

Abstract
Background & Aims:
Non-alcoholic fatty liver disease (NAFLD), type 2 diabetes (T2D) and obesity are epidemiologically correlated with each other but the causal inter-relationships among them remains incompletely understood. We aim to explore the causal relationships among the three diseases.
Methods:
Using both UK BioBank and the largest-to-date publicly available GWAS data, we performed a two-sample bidirectional Mendelian Randomization (MR) analysis to test the causal interrelationships among NAFLD, T2D, and obesity. Transgenic mice expressing the human PNPLA3-I148M isoforms (TghPNPLA3-I148M) were used as an example to validate the causal effects and explore the underlying mechanisms.
Results:
Genetically-instrumented NAFLD significantly increased the risk for T2D and central obesity but not insulin resistance or generalized obesity, while genetically-driven T2D, BMI and WHRadjBMI causally increase the NAFLD risk. The animal study focusing on PNPLA3 well-corroborated these causal effects: compared to the TghPNPLA3-I148I controls, the TghPNPLA3-I148M mice developed glucose intolerance and increased visceral fat, but normal insulin sensitivity, reduced body weight, and decreased circulating total cholesterol. Mechanistically, the TghPNPLA3-I148M mice demonstrated decreased pancreas insulin but increased glucagon secretion, which was associated with increased pancreatic inflammation. There were significantly suppressed transcription of hepatic cholesterol biosynthesis pathway genes while activation of thermogenic pathway genes in subcutaneous and brown adipose tissues but not in visceral fat of the TghPNPLA3-I148M mice compared to the TghPNPLA3-I148I mice.
Conclusions:
Our study suggests that lifelong, genetically-driven NAFLD causally promotes T2D with a late onset Type-1 like diabetic subphenotype and central obesity; while genetically-driven T2D, obesity, and central obesity all causally increase the NAFLD risk. This causal relationship revealed new insights into the nature and nurture of these diseases and provided novel hypotheses for disease subphenotyping.
Keywords: Mendelian Randomization, Non-alcoholic fatty liver disease, Type 2 diabetes, Obesity, PNPLA3
Graphical Abstract

LAY SUMMARY
Genetically caused nonalcoholic fatty liver disease (NAFLD) was found to cause diabetes mellitus and central obesity but protects against overall obesity; while diabetes and obesity also cause NAFLD. The study further suggested that each of the three closely related diseases, NAFLD, T2D and obesity should be further stratified to different subtypes. This is important for accurate diagnosis, prevention and treatment for these diseases.
INTRODUCTION
Non-alcoholic Fatty Liver Disease (NAFLD) is characterized by the presence of excess hepatic fat accumulation (≥ 5%) without significant alcohol use, hepatitis virus infection, or other secondary causes of hepatic fat accumulation[1]. The spectrum of NAFLD ranges from simple non-alcoholic fatty liver (NAFL) to non-alcoholic steatohepatitis (NASH), which over time can lead to cirrhosis, hepatocellular carcinoma, and organ failure[2]. Compelling observational epidemiological studies have shown that NAFLD is highly correlated with metabolic disorders such as type 2 diabetes (T2D)[3–5] and obesity[6, 7].
Dissecting the causal relationship between the three diseases is crucial for both understanding the disease etiology and developing effective diagnostic, therapeutic and preventive strategies. However, observational associations are limited in elucidating the causality due to various confounding factors (e.g. lifestyle, socioeconomic status) or the reverse causation bias[8]. As a result, the three diseases are often treated as comorbidities for each other in various biomedical research settings. Without clearly knowing the causality among the three diseases, treatments or preventive interventions may often lead to conflicting research findings and inconsistent responses among patients.
Mendelian randomization (MR) analysis, which uses genetic variants as proxies for the risk factors of interest, has been widely applied in understanding the causal relationship between various risks factors and human diseases, e.g. estimation of the causal effect of plasma HDL cholesterol on myocardial infarction risk[9]. Since during the process of meiosis the alleles of the parents are randomly segregated to the offspring, the MR method is considered to be analogous to a randomized controlled trial (RCT) but less likely to be influenced by confounding factors and reverse causation[10]. Bidirectional MR is an extension of traditional MR in which the exposure-outcome causal relationship was explored from both sides. The bidirectional framework provides an efficient way to ascertain the direction of a causal relationship, which helps alleviate the potential bias from reverse causation[11].
Recent MR studies have partially explored the causal relationships among the three diseases. Dongiovanni et al. showed genetically instrumented hepatic steatosis was associated with insulin resistance and a small increase in T2D risk[12]. A study by De Silva et al. indicated that genetically raised circulating ALT and AST increased the risk of T2D[13]. Stender et al. found that the genetic predictors of BMI were associated with NAFLD[14]. However, a systematic bidirectional MR study leveraging the latest GWAS data is particularly needed to elucidate the causal relationships among the three diseases under a uniformed setting. In addition, experimental analysis e.g. animal models with a characterized natural history under controlled conditions would also help further establish the causality.
In this study, we first aimed to explore whether NAFLD casually increases risks for T2D, obesity and their related intermediate traits. We then investigated the reverse relationships, i.e. whether T2D and obesity causally affect NAFLD risk. Further, we used a newly constructed transgenic mice model expressing human PNPLA3-I148M isoforms, a known genetic NAFLD model as a typical example to validate the causal effects of NAFLD on T2D and obesity identified in humans, as well as to explore the potential mechanisms underlying these causal relationships.
MATERIALS AND METHODS
Ethics statement
The summary-level GWAS data used for mendelian randomization analyses are publicly available[15–24] (Fig. 1 and Supplementary Table 1). Therefore, no specific ethical approval is required. The study of the transgenic mice experiments has been reviewed and approved by the IACUC of both Wayne State University and the Indiana University School of Medicine. This research has been conducted using the UK Biobank Resource under application number 24460.
Fig. 1. Flowchart of the study design.
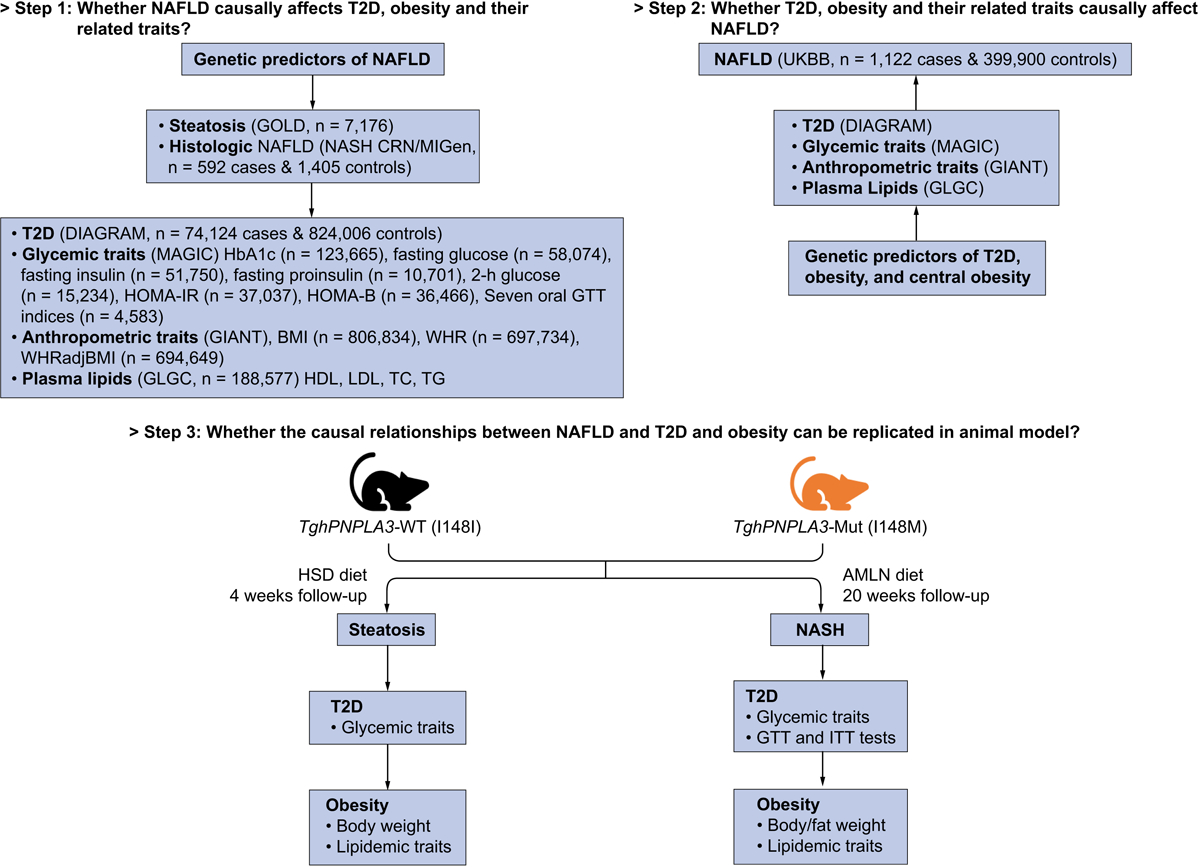
The summary-level associations were taken from the following genomics consortium: GOLD (Genetics of Obesity-related Liver Disease) for computerized tomography (CT) measured hepatic steatosis[15]; NASH Clinical Research Network (NASH CRN) and Myocardial Infarction Genetics Consortium (MIGen) for biopsy-proven NAFLD[15]; DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) for T2D[16]; Meta-Analyses of Glucose and Insulin-related traits (MAGIC) consortium for glycemic traits including HbA1c[17], fasting glucose[18], fasting insulin[18], fasting proinsulin[19], 2-h glucose[20], homeostatic model assessment of insulin resistance (HOMA-IR)[21] and β-cell function (HOMA-B)[21], and seven insulin secretion and action indices during oral glucose tolerance test (OGTT)[22]; The Genetic Investigation of ANthropometric Traits (GIANT) consortium for body mass index (BMI), waist-hip ratio (WHR), and WHR adjusted for BMI (WHRadjBMI)[23]; The Global Lipids Genetics Consortium (GLGC) for plasma high-density lipoprotein cholesterol (HDL), low-density lipoprotein cholesterol (LDL), total cholesterol (TC), and triglycerides (TG) levels[24].
MR analyses
GWAS summary data
We extracted the significant variants (p<5e-8) associated with hepatic steatosis and histologic NAFLD from the largest-to-date GWAS study[15]. For the purpose of estimating the reverse causal relationship, we generated the full GWAS summary statistics of NAFLD in UK Biobank (UKBB) samples consisting of 1,122 cases and 399,900 controls. The details of the GWAS of NAFLD in UKBB are described in the section below.
