

Abstract
Objective
To define the phenotypic spectrum of isolated sulfite oxidase (ISOD) and molybdenum cofactor deficiency (MoCD), aiming to promote timely diagnosis and assist in future clinical trial design.
Methods
We analyzed clinical, radiographic, biochemical, and genetic data from 146 patients reported in the literature.
Results
We stratified patients into 2 phenotypic subgroups based on clinical and radiographic characteristics. In the first (Class I), patients presented early in life (age 1–50 days) with acute onset of neurologic symptoms and development of diffuse brain injury with cystic leukomalacia. Patients in the second subgroup (Class II) presented later in life (age 30 days–23 years) with prominent movement abnormalities and selective injury of the basal ganglia and cerebellum. A significant difference in survival estimates correlated with milder disease severity among Class II patients. Substantial overlap in sulfur-containing metabolite levels prevented discrimination of subgroups based on diagnostic biomarkers, but genotype-phenotype correlations suggested that residual SUOX activity may contribute to milder phenotypes.
Conclusions
Patients with SUOX and MoCD gravitate toward 1 of 2 distinct clinicoradiographic profiles. Patient stratification may help promote accurate diagnosis, prognostication, and aid in the design of future clinical trials.
Isolated sulfite oxidase deficiency (ISOD) and molybdenum cofactor deficiency (MoCD) are inherited neurodegenerative conditions that comprise the known disorders of sulfite metabolism.1 Sulfite oxidase (SUOX) is a molybdenum cofactor–dependent enzyme, and loss of its activity is necessary and sufficient to cause the neurodegenerative phenotype of ISOD or MoCD. Although patients with ISOD carry biallelic pathogenic variants in SUOX, patients with MoCD harbor pathogenic variants in one of the molybdenum cofactor synthetic enzymes, MOCS1, MOCS2, gephyrin (GPHN), or MOCS3.
Classically, patients with ISOD and MoCD present in the first days of life with clinicoradiographic features that mimic neonatal hypoxic-ischemic encephalopathy.2–4 Milder phenotypic variants have also emerged in scattered case reports; however, a comprehensive analysis of phenotypic variants has not been performed, and the phenotypic spectrum of disease remains poorly defined.
Advances in therapeutic options for ISOD and MoCD have produced an urgent need to delineate the phenotypic spectrum of patients, which will be essential in guiding clinical trial design.5–7 Low patient numbers have been a major hurdle to studying these ultra-rare disorders. To overcome this limitation, we leveraged the clinicoradiographic congruency of ISOD and MoCD and systematically evaluated a cohort of 146 published patients from both disorders. We directed our efforts at defining the clinicoradiographic features that distinguish phenotypic subgroups and investigated subgroup associated differences in survival, age at neurologic symptom onset, diagnostic biomarker levels, and genotype-phenotype correlations.
Methods
Literature review and definition of variables
We performed literature searches on PubMed using the terms “sulfite oxidase deficiency” and “molybdenum cofactor deficiency,” encompassing the time frame up to July 2017. Searches were conducted in August of 2017. We excluded articles with insufficient data. We also excluded articles that only described MoCD type A patients treated with cyclic pyranopterin monophosphate (cPMP), as this treatment could alter the natural history of disease. In total, we identified 93 articles (published 1967–2017) for review (figure e-1, links.lww.com/NXG/A284, and table e-1, links.lww.com/NXG/A286). From those reports, we identified 146 patients for analysis after excluding duplicate reports of the same patient and additional MoCD type A patients treated with cPMP (table e-1). Patients with MoCD type B who received cPMP therapy were included as the treatment should not affect the underlying metabolic defect.
We extracted the following variables: age at onset of neurologic symptoms, age at the time of report, age at death, mode of diagnosis, ethnicity of the patient (only if explicitly stated), examination findings, brain MRI results, urine and plasma levels of sulfite, S-sulfocysteine (SSC), thiosulfate, taurine, l-cysteine/l-cystine, xanthine, and uric acid, and genetic diagnoses. We defined the age at onset of neurologic symptoms as the age at which seizures, infantile spasms, hyperekplexia, appendicular hypertonia, dystonia, opisthotonos, extrapyramidal signs not otherwise specified, ataxia, or acute hemiparesis were first observed or diagnosed or when the patient first presented with concerns for developmental delay. Plasma metabolite levels were all converted to μmol/L, and urine metabolite levels converted to mmol/molCr (urine). Quantitative measurements reported below the threshold of detection were recorded as a value of zero. Plasma metabolite levels normalized to creatinine or urine metabolite values not normalized to creatinine were excluded.
To minimize bias, brain MRI findings were only recorded as “present” if explicitly described or identifiable in published radiographic images. If information regarding time was semiquantitative, we took a conservative approach and used the following definitions: “at birth” = day 1, “first week” = day 7, and “first year” = 1 year.
