

SUMMARY
Lung alveolar epithelium is composed of alveolar type I (AT1) and type II (AT2) cells. AT1 cells mediate gas exchange, whereas AT2 cells act as progenitor cells to repair injured alveoli. Lung microvascular endothelial cells (LMVECs) play a crucial but still poorly understood role in regulating alveolar repair. Here, we studied the role of the LMVEC-derived bioactive lipid sphingosine-1-phosphate (S1P) in promoting alveolar repair using mice with endothelial-specific deletion of sphingosine kinase 1 (Sphk1), the key enzyme promoting S1P generation. These mutant lungs developed airspace-enlargement lesions and exhibited a reduced number of AT1 cells after Pseudomonas-aeruginosa-induced lung injury. We demonstrated that S1P released by LMVECs acted via its receptor, S1PR2, on AT2 cells and induced nuclear translocation of yes-associated protein (YAP), a regulator of AT2 to AT1 transition. Thus, angiocrine S1P released after injury acts via the S1PR2-YAP signaling axis on AT2 cells to promote AT2 to AT1 differentiation required for alveolar repair.
Graphical Abstract
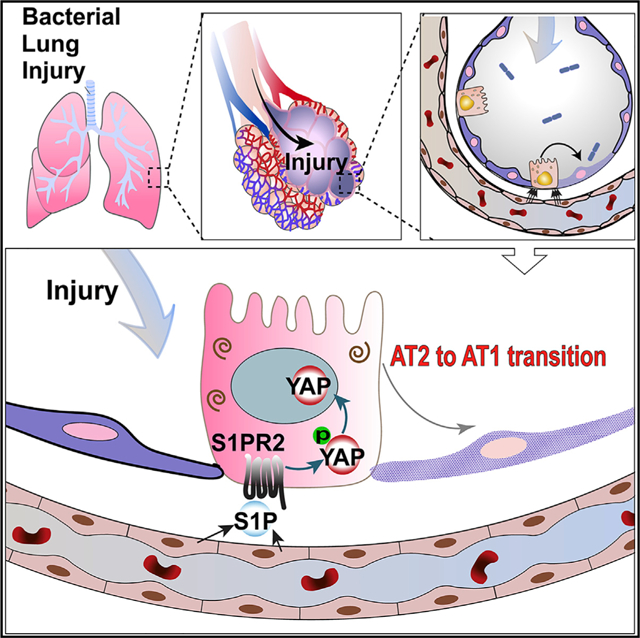
In Brief
Chen et al. showed that after bacterial lung injury, sphingosine-1-phosphate (S1P) is released from lung endothelial cells and regulates alveoli epithelial repair by promoting the progenitor function of alveolar type II cells via a S1P-S1PR2-YAP signaling axis. This work highlighted endothelial-epithelial interaction during lung repair mediated by bioactive lipid S1P.
INTRODUCTION
The lung alveolar epithelium is continuously exposed to particles and pathogens present in the air and is thus highly susceptible to inflammation and injury (Matthay et al., 2012). Alveolar epithelial cells (AECs) are normally quiescent with a slow turnover rate (Barkauskas et al., 2013; Desai et al., 2014), but upon injury, AECs are activated by signals released from a surrounding niche and start to repair the alveolar barrier. The alveolar epithelium is lined by two types of cells: alveolar type I cells (AT1) and alveolar type II cells (AT2) (Weibel, 2009). AT1 cells have a squamous and flat shape, covering 95% of the alveolar surface area, and provide a thin interface necessary for the efficient gas exchange (Schneeberger, 1997; Weibel, 2009). Cuboidal AT2 cells occupy only 5% of alveolar surface area and are responsible for secreting surfactant, transporting ions, and modulating immune responses (Mason, 2006). In addition, AT2 cells function as progenitor cells by self-renewal and differentiating into AT1 cells during homeostatic maintenance and alveolar repair/regeneration (Barkauskas et al., 2013; Desai et al., 2014; Evans et al., 1975).
As a highly vascularized organ, lung microvascular endothelial cells (LMVECs) comprise ~40% of the lung’s total cell population (Crapo et al., 1982). LMVECs and AECs are separated by a thin layer of extracellular matrix that is less than 2 μm (Weibel, 2009). LMVECs actively interact with inflammatory cells (Rao et al., 2007) and are able to sense micro-environmental changes after injury and serve as niches that direct the regenerative behavior of adjacent AECs. It has been shown that endothelial cells (ECs) can secrete paracrine factors (i.e., angiocrine factors) to stimulate repair/regeneration in various tissues (Rafii et al., 2016). In the lung, capillary ECs have been shown to modulate niche function and promote lung regeneration by producing matrix metal-loprotease MMP14 (Ding et al., 2011), HGF (hepatocyte growth factor) (Cao et al., 2017), and thrombospondin-1 (Lee et al., 2014).
Most studies on the trophic or regenerative properties of angiocrine factors have focused on proteins released by ECs. However, little is known about the role of bioactive lipid mediators such as sphingosine-1-phosphate (S1P) that could potentially regulate tissue regeneration. S1P is generated by phosphorylation of sphingosine, which is generated from the hydrolysis of ceramide, a degradation product from plasma membrane sphingomyelin (Proia and Hla, 2015; Pyne and Pyne, 2010; Spiegel and Milstien, 2003). ECs are major sources of S1P (Olivera et al., 2013; Proia and Hla, 2015; Venkataraman et al., 2008). After being synthesized by sphingosine kinase 1 (SPHK1), S1P is transported into the extracellular space and acts in an autocrine or paracrine fashion to regulate various cellular processes, such as proliferation, differentiation, adhesion, and migration (Kunkel et al., 2013; Proia and Hla, 2015; Spiegel and Milstien, 2003). Although most of the extracellular S1P is found in plasma, there are low levels of S1P in the tissue (Proia and Hla, 2015). Importantly, interstitial S1P levels are increased in response to inflammation and injury (Fukuhara et al., 2012).
The receptors of extracellular S1P are a family of G-protein-coupled receptors (GPCRs) named S1PR1–S1PR5 (Rosen et al., 2013). After being activated by S1P, the receptors can initiate a series of signaling events in the target cells mediated by G proteins (Kunkel et al., 2013; Pyne and Pyne, 2010). Importantly, S1PR2 acting through G12/13 can inhibit Hippo pathway kinases Lats1/2 and thus activate yes-associated protein (YAP) transcription coactivators (Miller et al., 2012; Yu et al., 2012). Here, we developed a mouse model in which the Sphk1 gene was specifically disrupted in ECs (Pappu et al., 2007) so that the mutant ECs were unable to produce lung interstitial S1P. We found that S1P is secreted from normal lung ECs into the bronchioalveolar space following Pseudomonas aeruginosa (PA) infection. The lack of angiocrine S1P in mutant lung results in an airspace-enlargement phenotype with a reduced number of AT1 cells. Our data also showed that S1P acts via S1PR2 and YAP and is required for the progenitor functions of AT2 cells during their transition into AT1 cells.
RESULTS
Angiocrine S1P Is Required for the Recovery of Lung Injury after PA Infection
To study the mechanisms of alveolar repair, we used a mouse lung injury model generated by intratracheal injection (i.t.) of PA (Liu et al., 2011; Sadikot et al., 2006), which is characterized by severe inflammation and alveolar damage 24 to 48 h post-injury and AT2-mediated repair 3–7 days post-injury (Finn et al., 2019; Liu et al., 2015). Based on our previously described endothelial barrier reparative properties of S1P (Natarajan et al., 2013; Tauseef et al., 2008), we further tested the role of S1P as an angiocrine mediator of alveolar epithelial repair in the PA model. We first assessed alveoli S1P levels by measuring S1P concentration in bronchoalveolar lavage (BAL) using LC-mass spectroscopy (Berdyshev et al., 2009). We used BAL instead of homogenized tissue to estimate the interstitial S1P level because it is technically difficult to completely remove the residual serum when isolating lung tissue, and the contaminating plasma could have a high concentration of S1P (Proia and Hla, 2015). We found that at 72 h after injury, BAL S1P levels were significantly increased (Figure 1A), consistent with its potential role as a regulator of alveolar repair, which is initiated at 72 h post-PA (Finn et al., 2019; Liu et al., 2015).
Figure 1. Endothelial-Specific Disruption of S1P Production Resulted in an Airspace-Enlargement Phenotype in Mice after PA Injury.
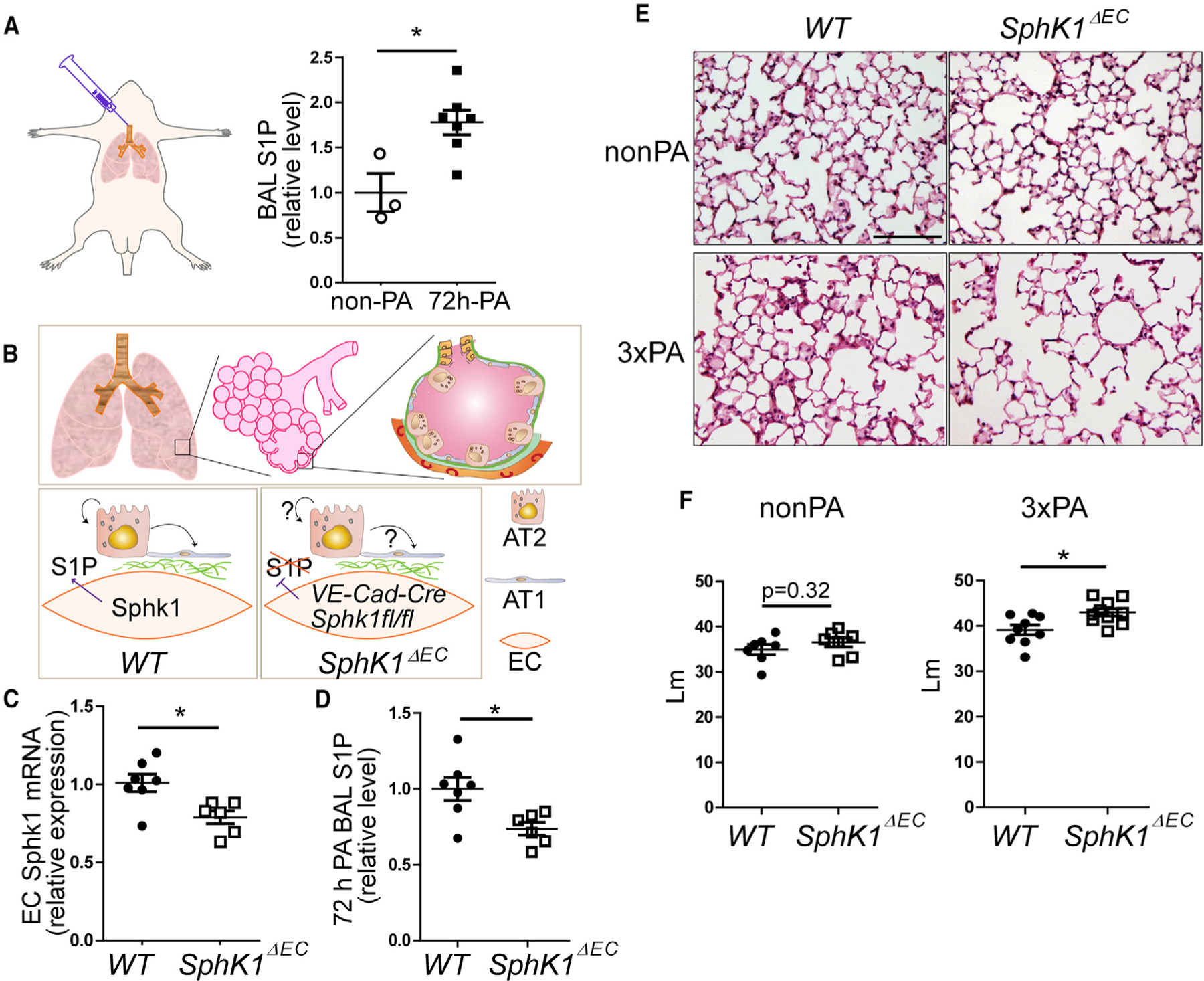
(A) Acute lung injury was induced by intratracheal (i.t.) injection of PA bacteria. BAL fluids were collected from uninjured and 72-h-post-injured lungs, S1P levels in the BAL were measured using LC-mass spectrometry.
(B) Lung alveoli are highly vascularized, with epithelial AT1 and AT2 cells residing closely to endothelial cells. To test the potential function of angiocrine S1P on AT2 cell progenitor function, we created VE-Cadherin-Cre/Sphk1fl/fl mice (SphK1ΔEC), in which Sphk1 expression was specifically disrupted in ECs.
(C) Sphk1 expression in ECs isolated from non-PA-treated WT and SphK1ΔEC mice was analyzed by qPCR.
(D) S1P levels in BAL were measured in WT and SphK1ΔEC mice at 72 h post-PA.
(E) Representative images of HE-stained lung sections of WT and SphK1ΔEC mice without injury (non-PA) or 7 days after the last injection of three repetitive PA injuries at 1-week intervals (33 PA). Scale bar, 100 μm.
(F) Mean linear intercept (Lm) were measured from HE-stained lung sections.
Mean ± SEM. *p < 0.05. See also Figure S1
Because SPHK1 generates extracellular S1P (Pyne and Pyne, 2010), we next generated VE-Cadherin-Cre; Sphk1fl/fl (SphK1ΔEC) mice, in which the gene encoding SPHK1 is specifically disrupted in ECs (Pappu et al., 2007) (Figure 1B), to investigate the role of endothelial S1P in lung epithelial repair. qPCR analysis confirmed the reduced expression of Sphk1 in SphK1ΔEC ECs (Figure 1C), whereas the Sphk1 expression in other cell types (CD45−CD31−), such as AT1 and AT2 cells, was not affected (Figures S1A–S1D). Also, EC expression of the transcript of SPHK2 (Ebenezer et al., 2019) enzyme did not change (Figure S1B). These data show the specificity of SPHK1 disruption in ECs in the mutants.