We downloaded full GWAS summary data of 22 glycemic and obesity traits from the largest published studies as of March 2019. Only association results from participants of European descent were used in the present study. The details on the phenotype information, sample size, and PubMed ID of the original study are summarized in Supplementary Table 1.
GWAS of NAFLD and T2D in UK Biobank
NAFLD was defined based on ICD-9 571.8 “Other chronic nonalcoholic liver disease”) and ICD-10 K76.0 [“Fatty (change of) liver, not elsewhere classified”] from inpatient hospital diagnosis within the UK Biobank dataset. Individuals with Hepatitis B or C or with other known liver diseases (e.g. liver transplant, hepatomegaly, jaundice, or abnormal liver function study results) were excluded from the analysis. It is noteworthy that, we recognized that this medical record-based phenotype may not exactly reflect the clinically/histologically characterized NAFLD. However, we found that the top genetic risk alleles identified in our GWAS well matched the patterns of previously identified loci for NAFLD/NASH, suggesting that the phenotypic definition in the UKBB reflect that of clinical NAFLD very well. The white British subset of UK Biobank was used for analysis in SAIGE[25] with sex, birth year, and 4 principle components as covariates.
Genetic predictors selection
We used the two strongest genetic predictors of NAFLD, Patatin-like phospholipase domain-containing protein 3 (PNPLA3) rs738409 and Transmembrane 6 superfamily member 2 (TM6SF2) rs58542926, as the proxy for hepatic steatosis and histologic NAFLD. We chose these two variants as the instruments based on the following rationale: 1) These two variants have been consistently shown to be associated with the whole spectrum of the NAFLD [15, 26–29]. Among numerous genetic association analyses, the association between these two variants and NAFLD and related traits are the most significant as compared to other genetic factors. Their associations with NAFLD are also much stronger as compared to those with other traits. 2) Mechanistic studies have clearly demonstrated that these two variants are causal to NAFLD [27, 30–32]. 3) Both genes are only highly expressed in limited tissue/organ, especially in the liver. According to the GTEX data, PNPLA3 is mainly expressed in the liver and skin/adipose tissue as oppose to other tissues, while TM6SF2 is almost exclusively expressed in the intestine and liver. 4) These two variants have been broadly accepted as genetic proxies to infer the causal relationship between NAFLD/liver fat/liver function enzyme and complex traits including liver damage and insulin resistance [12], ischaemic heart disease [33], and vitamin D intake and deficiency [34]. As TM6SF2 rs58542926 is not genotyped in most of the GWAS summary data used in this study, rs2228603 at the NCAN gene locus, which is in strong linkage disequilibrium with TM6SF2 rs58542926 (pairwise R2 = 0.76 based on the Phase 3 data of the 1000 Genomes Project in European individuals) and significantly associated with both hepatic fat content and NAFLD histology[35], was used as the proxy for TM6SF2 rs58542926. For the 22 glycemic and obesity traits, we selected the significant and independent genetic predictors in two steps: we first obtained all the variants that passed the genome-wide association significance level of p<5e-8. Then the independent genetic predictors were identified by clumping the top GWAS loci through PLINK 1.9 (https://www.cog-genomics.org/plink2) with the threshold of LD R2 < 0.1 in a 500-kb window. The linkage disequilibrium was estimated based on the European samples in phase 3 of the 1000 Genomes Project[36]. We omitted 10 traits including 2h glucose, HOMA-IR, HOMA-B, and seven OGTT traits due to lack of enough significant and independent genetic variants (number of valid variants < 3). Therefore, 12 traits were analyzed for the causal effects on NAFLD. The F statistics of all the genetic predictors in the present study were larger than the empirical threshold of 10[37] (Table 1 and Table 2).
Table 1.
MR estimate with hepatic steatosis or histologic NAFLD as the exposure.
| Hepatic steatosis (per SD) | Histologic NAFLD (per logOR) | |||
|---|---|---|---|---|
| T2D and glycemic traits | Effect (95% CI) | p | Effect (95% CI) | p |
| T2D (OR) | 1.30 (1.21, 1.39) | 8.30E-14 | 1.06 (1.03, 1.09) | 2.80E-04 |
| HbAlc (%) | −0.0072 (−0.025, 0.01) | 4.20E-01 | −0.0025 (−0.0074, 0.0024) | 3.10E-01 |
| Fasting glucose (mmol/L) | 0.026 (8.5E-05, 0.051) | 4.90E-02 | 0.0032 (−0.0031, 0.0096) | 3.20E-01 |
| Fasting insulin (pmol/L) | 0.025 (0.0035, 0.046) | 2.20E-02 | 0.0064 (0.00034, 0.012) | 3.80E-02 |
| Fasting proinsulin (pmol/L) | 0.032 (−0.026, 0.089) | 2.80E-01 | 0.0051 (−0.0091, 0.019) | 4.80E-01 |
| 2h glucose (mmol/L) | −0.066 (−0.21, 0.079) | 3.70E-01 | −0.015 (−0.051, 0.021) | 4.10E-01 |
| HOMA-IR ((mU/L)*(mmol/L)) | 0.03 (−0.0016, 0.061) | 6.30E-02 | 0.0066 (−0.0016, 0.015) | 1.10E-01 |
| HOMA-B ((mU/L)/(mmol/L)) | 0.007 (−0.019, 0.033) | 5.90E-01 | 0.0011 (−0.0052, 0.0074) | 7.30E-01 |
| AUCins (mU*min/L) | −0.012 (−0.18, 0.16) | 8.90E-01 | −0.0043 (−0.046, 0.038) | 8.40E-01 |
| AUCins/AUCgluc (mU/mmol) | −0.01 (−0.18, 0.16) | 9.10E-01 | −0.0037 (−0.046, 0.038) | 8.60E-01 |
| Incre30 (mU/L) | −0.0021 (−0.17, 0.16) | 9.80E-01 | −0.00033 (−0.041, 0.04) | 9.90E-01 |
| Ins30adjBMI | 0.017 (−0.15, 0.19) | 8.40E-01 | 0.00083 (−0.041, 0.043) | 9.70E-01 |
| ISI (mg/dL) | 0.011 (−0.18, 0.2) | 9.10E-01 | 0.017 (−0.032, 0.065) | 5.00E-01 |
| CIRadjBMI | −0.0055 (−0.18, 0.17) | 9.50E-01 | −0.0034 (−0.045, 0.039) | 8.80E-01 |
| DI | 0.078 (−0.082, 0.24) | 3.40E-01 | 0.022 (−0.018, 0.063) | 2.80E-01 |
| Obesity and lipid traits | ||||
| BMI (SD) | −0.027 (−0.041, −0.013) | 1.30E-04 | −0.0071 (−0.012, − 0.0023) | 3.40E-03 |
| WHR (SD) | 0.021 (0.0062, 0.036) | 5.40E-03 | 0.0029 (−0.00091, 0.0067) | 1.30E-01 |
| WHRadjBMI (SD) | 0.039 (0.023, 0.054) | 8.20E-07 | 0.0072 (0.0022, 0.012) | 4.50E-03 |
| HDL (SD) | −0.028 (−0.062, 0.0071) | 1.20E-01 | −0.0096 (−0.021, 0.0013) | 8.30E-02 |
| LDL (SD) | −0.098 (−0.14, −0.053) | 2.30E-05 | −0.014 (−0.026, −0.0012) | 3.10E-02 |
| TC (SD) | −0.12 (−0.17, −0.074) | 3.30E-07 | −0.019 (−0.033, −0.0054) | 6.80E-03 |
| TG (SD) | −0.02 (−0.06, 0.021) | 3.40E-01 | 0.0038 (−0.0057, 0.013) | 4.40E-01 |
Effect was estimated by a combined genetic vector of PNPLA3 and TM6SF2(NCAN) variants through inverse variance weighted (IVW) method. The F statistics of the genetic predictors of hepatic steatosis and histologic NAFLD are 110 and 10, respectively. P values less than Bonferroni–adjusted level of significance (p<0.05/20=2.5e–3) were considered as significant.
T2D: type 2 diabetes; HOMA–IR: The homeostatic model assessment (HOMA) insulin resistance; HOMA–B: HOMA beta cell function; AUCins: area under the curve (AUC) of insulin levels during oral glucose tolerance test (OGTT); AUCins/AUCgluc: ratio of AUC insulin and AUC glucose; Incre30: incremental insulin at 30 min; Ins30adjBMI: Insulin response to glucose during the first 30 min adjusted for BMI; ISI: insulin sensitivity index; CIRadjBMI: Corrected Insulin Response adjusted for ISI; DI: disposition index; BMI: body mass index; WHR: waist–hip ratio; WHRadjBMI: WHR adjusted for BMI; HDL: high–density lipoprotein cholesterol; LDL: low–density lipoprotein cholesterol; TC: total cholesterol; TG: triglycerides; OR: odds ratio; CI: confidence interval; SD: standard deviation.
Table 2.
MR estimate with NAFLD as the outcome.
| IVW | Weighted median | MR-Egger | Pleiotropy test | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| T2D and glycemic traits# | #SNPs | F | OR (95% CI) | p | OR (95% CI) | p | OR (95% CI) | p | MR-PRESSO global test p | modified Q’ p |
| T2D (logOR)* | 315 | 69 | 1.1 (1, 12) | 1.67E-03 | 1.1 (0.93, 1.2) | 3.54E-01 | 0.99 (0.84, 1.2) | 8.59E-01 | 3.11E-01 | 3.26E-01 |
| HbA1c (%) | 67 | 69 | 0.33 (0.17, 0.66) | 1.76E-03 | 0.33 (0.11, 1.11) | 7.44E-02 | 0.17 (0.04, 0.68) | 1.28E-02 | 3.85E-01 | 3.69E-01 |
| Fasting glucose (mmol/L) | 33 | 68 | 0.42 (0.25, 0.73) | 1.60E-03 | 0.33 (0.15, 0.80) | 1.34E-02 | 0.44 (0.12, 1.52) | 1.93E-01 | 8.78E-02 | 8.29E-02 |
| Fasting insulin (pmol/L)* | 9 | 36 | 7.7 (1.3, 46) | 2.40E-02 | 4.6 (0.42, 51) | 2.11E-01 | 4.2 (0.00035, 51000) | 7.64E-01 | 3.18E-01 | 2.82E-01 |
| Fasting proinsulin (pmol/L) | 15 | 72 | 1.13 (0.79, 1.62) | 4.94E-01 | 0.99 (0.58, 1.67) | 9.55E-01 | 1.57 (0.49, 4.95) | 4.51E-01 | 4.60E-02 | 4.74E-02 |
| Obesity and lipid traits | ||||||||||
| BMI (SD)* | 1839 | 60 | 2.3 (2, 2.7) | 1.41E-25 | 2.1 (16, 2.8) | 1.02E-07 | 1.7 (1.1, 2.7) | 1.58E-02 | 2.50E-03 | 3.89E-03 |
| WHR (SD) | 951 | 53 | 2.10 (1.72, 2.59) | 2.85E-12 | 2.34 (1.67, 3.32) | 6.91E-07 | 1.62 (0.94, 2.72) | 8.14E-02 | 3.62E-01 | 3.64E-01 |
| WHRadjB MI (SD) | 1156 | 63 | 1.54 (1.30, 1.82) | 1.11E-06 | 1.60 (1.20, 2.14) | 1.53E-03 | 1.57 (1.06, 2.34) | 2.37E-02 | 4.55E-01 | 4.54E-01 |
| HDL (SD)* | 226 | 123 | 0.88 (0.76, 1) | 8.78E-02 | 0.83 (0.64, 1.1) | 1.57E-01 | 0.93 (0.69, 1.3) | 6.62E-01 | 2.00E-03 | 1.26E-03 |
| LDL (SD)* | 192 | 148 | 0.96 (0.84, 1.1) | 5.47E-01 | 1 (0.79, 1.3) | 9.69E-01 | 0.84 (0.64, 1.1) | 2.11E-01 | <5.00E-04 | 7.29E-06 |
| TC (SD)* | 239 | 110 | 0.94 (0.82, 1.1) | 4.18E-01 | 0.95 (0.71, 1.3) | 7.13E-01 | 0.88 (0.66, 1.2) | 3.85E-01 | 1.00E-03 | 6.33E-04 |
| TG (SD)* | 149 | 119 | 1.6 (13, 1.9) | 5.10E-07 | 1.7 (12, 2.4) | 1.45E-03 | 1.7 (11, 2.4) | 8.92E-03 | 7.00E-03 | 7.83E-03 |
We considered as significant if the directions of the estimates by IVW, median, Egger were consistent, IVW method passed the Bonferroni corrected threshold (0.05/12=4.2e–3), and no significant pleiotropy tested by MR–PRESSO global test and modified Q’ statistics (both p>0.05).
