

Abstract
Epithelial cells require attachment to a support, such as the extracellular matrix, for survival. During cancer progression and metastasis, cancerous epithelial cells must overcome their dependence on adhesion signals. Dependence on glucose metabolism is a hallmark of cancer cells, but the nutrient requirements of cancer cells under anchorage-deficient conditions remain uncharacterized. Here, we report that cancer cells prioritize glutamine-derived tricarboxylic acid cycle energy metabolism over glycolysis to sustain anchorage-independent survival. Moreover, glutamine-dependent metabolic reprogramming is required not only to maintain ATP levels but also to suppress excessive oxidative stress through interaction with cystine. Mechanistically, AMPK, a central regulator of cellular responses to metabolic stress, participates in the induction of the expression of ASCT2, a glutamine transporter, and enhances glutamine consumption. Most interestingly, AMPK activation induces Nrf2 and its target proteins, allowing cancer cells to maintain energy homeostasis and redox status through glutaminolysis. Treatment with an integrin inhibitor was used to mimic the alterations in cell morphology and metabolic reprogramming caused by detachment. Under these conditions, cells were vulnerable to glutamine starvation or glutamine metabolism inhibitors. The observed preference for glutamine over glucose was more pronounced in aggressive cancer cell lines, and treatment with the glutaminase inhibitor, CB839, and cystine transporter inhibitor, sulfasalazine, caused strong cytotoxicity. Our data clearly show that anchorage-independent survival of cancer cells is supported mainly by glutaminolysis via the AMPK-Nrf2 signal axis. The discovery of new vulnerabilities along this route could help slow or prevent cancer progression.
Keywords: Extracellular matrix detachment, Metabolic reprogramming, Glutaminolysis, Anoikis, Metastasis
Graphical abstract

Highlights
-
•
AMPK-Nrf2 signaling is crucial in metabolic reprogramming during cancer progression.
-
•
Cancer metabolism does not preeminently depend on glycolysis.
-
•
Glutaminolysis mainly supports anchorage-independent cancer cell survival.
Abbreviations
- 2DG
2-deoxy-d-glucose
- AMPK
adenosine monophosphate-activated kinase
- ATP
adenosine triphosphate
- BSO
l-buthionine sulfoximine
- DM-αKG
dimethyl-α-ketoglutarate
- DMEM
Dulbecco's modified Eagle medium
- ECM
extracellular matrix
- FBS
fetal bovine serum
- GPNA
L-γ-glutamyl-p-nitroanilide
- NAC
N-acetyl-cysteine
- Nrf2
nuclear factor E2-related factor 2
- OXPHOS
oxidative phosphorylation
- PARP
poly (ADP-ribose) polymerase
- poly-HEMA
poly 2-hydroxyethyl methacrylate
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
- TCA
tricarboxylic acid
1. Introduction
Despite significant advances in diagnosing and treating cancer, metastasis limits cancer-free survival [1]. Given that metastases cause more than 90% of cancer-related deaths, it is of paramount importance to elucidate the mechanisms leading to their establishment [[2], [3], [4]]. One key restriction to metastasis is anoikis, a form of cell death elicited by loss of adhesion in cells that detach from the extracellular matrix (ECM) [5,6]. Anoikis is caused by caspase-dependent apoptosis or non-apoptotic cell death necroptosis [7]. Although the phenomenon is regulated by a complex web of events, disruption in glucose utilization, adenosine triphosphate (ATP) production, and redox homeostasis are hallmarks of cell death and suggest that cellular metabolism plays a critical role in ECM-independent survival of cancer cells [7]. Cancer cells that successfully metastasize acquire anchorage-independent survival mechanisms and dependence on metabolic alterations may provide effective therapeutic targets [5,8,9].
Cancer cells reprogram pathways of nutrient acquisition and metabolism to meet the bioenergetic, biosynthetic, and redox homeostasis necessary to sustain the malignant phenotype [10]. Unlike normal cells, cancer cells preferentially utilize the glycolytic pathway to produce large amounts of lactate even in the presence of oxygen, a phenomenon known as the “Warburg effect” [11]. Anoikis of mammary epithelial cells is rescued by forced expression of oncogenes as a result of enhanced glucose consumption and reduced oxidative stress [7]. Although numerous studies have demonstrated the importance of the Warburg effect for many cancer cell types [11], it is unclear whether protection from anoikis is determined solely by abnormal oncogene-mediated signaling pathways. In particular, it is unknown whether cancer cells detached from the ECM maintain a survival mechanism dependent on the Warburg effect.
Glutamine is the most abundant amino acid in the blood and plays an important role in providing both carbon and nitrogen for anabolic metabolism [12]. Like glucose, it is metabolized via the glycolytic pathway to produce ATP, but is also used to generate biosynthetic intermediates through the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) [12]. Recent studies have pointed to the importance of glutamine metabolism for the growth and survival of cancer cells, as it supports macromolecular biosynthesis and maintains bioenergetics and redox balance [13,14]. Furthermore, evidence suggests that the ability to counteract excessive reactive oxygen species (ROS) is necessary for effective metastasis because metastasis is augmented in animals treated with antioxidants [[15], [16], [17]]. Hence, metabolic changes other than the Warburg effect are likely essential to reduce oxidative stress and enable cancer cells to detach from the ECM and for tumors to spread to distant sites.
Cancer progression results from adaptation to various environmental stresses, in which major nutrients play a pivotal role in the development of malignant characteristics [4]. Although glucose and glutamine are essential for supporting cancer cells, the mechanism underlying cancer cell survival has not been studied by comparing auxotrophic alterations in adherent and non-adherent states. This gap prompted us to investigate the mechanisms enabling cancer progression and especially metabolic reprogramming induced by loss of anchorage.