Next, we compared the alveolar interstitial S1P levels in wild-type (WT) (littermates with the genotype of Sphk1fl/fl were designated as WT) and SphK1ΔEC lungs. In non-PA-treated mice, we detected similar levels of BAL S1P in SphK1ΔEC as in WT (Figure S1E). However, at 72 h post-PA, the BAL S1P concentrations in SphK1ΔEC mice were significantly lower than those of WT (Figure 1D). These data indicate that LMVECs are the main source of the increased alveolar interstitial S1P after PA injury. These results are consistent with the changes of SPHK1 protein expression in lung ECs after PA, as revealed by anti-SPHK1 antibody staining using ECs freshly isolated from WT and SphK1ΔEC lungs (Figures S1F and S1G). Without PA, most ECs from WT lungs expressed low levels of SPHK1. Compared with WT, while most ECs from SphK1ΔEC lungs expressed lower levels of SPHK1 that were almost undetectable, a few ECs from SphK1ΔEC lungs had relatively high levels of SPHK1. This might because the efficiency of Cre recombinase was <100%, and some ECs without Cre-dependent SPHK1 disruption expressed higher levels of SPHK1 due to compensation (Figures S1F and S1G). At 3 days post-PA, SPHK1 expression increased significantly in ECs of WT but not of SphK1ΔEC lungs (Figures S1F and S1G). While SPHK1 expression was disrupted in ECs of the mutant, the endothelial architectures in SphK1ΔEC lungs were similar to WT, as revealed by antibody staining using EC marker CD31 (Komarova and Malik, 2010) (Figure S1H).
To determine whether LMVEC-derived S1P is involved in the recovery of alveolar injury, we compared the morphology of the lung sections between SphK1ΔEC and WT. Without injury, no difference was observed between the two genotypes (Figures 1E and 1F). Next, we assessed whether SphK1ΔEC lungs would exhibit a phenotype in lung injury/repair. At 7 days post-PA, when alveolar repair is mostly completed in WT (Finn et al., 2019), SphK1ΔEC lungs showed a slight enlargement of air spaces (Figure S1I). We next used a repetitive injury model by giving the mice three PA injections at day 1, day 8, and day 15 (3× PA) to mimic prolonged bacterial pneumonia in patients. SphK1ΔEC and WT lungs were dissected at 7 days after the third PA injection. Repeated PA injections resulted in a marked enlargement of the air space in SphK1ΔEC lungs (Figure 1E). The fixed lung volumes, however, did not show changes between SphK1ΔEC and WT (Figure S1J). The airspace-enlargement phenotype was quantified by mean linear intercept (Lm) using images randomly selected from HE-stained lung sections by blinded scoring (Cooney and Thurlbeck, 1982; Dunnill, 1962). Our data showed that with 3× PA, the SphK1ΔEC lungs had 10% higher Lm values compared with WT (Figure 1F). Our Lm measurements could have limitations due to a lack of true random 3D sampling (Hyde et al., 2004; Knust et al., 2009; Weibel et al., 2007). To alleviate this limitation, we compared WT and SphK1ΔEC lungs in parallel and performed several independent experiments. Pairs of WT and SphK1ΔEC processed at the same time were matched to compare the same lobes with similar planes and orientations. While not an accurate quantification of the “true” values of alveolar size/ number, our data represent a valid comparison between WT and SphK1ΔEC lungs and indicate that endothelial S1P release is required for a complete restoration of the alveolar epithelium after PA injury.
Disrupted Angiocrine S1P Production Results in Reduced AT1 Cell Number after Injury
To further characterize the airspace-enlargement phenotype in SphK1ΔEC lungs, we quantified the numbers of AT1 and AT2 cells based on the immunostaining of AT1 marker HopX (homeodomain-only protein X) (Wang et al., 2018) and AT2 marker Sp-C (surfactant protein C; Sftpc) (Liu et al., 2011) (Figures 2A–2D). For each post-PA lung, 5–10 microscopic areas were randomly selected from the regions indicating signs of pneumonia, and >100 cells were scored blindly for each selected area. Without PA, no difference in AT1 or AT2 cell numbers was found between WT and SphK1ΔEC (Figure 2). At 3 days post-PA, the numbers of AT1 and AT2 cells and the AT1/AT2 cell ratio were also similar between WT and SphK1ΔEC (Figure S2A), suggesting similar degrees of AT1 cell damage and/or AT2 cell proliferation. At 7 days post-PA, the numbers of AT1 cells in SphK1ΔEC lungs were slightly lower than those of WT (Figure S2B). With 003× PA, SphK1ΔEC had significantly fewer AT1 cells compared with WT (Figure 2). In contrast, the numbers of AT2 cells in the SphK1ΔEC lungs were similar to WT, regardless of PA injection. While our quantification of AT1 and AT2 cells was not as accurate as that through stereology (Jansing et al., 2018), we carefully selected the same lobe and a similar plane and orientation between WT and SphK1ΔEC and performed several independent experiments to alleviate the limitation. Furthermore, the decreased AT1/AT2 ratio also supported the finding that the AT1 cell number was decreased in SphK1ΔEC lungs compared with WT.
Figure 2. SphK1ΔEC Mice Had Fewer AT1 Cells Than WT after PA.
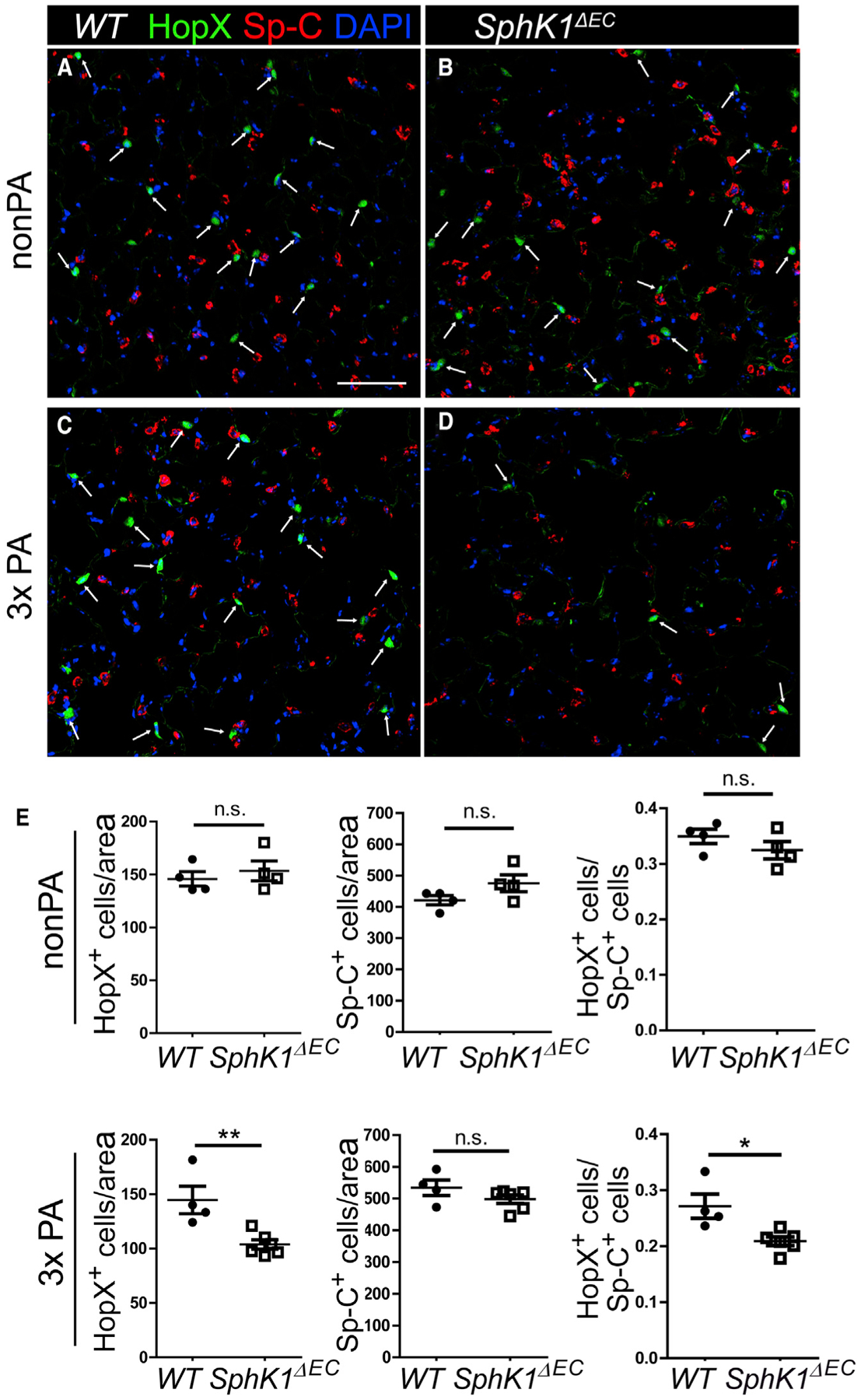
(A–D) Lung sections from uninjured or 33 PA injured WT and SphK1ΔEC mice were stained for Sp-C and HopX. Without injury (A and B), the numbers of HopX+ cells (arrow) were similar between WT (A) and SphK1ΔEC (B) lungs. After 3x PA injury (C and D), more HopX+ AT1 were present in WT (C) than SphK1ΔEC (D) lungs. Scale bar, 100 μm.
(E) The numbers of HopX+ and Sp-C+ cells were counted and normalized by area, and the ratios of HopX+ to Sp-C+ cells were calculated.
Mean ± SEM. *p < 0.05; **p < 0.01. n.s., not significant. See also Figures S2 and S3.
We next investigated the cause of the reduced AT1 cell number in SphK1ΔEC lungs after injury. In the 3× PA model, injuries in some areas of the lungs may not fully recover after one round (7 days) of injury/repair. With these lags, cells in different regions of lungs may be engaged in different cellular processes (e.g., cell death, proliferation, differentiation) during the second or third PA injury, and some cell behaviors may be masked by repetitive injury/repair. Thus, it is complicated to analyze cell behaviors at a certain time in this model. Therefore, to define the effects following each PA injury and analyze the sequential cellular changes that occur in WT and SphK1ΔEC lungs during injury/repair, we only challenged mice with a single PA inoculation in most of the following studies. We first analyzed cell death caused by PA. At 3 days after a single PA inoculation, there was a similar distribution of apoptotic cells in SphK1ΔEC as in WT. By 5 days post-PA, the apoptotic cells mostly disappeared in lungs of both genotypes (Figures S3A–S3D). Next, inflammation in WT and SphK1DEC lungs was accessed by analyzing BAL. At 3 days post-PA, which represents the inflammatory phase of this injury model (Finn et al., 2019), the total cell number, the fraction of neutrophils, and the protein concentration in BAL from SphK1ΔEC were all similar to those in WT (Figure S3E). Consistently, BAL cell counts, neutrophil percentage, and BAL protein at 7 days after 3× PA injection were also similar between WT and SphK1ΔEC (Figure S3F). Thus, the airspace-enlargement phenotype and the associated reduction in AT1 cell number of the mutant were not caused by the increase in lung damage or inflammation. Rather, it is likely that these phenotypes were caused by a disrupted repair process.
Abnormal AT2 Cell Progenitor Phenotype with Defective Angiocrine S1P Production
Since AT2 cells are the progenitor cells in alveolar repair after PA (Liu et al., 2011), the reduction of AT1 cell number and the AT1/ AT2 ratio in SphK1ΔEC suggests that EC-derived S1P affects the reparative function of AT2 cells. To test this, we first analyzed AT2 cell proliferation at the early repair phase (72 h post-PA) by bromodeoxyuridine (BrdU) and Sp-C double staining on lung sections (Figures 3A–3E). By scoring the ratio of BrdU+Sp-C+ cells versus total Sp-C+ cells, we found that the AT2 cell proliferation rate was similar in SphK1ΔEC lungs compared with WT. This result was further confirmed by qPCR analysis using AT2 cells isolated from non-PA SphK1ΔEC and WT lungs and lungs 72 h post-PA. For both genotypes, the expression of cell cycle genes CDC25C and CCNB1 was significantly increased after PA, but no differences were detected between SphK1ΔEC and WT at either time point (Figure 3F).
Figure 3. AT2 Cells Failed to Downregulate Sp-C Expression in SphK1ΔEC Mice after PA.
