

Abstract
γ-Secretase was initially defined as a proteolytic activity that cleaves within the transmembrane of the amyloid precursor protein (APP) to produce the amyloid β-peptide of Alzheimer’s disease. The discovery of mutations in APP and the presenilins associated with familial Alzheimer’s disease and their effects on APP processing dovetailed with pharmacological studies on γ-secretase, leading to the revelation that presenilins are unprecedented membrane-embedded aspartyl proteases. Other members of what became known as the γ-secretase complex were subsequently identified. In parallel with these advances, connections between presenilins and Notch receptors essential to metazoan development became evident, resulting in the concurrent realization that γ-secretase also carries out intramembrane proteolysis of Notch as part of its signaling mechanism. Substantial progress has been made toward elucidating how γ-secretase carries out complex processing of transmembrane domains, how it goes awry in familial Alzheimer’s disease, the scope of its substrates, and the atomic details of its structure. Critical questions remain for future study, toward further unraveling the complexity of this unique membrane-embedded proteolytic machine and its roles in biology and disease.
Keywords: protease, membrane proteins, amyloid, Notch, Alzheimer’s disease
1. Introduction
γ-Secretase is a proteolytic ensemble of four integral membrane proteins that carries out hydrolysis within the lipid bilayer, processing the transmembrane domain (TMD) of over 90 known substrates [1–3]. With so many substrates, γ-secretase is considered “the proteasome of the membrane” [4], clearing out membrane protein stubs remaining after ectodomain shedding. In certain cases, these TMD cleavage events are part of cell signaling pathways, including Notch signaling essential to development of all metazoans [5]. In one particular case, involving the amyloid precursor protein (APP), complex TMD cleavage to the amyloid β-peptide (Aβ) by γ-secretase is aberrant in dominantly inherited, early-onset familial Alzheimer’s disease (FAD) [6].
Thus, the complexity of γ-secretase can be appreciated on many levels. One level is structural and biochemical. The four membrane components of the enzyme assemble together to become an active protease capable of bringing water to the active site located within the hydrophobic environment of the lipid bilayer [7–9]. The protease complex must find, recognize, and unwind the α-helical TMD substrates to expose the scissile amide bond for hydrolysis. γ-Secretase then carries out processive proteolysis to trim down initially formed membrane-bound products to shorter secreted forms [10–14]. All these biochemical events are incompletely understood in mechanistic and structural detail.
A second level of complexity is the myriad and essential roles of γ-secretase in biology. Knockout of enzyme components results in embryonic lethal phenotypes that closely resemble those seen upon knockout of Notch receptors [15, 16], suggesting that the role of γ-secretase in Notch signaling is its most important biological function and likely the primary driver of evolution of the protease complex. Nevertheless, other key signaling pathways involve γ-secretase processing of TMD substrates [17–19].
A third level of complexity is the role of γ-secretase in human disease, particularly FAD. More than 20 years after the discovery of FAD missense mutations in presenilin [20, 21], the catalytic component of γ-secretase [22], the question of how these mutations alter γ-secretase activity to trigger FAD is still controversial. The issue is further inflamed by the many failures of candidate Alzheimer therapeutics in clinical trials, with no new drug approvals in the U.S. since 2003. As many of these clinical candidates have targeted Aβ, skepticism of the “amyloid hypothesis” of Alzheimer pathogenesis has grown [23].
This overview for the special issue on γ-secretase will recount the critical findings that ultimately led to the discovery of presenilin as the catalytic component of the protease, the identification of other members of the complex, and the realization of the connection between γ-secretase and Notch signaling. Advances in understanding the roles of γ-secretase in biology and disease, particularly FAD, will also be discussed, along with major progress in elucidating the atomic structure of the γ-secretase complex and how the enzyme recognizes TMD substrates.
2. Early Groundwork
γ-Secretase was initially defined as a proteolytic activity that cleaves within the APP TMD, after ectodomain shedding by α- or β-secretase, to produce either the 3-kDa p3 or the 4-kDa Aβ, respectively (Fig. 1) [24]. The Aβ peptide is amphipathic, highly aggregation prone, and found deposited in the brain in Alzheimer’s disease. That the proteolysis takes place in the TMD and the proteolytic product was observed pathologically led to the suggestion that this cleavage was aberrant, occurring after some disruption that pulls the APP fragments out of the membrane. The discovery that Aβ is secreted naturally from cultured cells changed this notion [25, 26], although the idea of proteolysis within the membrane still seemed implausible.
Figure 1.
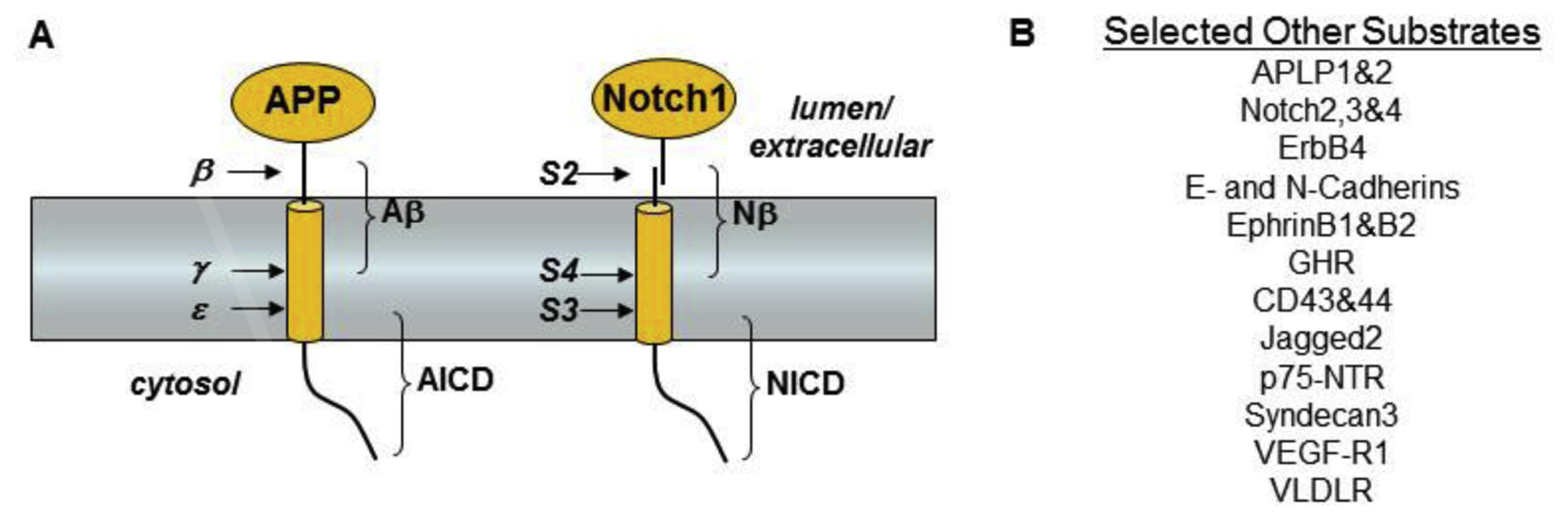
Substrates for γ-secretase. (A) Proteolytic processing of APP and Notch. The ectodomain of APP is first shed by β-secretase. Alternatively, APP can be cut within the Aβ region by α-secretases (not shown). The remaining membrane-associated stub is cleaved at least twice in the transmembrane region, at the γ site to produce Aβ and at the ε site to produce the intracellular domain (AICD). The Notch receptor is first processed in the secretory pathway at the S1 site by furin (not shown) to produce a heterodimer. Upon activation by ligand, this heterodimer is first cleaved at the S2 site by metalloproteases to shed the ectodomain, and the remaining membrane-associated stub is cleaved within the transmembrane domain at the S3 and S4 sites. The transmembrane cleavage events are carried out by the presenilin-containing γ-secretase complex. (B) A partial list of the ~90 other type I integral membrane proteins cleaved within their transmembrane regions by γ-secretase.
The discovery in 1991 of missense mutations in APP associated with FAD pointed to alterations in normal Aβ production as pathogenic [27, 28]. These mutations were found in and around the small Aβ region of APP, and they had the effect of either (1) increasing total Aβ production by increasing cleavage by β-secretase, (2) increasing the aggregation tendency of Aβ, or (3) shifting the preferred site of APP TMD cleavage by γ-secretase to increase the proportion of highly aggregation-prone 42-residue Aβ (Aβ42) over the more soluble 40-residue peptide (Aβ40) [29].
In 1995, the first missense mutations in genes encoding presenilin-1 and −2 (PSEN1, PSEN2) associated with FAD were discovered, providing the first important clue to the identity of the mysterious γ-secretase [20, 21]. The presenilins are multi-pass membrane proteins, of completely unknown function at that time. The only known homolog was an obscure gene in Caenorhabditis elegans called spe-4 involved in spermatogenesis [30]. The connection to Alzheimer’s disease was entirely unclear. However, the FAD presenilin mutations were soon found to increase the ratio of Aβ42 to Aβ40 [31–36], suggesting that pathogenesis is due to altering γ-secretase cleavage of APP substrate to increase the proportion of the aggregation-prone Aβ.
Presenilins were also quickly connected to signaling from the Notch family of cell-surface receptors. Through genetic screening, the C. elegans gene sel-12, related to spe-4 and human PSEN1 and PSEN2, was found to facilitate signaling from lin-12 and glp-1, worm Notch receptor orthologs [37]. Moreover, knockout of PSEN1 in mice resulted in an embryonic lethal phenotype closely resembling that seen upon knockout of Notch1 [15, 16]. Thus, presenilins were closely linked with a developmental signaling pathway critical to cell differentiation. Notch1 protein was then found to be cleaved within its single TMD to release an intracellular domain (NICD; Fig. 1) that translocates to the nucleus and activates transcription factors critical to development in metazoans [38]. NICD release and translocation was soon after found to be presenilin-dependent [39, 40].
