

Summary
Regulated cell death is intrinsically associated with inflammatory liver disease and is pivotal in governing outcomes of metabolic liver disease. Different types of cell death may coexist in the progression of metabolic liver disease to inflammation, fibrosis, and ultimately cirrhosis. In addition to apoptosis, lytic forms of hepatocellular death, such as necroptosis, pyroptosis and ferroptosis elicit strong inflammatory responses due to cell membrane permeabilization and release of cellular components, contributing to the recruitment of immune cells and activation of hepatic stellate cells. Controlling liver cell death, in turn, emerges with fundamental importance and offers novel opportunities for potential therapeutic intervention. This review summarizes the underlying mechanism of distinct lytic cell death modes and their commonalities, discusses its relevance to metabolic liver diseases of different aetiologies, and acknowledges the limitations of current knowledge in the field. We focus on the role of hepatocyte necroptosis, pyroptosis and ferroptosis in non-alcoholic fatty liver disease, alcohol-associated liver disease and other metabolic liver disorders, as well as potential of translation into human disease.
Keywords: Programmed cell death, Necroptosis, Pyroptosis, Ferroptosis, NAFLD, NASH, ASH, Hemochromatosis, Niemann-Pick disease, Gaucher’s disease
1. Introduction
Cells can be exposed to unrecoverable extra- or intra-cellular perturbations that disrupt cellular homeostasis and activate one of many signal transduction cascades, ultimately leading to cell death and liver injury. The regulated cell death (RCD) modalities are initiated and propagated by specific molecular mechanisms, with considerable interactivity. Moreover, each type of RCD is characterized by distinct morphological, biochemical, and molecular features with specific physiological consequences ranging from anti-inflammatory and tolerogenic to pro-inflammatory and immunogenic profiles.
Decades of research have revealed multiple forms of genetically encoded RCD pathways with increasingly recognized relevance in disease. Importantly, these novel RCD pathways can co-exist simultaneously in pathological contexts, and several share overlapping mechanisms that can act as a “backup” dying strategy to ensure organism homeostasis when a death-inducing cellular threshold is reached. In chronic liver diseases, viral, toxic, metabolic or autoimmune triggers cause hepatocellular death, followed by inflammation and compensatory proliferation, often closely linked to development of fibrosis, cirrhosis, and hepatocellular carcinoma [1]. Traditionally, apoptosis was considered as a highly regulated process, diametrically opposed to passive necrosis, itself considered as an accidental and uncontrolled form of cell death. Passive necrosis occurs when cells are irreparably damaged by external forces, resulting in rapid cytoplasmic and organelle swelling (oncosis), along with plasma membrane permeabilization and subsequent leakage of damage-associated molecular patterns (DAMPs) that trigger immune response. However, recent findings highlight multiple forms of regulated necrotic modalities, including necroptosis, pyroptosis and ferroptosis, that share key morphologic features with passive necrosis yet have well-defined and regulated causal mechanisms [2, 3]. Necroptosis is probably the best-understood form of regulated necrosis, since it shares multiple molecular components with the extrinsic apoptotic pathway and can act as a fail-safe mechanism to ensure cell death progression when apoptosis is abnormally inhibited, which may also be relevant in pathological conditions in the liver [4]. Nevertheless, other alternative forms of RCD, such as ferroptosis or pyroptosis may turn to be equally important as we gather experimental evidence for their relevance. Importantly, a better characterization of RCD in liver disease may lead to novel therapeutic opportunities. As the field continues to progress and novel signalling pathways that orchestrate RCD are still being characterized, this review summarizes current knowledge on the contribution of hepatocellular RCD modalities in metabolic liver disease. Experimental models and patient findings are addressed, as well as the pathophysiological relevance of each of the main types of RCD in hepatocytes, controversies and unsolved issues, and potential therapeutic applications. RCD of non-hepatocyte liver cells, immune cells and others will not be explored, although it may prove equally important in liver disease.
2. The Principles of Alternative Cell Death Modalities
2.1. Necroptosis
The first evidence of necroptosis was provided in 1996 by Ray et al., who observed a lytic mode of cell death in pig kidney cells infected with cow pox virus, which was governed by the expression of the viral cytokine response modifier A (CrmA), a caspase inhibitor [5]. In 2000, Holler et al. demonstrated that FAS, TRAIL and TNF receptors, the classical death receptors (DRs), initiated cell death by two alternative pathways, one relying on caspase-8 (i.e., the classical extrinsic apoptotic pathway) and the other dependent on the receptor interacting protein kinase 1 (RIPK1) (i.e. the necroptosis) [6]. However, the term used to describe this novel mode of cell death appeared only in 2005, when Degterev et al. showed that necrostatin-1, a chemical compound that blocks the kinase activity of RIPK1, was able to inhibit cell death in TNF-treated cell lines [7]. Later, the two downstream core components of the necroptotic machinery, namely RIPK3 and mixed lineage kinase domain like pseudokinase (MLKL), have been identified in less than a decade [8–11]. Morphologically, necroptosis, which is primarily triggered following microbial infections and physicochemical stressors (e.g., radiation or chemotherapy), exhibits the features of passive necrosis (e.g., in response to extreme external factors) with increased cell volume, swelling of organelles, loss of membrane integrity, cellular collapse, leading to the release of DAMPs (e.g. high-mobility group box 1 [HMGB1]; interleukin [IL]-1α, IL-33). Although no specific necroptotic DAMPs have been identified so far, their release during necroptosis provokes a strong inflammatory response that has been linked to the development of many diseases [12].
Necroptosis occurs typically upon DR activation (e.g., TNFR1, CD95, TRAIL-R), and only if apoptotic signalling components are inactive, absent or inhibited (e.g., caspase inhibition) (Figure 1). In this specific context, DR triggers the formation of a RIPK1/RIPK3 cell death platform, named the necrosome [13]. Inside, RIPK1 and RIPK3 adopt a hetero-amyloid structure through critical RIP homotypic interaction motif (RHIM) interactions [14]. Consequently, RIPK3 mediates the phosphorylation of MLKL, resulting in MLKL oligomerization and translocation to the plasma membrane [13]. Recent results suggest that casein kinase 1 (CK1) family proteins are necrosome components, which are required to directly phosphorylate serine 227 of human RIPK3 and induce necroptosis [15]. Finally, MLKL forms pore in the plasma membrane [16, 17], and increases permeability via the activation of A disintegrin and metalloproteinase (ADAM) proteases [18], Ca2+ influx by targeting the cation channel transient receptor potential melastatin related 7 (TRPM7) [19], and phosphatidylserine externalization [20]. Necroptosis activation requires MLKL phosphorylation, which is dependent on RIPK3 kinase activity, itself dependent on phosphorylation by RIPK1; hence, RIPK1 and/or 3 phosphorylation cannot be equated with necroptosis without MLKL phosphorylation. There are considerable crosstalk regulations between apoptosis and necroptosis in DR signalling pathways [21], and it seems that one cannot be achieved without inhibiting the other. On one hand, caspase-8 cleaves and inactivates RIPK1 and RIPK3, which has for consequence to suppress necroptosis [22, 23]. On the other hand, activity of RIPK3 determines whether cells die by necroptosis (i.e., the necrosome formed) or apoptosis (i.e., RIPK3 kinase activity inhibited) [24, 25].
Figure 1. RIPK3- and MLKL-dependent necroptosis mediated by various stimuli.
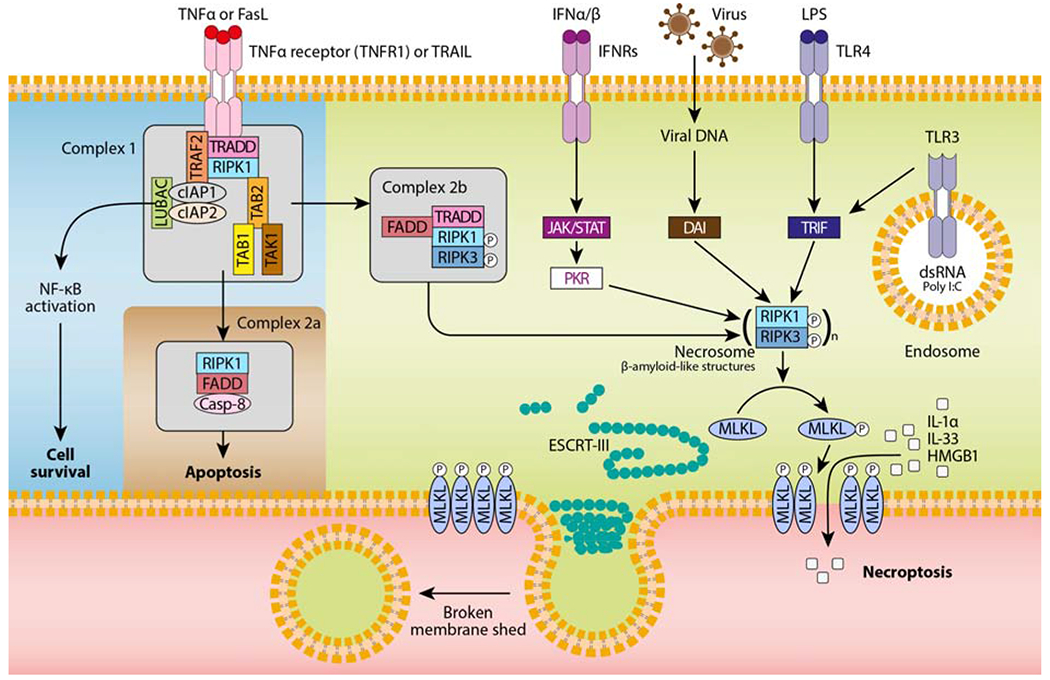
TNF-α or FasL binding to their receptor induces the formation of a receptor proximal complex (complex 1), which consists of adaptor proteins (e.g., TRADD, tumor necrosis factor receptor type 1-associated death domain; FADD, fas-associated protein with death domain; TAB, transforming growth factor beta-binding protein), ubiquitin ligases (e.g., TRAF2, TNF receptor associated factor 2; LUBAC, linear ubiquitin chain assembly complex; cIAPs, cellular inhibitors of apoptosis), and kinases (e.g., TAK, transforming growth factor beta-activated kinase 1; RIPK1, receptor interacting protein kinase 1). The ubiquitination of RIPK1 provides docking sites for the recruitment of key proteins leading to cell survival by nuclear factor κB (NF-κB)-dependent upregulation of pro-survival genes. Then, complex 1 internalization leads to the formation of complex 2a, which ultimately results in caspase-8-dependent apoptosis. Finally, when caspases or cIAPs are inhibited, RIPK1 associates with RIPK3 to form the necrosome (complex 2b), which in turn recruits the pseudokinase MLKL. RIPK3-mediated phosphorylation of MLKL results in MLKL translocation to the plasma membrane and pore formation. Other necroptotic stimuli such as IFNα/β, virus, pathogen-associated molecular patterns (e.g., LPS, poly I:C) are also depicted. Although the nature of these signalling complexes is not completely characterized, the main mediators activating the necrosome are shown. To antagonize necroptosis, ESCRT-III components are recruited to localized sites of MLKL-directed membrane damage to shed broken membrane into blebs.