Statistical analysis
Patient demographics, clinical characteristics, and radiographic findings were summarized descriptively. Survival was defined as the time elapsed between birth and time of death. Patients were censored at the age at the time of report if they were still alive. We used the Kaplan-Meier method to describe survival, and the log-rank test was used to assess differences in survival between patient subgroups. To compare metabolite levels between subgroups, we used single values from each patient. If multiple values were reported, we used the average. Measurements made during or after dietary restriction of cysteine and/or methionine or chelation therapy were excluded. Because of the low number of data points for each metabolite, normality of the sample populations could not be assessed. Median values for each metabolite were calculated for Class I and Class II subgroups. We calculated the power of a nonparametric Mann-Whitney test to detect the difference in median values and only considered the test acceptable if the power was ≥0.8. To calculate Spearman correlation coefficients between metabolite levels, we pooled concomitant measurements of multiple metabolites made in individual patients across the entire cohort. Multiple measurements from the same patient made during or after dietary restriction of methionine and/or cysteine were included to capture dynamic, synchronous changes between different metabolites. We performed all statistical analysis with Prism 8, GraphPad software.
Data availability
All data extracted from the literature that was used for the analyses reported in this article will be made freely available on request from the corresponding author. The data will be available for 5 years from the time of publication and may be requested by any clinician or researcher.
Results
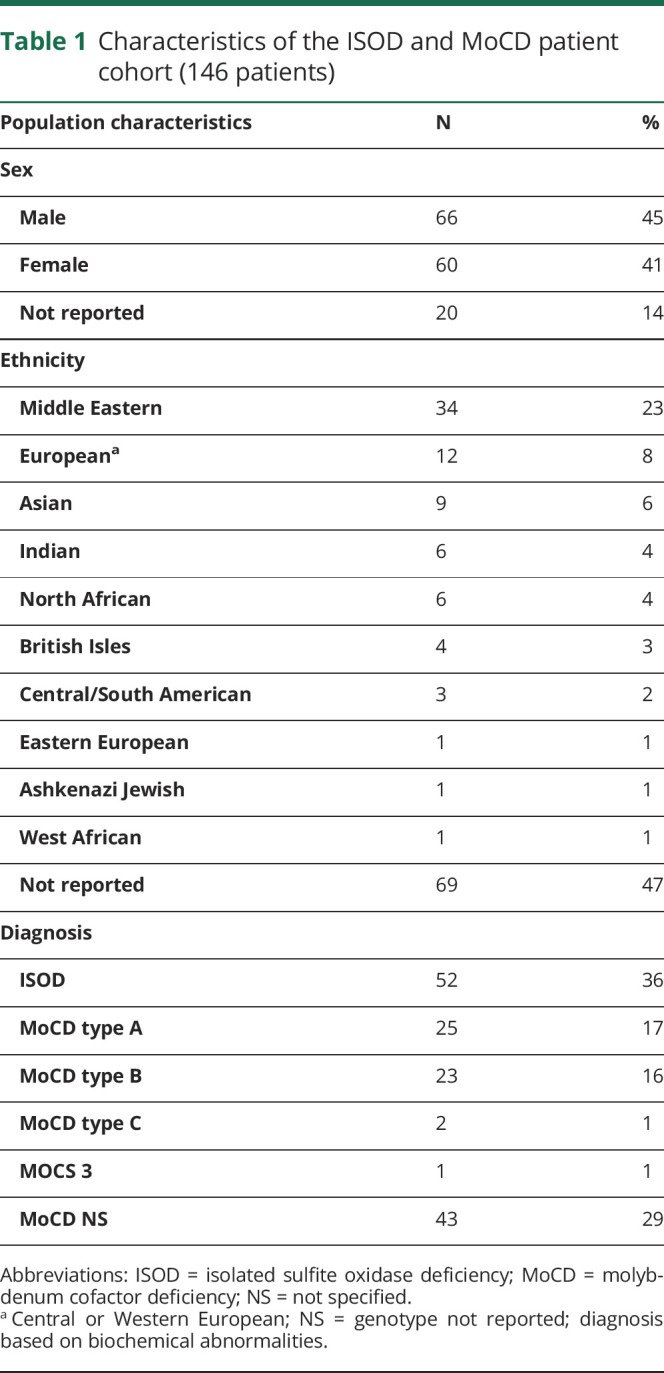
Demographic information and diagnoses for the patient cohort are listed in table 1. The prevalence of disease was roughly equal between sexes, and patients originated from a pan-ethnic distribution. Because available data varied between reports, patients had to be excluded from specific analyses. The corresponding sample size (N) is indicated with each analysis.
Table 1.
Characteristics of the ISOD and MoCD patient cohort (146 patients)
Confirmation of diagnosis
A diagnosis of ISOD or MoCD was confirmed by genetic sequencing of SUOX (ISOD; OMIM:606887), MOCS1 (MoCD type A; OMIM:603707), MOCS2 (MoCD type B; OMIM:603708), MOCS3, or GPHN (MoCD type C; OMIM:603930) in 62 (42%) patients, whereas diagnosis based on biochemical abnormalities was made in 84 (58%) patients. Additional biochemical characterization to determine MoCD subtype was not performed in 43 (29%) patients who were labeled MoCD not specified (NS). All MoCD NS patients had elevated levels of xanthine and hypoxanthine and/or low levels of uric acid to rule out ISOD. SUOX activity was assayed in fibroblasts from 49 patients and reported as undetectable in 47 samples. Fibroblasts from an additional 2 patients had measurable SUOX activity.8,9 Diagnosis in these patients was made by sequencing of the GPHN and MOCS3 genes and identifying a pattern of biochemical abnormalities consistent with MoCD.