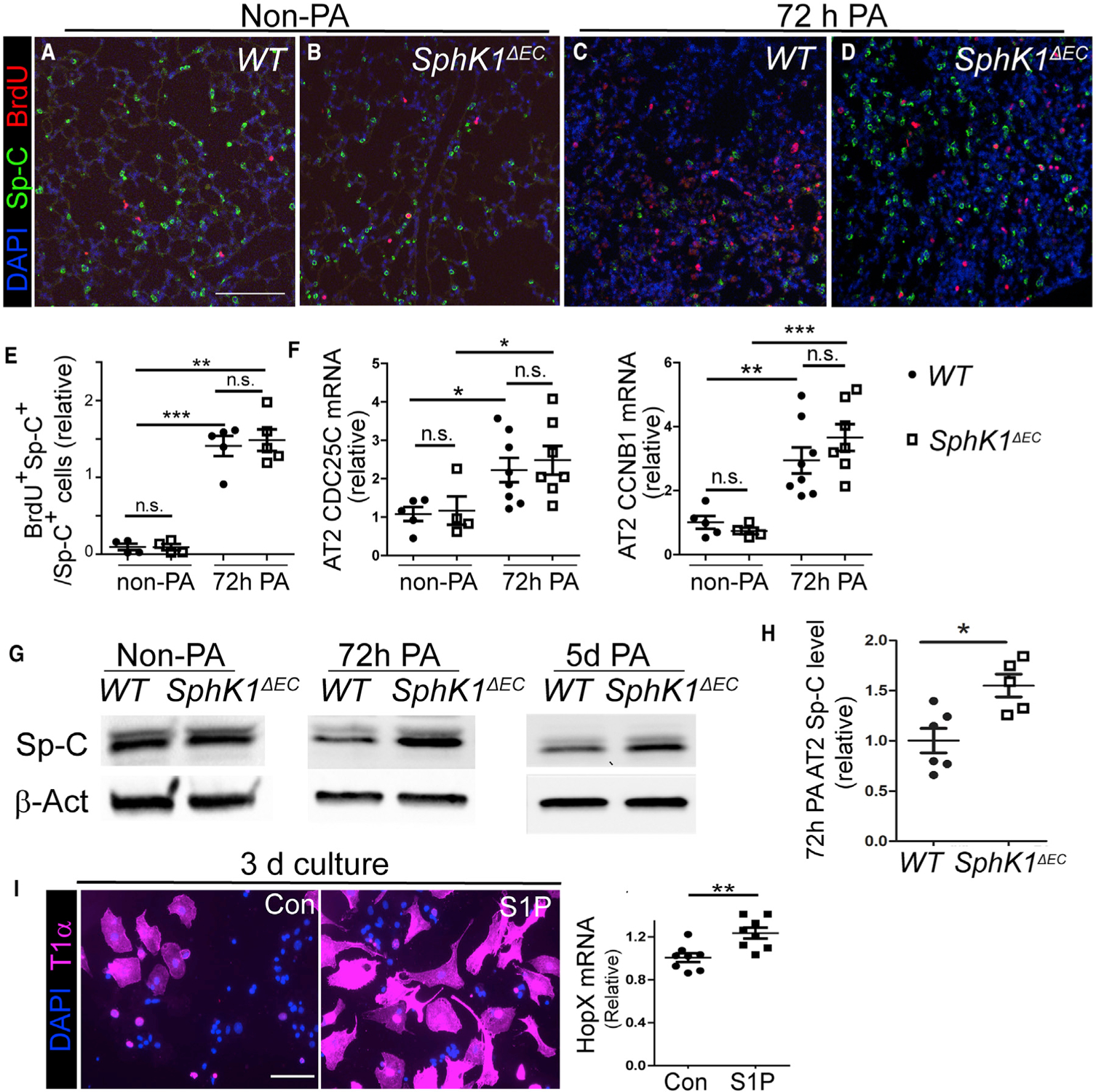
(A–D) Non-PA (A and B) and 72-h-post-PA (C and D) lung sections from WT (A and C) and SphK1ΔEC (B and D) mice were stained for Sp-C and BrdU. Images were taken randomly on non-PA lungs and at areas showing signs of pneumonia on injured lungs. Scale bar, 100 μm. Images are representative of 4–5 lungs for each genotype and condition. The number of Sp-C+BrdU+ cells were similar between WT and SphK1ΔEC without PA (A and B) or at 72-h-post-PA (C and D). In WT, Sp-C expression was reduced at 72-h-post-PA injury (C) compared with non-PA (A). Less reduction was detected in SphK1ΔEC comparing post PA (D) with non-PA (B).
(E) Percentage of BrdU+ AT2 cells that were quantified from lung sections stained with anti-BrdU and Sp-C antibodies.
(F) qPCR of CDC25C and CCNB1 expression in AT2 cells isolated from non-PA and 72-h-post-PA lungs of WT and SphK1ΔEC mice.
(G) AT2 cells were isolated from non-PA, 72-h-post-PA, and 5-day-post-PA lungs of WT and SphK1ΔEC mice and subjected to western blotting analysis for Sp-C.
(H) AT2 cell Sp-C levels normalized by b-actin were compared between WT and SphK1ΔEC at 72 h post-PA.
(I) Freshly isolated AT2 cells were cultured for 3 days and treated with 1 μM S1P or vehicle control. T1α expression in cells was assessed by immunostaining, and HopX expression was assessed by qPCR. Scale bar, 50 μm.
Mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant. See also Figure S4.
We next investigated another aspect of the AT2 cell progenitor functions: differentiation into AT1 cells. Before transitioning to AT1 cells, AT2 cells undergo partial de-differentiation by reducing their expression of AT2-specific proteins, such as Sp-C (Liu et al., 2015). By antibody staining, we observed in WT lungs a reduction of Sp-C expression at 3 days post-PA compared with non-PA lungs (Figures 3A and 3C). This reduction was more prominent at severely damaged areas (Figure S4A), consistent with the patchy nature of PA-induced pneumonia (Liu et al., 2015). The reduction of Sp-C in WT post-PA lungs was not due to cell death, as TUNEL staining did not detect a difference between areas with and without Sp-C reduction (Figure S4A). Moreover, we found that some cells with reduced Sp-C expression were positive for BrdU+ staining (BrdU+Sp-Clo) (Figure S4B), indicating these cells were involved in alveolar repair. In contrast, in SphK1ΔEC lungs, AT2 cells failed to down-regulate their Sp-C expression after PA (Figures 3B and 3D). Consistently, there were fewer BrdU+Sp-Clo AT2 cells in SphK1ΔEC lungs (Figure S4B). The lack of Sp-C reduction in AT2 cells of SphK1ΔEC lungs was quantified by measuring the fluorescence intensity of Sp-C staining of each Sp-C+ cell. At 3 days post-PA, the distributions of Sp-C intensities in SphK1ΔEC lungs were shifted to a higher range compared with WT (Figure S4C). To ensure that we had scored most of the AT2 cells, even if they had decreased Sp-C staining, we examined an AT2 cell lineage labeling mouse line SpC-CreER/ROSA-Tomato (SpC/Tomato) (Finn et al., 2019). In this line, AT2-derived cells can be visualized by Tomato even if they lose Sp-C staining (Finn et al., 2019). Examination using this line confirmed that at 3 days post-PA, there were more Sp-Chi AT2 cells in SphK1ΔEC lungs compared with WT (Figure S4D). These results were further confirmed by western blot, as the Sp-C level in isolated AT2 cells of SphK1ΔEC lungs was higher than WT at 3 and 5 days post-PA (Figures 3G and 3H). Since the loss of Sp-C is a prerequisite step for AT2 to AT1 cell differentiation, the failed AT2 Sp-C reduction at an early repair phase together with the decreased AT1 cell number at a later stage after PA in SphK1ΔEC lung indicates a defective AT2 to AT1 cell transition. Thus, it is likely that angiocrine S1P regulates the progenitor functions of AT2 cells by promoting the AT2 to AT1 cell transition.
Next, we tested whether S1P stimulatesAT2 cells to differentiate into AT1 cells in a 2D culture. AT2 cells were freshly isolated from non-injured WT lungs. The purity of AT2 cells was >90%, and the contamination of AT1 cells was <0.1% (Liu et al., 2015). Because regular fetal bovine serum (FBS) contains high levels of S1P (Proia and Hla, 2015), AT2 cells were seeded in media with 2% charcoal-stripped FBS. After 1 day, S1P was added at 1 μM, and cells were further cultured for 3 days. More AT2 cells in the S1P-treated culture changed into flat AT1-like cells than in controls (Figure 3I). This result was confirmed by qPCR of AT1 cell marker HopX (Figure 3I). These data show the potentially important role of S1P in promoting the AT2 to AT1 cell transition in repair.
S1P Promotes AT2 to AT1 Cell Transition through S1PR2
S1P acts through S1PRs, which are GPCRs located on the membrane of target cells (O’Sullivan and Dev, 2013). We therefore examined the dynamic expressions of each of the five S1PRs (S1PR1–S1PR5) (O’Sullivan and Dev, 2013) in AT2 cells during alveolar repair using AT2 cells isolated from uninjured and 72-h-post-PA lungs. qPCR analysis showed that among all five receptors, only S1PR2 expression was significantly increased after PA (Figure 4A). Western blot also showed that the protein level of S1PR2 was increased in AT2 cells at 5 days post-PA, while AT2 cells were undergoing transition into AT1 cells (Finn et al., 2019), and decreased at 7 days post-PA, when the transition was largely completed (Finn et al., 2019) (Figure 4B).
Figure 4. S1PR2 Expression in AT2 Cells Was Increased during Alveolar Repair and AT2 to AT1 Cell Transition.
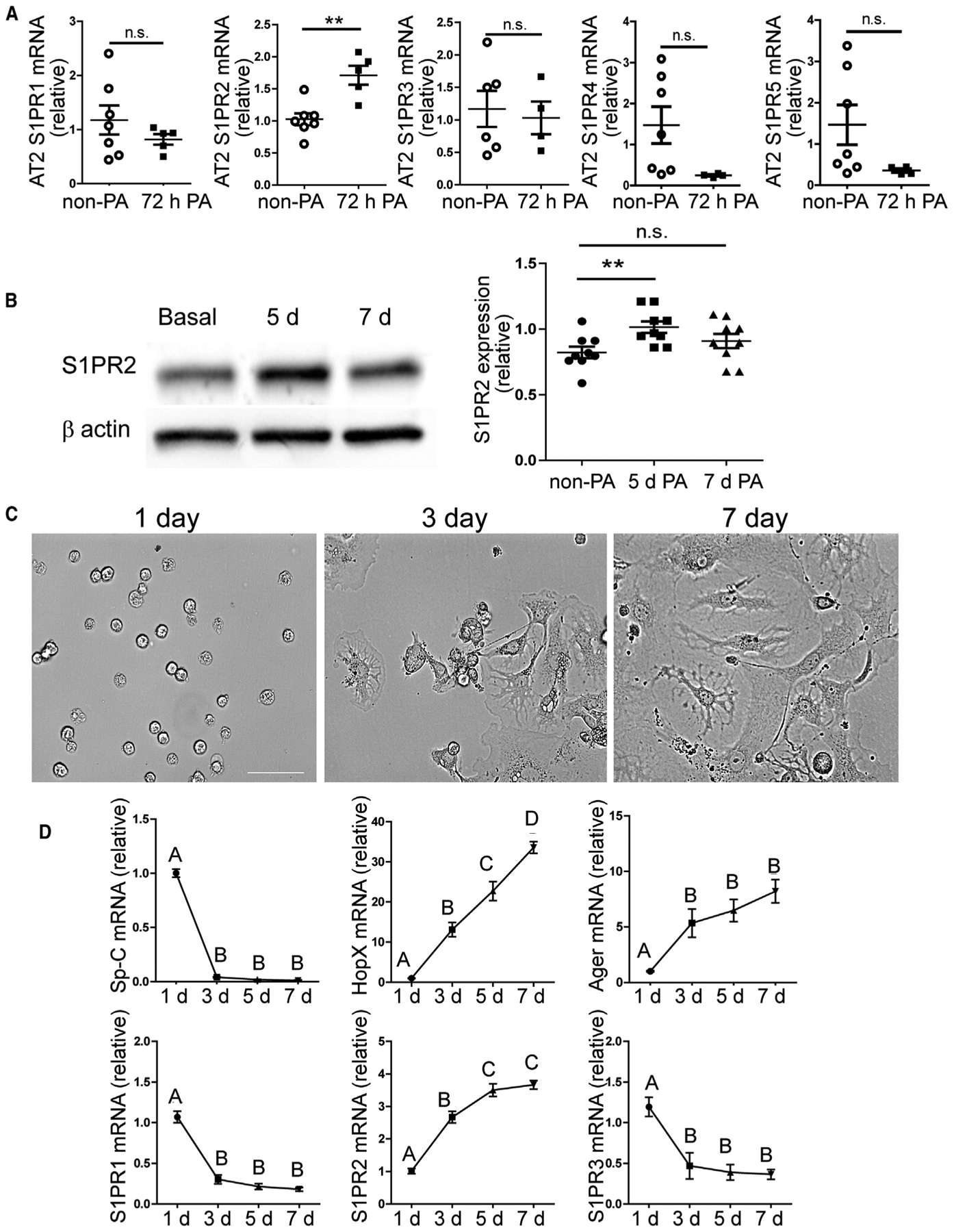
(A) AT2 cells were isolated from uninjured and 72-h-post-injured WT mice, and the expression levels of the five S1P receptors were analyzed by qPCR.
(B) WT AT2 cells isolated at indicated time points were subjected to western blotting analysis for S1PR2, and the S1PR2 levels normalized by β-actin were quantified. Mean ± SEM. **p < 0.01; n.s., not significant.
(C and D) In 2D culture, freshly isolated WT AT2 cells gradually changed from cuboidal shape to AT1-like squamous morphology within 7 days (C), and gene expression levels in cultured cells were measured at indicated time points by qPCR (D). Scale bar (C), 50 μm.
One-way ANOVA with Bonferroni correction was used to determine statistical differences. Groups labeled with different letters are significantly different. See also Figure S5.
To further correlate the temporal S1PR2 expression in AT2 cells with their transition to AT1 cells, we used an in vitro culture of freshly isolated WT AT2 cells. Within 7 days in culture, AT2 cells spontaneously converted from AT2- to AT1-like cells, as shown by the cell morphology changing from cuboidal to flattened squamous shape (Figure 4C). In addition, the expression of AT2 marker Sp-C decreased rapidly, while the expression of AT1 markers HopX and Ager (advanced glycosylation end product-specific receptor) (Yang et al., 2016) showed a steady increase (Finn et al., 2019) (Figure 4D). During this process, S1PR2 expression steadily increased as the cells adopted the AT1 cell phenotypes (Figure 4D). In contrast, S1PR1 and S1PR3 expression was decreased during the AT2 to AT1 cell transition (Figure 4D).