The activation of necroptosis is not limited to DRs, and it can be triggered by multiple pathways, including toll-like receptors (e.g., TLR3 and TLR4) [26], nucleic acid sensors (e.g., Z-DNA-binding protein 1 [ZBP1, also known as DAI]) [27], retinoic acid inducible gene 1 protein (RIG1) [28], transmembrane protein 173 (TMEM173, also known as STING) [29], and adhesion receptors [30]. These signalling pathways are often independent of RIPK1, but still have in common the phosphorylation of MLKL by RIPK3 and the MLKL-mediated pore formation in the plasma membrane [13]. Interestingly, the endosomal sorting complexes required for transport (ESCRT)-III complex, a membrane remodelling and scission machinery, limit MLKL-mediated necroptosis by promoting membrane repair [31]. As MLKL also regulates endosomal trafficking and extracellular vesicles generation [32], a delicate balance between membrane injury and repair exists and ultimately decides cell fate in necroptosis.
2.2. Pyroptosis
Pyroptosis was initially observed in 1992 by Zychlinsky et al., who described a lytic form of cell death in Shigella flexneri-infected macrophages [33]. However, the term has emerged nearly a decade later, in 2001, when Cookson and Brennan demonstrated that Salmonella-induced macrophage death was dependent on caspase-1 [34]. Since this discovery, the number of pyroptotic caspases (as opposed to apoptotic caspases) has considerably increased, including caspase-1, caspase-11 and its human orthologs caspase-4 and 5 [35, 36], and even more surprisingly the apoptotic effector caspase-3 under specific circumstances [37, 38]. Pyroptosis, which occurs primarily upon infection with intracellular pathogens, is characterized by caspase-dependent pore formation in the plasma membrane, swelling, rupture of the cell, and release of pro-inflammatory IL-1β and IL-18 [39]. In 2015, the pore-forming gasdermin D (GSDMD) was identified as the executioner of pyroptosis [40, 41]. Caspases-1 and -11 cleave GSDMD into a 31-kDa N-terminal GSDMDNT fragment, which exhibits intrinsic pore-forming activity, and a 22-kDa C-terminal GSDMDCT fragment that binds to GSDMDNT to maintain the protein in an inhibitory state [40, 41]. The overexpression of GSDMDNT alone results in induction of pyroptosis, whereas that of GSDMDCT blocks GSDMDNT-induced pyroptosis [41]. Interestingly, GSDMD belongs to a larger protein family, which consists of GSDMA, GSDMB, GSDMC, GSDMD, GSDME (also referred to as DFNA5 [deafness, autosomal dominant 5]), and DFNB59 [42]. Recently, GSDME was identified as an additional executioner of pyroptosis, able to switch caspase-3 mediated apoptosis induced by TNF or chemotherapy drugs to pyroptosis [38]. Although most of the gasdermins have been associated with the occurrence and development of various diseases, their precise function and molecular mechanism of activation remain largely unknown [43].
Pyroptosis is activated by canonical and non-canonical signalling pathways, which differ in the use of cytoplasmic multiprotein complexes named inflammasomes (Figure 2) [44, 45]. The canonical pathway begins with inflammasomes that recognize various exogenous and endogenous danger signals, including damage-associated molecular pattern molecules (DAMPs) and pathogen-associated molecular patterns (PAMPs). The canonical inflammasomes comprise a sensor protein belonging to the nucleotide-binding domain (NBD) and leucine-rich-repeat-(LRR)-containing (NLR) or the AIM2-like receptor (ALR) or pyrin family, an adaptor protein, apoptosis-associated speck-like protein containing a CARD (ASC), and an inactive zymogen, pro-caspase-1 [46]. When formed, canonical inflammasomes lead to the activation of caspase-1 that cleaves pro-IL-1β and pro-IL-18 into their active forms. Then, IL-1β and IL-18 are released extracellularly as a result of the pore formation in the plasma membrane by GSDMDNT. On the other side, the non-canonical pathway is dependent on caspase-11, which is able to cleave GSDMD independently of inflammasome priming. In this later case, GSDMDNT signals back to canonical NLRP3 inflammasome, which in turn activates the caspase-1-dependent pathway [44–46].
Figure 2. Caspase-1-dependent and -independent pyroptosis.
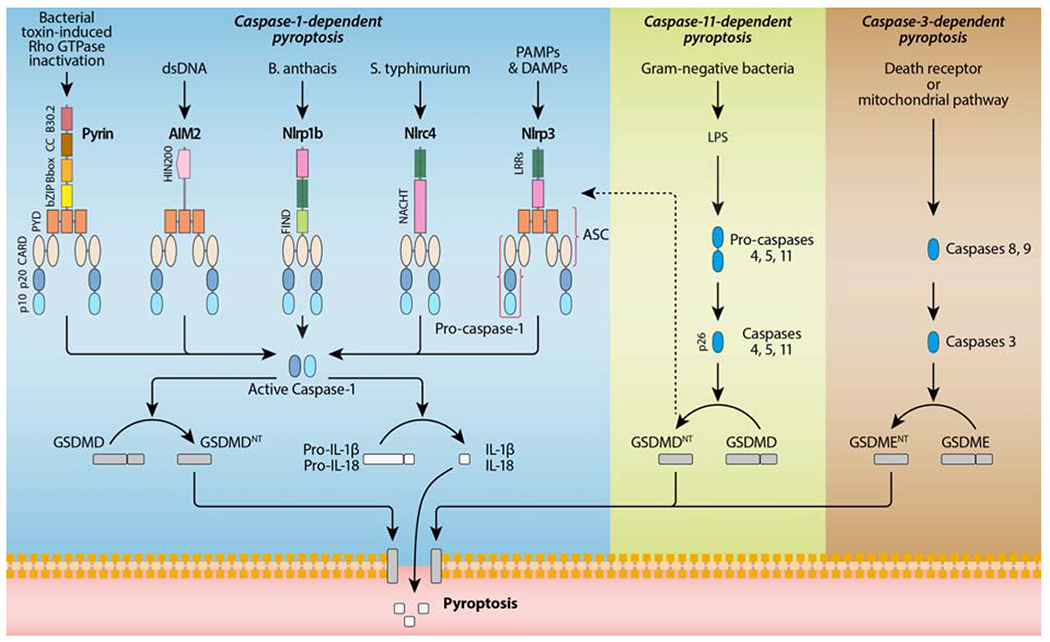
Caspase-1-dependent pyroptosis requires activation of the canonical inflammasomes, including NLRP1b, NLRP3, NLRC4, AIM2 and Pyrin. The NLRs are characterized by the combined presence of a nucleotide-binding and oligomerization domain (NACHT) and a variable number of LRRs. They also contain either a caspase recruitment domain (CARD) or pyrin domain (PYD) in their amino-terminus. The AIM2 protein is composed of a N-terminal PYD and a C-terminal DNA-binding HIN200 domain. The pyrin protein has a N-terminal PYD, a bZIP transcription factor domain, a B-box, a coiled-coil, and a C-terminal B30.2 domain. NLRP1b and NLRC4 recruit caspase-1 via their CARD domain, and the bipartite PYD-CARD adaptor protein ASC is required for assembly of the AIM2, NLRP3 and pyrin inflammasomes. The pyrin protein also binds directly to caspase-1 via its B30.2 domain. Caspase-1-independent pyroptosis requires the activation of caspase-11, which recognizes cytosolic LPS and cleaves GSDMD to initiate pyroptosis. In this pathway, GSDMDNT also activates the NLRP3 inflammasome and thereby caspase-1-dependent maturation of IL-1β and IL-18. Caspase-3-dependent pyroptosis requires the activation of Gasdermin E (GSDME). Caspase-3 can be activated by the mitochondrial and death receptor pathway. Active caspase-3 cleaves GSDME, to produce GSDME N-fragments (GSDMENT), which then forms pores in the plasma membrane.
2.3. Ferroptosis
Ferroptosis was originally observed in 2003 using erastin, a cell-permeable compound, which was only lethal to engineered human tumour cells expressing an oncogenic RAS mutation [47]. The term ferroptosis was coined in 2012 by the Stockwell lab to describe a non-apoptotic cell death caused by the accumulation of iron-dependent lipid peroxides, induced by erastin [48]. Ferroptosis, which principally exerts tumour-suppressor functions, is morphologically characterized by cell volume shrinkage and increased mitochondrial membrane density without typical apoptotic or necrotic manifestation [48]. Ferroptosis can be induced either in a canonical way by a direct or indirect inactivation of the glutathione peroxidase 4 (GPX4), the major protective mechanism of biological membranes against peroxidation damage, or in a non-canonical manner by increasing the labile iron pool [48]. The pathological relevance of ferroptosis has been evaluated using ferrostatin-1 (Fer-1), a lipid reactive oxygen species (ROS) scavenger that prevents ferroptosis induced by erastin [48–50].