Stratification of patients by clinicoradiographic presentation
We evaluated our entire cohort and observed that patients strongly gravitated toward 2 unique clinicoradiographic profiles. The first (Class I) embodied patients who presented acutely with abrupt onset of neurologic symptoms and evidence of diffuse brain injury on brain MRI or CT. The second class (Class II) embodied patients who presented with neurologic symptoms that emerged gradually or episodically against a background of normal or mildly delayed development and evidence of region-restricted brain injury on imaging.
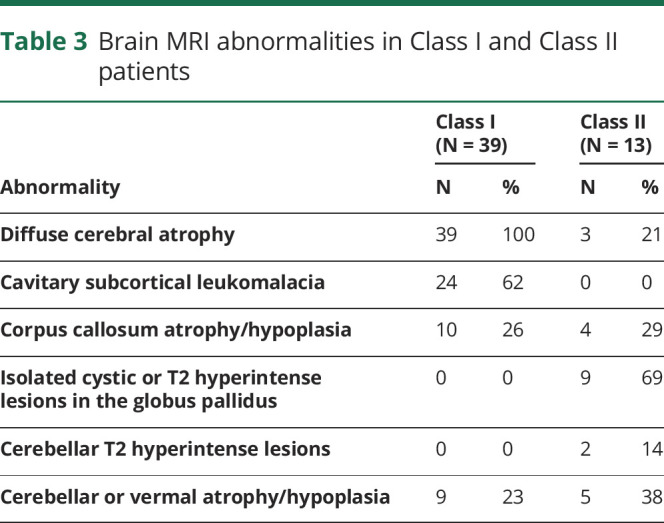
Based on the authors' consensus opinion, we stratified patients into the class that was most consistent with their available data (table e-1, links.lww.com/NXG/A286). A larger portion of patients with ISOD (25%) than patients with MoCD (13%) were stratified into Class II (table e-2, links.lww.com/NXG/A287). The most commonly reported clinical features in Class I and Class II patients are compared in table 2. Brain MRI abnormalities are compared in table 3; these only included MRI abnormalities in Class I patients reported >20 days after their initial neurologic symptom onset, as data from an equivalent acute or subacute phase of disease in Class II patients were lacking. Within each class, the frequency of clinical features and brain MRI abnormalities were similar between patients with ISOD and MoCD, except for atrophy/hypoplasia of the corpus callosum and cerebellum, which were more frequently reported in patients with ISOD (table e-3, links.lww.com/NXG/A288, and table e-4, links.lww.com/NXG/A289).
Table 2.
Clinical features in Class I and Class II patient subgroups
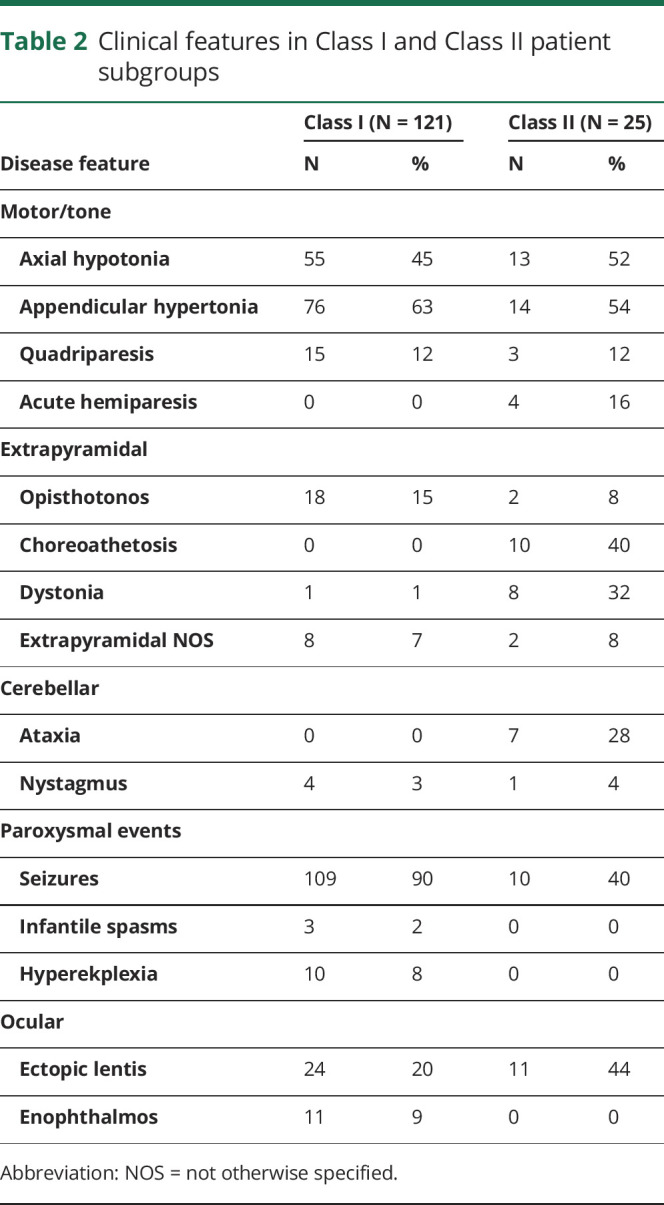
Table 3.