Further clues to the biochemical function of presenilin were provided by the findings that presenilin itself undergoes endoproteolytic processing into an N-terminal fragment (NTF) and C-terminal fragment (CTF), that the formation of these fragments is tightly regulated by limiting cellular factors, and that NTF and CTF are stable, remain associated, and assemble into a high-molecular-weight complex [41–48]. These findings together suggested that the presenilin holoprotein is a precursor and that the functional form of presenilin is the associated NTF and CTF. Subsequently, the culturing of embryonic cells from PSEN1 knockout mice revealed that APP TMD processing by γ-secretase to Aβ was dramatically deficient [49], with remaining Aβ production attributed to PSEN2 (later shown to be correct) [50, 51]. Thus, presenilin was found to be essential for γ-secretase activity. However, because it bore no resemblance to any known protease, presenilin was suggested as likely a regulator of γ-secretase activity [49], analogous to an activating factor involved in transmembrane processing of the sterol regulatory element binding protein [52].
In parallel with the discovery of the apparent requirement of presenilin in the cleavage of APP by γ-secretase were pharmacological studies aimed at characterizing this unidentified protease. Peptidomimetics based on the APP TMD were developed, containing a protease inhibitory motif, a difluoroketone [53]. This motif was installed in place of the amide bond that is cleaved to produce Aβ42 in short peptidomimetics, thereby creating the first reported substrate-based inhibitor of γ-secretase. While providing a useful chemical tool for the study of γ-secretase and its biological roles, this compound was not informative about the proteolytic mechanism. However, follow-up development of related difluoroalcohol peptidomimetics [54] provided the first clear evidence that γ-secretase is an aspartyl protease: the difluoroalcohol motif is only known to inhibit aspartyl proteases, not other mechanistic classes of proteases. A subsequent report of a hydroxyethyl-type peptidomimetic, a related type of transition-state analog, as an inhibitor of γ-secretase offered further evidence for this hypothesis [55].
3. Intramembrane Proteolysis by Presenilin
The evidence that γ-secretase is an aspartyl protease along with the finding that PSEN1 is critical for γ-secretase activity raised the question of whether presenilin might be an aspartyl protease responsible for the cleavage of the APP TMD [22]. Inspection of the sequences of the known presenilins led to the identification of two completely conserved aspartates predicted to reside in TMD 6 and TMD7 (Fig. 2). Mutation of either aspartate in PSEN1 resulted in diminished Aβ production and increased levels of APP CTF γ-secretase substrates upon stable expression in mammalian cells as well as in microsomes isolated from these cells. Even conservative mutation to glutamate led to inhibition of γ-secretase processing of APP CTF substrates.
Figure 2.
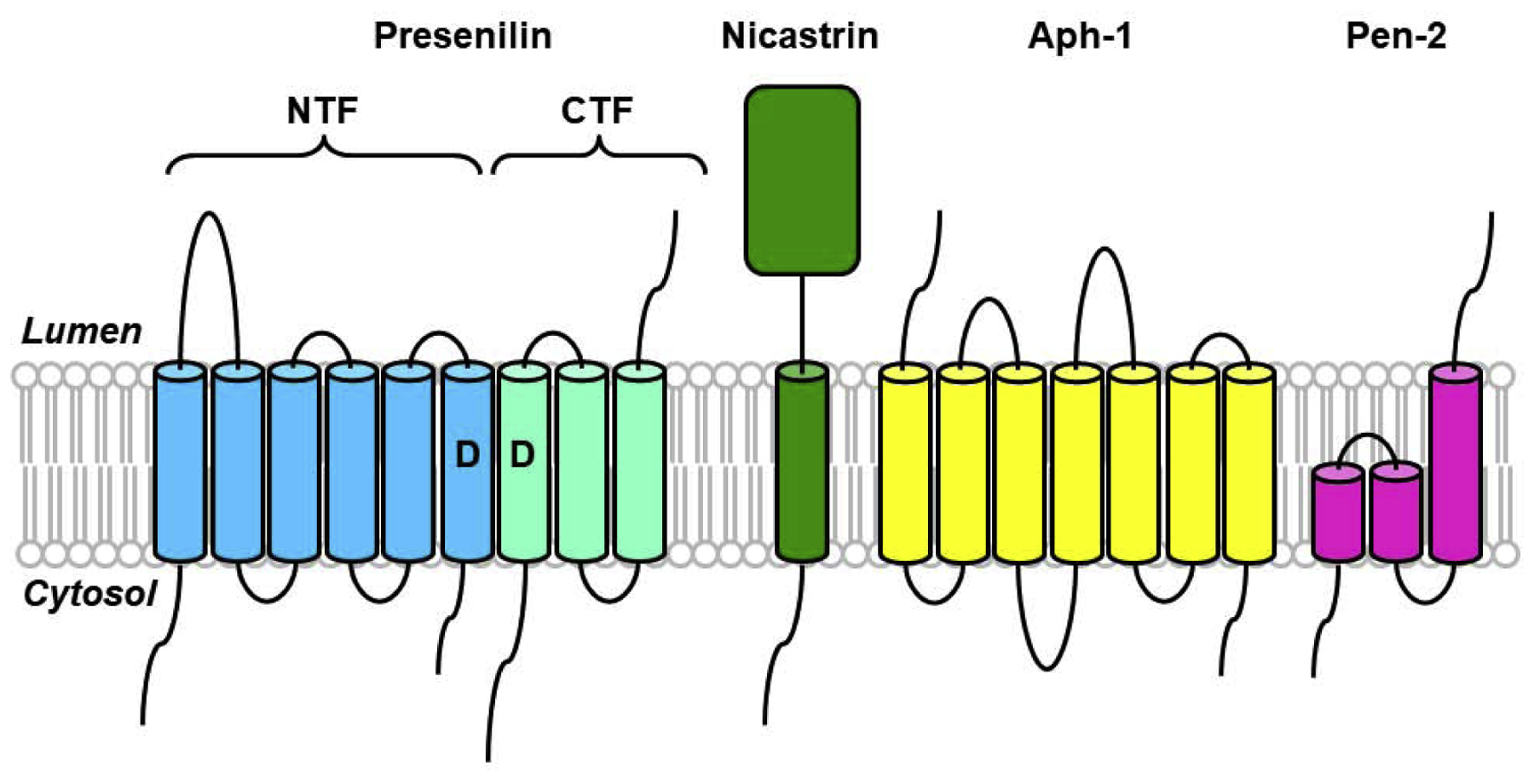
Components and assembly of the γ-secretase complex. γ-Secretase is composed of four different integral membrane proteins: presenilin, nicastrin, Aph-1, and Pen-2. Presenilin undergoes endoproteolysis into an N-terminal fragment (NTF) and a C-terminal fragment (CTF) that remain associated. Two conserved aspartates within adjacent transmembrane domains are essential for both presenilin endoproteolysis and γ-secretase activity.
Unexpectedly, mutation of either TMD aspartate also prevented normal endoproteolysis of PSEN1 into NTF and CTF. To test if the aspartates are critical for γ-secretase activity even without the need for PSEN1 NTF and CTF formation, these aspartates were mutated in the ΔE9 variant of PSEN1 [22]. This variant, associated with FAD, lacks exon 9, which encodes the endoproteolytic site within the large loop of PSEN1 between TMD 6 and TMD7. ΔE9 PSEN1 is not cleaved into NTF and CTF [41], yet still assembles into stable high-molecular-weight complexes [47] and acted as a functional presenilin (i.e., could substantially rescue an egg-laying deficiency in C. elegans caused by sel-12 mutation) [56]. Mutation of one of the TMD aspartates in ΔE9 PSEN1 likewise blocked γ-secretase cleavage of APP upon expression in mammalian cells, demonstrating the separate requirement of the aspartates for PSEN1 endoproteolysis and γ-secretase activity [22].
Taken together, these findings suggested that presenilin is a novel membrane-embedded aspartyl protease that cleaves the APP TMD to produce Aβ peptides and further suggested that full-length presenilin undergoes autoproteolysis into NTF and CTF. PSEN1 NTF and CTF each contribute one of the conserved TMD aspartates essential for proteolysis, suggesting that the active site of γ-secretase resides at the interface between these two subunits. Overexpression of wild-type PSEN1 did not lead to increased Aβ production, suggesting that the limiting cellular factors gating the formation of stable presenilin NTF/CTF heterodimers might also control the activation of γ-secretase.
This mutagenesis study was quickly confirmed in both PSEN1 and PSEN2 [57, 58]. Skepticism for the presenilin-as-protease hypothesis remained; however, this was soon largely allayed by identifying the interface between presenilin NTF and CTF as the binding site for aspartyl-protease transition-state analog inhibitors of γ-secretase [59, 60]. Such inhibitors were converted to biotinylated affinity reagents designed to covalently crosslink to their protein target containing the active site of γ-secretase. Detection of the biotinylated protein targets revealed presenilin NTF and CTF, suggesting that the active site of γ-secretase resides between these two subunits, consistent with the contribution by each subunit of one of the critical conserved TMD aspartates.
Concurrent with the inhibitor affinity labeling experiments was the discovery of another polytopic membrane protein serving as an aspartyl protease. The bacterial type 4 prepilin peptidases (TFPPs) were found to contain two conserved aspartic acid residues essential for cleavage of prepilin substrates and secretion of their ectodomains [61]. While the two essential aspartates reside outside predicted TMDs and the substrate cleavage site is likewise outside the membrane, TFPP provided important early support for presenilins as proteases, even though they are evolutionarily unrelated membrane proteins. This support was strengthened by the recognition that both TFPP and presenilins contain a GXGD active-site motif [62].
More convincing corroborating evidence came with the discovery of human signal peptide peptidase (SPP), responsible for the removal of remnant signal peptides left in the membrane by the action signal peptidase. Conversion of a peptidomimetic SPP inhibitor into a biotinylated affinity reagent led to the specific labeling of a presenilin homolog (PSH) [63]. Such PSHs, distantly related to presenilins, had been identified through searching of genomic databases and were found even in microbial organisms [64]. Cloning and expression in yeast of the identified target of the SPP inhibitor and isolation of yeast microsomes revealed that this human PSH had protease activity on its own, unlike the presenilins [63].