In the canonical pathway, erastin interacts directly with the transporter solute carrier family 7 member 5 (SLC7A5), which disrupts the transport of amino acids into the cell by the system Xc− (Figure 3) [48]. The Xc− transporter system is composed of a regulatory subunit solute carrier family 3 member 2 (SLC3A2) and a catalytic subunit solute carrier family 7 member 11 (SLC7A11), which promotes the cellular uptake of cystine, the plasma precursor of cysteine, by exchange with glutamate [51]. In cells, cysteine is required for the synthesis of glutathione (GSH), a major redox regulatory system [52]. By indirectly blocking the Xc− transporter, erastin inhibits GSH synthesis, which is used by GPX4 to eliminate the production of phospholipid hydroperoxides (PL-OOH), the major mediator of chain reactions in lipoxygenases [53]. System Xc− inhibitors (e.g., erastin, sulfasalazine, sorafenib and L-glutamate) are considered as class I ferroptosis inducers, whereas direct GPX4 inhibitors (e.g., RAS-selective lethality protein 3 [RSL3], ML162 and altretamine) are referred to class II inducers [50].
Figure 3. The core components controlling ferroptosis.
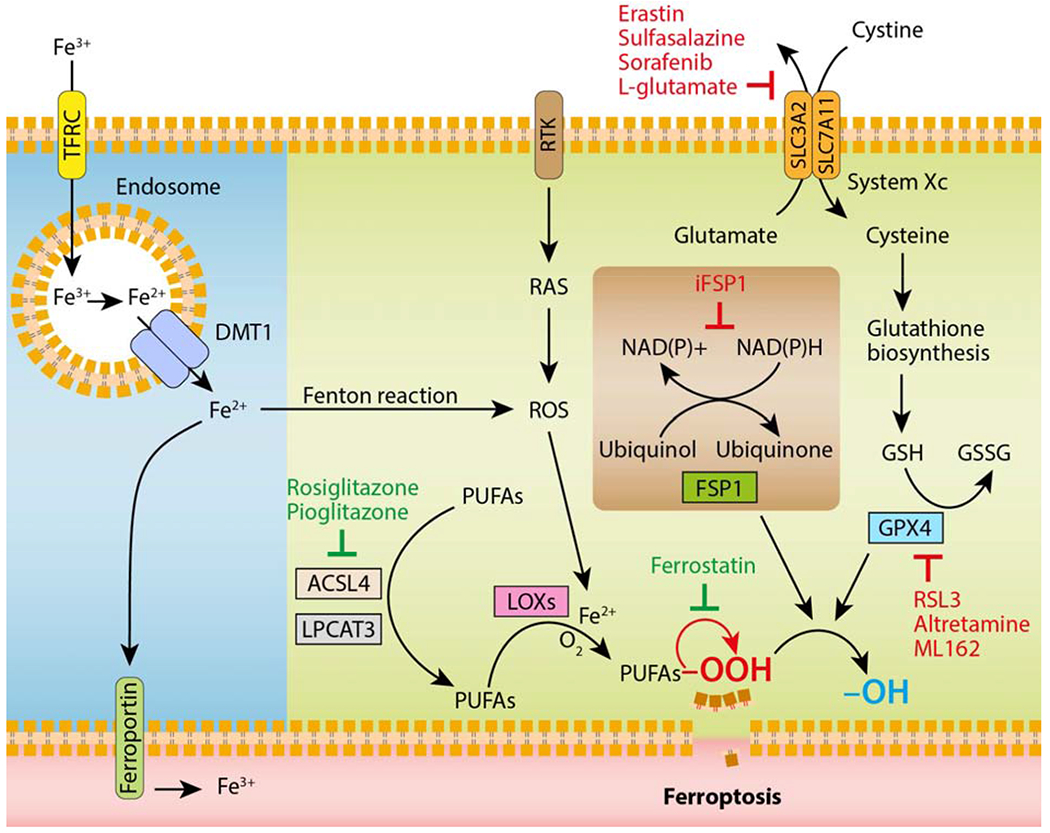
Glutathione (GSH) is a tripeptide essential for preventing damage caused by ROS, such as lipid peroxides. It is synthesized continuously from cysteine, glutamate and glycine. Cellular availability of cysteine is the limiting step for GSH synthesis. The Xc− transporter, which consists of two subunits belonging to the family of solute transporters (SLC3A2 and SLC7A11), is responsible for the uptake of extracellular cystine, the precursor of cysteine. GSH provides electrons to the key regulator of ferroptosis, GPX4, which reduces lipid peroxides (-OOH) in plasma membranes to the corresponding alcohols (-OH). Recently, the enzymes ACSL4 and LPCAT3 that are directly involved in shaping the cellular lipid composition, have been shown to sensitize cells to ferroptosis. Oxidation of lipid bilayers during ferroptosis occurs both in an enzymatic (i.e., LOXs, lipoxygenases) and non-enzymatic (i.e., radical-mediated autoxidative; symbolized by the red arrow) manner. Alternatively, FSP1 can suppress ferroptosis by ubiquinone, the reduced form of ubiquinol, which traps lipid peroxyl radicals. FSP1 catalyses the regeneration of ubiquinone using NAD(P)H. Cellular iron homeostasis, which is dependent on the coordination of iron uptake (i.e., TFRC) and export (i.e., ferroportin), directly regulates ferroptosis through Fenton reactions and generation of ROS. Fe3+ is reduced to Fe2+ by metalloreductases in the endosome, and the divalent metal transporter 1 (DMT1) mediates the transport of Fe2+ from the endosome into a labile iron pool in the cytoplasm. Receptor tyrosine kinase (RTK) activates the oncogene RAS, which is known to induce oxidative stress through generation of ROS. Ferroptosis-inducing drugs are depicted in red, whereas ferroptosis inhibitors are shown in green.
The main ROS produced by cellular metabolism or accumulated upon cell transformation (e.g., RAS-mediated) are the superoxide radical anion (O2.−) and hydrogen peroxide (H2O2). In the presence of free iron, these ROS are likely to be converted to hydroxyl radical (HO˙), which is highly reactive to macromolecules (e.g., PUFAs, polyunsaturated-fatty-acids). The reactions involving iron and leading to the hydroxyl or alkoxyl (RO˙) radical are traditionally called Fenton reactions [54]. The oxidation of PUFAs, including arachidonic acid, by a catalytic pathway involving acyl-CoA synthetase long-chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3), and lipoxygenases (LOXs), is required for lipotoxicity in ferroptosis [55–57]. Compared with other proteins of its family, GPX4 is the only member able to reduce membrane phospholipid hydroperoxides highlighting its key role against lipid peroxidation, which is prominent in ferroptosis and contributes to plasma membrane permeabilization and consequently release of DAMPs, even though the molecular mechanisms remain elusive [48]. Recently, the ferroptosis suppressor protein 1 (FSP1), previously known as apoptosis-inducing factor mitochondrial 2 (AIFM2), was identified as a key component of a non-mitochondrial CoQ10 (ubiquinone) antioxidant system that acts in parallel to the GPX4 canonical pathway to halt the progression of lipid peroxides [58, 59]. In the non-canonical pathway, increased iron uptake by transferrin receptor (TFRC) and reduced iron export by ferroportin promote oxidative damage and ferroptosis [60, 61].
2.4. Commonalities
Unlike necrosis, which is accidental and uncontrolled, lytic RCD, including necroptosis, pyroptosis and ferroptosis, are genetically defined and employ complex machinery specifically designed to perforate the plasma membrane. This common phenomenon plays an evolutionarily conserved role in systemic immunity, combining the killing of cells with alerting the immune system through the release of DAMPs. Although these three forms of lytic RCD are at first glance dedicated to eliminate intracellular pathogens (i.e., pyroptosis and necroptosis) or mutation-driven cancer cells (i.e., ferroptosis), their chronic stimulation can lead to an excessive inflammation that may be detrimental and start damaging healthy cells, tissues, and organs [62]. In fact, these three RCD forms have already been involved in the development of many inflammatory diseases from different aetiologies, including liver diseases [63].
Accumulating evidence indicates that a chronic low-level of inflammation (also known as sterile-inflammation) is mediated by DAMPs [64]. As DAMPs are hallmarks of lytic RCD, and are recognized by pattern recognition receptors (PRRs), which are also used to induce lytic RCD (e.g., TLRs and NLRs), DAMPs therefore appear to be the common denominators connecting lytic RCD pathways to each other [64]. A substantial number of DAMPs, including HMGB1, heat shock proteins (HSPs), ATP, and interleukins, have been differentially involved in pyroptosis, necroptosis or ferroptosis [65, 66]. However, further investigations are needed to identify which specific DAMPs might preferentially activate one lytic RCD or another.
Recent evidence suggests that caspase-8 may represent the molecular switch that controls apoptosis, necroptosis and pyroptosis, and prevents tissue damage [67, 68]. In addition to its enzymatic role in apoptosis and necroptosis, caspase-8 acts as a scaffolding protein that together with MLKL regulates NLRP3 inflammasome activation in macrophages [69]. These unexpected roles of caspase-8 involves the activation of the inflammasome and induction of pyroptosis under circumstances in which apoptosis and necroptosis are compromised, such as when the abundance of viral inhibitors may have driven the counteradaptation of pyroptosis as a host defence [70]. Questions that remain to be addressed include how the different modes of RCD influence disease progression and coordinate adaptive responses.
In the next section, we describe the potential roles of RCD in various metabolic liver diseases using an unexpurgated approach, in that the observations described as regulating RCD in these models are included regardless of whether they have been confirmed or even viewed skeptically.