Brain MRI abnormalities in Class I and Class II patients
Age at neurologic symptom onset
The median age at first symptom onset was 1 day (interquartile range: 1–2.5 days) for Class I (N = 113) and 11.5 months (interquartile range: 6 months to 1.5 years) for Class II (N = 25) subgroups. Because age at symptom onset did not affect patient stratification, we found a slight overlap between Class I and Class II patients (figure 1A).
Figure 1. Age at neurologic symptom onset and estimated survival distributions for Class I and Class II patients with isolated sulfite oxidase deficiency and molybdenum cofactor deficiency.
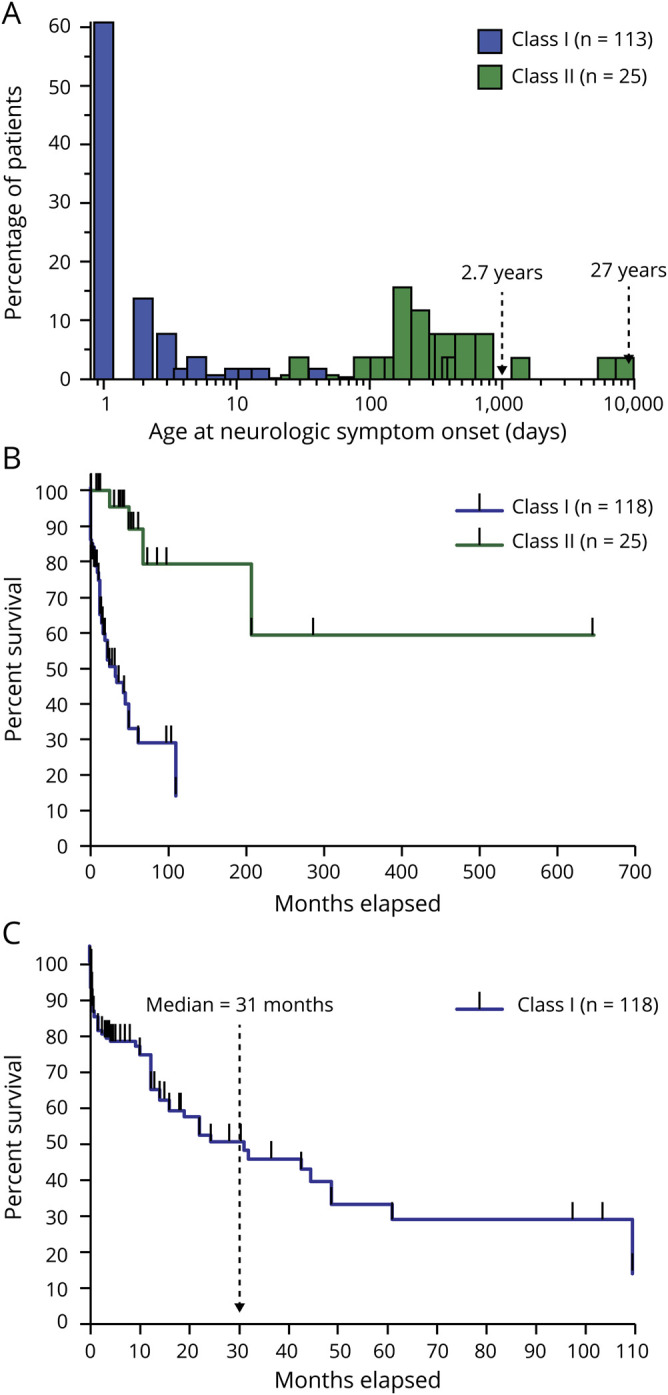
(A) Age at onset of neurologic symptoms for each patient subgroup. The percentage of patients was calculated separately for Class I and Class II. (B) Survival estimates for Class I and Class II subgroups. A tick mark represents censored patients. The median age of survival for Class I patients was 31 months (C). The median age of survival for Class II was not calculated as survival remained above 50%.
Survival estimations
In the Class I subgroup, 68 patients (58%) were alive, and 50 patients (42%) were deceased at the time of publication. In the Class II subgroup, 21 patients (84%) were alive, and 4 (16%) were deceased. The median age of survival for Class I was 2.5 years (N = 118), but it could not be calculated for Class II (N = 25) as survival exceeded 50% at the longest time point (figure 2, A and B). Survival was statistically shorter in the Class I subgroup (p < 0.0001, log-rank test).
Figure 2. Sulfur-containing metabolite levels reported in Class I and Class II patients with isolated sulfite oxidase deficiency and MoCD.

