

Abstract
Objective
To investigate the relationship between the ATN classification system (amyloid, tau, neurodegeneration) and risk of dementia and cognitive decline in individuals with subjective cognitive decline (SCD).
Methods
We classified 693 participants with SCD (60 ± 9 years, 41% women, Mini-Mental State Examination score 28 ± 2) from the Amsterdam Dementia Cohort and Subjective Cognitive Impairment Cohort (SCIENCe) project according to the ATN model, as determined by amyloid PET or CSF β-amyloid (A), CSF p-tau (T), and MRI-based medial temporal lobe atrophy (N). All underwent extensive neuropsychological assessment. For 342 participants, follow-up was available (3 ± 2 years). As a control population, we included 124 participants without SCD.
Results
Fifty-six (n = 385) participants had normal Alzheimer disease (AD) biomarkers (A–T–N–), 27% (n = 186) had non-AD pathologic change (A–T–N+, A–T+N–, A–T+N+), 18% (n = 122) fell within the Alzheimer continuum (A+T–N–, A+T–N+, A+T+N–, A+T+N+). ATN profiles were unevenly distributed, with A–T+N+, A+T–N+, and A+T+N+ containing very few participants. Cox regression showed that compared to A–T–N–, participants in A+ profiles had a higher risk of dementia with a dose–response pattern for number of biomarkers affected. Linear mixed models showed participants in A+ profiles showed a steeper decline on tests addressing memory, attention, language, and executive functions. In the control group, there was no association between ATN and cognition.
Conclusions
Among individuals presenting with SCD at a memory clinic, those with a biomarker profile A–T+N+, A+T–N–, A+T+N–, and A+T+N+ were at increased risk of dementia, and showed steeper cognitive decline compared to A–T–N– individuals. These results suggest a future where biomarker results could be used for individualized risk profiling in cognitively normal individuals presenting at a memory clinic.
The research framework for Alzheimer disease (AD) diagnosis, developed under the auspices of the National Institute on Aging and Alzheimer's Association, proposes to categorize individuals based on biomarker evidence of pathology using the so-called ATN classification system (amyloid, tau, neurodegeneration).1 According to the ATN system, each individual is rated for the presence of β-amyloid (CSF Aβ or amyloid PET: “A”), hyperphosphorylated tau (CSF p-tau or tau PET: “T”), and neurodegeneration (atrophy on structural MRI, FDG PET, or CSF total tau: “N”), resulting in 8 possible biomarker combinations.
Several former studies have applied the ATN classification scheme.2–8 Of these, 2 have used a cross-sectional design in cognitively unimpaired participants,2,3 and one focused on biomarker inconsistencies in healthy controls, patients with mild cognitive impairment (MCI), and patients with dementia.4 Three former studies had a longitudinal design, evaluating the association between ATN and cognitive decline in elderly patients without dementia (cognitively normal or MCI).5–7
Subjective cognitive decline (SCD) is characterized by self-perceived decline in cognition, but comparable cognitive performance to peers.9,10 In SCD, abnormal amyloid, abnormal tau, and signs of neurodegeneration are associated with an increased risk of cognitive decline.11–17 Longitudinal studies investigating the ATN classification scheme in relation to clinical progression in SCD, which has been described as stage 2 in the diagnostic framework,1 are not yet available.
We aimed to (1) examine the distribution and clinical correlates of the ATN biomarker profiles in individuals presenting with SCD at a memory clinic and (2) investigate the ATN predictive value for risk of clinical decline over time.
Methods
Population
We included 693 participants with SCD from the Amsterdam Dementia Cohort and the Subjective Cognitive Impairment Cohort (SCIENCe) project at the Alzheimer Center Amsterdam.18–20 All participants underwent a standardized diagnostic workup, which consisted of a neurologic, physical, and neuropsychological evaluation, and brain MRI.18,19 We used the Geriatric Depression Scale (GDS) to assess depressive symptoms.21,22 Participants were labeled SCD in a multidisciplinary consensus meeting when clinical and cognitive testing was normal and criteria for MCI, dementia, or other neurologic or psychiatric conditions (e.g., major depression, schizophrenia) were not met.10,23 Follow-up diagnoses were available for 342 participants (3 ± 2 years). These participants were on average 3 years older, but otherwise comparable to the entire sample of 693 participants. At follow-up, diagnoses were re-evaluated as SCD, MCI, AD dementia, or other types of dementia (frontotemporal dementia [FTD], primary progressive aphasia, vascular dementia, dementia with Lewy bodies).24–27 The clinical endpoints were (1) progression to dementia and (2) progression to MCI or dementia.
Participants were included for the current project when MRI and CSF were available within 1 year of the diagnosis.
Standard protocol approvals, registrations, and patient consents
The research is in accordance with ethical consent by VU University and the Helsinki Declaration of 1975. Written informed consent was available for all patients.
Neuropsychological assessment
All participants received an extensive standardized neuropsychological assessment.18 We used the Mini-Mental State Examination (MMSE) for global cognition. To asses memory, we used the Visual Association Test version A (VAT-A) and total immediate and delayed recall of the Dutch version of the Rey Auditory Verbal Learning Test (RAVLT). For language, we used category fluency (animals). To assess attention, we used the Trail-Making Test A (TMT-A), the forward condition of the Digit Span, and Stroop task I and II (naming and color naming). To assess executive functioning we used the TMT-B, Digit Span (backwards), and Stroop task III (color–word). Raw test scores for TMT and Stroop were log transformed, because the data were right-skewed, and subsequently inverted, such that a lower score implies worse performance. The proportion of missing tests ranged from 7.6% for the TMT-A to 19.1% for the Stroop III. In total, 1,424 neuropsychological investigations of 693 patients were available (299 ≥2; range 2–12, median 3).