3. Alternative Cell Death Modalities in Metabolic Liver Diseases
3.1. Non-alcoholic fatty liver disease and non-alcoholic steatohepatitis
Non-alcoholic fatty liver disease (NAFLD) is a spectrum of chronic liver disease that ranges from simple steatosis to non-alcoholic steatohepatitis (NASH) and is strongly associated with obesity and the metabolic syndrome. NAFLD is estimated to affect up to one-third of the general population worldwide [71]. Although metabolic alterations caused by free fatty acid overload is considered a major cause for hepatocyte damage, the mechanisms driving disease progression from simple steatosis to NASH remain incompletely understood [72]. Indeed, lipotoxicity, oxidative stress, organelle dysfunction and inflammatory response exacerbate ballooning and increase hepatocyte cell death [73]. Chronic liver cell death may then trigger immune cell recruitment and hepatic stellate cell activation, hepatocyte turnover and clonal expansion, in a feed-forward loop that drives NAFLD progression. While historically much emphasis was placed on apoptosis and necrosis, more recently other types of RCD, such as necroptosis or pyroptosis, controlled by overlapping molecular machineries, and capable of acting as a backup of each other, have been implicated in NAFLD and NASH pathogenesis.
3.1.1. Necroptosis
Hepatocellular necroptosis has been described in human NASH and expression of RIPK3 and MLKL is increased in liver of biopsy-proven NASH and other chronic liver disease patients [74–77]. The systemic release of necrosome proteins RIPK1 and MLKL in patients displaying disease activity further supports the clinical importance of this pathway in NASH [78]. RIPK3 and MLKL expression was also increased in visceral adipose tissue from obese and type II diabetes patients, while RIPK3 expression correlated with MLKL and metabolic serum markers [79]. High levels of core components of necroptosis were detected in liver, adipose tissue and muscle in diabetic mice, whereas targeting diabetic mice with RIPK1 inhibitors or depletion of MLKL prevented insulin resistance and glucose intolerance, despite no effect on inflammation [80]. Indeed, both specific knockdown of RIPK1, RIPK3 or MLKL and the use of chemical inhibitors activated insulin-stimulated AKT signalling, key in glucose homeostasis. Further evidence from animal studies confirmed that RIPK3 increases in the liver of experimental models of NASH [74, 75, 81]. JNK activation was implicated in RIPK3-dependent liver damage and fibrosis in methionine-choline deficient (MCD) diet-fed mice, since inhibition of JNK diminished RIPK3 levels in the liver [75]. Similarly, in common bile duct ligation hepatic injury, necroptosis was induced and contributed to fibrosis [82], although the effects of necroptosis inhibition on different hepatic fibrosis models are yet to be consistently determined. RIPK3-MLKL-mediated necroptosis contributed also to ischemia reperfusion injury of steatotic livers [83]. Mlkl−/− mice had decreased hepatic neutrophil infiltration and inflammation, while Ripk3−/− or RIPK3 kinase-dead knock-in mice were protected against late ischemia reperfusion injury.
Interestingly, RIPK3 deficiency protected from liver injury, inflammation and fibrosis in MCD-fed mice [74, 75], but exacerbated liver steatosis and apoptosis, as well as adipose tissue inflammation, insulin resistance and glucose intolerance induced by high-fat diet [79, 81]. While, RIPK3 global knockout mice exhibit no phenotype, the mechanism for the enhanced apoptosis in Ripk3−/− mice on high fat diet is uncertain, and contradictory to that reported earlier in an alcohol model [84]. Nevertheless, Ripk3−/− mice fed a choline-deficient high-fat diet as a mouse model of NASH developed insulin resistance as a result of increased compensatory apoptosis and inflammation in white adipose tissue [79]. A compartment protective function of RIPK3 in adipose tissue argues in favour of hepatocyte-specific RIPK3 inhibition in NASH. Moreover, if the increased hepatocellular apoptosis is indeed not directly determined by the absence of RIPK3 in hepatocytes, a nonparenchymal anti-inflammatory role of RIPK3 should be further dissected using conditional knockout models. Other functions of RIPK3 in hepatocarcinogenesis have been recognized [85], and its involvement in inflammatory signalling independent of necroptosis and mediation of cell death should not be neglected [86, 87]. RIPK3-deficient natural killer cells showed impaired immune responses to tumours and liver inflammation [88]. It is of interest that the strong induction of RIPK3 and MLKL in experimental models of NASH and in patients with chronic liver disease is required to sensitise to necroptosis. Epigenetic silencing of RIPK3, in contrast, has been associated with malignant transformation in hepatocellular carcinoma cells, while reestablishment of RIPK3 expression by promoter demethylation resensitized cells to chemotherapy-induced necroptosis [89], again implying that induction of RIPK3 is context-dependent.
Overall, RIP kinases are pleiotropic modulators of cell death and participate in the pathogenesis of many chronic diseases [90], suggesting that preventing the formation and/or activation of the necrosome might arrest disease progression in NASH. In a recent study, RIPA-56, a highly specific RIPK1 inhibitor, reduced hepatic inflammation and fibrosis in dietary obese mice, while reversing steatosis and dampening body weight gain [78]. Similarly, MLKL pharmacological inhibition decreased hepatic de novo fat synthesis and chemokine ligand expression in NASH [91]. Further studies in human NAFLD and NASH using robust hallmarks of necroptosis, as well as intervention studies in conditional knockouts and in models that better mimic the same degree of inflammation and fibrosis as human disease should provide additional insights on the therapeutic relevance of targeting necroptosis in the NAFLD context.
3.1.2. Pyroptosis
Lipotoxicity, innate immune response, cell death pathways, mitochondrial dysfunction and endoplasmic reticulum stress trigger chronic inflammation in the liver, potentially fuelling the transition from NAFL to NASH [92–94]. In this regard, activation of resident Kupffer cells and infiltrating macrophages is a remarkable feature of NASH pathogenesis [95]. While Kupffer cells are known to release TNF-α [96], further feeding a vicious cycle of inflammation and apoptosis leading to fibrosis, it is now apparent that inflammatory caspases, including caspase-1, murine caspase-11, and human caspase-4/5, play key important roles as mediators of inflammation [41]. This positions pyroptosis in non-alcoholic fatty liver development and progression to NASH.
Inflammasome activation triggers caspase activity and promotes inflammation and fibrosis in liver disease [97], while typical activators of inflammasomes such as fatty acids, DAMPs, and uric acid upregulate NLRP3 inflammasome components. Excessive activation of inflammatory caspases has been directly implicated in the pathogenesis of NAFLD in humans and mice, where pro-inflammatory cytokines released during pyroptosis are key effector molecules [97–99]. Indeed, activation of IL-1β signalling, downstream of inflammasomes, drives liver inflammation and fibrosis in experimental NASH [93] and amplifies the response of other cytokines [100]. Another generic substrate for inflammatory caspases, GSDMD, acts as a downstream effector of non-canonical inflammasome activation and plays a specific role in inflammatory caspase-mediated pyroptosis [43]. The GSDMDNT domain with intrinsic pyroptosis-inducing activity was positively correlated with the NAFLD activity score and fibrosis in patients with NASH [99]. Further, Gsdmd−/− mice fed an MCD diet showed significantly reduced steatohepatitis, noticeable improvement in liver inflammation, as well as reduced serum ALT levels and hepatic triglyceride content. In addition, liver fibrosis was strongly attenuated in Gsdmd−/− mice after MCD feeding [99]. Interestingly, Gsdmd−/− mice were protected from steatosis via downregulation of the lipogenic gene Srebp1c and induction of lipolytic genes, including Pparα and its downstream targets. Importantly, overexpression of the GSDMDNT domain, functionally responsible for pyroptosis, spontaneously induced liver injury even without MCD treatment, indicating that GSDMDNT-induced pyroptosis is a crucial mechanism involved in the pathogenesis of steatohepatitis.
PAMPs and DAMPs can directly induce pyroptotic cell death in hepatocytes or indirectly cause liver cell injury. Interestingly, compared to global NLRP3 knock-in mice, mice with myeloid-specific Nlrp3 mutations lack detectable pyroptotic hepatocyte cell death and have less severe liver inflammation, HSC activation, and fibrosis [101]. Thus, in addition to immune cells, hepatocyte pyroptosis resulting from intrinsic inflammasome activation exacerbates inflammation and fibrosis in the liver, indicating that both immune cell- and liver-specific NLRP3 inflammasome activation are essential for liver injury [101, 102]. Hepatocyte-specific NLRP3 mutant animals are nevertheless needed for direct evidence of the crosstalk between hepatocytes and the other cell types in the onset and progression of liver injury in NAFLD and NASH.
3.1.3. Ferroptosis
Evidence for a potential role of ferroptosis in NAFLD and NASH is scarce. Nevertheless, secondary products of lipid peroxidation, such as malondialdehyde and 4-hydroxinonenal are utilized as oxidative stress markers in NASH patients [103], and Vitamin E, an antioxidant that suppresses lipid peroxidation, reduces serum ALT in NASH patients [104]. Moreover, iron accumulation from metabolic dysfunction, aggravates NASH, such as in liver siderosis observed in some NASH patients [105], and primary hemochromatosis exacerbates NASH [106], while iron removal ameliorates liver damage and serum ALT [107]. In a recent study, ferroptosis inhibitors, trolox and deferiprone, suppressed necrotic cell death, infiltration of inflammatory cells, and inflammatory cytokine expression in the onset of steatohepatitis in the choline-deficient, ethionine-supplemented (CDE) diet model [108], in which hepatic cell death is an early event in the context of steatosis [109]. Oxygenated PE, implicated in the ferroptosis pathway, was increased in the liver of CDE-fed mice, but normalized after trolox treatment. Consistently, the hepatic PC/PE ratio is decreased in NASH patients [110, 111]. Overall, evidence of ferroptosis in NAFLD and NASH deserves further exploitation in meaningful models of disease and in patients, particularly since no therapeutic options are currently available for NASH.
3.2. Alcohol-associated liver disease and alcoholic steatohepatitis
Alcohol-associated liver disease (ALD) is a major health problem worldwide and a significant source of liver injury, characterized by a wide spectrum of hepatic lesions from steatosis, steatohepatitis and fibrosis that may progress to cirrhosis and even hepatocellular carcinoma [112]. Cell injury, inflammation, oxidative stress, regeneration and bacterial translocation are key drivers of alcohol-induced liver injury [113]. ALD develops via a complex process involving parenchymal and non-parenchymal cells, as well as recruitment of other cell types to the liver in response to damage and inflammation. Importantly, cross-regulation by the effectors of different cell death pathways most likely influences liver inflammation in ALD. It is possible that apoptosis occurs in early steatosis ALD, followed by necroptosis in early ASH and pyroptosis in alcoholic hepatitis, in agreement with neutrophilic inflammation and endotoxemia and septicaemia, the most common cause of death from alcoholic hepatitis.