All sulfur metabolite levels measured in urine (A, B, E, and F) and plasma (C and D) showed substantial overlap between Class I and Class II subgroups. Urine xanthine and plasma uric acid levels in patients with MoCD also showed substantial overlap between Class I and Class II patients (G and H). Horizontal bars represent the median value, and brackets encompass the 25th and 75th quartiles. Solid circles indicate a metabolite value outside of the reported reference range, whereas an open circle indicates a value within the reported reference range. Low sample numbers precluded assessment for a statistically significant difference in median values between subgroups. MoCD = molybdenum cofactor deficiency; SSC = S-sulfocysteine.
Biochemical abnormalities
SUOX is a molybdenum cofactor–dependent enzyme that oxidizes sulfite to sulfate in the terminal step of sulfur-containing amino acid and hydrogen sulfide metabolism (figure e-2, links.lww.com/NXG/A285). For a review on these pathways, please see Kohl et al.10 Commonly reported biochemical abnormalities in patients with ISOD and MoCD include elevated levels of urine and plasma sulfite, SSC, thiosulfate, and taurine, whereas l-cystine is diminished. Abnormalities unique to MoCD included elevated xanthine and decreased uric acid levels due to the inactivation of xanthine oxidase (a molybdenum cofactor–dependent enzyme). Across the entire cohort, all extracted laboratory values were consistent with the expected pattern of abnormalities.
Metabolite levels were compared between Class I and Class II subgroups to assess for an indirect correlation with the contrasting survival time. Urine and plasma metabolite levels from Class I and Class II subgroups demonstrated an appreciable overlap (figure 2, A–H). Because of the low number of reported values for Class II patients, we could not test for a significant difference between subgroup medians (for all metabolites, a nonparametric Mann-Whitney test with alpha = 0.05 would have a power <0.5).
Because sulfite and SSC exert direct neurotoxic effects on the CNS, these metabolites are potential biomarkers for assessing treatment efficacy in ISOD and MoCD. Unfortunately, aqueous sulfite is unstable, limiting its utility; however, if sulfite or SSC levels correlate with other sulfur-containing metabolites, additional biomarkers for monitoring treatment efficacy could be developed. To this end, we calculated Spearman correlation coefficients between metabolite levels concomitantly measured in individual patients across our entire cohort. SSC levels in urine and plasma correlated (r = 0.7, 95% confidence interval [CI] = 0.332–0.890, N = 17) as expected. Correlations were also identified between plasma cystine and SSC (urine SSC: r = −0.6, 95% CI = −0.851 to −0.178, N = 17; plasma SSC: r = −0.58, 95% CI = −0.840 to −0.102, N = 16), urine taurine and SSC (urine SSC: r = 0.54, 95% CI = 0.164–0.772, N = 25; plasma SSC: r = 0.88, 95% CI = 0.613–0.968, N = 12), and urine sulfite and urine thiosulfate (r = 0.5, 95% CI = 0.005–0.809, N = 16).
Genotype-phenotype correlations
Genotypes were reported for 52 Class I patients (43%) and 10 Class II patients (40%). Allelic variants uniquely segregated between patient subgroups with 1 exception (table e-5, links.lww.com/NXG/A290). The 38 allelic variants unique to Class I patients included 14 missense (37%), 10 frameshift (26%), 8 nonsense (21%), and 6 splice site (16%) mutations that were predicted to disrupt function of the associated gene product. The 10 allelic variants unique to Class II patients included 7 missense (70%), 1 frameshift (10%), 1 nonsense (10%), and 1 splice site (10%) mutation. The frameshift and nonsense mutations were both identified in patients compound heterozygous for a point mutation in the same gene.11,12 One allelic variant in MOCS1 (R222_G223insR) was found both in a Class I and a Class II patient who were homozygous for the mutation.
Discussion
We discerned 2 subgroups of patients with ISOD and MoCD based on their clinicoradiographic features. These subgroups roughly correspond to the “early-onset” or “severe” (Class I) and “late-onset” or “mild” (Class II) terms that have been variably used in the literature.1,13,14 The validity of our approach is supported by the contrasting survival estimates and genotype-phenotype associations that emerged. Dividing patients with ISOD and MoCD into 2 classes has clinical utility for several reasons. First, emphasizing the unique features of Class II patients will help raise awareness among clinicians and promote diagnosis. Because limited data suggest that dietary restriction of sulfur-containing amino acids may have an impact on neurologic symptoms severity or disease progression in mildly affected patients, an accurate diagnosis would make a potentially beneficial intervention available to patients.8,15–17 Second, clinicoradiographic features may predict neurologic outcomes better than the age at neurologic symptom onset. We identified Class I patients who first presented as late as 40–50 days of life.18,19 These patients exhibited an acute onset of neurologic symptoms and signs of severe forebrain injury that may not have been expected from their later onset. Finally, formal recognition of mild phenotypic variants will assist with patient stratification and identification of appropriate endpoints in future clinical trials.
We observed little phenotypic variation across patients in Class I, and severe neurologic impairment was reported in all patients with available follow-up. The narrow phenotypic spectrum and pattern of injury suggest a global susceptibility of the forebrain and cerebellum driven mainly by genotype. In contrast, we observed a higher degree of phenotypic variability across Class II patients. Reported outcomes ranged from static neurologic symptoms to progressive neurologic impairment with periodic decompensations in function. The wider phenotypic spectrum and pattern of neurologic injury in these patients suggest a selective susceptibility of the basal ganglia and cerebellum driven by a complex interaction of genotype, environment, and developmental context.