Outlier variants were identified and removed by MR–PRESSO at p<0.05. Initial estimates without removing outliers were shown in Supplementary Table 5.
Ten traits including 2h glucose, HOMA–IR, HOMA–B, and seven OGTT traits were omitted due to lack of enough significant and independent genetic variants (number of valid variants < 3).
IVW: inverse variance weighted; F: F statistics for the strength of correlation between instrument and exposure; MR–PRESSO: MR pleiotropy residual sum and outlier; Q’: Q’ statistics with modified second order weights; T2D: type 2 diabetes; BMI: body mass index; WHR: waist–hip ratio; WHRadjBMI: WHR adjusted for BMI; HDL: high–density lipoprotein cholesterol; LDL: low–density lipoprotein cholesterol; TC: total cholesterol; TG: triglycerides; OR: odds ratio; CI: confidence interval; SD: standard deviation.
MR estimation
For MR estimation with NAFLD as the exposure, we used the inverse variance weighted (IVW) method to estimate the combined causal effect of PNPLA3 and TM6SF2 (NCAN) variants by assuming a fixed-effect model[38]. As a sensitivity analysis, Wald’s method was used to estimate the causal effect with each of the genetic variant respectively. We considered a significant causal relationship if the directions of the estimates by PNPLA3, TM6SF2 (NCAN), and the two variants combined were consistent, and the combined estimate passed the Bonferroni-adjusted significance of p<0.05.
For the MR estimation with NAFLD as the outcome, besides the IVW method, we estimated the causal effects using additional methods including weighted median estimator[39] and MR-Egger[40] as a sensitivity analysis. To assess the heterogeneity and identify horizontal pleiotropic outliers, we used the Q’ statistics[41] with modified second order weights and MR-PRESSO global test[42]. If the horizontal pleiotropy is significant, MR-PRESSO was used to identify outliers at p<0.05. We then removed the outliers and retested if the causal relationship and pleiotropic effects were significant. We considered as significant if the directions of the estimates by three methods (IVW, weighted median, and MR-Egger) were consistent, IVW method passed the Bonferroni-adjusted significance of p<0.05, and no significant pleiotropy tested by MR-PRESSO global test and modified Q’ statistics. MR analyses were performed with “MendelianRandomization”[43], “MRPRESSO”[42], and “RadialMR”[44] packages in R version 3.5.0 (http://www.r-project.org/).
Sensitivity analysis
To test the robustness of the MR estimation, we incorporated more known but less significant GWAS-identified NAFLD risk variants as the genetic proxy. The sensitivity analysis was performed using two independent NAFLD GWAS summary data. Firstly, we re-estimated the causal effects of the hepatic steatosis and histologic NAFLD on T2D, obesity, and their related traits using all the 5 NAFLD risk variants (PNPLA3 rs738409, NCAN rs2228603, GCKR rs780094, PPP1R3B rs4240624, and LYPLAL1 rs12137855) identified in the GOLD cohort. Next, to incorporate other two known NAFLD risk variants (MBOAT-TMC4 rs641738 and HSD17B13 rs6834314), we extracted the summary statistics of all aforementioned 7 known variants from the UKBB NAFLD GWAS summary data. The MR estimation was performed using different combinations of the variants through the inverse variance weighted method.
Animal experiments
We constructed transgenic mouse model expressing human PNPLA3-I148I and PNPLA3-I148M isoforms, respectively. The mice were either fed with a high sucrose diet (HSD)[31] for 4 weeks to induce hepatosteatosis, and a high fat, high fructose and high cholesterol AMLN) diet[45–48] for 20 weeks to induce NASH. T2D and obesity phenotypes where characterized during the dietary feeding. Various experimental analyses were performed to explore the mechanism underlying the causal relationships between NAFLD, T2D and obesity, including histologic analyses of the mouse liver tissue, immunofluorescent staining of inflammation makers in both liver and pancreas tissue as well as the insulin and glucagon signaling in pancreas tissue, Western blotting analysis of the Akt signaling in the liver and skeletal muscle tissues, as well as real-time PCR to examine the gene expression levels of hepatic cholesterol biosynthesis and thermogenesis pathway genes in brown as well as subcutaneous and epidydimal white adipose tissues. Details on the transgenic mouse model construction and technical methods and materials for various phenotypic characterization and molecular assays were described in the Supplementary materials.
Statistical analyses
Power assessment of the animal study was described in the supplementary methods. Statistical significance of the experimental data on animal study were performed using ANOVA, Students’ t-test or Pearson correlation wherever appropriate. Tukey corrected p value<0.05 was considered significant. All statistical analyses for animal experiments were performed using GraphPad Prism Version 6.00 (GraphPad Software, CA, USA).
RESULTS
Study overview
The design of this study consists of three steps (Fig. 1). We first aimed to explore whether NAFLD (both CT-measured hepatic steatosis and biopsy-proven histologic NAFLD) casually affects T2D, obesity and their related intermediate traits (Step 1). To this end, we used the summary GWAS data for CT scan-measured steatosis (GOLD) as well as the histologic NAFLD (case and control) (NASH CRN/MIGen) presented in Speliotes et al.[15], which is thus far the largest GWAS comprehensively covering multiple phenotypes of NAFLD. We also use the summary GWAS meta-analysis data for T2D (DIAGRAM)[16], obesity (GIANT)[23], glycemic (MAGIC)[17–22] and lipid (GLGC)[24] traits as outcomes (Step 1), which are the largest-to-date GWAS data on these phenotypes as well. The causal role of NAFLD on T2D and obesity was conducted first by using two well-established NAFLD-associated polymorphisms in PNPLA3 and TM6SF2/NCAN loci as proxies for hepatic steatosis and histologic NAFLD. We then performed a GWAS on the NAFLD phenotype in the UK biobank dataset. Based on this new summary data, we further performed a sensitivity analysis by including other 5 GWAS-identified variants as a broad proxy for NAFLD to further examine the causal relationship between NAFLD and T2D or obesity. Second, using the aforementioned summary level data of UK Biobank, DIAGRAM and GIANT, we then investigated the reverse relationships, i.e. whether T2D or obesity causally affect NAFLD risk in the UK Biobank samples (Step 2). Finally, we constructed a transgenic mouse model expressing human PNPLA3-I148I or PNPLA3-I148M isoform. We aim to use this established typical genetic model for NAFLD as an example to further validate the causal effects of hepatic steatosis or NAFLD on T2D and obesity, as well as to explore the potential mechanisms underlying these causal effects (Step 3). To test the effect of steatosis and histologic NAFLD on the susceptibility to T2D and obesity, we used a high sucrose diet (HSD) known to induce steatosis in PNPLA3-I148M mice[31], as well as a high fat, high fructose and high cholesterol diet (known as the Amylin or AMLN diet) known to induce NASH in mice[45–48]. A schematic representation of the three assumptions for an MR analysis was shown in Supplementary Fig. 1A, and the MR methods and heterogeneity tests used in the study were listed in Supplementary Fig. 1B.
The causal effect of NAFLD on T2D risk and glycemic traits
We first used two well-established NAFLD-associated variants in PNPLA3 (rs738409) and TM6SF2/NCAN (rs2228603) gene loci as the genetic instruments to test the causal effect of NAFLD on T2D and glycemic traits in the latest publicly available GWAS data. As listed in Table 1, we observed a significant association between genetically instrumented hepatic steatosis and T2D risk, in which a one-standard deviation (SD) increase in CT measured hepatic steatosis caused a 30% increased risk of T2D (OR: 1.3, 95% CI: [1.2, 1.4], p=8.3e-14). As for glycemic traits, we detected nominal weak associations of steatosis with increased fasting glucose (β: 0.026 mmol/L, 95% CI: [8.5e-5, 0.051], p=0.049), fasting insulin (β: 0.025 pmol/L, 95% CI: [0.0035, 0.046], p=0.022). These results were not significant after adjusted with Bonferroni correction (p<2.5e-3, correction for 20 traits except HOMA-IR and HOMA-B due to the collinearity with glucose and insulin levels). There was no causal relationship between genetically instrumented hepatic steatosis and insulin resistance (HOMA-IR) (β: 0.03 (mU/L)*(mmol/L), 95% CI: [−0.0016, 0.061], p=0.063) levels. Other tested glycemic traits including HbA1c, fasting proinsulin, 2-h glucose, HOMA-B, and seven insulin secretion and action indices during OGTT did not show any significance as well.
We then tested if genetically increased risk for the histologic NAFLD also has causal effect on T2D susceptibility and glycemic traits. Consistent with the result of hepatic steatosis, there was evidence of a significant effect of histologically characterized NAFLD severity on an increased risk of T2D (OR: 1.06, 95% CI: [1.03, 1.09], p=2.8e-4). Again, no significant causal relationship was found between genetically driven NAFLD and glycemic traits especially when after adjusted for multiple testing (Table 1). Taken together, the genetically instrumented NAFLD causally increased risk for T2D, but not insulin resistance or other glycemic traits.