3.2.1. Necroptosis
The involvement of necroptosis has been suggested from studies where RIPK3 expression was induced in human ALD and in mice after ethanol exposure [84, 114]. RIPK3-mediated necroptosis and neutrophil-mediated liver inflammatory response were also highly correlated with poor prognosis in patients with end-stage alcoholic cirrhosis [115]. RIPK3 deficiency, in turn, effectively protected from ethanol-induced serum liver enzymes, steatosis and inflammation [84, 114]. Nevertheless, inhibition of RIP1 kinase activity blunted Gao-binge alcohol-induced hepatic inflammation but did not protect against chronic ethanol feeding-induced steatosis and liver injury, suggesting that alcohol-induced RIPK3-mediated necroptosis is independent of RIPK1 [114]. Other studies reported that ethanol-induced decrease of Nrf2 expression was abrogated by curcumin or gallic acid treatment, by suppressing RIPK3 and RIPK1 expression as well as DAMP release [116, 117]. Overall, RIPK1 involvement remains to be consistently demonstrated in ethanol-induced hepatic injury, RIPK3 depletion should be attempted in other relevant cell types, and MLKL targeting warrants further exploitation. While it appears clear that ethanol-induced necroptosis involves RIPK3, it is not yet known if MLKL-independent pathways are involved as reported in models of autoimmune arthritis [118]. Further, increased infiltration of inflammatory cells in the liver after alcohol exposure requires the inclusion of inflammatory cell-specific knockout mice in future studies to clarify the role of necroptosis in alcohol-induced liver pathogenesis.
3.2.2. Pyroptosis
Excessive and uncontrolled activation of inflammasomes has a damaging effect on the host, inducing uncontrolled inflammatory responses and the consequent cell death. Recent evidence shows that pyroptosis plays a prominent role in ALD pathogenesis. The NLRP3 inflammasome pathway is activated in hepatocytes in response to endotoxin challenge, a condition facilitated by alcohol intake [119]. Consistently, NLRP3 deficiency ameliorated alcohol-driven liver stetaosis and injury [120], while caspase-4/11 was upregulated in liver tissue from patients with alcoholic hepatitis and in mice [121]. Pyroptosis triggered by gut-derived PAMPS and by metabolic-derived DAMPs (ATP and uric acid) resulted in release of inflammasome-dependent cytokines from immune cells in ALD patients and in cells exposed to ethanol [120, 122]. Caspase-1 was also activated in hepatocytes of patients with alcoholic hepatitis [123]. Interestingly, a recent study demonstrated that alcohol elicits caspase-1-mediated pyroptosis through overexpression of thioredoxin-interacting protein (TXNIP), a member of the α-arrestin family, and this event is reversed by miR-148a [124]. MiR-148a levels, in turn, are deregulated in the liver of patients with alcoholic hepatitis or ethanol-fed animals. Consistently, hepatocyte-specific delivery of miR-148a alleviates alcoholic liver injury. These data together with the fact that serum ALT activities remained elevated after macrophage depletion supports the direct effect of alcohol on NLRP3 inflammasome activation in hepatocytes. Pyroptosis is thus fit for stimulating and effectively sustaining the inflammatory cycle in ALD, although the specific contribution of NLRP3 inflammasome in immune cells and hepatic stellate cells remains still debatable.
3.2.3. Ferroptosis
Iron overload and increased oxidative stress have been well documented in ALD [125]. Recent findings shed light on mechanisms underlying the alcohol-induced liver damage by describing that adipose-specific lipin-1 overexpression accelerated iron accumulation, caused lipid peroxidation, reduced GSH and GAPDH, and promoted ferroptotic liver damage in mice after ethanol administration [126]. Lipin-1 is a Mg2+-dependent phosphatidic acid phosphohydrolase involved in the generation of diacylglycerol during synthesis of phospholipids and triglycerides. Ethanol-mediated inhibitory effects on adipose-specific lipin-1 expression were associated with experimental steatohepatitis in rodents. The hepatic ferroptotic signalling in the liver damage induced by adipose lipin-1 overexpression in ethanol-fed mice remains, however, to be elucidated.
3.3. Other metabolic liver disorders
3.3.1. Hemochromatosis
Studies on cell death in other metabolic liver disorders, including iron-overload diseases, and genetic disorders such as Niemann-Pick disease or Gaucher’s disease are scarce. Iron is implicated in the pathogenesis of a number of human liver diseases. Hereditary haemochromatosis is the classical example of a liver disease caused by iron, but iron is an important contributor to the progression of other forms of chronic liver disease, including NAFLD. Iron-induced liver damage is likely potentiated by coexisting inflammation, which culminates with hepatocyte cell death and drives fibrogenesis [127]. Hereditary hemochromatosis is caused by mutations in genes whose protein products limit iron absorption. As a consequence, excess iron generates ROS thereby inducing cell death and global oxidative damage, ultimately leading to severe chronic complications, including hepatic cirrhosis. Nevertheless, the precise roles of iron and iron metabolism in cell death are largely unknown. A recent study investigated the role of iron homeostasis in SLC7A11-mediated ferroptosis and found that iron overload triggers ferroptosis both in vitro and in vivo [128]. Indeed, Slc7a11 knockout was not sufficient to induce ferroptosis under basal conditions but facilitated iron overload-induced ferroptosis due to impaired cystine uptake and increased ROS production. Ferroptosis is thus a potential therapeutic target for treating iron overload-associated diseases, including hemochromatosis. Other lytic forms of cell death need to be explored and extended further to other metal-related liver disorders.
3.3.2. Niemann-Pick disease
Niemann-Pick disease, type C1 (NPC1) is an inborn error of metabolism that results in endolysosomal accumulation of unesterified cholesterol. Clinically, NPC1 manifests as cholestatic liver disease in the newborn or as a progressive neurodegenerative condition characterized by cerebellar ataxia and cognitive decline. Neuroinflammation and necroptosis contribute to the NPC1 pathological cascade. Pharmacological and genetic inhibition of RIPK1 kinase activity increased lifespan in both Npc1−/− mice treated with RIPK1 inhibitors, and Npc1−/−/Ripk1 kinase death double mutant mice [129]. Nevertheless, the increase in lifespan was modest, suggesting that the therapeutic potential of RIPK1 inhibition, as a monotherapy, is limited. Conversely, no increased survival was noticed in Npc1−/−/Ripk3 mice compared to Npc1−/− mice. These data suggest that although necroptosis is occurring in NPC1, the effects of RIPK1 inhibition may be related to its RIPK3-independent role in neuroinflammation and cytokine production. Nevertheless, corroborating the role of alternative RCD sub-routines, cellular death of NPC1 fibroblasts was not mitigated by treatment with caspase inhibitors [129] or following neuronal overexpression of Bcl-2 [130]. More importantly, necroptosis inhibition and combination therapy with 2-hydroxypropyl-β-cyclodextrin has recently been suggested for NPC1 [131].
3.3.3. Gaucher’s disease
Necroptosis appears to have a role in the pathological cascade of other lysosomal storage diseases. Gaucher’s disease is an inherited metabolic disorder caused by mutations in the glucocerebrosidase gene (GBA), and the most common lysosomal storage disease. Neuronal cell death in mice with Gaucher’s disease is caspase-independent and nonapoptotic. In striking contrast, RIPK3-mediated necroptosis has been implicated in pathology of Gaucher’s disease [132]. In fact, levels of Ripk1 and Ripk3 were markedly upregulated in Gaucher’s disease brains, both in mice and in patients, but were unaltered in brains obtained from mouse models of NPC1. This indicates that although brain inflammation and microglial activation are shared features of many lysosomal storage diseases, pathways of neuroinflammation are disease specific. Moreover, RIPK3 deficiency improved the clinical course of Gaucher’s disease mice, with improved survival and motor coordination and beneficial effects on cerebral and hepatic injury. Ablation of Ripk3 resulted in fewer CD68-positive Kupffer cells and decreased ALT activity, suggesting that hepatocyte injury was also attenuated. Development of necroptosis inhibitors with improved pharmacokinetics may pave the way for alternative therapeutic approaches for Gaucher’s disease, for which innovative treatment is urgently required. The specific involvement of other RCD forms is yet to be determined.
4. Controversies and Unsolved Issues
4.1. Perspective of cell death in liver diseases
Cell death has been described as a clear and present danger for hepatocytes as they are directly exposed to portal blood from the intestines, participate in the xenobiotic metabolism of drugs and alcohol, play a central role in lipid, fatty acid, and bile acid metabolism, contact with prevalent hepatotropic viruses, and reside within a milieu of innate immune responding cells [133]. Yet elucidating the precise role for various modes of cell death in liver diseases has proven difficult, and unpredictable. For example, apoptosis as a driving force in liver injury and carcinogenesis has only recently been established despite intense study over the last two decades [134]. It will also take time and detailed analysis of relevant models to identify the potency and hierarchal relationship between apoptosis, necroptosis, pyroptosis and ferroptosis in liver diseases. Also, perturbations with therapeutic intent targeting one mode of cell death may potentiate or even inhibit additional modes of liver injury further complicating this analysis. For example, caspase inhibitors employed to prevent apoptosis, may promote necroptosis by preventing caspase-8 proteolytic degradation of RIP1 kinase, or inhibit pyroptosis by blocking caspase-1 or −8. In this section of this review, we will acknowledge these controversies providing nuanced insight and a critical appraisal of the information described above.