Furthermore, by analyzing recently published AT2 single-cell RNA sequencing (RNA-seq) datasets (Riemondy et al., 2019; Wang et al., 2018), we found that S1PR2 expression levels were markedly higher in AT2 cells than other S1PRs (Figure S5). Furthermore, S1PR2 transcripts were specifically enriched in an AT2 cell sub-population that appeared after LPS-induced lung injury and were undergoing differentiation into AT1 cells (Riemondy et al., 2019) (Figure S5). Thus, our results strongly indicate that S1PR2 mediates the S1P-dependent AT2 to AT1 cell transition in alveolar repair.
To test the function of S1PR2, we utilized an S1PR2-specific antagonist, JTE-013, to block S1P signaling during the AT2 to AT1 cell transition. We started from the in vitro AT2 cell culture (Figure 4C). In the vehicle control, after 3 days in the culture, more than half of the AT2 cells had adopted AT1-like phenotypes by expressing the AT1 cell marker Ager. However, when JTE-013 was included in the media at 10 μM, a significantly lower percentage of cells showed Ager expression (Figure 5A). Consistently, qPCR analysis of the 3-day-cultured cells showed that JTE-013-treated cells expressed a significantly smaller amount of Ager and HopX compared with control. On the other hand, the expression of AT2 marker Sp-C was similar between JTE-013-treated and control cells (Figure 5B). As a comparison, when AT2 cells were cultured for 3 days with 10 μM VPC23019, an antagonist of S1PR1 and S1PR3 (O’Sullivan and Dev, 2013), they expressed similar amounts of Ager and HopX as the vehicle control (Figure 5C). Furthermore, we expressed the short hairpin RNA (shRNA) targeting S1PR2 in cultured AT2 cells using an adenoviral vector. S1PR2 shRNA caused the same phenotype as JTE-013 compared with vector controls (Figure 5D). These results indicate the specificity of the JTE-013 effect.
Figure 5. Inhibition of S1PR2 Function Disrupted AT2 to AT1 Cell Transition In Vitro.
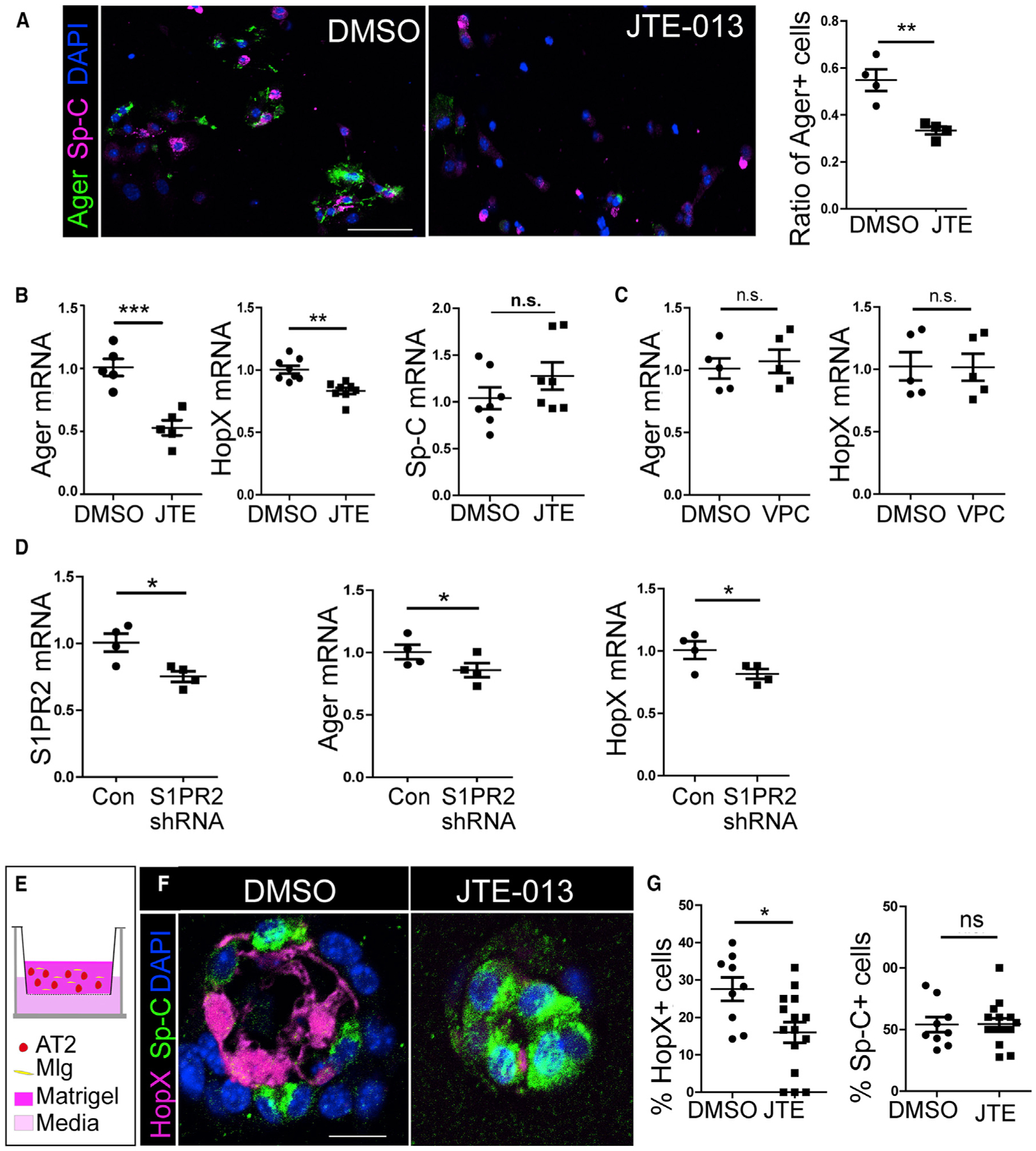
(A) Lineage labeled AT2 cells (from SpC/Tomato mice) were cultured in vitro and treated with DMSO or 10 μM JTE-013. Cells were stained for Ager and Sp-C after 3 days of culture, and the percentage of Ager+ cells over all lineage labeled Tomato+ cells was quantified. For each independent experiment, 200–300 cells were scored. Scale bar, 50 μm.
(B) AT2 cells were cultured for 3 days with DMSO or JTE-013, and expressions of Ager, HopX, and Sp-C were analyzed by qPCR.
(C) AT2 cells cultured with VPC-23019 for 3 days did not show altered expression of Ager and HopX by qPCR analysis.
(D) Freshly isolated AT2 cells were cultured with adenovirus expressing S1PR2 shRNA or vector control for 4 days. The expression of S1PR2, Ager, and HopX was analyzed by qPCR.
(E) Lineage labeled AT2 cells were co-cultured with Mlg fibroblast cells in a 3D organoid culture. JTE-013 or DMSO was added to the medium.
(F and G) After 14 days of culture, organoids were sectioned and stained for HopX and Sp-C (F), and the percentage of HopX+ and Sp-C+ cells over lineage labeled cells (Tomato+) in organoids were quantified (G). More than 200 cells from 10–15 organoids of each condition from two independent experiments were scored.
Scale bar, 10 μm. Mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant.
Next, we examined the effect of JTE-013 on the AT2 to AT1 cell transition using a 3D organoid culture. Tomato+ AT2 cells isolated from an AT2 cell lineage labeling mouse line SpC/Tomato (Liu et al., 2015; Rock et al., 2011) were co-cultured with mouse lung fibroblasts Mlg (ATCC) in Matrigel using transwell inserts (Chen et al., 2017) (Figure 5E). After 14 days, AT2 cells formed alveoli-like colonies. Matrigel plugs with colonies were then fixed and subjected to antibody staining for Sp-C and HopX. AT2 cells cultured with the vehicle control formed cyst-shaped colonies surrounded by AT2 and AT1 cells as described (Chen et al., 2017) (Figure 5F). However, if 10 μM JTE-013 was included in the medium, even though the colony forming efficiency was similar as the control (data not shown), the cysts contained significantly fewer HopX+ AT1 cells, while the percentage of Sp-C+ cells was similar as control (Figures 5F and 5G). These results further support that S1P promotes the AT2 cell transition into AT1 cells through S1PR2.
Next, we tested if JTE-013 also inhibits the AT2 to AT1 cell transition after lung injury in vivo using the AT2 cell lineage tracing mouse line SpC/Tomato (Figure 6A). In this line, tamoxifen (Tmx) induces Cre expression through AT2-specific Sp-C promoter, and Cre deletes the LoxP/Stop/LoxP locus, thus activating the expression of a fluorescent protein Tomato (Liu et al., 2015; Rock et al., 2011). In this way, AT2 cells and their progeny will be permanently labeled with Tomato. The efficiency and specificity of this system were shown by us previously (Finn et al., 2019; Liu et al., 2015). As shown in Figure 6B, SpC/Tomato mice were first injected with Tmx, and then the mice were challenged with PA. At 72 h post-PA, JTE-013 was introduced into these mice (1 mg/kg) via i.t. injection, and the lung sections were examined at 10 days post-PA. We observed that in DMSO-injected control lungs, the AT2 to AT1 cell transition was clearly detected, as shown by numerous Tomato+ lineage traced cells that have acquired AT1-like morphology (Figures 6C, 6E, and 6F). These cells expressed the AT1 marker T1α and HopX (Figures 6C′ and S6). In contrast, JTE-013-injected lungs showed significantly fewer Tomato+ squamous cells (Figures 6D, 6F, and S6). These data further indicate that the S1PR2-dependent AT2 to AT1 cell transition is essential for lung repair in vivo.
Figure 6. JTE-013 Inhibits AT2 to AT1 Cell Transition In Vivo.
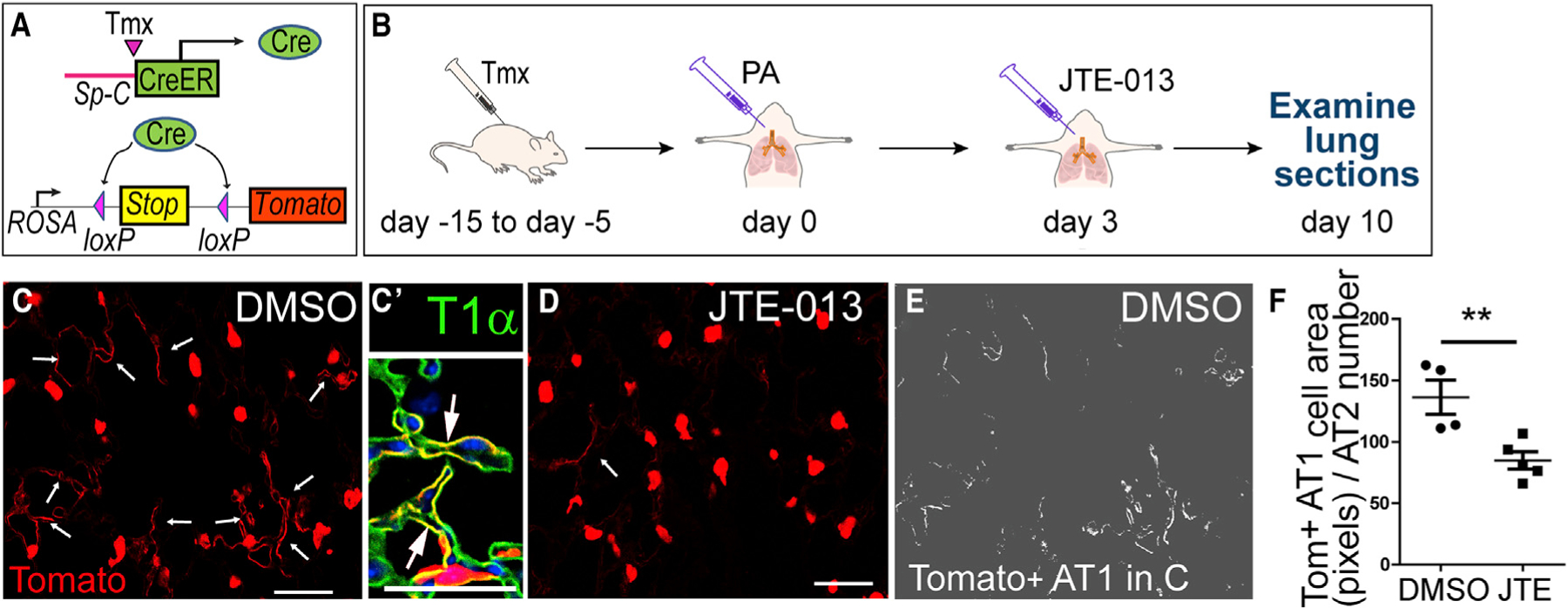
(A) Schematic of inducible AT2 cell lineage tracing mice SpC/Tomato.
(B) Four Tmx injections were given to SpC/Tomato mice at 15 to 5 days before PA injury. JTE-013 or DMSO was i.t. injected into the lungs 3 days after PA, and the lungs were harvested on day 10 after the PA.
(C and D) Lung sections were stained for Tomato to mark AT2 and AT2-derived cells. Fewer AT1-like Tomato+ cells (arrows) were detected in JTE-013-treated lungs (D) than control lungs (C). Scale bar, 30 μm. (C′) Enlarged image of a representative lung section showing squamous Tomato+ cells (arrows) co-expressing T1α. Scale bar, 30 μm.
(E and F) Pixel numbers of Tomato+AT1-like cells (E) were measured and normalized to the number of Tomato+ AT2 cells on the same section and plotted (F).
Mean ± SEM. **p < 0.01. See also Figure S6.