The causal impact of NAFLD on obesity
While obesity is a well-known risk factor for NAFLD, the reverse relationship, i.e. the causal effect of NAFLD on obesity, has not been explored before. Using the same genetic predictors of steatosis and histologic NAFLD, we implemented MR and observed a significant causal association of a one-SD increase in hepatic fat with a 0.027-SD decrease in BMI (β:−0.027, 95%CI: [−0.043, −0.013], p=1.3e-4), but a 0.039-SD increase in WHRadjBMI (WHR adjusted for BMI), an established marker for abdominal or central obesity (β:0.039, 95%CI: [0.023, 0.054], p=8.2e-7). Similar relationships were found between genetically raised histologic NAFLD with decreased BMI (β:−0.0071, 95%CI: [−0.012, −0.0023], p=3.4e-3) but increased WHRadjBMI (β:0.0072, 95%CI: [0.0022, 0.012], p=4.5e-3). Taken together, our analyses suggested a consistent negative causal relationship between NAFLD and generalized obesity (proxied by BMI), but a positive association with central or visceral obesity (proxied by WHRadjBMI).
We next investigated the causal effect of NAFLD on blood lipid levels including HDL, LDL, total cholesterol (TC), and triglycerides (TG). Our analyses demonstrated a significant negative correlation between genetically raised hepatic fat and TC levels (β:−0.12 SD, 95%CI: [−0.17, −0.074, p=3.3e-7) as well as the LDL levels (β: –0.098 SD, 95% CI [−0.14, −0.053], p=2.3e-5). These negative relationships also exist for histologic NAFLD (β:−0.019 SD, 95%CI: [−0.033, −0.0054], p=6.8e-3; and β: −0.014 SD, 95%CI: (–0.026, −0.0012), p=3.1e-02 for TC and LDL, respectively). No significant causal relationship was found with other lipids (Table 1). Taken together, our analysis demonstrated a causal role of genetically driven NAFLD on central fat accumulation, reduced circulating cholesterol, but not generalized obesity.
Sensitivity analysis using additional NAFLD genetic risk variants
It is known that horizontal pleiotropic effect of the genetic instruments may invalid the MR analysis. This risk can be reduced by using multiple genetic variants as instruments [49]. For this reason, we first performed a post-hoc analysis by separately examining the causal role of PNPLA3 and TM6SF2/NCAN polymorphisms separately, which generated similar results on the majority of phenotypes (Supplementary Table 4). Moreover, we tested if adding more NAFLD risk variants would change the results of the causal inference. As shown in Supplementary Tables 7–8, the MR estimates using all the 5 GWAS-identified NAFLD risk variants (PNPLA3 rs738409, NCAN rs2228603, GCKR rs780094, PPP1R3B rs4240624, and LYPLAL1 rs12137855) identified in the GOLD cohort were largely consistent with that using PNPLA3 and TM6SF2/NCAN alone. Similarly, we re-estimated the causal relationships by incorporating all the 7 known NAFLD risk variants (the aforementioned 5 variants plus MBOAT-TMC4 rs641738 and HSD17B13 rs6834314) using the UKBB samples. The results were still consistent with the original estimates using PNPLA3 and TM6SF2/NCAN only, in terms of the causal association between NAFLD and T2D, insulin resistance, BMI and WHRadjBMI (Supplementary Table 9). This result further confirmed that the causal relationship between NAFLD and T2D or obesity is highly likely a general mechanism rather than a biased association driven by a specific genetic variant.
Reverse MR testing the causal effects of T2D, obesity, and their related traits on NAFLD
To understand the causal relationships among the three diseases, we implemented MR analyses to test the existence of the reverse or bidirectional causal relationships between T2D or obesity and NAFLD. We used the data from our newly performed GWAS on NAFLD as an outcome, while the DIAGRAM and GIANT summary data for T2D and obesity as eposures, respectively. We found that genetic predictors of T2D exert positive effects on NAFLD (OR: 1.1, 95% CI: [1.0, 1.2], p=1.67e-3) without evidence of significant heterogeneity (PMR-PRESSOGlobal=0.31, Pmodified Q=0.33) after removing outlier variants identified by MR-PRESSO (Table 2). Initial MR estimates without removing outliers were shown in Supplementary Table 5. Consistent with the previous MR estimate[14], we found BMI causally increased the NAFLD risk (OR: 2.3, 95% CI: [2.0, 2.7], p=1.4e-25), but with remaining heterogeneity (PMR-PRESSOGlobal<2.5e-3, Pmodified Q=3.9e-3) after removing outliers. BMI adjusted WHR significantly aggravated NAFLD risk (OR: 1.5, 95% CI: [1.3, 1.8], p=1.1e-6) without significant heterogeneity (PMR-PRESSOGlobal=0.46, Pmodified Q=0.45). The circulating TG was also causally increase the NAFLD risk (OR: 1.6, 95% CI: [1.3, 1.9], p=5.1e-7), with a mild remaining heterogeneity (PMR-PRESSOGlobal=7.0e-3, Pmodified Q=7.8e-3). In summary, T2D, increased BMI and central obesity as well as circulating TG are likely causally increase the risk for NAFLD.
Transgenic mice study on the relationship between NAFLD and susceptibility to T2D and obesity.
To further examine the causal effect of NAFLD on T2D and obesity, we set out to induce hepatosteatosis and NASH using transgenic animal models expressing human PNPLA3 isoforms, and during which, to observe the development of T2D and obesity phenotypes. To do so, we constructed mice models transduced with the bacterial artificial chromosome (BAC)-containing the PNPLA3–148I isoform (TghPNPLA3-I148I) or that was engineered to the PNPLA3–148M (TghPNPLA3-I148M) isoform. The mice were then fed with a previously established high sucrose diet (HSD) for 4 weeks to induce hepatosteatosis[31]. To examine the effect of more severed NAFLD and NASH phenotypes on T2D or obesity phenotypes, we also fed the mice with the “Western diet” characterized with high fat, high fructose and high cholesterol (known as the AMLN diet) for 20 weeks, which has been an established NASH-inductive diet as demonstrated before[45–48].
After 4 weeks of HSD diet, the TghPNPLA3-I148M mice developed severe hepatosteatosis as compared to their TghPNPLA3-I148I littermates or the non-transgenic controls, characterized with significantly increased lipid droplets formation in the liver (Supplementary Fig. 2A) as well as hepatic triglycerides (TG) accumulation (Supplementary Fig. 2B). Meanwhile, as compared to the TghPNPLA3-I148I controls, the TghPNPLA3-I148M mice also demonstrated a trend of increased circulating glucose, but the insulin level remained unchanged (Supplementary Fig. 3A, B). After 4 weeks of HSD diet feeding, the TghPNPLA3-I148M mice demonstrated no significant change in total body weight (Supplementary Fig. 3C), but a marginal trend to a reduced total circulating cholesterol (Supplementary Fig. 3D, p=0.038), but not the circulating TG levels (Supplementary Fig. 3E), as compared to their TghPNPLA3-I148I littermates.
To examine the effect of NASH progression on the susceptibility to T2D and obesity, we fed the mice with the NASH-inducing AMLN diet for 20 weeks. The TghPNPLA3-I148M mice developed significantly more severe NAFLD/NASH phenotypes as compared to the TghPNPLA3-I148I littermates, as characterized by increased inflammation and fibrosis (Supplementary Figures 4–7), confirming that PNPLA3 I148M possesses a strong genetic predisposition to NAFLD and NASH. H&E staining results indicated that all the three groups have developed hepatosteatosis after 20 weeks of AMLN diet (Supplementary Fig. 7).
We next evaluated the effect of PNPLA3 I148M on glucose homeostasis over a 20-week follow-up in the cohort fed with AMLN diet. As shown in Fig. 2A, we observed a significant genotype-time interaction on the fasting glucose level (two-way repeated measure ANOVA, p<0.0001), suggesting that the effect of PNPLA3 148M on glucose levels depended on disease progression. At week 16 and 18, the TghPNPLA3-I148M mice displayed significantly higher fasting glucose levels than their TghPNPLA3-I148I littermates (p=0.0048 and 0.0082, respectively). There is also a significant interaction between genotype and time on fasting insulin levels between the TghPNPLA3-I148I and TghPNPLA3-I148M groups (p=0.0009). However, there is no significant difference between the TghPNPLA3-I148M and non-transgenic wildtype mice. Beginning at week 12, the insulin levels between the latter two groups remain unchanged (Fig. 2B). Results of the glucose tolerance test (GTT) showed that TghPNPLA3-I148M mice experienced a reduced clearance of blood glucose as compared to the TghPNPLA3-I148I controls (p=0.012) (Fig. 2C). However, we did not observe a significant genotypic difference in response to insulin challenge in the insulin tolerance test (ITT) between (Fig. 2D).
Fig. 2. Phenotypic changes of the mice fed with an AMLN diet.