Here, we report that metabolic reprogramming favoring glutamine over glucose is indispensable for survival of cancer cells under detachment conditions. Intervention in these metabolic processes could foster novel approaches to improve cancer therapy.
2. Materials and methods
2.1. Reagents and antibodies
The chemical and biochemical reagents were obtained from the following sources: ascorbic acid, l-buthionine sulfoximine (BSO), cystine, and L-γ-glutamyl-p-nitroanilide (GPNA) were purchased from Wako Pure Chemicals (Osaka, Japan); oligomycin and 3PO were from Calbiochem-Merck (Darmstadt, Germany); antimycin A1, bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES), cilengitide, dimethyl-α-ketoglutarate (DM-αKG), d-glucose, Dulbecco's modified Eagle's medium (DMEM), dimethylsulfoxide, l-glutamine, MEM amino acids solution, MEM non-essential amino acids solution, MEM vitamin solution, N-acetyl-cysteine (NAC), poly 2-hydroxyethyl methacrylate (poly-HEMA), proteinase and phosphatase inhibitor cocktail, sulfasalazine, and 2-deoxy-d-glucose (2DG) were from Sigma-Aldrich (St. Louis, MO, USA); CB839 and UK5099 were from Cayman Chemical (Ann Arbor, MI, USA); etomoxir was from Selleck Chemical (Houston, TX, USA). Antibodies against AMPKα, phosphorylated (p)-AMPKα (Thr172), ASCT2, FAK (Tyr397), p-FAK, IDH1, LKB1, PARP, p-H2AX (Ser139), and xCT were obtained from Cell Signaling (Beverly, MA). Antibodies against GCLC, GCLM, and ME1 were from Abcam (Cambridge, UK). Antibodies against α-tubulin and β-actin were from Wako Pure Chemicals. Antibodies against lamin B1 and Nrf2 were from MBL (Nagoya, Japan) and GeneTex (San Antonio, TX, USA), respectively.
2.2. Cell cultures and medium composition
Cells were obtained from the following sources: HeLa and HepG2 cells were purchased from JCRB (Osaka, Japan); BxPC-3, HT1080, and G361 cells were from ECACC (Salisbury, Scotland); A375-P, A375-MA2, and MDA-MB-231 cells were from ATCC (Manassas, VA, USA). HaCaT cells were provided by Dr. Fusenig (German Cancer Research Center, Heidelberg). MCF-7 cells were a kind gift from Dr. Kametani (Tokai University School of Medicine, Kanagawa, Japan). All cell lines were maintained in DMEM supplemented with 10% fetal bovine serum (FBS; HyClone, GE Healthcare, South Logan, UT, USA), 50 U/mL penicillin, 50 mg/mL streptomycin, and non-essential amino acids (Life Technologies, Carlsbad, CA) at 37 °C and 5% CO2. For detachment experiments, tissue culture dishes or plates were coated with 3 mg/mL poly-HEMA in 95% ethanol as mentioned previously [18] with some modifications, and were then incubated at 37 °C for more than a day until dry. Media deficient for glucose, glutamine, or both were made based on powdered glucose- and glutamine-free DMEM (Sigma-Aldrich) that contained neither phenol red nor pyruvate. When these nutrients had to be present, glucose or glutamine were added into the nutrient-deficient medium to a final concentration of 5.5 mM and 2 mM, respectively. All nutrient-deficient DMEM was prepared by dissolving inorganic salts (CaCl2 265 mg/L, MgSO4 97.67 g/L, KCl 400 mg/L, NaHCO3 3.7 g/L, NaH2PO4 109 mg/L, NaCl 6.8 g/L, Fe(NO3) 3·9H2O 0.1 mg/L) in ultrapure water, with the final pH adjusted to pH 7.4. To examine the nutrient requirements, nutrient-deficient culture media with or without a specific nutrient were changed during detachment culture conditions. To test for auxotrophy under the adherent state, the intended medium was exchanged after the cells were sufficiently attached. Media for all experiments were supplemented with 10% dialyzed FBS.
2.3. Stably expressing cell lines
Cell lines stably expressing short hairpin RNA (shRNA) were established as described previously [19,20]. HepG2 cells were transfected with a shRNA plasmid that targeted human AMPKα1 (sh-AMPK) or human NFE2L2/Nrf2 (sh-Nrf2). A non-targeting control shRNA plasmid served as a negative control (sh-con).
2.4. Transient transfection
The mammalian expression vector, pcDNA3-EGFP-C4-Nrf2, encoding human Nrf2 (EGFP-Nrf2) was obtained from Addgene (#21549, Watertown, MA, USA). The control expression vector was from Clontech (Mountain View, CA, USA). Cells stably expressing sh-con or AMPK shRNA were seeded and grown in 10-cm dishes to 70% confluence before transfection with plasmids in the presence of Lipofectamine LTX and PLUS Reagent (Life Technologies) according to the manufacturer's instructions. After 24 h, the cells were trypsinized, collected by centrifugation, washed with DMEM, and seeded in normal culture dishes or poly-HEMA-coated dishes at a density of 2.5 × 105 cells/mL. After 24 h, they were harvested and analyzed by western blotting.
2.5. Western blot analysis
Western blot analysis was performed as described previously [21,22]. Adherent cultured cells were trypsinized into a single-cell suspension in medium and then seeded in monolayer culture dishes or poly-HEMA-coated dishes at a density of 2.5 × 105 cells/mL, followed by a 24-h incubation. Scraped attached or detached cells were collected by centrifugation at 300 × g for 3 min, washed twice with ice-cold phosphate-buffered saline, and whole protein lysates were prepared using a radioimmunoprecipitation assay buffer (Wako Pure Chemicals) containing a complete protease and phosphatase inhibitor cocktail. Nuclear proteins were extracted using the NE-PER nuclear and cytoplasmic extraction kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's protocol. Protein concentration was measured by the BCA protein assay kit (Wako Pure Chemicals). Equal amounts of protein were then resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. The membranes were developed by chemiluminescence using Immobilon Western Chemiluminescent HRP Substrate (Millipore, Billerica, MA, USA).