The evidence for presenilins as proteases was compelling. However, the absence of proteolytic activity on their own, the assembly into high-molecular-weight complexes, and conversion into stable NTF/CTF heterodimers together indicated that presenilins were the catalytic component of a larger γ-secretase complex [41–48]. The search for cofactors, protein partners for human presenilins, first identified a 709-residue type I transmembrane protein dubbed nicastrin by immunoprecipitation [65]. Presenilin and nicastrin were still insufficient to reconstitute γ-secretase activity, so apparently other components remained to be identified.
Genetic screening in C. elegans for novel components of the Notch signaling pathway led to the discovery of anterior pharynx-defective 1 (Aph-1) and presenilin enhancer 2 (Pen-2) [66–70]. Aph-1 was predicted to be a ~25 kDa seven-TMD protein, and Pen-2 as a small (101 amino acid) protein with two predicted TMDs. Expression of all four proteins (PSEN1, nicastrin, Aph-1, and Pen-2) resulted in PSEN1 endoproteolysis to NTF and CTF and reconstitution of γ-secretase activity in mammalian cells and yeast [7–9], and all four proteins (presenilin as NTF and CTF) copurified by affinity isolation using an immobilized transition-state analog inhibitor of γ-secretase [8].
With the identification of all the essential components of γ-secretase (Fig. 2), the question of the size and stoichiometry of the protease complex remained. Native gel electrophoresis gave varying results from 250–900 kDa [71–74], and some studies suggested the possibility of two presenilin molecules per complex [73, 75, 76]. Differential epitope labeling and co-immunoprecipitation, however, supported only one of each component in proteolytically active γ-secretase [77]. Furthermore, purification of the active protease allowed scanning transmission electron microscopy and determination of a ~230 kDa complex, again consistent with one of each component [78]. Initial elucidation of a detailed structure of the γ-secretase complex by cryo-electron microscopy (cryoEM) further confirmed a 1:1:1:1 stoichiometry [79]. Although this issue seemed settled, a more recent study has suggested the possibility that the protease may consist of two interacting 1:1:1:1 complexes [80].
An interesting mechanistic question is how TMD substrates gain access to what was presumed to be an active site sequestered inside presenilin, thereby avoiding interaction of the TMD aspartates and water with the hydrophobic lipid tails. The first clue was the copurification of an endogenous APP substrate upon isolation of γ-secretase using an immobilized transition-state analog inhibitor [81]. The substrate was capable of stably interacting with the protease even though the active site was occupied by the immobilized inhibitor, suggesting the existence of an initial substrate docking site on the outside of presenilin. Subsequent movement of the substrate TMD in whole or in part into the internal active site would allow proteolysis. Further evidence for a docking site came from enzyme inhibition kinetics [82] and designed helical peptide inhibitors based on the APP TMD [83, 84]. Conversion of these inhibitors to affinity reagents led to labeling of PS1 NTF and CTF, as seen with transition-state analog inhibitors [85]. However, the two types of inhibitors could not compete with each other, indicating distinct binding sites. Lengthening the helical peptide inhibitors from 10 to 13 amino acids resulted in competition with the transition-state analog inhibitors, suggesting that the active site and docking sites were proximal, within the length of three amino acids.
A remarkable recent study involved the systematic installation of a photoreactive phenylalanine analog into APP substrate and analyzing the labeling of γ-secretase subunits at each position [86]. Labeling was strongest in the C-terminal region of the substrate TMD, specifically at positions 42, 44, 45, and 49 (which labeled PS1 NTF) and positions 51 and 52 (which labeled PS1 CTF). This finding supports the model for substrate TMD interacting at the interface between PS1 NTF and CTF [85, 87]. Very little binding to other γ-secretase subunits were observed, although low-level labeling was seen to nicastrin with the photoreactive residue in the lumen/extracellular N-terminal region of the substrate, and to Pen-2 with the reactive residue in the juxtamembrane regions [86]. (A new report also suggests some interaction of the N-terminal region of APP substrate with nicastrin [88].) Interestingly, two FAD mutations in PS1 resulted in a shift in the labeling patterns with the photoreactive substrates [86]. This altered interaction of APP substrate with FAD-mutant γ-secretase may result in the observed altered patterns of Aβ production.
4. Proteolytic Function and Dysfunction
Substrate requirements for γ-secretase appear to be quite broaden, with many type I integral membrane proteins serving as substrates [1–3]. However, this is generally only after lumenal/extracellular ectodomain shedding, suggesting that substrate must have a relatively short juxtamembrane region to access the γ-secretase active site. An early report using an in vivo genetic reporter assay in Drosophila with Notch as substrate concluded that presenilin-dependent processing did not depend on the specific TMD sequence [89]. Interestingly, the TMD of glycophorin A, known to dimerize as a coiled coil, did not appear to serve as a substrate, but a single point mutation that disrupts dimerization apparently allowed presenilin-dependent cleavage. The length of the extracellular domain was also found to be critical: increasing the excellular domain beyond ~50 amino acids hindered processing in a length-dependent manner. Subsequent biochemical studies have confirmed and extended these early in vivo findings [90, 91]. Interestingly, a substrate with a naturally short ectodomain was identified recently [92].
The lack of TMD specificity apparently allows γ-secretase to process many integral membrane protein stubs. A proteomic analysis was conducted using stable isotope labeling with amino acids in cell culture (SILAC) in the presence and absence of a γ-secretase inhibitor, revealing increases in putative membrane-bound CTF substrates [3]. Follow-up genetic and pharmacological experiments validated genuine substrates. Not all membrane protein stubs were processed by γ-secretase, suggesting that non-substrates may have inhibitory domains, a concept supported by a study of the role of the juxtamembrane region in APP processing [93]. Over 90 substrates for γ-secretase have been identified (selected examples listed in Fig. 1). Indeed, one of its biological functions may be to clear out membrane protein stubs, leading to γ-secretase being dubbed “the proteasome of the membrane”[4]. However, inhibition or knockout of γ-secretase does not generally lead to toxicity in cell culture, suggesting that general removal of protein stubs is not essential or that other means of removal exists.
In whole organisms, however, γ-secretase is an essential enzyme. Genetic knockout of γ-secretase is embryonic lethal, due to inhibition of the Notch1 signaling pathway [15, 16]. Beyond development, inhibition of γ-secretase in human clinical trials also resulted in severe toxic effects such as gastrointestinal bleeding, immunosuppression, and skin lesions, likewise due to inhibition of Notch signaling needed for regulation of cell differentiation [94, 95]. The central importance of Notch signaling often masks effects of inhibiting the proteolysis of other critical γ-secretase substrates. These include N- and E-cadherin (cell-cell adhesion) [17, 18], ErbB4 (receptor tyrosine kinase signaling) [19], p75 NTR (neurotrophin signaling) [96], and the β2 subunit of the voltage-gated sodium channel (cell-cell adhesion and migration) [97]. The essential nature of γ-secretase notwithstanding, PSEN2 can be knocked out in mice, resulting in only a mild pulmonary phenotype with age [98]. PSEN2 is incorporated into distinct γ-secretase complexes [99], and while there are few FAD-linked PSEN2 mutations, these also can increase Aβ42 production (e.g., [32, 33]).
Proteolytic processing by γ-secretase has been most comprehensively studied with APP substrate. Sequence analysis revealed that the secreted Aβ peptides and AICD proteolytic products were not generated from a single proteolytic event. The AICD-generating cut site was dubbed the ε cleavage site, 3–4 residues into the TMD on the cytosolic side [100–104]. Longer AICD fragments complementary to the secreted Aβ peptides were not observed. The identification of Aβ peptides produced by so-called ζ cleavage, Aβ45 and Aβ46, were then identified [11, 105]. The expression in mammalian cells of Aβ48 and Aβ49 peptides complementary to the AICD products—fused to a signal peptide for membrane insertion—led to production of Aβ40 and Aβ42 that could be blocked by inhibition of γ-secretase, suggesting a precursor-product relationship [10]. Aβ49 gave primarily Aβ40, while the poorly expressed Aβ48 gave equal but low levels of Aβ40 and Aβ42.
Taken together, these findings led to the hypothesis that γ-secretase initially cleaves at the ε site to release AICD and generate Aβ48 and Aβ49, which are then trimmed generally every three amino acids along two pathways: Aβ49→Aβ46→Aβ43→Aβ40 and Aβ48→Aβ45→Aβ42→Aβ38 (Fig. 3) [11]. The detection of the small peptide by-products by mass spectrometry from enzyme assays supported this model [13], which was subsequently validated using purified γ-secretase preparations and synthetic Aβ45 and Aβ48 (converted primarily to Aβ42 over Aβ40) and Aβ46 and Aβ49 (converted primarily to Aβ40 over Aβ42) [14]. Whether all γ-secretase substrates are processed in this manner is not known, although proteolysis of the Notch1 TMD likewise leads to generation of NICD with only 3–4 TMD residues [38] and secreted “Nβ” peptides of varying lengths [106] that cannot be explained by a single proteolytic event. Moreover, PSEN1 autoproteolysis also apparently involves tripeptide trimming [107], and both Notch and PSEN1 processing are affected by PSEN1 FAD mutations [106, 107].
Figure 3.