Despite a limited sample size, we were able to appreciate an association between genotypes and patient subgroups. Extensive analyses of disease-associated missense mutations in SUOX have been performed.20–24 Karakas and Kisker place these variants into 3 categories: (1) mutations affecting the active site, (2) mutations destabilizing coordination of the molybdenum cofactor, and (3) mutations disrupting dimerization. The mutations associated with Class I patients with ISOD fit into these categories and are predicted to abolish SUOX activity. In contrast, the L61P25 and H143N15 mutations found in 2 Class II patients with ISOD lie in the hydrophobic region of the mitochondrial targeting domain and cytochrome-b5 heme-binding domain, respectively. These variants may retain some enzymatic activity explaining the milder associated phenotypes. This notion is supported by the identification of the R217Q (previously R160Q) mutation in 2 Class II patients with ISOD. The R217Q mutation in SUOX modifies the binding site for sulfite,26 but retains residual activity.27
Similar structural analyses have been performed for the molybdenum cofactor synthetic enzymes, and data suggest that variants associated with Class I patients with MoCD abolish enzymatic activity.28–34 While this article was in preparation, an atypical a case of MoCD type A was reported in which a novel MOCS1 mutation (S442 fs) resulted in the translation of a truncated peptide that was expressed at low levels but retained catalytic activity.35 This patient had a mild clinical course that could have resulted from SUOX activity supported by residual molybdenum cofactor production.
Despite the observed associations between genotype and patient subgroups, it is important to note that genotype does not fully explain reported phenotypes. Disease severity can significantly vary between affected siblings, highlighting the contribution of the environment or unappreciated differences in genetic background.11,14,36
We found a large degree of overlap in metabolite levels between subgroups. Low sample numbers precluded our ability to test for significant differences between median metabolite values; however, the overlap observed is sufficient to suggest that metabolite levels alone may not directly correlate with disease severity. Data currently support a disease model in which sulfite and SSC are mainly produced in peripheral tissues and then accumulate in the CNS, where they directly injury neural tissue.6,7 To interpret the prognostic significance of circulating metabolite levels, a better understanding of their origin(s), distribution kinetics, and quantitative relationship to CNS damage are still needed.
Despite a limited understanding of how plasma and urine levels of sulfite and SSC relate to the degree of CNS injury, their utility as biomarkers to assess treatment efficacy is strongly implied by their direct role in disease pathogenesis. SSC is a stable in solution, while aqueous sulfite rapidly oxidizes to sulfate, limiting its utility as a clinical biomarker. Our analysis showed that urine sulfite correlated with plasma cystine and urine thiosulfate levels. As these molecules are stable in solution, they may act as proxy measures of sulfite. Moreover, analyzing combinations of metabolites in relation to disease severity may prove more efficacious in predicting treatment efficacy than single metabolites alone. Clinical trials with cPMP substitution therapy should generate large data sets of sulfur metabolite levels necessary for deeper analysis in the future.
Several important limitations are inherent in the design of our study. First, it was not possible to measure the true frequency of a clinical or radiographic finding based on a collection of reports originating from different institutions. Variations in clinical examinations or the specific focus of a case report could influence which disease features were reported, thereby introducing an ascertainment bias. Different imaging protocols and MRI machines between institutions and the lack of blinded interpretation also obscure the true frequency of radiographic abnormalities. Furthermore, the quality and completeness of clinical data varied between case reports, and it was often not possible to conclude whether a clinical or radiographic feature was absent vs not reported in the article. As such, we analyzed the frequency with which a symptom or radiographic finding was reported, although we believe that this still holds value as a proxy measure of true frequency.
Several factors could have influenced the accuracy of survival estimates. First, the publications included in our cohort ranged from 1967 to 2017, a period over which technology and the quality of medical substantially evolved, potentially improving outcomes in more recently reported patients. Second, our cohort consisted of patients around the world. Although this enriched our data pool, regional differences in standard of care may have influenced outcomes. Finally, patient deaths were not frequently reported. This particularly affected analysis of the Class II subgroup where patient survival remained above 50%, precluding estimation of median survival time.
Because we pooled sulfur metabolite values that were measured in different laboratories, there is an element of interlaboratory variability in our analysis that we are unable to quantify. Future prospective studies centered on assessing the efficacy of experimental treatments will be well poised to centralize the measurement of metabolite levels and provide a more accurate comparison of levels between patient subgroups.
In this report, we provide a quantitative analysis of phenotypic subgroups in ISOD and MoCD. These results have diagnostic and prognostic implications that may help guide clinical care and inform the design of future clinical trials.
Acknowledgment
The authors thank Dr. Guenter Schwarz for his insight and guidance on this project and Alexa Puleio for her help with data extraction. The research reported in this publication was supported by the NINDS of the NIH under awards 1K12NS098482-01 and 5R25NS065743-09. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Glossary
- CI
confidence interval
- cPMP
cyclic pyranopterin monophosphate
- GPHN
gephyrin
- ISOD
isolated sulfite oxidase deficiency
- MoCD
molybdenum cofactor deficiency
- NS
not specified
- SSC
S-sulfocysteine
- SUOX
sulfite oxidase
Appendix. Authors
Study funding
Research reported in this publication was supported by the NINDS of the NIH under awards 1K12NS098482-01 and 5R25NS065743-09.