2.6. Determination of ATP content
To measure intracellular ATP levels, the CellTiter-Glo 2.0 Luminescent Cell Viability Assay (Promega, Madison, WI, USA) was used as described previously [20]. Briefly, 1 × 104 cells/100 μL were loaded into each well of a 96-well plate. After addition of 100 μL of CellTiter-Glo reagents, relative luminescence units were measured using the GloMax96 microplate luminometer (Promega). The ATP content of cells cultured in complete medium (control) was set to 100%, and each batch of ATP measurements was calculated based on the control group. All values were normalized to protein concentrations.
2.7. ROS assays
The ROS-Glo H2O2 assay (Promega) was used to measure the level of hydrogen peroxide (H2O2) in the culture according to the manufacturer's instructions. The ROS assay was carried out by plating 1 × 104 cells/100 μL into each well of a 96-well plate. Cells were incubated in culture medium with or without the intended nutrient and H2O2 substrate solution for 4 h, following which the ROS-Glo detection solution was added. Luminescence units were measured using the GloMax96 microplate luminometer and expressed as fold changes. All values were normalized to protein concentrations.
2.8. Cell viability assays
To assess cell viability, the CellTiter 96 AQueous One Solution Cell Proliferation Assay System (Promega) was used according to the manufacturer's instructions. Briefly, 5 × 103 cells were seeded into each well in adherent or poly-HEMA-coated 96-well plates and 10 μL per well of CellTiter 96 AQueous One Solution reagent was added. After 4 h of incubation in a humidified 5% CO2 atmosphere, absorbance at 490 nm was measured using a SpectraMax I3 microplate reader (Molecular Devices, Sunnyvale, CA, USA). Five replicate wells per indicated group were used to estimate cell viability. The viability of cells cultured in complete medium was set to 100% and each batch of measurements was calculated based on the control group.
2.9. Glucose and glutamine determination
Glucose levels were determined using a Glucose Colorimetric assay kit II (BioVision, Milpitas, CA, USA). Glutamine levels were determined using a Glutamine Detection Assay Kit (Abcam) in accordance with the manufacturer's instructions. Glucose or glutamine consumption was calculated by subtracting the detected concentration of each compound in the medium from the original glucose or glutamine concentration, and was expressed as fold change. All values were normalized to protein concentrations.
2.10. Lactate production assays
Conditioned medium derived from attached or detached cells was collected and deproteinized with a 10-kDa MWCO spin filter (Amicon Ultra-4; Merck-Millipore) to remove lactate dehydrogenase. Lactate levels in culture medium were measured using a Lactate Colorimetric/Fluorometric Assay Kit (BioVision) according to the manufacturer's protocol, and were expressed as fold changes. All values were normalized to protein concentrations.
2.11. Statistical analysis
Data are expressed as the mean ± SD. Differences between treatment groups were determined either by Student's t-tests (two-tailed) for a comparison between two groups or by one-way analysis of variance (ANOVA) for multiple groups, followed by Dunnett's or two-way ANOVA with Tukey's post hoc statistical test using GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA, USA). Test results with P < 0.05 were considered statistically significant.
3. Results
3.1. Metabolic reprogramming enables cancer cell survival in response to ECM detachment
To investigate whether anchorage regulated the energy metabolism, we first examined intracellular ATP levels in hepatoma HepG2 cells cultured under adherent or non-adherent conditions. We detected a negligible reduction of ATP in HepG2 cells and cancer cell lines of various origin cultured under detached conditions for 24 h (Fig. 1A, Fig. S1A). Whereas net uptake of glucose and production of lactate were approximately 20% lower in HepG2 cells after ECM detachment (Fig. 1B and C), glutamine consumption was increased by a similar extent (Fig. 1D). These results suggest that ECM detachment caused metabolic reprogramming, favoring glutamine-dependent energy metabolism over the Warburg effect. Treatment with UK5099, an inhibitor of the mitochondrial pyruvate carrier [23,24], did not increase cleavage of the apoptotic marker, poly (ADP-ribose) polymerase-1 (PARP), in attached cells, but cleaved PARP was clearly visible in detached cells (Fig. 1E). This finding indicates a shift in energy production from aerobic glycolysis to TCA cycle-dependent OXPHOS. To examine the OXPHOS dependence of each condition, cells were treated either with the electron transport chain complex III inhibitor, antimycin, or the complex V (ATPase) inhibitor, oligomycin. Detached cells were more sensitive to PARP cleavage by these inhibitors (Fig. 1F), indicating an increased reliance on ATP generation by glutamine-based mitochondrial OXPHOS. Consistent with these results, treatment with either 2DG [25] or 3PO [26], two glycolytic inhibitors, dramatically induced PARP cleavage under attached conditions (Fig. 1G). Similarly, PARP cleavage was stimulated upon addition of the glutamine transport inhibitor, GPNA, or glutamine metabolism inhibitor, BPTES [27], under detached conditions in HepG2, HeLa, and HT1080 cells (Fig. 1G). No dependence on fatty acid oxidation was observed when detached cells were cultured (Fig. S1B). In addition, the plasma glutamine concentration of 0.6–0.9 mmol/L [28] was ideal for survival of detached cancer cells (Fig. S1C).
Fig. 1.