MRI studies
All participants underwent an MRI scan of the brain (Siemens Avanto, n = 7; GE Discovery MR750, n = 14; Impax, n = 119; 3T Philips Ingenuity TF PET/MR system, n = 123; 1.5T GE Signa HDxt, n = 21; 3.0T GE Signa HDxt, n = 262; 1.5T Siemens Sonata, n = 27; 3T Toshiba Vantage Titan, n = 119; Vision, n = 1). The protocol included 3D T1-weighted images, 3D T2-weighted images, and 3D T2-weighted fluid-attenuated inversion-recovery (FLAIR) images.18 Visual rating of medial temporal lobe atrophy (MTA) was performed on coronal T1-weighted images averaging scores for the left and right sides (range 0–4).28 Posterior atrophy was rated using sagittal, axial, and coronal planes of T1 and FLAIR-weighted images averaging scores for the left and right sides (range 0–3).29 Global cortical atrophy (GCA) was rated using axial FLAIR images (range 0–3).30 The severity of white matter hyperintensities was determined on the FLAIR sequence using the Fazekas scale (range 0–3).31 Lacunes were defined as deep lesions (3–15 mm) with CSF-like signal on all sequences. They were counted and dichotomized into absent (0) of present (≥1 lacune). Microbleeds were defined as small dot-like hypointense lesions on T2-weighted images. They were also counted and dichotomized into absent (0) or present (≥1 microbleed). An experienced neuroradiologist reviewed all scans.
CSF studies
CSF was obtained by lumbar puncture between the L3/L4, L4/L5, or L5/S1 intervertebral space by a 25-gauge needle and syringe and collected in polypropylene tubes.32 Aβ1-42, total tau, and tau phosphorylated threonine 181 (p-tau) were measured using sandwich ELISAs (Innotest β-amyloid1-42, n = 579; Innotest hTAU-Ag and Innotest PhosphoTAU-181p).33 CSF Aβ levels were adjusted for the drift in CSF biomarker analyses that occurred over the years.34 For 9 participants, we used Elecsys for analyses of Aβ. These values were transformed to the Innotest-equivalent values by the following formula: Elecsys Aβ (pg/mL) = −365 + 1.87 × Innotest Aβ (pg/mL).35
PET studies
For 105 participants, amyloid PET was performed in research context using the tracers [18F]Florbetapir (n = 19), [18F]Florbetaben (n = 65), [18F]Flutemetamol (n = 10), or [11C]Pittsburgh compound B (PiB, n = 11). The tracers were administered intravenously through a cannula. PET scans were acquired on the following systems: Gemini TF PET-CT, Ingenuity TF PET-CT, and Ingenuity PET/MRI (Philips Healthcare, Best, the Netherlands). During scans, laser beams were used to monitor head movement. For [18F]Florbetapir imaging, participants were injected with a tracer dose of approximately 370 megabecquerel (MBq) [18F]Florbetapir.20 Ninety-minute dynamic PET emission scans were obtained simultaneously starting with tracer injection. For [18F]Florbetaben imaging, participants were injected with a tracer dose of approximately 300 MBq [18F]Florbetaben.36 The image acquisition window extended from 90 to 110 minutes (4 × 5 minutes frames) after dose injection. For [18F]Flutemetamol imaging, participants were injected with approximately 191 MBq [18F]Flutemetamol.37 The image acquisition window extended from 90 minutes to 110 minutes (4 × 5 minutes frames) after dose injection. For [11C]PiB imaging, participants were injected with a tracer dose of approximately 365 MBq [11C]PiB in younger participants and approximately 382 MBq [11C]PiB in older participants.38,39 Ninety-minute dynamic PET emission scans were obtained immediately starting with tracer injection.
All scans were visually rated as positive or negative by a trained nuclear medicine physician.
ATN classification
We used amyloid PET (n = 105) or CSF Aβ (n = 588) to determine whether a participant was A– or A+. If both measures were available, the PET result was used. CSF concentrations were considered amyloid positive <813 pg/mL.34 For tau (T), we used CSF p-tau concentrations. Values were considered p-tau positive >52 pg/mL.40 We used the average MTA to determine neurodegeneration (N). For participants <65 years of age, an average MTA score ≥1 was considered positive; for participants ≥65 years of age, an average MTA score ≥1.5 was considered positive.41 Because a number of ATN profiles contained very few participants, we also clustered the 8 biomarker profiles into 3 categories. The A–T–N– profile was labeled as the “normal AD biomarker” category. We clustered the remaining A– profiles (A–T–N+, A–T+N–, and A–T+N+) as “non-AD pathologic change” and we clustered all A+ profiles (A+T–N–, A+T–N+, A+T+N–, A+T+N+) as “Alzheimer continuum.”1
Control group
For comparison, we also included a control group without subjective cognitive decline, recruited from the EMIF-AD PreclinAD study.42 We included 124 participants, including 53 monozygotic twin pairs and 18 singletons. For 119 participants, A was determined by visual read of [18F]Flutemetamol PET. Levels of CSF Aβ40 and Aβ42 and p-tau were analyzed using kits from ADx Neurosciences/Euroimmun. CSF concentrations were considered amyloid positive when the CSF Aβ42/40 ratio was <0.065 (n = 5 for whom amyloid PET was not available). To determine T, we used the 75th percentile of p-tau (≥86 pg/mL). N was determined using average MTA (<65 years, MTA ≥1; ≥65 years, MTA ≥1.5 considered positive). Neuropsychological testing procedures for the control group were largely similar to procedures for participants with SCD. For 121 participants, follow-up assessments were available (2 ± 0 years).