4.2. Unresolved issues regarding necroptosis in liver diseases
The role of necroptosis in liver diseases remains to be firmly established. This uncertanty relates to current tools, models, and evolving information that permits a reinterpretation of prior studies. First, there is vigorous controversy regarding the expression of RIPK3 by hepatocytes, but under basal conditions expression appears to be quite limited [135, 136]. Whether they can be induced in various liver diseases remains unclear, as the antibodies employed for immunoblot analysis and immunohistochemistry are fraught with nonspecificity. We are not aware of any detailed proteomic studies which have been performed to address these questions, although the technology to examine phosphoproteins by this approach is widely available. Moreover, activating phosphorylation of RIPK3 and MLKL have been identified in the absence of cell death questioning the use of these phosphoproteins as biomarkers for necroptosis [135]. Second, RIPK3 deficiency does not phenocopy MLKL deficiency, and hence studies with RIPK3 knockout or kinase dead knock-in mice, are not sufficient to invoke necroptosis as a cause of cell death, as RIPK3 has diverse functions [137]. Moreover, inhibition of RIPK3 kinase often provokes apoptosis further confusing this approach in examining necroptosis in disease pertinent models [25]. The multifunctional aspect of RIPK3 biology in different cell types certainly needs to be taken into account when germ line vs. cell type specific knockout or kinase dead knock-in paradigms are employed. Third, caspase-8 limits necroptosis by proteolytically cleaving RIPK1 [138], and the genetic absence of caspase-8 in hepatocytes does not trigger wide-spread necroptosis [134]. Finally, genetic studies employing RIPK3 knockout mice suggest cholangiocyte, but not hepatocyte injury is ameliorated suggesting necroptosis mediates cholangiocyte but not hepatocyte injury [134]. The above observations will need to be reconciled with those presented above implicating necroptosis in liver disease (see future directions below).
4.3. Unresolved issues regarding pyroptosis in liver diseases
Pyroptosis requires caspase-dependent cleavage of GSDMD to plasma membrane pore-forming fragments [139]. Cleave of GSDMD occurs via a canonical inflammasome caspase-1 dependent mechanism, a non-canonical inflammasome pathway involving caspases 4, 5 or 11 [139], or in an inflammasome-independent caspase-8 mediated pathway [140]. Although all cell types appear to be inflammasome competent, the best examples are in cells of the innate immune system, especially those interacting with intracellular organisms. Cell type specific GSDMD will be requisite to examine this form of cell death in hepatocytes. Interestingly, in shock models of liver injury, GSDMD germ line deficient mice display increased caspase-8 activation and apoptosis in the liver [141]. This is another cautionary example where inhibiting one mode of cell death enhances a different mechanism of liver injury.
4.4. Unresolved issues regarding ferroptosis in liver diseases
Oxidative stress has long-been implicated in liver injury. Ferroptosis which is regulated by the availability of cytosolic free iron, cystine transport into the cell, GPX4 activity, and glutathione levels, leads to cell death by lipid peroxidation. A specific linchpin molecular executioner of ferroptosis has not been described, and hence genetic deletion of a final common pathway mediator is not yet feasible. In fact, propagation of lipid peroxidation is non-enzymatic and may prove difficult to target. In this regard, the role of ferroptosis in liver disease will be largely circumstantial, and we note that to date, antioxidant therapy has not been impactful in human liver diseases. Careful work will be necessary to dissect the ultimate role of ferroptosis in liver diseases.
5. Future directions and therapeutic implications
The contribution of different modes of cell death to various liver diseases will require carefully defined preclinical studies that address causality. These studies will need to focus on the terminal executioners of cell death, as upstream pathway mediators have multiple functions. For example, RIP kinases have diverse functions in addition to promoting necroptosis [90]. Such studies need to focus initially on hepatocyte cell death employing hepatocyte conditional knockout models of MLKL, GSDMD, and Bax plus Bak, to discern the interrelationships between necroptosis, pyroptosis and apoptosis. These studies will require development of genetic mouse models and hence will take time. Also, conditional knockouts in other cell types (e.g., cholangiocytes, immune cells such as macrophages) will ultimately be necessary to complete this puzzle of cell death in liver injury. Finally, we note that cell death per se may be a biomarker for stressed, proinflammatory hepatocytes as recently suggested in NASH [142], and therefore, precluding cell death may not have the major impact on liver injury that we anticipate.
These various modes of cell death also represent unique therapeutic avenues for the treatment of cell death in human liver diseases. However, inhibiting one mode of cell death may potentiate other modes of cell death as noted above, negating a therapeutic benefit. Recently reported, caspase inhibition with Emricasan treatment did not improve clinical outcomes nor liver histology in patients with NASH fibrosis, resulting in more liver fibrosis and hepatocyte ballooning [143, 144]. Caspase inhibition may have directed cells to alternative, more inflammatory mechanisms of cell death, such as necroptosis. Indeed, Emricasan has previously been shown to shift cell death from apoptosis towards necroptosis in acute myeloid leukemia cells treated with birinapant, a second mitochondrial derived activator of caspases mimetic, which enhances cancer cell apoptosis [145]. MLKL inhibitors are being developed such as necrosulfonamide, which binds to the N-terminal 4 helical bundle of MLKL to prevent necroptosis [146, 147]. Indeed, a variety of targets and inhibitors are being investigated to block necroptosis, and we refer the reader to this literature [147]. Interestingly, necrosulfonamide also binds GSDMD and blocks pyroptosis [148]. As these compounds and others become further developed as pharmacologic agents, we look forward to future human clinical trials with anticipation.
In summary, we now recognize several different forms of cell death, executed by different pathways and mediators. The mechanisms are being unraveled and their contribution to human liver diseases is being carefully examined. These insights provide further complexity to understanding liver injury, but these challenges also provide new and exciting therapeutic opportunities.
Key points.
Regulated hepatocyte cell death drives metabolic liver disease progression to liver inflammation, fibrosis and cirrhosis.
Accumulating evidence suggests that lytic cell death modalities such as hepatocyte necroptosis, pyroptosis and ferroptosis play pathophysiological roles in metabolic liver disease, while other functions in non-hepatocyte liver cells, immune cells and others have been suggested.
Functional characterization of lytic cell death in appropriate models of metabolic liver disease and in patients might result in novel therapeutic strategies that target regulated hepatocyte cell death to prevent disease progression.
Acknowledgments:
The authors would like to thank Yves Chrétien for expert artwork.
Financial support: JG is supported by grants from the Mairie de Paris (Emergences), the Société Francophone du Diabète (SFD), the Institute of Cardiometabolism and Nutrition (ICAN), and the Fondation pour la Recherche Médicale (FRM grant number ARF20170938613). GJG is supported by NIH grants DK 124182 and the Digestive Disease Research Center for Cell Signaling in Gastroenterology P30DK084567, the Chris M Carlos and Catharine Nicole Jockisch Carlos Foundation for PSC, and the Mayo Clinic. CMPR is supported by the Fundação para a Ciência e a Tecnologia (FCT) and European Structural & Investment Funds through the COMPETE Programme (grants UID/DTP/04138/2019, PTDC/MED-FAR/29097/2017 and SAICTPAC/0019/2015 - LISBOA-01-0145-FEDER-016405), and the EU H2020 Marie Sklodowska-Curie Project Foie Gras (grant 722619).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: The authors declare no conflicts of interest that pertain to this work.