ECM detachment causes a switch from glucose-to glutamine-dependent survival mechanisms. (A) Intracellular ATP levels in HepG2 cells 24 h after plating in adherent (AT) or non-adherent (poly-HEMA-coated) (DT) plates. (B–D) Changes in glucose, lactate, and glutamine concentration in the medium of HepG2 cells after 24 h under attached or detached conditions. Data are presented as mean ± SD of three independent experiments. Full-length PARP (F-PARP) and cleaved PARP (C-PARP) expression in protein extracts obtained from HepG2 cells incubated in the presence of (E) 50 μM UK5099 and (F) 1 μM antimycin A1 or oligomycin, or in their absence (Cont) for 24 h under attached or detached conditions. (G) F-PARP and C-PARP expression in protein extracts obtained from three different cancer cell lines (HepG2, HeLa, and HT1080) incubated with 25 mM 2DG, 20 μM 3PO, 1 mM GPNA, or 20 μM BPTES, or without these chemicals (Cont) for 24 h under attached or detached conditions. α-tubulin served as a loading control. Representative western blots are shown. *P < 0.05; **P < 0.01.
Glutamine metabolism plays a role not only in ATP production but also in suppressing oxidative stress and supports survival upon ECM detachment.
Adherent or non-adherent cancer cells were found to be dependent on glucose or glutamine to survive. As expected, removing glucose or glutamine resulted in a more dramatic reduction in intracellular ATP levels under adherent or non-adherent conditions, respectively, in all cell lines and compared to complete nutrient medium (Fig. 2A). Similar results were obtained in Western blot analysis for the expression of H2AX phosphorylated on Ser139, a marker of DNA damage, and PARP cleavage (Fig. 2B). Reflecting these findings, cell viability was markedly reduced in both glucose deficiency under attached conditions and in glutamine deficiency under detached conditions (Fig. 2C). To elucidate the nutritional requirements for cell survival in more detail, we investigated ATP levels and cell death during attached or detached conditions using a medium without specific nutrients, or nutrient-free medium supplemented with those nutrients (Fig. 2D and E). Under attached conditions, glucose supplementation was sufficient for ATP production and survival. Glutamine deficiency or glutamine supplementation had the greatest effect on ATP levels under detached conditions. As with media devoid of all nutrients, glutamine deficiency also caused cell death, suggesting that glutamine utilization plays a crucial role in ATP production and survival of detached cancer cells. Surprisingly, whereas glutamine addition to the medium lacking all nutrients was a prerequisite for ATP production, it could not completely rescue PARP cleavage, indicating that glutamine is responsible for cancer cell survival in coordination with other nutrients.
Fig. 2.


Glutamine is indispensable for cancer cell survival under ECM detachment. (A) ATP levels in HepG2, HeLa, and HT1080 cells cultured in complete medium, glucose-deficient (Glc (-)), glutamine-deficient (Gln (-)), or glucose and glutamine-deficient (Neither) medium for 24 h under attached (AT) or detached (DT) conditions. (B) Full-length PARP (F-PARP) and cleaved PARP (C-PARP) expression in HepG2 cells cultured as indicated in panel A for 24 h. β-actin served as a loading control. (C) Viability of HepG2 cells cultured for 24 h in the indicated medium on adherent or poly-HEMA-coated plates. (D) ATP levels in HepG2 cells cultured in the indicated nutrient-deficient or supplemented medium on attachment or detachment plates for 24 h. Complete: DMEM-based complete nutrient medium; AA: DMEM-amino acid; Vit: DMEM-vitamin; All deficient: deficient for all nutrients. (E) F-PARP and C-PARP expression in HepG2 cells cultured in the indicated nutrient-deficient or supplemented medium on attachment or detachment plates for 24 h. (F) F-PARP or C-PARP expression in HepG2 cells cultured in complete medium or in all nutrients-free medium supplemented with 2 mM glutamine and the indicated doses of cystine for 24 h under detached conditions. α-tubulin served as a loading control. (G) ROS (H2O2) and (H) ATP levels in HepG2 cells cultured in complete medium or all nutrients-free medium supplemented with 2 mM glutamine and the indicated dose of cystine for 24 h in detached conditions. (I) H2AX phosphorylation (p-H2AX) and PARP expression in whole-cell extracts of HepG2 cells incubated with or without 1 mM BSO for 24 h under attached or detached conditions. β-actin served as a loading control. (J) ATP levels, (K) ROS levels, (L) H2AX phosphorylation and PARP expression in HepG2 cells cultured in glutamine-free medium in the presence (Cont) or absence (-) of glutamine, with or without 4 mM DM-αKG under detached conditions. β-actin served as a loading control. Representative western blots are shown. Data are presented as mean ± SD of three independent sets of experiments. NS, not significant; **P < 0.01; ***P < 0.001.
Products of glutamine metabolism are essential in promoting cellular anti-oxidative defenses [13]. Glutamine-derived glutamate contributes to glutathione biosynthesis and is thought to facilitate the uptake of cystine via the xCT antiporter [29], which is coupled to the efflux of glutamate. Cystine is converted to cysteine, which can then be incorporated into glutathione [29,30]. Thus, we hypothesized that glutamine and cystine reduced excess intracellular ROS generated by ECM detachment. As expected, PARP cleavage was almost completely rescued by supplementation with both glutamine and cystine under detached conditions (Fig. 2F), but not by addition of cystine alone or cystine with glucose (Figs. S2A and B). Consistent with these results, supplementation with glutamine and cystine suppressed ROS to about the same level as in complete nutrient medium (Fig. 2G); however, it failed to restore ATP levels (Fig. 2H) even in the presence of glucose (Fig. S2C). Treatment with BSO, a glutamyl cysteine synthetase inhibitor, inhibited glutathione synthesis and induced PARP cleavage under detached conditions (Fig. 2I). In addition, non-tumorigenic HaCaT cells showed a marked decrease in ATP and cleavage of PARP under non-adhesive conditions (Figs. S2D and E). Treatment with the antioxidants, NAC and ascorbic acid, completely inhibited the cleavage of PARP and the generation of excess ROS, but could not rescue ATP levels (Figs. S2G and F). These results suggest that, under detached conditions, glutamine played a central role not only in ATP production but also in the suppression of oxidative stress through glutathione synthesis and cystine consumption. Supporting this hypothesis, addition of cell-permeable DM-αKG, a source of fuel for the TCA cycle or precursor of glutamate [13], was sufficient to produce ATP and to reduce excess ROS during glutamine deprivation under detached conditions, leading to complete protection against DNA damage and cell death (Fig. 2J, K, L).