Statistics
We compared demographic and clinical variables among the 8 ATN biomarker profiles. For continuous variables, we used analysis of variance and Kruskal-Wallis where appropriate and post hoc Tukey test if the assumption for homogeneity of variances was met and Games-Howell if the assumption was not met. For dichotomous variables, we used Fisher exact test, and post hoc looked at standardized residuals with values <−1.96 or >1.96 considered significant. We used χ2 test to compare the distribution of ATN profiles between the SCD and control group.
We performed Cox proportional hazards analyses to evaluate the association between 8-profile ATN classification (A–T–N– reference) and clinical progression to dementia. Analyses were adjusted for age, sex, and education. In additional analyses, we used progression to MCI or dementia as outcome.
To explore the additive effect of A-status and memory function, we ran an additional analysis after constructing a new, 4-level variable: (1) A–, high memory (baseline RAVLT delayed recall z score ≥ −1.0); (2) A–, low memory (z score < −1.0); (3) A+, high memory; (4) A+, low memory. Analyses were adjusted for age, sex, and education. As reference category we used (1) A–, high memory.
Subsequently, we assessed the associations between ATN classification and cognitive decline using linear mixed models (LMMs). ATN profiles (included as dummies, with the A–T–N– profile as reference), time, and the interactions between ATN profiles and time were included as independent variables; age, sex, and education were included as covariates; and cognitive test scores were used as dependent variables. Intercept and time were included as random factors. Separate models were run for 11 individual neuropsychological tests. We used the false discovery rate method to correct for multiple testing with q set at 0.05.
For the analyses in the control group without SCD, we added family as a random factor to account for within-twin pair dependence.
All analyses were done using SPSS version 22. p Values <0.05 were considered significant. Kaplan-Meier curves and figures showing association between ATN and cognitive decline were made with R studio 3.4.2.
Data availability
Any data not published within the article may be shared upon request.
Results
Baseline demographics and clinical features
At baseline, the 693 participants with SCD were on average 60 ± 9 years old, 283 (41%) were female, and MMSE was 28 ± 2. With 385 (56%) participants, the majority of the participants was negative for each of the 3 biomarkers (A–T–N–, normal AD biomarkers). Figure 1 and table 1 show the distribution, demographics, and clinical characteristics of all biomarker profiles (3-category comparison provided in table e-1, doi.org/10.5061/dryad.bg79cnp71). Participants were older in A+ profiles and in profiles with a higher number of biomarkers affected. There were no significant differences in sex, education, MMSE, or GDS scores among ATN profiles. APOE ε4 varied by ATN biomarker profiles, with A+ profiles containing the highest number of APOE ε4 carriers.
Figure 1. Distribution of the ATN biomarker profiles in subjective cognitive decline.
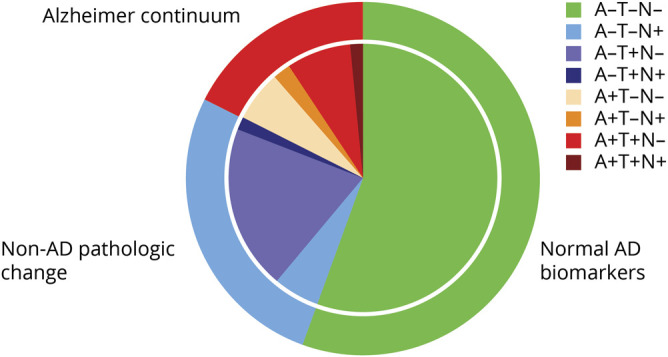
Pie chart illustrates the distribution of the 8-profile and 3-category ATN classification. AD = Alzheimer disease.
Table 1.
Baseline demographics, PET, CSF, and MRI values in 8-profile ATN classification

Comparing biomarker values among ATN profiles, we found that total tau (not included in our ATN definition) was lowest in A–T–N– and was higher in T+ groups, but not in N+ groups. P-tau and total tau strongly correlated with each other (Spearman ρ 0.90, p ≤ 0.00). GCA scores were higher in N+ profiles (Spearman ρ MTA ∼ GCA: 0.35, p ≤ 0.00). There was no difference in any of the MRI measures of small vessel disease among groups.
Participants in the control group without SCD were on average 9 years older than our SCD sample and more often female (52% vs 41%; table e-2, doi.org/10.5061/dryad.bg79cnp71). The distribution of ATN profiles differed between the 2 cohorts (p = 0.00). The prevalence of A–T–N– was similar in both cohorts, but N+ biomarker profiles were more prevalent in the control group (23% vs 11%), while the A+ was somewhat more common in our SCD sample (18% vs 14%). In the control group, age differed across ATN profiles, with on visual inspection a stronger age effect than in the SCD sample. ATN profiles also differed on MMSE.