References
- [1].Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 2014;147:765–783 e764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018;25:486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 2014;15:135–147. [DOI] [PubMed] [Google Scholar]
- [4].Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol 2018;15:738–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ray CA, Pickup DJ. The mode of death of pig kidney cells infected with cowpox virus is governed by the expression of the crmA gene. Virology 1996;217:384–391. [DOI] [PubMed] [Google Scholar]
- [6].Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 2000;1:489–495. [DOI] [PubMed] [Google Scholar]
- [7].Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005;1:112–119. [DOI] [PubMed] [Google Scholar]
- [8].He S, Wang L, Miao L, Wang T, Du F, Zhao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009;137:1100–1111. [DOI] [PubMed] [Google Scholar]
- [9].Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012;148:213–227. [DOI] [PubMed] [Google Scholar]
- [10].Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009;325:332–336. [DOI] [PubMed] [Google Scholar]
- [11].Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A 2012;109:5322–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 2017;18:127–136. [DOI] [PubMed] [Google Scholar]
- [13].Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res 2019;29:347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mompean M, Li W, Li J, Laage S, Siemer AB, Bozkurt G, et al. The Structure of the Necrosome RIPK1-RIPK3 Core, a Human Hetero-Amyloid Signaling Complex. Cell 2018;173:1244–1253 e1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hanna-Addams S, Liu S, Liu H, Chen S, Wang Z. CK1alpha, CK1delta, and CK1epsilon are necrosome components which phosphorylate serine 227 of human RIPK3 to activate necroptosis. Proc Natl Acad Sci U S A 2020;117:1962–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, Bruggeman I, et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 2014;7:971–981. [DOI] [PubMed] [Google Scholar]
- [17].Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, Sharma P, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A 2014;111:15072–15077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cai Z, Zhang A, Choksi S, Li W, Li T, Zhang XM, et al. Activation of cell-surface proteases promotes necroptosis, inflammation and cell migration. Cell Res 2016;26:886–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, et al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol 2014;16:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zargarian S, Shlomovitz I, Erlich Z, Hourizadeh A, Ofir-Birin Y, Croker BA, et al. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol 2017;15:e2002711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vanden Berghe T, Kaiser WJ, Bertrand MJ, Vandenabeele P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol Cell Oncol 2015;2:e975093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 1999;13:2514–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Feng S, Yang Y, Mei Y, Ma L, Zhu DE, Hoti N, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal 2007;19:2056–2067. [DOI] [PubMed] [Google Scholar]
- [24].Newton K, Dugger DL, Wickliffe KE, Kapoor N, de Almagro MC, Vucic D, et al. Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 2014;343:1357–1360. [DOI] [PubMed] [Google Scholar]
- [25].Mandal P, Berger SB, Pillay S, Moriwaki K, Huang C, Guo H, et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol Cell 2014;56:481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci U S A 2011;108:20054–20059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 2012;11:290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schock SN, Chandra NV, Sun Y, Irie T, Kitagawa Y, Gotoh B, et al. Induction of necroptotic cell death by viral activation of the RIG-I or STING pathway. Cell Death Differ 2017;24:615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Brault M, Olsen TM, Martinez J, Stetson DB, Oberst A. Intracellular Nucleic Acid Sensing Triggers Necroptosis through Synergistic Type I IFN and TNF Signaling. J Immunol 2018;200:2748–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wang X, He Z, Liu H, Yousefi S, Simon HU. Neutrophil Necroptosis Is Triggered by Ligation of Adhesion Molecules following GM-CSF Priming. J Immunol 2016;197:4090–4100. [DOI] [PubMed] [Google Scholar]
- [31].Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, et al. ESCRT-III Acts Downstream of MLKL to Regulate Necroptotic Cell Death and Its Consequences. Cell 2017;169:286–300 e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Yoon S, Kovalenko A, Bogdanov K, Wallach D. MLKL, the Protein that Mediates Necroptosis, Also Regulates Endosomal Trafficking and Extracellular Vesicle Generation. Immunity 2017;47:51–65 e57. [DOI] [PubMed] [Google Scholar]
- [33].Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992;358:167–169. [DOI] [PubMed] [Google Scholar]
- [34].Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol 2001;9:113–114. [DOI] [PubMed] [Google Scholar]
- [35].Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011;479:117–121. [DOI] [PubMed] [Google Scholar]
- [36].Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014;514:187–192. [DOI] [PubMed] [Google Scholar]
- [37].Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 2017;8:14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017;547:99–103. [DOI] [PubMed] [Google Scholar]
- [39].Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 2009;7:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015;526:666–671. [DOI] [PubMed] [Google Scholar]
- [41].Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015;526:660–665. [DOI] [PubMed] [Google Scholar]
- [42].Orning P, Lien E, Fitzgerald KA. Gasdermins and their role in immunity and inflammation. J Exp Med 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Broz P, Pelegrin P, Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat Rev Immunol 2019. [DOI] [PubMed] [Google Scholar]
- [44].Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 2016;16:407–420. [DOI] [PubMed] [Google Scholar]
- [45].Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell 2014;157:1013–1022. [DOI] [PubMed] [Google Scholar]
- [46].Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol 2015;25:308–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003;3:285–296. [DOI] [PubMed] [Google Scholar]
- [48].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 2014;136:4551–4556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ 2016;23:369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lo M, Wang YZ, Gout PW. The x(c)- cystine/glutamate antiporter: a potential target for therapy of cancer and other diseases. J Cell Physiol 2008;215:593–602. [DOI] [PubMed] [Google Scholar]
- [52].Lu SC. Glutathione synthesis. Biochim Biophys Acta 2013;1830:3143–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014;156:317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Feng H, Stockwell BR. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol 2018;16:e2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 2017;13:91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 2017;13:81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 2016;113:E4966–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019;575:693–698. [DOI] [PubMed] [Google Scholar]
- [59].Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019;575:688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol Cell 2015;59:298–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Geng N, Shi BJ, Li SL, Zhong ZY, Li YC, Xua WL, et al. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur Rev Med Pharmacol Sci 2018;22:3826–3836. [DOI] [PubMed] [Google Scholar]
- [62].Kolb JP, Oguin TH 3rd, Oberst A, Martinez J Programmed Cell Death and Inflammation: Winter Is Coming. Trends Immunol 2017;38:705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Aizawa S, Brar G, Tsukamoto H. Cell Death and Liver Disease. Gut Liver 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 2010;10:826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol 2019;12:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ 2019;26:99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Fritsch M, Gunther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP, et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019;575:683–687. [DOI] [PubMed] [Google Scholar]
- [68].Newton K, Wickliffe KE, Maltzman A, Dugger DL, Reja R, Zhang Y, et al. Activity of caspase-8 determines plasticity between cell death pathways. Nature 2019;575:679–682. [DOI] [PubMed] [Google Scholar]
- [69].Kang S, Fernandes-Alnemri T, Rogers C, Mayes L, Wang Y, Dillon C, et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun 2015;6:7515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Mocarski ES, Upton JW, Kaiser WJ. Viral infection and the evolution of caspase 8-regulated apoptotic and necrotic death pathways. Nat Rev Immunol 2011;12:79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Tsochatzis EA, Newsome PN. Non-alcoholic fatty liver disease and the interface between primary and secondary care. Lancet Gastroenterol Hepatol 2018;3:509–517. [DOI] [PubMed] [Google Scholar]
- [72].Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Afonso MB, Castro RE, Rodrigues CMP. Processes exacerbating apoptosis in non-alcoholic steatohepatitis. Clin Sci (Lond) 2019;133:2245–2264. [DOI] [PubMed] [Google Scholar]
- [74].Afonso MB, Rodrigues PM, Carvalho T, Caridade M, Borralho P, Cortez-Pinto H, et al. Necroptosis is a key pathogenic event in human and experimental murine models of non-alcoholic steatohepatitis. Clin Sci (Lond) 2015;129:721–739. [DOI] [PubMed] [Google Scholar]
- [75].Gautheron J, Vucur M, Reisinger F, Cardenas DV, Roderburg C, Koppe C, et al. A positive feedback loop between RIP3 and JNK controls non-alcoholic steatohepatitis. EMBO Mol Med 2014;6:1062–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Afonso MB, Rodrigues PM, Simao AL, Ofengeim D, Carvalho T, Amaral JD, et al. Activation of necroptosis in human and experimental cholestasis. Cell Death Dis 2016;7:e2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Gautheron J, Vucur M, Luedde T. Necroptosis in Nonalcoholic Steatohepatitis. Cell Mol Gastroenterol Hepatol 2015;1:264–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Majdi A, Aoudjehane L, Ratziu V, Islam T, Afonso MB, Conti F, et al. Inhibition of receptor-interacting protein kinase 1 improves experimental non-alcoholic fatty liver disease. J Hepatol 2019. [DOI] [PubMed] [Google Scholar]
- [79].Gautheron J, Vucur M, Schneider AT, Severi I, Roderburg C, Roy S, et al. The necroptosis-inducing kinase RIPK3 dampens adipose tissue inflammation and glucose intolerance. Nat Commun 2016;7:11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Xu H, Du X, Liu G, Huang S, Du W, Zou S, et al. The pseudokinase MLKL regulates hepatic insulin sensitivity independently of inflammation. Mol Metab 2019;23:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Roychowdhury S, McCullough RL, Sanz-Garcia C, Saikia P, Alkhouri N, Matloob A, et al. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology 2016;64:1518–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Afonso MB, Rodrigues PM, Simao AL, Gaspar MM, Carvalho T, Borralho P, et al. miRNA-21 ablation protects against liver injury and necroptosis in cholestasis. Cell Death Differ 2018;25:857–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Ni HM, Chao X, Kaseff J, Deng F, Wang S, Shi YH, et al. Receptor-Interacting Serine/Threonine-Protein Kinase 3 (RIPK3)-Mixed Lineage Kinase Domain-Like Protein (MLKL)-Mediated Necroptosis Contributes to Ischemia-Reperfusion Injury of Steatotic Livers. Am J Pathol 2019;189:1363–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 2013;57:1773–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Vucur M, Reisinger F, Gautheron J, Janssen J, Roderburg C, Cardenas DV, et al. RIP3 inhibits inflammatory hepatocarcinogenesis but promotes cholestasis by controlling caspase-8- and JNK-dependent compensatory cell proliferation. Cell Rep 2013;4:776–790. [DOI] [PubMed] [Google Scholar]