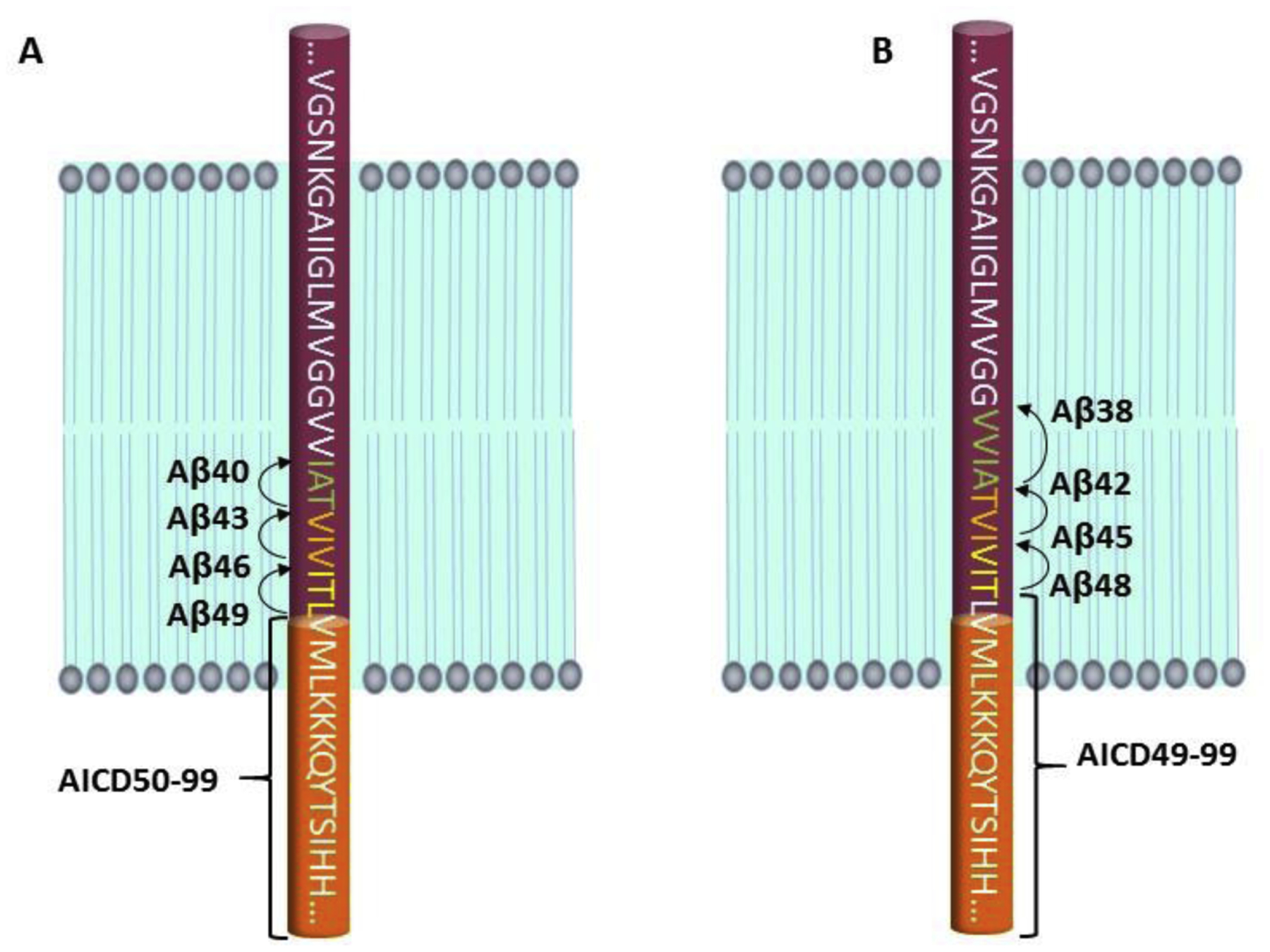
Processive proteolysis of the APP transmembrane domain by γ-secretase. An endoproteolytic activity of the enzyme cleaves at the ε site within the transmembrane domain close to the membrane-cytosol interface to give long Aβ peptides Aβ48 and Aβ49 and the APP intracellular domain (AICD). The carboxypeptidase activity of γ-secretase then trims Aβ48 and Aβ49 in 3–4 amino acid increments along two pathways: (A) Aβ49→Aβ46→Aβ43→Aβ40 and (B) Aβ48→Aβ45→Aβ42→Aβ38.
As noted earlier, FAD mutations in the presenilins were found to increase the proportion of the much more aggregation-prone Aβ42 over the predominant secreted product Aβ40. Thus, Aβ42 has been presumed as the pathogenic form of Aβ and the major focus of studies aimed at determining the role of Aβ in Alzheimer’s disease. However, this “toxic gain-of-function” hypothesis to explain the pathogenicity of PSEN FAD mutations has been called into question, including in a recent report showing that many PSEN1 FAD mutations do not increase the Aβ42/Aβ40 ratio in isolated enzyme reactions [108]. (Note: This study itself has been called into question [109].) An alternative hypothesis posits that presenilin FAD mutations lead to a loss of function [110, 111], pointing to how most FAD mutations reduce NICD release from Notch receptors [112] and that two mutations (L435F, C410Y) cause complete loss of production of secreted Aβ [113].
The loss-of-function hypothesis, however, has inconsistencies as well: (1) Not all PSEN1 FAD mutations result in a reduction of ε-site cleavage [114, 115], (2) no FAD mutations lead to nonsense-mediated decay (NMD); remarkably, NMD mutations in γ-secretase components lead to familial acne inversa, not neurodegeneration [116] (3) C410Y and L435F PSEN1 do in fact support Aβ production, and L435F primarily produces Aβ43 that deposits in the form of cerebral plaques in mutation carriers [117, 118]. Lost in the debate over gain-of-function versus loss-of-function is that PSEN/γ-secretase has multiple proteolytic functions. A comprehensive analysis of all proteolytic products—including small peptide byproducts—generated by PSEN FAD-mutant protease complexes should help resolve this question. At present, evidence suggests that PSEN1 FAD mutations cause a reduction in the carboxypeptidase (aka trimming) function of γ-secretase, rather than in its endoproteolytic function [14, 114, 115, 119, 120].
5. Structure of γ-secretase
Perhaps the most remarkable breakthrough in the study of γ-secretase has been structure elucidation. Advances in cryo-EM technology have revolutionized structural biology, allowing the determination of high-resolution structures of large complexes [121]. The application of cutting-edge cryo-EM approaches led to the first detailed structure of the γ-secretase complex, showing a horseshoe-shaped arrangement of its 19 total TMDs and the location and orientation of the nicastrin ectodomain [79]. A follow-up study provided full assignment of the TMDs, revealing that the active site resides just within the convex side of the horseshoe-shaped arrangement (Fig. 4) [122]. Jutting out over the active site was the nicastrin ectodomain, apparently preventing the approach of substrates with long lumenal/extracellular domains. The new structure was consistent with a concurrent biochemical study demonstrating that shorter substrate luminal/extracellular domains were processed faster, that the substrate TMD is sufficient for high-affinity binding and processing by γ-secretase, and that reducing the disulfide bonds in nicastrin allowed longer substrates to be processed [91]. The new structure was also consistent with previous biochemical reports on the arrangement of the subunits within the complex [72, 123]. Unexpectedly, TMD 1 of Pen-2 was found not to pass through the membrane; rather, it appears to dip in and out of the membrane (This finding was concurrently confirmed through cellular/biochemical experiments [124]). Pen-2 interacted directly with TMD 4 of PSEN1, consistent with mutagenesis studies [125, 126].
Figure 4.
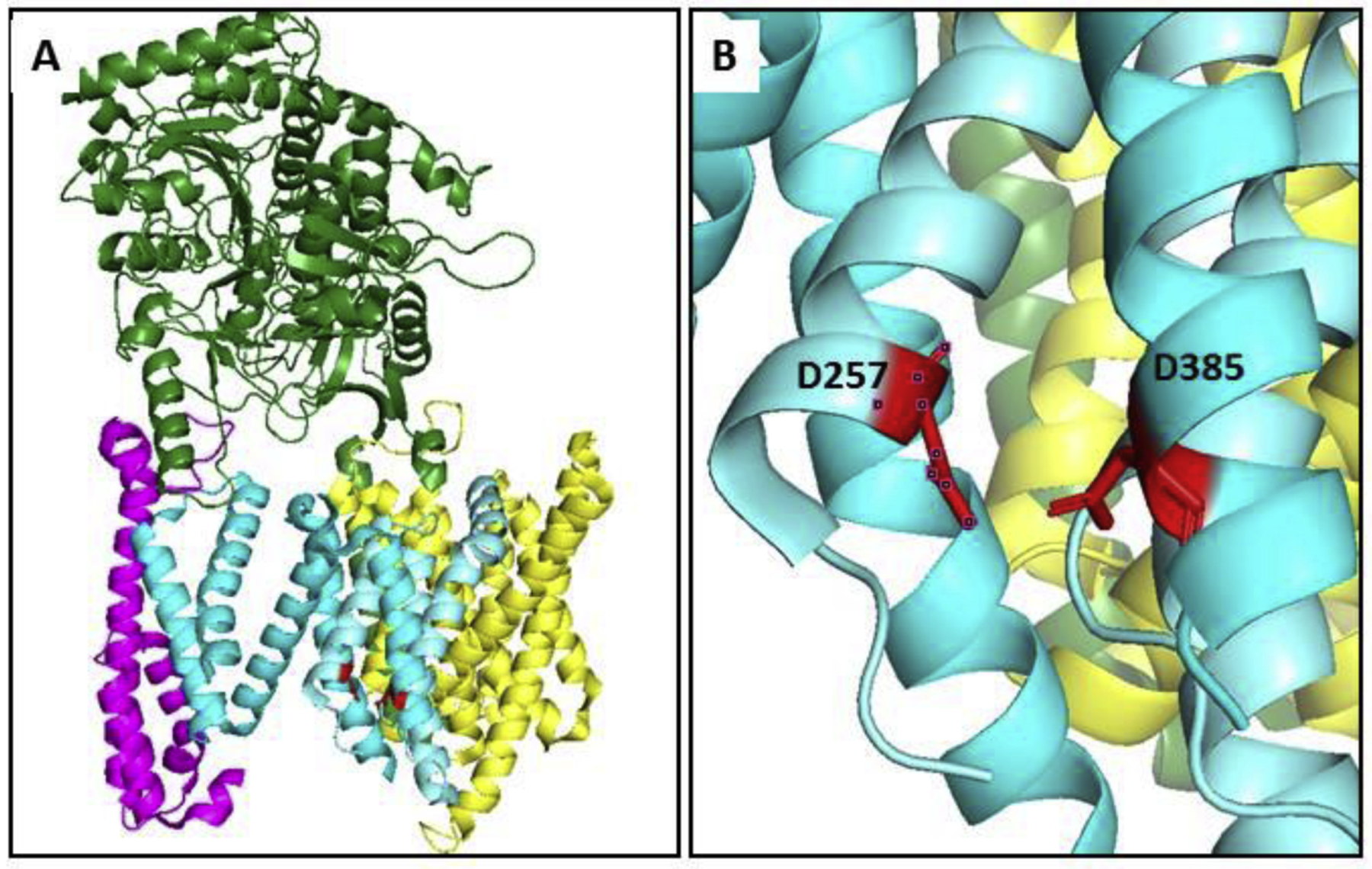
First detailed structure of the γ-secretase complex determined by cryo-EM, single-particle analysis, and image reconstruction. (A) Nicastrin: green; Aph-1: yellow; Pen-2: magenta; PSEN1: cyan (NTF) and aquamarine (CTF), with catalytic aspartates in red. PSEN1 TMD 2 was not resolved. (B) The active site on PSEN1, with D257 in TMD 6 and D385 in TMD 7 in close proximity. PDB: 5A63.