Risk of dementia
Table 2 shows that after 3 ± 2 years of follow-up, 16 (21%) participants in A+ profiles showed incident dementia (AD, n = 14; non-AD, n = 2), compared to 2 (1%) participants in A–T–N– (non-AD n = 2). The supplemental e-Box, doi.org/10.5061/dryad.bg79cnp71, provides a case description of the 2 participants in A–T+N– and A–T+N+ who progressed to AD dementia and 2 participants in A–T–N– who progressed to dementia. Both A– participants who progressed to AD dementia became A+T+N+ on follow-up. The participants who initially were A–T–N– progressed to primary progressive aphasia and to possible FTD.
Table 2.
Clinical progression in 8 ATN biomarker profiles

Cox proportional hazard analyses showed that compared to A–T–N–, participants in the A+ profiles were at increased risk of dementia with an incremental increase in hazard ratio (HR) (A+T–N–, HR 9.7 [1.6–59.3]; A+T+N–, HR 20.2 [3.7–110.2]; and A+T+N+, HR 62.3 [9.5–408.4]; figure 2). Within the A– profiles, participants in A–T+N+ were at increased risk of dementia (HR 18.5 [1.6–211.4]), but participants in A–T+N– and A–T–N+ were not. When we repeated the analyses with clinical progression to MCI or dementia as outcome, a similar pattern emerged, although the HRs were lower, caused by a higher number of participants progressing to MCI in the reference profile.
Figure 2. Kaplan-Meier curves illustrating clinical progression in subjective cognitive decline.

Kaplan-Meier curves illustrate clinical progression to dementia (A) and to mild cognitive impairment or dementia (B). Separate lines represent the 8 ATN biomarker profiles. The numbers at risk for time points 0, 1, 2, 3, 4, and 5 years are depicted below the figure.
As an additional analysis, we performed Cox analyses based on the 3 clustered ATN categories, as shown in table e-3 and figure e-1 (doi.org/10.5061/dryad.bg79cnp71). We found that compared to participants with normal AD biomarkers, Alzheimer continuum participants had a strongly increased risk of progression to dementia (HR, 17 [95% confidence interval (CI), 3.6–79]). The risk associated with non-AD pathologic change was 3.2 (95% CI, 0.6–17.8), but did not reach significance.
Subsequently, we ran an additional analysis to assess the putative combined effect of memory impairment and amyloid status. We found that the effects of memory and amyloid positivity seem additive, as—compared to individuals with a high RAVLT delayed recall score and no evidence of amyloid—those with both a low RAVLT delayed recall score and positive amyloid were at the highest risk of dementia (HR, 18.9 [5.5–64.6]; table 3). Participants with only low baseline memory were not at increased risk of dementia (HR, 1.2 [0.1–11.0]), but participants with only a positive A biomarker alone were (HR, 8.8 [2.3–34.2]).
Table 3.
Association between baseline Rey Auditory Verbal Learning Test (RAVLT) delayed recall score and clinical progression

Cognitive decline
We used LMMs to assess the association between ATN classification and cognitive test performance. Figure 3 shows the observed raw neuropsychological test scores and trajectories over time; table 4 shows estimated baseline cognitive tests scores and annual change by ATN profile. There were a few modest associations between ATN profile and baseline cognitive test performance. By contrast, we found significant interactions of ATN profile with time for a large number of tests. Participants in A+T+N– and A+T+N+ showed a steeper decline over time than A–T–N– on all memory tests. Other A+ profiles also showed a steeper decline on tests for memory, attention, and executive function (A+T–N– on VAT-A and TMT-B; A+T+N– on TMT-A, TMT-B, and Stroop III). A–T+N+ was the only A– profile associated with steeper decline over time, namely on 2 memory tests (RAVLT immediate and delayed recall).
Figure 3. Spaghetti plots illustrating raw neuropsychological test performance over time in subjective cognitive decline.

Spaghetti plots show individual neuropsychological trajectories on 6 neuropsychological tests: (A) Visual Association Test version A (VAT-A); (B) Rey Auditory Verbal Learning Test (RAVLT) delayed recall; (C) animal fluency; (D) Trail-Making Test, part A (TMT-A); (E) Stroop I; (F) Stroop III. Separate lines represent the unadjusted mean trajectory of the 8 ATN profiles with 95% confidence intervals. Figures represent raw test scores.
Table 4.
Relationship between ATN biomarker profiles and baseline and longitudinal cognition

Table e-4 and figure e-2 (doi.org/10.5061/dryad.bg79cnp71) show the results of the additional analyses based on 3 clustered ATN categories. There was a main effect of ATN category on RAVLT delayed recall, implying that participants classified in the Alzheimer continuum had a lower baseline memory performance than participants with normal AD biomarkers. There were no associations with baseline score for any of the other cognitive tests. Participants in the Alzheimer continuum showed a steeper decline over time in tests for memory, attention, language, and executive functioning compared to the normal AD biomarker category. Participants with non-AD pathologic change only showed a steeper decline over time on RAVLT delayed recall.