AMPK-Nrf2 regulates glutamine metabolism-mediated energy and redox homeostasis in detached cancer cells.
Adenosine monophosphate-activated kinase (AMPK) is a central regulator of cellular responses to metabolic stress [31] and is involved in cancer cell survival under glucose-limiting conditions and anchorage-independent growth [32,33]. We previously reported that glucose starvation induced AMPK activation [20]. In the current study, we hypothesized that reduced glucose consumption under non-adhesion conditions (Fig. 1B) implicated AMPK activation in cancer cells survival. Indeed, AMPK knockdown resulted in a marked induction of phosphorylated H2AX and PARP cleavage following ECM detachment (Fig. 3A). Detached AMPK knockdown cells exhibited decreased glutamine consumption, obvious suppression of the induced glutamine transporter, ASCT2 [34] (Fig. 3B and C), and lower ATP levels (Fig. 3D).
Fig. 3.

AMPK is involved in glutamine metabolism upon ECM detachment. HepG2 cells stably expressing control-shRNA (sh-con) or AMPKα1-shRNA (sh-AMPK) were cultured in attachment (AT) or detachment (DT) dishes for 24 h. (A) Full-length PARP (F-PARP) and cleaved PARP (C-PARP), phosphorylated H2AX (p-H2AX) expression, and (C) ASCT2 expression in protein extracts of the above cells. β-actin served as a loading control. (B) Glutamine in culture medium and (D) intracellular ATP levels in the above cells. Representative western blots are shown. The expression levels of p-H2AX, C-PARP, and ASCT2 were measured by densitometric analysis. Data are presented as mean ± SD of three independent sets of experiments. NS, not significant; *P < 0.05; **P < 0.01; ***P < 0.001.
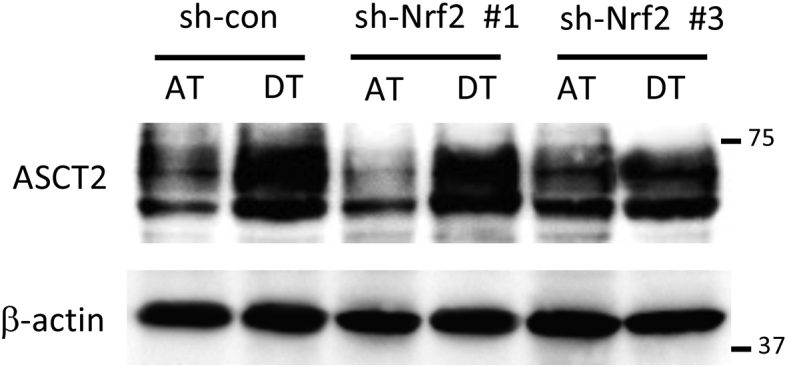
We previously reported that AMPK activation induced the expression of nuclear factor E2-related factor 2 (Nrf2), a master regulator of redox homeostasis, in cancer cells under glucose-starved conditions [20]. Here, Nrf2 expression induced by cell detachment was inhibited by sh-AMPK treatment (Fig. 4A). Given that Nrf2 contributes to cell survival by suppressing oxidative stress [35,36], Nrf2 knockdown made cells more sensitive to ECM detachment-induced cell damage, leading to markedly higher H2AX phosphorylation and PARP cleavage (Fig. 4B). To investigate the role of AMPK and Nrf2 in signal transduction in more detail, we examined detached cells treated with sh-AMPK following transient transfection with EGFP-tagged-Nrf2. H2AX phosphorylation and PARP cleavage were completely rescued by Nrf2 overexpression (Fig. 4C), indicating that AMPK activation plays a critical role in Nrf2-mediated protection from metabolic stress incurred following ECM detachment. Nrf2 is the sole controller of GCLC and GCLM, the enzymes responsible for producing glutathione [37]. Nrf2 can also modulate energy metabolism by directly increasing the transcription of all NADPH-generating enzymes, which links Nrf2 to NADPH production and subsequent glutathione maintenance [38,39]. In addition, Nrf2 controls the abundance of cysteine within cells, which is the rate-limiting substrate in glutathione synthesis via Nrf2-mediated expression of the cystine/glutamate antiporter, xCT [40]. Here, sh-Nrf2 suppressed the induction of Nrf2-targeted NADPH-producing enzymes, ME1 and IDH1 [38,39], as well as glutathione synthesis-related enzymes, GCLC, GCLM, and xCT, under detached conditions (Fig. 4D). Knockdown of AMPK also suppressed those Nrf2-targeted enzymes induced by ECM deprivation, confirming that Nrf2 functioned downstream of AMPK (Fig. 4E). Unexpectedly, detachment-induced ASCT2 expression was not affected by Nrf2 inhibition, suggesting that AMPK was responsible for glutamine uptake, whereas Nrf2 regulated glutamine metabolism (Fig. S3). Consequently, the substantial ROS generation induced by ECM detachment was further exacerbated by AMPK or Nrf2 knockdown (Fig. 4F).
Fig. 4.