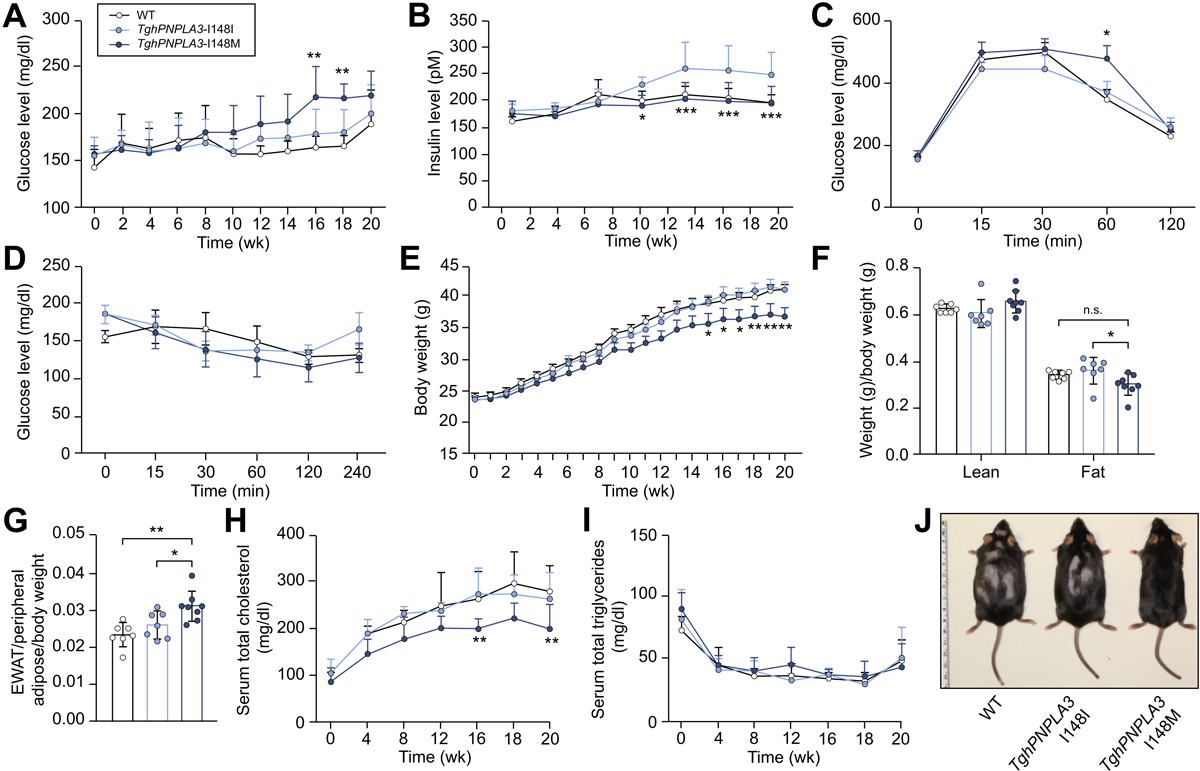
(A) Change in glucose levels over time of TghPNPLA3-I148I, TghPNPLA3-I148M, and non-transgenic wide type mice fed with an AMLN diet for 20 weeks. Error bar represents standard deviation (SD). The significance level of the comparison between TghPNPLA3-I148I and TghPNPLA3-I148M was indicated as follows: **: p<0.01, ANOVA with Tukey’s multiple comparison test; (B) Change in insulin levels for 20 weeks. Error bar represents SD. The significance level of the comparison between TghPNPLA3-I148I and TghPNPLA3-I148M was indicated as follows: *: p<0.05, ***: p<0.001, ANOVA with Tukey’s multiple comparison test; (C) glucose tolerance test (GTT), Error bar represents SD. The significance level of the comparison between TghPNPLA3-I148I and TghPNPLA3-I148M was indicated as follows: *: p<0.05, ANOVA with Tukey’s multiple comparison test; and (D) insulin tolerance test (ITT) were performed at the 16th week of HFFC diet feeding. Error bar represents SD, ANOVA with Tukey’s multiple comparison test, all p>0.05; (E) Change in body weight of TghPNPLA3-I148I, TghPNPLA3-I148M, and non-transgenic wide type mice fed with the AMLN diet for 20 weeks. Error bar represents SD. The significance level of the comparison between TghPNPLA3-I148I and TghPNPLA3-I148M was indicated as follows: *: p<0.05, **: p<0.01, ANOVA with Tukey’s multiple comparison test; (F) Body composition analysis by magnetic resonance imaging (MRI). Fat tissue weight and non-fat lean mass were normalized by the body weight. Error bar represents SD. The significance level was indicated as follows: *: p<0.05, ns: not significant, ANOVA with Tukey’s multiple comparison test; (G) Epididymal white adipose tissue (EWAT) accumulation at the 20th week of AMLN diet feeding. EWAT accumulation was calculated as weight of EWAT normalized by the total peripheral adipose tissue weight and then further normalized by the body weight. Error bar represents SD. The significance level was indicated as follows: *: p<0.05, **: p<0.01, ANOVA with Tukey’s multiple comparison test; (H) Change in serum total cholesterol levels over 20 weeks. Error bar represents SD. The significance level of the comparison between TghPNPLA3-I148I and TghPNPLA3-I148M was indicated as follows: **: p<0.01, ANOVA with Tukey’s multiple comparison test; (I) Change in serum total triglycerides levels over 20 weeks. Error bar represents SD. ANOVA with Tukey’s multiple comparison test, all p>0.05; (J) The representative image of TghPNPLA3-I148I, TghPNPLA3-I148M, and non-transgenic wide type mouse after 20 weeks of AMLN diet feeding. Sample size: TghPNPLA3-I148I (n=7), TghPNPLA3-I148M (n=8), and non-transgenic wide type (n=7).
The body weight change of the mice on AMLN diet over time was shown in Fig. 2E. The TghPNPLA3-I148M mice were significantly lighter than the TghPNPLA3-I148I controls beginning at week 15 (all p<0.05). Magnetic resonance imaging (MRI) examination at week 20 showed that less total fat was accumulated in the TghPNPLA3-I148M mice than the TghPNPLA3-I148I controls (p=0.012), while the lean mass of non-fat tissues was not significantly different (Fig. 2F). Further investigation on the composition of the isolated fat showed that there was a significantly more epidydimal white adipose tissue (eWAT) accumulation relative to the total peripheral adipose tissue in the TghPNPLA3-I148M mice than their TghPNPLA3-I148I littermates or non-transgenic controls (p=0.034 and 0.0015, respectively) (Fig. 2G). Examining the plasma lipid profile showed a significant decrease in total cholesterol levels beginning at week 16 in TghPNPLA3-I148M mice as compared to the TghPNPLA3-I148I controls (Fig. 2H). No significant difference in circulating TG levels between the three groups was observed (Fig. 2I). The representative image of the mice after 20 weeks of AMLN diet feeding was shown in Fig. 2J.
Assessment of insulin and glucagon signals and inflammatory status of the mouse pancreas
The increased glucose level, reduced glucose clearance and the lower level of insulin in the TghPNPLA3-I148M mice as compared to the TghPNPLA3-I148I controls suggested a potentially lower pancreatic secretion of insulin in the TghPNPLA3-I148M mice. To verify this. we next stained the insulin and glucagon signals in the pancreas tissue of all three groups of mice. Immunofluorescence (IF) assay revealed a significantly lower staining signal of insulin but higher glucagon in the pancreas of TghPNPLA3-I148M mice than in the TghPNPLA3-I148I mice (p=0.0149 and p=0.0197, respectively) (Fig. 3A).
Fig. 3. Underlying mechanism of PNPLA3 I148M in regulating pancreas function and inflammation, as well as adipose thermogenic and hepatic cholesterol metabolism pathways.
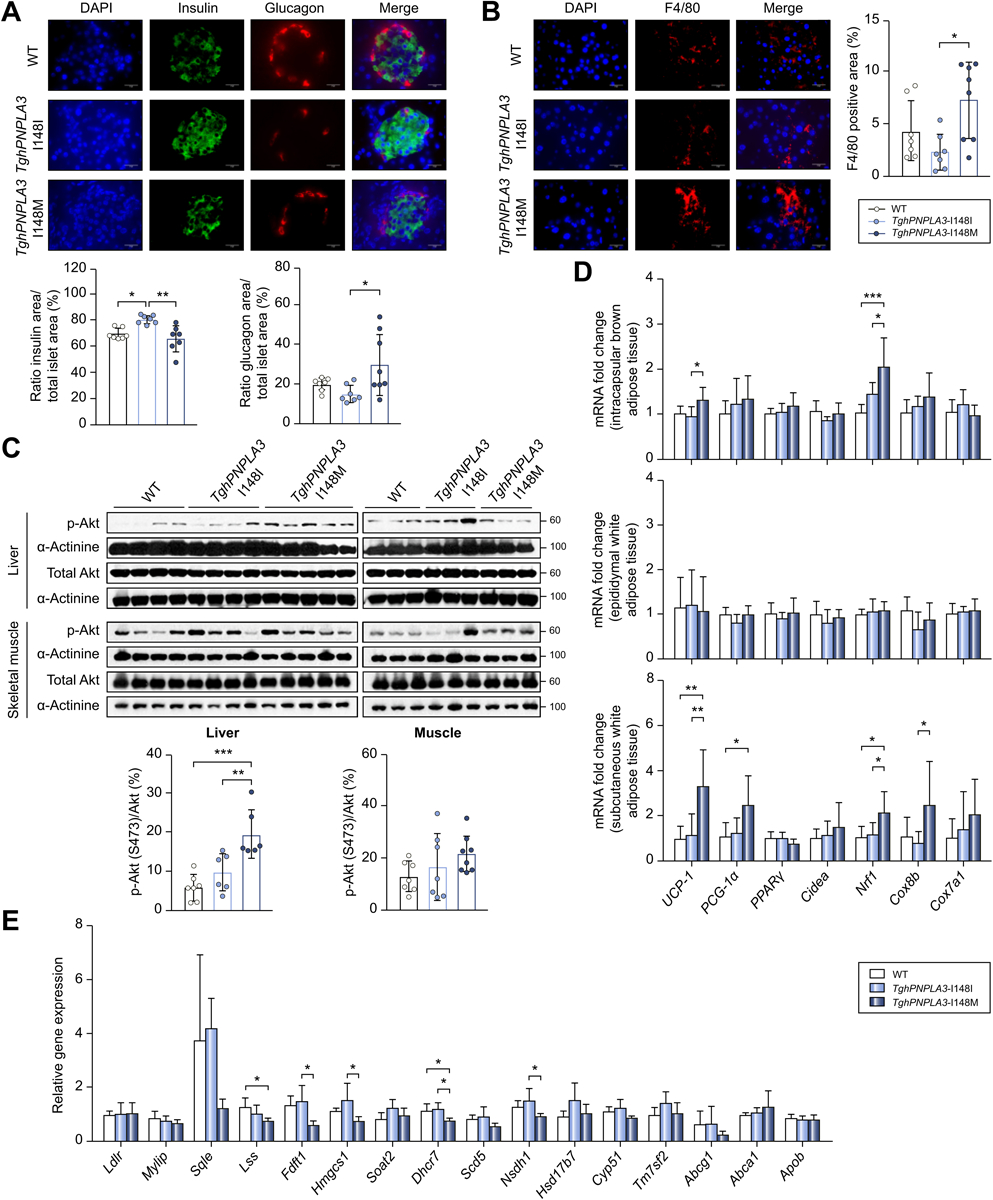
(A) Immunofluorescence double staining to identify insulin and glucagon secretion with antibodies against insulin (green) and glucagon (red) in non-transgenic wide type, TghPNPLA3-I148I or TghPNPLA3-I148M mouse pancreas. Nuclei were labeled with DAPI (blue). Scale bars: 20 m, 630X magnification. The quantifications of area of stained insulin and glucagon signals were shown below. Error bar represents standard deviation (SD). The significance level was indicated as follows: *: p<0.05, **: p<0.01, ANOVA with Tukey’s multiple comparison test; (B) Immunofluorescence staining for macrophage marker F4/80 (red) in non-transgenic wide type, TghPNPLA3-I148I and TghPNPLA3-I148M mouse pancreas. Nuclei were labeled with DAPI (blue). Scale bars: 20 μm, 630X magnification. The quantification of F4/80-positive area was shown in the right. The positive area was measured in randomly selected fields (three fields per section). Error bar represents SD. The significance level was indicated as follows: *: p<0.05, ANOVA with Tukey’s multiple comparison test; (C) Western blot analysis of the level of total and phospho-Akt (Ser473) proteins in liver and skeletal muscle tissue. α-Actinin was used as the loading control. The densitometry ratio of the expression of p-Akt and total Akt was demonstrated below. Error bar represents SD. The significance level was indicated as follows: **: p<0.01, ***: p<0.001, ANOVA with Tukey’s multiple comparison test; (D) Fold change in expression of thermogenic genes in interscapular brown adipose tissue, epididymal white adipose tissue and subcutaneous white adipose tissue. Error bar represents SD. The significance level was indicated as follows: *: p<0.05, **: p<0.01, ***: p<0.001, ANOVA with Tukey’s multiple comparison test; (E) Fold change in the cholesterol metabolism-related gene expression in liver. Error bar represents SD. The significance level was indicated as follows: *: p<0.05, ANOVA with Tukey’s multiple comparison test. Sample size: TghPNPLA3-I148I (n=7), TghPNPLA3-I148M (n=8), and non-transgenic wide type (n=7).