YAP Mediates S1P/S1PR2 Signaling during AT2 to AT1 Cell Transition
Since the transcriptional co-activator YAP plays an important role in regulating AT2 cell progenitor functions (LaCanna et al., 2019; Liu et al., 2016), we tested whether S1P acts through an S1PR2-YAP axis to regulate the AT2 to AT1 cell transition. We found that after isolated AT2 cells were treated with 1 μM S1P for 30 min to 1 h, phospho-YAP (p-YAP) levels were decreased (Figure 7A), indicative of YAP activation (Miller et al., 2012; Yu et al., 2012). Moreover, in 2D AT2 cell culture, we observed that in most control cells, YAP translocated into nucleus (nu-YAP) as the cells transitioned from AT2 to AT1. However, after treated with JTE-013, a significant number of cells had YAP retained in the cytoplasm, even if they adopted a flattened morphology (Figures 7B and 7C). Next, we expressed an active form of YAP (YAP2–5SA) (Zhao et al., 2007) in AT2 cells using adenoviral vectors. Activated YAP rescued the phenotype of JTE-013 treatment, while the vector control had no effect (Figure 7D). Without JTE-013, YAP2–5SA did not show an effect that might reflect that the YAP activity or HopX/Ager expression was saturated in this culture condition (Figure 7D).
Figure 7. YAP Is Downstream of S1P/S1PR2 Signaling in AT2 Cells.
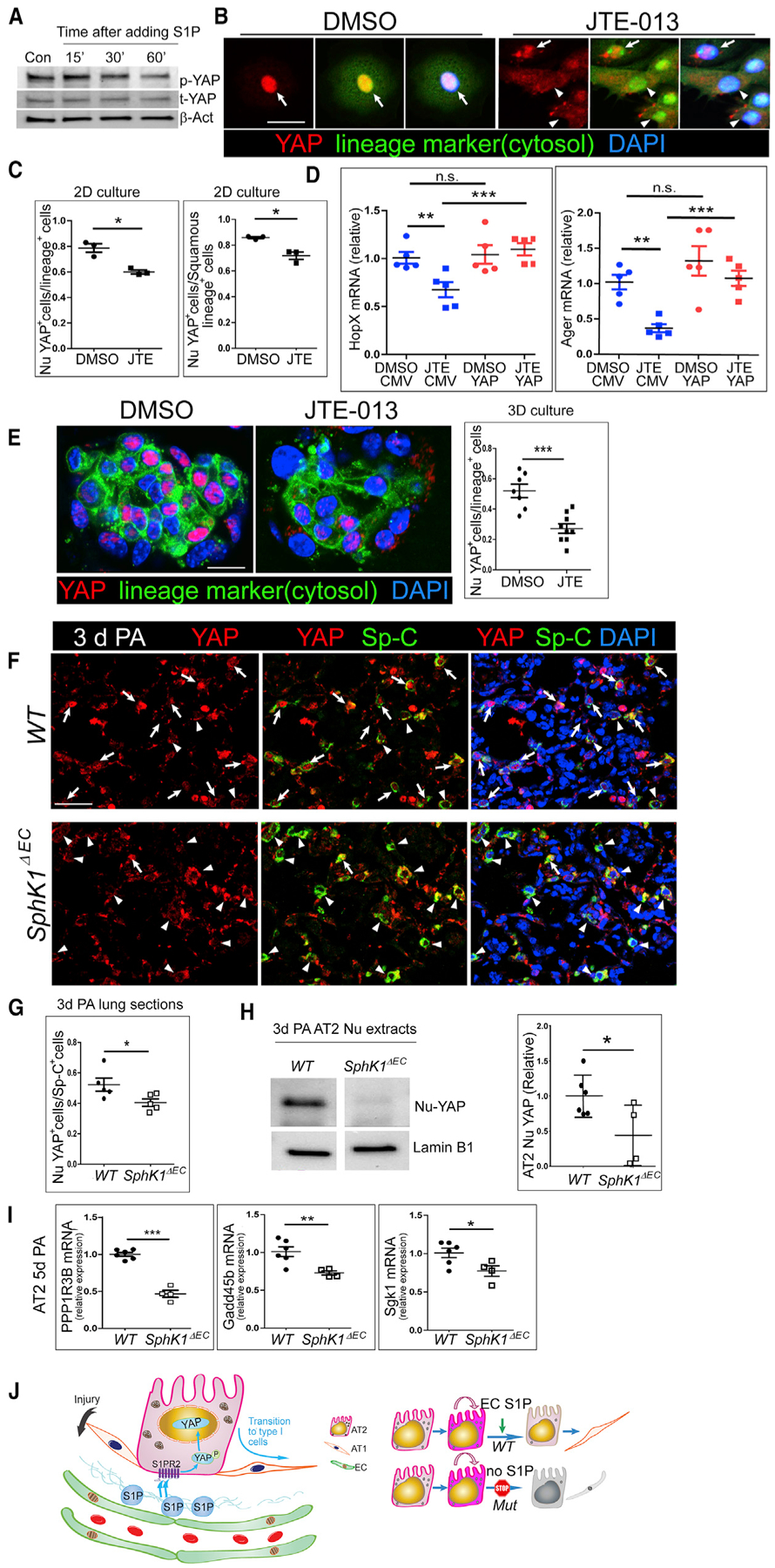
(A) Freshly isolated AT2 cells were cultured overnight and serum starved before treated with 1 μM S1P for the indicated times. Phospho-YAP and total YAP levels in cells were measured by western blot.
(B) Lineage labeled AT2 cells (i.e., GFP+ cells from SpC
CreER/ROSA-mTmG [SpC/mTmG] mice) were isolated and treated with 10 μM JTE-013 or DMSO in culture. Cells were stained for YAP after 3 days of culture. Arrows: nuclear YAP (nu-YAP). Arrowheads: cells with YAP presence in the cytosol. Scale bar, 20 μm.
(C) The percentages of cells with nu-YAP localization among total lineage labeled cells and those among squamous lineage labeled cells were quantified in JTE-013-treated and control cells. More than 100 cells were scored for each condition in each of the three independent experiments.
(D) DMSO and 10-μM JTE-013-treated cells were infected with adenovirus expressing YAP-5SA (YAP) or the vector control (CMV). Cells were collected after 3 days of culture, and expressions of HopX and Ager were analyzed by qPCR.
(E) Lineage labeled (GFP+) AT2 cells were co-cultured with Mlg in a 3D organoid culture and treated with JTE-013 or DMSO. Organoids were sectioned and stained for YAP after 14 days of culture. Scale bar, 10 μm. The percentage of lineage labeled cells showing nu-YAP staining was quantified. More than 100 cells were scored for each condition in three independent experiments.
(F) Lung sections of WT and SphK1ΔEC mice at 72 h post-PA were stained for YAP and Sp-C. Arrow, Nu-YAP; arrowheads, cytosolic YAP. Scale bar, 30 μm.
(G) SphK1ΔEC lungs showed a lower percentage of cells with nu-YAP among the Sp-C+ cells compared with that of WT. For each lung, 5–10 microscopic areas were randomly selected from the regions indicating signs of pneumonia, and at least 100 AT2 cells were scored for each selected area.
(H) Nuclear proteins were extracted from AT2 cells isolated from 72-h-post-PA lungs. The representative image shows protein samples run on the same blot. Relative YAP levels in the nucleus were quantified comparing with Lamin B1.
(I) Ppp1r3b, Gadd45b, and Sgk1 expression in AT2 cells at 5 days post-PA was compared between WT and SphK1ΔEC by qPCR. Mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; n.s., not significant.
(J) Schematic of a model for the role of angiocrine S1P in regulating the progenitor function of AT2 cells. In response to injury, S1P is secreted from ECs and acts through an S1PR2-YAP axis to promote AT2 to AT1 cell transition. In SphK1ΔEC mice, the angiocrine production of S1P is blocked, and AT2 cells proliferate normally but are unable to differentiate into AT1 cells.
See also Figure S7.
Similarly, in 3D organoid culture, significantly fewer JTE-013-treated AT2 cells showed nu-YAP compared with the vehicle control (Figure 7E). This result, together with the data that JTE-013 treatment led to less AT1-like cell formation in 3D organoid (Figures 5E–5G), further shows that YAP mediates the S1P/ S1PR2-induced AT2 to AT1 cell transition. Next, we examined lung sections of WT and SphK1ΔEC mice at 3 days post-PA. In WT, numerous AT2 cells showed nu-YAP, indicating that these cells had active YAP signaling and likely initiated the AT2 to AT1 cell transition. However, in the mutant lung, significantly fewer AT2 cells showed nu-YAP, while the majority of AT2 cells retained cytoplasmic YAP (Figures 7F and 7G). To further quantify the number of AT2 nu-YAP cells, AT2 nuclear extracts were isolated from WT and SphK1ΔEC mice at 3 days post-PA and proceeded to western blot analysis. Our results show that AT2 cells from WT had a significantly higher level of nu-YAP than those of SphK1ΔEC (Figures 7H and S7A). Furthermore, AT2 cells isolated from SphK1ΔEC mice at 5 days post-PA expressed significantly fewer YAP targets such as Ppp1r3b, Gadd45b, and Sgk1 (Dupont et al., 2011; Yu et al., 2012) compared with WT (Figure 7I). Thus, our data showed that S1P acts through S1PR2 and then activates YAP transcription activity to promote the AT2 to AT1 cell transition (Figure 7J).
DISCUSSION
AT2 cells are normally quiescent with a slow turnover rate (Barkauskas et al., 2013; Desai et al., 2014). After injury, they can be activated by signals sent from surrounding niche cells and start to proliferate and further differentiate into AT1 cells to repair the injured alveoli (Zepp et al., 2017). However, the signals that activate the AT2 to AT1 cell transition are poorly defined. Recent studies suggest that ECs can act as a niche to regulate epithelial progenitor functions by releasing protein growth factors (Rafii et al., 2016). Here, we have shown that the LMVEC niche function also requires the release of the bioactive lipid S1P, which serves as an essential trophic signal from ECs to alveolar epithelium in directing the regenerative function of AT2 cells.
Our datashowedthatS1P isnormallymaintainedatlow concentrations in the interstitial space between lung ECs and the epithelial barrier. However, in response to PA-induced inflammatory lung injury, the alveolar interstitial S1P level was significantly increased, consistent with what has been reported by another recent study (Ebenezer et al., 2019). S1P levels are under tight control through the balance between synthesis and degradation (Olivera et al., 2013; Proia and Hla, 2015). ECs account for ~40% of total lung cells (Crapo et al., 1982) and play a major role in producing extracellular S1P (Fukuhara et al., 2012; Olivera et al., 2013; Venkataraman et al., 2008). Using EC-specific Sphk1 knock-out mice SphK1ΔEC, we found that the S1P concentration in mutant BAL, which reflects its interstitial level, was significantly lower than control after PA. Thus, SPHK1 expressed in LMVECs plays a major role in producing the increased level of interstitial S1P after PA. Other than EC-derived S1P, other cells including platelets, erythrocytes, neutrophils, mast cells, and fibroblasts can be sources of S1P (Proia and Hla, 2015). Edema caused by vasculature leakage might also cause an increased tissue S1P (Tauseef et al., 2008). The S1P level is also controlled by S1P phosphatase (Venkataraman et al., 2008), S1P lysase (Kunkel et al., 2013; Spiegel and Milstien, 2011), S1P lyase, and S1P transporter Spns2 (Pyne and Pyne, 2010). Thus, it is important to further study the mechanisms that regulate the alveoli interstitial S1P level post-injury.
Our studies have identified an important role of S1P signaling in maintaining the integrity and homeostasis of the alveolar epithelial barrier by regulating the regenerative function of AT2 cells. We did not detect abnormalities in immune responses and cell survival in SphK1ΔEC lungs after PA injury; these results are consistent with the report that SPHK2, but not SPHK1, regulates lung inflammatory responses after PA infection (Ebenezer et al., 2019). Importantly, we found that the AT2 cell progenitor function was impaired when ECs’ production of S1P was disrupted. We found that in SphK1ΔEC mice, while AT2 cell proliferation was not affected, the transition from AT2 to AT1 cells was blocked. AT2 cells of the mutant lungs failed to downregulate their Sp-C expression at the repair phase after injury, which is a prerequisite step for AT2 cells to differentiate into AT1 cells. Consistently, SphK1ΔEC lungs exhibited a reduced number of AT1 cells post-PA injury. Even though we cannot rule out that other cells such as fibroblasts or ECMs (extracellular matrix) could also be affected by reduced alveoli S1P level after injury (Petrache and Berdyshev, 2016), we believe that the reduced AT1 cell number is a main contributor to the airspace-enlargement phenotype we observed in the mutant lungs.
In search of the S1P receptors that mediate the AT2 to AT1 cell transition, we found that S1PR2 expression was increased in AT2 cells during their transition to AT1 cells. Moreover, by analyzing recently published single-cell RNA-seq data (Riemondy et al., 2019), we found that S1PR2 transcripts are enriched in the subpopulation of AT2 cells that undergo the transition into AT1 cells (Riemondy et al., 2019). Furthermore, we have demonstrated that blocking S1PR2 using the JTE-013 inhibitor as well as shRNA targeting S1PR2 specifically impaired the AT2 to AT1 cell transition. Thus, our data established an important role of S1PR2 in transducing the S1P signaling in AT2 cells and mediating the AT2 to AT1 cell differentiation.
Our data also show that YAP mediates S1P-S1PR2 signaling in AT2 cells. YAP is a central component of the Hippo signaling and is an important regulator of development and homeostasis in multiple tissues (Misra and Irvine, 2018; Yu et al., 2015). It has been shown in several tissues that Hippo/YAP signaling can regulate regenerative cell functions such as proliferation, pluripotency, and cell fate determination (Misra and Irvine, 2018; Yu et al., 2015). Other core components of the Hippo/ YAP pathway include Hippo and Lats1/2 kinase (Misra and Irvine, 2018; Yu et al., 2015). When Hippo is activated, YAP is phosphorylated by Lats1/2 and remains in the cytoplasm. When dephosphorylated, YAP is activated and translocated into the nucleus, where it interacts with other transcriptional co-factors to regulate downstream targets (Misra and Irvine, 2018; Yu et al., 2015). Hippo pathway genes play key roles during lung development and remodeling (Lange et al., 2015; Mahoney et al., 2014; Nantie et al., 2018); YAP activation is also required for the AT2 to AT1 cell transition after lung injury (LaCanna et al., 2019; Liu et al., 2016; Zhou et al., 2018). However, the upstream signals that activate YAP in AT2 cells in response to injury are unknown. Here, we showed that EC-niche-generated S1P is an important signal to activate AT2 cell YAP through S1PR2.
