2007;81:228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Strobbe D, Caporali L, Iommarini L, et al. Haplogroup J mitogenomes are the most sensitive to the pesticide rotenone: relevance for human diseases. Neurobiol Dis 2018;114:129–139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Any data not published within the article will be shared, as anonymized data, by request from any qualified investigator.