In the control group without SCD, only one participant (A+T+N+) progressed to dementia, precluding any formal testing. In the control group, we found no significant associations between ATN profiles and (cross-sectional or longitudinal) cognitive test score (table e-5, doi.org/10.5061/dryad.bg79cnp71).
Discussion
In a sample of individuals with SCD presenting at a memory clinic, we found that Alzheimer continuum profiles (A+T–N–, A+T–N+, A+T+N–, A+T+N+) together make up one fifth and non-AD pathologic change profiles (A–T–N+, A–T+N–, A–T+N+) one third of cases. Most frequent is the profile with all biomarkers normal, observed in more than half of participants. Compared to A–T–N–, participants in A+ profiles were at increased risk of dementia and showed a steeper subsequent cognitive decline over the years on tests for memory, attention, and executive functioning. Participants in A–T+N+ showed steeper decline on 2 memory tests, but other A– profiles were comparable to A–T–N– in terms of their cognitive trajectories.
We extend on former findings by longitudinally evaluating the ATN model in a population of cognitively normal individuals with SCD. In this clinically relevant population of individuals with SCD, particularly the A+ profiles defined in the ATN model have predictive value in terms of risk of future dementia and rate of cognitive decline; however, not all profiles were sufficiently populated to warrant reliable risk estimates. This illustrates that biomarker information helps to identify those at risk, but also shows that the 8 different profiles as defined in the ATN model are somewhat difficult to implement. We therefore supplemented our results with a more robust categorization in 3 clustered ATN categories (normal AD biomarkers, Alzheimer continuum, non-AD pathologic change). The results of the analyses using the 3 clustered categories confirm that of all biomarkers assessed, especially the A biomarker is strongly associated with clinical progression and cognitive decline.
We found an uneven distribution of ATN profiles. This observation is largely consistent with former studies in individuals without dementia, although the exact prevalence of specific profiles somewhat differs between studies.2–4,6,7 Compared to earlier studies, most noteworthy is that more than half of our participants in both the SCD and control group were negative for all biomarkers (56% and 57%, respectively), while other studies found a much lower prevalence (29%–39%).2,6,7 On the other end of the profile spectrum, we found a very low prevalence of A+T+N+ in our SCD sample (2%), while other studies found higher percentages (9%–12%).2,6,7 The fact that our SCD sample is 10–15 years younger than other cohorts and was carefully selected to be cognitively normal at baseline probably explains this effect and explains our apparently lower amyloid rate, as age has been shown to play a large role in biomarker positivity.2,43,44 At the same time, our results show that in this population, the observation of amyloid is not benign, as it is strongly associated with future dementia. Discrepancies with other studies can be explained by their defining both T and N based on CSF (p-tau and total tau). Due to the high correlation between these 2 measures (also shown in the current study), profiles like A+T+N– and A–T+N– become highly improbable when both T and N are defined based on CSF. When this approach was chosen, none of the participants falls into these mutually exclusive profiles.3 In our study, we used CSF p-tau to define T and MRI-based medial temporal lobe atrophy for N, because they are both widely available and clearly measure different aspects of the disease, as evidenced by the fact that both categories were populated by a considerable fraction of the sample (A–T+N–: 19.8%; A–T–N+: 5.6%). When we compare our SCD sample to our own control group without SCD, we found a larger number of participants who were positive for N in our control group, resulting in different percentages in A–T–N+ (6% vs 17%) and A–T+N– (20% vs 10%). In addition, A+ seemed somewhat less prevalent in the controls than in individuals with SCD (14% vs 18%), particularly when taking into account that controls were older than individuals with SCD. It should be noted our study was not designed to study the effect of SCD on risk of progression, but rather to evaluate the clinical usefulness of ATN profiles in this clinically relevant population. Adding SCD as an additional factor to the ATN scheme (“ATN-S”) would increase the number of categories even more, and our current results show that the high number of categories is already a limitation to the clinical applicability of the ATN scheme.
We observed that participants in 3 A+ profiles showed a faster cognitive decline over the years on tests for memory, attention, and executive functioning. This is largely in line with a study conducted by the Mayo Clinic that found especially A+T+N–, A+T–N+, and A+T+N+ show a steeper decline on memory tests compared to A–T–N–.6 Another study found that only A+T+N+ had a greater decline on a cognitive factor score.7 Our results provide further support for the observation that effect sizes of cognitive decline seem to increase with the number of biomarkers affected.
We found participants in A+ profiles to be at increased risk of dementia. Of note, within the 3 A+ profiles, we observed risk estimates to increase with the number of biomarkers affected, illustrating the added value of having biomarker information on all 3 biomarkers available, and—conversely—that lumping all A+ profiles together may obscure differences in outcomes of the different biomarker categories. The 2 previous longitudinal studies investigating ATN in cognitively unimpaired participants assessed cognitive trajectories, but did not assess risk of clinical progression to MCI or dementia.6,7 There is only one longitudinal study evaluating the risk of dementia associated with the ATN classification, using an MCI sample, that found amyloid positivity was associated with a higher risk of dementia.5 In addition, we found a higher risk of dementia in A–T+N+. The A–T+N+ category contained only 7 individuals, one of whom progressed to AD dementia. Upon scrutinizing this patient's chart, this diagnosis was supported by a change in biomarker status from A– to A+ (supplemental e-Box, doi.org/10.5061/dryad.bg79cnp71). This suggests an unusual order of pathologic events, which has been described before.6,45 In line with the proposed model of amyloid changes being among the first changes in the cascade of events eventually leading to AD dementia, earlier studies have also found amyloid positivity to be a strong risk factor for clinical progression in the clinically relevant population of individuals with SCD.11–13,15,46–48 In addition, we found that the effects of memory and amyloid positivity seem additive, as individuals with a low baseline memory score and positive amyloid were at the highest risk of dementia.