Glutamine metabolism-mediated cancer cell survival is regulated by AMPK-Nrf2 under ECM detachment. (A, B, D, E) HepG2 cells expressing sh-con or sh-AMPK were cultured in attachment (AT) or poly-HEMA-coated (DT) dishes for 24 h. Western blot analysis was performed on protein extracts of these cells with antibodies against the indicated proteins, with either β-actin or lamin B1 as loading controls. (A, D) The asterisk indicates a nonspecific band. The expression levels of protein were measured by densitometric analysis. (C) Cells stably expressing sh-con were transfected with an empty vector. Cells stably expressing sh-AMPK were transfected with either an empty vector or the EGFP-Nrf2 expression vector. Western blot analysis was performed on protein extracts of these cell lines with antibodies against the indicated proteins after culturing in adherent or poly-HEMA-coated dishes for 24 h. Representative image of western blots are shown. (F) ROS levels in HepG2 cells stably expressing sh-con, sh-AMPK, or sh-Nrf2 for 24 h after plating in adherent or poly-HEMA-coated pates for 24 h. Data are presented as mean ± SD of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
Suppression of integrin causes a shift to glutamine metabolism even in an adhesive environment.
Anoikis is caused by loss of interaction between adherent cells and the ECM and is triggered by the absence of certain adhesion molecules [8]. Disruption of integrin ligation has been shown to promote cell death by anoikis, which can be prevented by integrin-mediated adhesion [[41], [42], [43]]. To elucidate the mechanism by which loss of adhesion molecules causes metabolic reprogramming, we treated HepG2 cells with the integrin inhibitor, cilengitide [44], under adherent monolayer culture conditions. As expected, dose-dependent treatment with cilengitide resulted in cells whose morphology resembled that of cells cultured in a poly-HEMA-coated culture dish even if a normal culture dish for adherent cells was used (Fig. 5A). ASCT2, xCT, nuclear Nrf2, and phosphorylation of AMPK were substantially induced under attached conditions in the presence of cilengitide. Phosphorylation of FAK, an established marker of integrin-ECM interaction [18], decreased with increasing exposure to cilengitide (Fig. 5B). To investigate whether the integrin signal triggered a metabolic shift associated with changes in nutrient demand, we examined the effect of cilengitide treatment under depletion of glucose, glutamine, or both. In the presence of cilengitide, glucose deprivation-induced PARP cleavage was suppressed under attached conditions, whereas glutamine-starved PARP cleavage was dramatically induced (Fig. 5C). Reduction in ATP levels by cilengitide treatment was greater with glutamine starvation than with glucose starvation (Fig. 5D), thus, mimicking the results shown in Fig. 2A. In addition, supplementation of DM-αKG successfully rescued glutamine starvation-induced PARP cleavage in the presence of cilengitide under adhesion culture conditions (Fig. 5E). Intracellular ATP levels were hardly altered by the addition of cilengitide alone, but were markedly lower when combining cilengitide and glutamine deficiency (Fig. 5F). Hence, the cooperative function of AMPK and Nrf2 seemed to play an essential role in cancer cell survival through glutamine metabolism in a non-adhesive environment. In AMPK- or Nrf2-knockdown cells, treatment with cilengitide significantly reduced intracellular ATP, which was further decreased by glutamine starvation (Fig. 5G and H). Consistent with these results, cilengitide treatment alone caused only minor PARP cleavage, whereas its combination with glutamine deficiency led to pronounced PARP cleavage in sh-con cells (Fig. 5I). Notably, PARP was cleaved in sh-AMPK and sh-Nrf2 cells even in the absence of cilengitide (Fig. 5I). These results clearly suggest that inhibition of integrin by cilengitide was the most important trigger for glutaminolysis due to changes in nutritional requirements. Considering that this occurred under attached conditions, substantial ROS generation was induced by treatment with small-molecule glutaminase inhibitors, BPTES and CB839 [27] (Fig. 5J), further exacerbating PARP cleavage (Fig. 5K). Taken together, these results reveal that metabolic reprogramming was caused by the synergistic action of integrin suppression-triggered AMPK activation and Nrf2 expression in cancer cells, thereby favoring adaptation and survival under the severe conditions elicited by ECM detachment.
Fig. 5.

Loss of integrin regulates glutaminolysis in cancer cells. (A) Images obtained by phase-contrast microscopy of HepG2 cells cultured in the presence of different cilengitide (Cil) doses for 24 h in adherent culture dishes. Control cells were cultured using a poly-HEMA-coated dish without cilengitide treatment. Scale bar = 100 μm. (B) Western blot analysis of HepG2 cells treated with various doses (0–20 μM) of cilengitide for 24 h under attached conditions (unless otherwise indicated) and probed with antibodies against the indicated proteins, with either α-tubulin or lamin B1 as loading controls. (C) Full-length PARP (F-PARP) and cleaved PARP (C-PARP) expression in whole-cell lysates of HepG2 cells cultured in complete medium, glucose-free medium (Glc (-)), glutamine-free medium (Gln (-)), glucose- and glutamine-free medium (Neither), and the latter supplemented with either glucose (Glc (+)) or glutamine (Gln (+)) in the absence or presence of 20 μM cilengitide for 24 h. α-tubulin served as loading control. (D) ATP levels in HepG2 cells cultured in complete medium, glucose-deficient (Glc (-)), glutamine-deficient (Gln (-)), or glucose- and glutamine-deficient (Neither) medium in the presence of 20 μM cilengitide for 24 h. (E) F-PARP and C-PARP expression in whole-cell lysates of HepG2 cells cultured in glutamine-free medium (-) or control medium (+) for 24 h in the presence or absence of 20 μM cilengitide and 4 mM DM-αKG. α-tubulin served as loading control. (F–H) ATP levels and (I) F-PARP and C-PARP expression in HepG2 cells stably expressing sh-con, sh-AMPK, or sh-Nrf2 incubated in vehicle control medium (Cont) or glutamine-free medium (-) supplemented with 20 μM cilengitide for 24 h. β-actin served as a loading control. (J) ROS levels and (K) F-PARP and C-PARP expression in HepG2 cells treated or not with 20 μM BPTES or 10 μM CB839 in the presence of 20 μM cilengitide or in vehicle control medium (Cont) for 24 h. α-tubulin served as a loading control. Representative western blots are shown. Data are presented as mean ± SD of three independent experiments. *P < 0.05; **P < 0.01.