In order to further understand the reason underlying this imbalanced insulin and glucagon signals in the islets, we hypothesized that the central organ fat accumulation promoted by PNPLA3–148M may increase the chronic inflammation in pancreas tissue. To test this hypothesis, we stained the murine macrophage marker F4/80 using IF in the pancreas. Indeed, the analysis revealed that the F4/80+ staining signals were significantly more abundant in the pancreas of the TghPNPLA3-I148M mice than that of the TghPNPLA3-I148I littermates (p=0.0111) (Fig. 3B), though no significant distribution of the F4/80+ signals within or around the islets were observed (data not shown). Taken together, these data indicated that increased chronic pancreatic inflammation may lead to decreased insulin but increased glucagon secretion in the TghPNPLA3-I148M mice as compared to the TghPNPLA3-I148I controls.
Akt signaling in mouse liver and muscle tissues
In order to further investigate the insulin signaling in these three groups mice, hepatic and muscle Akt phosphorylation levels were examined using Western blotting. The results showed that phosphorylation of Akt (pSer473) signal in TghPNPLA3-I148M group was marginally increased in the liver (p=0.0079) but not in the muscle tissues as compared to their TghPNPLA3-I148I littermates (Fig. 3C), suggesting that TghPNPLA3-I148M mice did not develop tissue insulin resistance as compared to the TghPNPLA3-I148I mice on the AMLN diet feeding.
Transcriptional changes of the thermogenesis and cholesterol metabolism pathways in adipose and liver tissues, respectively
We further set out to understand the potential mechanism underlying the reduced body weight while increased central fat accumulation in the TghPNPLA3-I148M mice. Previous studies have demonstrated that thermogenesis is a physiological defense against obesity by limiting weight gain in response to metabolic stress [50, 51]. We thus measured and compared the expression level of key thermogenic pathway genes in the intrascapular brown adipose tissue (iBAT), epidydimal white adipose tissue (eWAT) and subcutaneous white adipose tissue (scWAT) of the three groups mice. The results showed that there was significantly increased expression of thermogenic marker genes in iBAT (Ucp-1 and Nrf1) and scWAT (Ucp-1, Nrf1 and cox8b) (p<0.05 for all tests) but not in eWAT of the TghPNPLA3-I148M mice as compared to their TghPNPLA3-I148I littermates (Fig. 3D). This result suggested that the body weight loss in TghPNPLA3-I148M mice may be attributed to, at least in part, the activation of thermogenesis in both iBAT and scWAT.
To further understand the mechanism underlying the lower circulating total cholesterol in the TghPNPLA3-I148M mice, we measured the expression of genes involved in hepatic cell transporting (Ldlr, Mylip), cholesterol biosynthesis (Sqle, Lss, Fdft1, Hmgcs1, Soat2, Dhcr7, Scd5, Nsdh1, Hsd17b7, and Cyp51) and cholesterol secretion (Tm7sf2, Abcg1, Abca1 and Apob). We found that while there is no significant change in the expression of genes involved in cholesterol transporting and secretion, the expression of multiple genes involved in cholesterol biosynthesis were significantly downregulated in the TghPNPLA3-I148M mice compared to the wild-type groups (p<0.05 for all tests) (Fig. 3E).
Discussion
This is the first large-scale study to simultaneously delineate the causal inter-relationship between NAFLD, T2D, and obesity using a two-sample and bidirectional MR analysis. The findings were also in part, experimentally validated using an established genetic murine model of NAFLD, which largely corroborated the causal relationship between NAFLD and the susceptibility to T2D and obesity observed in humans. Overall, we found that genetically driven NAFLD is a causal risk factor for T2D but with a unique phenotype characterized as normal insulin sensitivity that is further associated with reduced insulin secretion, thus likely a late-onset type I-like diabetic phenotype. The genetically instrumented NAFLD also protects against overall or generalized obesity (indexed by BMI), but increases the risk for central obesity. On the other hand, genetically driven T2D and central or generalized obesity all causally increase risk for NAFLD. Our study thus suggests that genetically driven NAFLD while is likely “lean” given the low BMI, actually a “lipodystrophic NAFLD” phenotype characterized as reduced peripheral but increased central fat accumulation with normal or low circulating lipid profile. This phenotype may be distinct from the NAFLD that is metabolically driven by T2D, obesity and/or increased TG, thus a “metabolic NAFLD” subphenotype. Our findings hence reveal new insights into the nature and nurture of these three diseases, and provided strong evidence for disease subphenotyping.
It has been long regarded that NAFLD and NASH are the central manifestations of T2D and obesity. Due to the lack of data in the natural history of NAFLD/NASH in humans, it remains largely unclear with regard to the causal relationship between NAFLD and T2D or obesity. Addressing this issue is of critical importance to patient stratification, clinical management, as well as guiding various research for drug discovery and development. Our study for the first time, under a uniformed setting dissected the three diseases into important subtypes. First, we observed that genetically driven NAFLD is a causal risk factor for T2D. Our study confirmed the findings of a previous small-scale MR analysis[12], and is in line with the results from numerous observational studies, including a recent meta-analysis of 19 observational studies with 296,439 individuals which indicated that patients with NAFLD had a two-fold risk of developing T2D than those without NAFLD[3]. Interestingly, while a strong causal effect between NAFLD and T2D was observed, there was no significant causal relationship between genetic NAFLD and insulin resistance. This observation in human data was confirmed in the animal study: there is no genotypic difference in the glucose level in response to insulin as demonstrated in the ITT assay (Fig. 2D). Also, no evidence of tissue insulin resistance was observed as demonstrate by the Akt signaling in the liver and skeletal muscle tissues. While in humans there is no strong causal association between PNPLA3–148M and other glycemic traits, the PNPLA3–148M mice demonstrated a hyperglycemia and prolonged glucose clearance in the GTT assay. Why there is such a dissociation between T2D and insulin resistance, and why there are discrepancies between human and mouse? There are a few possibilities: First, the glucose disturbance associated with PNPLA3-driven NAFLD may be an atypical T2D phenotype, thus a disease subtype. Indeed, in our animal study, we found that the circulatory insulin level showed a significant decrease in the 148M mice as compared to the 148I mice after 12 weeks (Fig. 2B). After staining the insulin and glucagon signal in the pancreas, we found that the stained insulin signal was significantly lower in the 148M than the 148I mice, while glucagon signal was higher in 148M mice than the 148I mice (Fig. 3A), suggesting an imbalanced pancreatic production of insulin and glucagon in the 148M mice. We further hypothesize that this may be due to chronic pancreatic inflammation given the increased central organ fat accumulation associated with this model. By staining the F4/80, a murine macrophage marker in the pancreas, we found that the pancreatic inflammation level of the 148M mice was indeed significantly higher than that of the 148I mice (Fig. 3B). Therefore, it seems that PNPLA3–148M may lead to increased chronic pancreatic inflammation, which further alters the insulin/glucagon secretion balance. This resembles a Type I-like diabetes phenotype. This finding is consistent with the normal insulin sensitivity of the PNPLA3–148M mice as compared to the wild-type littermates in the ITT assay (Fig. 2D). In fact, the dissociation between the PNPLA3–148M-driven hepatic steatosis and insulin resistance has been consistently observed among human populations but lacks a clear mechanism [26, 52–54]. Our animal study provides new evidence to this question. Second, in contrast to a well-controlled animal study, in human populations, as compared to the disease diagnosis (here means T2D yes/no), metabolic markers e.g. glucose, insulin, or HbA1c are more likely modified by other factors, e.g. medications, co-morbidities or other disease conditions. Medication intake may likely normalize these markers thus weaken the genetic association. Meanwhile, insulin levels and insulin resistance status could be further modified by other disease conditions, e.g. systemic obesity. Co-existence of various risk factors in PNPLA3–148M individuals may also increase susceptibility to obesity, which may further lead to elevated insulin resistance thus compromises the normal insulin sensitivity conferred by PNPLA3–148M. This may be the reason underlying the mixed results in the association between PNPLA3–148M and insulin resistance. If this is true, then individuals carrying 148M without obesity or metabolic stresses would maintain a normal response to insulin. Indeed, in a large-scale study by Palmer et al [55], the association between 148M allele and insulin resistance disappeared among individuals after bariatric surgery, and no association was observed among obese individuals with BMI<35. Taken together, these data suggest that the PNPLA3-driven NAFLD may lead to a late-onset Type I-like diabetes that is distinct to the typical T2D characterized with insulin resistance. Increasing evidence has demonstrated that the Type-I diabetes (T1D) may commonly (40–50% of all T1D) occur in adulthood, and these patients are often misdiagnosed and treated as T2D, which can lead to serious clinical consequences[56, 57]. Interestingly, in humans late onset T1D is significantly associated with low BMI[56], similar to what we observed in our study. However, whether the phenotypes we have observed in our study match the typical late onset T1D defined in humans remains further confirmation. Upon confirmed, this would help stratify the T2D patients with different clinical managements. Our data again highlights the importance of disease subphenotyping.
Our study also revealed an interesting relationship between NAFLD and obesity. Interestingly, although epidemiologically obesity is highly correlated with NAFLD, the genetically instrumented NAFLD is actually not a causal risk factor for overall obesity. Rather, it protects against the overall BMI elevation. However, genetically driven NAFLD causally increases risks for central obesity, characterized as an increased waist-to-hip ratio in humans, thus a lipodystrophy. Our analysis suggests that genetically driven NAFLD may promote the remodeling of the fat distribution in the whole body. This finding is consistent with numerous observational studies on the associations between NAFLD, visceral fat or WHR and BMI[58, 59], and corroborates the widely discussed hypothesis about “lean NAFLD” and “obese NAFLD” [60, 61]. Our study echoes this observation and indicates that genetically driven NAFLD may be more likely progressed to “lean” (given the low BMI) NAFLD while does not promote the development of overall obesity, therefore actually a “lipodystrophic NAFLD”. This is also consistent with the observation that lean NAFLD is associated with adipose tissue disfunction, impaired glucose tolerance, and the PNPLA3 I148M allele [60, 61]. Our mouse study accurately recapitulated these relationships. Further mechanistic analyses revealed that the thermogenic pathway gene expression was significantly upregulated especially in subcutaneous white adipose tissue and brown adipose tissue but not in the epidydimal fat, suggesting that the TghPNPLA3-I148M mice may have an increased thermogenesis by turning the subcutaneous fat into heat. This is the first time to observe the effect of PNPLA3–148M on adipose tissue browning or thermogenesis. We postulate that this increased visceral fat accumulation and activated thermogenesis in the subcutaneous fat may reflect a genetic adaption to cold stress during human evolution and migration, given the dramatically increased PNPLA3 148M allele frequency from Africans (<10%) to Eurasians (20–40%) and Native Americans (70%−80%) [62]. The underlying mediator for this genotype-specific activation of thermogenesis remains unclear thus warranting continued investigation.