Another cryo-EM study employed an image classification technique to reveal three different conformations of the protease complex [127]. In one of these structures, an unidentified electron density consistent with a transmembrane helix was observed, and the position of this helix within the PSEN1 subunit strongly suggested that endogenous membrane proteins copurified with the γ-secretase complex. The helical-shaped electron density disappeared as it approached the catalytic aspartates, and none of the identified copurifying proteins were previously known to be substrates. Nevertheless, the observed density provided evidence for where substrate might bind to the protease. Moreover, comparison with the other two observed conformations suggested that TMD 2 and the cytosolic end of TMD 6 undergo substantial changes upon substrate binding. In this same study, the protease complex was also analyzed in the presence of an inhibitor called DAPT. The enzyme assumed a similar conformation to that seen with the unidentified helical density (putative endogenous substrate that copurified), suggesting that DAPT inhibits γ-secretase by inducing the protease into a conformation normally formed upon substrate binding.
Most recently, two structures of γ-secretase bound to recombinant substrates, APP [88] and Notch [128], have been reported (shown with APP in Fig. 5). To capture these substrates with γ-secretase, the investigators mutated one of the active site aspartates to alanine, thereby disabling protease function, and cysteine-crosslinked the substrate to Loop 1 of PSEN1. The N-terminal region of substrate was α-helical and in a closely similar location inside PSEN1 as the unidentified helical density in the earlier study [127]. However, for both APP and Notch substrates, as the helical TMD of substrate approaches the active site, unwinding and extension occurs, forming a β-sheet with the cytosolic end PSEN1 TMD 7. Although the enzyme is in an inactivated state due to mutation of a catalytic aspartate, the new structures revealed that substrate unwinding and extension is critical for setting up intramembrane proteolysis. Remarkably, the majority of FAD mutations in PSEN1 appear to interact with the substrate-binding cavity, either directly or indirectly, an important clue to the pathogenic mechanism of these dominant mutations.
Figure 5.
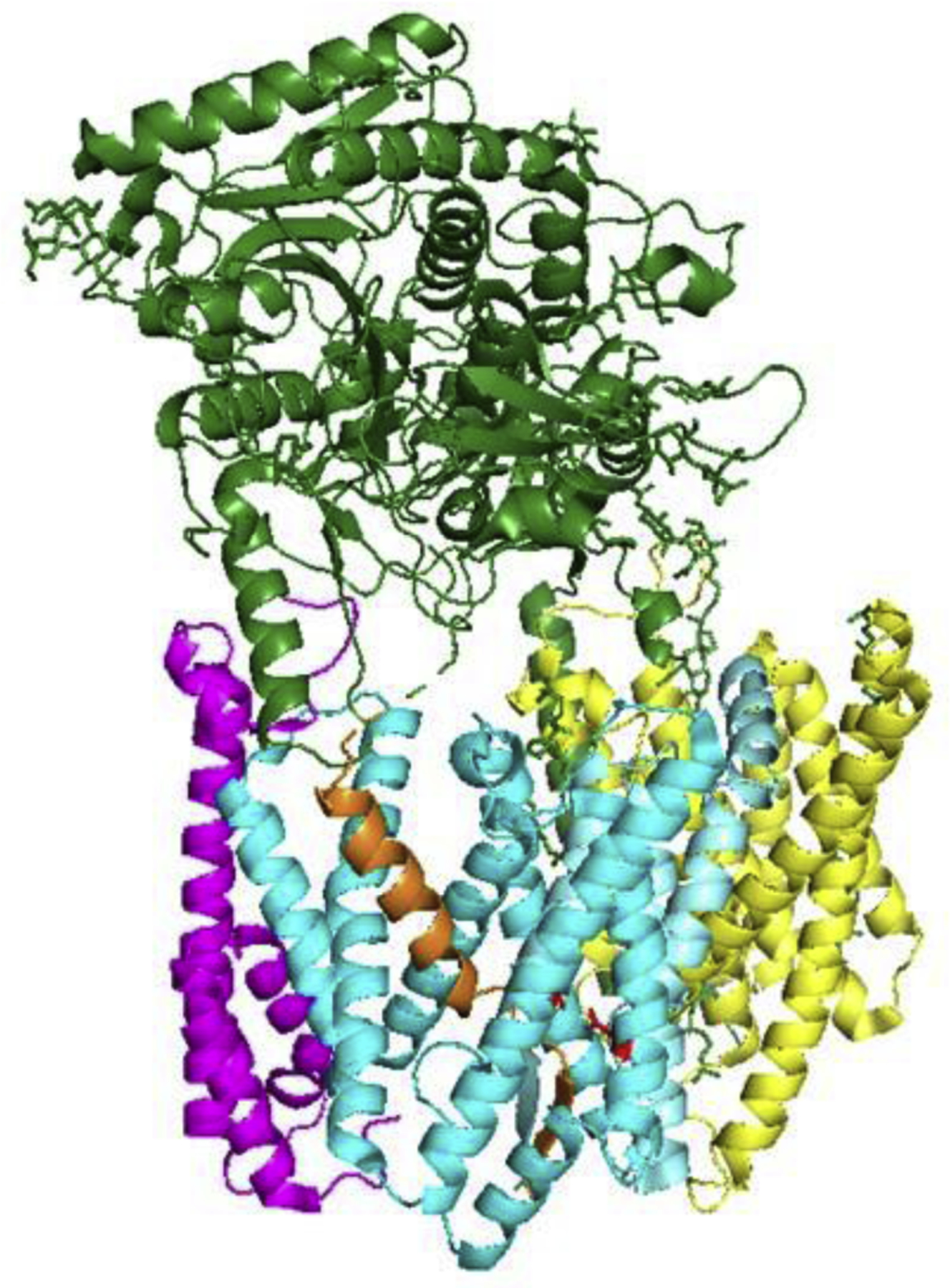
γ-Secretase bound to APP-derived substrate. APP substrate is located inside PSEN1. The substrate TMD assumes a β-strand conformation near the cytoplasmic side as it interacts with the active site. PSEN1 TMD 2 is now visible (foreground), as is the cytoplasmic side of TMD 6 (cf. Figure 4). PDB: 6IYC.
6. Perspective
Since the initial discovery of presenilin as a novel membrane-embedded aspartyl protease, substantial progress has been made in understanding the structure, mechanism, and biology of the γ-secretase complex. Molecular biology and chemical biology have been critical to this progress, and most recently structural biology has provided atomic-level details of the protease in several different conformations, include inhibitor-bound and substrate-bound states. Nevetheless, important steps in substrate recognition and processive proteolysis by γ-secretase remain to be elucidated. Also critical is deciphering the effects of FAD mutations—both in presenilins and APP—on the various proteolytic events carried out by the enzyme complex. It seems more than merely coincidental that FAD mutations have been found only in the substrate and enzyme that directly produce Aβ. A comprehensive understanding of the effects of FAD mutations on the processing of APP substrate by γ-secretase should expedite the unraveling of molecular details of pathogenesis and the discovery of therapeutic agents for the prevention and treatment of Alzheimer’s disease.
Acknowledgment:
M.S.W. is supported for this work through grant GM122894 from the National Institutes of Health.
Abbreviations:
- Aβ
amyloid β-peptide
- APP
amyloid precursor protein
- AICD
APP intracellular domain
- Aph-1
anterior pharynx-defective 1
- cryoEM
cryo-electron microscopy
- CTF
C-terminal fragment
- FAD
familial Alzheimer’s disease
- NTF
N-terminal fragment
- Pen-2
presenilin enhancer 2
- PSEN
presenilin
- PSH
presenilin homolog
- SPP
signal peptide peptidase
- TMD
transmembrane domain
- TFPPs
type 4 prepilin peptidases
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: The author declares no conflicts of interest.