This study has important clinical implications. There is an increasing interest in SCD as a clinically relevant construct, because these are individuals who seek help at a memory clinic. We demonstrated that within SCD, the ATN classification is associated with clinical progression and cognitive decline at a group level. Although it originally emerged as a research framework, applying ATN to participants with SCD presenting to a memory clinic is a first step in evaluating the potential for translation of these criteria to clinical practice. Our results show that ATN biomarker profiles help to identify which individuals with SCD are at risk of clinical progression. Just as important, the ATN classification can help to avoid misjudging SCD as a group, because most individuals with SCD have normal biomarkers and our results show that they are highly unlikely to show cognitive decline over time.
This study has some major strengths. First, we included a large cohort with almost 700 participants with SCD. We had follow-up for many, which enabled us to analyze dementia as an outcome in this initially cognitively normal sample. We used a combination of modalities to define ATN categories, which resulted in a robust classification. Among the potential limitations of this study is that we found there is an important relationship between age and biomarker abnormality. Although we corrected for age in all models used, this might not have been sufficient to eliminate this factor. Second, despite the fact that we had a large cohort, some groups were very small. This hampered the feasibility to investigate the true value of each of the 8 biomarker profiles, and furthermore illustrates that 8 categories may be too large a number for practical use, while simultaneously not even capturing all heterogeneity between patients. As an additional analysis, we used the 3 clustered categories, clearly showing that the A+ categories are associated with an increased risk of cognitive decline and dementia. Third, inherent to using the ATN framework, we dichotomized all biomarker data. This means we used cutoff values, which implies loss of information. As an alternative, it would be worth considering each of the biomarkers as a continuous variable, especially because recent data show that there may be information in subthreshold amyloid deposition.49
We found that participants with SCD in A+ profiles show an increased risk of clinical progression to dementia and steeper decline in the cognitive domains of memory, attention, language, and executive functions compared to participants with normal biomarkers. These findings implicate that within SCD, the ATN classification helps to identify individuals at risk of dementia, and—maybe even more importantly—which individuals are highly unlikely to show progression over time.
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- CI
confidence interval
- FLAIR
fluid-attenuated inversion-recovery
- FTD
frontotemporal dementia
- GCA
global cortical atrophy
- GDS
Geriatric Depression Scale
- HR
hazard ratio
- LMM
linear mixed model
- MBq
megabecquerel
- MCI
mild cognitive impairment
- MMSE
Mini-Mental State Examination
- MTA
medial temporal lobe atrophy
- p-tau
phosphorylated threonine 181
- PiB
Pittsburgh compound B
- RAVLT
Rey Auditory Verbal Learning Test
- SCD
subjective cognitive decline
- TMT
Trail-Making Test
- VAT-A
Visual Association Test version A
Appendix. Authors
Study funding
The Alzheimer Center Amsterdam is supported by Alzheimer Nederland and Stichting VUMC funds. Research of the Alzheimer Center Amsterdam is part of the neurodegeneration research program of Amsterdam Neuroscience. The clinical database structure was developed with funding from Stichting Dioraphte. The SCIENCe project is supported by a research grant from Gieskes-Strijbis fonds. W.M.v.d.F. holds the Pasman chair. W.M.v.d.F. and P.J.V. received funding in the context of Deltaplan Dementie from ZonMW Memorabel (Netherlands Consortium of Dementia Cohorts [NCDC] project 73305095005) and Alzheimer Nederland. PET scans were funded by research grants from AVID and Piramal Neuroimaging. F.B. is supported by the University College London Hospitals NHS Foundation Trust Biomedical Research Center.