Disclosure
A.L. Misko consults for Origin Biosciences, a pharmaceutical company developing cPMP substitution therapy for patients with molybdenum cofactor deficiency type A. This study does not address the effects or efficacy of cPMP therapy and was not supported by commercial funding. Y. Liang and J.B. Kohl do not have any conflicts to disclose. F. Eichler has participated in an advisory board for Origin Biosciences. Go to Neurology.org/NG for full disclosure.
References
- 1.Johnson JL, Duran M. Molybdenum cofactor deficiency and isolated sulfite oxidase deficiency. In: Beaudet AL, Vogelstein B, Kinzler KW, et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: The McGraw-Hill Companies, Inc.; 2014. Available at: ommbid.mhmedical.com/content.aspx?aid=1102891947. [Google Scholar]
- 2.Eichler F, Tan W-H, Shih VE, Grant PE, Krishnamoorthy K. Proton magnetic resonance spectroscopy and diffusion-weighted imaging in isolated sulfite oxidase deficiency. J Child Neurol 2006;21:801–805. [DOI] [PubMed] [Google Scholar]
- 3.Bakker HD, Abeling NG, ten Houten R, et al. Molybdenum cofactor deficiency can mimic postanoxic encephalopathy. J Inherit Metab Dis 1993;16:900–901. [DOI] [PubMed] [Google Scholar]
- 4.Hobson EE, Thomas S, Crofton PM, Murray AD, Dean JCS, Lloyd D. Isolated sulphite oxidase deficiency mimics the features of hypoxic ischaemic encephalopathy. Eur J Pediatr 2005;164:655–659. [DOI] [PubMed] [Google Scholar]
- 5.Veldman A, Santamaria-Araujo JA, Sollazzo S, et al. Successful treatment of molybdenum cofactor deficiency type A with cPMP. Pediatrics 2010;125:e1249–e1254. [DOI] [PubMed] [Google Scholar]
- 6.Kügler S, Hahnewald R, Garrido M, Reiss J. Long-term rescue of a lethal inherited disease by adeno-associated virus-mediated gene transfer in a mouse model of molybdenum-cofactor deficiency. Am J Hum Genet 2007;80:291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hahnewald R, Wegner W, Reiss J. AAV-mediated gene therapy for metabolic diseases: dosage and reapplication studies in the molybdenum cofactor deficiency model. Genet Vaccines Ther 2009;7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huijmans JGM, Schot R, de Klerk JBC, et al. Molybdenum cofactor deficiency: identification of a patient with homozygote mutation in the MOCS3 gene. Am J Med Genet A 2017;173:1601–1606. [DOI] [PubMed] [Google Scholar]
- 9.Reiss J, Lenz U, Aquaviva-Bourdain C, Joriot-Chekaf S, Mention-Mulliez K, Holder-Espinasse M. A GPHN point mutation leading to molybdenum cofactor deficiency. Clin Genet 2011;80:598–599. [DOI] [PubMed] [Google Scholar]
- 10.Kohl JB, Mellis A-T, Schwarz G. Homeostatic impact of sulfite and hydrogen sulfide on cysteine catabolism. Br J Pharmacol 2019;176:554–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alkufri F, Harrower T, Rahman Y, et al. Molybdenum cofactor deficiency presenting with a Parkinsonism-dystonia syndrome. Mov Disord 2013;28:399–400. [DOI] [PubMed] [Google Scholar]
- 12.Johnson JL, Coyne KE, Rajagopalan KV, et al. Molybdopterin synthase mutations in a mild case of molybdenum cofactor deficiency. Am J Med Genet 2001;104:169–173. [DOI] [PubMed] [Google Scholar]
- 13.Bindu PS, Nagappa M, Bharath RD, Taly AB. Isolated sulfite oxidase deficiency. In: GeneReviews®. Seattle: University of Washington, Seattle; 1993–2020. [PubMed] [Google Scholar]
- 14.Vijayakumar K, Gunny R, Grunewald S, et al. Clinical neuroimaging features and outcome in molybdenum cofactor deficiency. Pediatr Neurol 2011;45:246–252. [DOI] [PubMed] [Google Scholar]
- 15.Del Rizzo M, Burlina AP, Sass JO, et al. Metabolic stroke in a late-onset form of isolated sulfite oxidase deficiency. Mol Genet Metab 2013;108:263–266. [DOI] [PubMed] [Google Scholar]
- 16.Touati G, Rusthoven E, Depondt E, et al. Dietary therapy in two patients with a mild form of sulphite oxidase deficiency. Evidence for clinical and biological improvement. J Inherit Metab Dis 2000;23:45–53. [DOI] [PubMed] [Google Scholar]
- 17.Bamforth FJ, Johnson JL, Davidson AGF, Wong LTK, Lockitch G, Applegarth DA. Biochemical investigation of a child with molybdenum cofactor deficiency. Clin Biochem 1990;23:537–542. [DOI] [PubMed] [Google Scholar]
- 18.Sass JO, Gunduz A, Araujo Rodrigues Funayama C, et al. Functional deficiencies of sulfite oxidase: differential diagnoses in neonates presenting with intractable seizures and cystic encephalomalacia. Brain Dev 2010;32:544–549. [DOI] [PubMed] [Google Scholar]