- [86].Moriwaki K, Chan FK. Necrosis-dependent and independent signaling of the RIP kinases in inflammation. Cytokine Growth Factor Rev 2014;25:167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Newton K RIPK1 and RIPK3: critical regulators of inflammation and cell death. Trends Cell Biol 2015;25:347–353. [DOI] [PubMed] [Google Scholar]
- [88].Kang YJ, Bang BR, Han KH, Hong L, Shim EJ, Ma J, et al. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat Commun 2015;6:8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res 2015;25:707–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].He S, Wang X. RIP kinases as modulators of inflammation and immunity. Nat Immunol 2018;19:912–922. [DOI] [PubMed] [Google Scholar]
- [91].Saeed WK, Jun DW, Jang K, Oh JH, Chae YJ, Lee JS, et al. Decrease in fat de novo synthesis and chemokine ligand expression in non-alcoholic fatty liver disease caused by inhibition of mixed lineage kinase domain-like pseudokinase. J Gastroenterol Hepatol 2019. [DOI] [PubMed] [Google Scholar]
- [92].Farrell GC, van Rooyen D, Gan L, Chitturi S. NASH is an Inflammatory Disorder: Pathogenic, Prognostic and Therapeutic Implications. Gut Liver 2012;6:149–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Mridha AR, Wree A, Robertson AAB, Yeh MM, Johnson CD, Van Rooyen DM, et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J Hepatol 2017;66:1037–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Schuster S, Cabrera D, Arrese M, Feldstein AE. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol 2018;15:349–364. [DOI] [PubMed] [Google Scholar]
- [95].Syn WK, Oo YH, Pereira TA, Karaca GF, Jung Y, Omenetti A, et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 2010;51:1998–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Tosello-Trampont AC, Landes SG, Nguyen V, Novobrantseva TI, Hahn YS. Kuppfer cells trigger nonalcoholic steatohepatitis development in diet-induced mouse model through tumor necrosis factor-alpha production. J Biol Chem 2012;287:40161–40172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Szabo G, Petrasek J. Inflammasome activation and function in liver disease. Nat Rev Gastroenterol Hepatol 2015;12:387–400. [DOI] [PubMed] [Google Scholar]
- [98].Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012;482:179–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Xu B, Jiang M, Chu Y, Wang W, Chen D, Li X, et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J Hepatol 2018;68:773–782. [DOI] [PubMed] [Google Scholar]
- [100].Tilg H, Moschen AR, Szabo G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2016;64:955–965. [DOI] [PubMed] [Google Scholar]
- [101].Wree A, Eguchi A, McGeough MD, Pena CA, Johnson CD, Canbay A, et al. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology 2014;59:898–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Wree A, McGeough MD, Inzaugarat ME, Eguchi A, Schuster S, Johnson CD, et al. NLRP3 inflammasome driven liver injury and fibrosis: Roles of IL-17 and TNF in mice. Hepatology 2018;67:736–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Loguercio C, De Girolamo V, de Sio I, Tuccillo C, Ascione A, Baldi F, et al. Non-alcoholic fatty liver disease in an area of southern Italy: main clinical, histological, and pathophysiological aspects. J Hepatol 2001;35:568–574. [DOI] [PubMed] [Google Scholar]
- [104].Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 2010;362:1675–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Nelson JE, Wilson L, Brunt EM, Yeh MM, Kleiner DE, Unalp-Arida A, et al. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011;53:448–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Bonkovsky HL, Jawaid Q, Tortorelli K, LeClair P, Cobb J, Lambrecht RW, et al. Non-alcoholic steatohepatitis and iron: increased prevalence of mutations of the HFE gene in non-alcoholic steatohepatitis. J Hepatol 1999;31:421–429. [DOI] [PubMed] [Google Scholar]
- [107].Valenti L, Moscatiello S, Vanni E, Fracanzani AL, Bugianesi E, Fargion S, et al. Venesection for non-alcoholic fatty liver disease unresponsive to lifestyle counselling--a propensity score-adjusted observational study. QJM 2011;104:141–149. [DOI] [PubMed] [Google Scholar]
- [108].Tsurusaki S, Tsuchiya Y, Koumura T, Nakasone M, Sakamoto T, Matsuoka M, et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis 2019;10:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Kohn-Gaone J, Dwyer BJ, Grzelak CA, Miller G, Shackel NA, Ramm GA, et al. Divergent Inflammatory, Fibrogenic, and Liver Progenitor Cell Dynamics in Two Common Mouse Models of Chronic Liver Injury. Am J Pathol 2016;186:1762–1774. [DOI] [PubMed] [Google Scholar]
- [110].Li Z, Agellon LB, Allen TM, Umeda M, Jewell L, Mason A, et al. The ratio of phosphatidylcholine to phosphatidylethanolamine influences membrane integrity and steatohepatitis. Cell Metab 2006;3:321–331. [DOI] [PubMed] [Google Scholar]
- [111].Hernandez-Alvarez MI, Sebastian D, Vives S, Ivanova S, Bartoccioni P, Kakimoto P, et al. Deficient Endoplasmic Reticulum-Mitochondrial Phosphatidylserine Transfer Causes Liver Disease. Cell 2019;177:881–895 e817. [DOI] [PubMed] [Google Scholar]
- [112].Bataller R, Gao B. Liver fibrosis in alcoholic liver disease. Semin Liver Dis 2015;35:146–156. [DOI] [PubMed] [Google Scholar]
- [113].Louvet A, Mathurin P. Alcoholic liver disease: mechanisms of injury and targeted treatment. Nat Rev Gastroenterol Hepatol 2015;12:231–242. [DOI] [PubMed] [Google Scholar]
- [114].Wang S, Ni HM, Dorko K, Kumer SC, Schmitt TM, Nawabi A, et al. Increased hepatic receptor interacting protein kinase 3 expression due to impaired proteasomal functions contributes to alcohol-induced steatosis and liver injury. Oncotarget 2016;7:17681–17698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Zhang Z, Xie G, Liang L, Liu H, Pan J, Cheng H, et al. RIPK3-Mediated Necroptosis and Neutrophil Infiltration Are Associated with Poor Prognosis in Patients with Alcoholic Cirrhosis. J Immunol Res 2018;2018:1509851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Lu C, Xu W, Zhang F, Shao J, Zheng S. Nrf2 Knockdown Disrupts the Protective Effect of Curcumin on Alcohol-Induced Hepatocyte Necroptosis. Mol Pharm 2016;13:4043–4053. [DOI] [PubMed] [Google Scholar]
- [117].Zhou Y, Jin H, Wu Y, Chen L, Bao X, Lu C. Gallic acid protects against ethanol-induced hepatocyte necroptosis via an NRF2-dependent mechanism. Toxicol In Vitro 2019;57:226–232. [DOI] [PubMed] [Google Scholar]
- [118].Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun 2015;6:6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Boaru SG, Borkham-Kamphorst E, Tihaa L, Haas U, Weiskirchen R. Expression analysis of inflammasomes in experimental models of inflammatory and fibrotic liver disease. J Inflamm (Lond) 2012;9:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Petrasek J, Iracheta-Vellve A, Saha B, Satishchandran A, Kodys K, Fitzgerald KA, et al. Metabolic danger signals, uric acid and ATP, mediate inflammatory cross-talk between hepatocytes and immune cells in alcoholic liver disease. J Leukoc Biol 2015;98:249–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Khanova E, Wu R, Wang W, Yan R, Chen Y, French SW, et al. Pyroptosis by caspase11/4-gasdermin-D pathway in alcoholic hepatitis in mice and patients. Hepatology 2018;67:1737–1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Shulga N, Pastorino JG. Hexokinase II binding to mitochondria is necessary for Kupffer cell activation and is potentiated by ethanol exposure. J Biol Chem 2014;289:26213–26225. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [123].Peng Y, French BA, Tillman B, Morgan TR, French SW. The inflammasome in alcoholic hepatitis: Its relationship with Mallory-Denk body formation. Exp Mol Pathol 2014;97:305–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Heo MJ, Kim TH, You JS, Blaya D, Sancho-Bru P, Kim SG. Alcohol dysregulates miR-148a in hepatocytes through FoxO1, facilitating pyroptosis via TXNIP overexpression. Gut 2019;68:708–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Nagy LE, Ding WX, Cresci G, Saikia P, Shah VH. Linking Pathogenic Mechanisms of Alcoholic Liver Disease With Clinical Phenotypes. Gastroenterology 2016;150:1756–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Zhou Z, Ye TJ, Bonavita G, Daniels M, Kainrad N, Jogasuria A, et al. Adipose-Specific Lipin-1 Overexpression Renders Hepatic Ferroptosis and Exacerbates Alcoholic Steatohepatitis in Mice. Hepatol Commun 2019;3:656–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Bloomer SA, Brown KE. Iron-Induced Liver Injury: A Critical Reappraisal. Int J Mol Sci 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017;66:449–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Cougnoux A, Cluzeau C, Mitra S, Li R, Williams I, Burkert K, et al. Necroptosis in Niemann-Pick disease, type C1: a potential therapeutic target. Cell Death Dis 2016;7:e2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Erickson RP, Bernard O. Studies on neuronal death in the mouse model of Niemann-Pick C disease. J Neurosci Res 2002;68:738–744. [DOI] [PubMed] [Google Scholar]
- [131].Cougnoux A, Clifford S, Salman A, Ng SL, Bertin J, Porter FD. Necroptosis inhibition as a therapy for Niemann-Pick disease, type C1: Inhibition of RIP kinases and combination therapy with 2-hydroxypropyl-beta-cyclodextrin. Mol Genet Metab 2018;125:345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Vitner EB, Salomon R, Farfel-Becker T, Meshcheriakova A, Ali M, Klein AD, et al. RIPK3 as a potential therapeutic target for Gaucher’s disease. Nat Med 2014;20:204–208. [DOI] [PubMed] [Google Scholar]
- [133].Malhi H, Guicciardi ME, Gores GJ. Hepatocyte death: a clear and present danger. Physiol Rev 2010;90:1165–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Krishna-Subramanian S, Singer S, Armaka M, Banales JM, Holzer K, Schirmacher P, et al. RIPK1 and death receptor signaling drive biliary damage and early liver tumorigenesis in mice with chronic hepatobiliary injury. Cell Death Differ 2019;26:2710–2726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Dara L, Liu ZX, Kaplowitz N. Questions and controversies: the role of necroptosis in liver disease. Cell Death Discov 2016;2:16089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Dara L The Receptor Interacting Protein Kinases in the Liver. Semin Liver Dis 2018;38:73–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev 2013;27:1640–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Newton K, Wickliffe KE, Dugger DL, Maltzman A, Roose-Girma M, Dohse M, et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 2019;574:428–431. [DOI] [PubMed] [Google Scholar]
- [139].Kayagaki N, Dixit VM. Rescue from a fiery death: A therapeutic endeavor. Science 2019;366:688–689. [DOI] [PubMed] [Google Scholar]
- [140].Orning P, Lien E, Fitzgerald KA. Gasdermins and their role in immunity and inflammation. J Exp Med 2019;216:2453–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Yang C, Sun P, Deng M, Loughran P, Li W, Yi Z, et al. Gasdermin D protects against noninfectious liver injury by regulating apoptosis and necroptosis. Cell Death Dis 2019;10:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Ibrahim SH, Hirsova P, Gores GJ. Non-alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018;67:963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Garcia-Tsao G, Bosch J, Kayali Z, Harrison SA, Abdelmalek MF, Lawitz E, et al. Randomized Placebo-Controlled Trial of Emricasan in Non-alcoholic Steatohepatitis (NASH) Cirrhosis with Severe Portal Hypertension. J Hepatol 2019. [DOI] [PubMed] [Google Scholar]
- [144].Harrison SA, Goodman Z, Jabbar A, Vemulapalli R, Younes ZH, Freilich B, et al. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J Hepatol 2019. [DOI] [PubMed] [Google Scholar]
- [145].Brumatti G, Ma C, Lalaoui N, Nguyen NY, Navarro M, Tanzer MC, et al. The caspase-8 inhibitor emricasan combines with the SMAC mimetic birinapant to induce necroptosis and treat acute myeloid leukemia. Sci Transl Med 2016;8:339ra369. [DOI] [PubMed] [Google Scholar]
- [146].Bansal N, Sciabola S, Bhisetti G. Understanding allosteric interactions in hMLKL protein that modulate necroptosis and its inhibition. Sci Rep 2019;9:16853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Molnar T, Mazlo A, Tslaf V, Szollosi AG, Emri G, Koncz G. Current translational potential and underlying molecular mechanisms of necroptosis. Cell Death Dis 2019;10:860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]