Disclosure
J.L. Ebenau, T. Timmers, and L.M.P. Wesselman report no relevant disclosures. I.M.W. Verberk received research funding from Health ∼ Holland for collaborating with Crossbeta Biosciences (grant paid directly to her institution). S.C.J. Verfaillie, R.E.R. Slot, and A.C. van Harten report no relevant disclosures. C.E. Teunissen serves on the advisory board of Roche; performed contract research for Boehringer, Roche, Toyama Fujifilm, Eisai, and Probiodruk; obtained a grant with ADxNeurosciences; and received lecture fees from Biogen and Axon Neurosciences. F. Barkhof is a consultant for Biogen-Idec, Janssen Alzheimer Immunotherapy, Bayer-Schering, Merck-Serono, Roche, Novartis, Genzyme, and Sanofi-Aventis; has received sponsoring from European Commission–Horizon 2020, National Institute for Health Research–University College London Hospitals Biomedical Research Centre, Scottish Multiple Sclerosis Register, TEVA, Novartis, and Toshiba; and serves on the editorial boards of Radiology, Brain, Neuroradiology, Multiple Sclerosis Journal, and Neurology®. K.A. van den Bosch, M. van Leeuwenstijn, J. Tomassen, and A. den Braber report no relevant disclosures. P.J. Visser is recipient of NCDC, which is funded in the context of Deltaplan Dementie from ZonMW Memorabel (project 73305095005) and Alzheimer Nederland. N.D. Prins reports consulting, advisory, and speaker fees from Boehringer Ingelheim, Envivo, Janssen, Novartis, Probiodrug, Sanofi, Takeda, Kyowa Kirin Pharmaceutical Development, DSMB of AbbVie M15-566, and grants from Alzheimer Nederland (all paid directly to his institution) outside the submitted work. Dr. Prins is CEO and co-owner of the Alzheimer Research Center, Amsterdam, the Netherlands. S.A.M. Sikkes provided consultancy services for Boehringer Ingelheim, Takeda, and Nutrica; her work is supported by grants from the Dutch Research Council (ZonMW) and the EU Joint Programme—Neurodegenerative Disease Research (all funds paid to her institution). P. Scheltens has acquired grant support (for the institution) from Biogen; in the past 2 years, he has received consultancy/speaker fees (paid to the institution) from Probiodrug Biogen, EIP Pharma, and Merck AG. B.N.M. van Berckel reports no relevant disclosures. W.M. van der Flier's research programs have been funded by ZonMW, the Netherlands Organization of Scientific Research, Alzheimer Nederland, Cardiovascular Onderzoek Nederland, Stichting Dioraphte, Gieskes-Strijbis fonds, Pasman stichting, Boehringer Ingelheim, Life-MI, AVID, Biogen MA, and Combinostics (all funding paid to her institution). Go to Neurology.org/N for full disclosures.
References
- 1.Jack CR, Bennett DA, Blennow K, et al. NIA-AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jack CR Jr, Wiste HJ, Weigand SD, et al. Age-specific and sex-specific prevalence of cerebral beta-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50-95 years: a cross-sectional study. Lancet Neurol 2017;16:435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kern S, Zetterberg H, Kern J, et al. Prevalence of preclinical Alzheimer disease: comparison of current classification systems. Neurology 2018;90:e1682–e1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Illan-Gala I, Pegueroles J, Montal V, et al. Challenges associated with biomarker-based classification systems for Alzheimer's disease. Alzheimers Dement 2018;10:346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ekman U, Ferreira D, Westman E. The A/T/N biomarker scheme and patterns of brain atrophy assessed in mild cognitive impairment. Sci Rep 2018;8:8431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jack CR Jr, Wiste HJ, Therneau TM, et al. Associations of amyloid, tau, and neurodegeneration biomarker profiles with rates of memory decline among individuals without dementia. JAMA 2019;321:2316–2325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soldan A, Pettigrew C, Fagan AM, et al. ATN profiles among cognitively normal individuals and longitudinal cognitive outcomes. Neurology 2019;92:e1567–e1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Altomare D, de Wilde A, Ossenkoppele R, et al. Applying the ATN scheme in a memory clinic population: the ABIDE project. Neurology 2019;93:e1635–e1646. [DOI] [PubMed] [Google Scholar]
- 9.Molinuevo JL, Rabin LA, Amariglio R, et al. Implementation of subjective cognitive decline criteria in research studies. Alzheimers Dement 2017;13:296–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jessen F, Amariglio RE, van Boxtel M, et al. A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer's disease. Alzheimers Dement 2014;10:844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Harten AC, Smits LL, Teunissen CE, et al. Preclinical AD predicts decline in memory and executive functions in subjective complaints. Neurology 2013;81:1409–1416. [DOI] [PubMed] [Google Scholar]
- 12.van Harten AC, Visser PJ, Pijnenburg YA, et al. Cerebrospinal fluid Abeta42 is the best predictor of clinical progression in patients with subjective complaints. Alzheimers Dement 2013;9:481–487. [DOI] [PubMed] [Google Scholar]
- 13.Burnham SC, Bourgeat P, Doré V, et al. Clinical and cognitive trajectories in cognitively healthy elderly individuals with suspected non-Alzheimer's disease pathophysiology (SNAP) or Alzheimer's disease pathology: a longitudinal study. Lancet Neurol 2016;15:1044–1053. [DOI] [PubMed] [Google Scholar]
- 14.Mormino EC, Betensky RA, Hedden T, et al. Synergistic effect of beta-amyloid and neurodegeneration on cognitive decline in clinically normal individuals. JAMA Neurol 2014;71:1379–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vos SJB, Xiong C, Visser PJ, et al. Preclinical Alzheimer's disease and its outcome: a longitudinal cohort study. Lancet Neurol 2013;12:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verfaillie SC, Tijms B, Versteeg A, et al. Thinner temporal and parietal cortex is related to incident clinical progression to dementia in patients with subjective cognitive decline. Alzheimers Dement 2016;5:43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruun M, Rhodius-Meester HFM, Koikkalainen J, et al. Evaluating combinations of diagnostic tests to discriminate different dementia types. Alzheimers Dement 2018;10:509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Flier WM, Pijnenburg YA, Prins N, et al. Optimizing patient care and research: the Amsterdam dementia cohort. J Alzheimers Dis 2014;41:313–327. [DOI] [PubMed] [Google Scholar]