References
- 1.Beel AJ, and Sanders CR (2008). Substrate specificity of γ-secretase and other intramembrane proteases. Cell Mol Life Sci 65, 1311–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haapasalo A, and Kovacs DM (2011). The many substrates of presenilin/γ-secretase. J Alzheimer Dis 25, 3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hemming ML, Elias JE, Gygi SP, and Selkoe DJ (2008). Proteomic profiling of γ-secretase substrates and mapping of substrate requirements. PLoS Biol 6, e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kopan R, and Ilagan MX (2004). γ-Secretase: proteasome of the membrane? Nature reviews. Mol Cell Biol 5, 499–504. [DOI] [PubMed] [Google Scholar]
- 5.Kopan R, and Ilagan MX (2009). The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolfe MS (2019). Dysfunctional γ-secretase in familial Alzheimer’s disease. Neurochem Res 44, 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takasugi N, Tomita T, Hayashi I, Tsuruoka M, Niimura M, Takahashi Y, Thinakaran G, and Iwatsubo T (2003). The role of presenilin cofactors in the γ-secretase complex. Nature 422, 438–441. [DOI] [PubMed] [Google Scholar]
- 8.Kimberly WT, LaVoie MJ, Ostaszewski BL, Ye W, Wolfe MS, and Selkoe DJ (2003). γ-Secretase is a membrane protein complex comprised of presenilin, nicastrin, aph-1, and pen-2. Proc Natl Acad Sci USA 100, 6382–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edbauer D, Winkler E, Regula JT, Pesold B, Steiner H, and Haass C (2003). Reconstitution of γ-secretase activity. Nat Cell Biol 5, 486–488. [DOI] [PubMed] [Google Scholar]
- 10.Funamoto S, Morishima-Kawashima M, Tanimura Y, Hirotani N, Saido TC, and Ihara Y (2004). Truncated carboxyl-terminal fragments of β-amyloid precursor protein are processed to amyloid β-proteins 40 and 42. Biochemistry 43, 13532–13540. [DOI] [PubMed] [Google Scholar]
- 11.Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, et al. (2005). Longer forms of amyloid β protein: implications for the mechanism of intramembrane cleavage by γ-secretase. J Neurosci 25, 436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yagishita S, Morishima-Kawashima M, Ishiura S, and Ihara Y (2008). Aβ46 is processed to Aβ40 and Aβ43, but not to Aβ42, in the low density membrane domains. J Biol Chem 283, 733–738. [DOI] [PubMed] [Google Scholar]
- 13.Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S, and Ihara Y (2009). γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J Neurosci 29, 13042–13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandez MA, Klutkowski JA, Freret T, and Wolfe MS (2014). Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by β-secretase to increase 42-to-40-residue Aβ. J Biol Chem 289, 31043–31052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, and Sisodia SS (1997). Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature 387, 288–292. [DOI] [PubMed] [Google Scholar]
- 16.Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, and Tonegawa S (1997). Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 89, 629–639. [DOI] [PubMed] [Google Scholar]
- 17.Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, et al. (2002). A presenilin-1/γ-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 21, 1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, and Robakis NK (2003). A CBP binding transcriptional repressor produced by the PS1/ε-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 114, 635–645. [DOI] [PubMed] [Google Scholar]
- 19.Sardi SP, Murtie J, Koirala S, Patten BA, and Corfas G (2006). Presenilin-dependent ErbB4 nuclear signaling regulates the timing of astrogenesis in the developing brain. Cell 127, 185–197. [DOI] [PubMed] [Google Scholar]
- 20.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, et al. (1995). Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375, 754–760. [DOI] [PubMed] [Google Scholar]
- 21.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, et al. (1995). Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376, 775–778. [DOI] [PubMed] [Google Scholar]
- 22.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, and Selkoe DJ (1999). Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 398, 513–517. [DOI] [PubMed] [Google Scholar]
- 23.Mullane K, and Williams M (2018). Alzheimer’s disease (AD) therapeutics - 1: Repeated clinical failures continue to question the amyloid hypothesis of AD and the current understanding of AD causality. Biochem Pharmacol 158, 359–375. [DOI] [PubMed] [Google Scholar]
- 24.Selkoe DJ (1994). Cell biology of the amyloid β-protein precursor and the mechanism of Alzheimer’s disease. Annu Rev Cell Biol 10, 373–403. [DOI] [PubMed] [Google Scholar]
- 25.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB, et al. (1992). Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 359, 322–325. [DOI] [PubMed] [Google Scholar]
- 26.Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B, et al. (1992). Production of the Alzheimer amyloid βprotein by normal proteolytic processing. Science 258, 126–129. [DOI] [PubMed] [Google Scholar]
- 27.Chartier-Harlin MC, Crawford F, Houlden H, Warren A, Hughes D, Fidani L, Goate A, Rossor M, Roques P, Hardy J, et al. (1991). Early-onset Alzheimer’s disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature 353, 844–846. [DOI] [PubMed] [Google Scholar]
- 28.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, et al. (1991). Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349, 704–706. [DOI] [PubMed] [Google Scholar]
- 29.Tanzi RE, and Bertram L (2005). Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell 120, 545–555. [DOI] [PubMed] [Google Scholar]
- 30.L’Hernault SW, and Arduengo PM (1992). Mutation of a putative sperm membrane protein in Caenorhabditis elegans prevents sperm differentiation but not its associated meiotic divisions. J Cell Biol 119, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, et al. (1996). Familial Alzheimer’s disease-linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron 17, 1005–1013. [DOI] [PubMed] [Google Scholar]
- 32.Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, et al. (1997). Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat Med 3, 67–72. [DOI] [PubMed] [Google Scholar]
- 33.Tomita T, Maruyama K, Saido TC, Kume H, Shinozaki K, Tokuhiro S, Capell A, Walter J, Grunberg J, Haass C, et al. (1997). The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid β protein ending at the 42nd (or 43rd) residue. Proc Natl Acad Sci USA 94, 2025–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xia W, Zhang J, Kholodenko D, Citron M, Podlisny MB, Teplow DB, Haass C, Seubert P, Koo EH, and Selkoe DJ (1997). Enhanced production and oligomerization of the 42-residue amyloid β-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem 272, 7977–7982. [DOI] [PubMed] [Google Scholar]
- 35.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, et al. (1996). Increased amyloid-β 42(43) in brains of mice expressing mutant presenilin 1. Nature 383, 710–713. [DOI] [PubMed] [Google Scholar]
- 36.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, et al. (1996). Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 2, 864–870. [DOI] [PubMed] [Google Scholar]
- 37.Levitan D, and Greenwald I (1995). Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature 377, 351–354. [DOI] [PubMed] [Google Scholar]
- 38.Schroeter EH, Kisslinger JA, and Kopan R (1998). Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393, 382–386. [DOI] [PubMed] [Google Scholar]
- 39.De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, et al. (1999). A presenilin-1-dependent γ-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522. [DOI] [PubMed] [Google Scholar]
- 40.Struhl G, and Greenwald I (1999). Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature 398, 522–525. [DOI] [PubMed] [Google Scholar]
- 41.Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, et al. (1996). Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17, 181–190. [DOI] [PubMed] [Google Scholar]
- 42.Ratovitski T, Slunt HH, Thinakaran G, Price DL, Sisodia SS, and Borchelt DR (1997). Endoproteolytic processing and stabilization of wild-type and mutant presenilin. J Biol Chem 272, 24536–24541. [DOI] [PubMed] [Google Scholar]
- 43.Podlisny MB, Citron M, Amarante P, Sherrington R, Xia W, Zhang J, Diehl T, Levesque G, Fraser P, Haass C, et al. (1997). Presenilin proteins undergo heterogeneous endoproteolysis between Thr291 and Ala299 and occur as stable N- and C-terminal fragments in normal and Alzheimer brain tissue. Neurobiol Dis 3, 325–337. [DOI] [PubMed] [Google Scholar]
- 44.Thinakaran G, Harris CL, Ratovitski T, Davenport F, Slunt HH, Price DL, Borchelt DR, and Sisodia SS (1997). Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J Biol Chem 272, 28415–28422. [DOI] [PubMed] [Google Scholar]
- 45.Steiner H, Capell A, Pesold B, Citron M, Kloetzel PM, Selkoe DJ, Romig H, Mendla K, and Haass C (1998). Expression of Alzheimer’s disease-associated presenilin-1 is controlled by proteolytic degradation and complex formation. J Biol Chem 273, 32322–32331. [DOI] [PubMed] [Google Scholar]
- 46.Seeger M, Nordstedt C, Petanceska S, Kovacs DM, Gouras GK, Hahne S, Fraser P, Levesque L, Czernik AJ, George-Hyslop PS, et al. (1997). Evidence for phosphorylation and oligomeric assembly of presenilin 1. Proc Natl Acad Sci USA 94, 5090–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Capell A, Grunberg J, Pesold B, Diehlmann A, Citron M, Nixon R, Beyreuther K, Selkoe DJ, and Haass C (1998). The proteolytic fragments of the Alzheimer’s disease-associated presenilin-1 form heterodimers and occur as a 100–150-kDa molecular mass complex. J Biol Chem 273, 3205–3211. [DOI] [PubMed] [Google Scholar]