The combination of xCT inhibitor, sulfasalazine, and CB839 synergistically damages cancer cells under detached conditions.
Based on the present results, we hypothesized that ECM detachment-induced glutamine metabolism addiction was an essential mechanism for suppression of oxidative stress and this was necessary for anoikis resistance exhibited by cancer cells. As the expression of xCT is required for maintaining glutaminolysis-mediated redox balance [29,30], a marked induction of xCT expression was observed in HepG2, HeLa, and HT1080 cells under detached conditions (Fig. 6A). In HaCaT cells, xCT expression was not induced during ECM detachment and the transport of glutamate and cystine was likely inadequate; thereby PARP cleavage could not be rescued even when DMα-KG was added (Fig. S4A). Induction of AMPK activation and nuclear Nrf2 expression were also not detected in HaCaT cells (Fig. S4B). To further confirm the significance of xCT expression in cancer cell survival under non-adherent conditions, we examined the effect of treatment with sulfasalazine, a specific inhibitor of xCT-mediated cystine transport [45], on cell damage. Intriguingly, induction of PARP cleavage and H2AX phosphorylation by sulfasalazine treatment was more evident under detached than adherent conditions (Fig. 6B). In addition, effective and synergistic cell damage was observed only during detached conditions when CB839 and sulfasalazine were utilized alone or together at a concentration that had little effect in attached cells (Fig. 6C). Consequently, whether alone or in combination, these inhibitors caused a significant increase in ROS levels during detached conditions (Fig. 6D). From these results, it appears that xCT expression cooperated with glutaminolysis to ensure cell survival under detached conditions via oxidative stress neutralization and a predominantly glutamine-oriented metabolism.
Fig. 6.

A combination of sulfasalazine and CB839 causes cell damage by increasing oxidative stress in cancer cells following ECM detachment. (A) xCT expression in HepG2, HeLa, HT1080, and HaCaT cells cultured for 24 h under attached (AT) or detached (DT) conditions. β-actin served as loading control. The expression levels of xCT were measured by densitometric analysis. (B) Full-length PARP (F-PARP), cleaved PARP (C-PARP), and phosphorylated H2AX (p-H2AX) expression in HepG2 cells treated with various doses (0–500 μM) of sulfasalazine (SAS) for 24 h under attached or detached conditions. α-tubulin served as a loading control. (C) F-PARP, C-PARP, and p-H2AX expression and (D) ROS levels in HepG2 cells treated with either 10 μM CB839 or 250 μM sulfasalazine, or both. α-tubulin served as a loading control. Representative western blots are shown. Data are presented as mean ± SD of three independent experiments. *P < 0.05; **P < 0.01.
Dependence on glutamine metabolism in highly metastatic cancer cells under detachment conditions contributes to therapeutic vulnerability.
To investigate the significance of glutamine metabolism on factors other than ATP production or oxidative stress neutralization during cancer cell survival under detached conditions, we focused on the cells’ ability to metastasize. We examined two existing melanoma cell lines whose parental cells had been established by in vivo selection for more metastatic variants: poorly metastatic A375-P cells and their more metastatic A375-MA2 variant [46]. In A375-P cells, glutamine starvation caused more PARP cleavage and H2AX phosphorylation compared to glucose starvation under both attached and detached conditions (Fig. 7A). In contrast, in A375-MA2 cells, both glucose or glutamine starvation caused damage under adherent conditions; however, pronounced PARP cleavage and H2AX phosphorylation were enhanced only during glutamine starvation under detached conditions (Fig. 7A). Based on these findings, we hypothesized that combined treatment with inhibitors of glutamine metabolism and cystine transport could hinder survival of A375-MA2 cells under non-adhesion conditions due to a greater dependence on glutamine metabolism. As expected, whereas damage to A375-MA2 cells was caused by exposure to CB839, but not sulfasalazine, in attached cells, a combination of these inhibitors displayed a synergistic damaging effect in detached cells (Fig. 7B).
Fig. 7.

Cancer cells with high metastatic potential have an increased dependence on glutamine metabolism under detached conditions. (A, C) Full-length PARP (F-PARP), cleaved PARP (C-PARP), and phosphorylated H2AX (p-H2AX) expression in whole-cell lysates of A375P, A375-MA2, MCF-7, and MDA-MB-231 cells cultured in complete medium, glucose-deficient (Glc (-)), glutamine-deficient (Gln (-)), or glucose and glutamine-deficient (Neither) medium for 24 h under attached (AT) or detached (DT) conditions. (B, D) F-PARP, C-PARP, and p-H2AX expression in whole-cell lysates of HepG2 cells treated with either 10 μM CB839 or 250 μM sulfasalazine, or both for 24 h. α-tubulin served as loading control. Data are representative of three independent sets of experiments.