- 19.van der Flier WM, Scheltens P. Amsterdam dementia cohort: performing research to optimize care. J Alzheimers Dis 2018;62:1091–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slot RER, Verfaillie SCJ, Overbeek JM, et al. Subjective Cognitive Impairment Cohort (SCIENCe): study design and first results. Alzheimers Res Ther 2018;10:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res 1982;17:37–49. [DOI] [PubMed] [Google Scholar]
- 22.Sheikh JI, Yesavage JA. Geriatric Depression Scale (GDS): recent evidence and development of a shorter version. Clin Gerontologist 1986;5:165–173. [Google Scholar]
- 23.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011;42:2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology 2017;89:88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheltens P, Leys D, Barkhof F, et al. Atrophy of medial temporal lobes on MRI in “probable” Alzheimer's disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol Neurosurg Psychiatry 1992;55:967–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koedam EL, Lehmann M, van der Flier WM, et al. Visual assessment of posterior atrophy development of a MRI rating scale. Eur Radiol 2011;21:2618–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pasquier F, Leys D, Weerts JGE, Mounier-Vehier F, Barkhof F, Scheltens P. Inter-and intraobserver reproducibility of cerebral atrophy assessment on MRI scans with hemispheric infarcts. Eur Neurol 1996;36:268–272. [DOI] [PubMed] [Google Scholar]
- 31.Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer's dementia and normal aging. AJR Am J Roentgenol 1987;149:351–356. [DOI] [PubMed] [Google Scholar]
- 32.Teunissen CE, Tumani H, Engelborghs S, Mollenhauer B. Biobanking of CSF: international standardization to optimize biomarker development. Clin Biochem 2014;47:288–292. [DOI] [PubMed] [Google Scholar]
- 33.Duits FH, Prins ND, Lemstra AW, et al. Diagnostic impact of CSF biomarkers for Alzheimer's disease in a tertiary memory clinic. Alzheimers Dement 2015;11:523–532. [DOI] [PubMed] [Google Scholar]
- 34.Tijms BM, Willemse EAJ, Zwan MD, et al. Unbiased approach to counteract upward drift in cerebrospinal fluid amyloid-beta 1-42 analysis results. Clin Chem 2018;64:576–585. [DOI] [PubMed] [Google Scholar]
- 35.Willemse EAJ, van Maurik IS, Tijms BM, et al. Diagnostic performance of Elecsys immunoassays for cerebrospinal fluid Alzheimer's disease biomarkers in a nonacademic, multicenter memory clinic cohort: the ABIDE project. Alzheimers Dement 2018;10:563–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de Wilde A, van Maurik IS, Kunneman M, et al. Alzheimer's biomarkers in daily practice (ABIDE) project: rationale and design. Alzheimers Dement 2017;6:143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zwan MD, Bouwman FH, Konijnenberg E, et al. Diagnostic impact of [(18)F]flutemetamol PET in early-onset dementia. Alzheimers Res Ther 2017;9:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ossenkoppele R, van der Flier WM, Verfaillie SC, et al. Long-term effects of amyloid, hypometabolism, and atrophy on neuropsychological functions. Neurology 2014;82:1768–1775. [DOI] [PubMed] [Google Scholar]
- 39.Ossenkoppele R, Zwan MD, Tolboom N, et al. Amyloid burden and metabolic function in early-onset Alzheimer's disease: parietal lobe involvement. Brain 2012;135:2115–2125. [DOI] [PubMed] [Google Scholar]
- 40.Mulder C, Verwey NA, van der Flier WM, et al. Amyloid-beta(1-42), total tau, and phosphorylated tau as cerebrospinal fluid biomarkers for the diagnosis of Alzheimer disease. Clin Chem 2010;56:248–253. [DOI] [PubMed] [Google Scholar]
- 41.Rhodius-Meester HFM, Benedictus MR, Wattjes MP, et al. MRI visual ratings of brain atrophy and white matter hyperintensities across the spectrum of cognitive decline are differently affected by age and diagnosis. Front Aging Neurosci 2017;9:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Konijnenberg E, Carter SF, Ten Kate M, et al. The EMIF-AD PreclinAD study: study design and baseline cohort overview. Alzheimers Res Ther 2018;10:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Snitz BE, Wang T, Cloonan YK, et al. Risk of progression from subjective cognitive decline to mild cognitive impairment: the role of study setting. Alzheimers Dement 2018;14:734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slot RER, Sikkes SAM, Berkhof J, et al. Subjective cognitive decline and rates of incident Alzheimer's disease and non-Alzheimer's disease dementia. Alzheimers Dement 2019;15:465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jack CR, Wiste HJ, Weigand SD, et al. Amyloid-first and neurodegeneration-first profiles characterize incident amyloid PET positivity. Neurology 2013;81:1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Villemagne VL, Pike KE, Chetelat G, et al. Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol 2011;69:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morris JC, Roe CM, Grant EA, et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol 2009;66:1469–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knopman DS, Jack CR Jr, Wiste HJ, et al. Short-term clinical outcomes for stages of NIA-AA preclinical Alzheimer disease. Neurology 2012;78:1576–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Landau SM, Horng A, Jagust WJ. Memory decline accompanies subthreshold amyloid accumulation. Neurology 2018;90:e1452–e1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verhage F, Van Der Werff JJ. An analysis of variance based on the Groninger Intelligence Test Scores [in Dutch]. Nederlands Tijdschrift Psychologie Haar Grensgebieden 1964;19:497–509. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Any data not published within the article may be shared upon request.