- 48.Yu G, Chen F, Levesque G, Nishimura M, Zhang DM, Levesque L, Rogaeva E, Xu D, Liang Y, Duthie M, et al. (1998). The presenilin 1 protein is a component of a high molecular weight intracellular complex that contains β-catenin. J Biol Chem 273, 16470–16475. [DOI] [PubMed] [Google Scholar]
- 49.De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, and Van Leuven F (1998). Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390. [DOI] [PubMed] [Google Scholar]
- 50.Herreman A, Serneels L, Annaert W, Collen D, Schoonjans L, and De Strooper B (2000). Total inactivation of γ-secretase activity in presenilin-deficient embryonic stem cells. Nat Cell Biol 2, 461–462. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Z, Nadeau P, Song W, Donoviel D, Yuan M, Bernstein A, and Yankner BA (2000). Presenilins are required for γ-secretase cleavage of β-APP and transmembrane cleavage of Notch-1. Nat Cell Biol 2, 463–465. [DOI] [PubMed] [Google Scholar]
- 52.Brown MS, and Goldstein JL (1997). The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340. [DOI] [PubMed] [Google Scholar]
- 53.Wolfe MS, Citron M, Diehl TS, Xia W, Donkor IO, and Selkoe DJ (1998). A substrate-based difluoro ketone selectively inhibits Alzheimer’s γ-secretase activity. J Med Chem 41, 6–9. [DOI] [PubMed] [Google Scholar]
- 54.Wolfe MS, Xia W, Moore CL, Leatherwood DD, Ostaszewski B, Donkor IO, and Selkoe DJ (1999). Peptidomimetic probes and molecular modeling suggest Alzheimer’s γ-secretases are intramembrane-cleaving aspartyl proteases. Biochemistry 38, 4720–4727. [DOI] [PubMed] [Google Scholar]
- 55.Shearman MS, Beher D, Clarke EE, Lewis HD, Harrison T, Hunt P, Nadin A, Smith AL, Stevenson G, and Castro JL (2000). L-685,458, an aspartyl protease transition state mimic, is a potent inhibitor of amyloid β-protein precursor γ-secretase activity. Biochemistry 39, 8698–8704. [DOI] [PubMed] [Google Scholar]
- 56.Levitan D, Doyle TG, Brousseau D, Lee MK, Thinakaran G, Slunt HH, Sisodia SS, and Greenwald I (1996). Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc Natl Acad Sci USA 93, 14940–14944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Steiner H, Duff K, Capell A, Romig H, Grim MG, Lincoln S, Hardy J, Yu X, Picciano M, Fechteler K, et al. (1999). A loss of function mutation of presenilin-2 interferes with amyloid β-peptide production and notch signaling. J Biol Chem 274, 28669–28673. [DOI] [PubMed] [Google Scholar]
- 58.Kimberly WT, Xia W, Rahmati T, Wolfe MS, and Selkoe DJ (2000). The transmembrane aspartates in presenilin 1 and 2 are obligatory for γ-secretase activity and amyloid β-protein generation. J Biol Chem 275, 3173–3178. [DOI] [PubMed] [Google Scholar]
- 59.Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, et al. (2000). Photoactivated γ-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405, 689–694. [DOI] [PubMed] [Google Scholar]
- 60.Esler WP, Kimberly WT, Ostaszewski BL, Diehl TS, Moore CL, Tsai J-Y, Rahmati T, Xia W, Selkoe DJ, and Wolfe MS (2000). Transition-state analogue inhibitors of γ-secretase bind directly to presenilin-1. Nat Cell Biol 2, 428–434. [DOI] [PubMed] [Google Scholar]
- 61.LaPointe CF, and Taylor RK (2000). The type 4 prepilin peptidases comprise a novel family of aspartic acid proteases. J Biol Chem 275, 1502–1510. [DOI] [PubMed] [Google Scholar]
- 62.Steiner H, Kostka M, Romig H, Basset G, Pesold B, Hardy J, Capell A, Meyn L, Grim ML, Baumeister R, et al. (2000). Glycine 384 is required for presenilin-1 function and is conserved in bacterial polytopic aspartyl proteases. Nat Cell Biol 2, 848–851. [DOI] [PubMed] [Google Scholar]
- 63.Weihofen A, Binns K, Lemberg MK, Ashman K, and Martoglio B (2002). Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science 296, 2215–2218. [DOI] [PubMed] [Google Scholar]
- 64.Ponting CP, Hutton M, Nyborg A, Baker M, Jansen K, and Golde TE (2002). Identification of a novel family of presenilin homologues. Hum Mol Genet 11, 1037–1044. [DOI] [PubMed] [Google Scholar]
- 65.Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song YQ, Rogaeva E, Chen F, Kawarai T, et al. (2000). Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature 407, 48–54. [DOI] [PubMed] [Google Scholar]
- 66.Goutte C, Tsunozaki M, Hale VA, and Priess JR (2002). APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA 99, 775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gu Y, Chen F, Sanjo N, Kawarai T, Hasegawa H, Duthie M, Li W, Ruan X, Luthra A, Mount HT, et al. (2002). APH-1 interacts with mature and immature forms of presenilins and nicastrin and may play a role in maturation of presenilin-nicastrin complexes. J Biol Chem 278, 7374–7380. [DOI] [PubMed] [Google Scholar]
- 68.Lee SF, Shah S, Li H, Yu C, Han W, and Yu G (2002). Mammalian APH-1 interacts with presenilin and nicastrin and is required for intramembrane proteolysis of amyloid-βprecursor protein and Notch. J Biol Chem 277, 45013–45019. [DOI] [PubMed] [Google Scholar]
- 69.Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, et al. (2002). aph-1 and pen-2 are required for Notch pathway signaling, γ-secretase cleavage of βAPP, and presenilin protein accumulation. Dev Cell 3, 85–97. [DOI] [PubMed] [Google Scholar]
- 70.Steiner H, Winkler E, Edbauer D, Prokop S, Basset G, Yamasaki A, Kostka M, and Haass C (2002). PEN-2 is an integral component of the γ-secretase complex required for coordinated expression of presenilin and nicastrin. J Biol Chem 277, 39062–39065. [DOI] [PubMed] [Google Scholar]
- 71.Edbauer D, Winkler E, Haass C, and Steiner H (2002). Presenilin and nicastrin regulate each other and determine amyloid β-peptide production via complex formation. Proc Natl Acad Sci USA 99, 8666–8671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fraering PC, LaVoie MJ, Ye W, Ostaszewski BL, Kimberly WT, Selkoe DJ, and Wolfe MS (2004). Detergent-dependent dissociation of active γ-secretase reveals an interaction between Pen-2 and PS1-NTF and offers a model for subunit organization within the complex. Biochemistry 43, 323–333. [DOI] [PubMed] [Google Scholar]
- 73.Cervantes S, Saura CA, Pomares E, Gonzalez-Duarte R, and Marfany G (2004). Functional implications of the presenilin dimerization. Reconstitution of γ-secretase activity by assembly of a catalytic site at the dimer interface of two catalytically inactive presenilins. J Biol Chem 279, 36519–36529. [DOI] [PubMed] [Google Scholar]
- 74.Evin G, Canterford LD, Hoke DE, Sharples RA, Culvenor JG, and Masters CL (2005). Transition-state analogue γ-secretase inhibitors stabilize a 900 kDa presenilin/nicastrin complex. Biochemistry 44, 4332–4341. [DOI] [PubMed] [Google Scholar]
- 75.Schroeter EH, Ilagan MX, Brunkan AL, Hecimovic S, Li YM, Xu M, Lewis HD, Saxena MT, De Strooper B, Coonrod A, et al. (2003). A presenilin dimer at the core of the γ-secretase enzyme: insights from parallel analysis of Notch 1 and APP proteolysis. Proc Natl Acad Sci USA 100, 13075–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Clarke EE, Churcher I, Ellis S, Wrigley JD, Lewis HD, Harrison T, Shearman MS, and Beher D (2006). Intra- or intercomplex binding to the γ-secretase enzyme. A model to differentiate inhibitor classes. J Biol Chem 281, 31279–31289. [DOI] [PubMed] [Google Scholar]
- 77.Sato T, Diehl TS, Narayanan S, Funamoto S, Ihara Y, De Strooper B, Steiner H, Haass C, and Wolfe MS (2007). Active γ-secretase complexes contain only one of each component. J Biol Chem 282, 33985–33993. [DOI] [PubMed] [Google Scholar]
- 78.Osenkowski P, Li H, Ye W, Li D, Aeschbach L, Fraering PC, Wolfe MS, Selkoe DJ, and Li H (2009). Cryoelectron microscopy structure of purified γ-secretase at 12 Å resolution. J Mol Biol 385, 642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, Yang G, Zhao Y, Zhou R, Scheres SH, et al. (2014). Three-dimensional structure of human γ-secretase. Nature 512, 166–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhou R, Yang G, and Shi Y (2017). Dominant negative effect of the loss-of-function γ-secretase mutants on the wild-type enzyme through heterooligomerization. Proc Natl Acad Sci USA 114, 12731–12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Esler WP, Kimberly WT, Ostaszewski BL, Ye W, Diehl TS, Selkoe DJ, and Wolfe MS (2002). Activity-dependent isolation of the presenilin/γ-secretase complex reveals nicastrin and a γ substrate. Proc Natl Acad Sci U.S.A. 99, 2720–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tian G, Sobotka-Briner CD, Zysk J, Liu X, Birr C, Sylvester MA, Edwards PD, Scott CD, and Greenberg BD (2002). Linear non-competitive inhibition of solubilized human γ-secretase by pepstatin A methylester, L685458, sulfonamides, and benzodiazepines. J Biol Chem 277, 31499–31505. Epub 32002 Jun 31418. [DOI] [PubMed] [Google Scholar]
- 83.Das C, Berezovska O, Diehl TS, Genet C, Buldyrev I, Tsai JY, Hyman BT, and Wolfe MS (2003). Designed helical peptides inhibit an intramembrane protease. J Am Chem Soc 125, 11794–11795. [DOI] [PubMed] [Google Scholar]
- 84.Bihel F, Das C, Bowman MJ, and Wolfe MS (2004). Discovery of a subnanomolar helical D-tridecapeptide inhibitor of γ-secretase. J Med Chem 47, 3931–3933. [DOI] [PubMed] [Google Scholar]
- 85.Kornilova AY, Bihel F, Das C, and Wolfe MS (2005). The initial substrate-binding site of γ-secretase is located on presenilin near the active site. Proc Natl Acad Sci USA 102, 3230–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fukumori A, and Steiner H (2016). Substrate recruitment of γ-secretase and mechanism of clinical presenilin mutations revealed by photoaffinity mapping. EMBO J 35, 1628–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wolfe MS, De Los Angeles J, Miller DD, Xia W, and Selkoe DJ (1999). Are presenilins intramembrane-cleaving proteases? Implications for the molecular mechanism of Alzheimer’s disease. Biochemistry 38, 11223–11230. [DOI] [PubMed] [Google Scholar]
- 88.Zhou R, Yang G, Guo X, Zhou Q, Lei J, and Shi Y (2019). Recognition of the amyloid precursor protein by human γ-secretase. Science 363, eaaw0930 [DOI] [PubMed] [Google Scholar]