- 19.Zaki MS, Selim L, El-Bassyouni HT, et al. Molybdenum cofactor and isolated sulphite oxidase deficiencies: clinical and molecular spectrum among Egyptian patients. Eur J Paediatr Neurol 2016;20:714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kisker C, Schindelin H, Pacheco A, et al. Molecular basis of sulfite oxidase deficiency from the structure of sulfite oxidase. Cell 1997;91:973–983. [DOI] [PubMed] [Google Scholar]
- 21.Johnson JL, Coyne KE, Garrett RM, et al. Isolated sulfite oxidase deficiency: identification of 12 novel SUOX mutations in 10 patients. Hum Mutat 2002;20:74. [DOI] [PubMed] [Google Scholar]
- 22.Karakas E, Kisker C. Structural analysis of missense mutations causing isolated sulfite oxidase deficiency. Dalton Trans 2005:3459–3463. [DOI] [PubMed] [Google Scholar]
- 23.Rajapakshe A, Tollin G, Enemark JH. Kinetic and thermodynamic effects of mutations of human sulfite oxidase. Chem Biodivers 2012;9:1621–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feng C, Wilson HL, Tollin G, et al. The pathogenic human sulfite oxidase mutants G473D and A208D are defective in intramolecular electron transfer. Biochemistry 2005;44:13734–13743. [DOI] [PubMed] [Google Scholar]
- 25.Rocha S, Ferreira AC, Dias AI, Vieira JP, Sequeira S. Sulfite oxidase deficiency—an unusual late and mild presentation. Brain Dev 2014;36:176–179. [DOI] [PubMed] [Google Scholar]
- 26.Karakas E, Wilson HL, Graf TN, et al. Structural insights into sulfite oxidase deficiency. J Biol Chem 2005;280:33506–33515. [DOI] [PubMed] [Google Scholar]
- 27.Garrett RM, Johnson JL, Graf TN, Feigenbaum A, Rajagopalan KV. Human sulfite oxidase R160Q: identification of the mutation in a sulfite oxidase-deficient patient and expression and characterization of the mutant enzyme. Proc Natl Acad Sci USA 1998;95:6394–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hänzelmann P, Schindelin H. Crystal structure of the S-adenosylmethionine-dependent enzyme MoaA and its implications for molybdenum cofactor deficiency in humans. Proc Natl Acad Sci USA 2004;101:12870–12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wuebbens MM, Liu MT, Rajagopalan K, Schindelin H. Insights into molybdenum cofactor deficiency provided by the crystal structure of the molybdenum cofactor biosynthesis protein MoaC. Structure 2000;8:709–718. [DOI] [PubMed] [Google Scholar]
- 30.Rudolph MJ, Wuebbens MM, Rajagopalan KV, Schindelin H. Crystal structure of molybdopterin synthase and its evolutionary relationship to ubiquitin activation. Nat Struct Biol 2001;8:42–46. [DOI] [PubMed] [Google Scholar]
- 31.Daniels JN, Wuebbens MM, Rajagopalan KV, Schindelin H. Crystal structure of a molybdopterin synthase-precursor Z complex: insight into its sulfur transfer mechanism and its role in molybdenum cofactor deficiency. Biochemistry 2008;47:615–626. [DOI] [PubMed] [Google Scholar]
- 32.Liu MT, Wuebbens MM, Rajagopalan KV, Schindelin H. Crystal structure of the gephyrin-related molybdenum cofactor biosynthesis protein MogA from Escherichia coli. J Biol Chem 2000;275:1814–1822. [DOI] [PubMed] [Google Scholar]
- 33.Schwarz G, Schrader N, Mendel RR, Hecht HJ, Schindelin H. Crystal structures of human gephyrin and plant Cnx1 G domains: comparative analysis and functional implications. J Mol Biol 2001;312:405–418. [DOI] [PubMed] [Google Scholar]
- 34.Xiang S, Nichols J, Rajagopalan KV, Schindelin H. The crystal structure of Escherichia coli MoeA and its relationship to the multifunctional protein gephyrin. Structure 2001;9:299–310. [DOI] [PubMed] [Google Scholar]
- 35.Mayr SJ, Sass JO, Vry J, et al. A mild case of molybdenum cofactor deficiency defines an alternative route of MOCS1 protein maturation. J Inherit Metab Dis 2018;41:187–196. [DOI] [PubMed] [Google Scholar]
- 36.Hughes EF, Fairbanks L, Smmonds HA, Robinson RO. Molybdenum cofactor deficiency—phenotypic variability in a family with a late-onset variant. Dev Med Child Neurol 1998;40:57–61. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data extracted from the literature that was used for the analyses reported in this article will be made freely available on request from the corresponding author. The data will be available for 5 years from the time of publication and may be requested by any clinician or researcher.