A number of cues, such as change in cell adhesion, mechanical tension, and metabolic status, are known to regulate the Hippo-YAP signaling (Misra and Irvine, 2018; Yu et al., 2015). The S1P/S1PR2-dependent YAP activation has been shown in several other cell types and may represent a specific branch of the YAP activation pathway (Miller et al., 2012; Yu et al., 2012). During pancreas development, S1PR2 expressed on pancreatic multipotent progenitor cells transduces signals that result in the stabilization of YAP and the differentiation of these cells toward the endocrine and acinar lineages (Serafimidis et al., 2017). S1PS1PR2-YAP signaling also plays important roles in the development of zebra fish endoderm (Fukui et al., 2014). These findings and our study indicate that the S1P/S1PR2/YAP axis represents a conserved but poorly characterized signaling pathway in regulating the functions of progenitor cells during development, repair, and regeneration.
Our data show that the S1P-S1PR2-YAP signaling axis plays an essential role in the differentiation of AT2 cells to AT1 cells. We did not find a role of this signaling axis in AT2 cell proliferation or in modulating the inflammatory response. In a pneumonectomy (PNX) model, YAP appears to also mediate AT2 cell proliferation in addition to the AT2 to AT1 cell transition during alveolar regeneration (Liu et al., 2016). Another study suggested that YAP might also regulate inflammatory response through NFκB/IκB (LaCanna et al., 2019). The differences between these reports and ours may have arisen because in those studies, YAP was completely disrupted in AT2 cells, whereas in our study, YAP function was repressed by the lack of S1P/S1PR2 signaling. In fact, we observed a significant decrease of the YAP targets PPP1R3B, Gadd45b, and Sgk1 but not of the other YAP target, Ctgf, that had been shown by these studies (Figure S7B) (LaCanna et al., 2019; Liu et al., 2016). Therefore, it appears that depending on how it is activated, YAP may turn on different sets of downstream targets and play different roles. This may reflect different doses of YAP that translocate into the nucleus or different transcriptional co-activators that were mobilized. Thus, it is important to further dissect the YAP pathways during alveolar repair using various injury models.
In conclusion, we have identified the bioactive lipid S1P as an angiocrine factor that plays an essential role in regulating the progenitor functions of AT2 cells during alveolar epithelial repair. Our finding indicates that LMVECs and AECs are interconnected during the injury/repair process; thus, future studies on lung repair should consider both components. Furthermore, we have identified a distinct biolipid-mediated mechanism in lung repair that could lead to approaches for regeneration therapies. Although S1P itself has pleiotropic effects on various tissues and cell types, specific branches of S1P signaling can be manipulated pharmacologically by S1P receptor antagonists and agonists (Kunkel et al., 2013), as well as by specific small molecules targeting SPHK1 or SPHK2 (Kunkel et al., 2013), which provide opportunities for therapeutic intervention. Thus, characterizing and dissecting the S1P/S1PR2/YAP pathway during alveolar repair is likely to result in additional targets for therapeutic intervention.
STAR+METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information as well as requests for reagents can be directed to the Lead Contact, Dr. Yuru Liu (yuruliu@uic.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate datasets/code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal studies were approved by the Institutional Animal Care Committee of the University of Illinois at Chicago (UIC). All mice used in this study were maintained in the UIC barrier facility with free access to water and food. Male and female mice of 6–12 wk old were used. VE-Cadherin-Cre; Sphk1fl/fl (SphK1ΔEC) mice were generated by crossing B6.FVB-Tg(Cdh5-cre)7Mlia/J; (VE-Cre) mice (stock 006137, The Jackson Laboratory) with Sphk1fl/fl mice (B6;129S-Sphk1tm2Cgh/Mmucd, MMRRC Stock Number: 030038-UCD) (Pappu et al., 2007). SphK1ΔEC mice can be raised into adults with no obvious defect. AT2 lineage labeling lines SpC /Tomato or SpC/mTmG mice were generated by crossing Sftpc-CreERT2 mice (abbreviated SpC-CreER and kindly provided by Dr. Brigid Hogan at Duke University) with B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J (stock 007909, The Jackson Laboratory) or B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J (stock 007976, The Jackson Laboratory) mice. To induce Cre activation, mice were injected 4 times of tamoxifen at every other day for a total dose of 1 mg/g body weight. Tamoxifen-injected mice were then maintained for 1–4 weeks before use in experiments unless otherwise described in Results. Bromodeoxyuridine (BrdU; Sigma-Aldrich) was intraperitoneally injected at 75 mg/kg body weight 5 hours before lung collection.
METHOD DETAILS
Intratracheal injection of PA 103 and JTE-013
Live PA (strain 103) was kindly provided by Dr. Ruxana T Sadikot at Emory University School of Medicine. Live PA 103 was tittered by grown on blood agar plates for 18 h. 2×104 live and 5×106 freeze-killed dead PA was mixed in 20 μl of sterile PBS and administered into each mouse. Mice were anesthetized by injection with ketamine (100mg/kg), xylazine (5mg/kg) and lidocaine (1mg/kg) and the neck surgical site was prepared by shaving and sterilizing with 70% ethanol before PA injection. An approximately 1 cm incision was made to expose the trachea and bacteria was slowly injected. Buprenorphine (0.1mg/kg) was administered as post-operative anal-gesia. JTE-013 (Cayman Chemical) diluted in PBS was injected at 1 μg/g of mice bodyweight at 3 days after PA injection following the same surgical procedure. Same amount of DMSO was injected into control mice.
BAL fluid analysis
Mice were anesthetized with ketamine/xylazine. An incision was made to expose the trachea and a blunt needle was inserted into and tied on the trachea. 0.8ml of ice-cold sterile PBS was gently injected into the lungs and 0.5ml of BAL fluids was withdrawn while gently massaging the thorax of the mice. BAL fluids contaminated with blood were discarded. BAL fluid was centrifuged at 600 × g for 5 min to separate cells from supernatant. S1P levels were measured by LC-mass spectrometry at National Jewish Health. BAL protein concentrations were determined by BCA protein assay (Thermo Fisher Scientific). Neutrophil cell counts were determined by staining cytocentrifuge slides with Hema3 stain (Thermo Fisher Scientific).
Isolation of lung EC and AT2
Lung EC were isolated as described (Cheng et al., 2017; Kovacs-Kàsa et al., 2017; Niethamer et al., 2020; Vila Ellis et al., 2020). Briefly, PBS perfused lungs were chopped into small pieces and digested in 2 mg/ml collagenase I (Life technologies) for 1 h at 37°C. Cells suspension were filtered through a 70 μm cell strainer and pelleted by centrifugation at 1500 rpm for 8 minutes. Red blood cells were removed by RBC lysis buffer (eBioscience). Cells were resuspended in isolation buffer (0.5% BSA, 2mM EDTA, 4.5mg/ml D-glucose in PBS) and incubated with rat anti-CD45 antibodies (Biolegend) followed by anti-rat magnetic Dynabeads (Thermo Fisher Scientific). Negative selected CD45− cells were further incubated with anti-CD31 antibodies (eBioscience) and subsequently with magnetic Dynabeads (Thermo Fisher Scientific). CD45−CD31+ EC and CD45−CD31− nonEC mesenchymal cells were collected. Most of the EC collected should be microvascular ECs (Cheng et al., 2017; Kovacs-Kàsa et al., 2017; Niethamer et al., 2020; Vila Ellis et al., 2020).
For AT2 isolation, 2 mL dispase (Corning) was instilled into perfused lungs through trachea followed by 0.5 mL 1% agarose to seal the upper airway. The lungs were removed and incubate in additional 0.5 mL dispase at room temperature for 45 min. Cells were dissociated and treated with 100 U/ml DNase I in 5 ml culture medium (DMEM+10% FBS + 25mMHEPES + Penicillin/streptomycin+ Gentamicin/Amphotericin+ Antibiotic-Antimytotic) for 10 minutes at room temperature and sequentially filtered through a 70 μm cell strainer (BD Bioscience) and 20 μm nylon gauze (Small Parts). Contamination of other cells was minimized by panning cells on plates coated with anti-CD45 and anti-CD32 antibodies (BioLegend) for 1 hour in a cell culture incubator. Non-adherent AT2 cells were collected and red blood cells were lysed with RBC lysis buffer. Cells were washed and then sequentially incubated with Biotin-Ep-CAM antibodies (Biolegend) and Streptavidin conjugated magnetic beads (BD Biosciences). EpCAM+ cells were selected using a magnetic rack (GE Healthcare). For some experiments, AT2 were isolated by FACS sorting using high speed Beckman Coulter MoFlo sorter in flow cytometry core in UIC.
Harvest and fixation of lungs
Lungs were perfused with 10 mL PBS through the pulmonary artery. 1.5 to 2 mL of 4% paraformaldehyde in PBS was instilled into the lungs through trachea which was later tied and sealed with thread. The lungs were removed and fixed in 4% paraformaldehyde/PBS overnight at 4°C. Paraffin embedding and sectioning was performed by the UIC Histology Core.
Immunofluorescence staining
Paraffin embedded slides were dewaxed and rehydrated. Slides were treated with antigen retrieval buffer (Vector) for 15 min in pressure cooker and permeablized with 0.2% Triton X-100 in PBS before blocked with 10% normal serum for 1 hr at room temperature. When mouse primary antibody was used, sections were also treated with M.O.M mouse IgG blocking reagent (Vector). Cells fixed on coverslips were permeablized and blocked following the same protocol. Slides were incubated with primary antibody overnight at 4°C, followed by fluorescence conjugated secondary antibody incubation. For YAP staining, samples were incubated with a biotin-conjugated secondary antibody and ABC reagent (VECTASTAIN), followed by TSA amplification (PerkinElmer). Images were taken using an LSM-880 Zeiss confocal microscope (Carl Zeiss) or Zeiss ApoTome microscope (Carl Zeiss). Images of the control and treated/mutant samples of same experiment were taken under the same exposure parameters. When images were adjusted for brightness and/or contrast for better demonstration, the same adjustment was applied to control and experimental images.
Cells with positive staining of HopX, BrdU or YAP were scored from stained lung sections. For the non-PA lungs, 5–10 microscopic areas were randomly selected and at least 100 cells were scored for each selected area. For each post PA lung, 5–10 microscopic areas were randomly selected from the regions indicating signs of pneumonia and at least 100 cells were scored blindly for each selected area. For measurement of fluorescence staining intensity, Mean fluorescence intensity (mean gray value) and total fluorescence intensity (integrated density) were measured by ImageJ. At least 100 cells were measured per animal.
Quantification of Lung volume, mean linear intercept (Lm) and numbers of AT1 and AT2 cells from lung sections
WT and SphK1ΔEC mouse lungs were processed in parallel and perfused by PBS as described above. Perfused lungs were then fixed under constant 25 cm water pressure for 20 minutes at room temperature before tied the trachea. The lungs were further fixed at 4°C overnight, washed in PBS and lung volume was measured using a water replacement method. The lung was put in a beaker with ~100 mL of water on a balance, and a looped wired was used to keep the lung fully submerged under water. The weight shown on the balance, which reflected the volume of water displaced by the lung, was recorded. The measurement was repeated twice for each mouse and the average was calculated. To quantify Lm, lungs were further paraffin-embedded, and sections were stained by H&E and images were taken by an Olympus BX51 microscope. For each independent experiment, WT and SphK1ΔEC lungs were processed in parallel, same lobe and similar plane and orientation were selected to compare between each pair of WT and SphK1ΔEC lungs. At least 7 images were randomly taken from each mouse. Grids with random offset were applied to WT and SphK1ΔEC images using ImageJ and Lm was calculated as the sum of the lengths of all lines divided by the number of intersections between alveolar walls (Liu et al., 2017). Both horizontal and vertical lines were analyzed. The investigators were blinded to the genotypes when took the images and quantified the Lm values. To quantify the numbers of AT1 and AT2 cells, paraffin sections were stained with Sp-C and HopX antibodies and 5 to 10 images were taken from each sample within regions showing signs of pneumonia. At least 100 cells were counted on each image and were scored blindly.
Quantitative PCR
Freshly isolated or cultured cells were lysed in TriZol reagent (Life Technologies) and RNA was isolated using a RNeasy kit (QIAGEN). The result RNA was reverse transcribed using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and qPCR was performed on an ABI Viia7 system (Applied Biosystems) using SYBR Green (Roche Life Science). Data is expressed as fold change and Cyclophilin (CLO) was used as housekeeping reference. For Ager expression, the FAM-MGB labeled Taqman assays were used. See Table S1 for primer sequences.
Nucleus Protein Extraction
Fleshly isolated AT2 were incubated in hypotonic buffer (20mM Tris-HCL pH 7.4, 10mM NaCl, 3mM MgCl2) on ice for 15 mins. Cells were gently lysed with 0.5% Nonidet P 40 (Sigma-Aldrich), vortexed for 30 s, followed by centrifugation at 1,000 g at 4°C. Pellets were collected and re-suspended in RIPA buffer supplemented with protease inhibitor, incubated on ice for 30 min, with 30 s vortex in 10 min intervals. Samples were then centrifuged at 14,000 g at 4°C for 10 min. Supernatant was transferred to centrifugal filter tube (Amicon® Ultra 0.5 mL Centrifugal Filters, Sigma-Aldrich), centrifuged at 1,4000 g for 10–20 min to collect concentrated nucleus proteins. Protein concentrations were determined using Pierce Micro BCA assay (Micro BCA Protein Assay Kit, Thermo Scientific).