- 89.Struhl G, and Adachi A (2000). Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol Cell 6, 625–636. [DOI] [PubMed] [Google Scholar]
- 90.Funamoto S, Sasaki T, Ishihara S, Nobuhara M, Nakano M, Watanabe-Takahashi M, Saito T, Kakuda N, Miyasaka T, Nishikawa K, et al. (2013). Substrate ectodomain is critical for substrate preference and inhibition of γ-secretase. Nat Commun 4, 2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bolduc DM, Montagna DR, Gu Y, Selkoe DJ, and Wolfe MS (2016). Nicastrin functions to sterically hinder γ-secretase-substrate interactions driven by substrate transmembrane domain. Proc Natl Acad Sci USA 113, E509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Laurent SA, Hoffmann FS, Kuhn PH, Cheng Q, Chu Y, Schmidt-Supprian M, Hauck SM, Schuh E, Krumbholz M, Rubsamen H, et al. (2015). γ-Secretase directly sheds the survival receptor BCMA from plasma cells. Nat Commun 6, 7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tian Y, Bassit B, Chau D, and Li YM (2010). An APP inhibitory domain containing the Flemish mutation residue modulates γ-secretase activity for Aβ production. Nat Struct Mol Biol 17, 151–158. [DOI] [PubMed] [Google Scholar]
- 94.Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, et al. (2013). A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369, 341–350. [DOI] [PubMed] [Google Scholar]
- 95.Coric V, Salloway S, van Dyck CH, Dubois B, Andreasen N, Brody M, Curtis C, Soininen H, Thein S, Shiovitz T, et al. (2015). Targeting prodromal Alzheimer disease With avagacestat: a randomized clinical trial. JAMA Neurol 72, 1324–1333. [DOI] [PubMed] [Google Scholar]
- 96.Kanning KC, Hudson M, Amieux PS, Wiley JC, Bothwell M, and Schecterson LC (2003). Proteolytic processing of the p75 neurotrophin receptor and two homologs generates C-terminal fragments with signaling capability. J Neurosci 23, 5425–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim DY, Ingano LA, Carey BW, Pettingell WH, and Kovacs DM (2005). Presenilin/γ-secretase-mediated cleavage of the voltage-gated sodium channel β2-subunit regulates cell adhesion and migration. J Biol Chem 280, 23251–23261. [DOI] [PubMed] [Google Scholar]
- 98.Herreman A, Hartmann D, Annaert W, Saftig P, Craessaerts K, Serneels L, Umans L, Schrijvers V, Checler F, Vanderstichele H, et al. (1999). Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci USA 96, 11872–11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lai MT, Chen E, Crouthamel MC, DiMuzio-Mower J, Xu M, Huang Q, Price E, Register RB, Shi XP, Donoviel DB, et al. (2003). Presenilin-1 and presenilin-2 exhibit distinct yet overlapping γ-secretase activities. J Biol Chem 278, 22475–22481. [DOI] [PubMed] [Google Scholar]
- 100.Gu Y, Misonou H, Sato T, Dohmae N, Takio K, and Ihara Y (2001). Distinct intramembrane cleavage of the β-amyloid precursor protein family resembling γ-secretase-like cleavage of Notch. J Biol Chem 276, 35235–35238. [DOI] [PubMed] [Google Scholar]
- 101.Yu C, Kim SH, Ikeuchi T, Xu H, Gasparini L, Wang R, and Sisodia SS (2001). Characterization of a presenilin-mediated amyloid precursor protein carboxyl-terminal fragment γ. Evidence for distinct mechanisms involved in γ-secretase processing of the APP and Notch1 transmembrane domains. J Biol Chem 276, 43756–43760. [DOI] [PubMed] [Google Scholar]
- 102.Weidemann A, Eggert S, Reinhard FB, Vogel M, Paliga K, Baier G, Masters CL, Beyreuther K, and Evin G (2002). A novel var ε-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry 41, 2825–2835. [DOI] [PubMed] [Google Scholar]
- 103.Sato T, Dohmae N, Qi Y, Kakuda N, Misonou H, Mitsumori R, Maruyama H, Koo EH, Haass C, Takio K, et al. (2003). Potential link between amyloid β-protein 42 and C-terminal fragment gamma 49–99 of β-amyloid precursor protein. J Biol Chem 278, 24294–24301. [DOI] [PubMed] [Google Scholar]
- 104.Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, and Haass C (2001). Presenilin-dependent γ-secretase processing of β-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep 2, 835–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhao G, Cui MZ, Mao G, Dong Y, Tan J, Sun L, and Xu X (2005). gamma-Cleavage is dependent on ζ-cleavage during the proteolytic processing of amyloid precursor protein within its transmembrane domain. J Biol Chem 280, 37689–37697. [DOI] [PubMed] [Google Scholar]
- 106.Okochi M, Steiner H, Fukumori A, Tanii H, Tomita T, Tanaka T, Iwatsubo T, Kudo T, Takeda M, and Haass C (2002). Presenilins mediate a dual intramembranous γ-secretase cleavage of Notch-1. EMBO J 21, 5408–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fukumori A, Fluhrer R, Steiner H, and Haass C (2010). Three-amino acid spacing of presenilin endoproteolysis suggests a general stepwise cleavage of γ-secretase-mediated intramembrane proteolysis. J Neurosci 30, 7853–7862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sun L, Zhou R, Yang G, and Shi Y (2017). Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc Natl Acad Sci USA 114, E476–E485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tang N, and Kepp KP (2018). Aβ42/Aβ40 ratios of presenilin 1 mutations correlate with clinical onset of Alzheimer’s disease. J Alzheimer Dis 66, 939–945. [DOI] [PubMed] [Google Scholar]
- 110.Shen J, and Kelleher RJ 3rd (2007). The presenilin hypothesis of Alzheimer’s disease: evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci USA 104, 403–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kelleher RJ 3rd, and Shen J (2017). Presenilin-1 mutations and Alzheimer’s disease. Proc Natl Acad Sci USA 114, 629–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Song W, Nadeau P, Yuan M, Yang X, Shen J, and Yankner BA (1999). Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc Natl Acad Sci USA 96, 6959–6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xia D, Watanabe H, Wu B, Lee SH, Li Y, Tsvetkov E, Bolshakov VY, Shen J, and Kelleher RJ 3rd (2015). Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron 85, 967–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Quintero-Monzon O, Martin MM, Fernandez MA, Cappello CA, Krzysiak AJ, Osenkowski P, and Wolfe MS (2011). Dissociation between the processivity and total activity of γ-secretase: implications for the mechanism of Alzheimer’s disease-causing presenilin mutations. Biochemistry 50, 9023–9035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chavez-Gutierrez L, Bammens L, Benilova I, Vandersteen A, Benurwar M, Borgers M, Lismont S, Zhou L, Van Cleynenbreugel S, Esselmann H, et al. (2012). The mechanism of γ-secretase dysfunction in familial Alzheimer disease. EMBO J 31, 2261–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang B, Yang W, Wen W, Sun J, Su B, Liu B, Ma D, Lv D, Wen Y, Qu T, et al. (2010). γ-Secretase gene mutations in familial acne inversa. Science 330, 1065. [DOI] [PubMed] [Google Scholar]
- 117.Kretner B, Trambauer J, Fukumori A, Mielke J, Kuhn PH, Kremmer E, Giese A, Lichtenthaler SF, Haass C, Arzberger T, et al. (2016). Generation and deposition of Aβ43 by the virtually inactive presenilin-1 L435F mutant contradicts the presenilin loss-of-function hypothesis of Alzheimer’s disease. EMBO Mol Med 8, 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Veugelen S, Saito T, Saido TC, Chavez-Gutierrez L, and De Strooper B (2016). Familial Alzheimer’s disease mutations in presenilin generate amyloidogenic Aβ peptide seeds. Neuron 90, 410–416. [DOI] [PubMed] [Google Scholar]
- 119.Szaruga M, Veugelen S, Benurwar M, Lismont S, Sepulveda-Falla D, Lleo A, Ryan NS, Lashley T, Fox NC, Murayama S, et al. (2015). Qualitative changes in human γ-secretase underlie familial Alzheimer’s disease. J Exp Med 212, 2003–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Szaruga M, Munteanu B, Lismont S, Veugelen S, Horre K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, et al. (2017). Alzheimer’s-causing mutations shift Aβ Length by destabilizing γ-secretase-Aβn interactions. Cell 170, 443–456 e414. [DOI] [PubMed] [Google Scholar]
- 121.Quentin D, and Raunser S (2018). Electron cryomicroscopy as a powerful tool in biomedical research. J Mol Med 96, 483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bai XC, Yan C, Yang G, Lu P, Ma D, Sun L, Zhou R, Scheres SH, and Shi Y (2015). An atomic structure of human γ-secretase. Nature 525, 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Steiner H, Winkler E, and Haass C (2008). Chemical crosslinking provides a model of the γ-secretase complex subunit architecture and evidence for close proximity of the C-terminal fragment of presenilin with APH-1. J Biol Chem 283, 34677–34686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang X, Yu CJ, and Sisodia SS (2015). The topology of pen-2, a γ-secretase subunit, revisited: evidence for a reentrant loop and a single pass transmembrane domain. Mol Neurodegener 10, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kim SH, and Sisodia SS (2005). Evidence that the “NF” motif in transmembrane domain 4 of presenilin 1 is critical for binding with PEN-2. J Biol Chem 280, 41953–41966. [DOI] [PubMed] [Google Scholar]
- 126.Watanabe N, Tomita T, Sato C, Kitamura T, Morohashi Y, and Iwatsubo T (2005). Pen-2 is incorporated into the γ-secretase complex through binding to transmembrane domain 4 of presenilin 1. J Biol Chem 280, 41967–41975. [DOI] [PubMed] [Google Scholar]
- 127.Bai XC, Rajendra E, Yang G, Shi Y, and Scheres SH (2015). Sampling the conformational space of the catalytic subunit of human γ-secretase. eLife 4, pii: e11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang G, Zhou R, Zhou Q, Guo X, Yan C, Ke M, Lei J, and Shi Y (2019). Structural basis of Notch recognition by human γ-secretase. Nature 565, 192–197. [DOI] [PubMed] [Google Scholar]