Another interesting observation in our study is that, both the MR and animal data indicated that genetic NAFLD causally decrease circulating cholesterol (TC and LDL). This phenotype was similar in the mice fed with either HSD or the AMLN diet, suggesting a diet-independent mechanism. Mechanistically, this may be related to the hepatic VLDL retention associated with PNPLA3–148M as previously observed [63]. After profiling the key genes involved in cholesterol metabolism in the liver, we found that the cholesterol biosynthesis pathway genes were particularly downregulated. Therefore, central obesity, at least in part, may possess a subtype attributed to genetically-driven NAFLD. This is particularly significant, as both TC and TG are generally correlated with visceral fat accumulation as demonstrated in many studies[64, 65]. The dissociation observed in our study suggests that the NAFLD-driven central obesity is likely a unique subphenotype as well.
On the other hand, our findings also demonstrated that T2D, obesity (both generalized and central) and increased TG are likely causal risk factors for NAFLD, though certain pleiotropic effects may remain. This is not surprising. First, hyperglycemia and adiposity could amplify or modify the effect of genetic risk factors to promote the development of NAFLD. High glucose level can increase the expression of PNPLA3 via regulating carbohydrate-response element-binding protein (ChREBP)[66], which is a necessary step for the accumulation of PNPLA3–148M protein on the surface of lipid droplet in hepatocytes [30, 31, 67]. This accumulation further alters the dynamics of hydrolysis of triglycerides and lead to hepatic steatosis[68]. Similarly, adiposity has also been demonstrated to synergize the effects of genetic risk factors on the development of NASH or more severe liver injuries[14]. Second, impaired glycose perturbation and adiposity can also increase the risk for NAFLD independent of genetic risk factors. Numerous studies in humans have shown that NAFLD and NASH can be developed among T2D or obese individuals carrying wildtype genotypes of PNPLA3 or other genetic variants [69]. And many diet-induced animal models without genetic modification also develop NAFLD/NASH [45–48]. In this case, NAFLD/NASH should be a co-morbidity of T2D or obesity. This highlights the impact of “nurture” on the development of NAFLD.
Taken together, it is now clear that NAFLD should be re-classified into at least two subtypes: 1) “genetic NAFLD” (nature) which is characterized with central fat deposition (lipodystrophy), type-1 like diabetic symptom without insulin resistance as well as normal or low lipid profile; and 2) “metabolic NAFLD” (nurture) which is a comorbidity of diabetes mellitus and/or obesity. Distinguishing these two subtypes may have important clinical implication. First, as the genetically driven NAFLD generally has a low or normal BMI (thus generally “lean”) and lipid profile, Individuals with this subtype may seem to be “healthier” than the “metabolic NAFLD”. For this reason, they may be under diagnosed for their liver injuries. This is especially an issue as “lean” NAFLD patients have demonstrated even a more advanced liver histology as compared to that of obese NAFLD patients [70]. Second, distinguishing the two subtypes would be critical for a precision treatment of NAFLD/NASH given the currently limited therapeutic strategy. Given the different causes for the two subtypes, designing of the treatment for genetic NAFLD should be focused on the causal genes and underlying pathways. To this end, to develop genotype-based, targeted therapies would be an intriguing direction [71]. Indeed, recent PNPLA3-targeting strategy has demonstrated promising results in ameliorating NASH in mouse [72]. Similar investigation is currently ongoing in our group as well. On the other hand, metabolic NAFLD as a comorbidity for diabetes or obesity should be managed by focusing on the metabolic disease, e.g. to reduce the body weight. These new hypotheses should be examined in future studies.
It is worth noting that our MR analyses using various combinations of multiple genetic variants as instruments generated similar results about the causal relationships between NAFLD, T2D, obesity as well as insulin resistance. This strongly suggests that these causal relationships are general to all genetically driven NAFLD rather than a biased association caused by a specific gene (e.g. due to horizontal pleiotropy). Of course, it is still possible that the mechanisms underlying these relationships observed in our animal study is specific to PNPLA3. Notably, our collaborator’s (co-author Y.E.C) unpublished work on a mouse model expressing the TM6SF2 variant also demonstrated a similar phenotype. In addition, although we used the largest-to-date dataset for our analysis, we only focused on using hepatic steatosis or binary histologic NAFLD as exposures. Previous studies demonstrated that severity of liver damage may lead to different causal impact on other metabolic diseases. Therefore, it would be important to dissect the severity of NAFLD to further delineate the potential causal impact on other perturbations. Unfortunately, there currently lacks sufficient high quality GWAS data on well-characterized NAFLD/NASH especially in large cohorts. Future studies should further explore this question.
In summary, our study combined both a large-scale bidirectional MR analysis and animal models to delineate the causal relationships between NAFLD, T2D, and obesity. Our findings provided strong evidence for disease subphenotyping and fostered new hypotheses to the development of precision diagnostic, preventive and therapeutic strategies for the three diseases. The PNPLA3-specific study demonstrated that the 148M variant leads to NAFLD and central organ fat accumulation, which may result in chronic tissue inflammation and alters insulin/glucagon signaling, further reducing glucose tolerance. Meanwhile, the genetic NAFLD may also promote browning and thermogenesis of peripheral adipose tissue but maintains a normal circulating lipid profile. Together with the central fat deposition, this pattern may reflect a general defensive mechanism against the cold stress during human evolution and migration. A schematic about this mechanism was summarized in Fig. 4.
Fig. 4. Schematic summary of the causal relationship between PNPLA3–148M driven NAFLD and obesity, diabetes and cholesterol metabolism.

Chol=cholesterol.
Supplementary Material
HIGHLIGHTS.
Genetic NAFLD promotes risks for a late onset Type 1-like diabetes
Genetic NAFLD promotes central obesity but protects against overall obesity
Genetic NAFLD leads to lower circulating cholesterol
Genetically driven T2D and obesity causally increase the NAFLD risk
PNPLA3-148M-driven NAFLD impairs insulin secretion but not insulin resistance
PNPLA3-148M-driven NAFLD activates thermogenesis pathway in peripheral fat tissue
Acknowledgements
We thank all the genetics consortiums for making the GWAS summary data publicly available. We would also like to thank Drs. Anjaneyulu Kowluru and James Granneman, as well as Jean-Christophe Rochet and his group for their insightful discussion and comments on this study.
Financial support statement
This study is supported in part by the NIH/NIDDK grant (R01DK106540) (W.L.), and the start-up fund of the Office of Vice President for Research of Wayne State University (W.L.), NIH R21AA024550 (X.C.D.), R01DK091592 (X.C.D), R56DK091592 (X.C.D), Indiana Diabetes Research Center grant NIH P30DK097512, and the Indiana Clinical and Translational Sciences Institute funded from the NIH NCATS CTSA UL1TR002529. D.C is a visiting scholar supported by the Shenzhen Children’s Hospital, Shenzhen, China.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement
All authors have reviewed the manuscript. C.W’s spouse works at Regeneron Pharmaceuticals, and all other co-authors declared no conflict of interest. The sponsor of the study has no role in the study design, collection, analysis, and interpretation of data.