Western blotting
Cells were lysed in RIPA buffer supplemented with protease inhibitors and protein concentration was determined by BCA assay (Thermo Fisher Scientific). Equal amounts of protein were run on 4% −20% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. Membrane was blocked in 5% skim milk or 5% BSA. Blots were incubated with primary antibodies overnight at 4°C followed by HRP conjugated-secondary antibody for 1–2 h at room temperature. Signals were developed with chemiluminescent substrate (Pierce) and digital images were obtained on ImageQuant LAS 4000 imager (GE Healthcare). Protein band intensities were quantified using the ImageJ program (National Institutes of Health) and values were normalized to β-actin bands.
For western blot using nuclear extracts, equal amounts of protein were mixed in laemmli sample buffer (BRO-RAD, USA) with beta-mercaptoethanol. After boiled at 95°C for 5 mins, protein was separated on 4%–20% SDS-PAGE gels and transferred to PVDF membrane for immunoblotting. Membrane was blocked by incubation in 5% skim milk in Tris buffered saline with 1% Tween-20 (Fisher Scientific). Blots were then probed with YAP (4912S, Cell signaling Technology) and Lamin B1 (ab16048, Abcam) as primary antibody followed by appropriate horseradish peroxidase-conjugated secondary antibody. Blots were developed with chemiluminescent substrate (SuperSignal West Femto Maximum Sensitivity Substrate, Thermo Scientific) and images were obtained with Western Blot Imaging Systems (iBright FL1500 Imaging System, Invitrogen). Quantification of immunoblotting band intensity was performed using iBright analysis software (Invitrogen).
Cell culture
For 2D culture, AT2 cells were isolated from lineage labeling mouse lines using above protocols and seeded in 24-well plates coated with collagen. In experiments in which cells were stained after culture, cells were seeded on coverslips coated with collagen in 24-well plates. In experiments with the inhibitors, JTE-013 and VPC23019 (Cayman Chemical) were dissolved in DMSO according to the manufacturer’s instruction and 10 μM of inhibitors or DMSO of the same volume was added to the culture medium (DMEM + 10% FBS + 25mMHEPES + Penicillin/streptomycin+ Gentamicin/Amphotericin + Antibiotic-Antimytotic). Medium was changed with added inhibitors or DMSO every other day.
For S1P treatment of cultured cells, solid S1P (Cayman chemical) was dissolved and kept in ethanol. Immediately before treatment, ethanol was removed through evaporation and S1P was re-suspended in 4 mg/ml fat-free BSA to make a 125 μM stock. S1P was further diluted from stock to 1 μM in DMEM + 2% charcoal stripped FBS and used to treat the cells. For preparing the charcoal-stripped FBS, 1 g of dextran-coated charcoal (Sigma) was incubated with 50 mL of FBS at 4°C overnight with gentle shaking. The mixture was then centrifuged at 2000 x g for 15 min at 4°C and filtered through 0.22 μm syringe filters to remove the charcoal.
For 3D culture, AT2 cells isolated from SPC/Tomato or SpC/mTmG mice were stained for DAPI after panned on the CD64/CD32 plates. Tomato+DAPI− or GFP+DAPI− cells were sorted using a MoFLo cell sorter (Beckman) located in the Flow Cytometry Core of UIC. 5,000 sorted AT2 cells combined with 1×105 Mlg fibroblast cells were resuspended in 200 μl of medium containing 100 μl growth factor reduced Matrigel (Corning). The cell/Matrigel mixture was seeded in 24-well inserts (pore size 0.4 μm) (Corning) placed in 24-well plates. 600 μl of medium was added to the outside of the insert. DMEM/F-12 supplemented with 10% FBS, penicillin/streptomycin, 1 mM HEPES, and insulin/transferrin/selenium was used in 3D culture. 10 μM JTE-013 or the same amount of DMSO was added to the medium and medium was changed every other day for 2 weeks.
Analysis of published scRNA-seq data
GSM2858342 from the GSE106960 containing postnatal day 60 (P60) AT2 cell data was downloaded from GEO. The data was already filtered and normalized for each cell as indicated on the GEO website therefore the numbers were directly used for generating the scatterplot. GSE113049 was downloaded from GEO and processed in Seurat (Version 2.0) R package for quality control filtering, normalization and clustering. For analyzing S1P receptor expression in uninjured AT2 cells, naive type II cells as identified in the meta-data were extracted out to proceed for analysis. For tSNE clustering and analyzing S1PR2 expression in the subpopulations of regenerating AT2 cells, the whole dataset was used. Genes were removed if expressed in less than 3 cells. Cells were removed if less than 100 genes were detected, nUMI was less than 1024 or greater than 3 standard deviations above the mean of each sample, or the percentage of mitochondria gene was greater than 5%. Gene expression was normalized for each cell by the total expression, multiply by 10,000 and log transformed. The normalized data was used for generating the scatterplot for S1P receptor expression in uninjured AT2 cells. For clustering, normalized expression was scaled and subsequently subjected to principle component analysis. The first 15 PCA dimensions were selected for tSNE projection, with 30 nearest neighbors and a resolution of 1.2. Cell identities were determined by the metadata.
Adenovirus generation
Oligo of S1PR2 shRNA (Mission shRNA TRCN0000028917) was synthesized by IDT and sub-cloned into pShuttle-H1-GFP vector (obtained from the Viral Vector Core at UIC). pCMV-flag-YAP2–5SA plasmid was from Dr. Kunliang Guan (Addgene plasmid #27371) (Zhao et al., 2007) and the YAP2–5SA sequence was sub-cloned into pShuttle-CMV vector. Adenovirus was generated using the AdEasy system (Agilent) following manufacturer’s instruction. Briefly, shuttle vectors carrying genes of interest (or empty vectors controls) were transformed into BJ5183-AD-1 cells (Agilent), which were pre-transformed with pAdEasy-1 plasmids. Correct recombinant clones were amplified in recombinant-deficient strains of cells, XL10-Gold cells (Agilent) (for YAP2–5SA and its control) or DH10B cells (NEB, for S1PR2 shRNA and its control). Plasmids prepared after amplification were linearized and transfected into AD-293 cells using lipofectamine 3000 (Thermo Fisher Scientific). Adenovirus collected from AD-293 cells were re-suspended in PBS. S1PR2 shRNA-expressing adenovirus were tittered based on GFP expression and YAP2–5SA-expressing adenovirus were tittered using an AdEasy viral titer kit (Agilent). S1PR2 shRNA-expressing adenovirus or control adenovirus (MOI = 50) were incubated with AT2 cells for 4 days to knockdown the expression of S1PR2. YAP2–5SA or control adenovirus (MOI = 10) were incubated with cells for 3 days.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are expressed as mean ± SE. Student’s t test was used to determine the statistical significance unless otherwise indicated in the text. A p value of less than 0.05 was considered significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| b-Actin (mouse, Clone AC-74, 1:5000) | Sigma Aldrich | Cat# A2228 RRID AB_476697 |
| pro-Sp-C (rabbit, 1:500) | Millipore | Cat # AB3786, RRID AB_91588 |
| T1a (hamster, 1:50) | Developmental Studies Hybridoma Bank | Cat# 8.1.1, RRID:AB_531893 |
| RFP (rabbit, 1:200) | Rockland Immunochemicals | Cat# 600-401-379, RRIC11182807 |
| GFP (chicken, 1:500) | Aves Laboratories | Cat# GFP-1020, RRID AB_10000240 |
| HopX (mouse, 1:50) | Santa Cruz Biotechnology | Cat# sc-398703, RRID:AB_2687966 |
| Ager (rat, 1:50) | R&D Systems | Cat# MAB1179, RRID:AB_2289349 |
| BrdU (mouse, 1:5) | BD Biosciences | Cat# 347583, RRID:AB_400327 |
| YAP (rabbit, 1:100) | Cell Signaling Technology | Cat# 4912, RRID:AB_2218911 |
| Phospho-Yap (1:100) | Cell Signaling Technology | Cat# 4911, RRID:AB_2218913 |
| YAP (mouse, 1:50) | Santa Cruz Biotechnology | Cat# sc-101199, RRID:AB_1131430 |
| EpCAM-APC (CD326, 1:250) | BioLegend | Cat# 118213, RRID:AB_1134105 |
| Lamin B1 (rabbit, 1:1000) | Abcam | Cat# ab16048, RRID:AB_10107828 |
| S1PR2 (EDG5, mouse, 1:250) | Santa Cruz Biotechnology | Cat# sc-365963, RRID:AB_10844763 |
| SPHK1 (mouse, 1:50) | Santa Cruz Biotechnology | Cat# sc-365401, RRID:AB_10859210 |
| CD31 (rat, 1:50) | BD Bioscience | Cat# 550274, RRID:AB_393571 |
| CD31 (For cell isolation, rat, 1:100) | BD Bioscience | Cat# 557355, RRID:AB_396660 |
| Biotin-EpCAM(CD326, for cell isoaltion, rat, 1:50) | Thermo Fisher Scientific | Cat# 13-5791-82, RRID:AB_1659713 |
| Bacterial and Virus Strains | ||
| Pseudomonas aeruginosa P103 | From Dr. Ruxana Sadikot, Emory University | N/A |
| BJ5183-AD-1 electroporation competent cell | Agilent | Cat# 200157 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma Aldrich | Cat# T5648 |
| Dispase | Corning | Cat# 354235 |
| JTE-013 | Cayman Chemical | Cat# 10009458 |
| VPC23019 | Cayman Chemical | Cat# 13240 |
| RIPA buffer | Cell Signaling Technology | Cat# 9803 |
| Complete Protease Inhibitor Cocktail | Roche | Cat# 11-836-145-001 |
| Matrigel | Corning | Cat# 354230 |
| SYBR Green | Roche | Cat# 04913914001 |
| Sphingosine-1-phosphate | Cayman Chemical | Cat# 62570 |
| Critical Commercial Assays | ||
| VECTASTAIN Elite ABC-Peroxidase Kit | Vector Laboratories | Cat# PK-6200, RRID:AB_2336826 |
| TSA Cyanine 3 System | Perkin Elmer | Cat# NEL704A001KT, RRID:AB_2572409 |
| RNeasy mini kit | QIAGEN | Cat# 74104 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 43–688-14 |
| Micro BCA Protein Assay kit | Thermo Scientific | Cat# 23235 |
| in situ Cell Death Kit TMR | Sigma Aldrich | Cat# 12156792910 |
| AdEasy Viral Titer Kit | Agilent | Cat# 972500 |
| Deposited Data | ||
| scRNA sequencing | NCBI Gene Expression Omnibus (GEO) database | GEO: GSE 113049 |
| scRNA sequencing | NCBI Gene Expression Omnibus (GEO) database | GEO: GSE 106960 |
| Experimental Models: Cell Lines | ||
| Mlg mouse lung fibroblast cells | ATCC | CCL-206 |
| AD-293 | From Dr. Jody Martin, UIC Viral Vector Core | N/A |
| Experimental Models: Organisms/Strains | ||
| SpC-CreER (Sfptctm(cre/ERT2)Blh) | From Dr. Brigid Hogan, Duke University | N/A |
| Rosa-Tomato (B6.Cg-Gt(ROSA) 26Sortm14(CAG-tdTomato)Hze/j) | The Jackson Laboratory | Stock# 007914 |
| mTmG (Gt(ROSA) 26sortm4(ACTB-tdTomato,-EGFP)Luo/j) | The Jackson Laboratory | Stock# 007576 |
| B6.FVB-Tg(Cdh5-cre)7Mlia/J; (VE-Cre) | The Jackson Laboratory | Stock# 006137 |
| Sphk1fl/fl (B6;129S-Sphk1 tm2Cgh/Mmucd) | MMRRC | Stock# 030038-UCD |
| Oligonucleotides | ||
| qPCR primers | Table S1 | IDT, QIAGEN or Thermo Fisher |
| Recombinant DNA | ||
| pShuttle-CMV | From Dr. Jody Martin, UIC Viral Vector Core | N/A |
| pCMV-flag YAP2 5SA | Addgene | Addgene_27371 |
| pShuttle-H1-GFP | From Dr. Jody Martin, UIC Viral Vector Core | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| iBright Analysis Software | Thermo Fisher Sicentific | https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-assays-analysis/western-blotting/detect-proteins-western-blot/western-blot-imaging-analysis/ibright-western-blot-imaging-systems/ibright-analysis-software-connectivity.html |
| Prism 5.01 | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| R 3.6 | R project | https://www.r-project.org |
| Seurat 2.0 | Satija lab | https://satijalab.org/seurat/ |
| ZEN v2.3 | Zeiss Microscopy | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| QuantStudio v1.3 | Applied Biosystems | https://www.thermofisher.com/us/en/home/global/forms/life-science/quantstudio-6–7-flex-software.html |
Highlights.
S1P is released from lung endothelial cells upon Pseudomonas aeruginosa injury
Angiocrine S1P promotes alveolar type II to type I cell transition
S1P regulates type II cell progenitor functions via a S1PR2-YAP signaling axis
ACKNOWLEDGMENTS
This work was supported by grants NHLBI R01 HL105947 (Y.L.), AHA17GRNT33700246 (Y.L.), AHA19POST34380566 (Q.C.), NHLBI P01 HL060678 (A.B.M., J.R., and V.N.), NHLBI R01 HL090152 (A.B.M. and J.R.), and NHLBI R01 HL127342 (V.N.). We thank Dr. Evgeny Berdyshev (National Jewish) for help in measuring the BAL S1P level.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107828.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Barkauskas CE, Cronce MJ, Rackley CR, Bowie EJ, Keene DR, Stripp BR, Randell SH, Noble PW, and Hogan BL (2013). Type 2 alveolar cells are stem cells in adult lung. J. Clin. Invest 123, 3025–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdyshev EV, Gorshkova I, Skobeleva A, Bittman R, Lu X, Dudek SM, Mirzapoiazova T, Garcia JG, and Natarajan V (2009). FTY720 inhibits ceramide synthases and up-regulates dihydrosphingosine 1-phosphate formation in human lung endothelial cells. J. Biol. Chem 284, 5467–5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Ye T, Sun Y, Ji G, Shido K, Chen Y, Luo L, Na F, Li X, Huang Z, et al. (2017). Targeting the vascular and perivascular niches as a regenerative therapy for lung and liver fibrosis. Sci. Trans. Med 9, eaai8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Suresh Kumar V, Finn J, Jiang D, Liang J, Zhao YY, and Liu Y (2017). CD44high alveolar type II cells show stem cell properties during steady-state alveolar homeostasis. Am. J. Physiol. Lung Cell. Mol. Physiol 313, L41–L51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, Gao X, An S, Mittal M, Vogel SM, et al. (2017). Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J. Clin. Invest 127, 4124– 4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney TP, and Thurlbeck WM (1982). The radial alveolar count method of Emery and Mithal: a reappraisal 1–postnatal lung growth. Thorax 37, 572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crapo JD, Barry BE, Gehr P, Bachofen M, and Weibel ER (1982). Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis 126, 332–337. [DOI] [PubMed] [Google Scholar]
- Desai TJ, Brownfield DG, and Krasnow MA (2014). Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 507, 190–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding BS, Nolan DJ, Guo P, Babazadeh AO, Cao Z, Rosenwaks Z, Crystal RG, Simons M, Sato TN, Worgall S, et al. (2011). Endothelial-derived angiocrine signals induce and sustain regenerative lung alveolarization. Cell 147, 539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnill MS (1962). Quantitative methods in the study of pulmonary pathology. Thorax 17, 320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, et al. (2011). Role of YAP/ TAZ in mechanotransduction. Nature 474, 179–183. [DOI] [PubMed] [Google Scholar]
- Ebenezer DL, Berdyshev EV, Bronova IA, Liu Y, Tiruppathi C, Komarova Y, Benevolenskaya EV, Suryadevara V, Ha AW, Harijith A, et al. (2019). Pseudomonas aeruginosa stimulates nuclear sphingosine-1-phosphate generation and epigenetic regulation of lung inflammatory injury. Thorax 74, 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans MJ, Cabral LJ, Stephens RJ, and Freeman G (1975). Transformation of alveolar type 2 cells to type 1 cells following exposure to NO2. Exp. Mol. Pathol 22, 142–150. [DOI] [PubMed] [Google Scholar]
- Finn J, Sottoriva K, Pajcini KV, Kitajewski JK, Chen C, Zhang W, Malik AB, and Liu Y (2019). Dlk1-Mediated Temporal Regulation of Notch Signaling Is Required for Differentiation of Alveolar Type II to Type I Cells during Repair. Cell Rep. 26, 2942–2954.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara S, Simmons S, Kawamura S, Inoue A, Orba Y, Tokudome T, Sunden Y, Arai Y, Moriwaki K, Ishida J, et al. (2012). The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J. Clin. Invest 122, 1416–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui H, Terai K, Nakajima H, Chiba A, Fukuhara S, and Mochizuki N (2014). S1P-Yap1 signaling regulates endoderm formation required for cardiac precursor cell migration in zebrafish. Dev. Cell 31, 128–136. [DOI] [PubMed] [Google Scholar]
- Hyde DM, Tyler NK, Putney LF, Singh P, and Gundersen HJ (2004). Total number and mean size of alveoli in mammalian lung estimated using fractionator sampling and unbiased estimates of the Euler characteristic of alveolar openings. Anat. Rec. A Discov. Mol. Cell. Evol. Biol 277, 216–226. [DOI] [PubMed] [Google Scholar]
- Jansing NL, Patel N, McClendon J, Redente EF, Henson PM, Tuder RM, Hyde DM, Nyengaard JR, and Zemans RL (2018). Flow Cytometry Underestimates and Planimetry Overestimates Alveolar Epithelial Type 2 Cell Expansion after Lung Injury. Am. J. Respir. Crit. Care Med 198, 390–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knust J, Ochs M, Gundersen HJ, and Nyengaard JR (2009). Stereological estimates of alveolar number and size and capillary length and surface area in mice lungs. Anat. Rec. (Hoboken) 292, 113–122. [DOI] [PubMed] [Google Scholar]
- Komarova Y, and Malik AB (2010). Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol 72, 463–493. [DOI] [PubMed] [Google Scholar]
- Kovacs-Kàsa A, Varn MN, Verin AD, and Gonzales JN (2017). Method for the Culture of Mouse Pulmonary Microvascular Endothelial Cells. Sci. Pages Pulmonol 1, 7–18. [PMC free article] [PubMed] [Google Scholar]
- Kunkel GT, Maceyka M, Milstien S, and Spiegel S (2013). Targeting the sphingosine-1-phosphate axis in cancer, inflammation and beyond. Nat. Rev. Drug Discov 12, 688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCanna R, Liccardo D, Zhang P, Tragesser L, Wang Y, Cao T, Chapman HA, Morrisey EE, Shen H, Koch WJ, et al. (2019). Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J. Clin. Invest 129, 2107–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange AW, Sridharan A, Xu Y, Stripp BR, Perl AK, and Whitsett JA (2015). Hippo/Yap signaling controls epithelial progenitor cell proliferation and differentiation in the embryonic and adult lung. J. Mol. Cell Biol 7, 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Bhang DH, Beede A, Huang TL, Stripp BR, Bloch KD, Wagers AJ, Tseng YH, Ryeom S, and Kim CF (2014). Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis. Cell 156, 440–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sadikot RT, Adami GR, Kalinichenko VV, Pendyala S, Natarajan V, Zhao YY, and Malik AB (2011). FoxM1 mediates the progenitor function of type II epithelial cells in repairing alveolar injury induced by Pseudomonas aeruginosa. J. Exp. Med 208, 1473–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Kumar VS, Zhang W, Rehman J, and Malik AB (2015). Activation of type II cells into regenerative stem cell antigen-1(+) cells during alveolar repair. Am. J. Respir. Cell Mol. Biol 53, 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Wu H, Jiang K, Wang Y, Zhang W, Chu Q, Li J, Huang H, Cai T, Ji H, et al. (2016). MAPK-Mediated YAP Activation Controls Mechanical-Tension-Induced Pulmonary Alveolar Regeneration. Cell Rep. 16, 1810–1819. [DOI] [PubMed] [Google Scholar]
- Liu Z, Fu S, and Tang N (2017). A Standardized Method for Measuring Internal Lung Surface Area via Mouse Pneumonectomy and Prosthesis Implantation. J. Vis. Exp, 56114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney JE, Mori M, Szymaniak AD, Varelas X, and Cardoso WV (2014). The hippo pathway effector Yap controls patterning and differentiation of airway epithelial progenitors. Dev. Cell 30, 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason RJ (2006). Biology of alveolar type II cells. Respirology 11, S12–S15. [DOI] [PubMed] [Google Scholar]
- Matthay MA, Ware LB, and Zimmerman GA (2012). The acute respiratory distress syndrome. J. Clin. Invest 122, 2731–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller E, Yang J, DeRan M, Wu C, Su AI, Bonamy GM, Liu J, Peters EC, and Wu X (2012). Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem. Biol 19, 955–962. [DOI] [PubMed] [Google Scholar]
- Misra JR, and Irvine KD (2018). The Hippo Signaling Network and Its Biological Functions. Annu. Rev. Genet 52, 65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nantie LB, Young RE, Paltzer WG, Zhang Y, Johnson RL, Verheyden JM, and Sun X (2018). Lats inactivation reveals hippo function in alveolar type I cell differentiation during lung transition to air breathing. Development 145, dev163105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan V, Dudek SM, Jacobson JR, Moreno-Vinasco L, Huang LS, Abassi T, Mathew B, Zhao Y, Wang L, Bittman R, et al. (2013). Sphingo-sine-1-phosphate, FTY720, and sphingosine-1-phosphate receptors in the pathobiology of acute lung injury. Am. J. Respir. Cell Mol. Biol 49, 6–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethamer TK, Stabler CT, Leach JP, Zepp JA, Morley MP, Babu A, Zhou S, and Morrisey EE (2020). Defining the role of pulmonary endothelial cell heterogeneity in the response to acute lung injury. eLife 9, e53072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan C, and Dev KK (2013). The structure and function of the S1P1 receptor. Trends Pharmacol. Sci 34, 401–412. [DOI] [PubMed] [Google Scholar]
- Olivera A, Allende ML, and Proia RL (2013). Shaping the landscape: metabolic regulation of S1P gradients. Biochim. Biophys. Acta 1831, 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappu R, Schwab SR, Cornelissen I, Pereira JP, Regard JB, Xu Y, Camerer E, Zheng YW, Huang Y, Cyster JG, and Coughlin SR (2007). Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 316, 295–298. [DOI] [PubMed] [Google Scholar]
- Petrache I, and Berdyshev EV (2016). Ceramide Signaling and Metabolism in Pathophysiological States of the Lung. Annu. Rev. Physiol 78, 463–480. [DOI] [PubMed] [Google Scholar]
- Proia RL, and Hla T (2015). Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J. Clin. Invest 125, 1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne NJ, and Pyne S (2010). Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 10, 489–503. [DOI] [PubMed] [Google Scholar]
- Rafii S, Butler JM, and Ding BS (2016). Angiocrine functions of organ-specific endothelial cells. Nature 529, 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RM, Yang L, Garcia-Cardena G, and Luscinskas FW (2007). Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ. Res 101, 234–247. [DOI] [PubMed] [Google Scholar]
- Riemondy KA, Jansing NL, Jiang P, Redente EF, Gillen AE, Fu R, Miller AJ, Spence JR, Gerber AN, Hesselberth JR, et al. (2019). Single cell RNA sequencing identifies TGFbeta as a key regenerative cue following LPS-induced lung injury. JCI Insight 5, e123637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, and Hogan BL (2011). Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 108, E1475–E1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen H, Stevens RC, Hanson M, Roberts E, and Oldstone MB (2013). Sphingosine-1-phosphate and its receptors: structure, signaling, and influence. Annu. Rev. Biochem 82, 637–662. [DOI] [PubMed] [Google Scholar]
- Sadikot RT, Zeng H, Joo M, Everhart MB, Sherrill TP, Li B, Cheng DS, Yull FE, Christman JW, and Blackwell TS (2006). Targeted immuno-modulation of the NF-kappaB pathway in airway epithelium impacts host defense against Pseudomonas aeruginosa. J. Immunol 176, 4923–4930. [DOI] [PubMed] [Google Scholar]
- Schneeberger EE (1997). Alveolar Type I Cells In The Lung: Scientific foundations, Crystal RG, West JB, Weibel ER, and Barnes PJ, eds. (Lippincott Williams & Wilkins; ), pp. 535–542. [Google Scholar]
- Serafimidis I, Rodriguez-Aznar E, Lesche M, Yoshioka K, Takuwa Y, Dahl A, Pan D, and Gavalas A (2017). Pancreas lineage allocation and specification are regulated by sphingosine-1-phosphate signalling. PLoS Biol. 15, e2000949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S, and Milstien S (2003). Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat. Rev. Mol. Cell Biol 4, 397–407. [DOI] [PubMed] [Google Scholar]
- Spiegel S, and Milstien S (2011). The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol 11, 403–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauseef M, Kini V, Knezevic N, Brannan M, Ramchandaran R, Fyrst H, Saba J, Vogel SM, Malik AB, and Mehta D (2008). Activation of sphingo-sine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ. Res 103, 1164–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman K, Lee YM, Michaud J, Thangada S, Ai Y, Bonkovsky HL, Parikh NS, Habrukowich C, and Hla T (2008). Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res 102, 669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila Ellis L, Cain MP, Hutchison V, Flodby P, Crandall ED, Borok Z, Zhou B, Ostrin EJ, Wythe JD, and Chen J (2020). Epithelial Vegfa Spec-ifies a Distinct Endothelial Population in the Mouse Lung. Dev. Cell 52, 617– 630.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tang Z, Huang H, Li J, Wang Z, Yu Y, Zhang C, Li J, Dai H, Wang F, et al. (2018). Pulmonary alveolar type I cell population consists of two distinct subtypes that differ in cell fate. Proc. Natl. Acad. Sci. USA 115, 2407– 2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel ER (2009). What makes a good lung? Swiss Med. Wkly 139, 375–386. [DOI] [PubMed] [Google Scholar]
- Weibel ER, Hsia CC, and Ochs M (2007). How much is there really? Why stereology is essential in lung morphometry. J. Appl. Physiol 102, 459–467. [DOI] [PubMed] [Google Scholar]
- Yang J, Hernandez BJ, Martinez Alanis D, Narvaez del Pilar O, Vila-Ellis L, Akiyama H, Evans SE, Ostrin EJ, and Chen J (2016). The development and plasticity of alveolar type 1 cells. Development 143, 54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, Zhao J, Yuan H, Tumaneng K, Li H, et al. (2012). Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 150, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, and Guan KL (2015). Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 163, 811–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zepp JA, Zacharias WJ, Frank DB, Cavanaugh CA, Zhou S, Morley MP, and Morrisey EE (2017). Distinct Mesenchymal Lineages and Niches Promote Epithelial Self-Renewal and Myofibrogenesis in the Lung. Cell 170, 1134–1148.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. (2007). Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 21, 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, Flodby P, Luo J, Castillo DR, Liu Y, Yu FX, McConnell A, Varghese B, Li G, Chimge NO, et al. (2018). Claudin-18-mediated YAP activity regulates lung stem and progenitor cell homeostasis and tumorigenesis. J. Clin. Invest 128, 970–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate datasets/code.