References
- [1].Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328–357. [DOI] [PubMed] [Google Scholar]
- [2].Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu Rev Pathol 2016;11:451–496. [DOI] [PubMed] [Google Scholar]
- [3].Mantovani A, Byrne CD, Bonora E, Targher G. Nonalcoholic Fatty Liver Disease and Risk of Incident Type 2 Diabetes: A Meta-analysis. Diabetes Care 2018;41:372–382. [DOI] [PubMed] [Google Scholar]
- [4].Hu M, Phan F, Bourron O, Ferre P, Foufelle F. Steatosis and NASH in type 2 diabetes. Biochimie 2017;143:37–41. [DOI] [PubMed] [Google Scholar]
- [5].Lonardo A, Lugari S, Ballestri S, Nascimbeni F, Baldelli E, Maurantonio M. A round trip from nonalcoholic fatty liver disease to diabetes: molecular targets to the rescue? Acta Diabetol 2019;56:385–396. [DOI] [PubMed] [Google Scholar]
- [6].Loomis AK, Kabadi S, Preiss D, Hyde C, Bonato V, St Louis M, et al. Body Mass Index and Risk of Nonalcoholic Fatty Liver Disease: Two Electronic Health Record Prospective Studies. J Clin Endocrinol Metab 2016;101:945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology 2010;51:679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Smith GD. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Statistics in Medicine 2008;27:1133–1163. [DOI] [PubMed] [Google Scholar]
- [9].Voight BF, Peloso GM, Orho-Melander M, Frikke-Schmidt R, Barbalic M, Jensen MK, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet 2012;380:572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Paternoster L, Tilling K, Smith GD. Genetic epidemiology and Mendelian randomization for informing disease therapeutics: Conceptual and methodological challenges. Plos Genetics 2017;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Welsh P, Polisecki E, Robertson M, Jahn S, Buckley BM, de Craen AJM, et al. Unraveling the Directional Link between Adiposity and Inflammation: A Bidirectional Mendelian Randomization Approach. J Clin Endocr Metab 2010;95:93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Dongiovanni P, Stender S, Pietrelli A, Mancina RM, Cespiati A, Petta S, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med 2018;283:356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].De Silva NMG, Borges MC, Hingorani A, Engmann J, Shah T, Zhang X, et al. Liver Function and Risk of Type 2 Diabetes: Bidirectional Mendelian Randomization Study. Diabetes 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Stender S, Kozlitina J, Nordestgaard BG, Tybjaerg-Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet 2017;49:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Speliotes EK, Yerges-Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 2011;7:e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 2018;50:1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wheeler E, Leong A, Liu CT, Hivert MF, Strawbridge RJ, Podmore C, et al. Impact of common genetic determinants of Hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: A transethnic genome-wide meta-analysis. PLoS Med 2017;14:e1002383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia-Naji N, Chen H, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 2012;44:659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Strawbridge RJ, Dupuis J, Prokopenko I, Barker A, Ahlqvist E, Rybin D, et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 2011;60:2624–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Saxena R, Hivert MF, Langenberg C, Tanaka T, Pankow JS, Vollenweider P, et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet 2010;42:142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 2010;42:105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Prokopenko I, Poon W, Magi R, Prasad BR, Salehi SA, Almgren P, et al. A central role for GRB10 in regulation of islet function in man. PLoS Genet 2014;10:e1004235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Pulit SL, Stoneman C, Morris AP, Wood AR, Glastonbury CA, Tyrrell J, et al. Meta-analysis of genome-wide association studies for body fat distribution in 694 649 individuals of European ancestry. Hum Mol Genet 2019;28:166–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet 2013;45:1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhou W, Nielsen JB, Fritsche LG, Dey R, Gabrielsen ME, Wolford BN, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat Genet 2018;50:1335–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjaerg-Hansen A, et al. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liu YL, Reeves HL, Burt AD, Tiniakos D, McPherson S, Leathart JB, et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat Commun 2014;5:4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang X, Liu Z, Wang K, Wang Z, Sun X, Zhong L, et al. Additive Effects of the Risk Alleles of PNPLA3 and TM6SF2 on Non-alcoholic Fatty Liver Disease (NAFLD) in a Chinese Population. Front Genet 2016;7:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest 2012;122:4130–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015;61:108–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ehrhardt N, Doche ME, Chen S, Mao HZ, Walsh MT, Bedoya C, et al. Hepatic Tm6sf2 overexpression affects cellular ApoB-trafficking, plasma lipid levels, hepatic steatosis and atherosclerosis. Hum Mol Genet 2017;26:2719–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lauridsen BK, Stender S, Kristensen TS, Kofoed KF, Kober L, Nordestgaard BG, et al. Liver fat content, non-alcoholic fatty liver disease, and ischaemic heart disease: Mendelian randomization and meta-analysis of 279 013 individuals. Eur Heart J 2018;39:385–393. [DOI] [PubMed] [Google Scholar]
- [34].Wang N, Chen C, Zhao L, Chen Y, Han B, Xia F, et al. Vitamin D and Nonalcoholic Fatty Liver Disease: Bi-directional Mendelian Randomization Analysis. EBioMedicine 2018;28:187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gorden A, Yang RZ, Yerges-Armstrong LM, Ryan KA, Speliotes E, Borecki IB, et al. Genetic Variation at NCAN Locus Is Associated with Inflammation and Fibrosis in Non-Alcoholic Fatty Liver Disease in Morbid Obesity. Human Heredity 2013;75:34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Staiger D, Stock JH. Instrumental variables regression with weak instruments. Econometrica 1997;65:557–586. [Google Scholar]
- [38].Burgess S, Butterworth A, Thompson SG. Mendelian Randomization Analysis With Multiple Genetic Variants Using Summarized Data. Genet Epidemiol 2013;37:658–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent Estimation in Mendelian Randomization with Some Invalid Instruments Using a Weighted Median Estimator. Genet Epidemiol 2016;40:304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol 2015;44:512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bowden J, Del Greco MF, Minelli C, Zhao Q, Lawlor DA, Sheehan NA, et al. Improving the accuracy of two-sample summary-data Mendelian randomization: moving beyond the NOME assumption. Int J Epidemiol 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet 2018;50:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yavorska OO, Burgess S. MendelianRandomization: an R package for performing Mendelian randomization analyses using summarized data. Int J Epidemiol 2017;46:1734–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bowden J, Spiller W, Del Greco MF, Sheehan N, Thompson J, Minelli C, et al. Improving the visualization, interpretation and analysis of two-sample summary data Mendelian randomization via the Radial plot and Radial regression. Int J Epidemiol 2018;47:2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Denk H, Abuja PM, Zatloukal K. Animal models of NAFLD from the pathologist’s point of view. Biochim Biophys Acta Mol Basis Dis 2018. [DOI] [PubMed] [Google Scholar]
- [46].Van Herck MA, Vonghia L, Francque SM. Animal Models of Nonalcoholic Fatty Liver Disease-A Starter’s Guide. Nutrients 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Clapper JR, Hendricks MD, Gu G, Wittmer C, Dolman CS, Herich J, et al. Diet-induced mouse model of fatty liver disease and nonalcoholic steatohepatitis reflecting clinical disease progression and methods of assessment. Am J Physiol Gastrointest Liver Physiol 2013;305:G483–495. [DOI] [PubMed] [Google Scholar]
- [48].Panasevich MR, Meers GM, Linden MA, Booth FW, Perfield JW 2nd, Fritsche KL, et al. High-fat, high-fructose, high-cholesterol feeding causes severe NASH and cecal microbiota dysbiosis in juvenile Ossabaw swine. Am J Physiol Endocrinol Metab 2018;314:E78–E92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hemani G, Bowden J, Davey Smith G. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum Mol Genet 2018;27:R195–R208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ho KKY. Diet-induced thermogenesis: fake friend or foe? J Endocrinol 2018;238:R185–R191. [DOI] [PubMed] [Google Scholar]
- [51].Liao WH, Henneberg M, Langhans W. Immunity-Based Evolutionary Interpretation of Diet-Induced Thermogenesis. Cell Metab 2016;23:971–979. [DOI] [PubMed] [Google Scholar]
- [52].Kantartzis K, Peter A, Machicao F, Machann J, Wagner S, Konigsrainer I, et al. Dissociation between fatty liver and insulin resistance in humans carrying a variant of the patatin-like phospholipase 3 gene. Diabetes 2009;58:2616–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Peter A, Kovarova M, Nadalin S, Cermak T, Konigsrainer A, Machicao F, et al. PNPLA3 variant I148M is associated with altered hepatic lipid composition in humans. Diabetologia 2014;57:2103–2107. [DOI] [PubMed] [Google Scholar]
- [54].Franko A, Merkel D, Kovarova M, Hoene M, Jaghutriz BA, Heni M, et al. Dissociation of Fatty Liver and Insulin Resistance in I148M PNPLA3 Carriers: Differences in Diacylglycerol (DAG) FA18:1 Lipid Species as a Possible Explanation. Nutrients 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Palmer CN, Maglio C, Pirazzi C, Burza MA, Adiels M, Burch L, et al. Paradoxical lower serum triglyceride levels and higher type 2 diabetes mellitus susceptibility in obese individuals with the PNPLA3 148M variant. PLoS One 2012;7:e39362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Thomas NJ, Jones SE, Weedon MN, Shields BM, Oram RA, Hattersley AT. Frequency and phenotype of type 1 diabetes in the first six decades of life: a cross-sectional, genetically stratified survival analysis from UK Biobank. Lancet Diabetes Endocrinol 2018;6:122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bao YK, Ma J, Ganesan VC, McGill JB. Mistaken Identity: Missed Diagnosis of Type 1 Diabetes in an Older Adult. Med Res Arch 2019;7. [PMC free article] [PubMed] [Google Scholar]
- [58].Radmard AR, Rahmanian MS, Abrishami A, Yoonessi A, Kooraki S, Dadgostar M, et al. Assessment of Abdominal Fat Distribution in Non-Alcoholic Fatty Liver Disease by Magnetic Resonance Imaging: a Population-based Study. Arch Iran Med 2016;19:693–699. [PubMed] [Google Scholar]
- [59].Pang Y, Kartsonaki C, Turnbull I, Guo Y, Chen Y, Clarke R, et al. Adiposity in relation to risks of fatty liver, cirrhosis and liver cancer: a prospective study of 0.5 million Chinese adults. Sci Rep 2019;9:785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Feldman A, Eder SK, Felder TK, Kedenko L, Paulweber B, Stadlmayr A, et al. Clinical and Metabolic Characterization of Lean Caucasian Subjects With Non-alcoholic Fatty Liver. Am J Gastroenterol 2017;112:102–110. [DOI] [PubMed] [Google Scholar]
- [61].Lu FB, Hu ED, Xu LM, Chen L, Wu JL, Li H, et al. The relationship between obesity and the severity of non-alcoholic fatty liver disease: systematic review and meta-analysis. Expert Rev Gastroenterol Hepatol 2018;12:491–502. [DOI] [PubMed] [Google Scholar]
- [62].Tepper CG, Dang JHT, Stewart SL, Fang DM, Wong KA, Liu SY, et al. High frequency of the PNPLA3 rs738409 [G] single-nucleotide polymorphism in Hmong individuals as a potential basis for a predisposition to chronic liver disease. Cancer 2018;124 Suppl 7:1583–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Pirazzi C, Adiels M, Burza MA, Mancina RM, Levin M, Stahlman M, et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J Hepatol 2012;57:1276–1282. [DOI] [PubMed] [Google Scholar]
- [64].Sadeghi M, Pourmoghaddas Z, Hekmatnia A, Sanei H, Tavakoli B, Tchernof A, et al. Abdominal fat distribution and serum lipids in patients with and without coronary heart disease. Arch Iran Med 2013;16:149–153. [PubMed] [Google Scholar]
- [65].Luo Y, Ma X, Shen Y, Hao Y, Hu Y, Xiao Y, et al. Positive relationship between serum low-density lipoprotein cholesterol levels and visceral fat in a Chinese nondiabetic population. PLoS One 2014;9:e112715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Huang Y, He S, Li JZ, Seo YK, Osborne TF, Cohen JC, et al. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc Natl Acad Sci U S A 2010;107:7892–7897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Liu W, Anstee QM, Wang X, Gawrieh S, Gamazon ER, Athinarayanan S, et al. Transcriptional regulation of PNPLA3 and its impact on susceptibility to nonalcoholic fatty liver Disease (NAFLD) in humans. Aging (Albany NY) 2016;9:26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wang Y, Kory N, BasuRay S, Cohen JC, Hobbs HH. PNPLA3, CGI-58, and Inhibition of Hepatic Triglyceride Hydrolysis in Mice. Hepatology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Lee YH, Cho Y, Lee BW, Park CY, Lee DH, Cha BS, et al. Nonalcoholic Fatty Liver Disease in Diabetes. Part I: Epidemiology and Diagnosis. Diabetes Metab J 2019;43:31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Denkmayr L, Feldman A, Stechemesser L, Eder SK, Zandanell S, Schranz M, et al. Lean Patients with Non-Alcoholic Fatty Liver Disease Have a Severe Histological Phenotype Similar to Obese Patients. J Clin Med 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Colombo M, Pelusi S. Towards Precision Medicine in Nonalcoholic Fatty Liver Disease with PNPLA3 as a Therapeutic Target. Gastroenterology 2019;157:1156–1157. [DOI] [PubMed] [Google Scholar]
- [72].Linden D, Ahnmark A, Pingitore P, Ciociola E, Ahlstedt I, Andreasson AC, et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol Metab 2019;22:49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.