Finally, we investigated ECM deprivation-induced metabolic changes using breast cancer-derived cell lines of different grades and molecular subtypes. The luminal ER+, HER2- MCF-7 cell line [47] was vulnerable to both glucose or glutamine deficiency under attached conditions, but was more susceptible during glutamine starvation under detached conditions. In the basal-like triple-negative MDA-MB-231 cell line [47], PARP cleavage changed dramatically upon switching from glucose-to glutamine-dependent metabolism, indicating that a change from adherent to non-adherent cultures had a strong impact on cell survival (Fig. 7C). Although treatment with CB839 had a minor effect on H2AX phosphorylation, effective induction of H2AX phosphorylation and PARP cleavage by combined use of sulfasalazine could not be observed under attached conditions, suggesting that glutamine was not involved in redox homeostasis and thus did not cause cell death in adherent MDA-MB-231 cells. Importantly, similar to the results obtained with A375 cells, exposure of MDA-MB-231 cells to CB839 and sulfasalazine caused remarkable PARP cleavage, and their combination exerted a synergistic effect under detached conditions (Fig. 7D). It was unsurprising that activation of AMPK, as well as induction of nuclear Nrf2 and xCT, were caused by non-adherent cultures. These changes were consistent with switching to glutamine-dependent metabolism following detachment, and they were more clearly observed in aggressive cancer cell lines, such as A375-MA2 or MDA-MB-231 (Figs. S5A and B). Taken together, our observations strongly indicate that glutamine demand and its metabolism are not only indispensable adaptation mechanisms for survival in a non-adherent environment, but may also play an important role in acquisition of metastasizing capacity during cancer progression.
4. Discussion
Escape from anoikis, particularly during the complex cascade of invasion and metastasis, has long been recognized as a hallmark of cancer [[2], [3], [4]]. During the spread and circulation of metastatic cancer cells, avoiding the severe environmental stress caused by separation from the ECM is an essential prerequisite for successful metastasis [5,6,8]. To this end, anti-anoikis, or anchorage-independent survival, is one of the most important adaptations. However, nutrient demand and related mechanisms for sustained survival of cancer cells following ECM detachment have not been sufficiently elucidated.
Here, we show that cancer cells survive under detached conditions by switching from glucose-to glutamine-dependent energy metabolism and, therefore, do not depend on the Warburg effect. We report that glutamine serves to generate ATP, as well as counteract oxidative stress by acting in concert with cystine. Detachment-induced AMPK activation increases glutamine consumption and induces Nrf2, as well as its target enzymes, to maintain ATP levels and ROS homeostasis. Detachment was explained by the suppression of integrin, as activation of the AMPK-Nrf2 signal, changes in nutrient demand, and oxidative stress response were almost completely mimicked by integrin inhibitors. Therefore, the AMPK-Nrf2 signal could serve as a base for metabolic reprogramming in cells under severe anchorage-deficient stress. Our findings could also explain why inhibition of excess oxidative stress can cause the development of metastatic capacity in ECM-deprived tumor cells.
Because some metabolic features are altered across many types of cancer cells, metabolic reprogramming is considered a hallmark of cancer [10,11]. These metabolic alterations play a prominent role in the acquisition and maintenance of malignant properties [[48], [49], [50]]. Although the enhanced glycolytic metabolism of the Warburg effect is considered essential for cell growth and proliferation, aggressive growth is suppressed by loss of attachment to the ECM in non-adherent cultures and tumor spheres [51,52]. Accordingly, glucose is expected to be less important for survival following detachment. In our study, we show that cancer cells favor glutamine over glucose under detached conditions. Indeed, ECM detachment is now well-understood to cause some drastic metabolic alterations, including defective glucose uptake, diminished pentose phosphate pathway, reduced cellular ATP levels, and a substantial increase in ROS in the non-tumorigenic MCF-10A cell line, which is restored by overexpression of the oncogene, ErbB2 [[53], [54], [55]]. Recent studies have revealed that cancerous metabolic reprogramming is induced by aberrant constitutive activation of various oncogenes, transcription factors, and signaling pathways, such as c-Myc, p53, HIF1α, PI3K/AKT, and mTOR [[10], [11], [12]]. However, because cell lines with different genetic backgrounds and tissues of origin were used in our experiments, we believe that metabolic reprogramming from glycolysis-to glutaminolysis-based survival mechanisms is regulated by distinct molecules and/or signaling pathways during ECM detachment.
AMPK is a key energy sensor in cancerous and non-cancerous cells [31]. During nutrient deprivation, including glucose deprivation, AMPK is activated by decreased intracellular ATP levels. Because ECM detachment impairs the uptake of glucose as the preferred nutrient, it is possible that AMPK prevents ATP depletion by metabolic reprogramming. Recently, Blagih et al. (2015) reported that AMPK played a crucial role in metabolic reprogramming of effector T cells in response to glucose insufficiency by enhancing glutaminolysis [56]. Here, we report that expression of glutamine transporter, ASCT2, was induced by AMPK activation under detached conditions. In fact, the most important role played by glutamine metabolism was to reduce oxidative stress by promoting cystine uptake. When glucose metabolism is impaired in glioblastoma cells, the activity of glutamate dehydrogenase is increased to enable energy production from glutamine via the TCA cycle [57]. Thus, in a reciprocal manner, glutamine metabolism in tumor cells may influence glucose metabolism. At present, the mutual regulation between these two metabolic modes under ECM detachment remains to be elucidated. Our findings indicate that AMPK-mediated glutamine metabolism represents a reasonable strategy in a non-adherent environment that prioritizes cancer cell survival over proliferation when initiating metastasis.
Integrin-mediated signaling plays a central role in suppressing apoptosis in adherent cells; loss of adhesion, and consequent suppression of its downstream FAK signal, has been implicated as a major cause of anoikis [6,58]. In pancreatic cancer cells, FAK promotes glucose consumption and glycolysis while decreasing mitochondrial respiration. However, detachment-induced suppression of FAK reverts the balance from glycolysis to OXPHOS [59], suggesting that suppression of integrin-FAK adhesion signaling causes a shift to glutaminolysis to avoid cell death elicited by disruption of the glucose-derived survival mechanism. Consistent with this hypothesis, our results clearly show that the integrin inhibitor causes glutaminolysis to prevent energy loss due to suppressed glycolysis and to protect cells from oxidative stress through masterfully orchestrated AMPK-Nrf2 activation.
The ability to cope with oxidative stress may be essential in some cancer types, as inhibition of folate pathway-mediated anti-oxidative defenses decreases the metastatic potential of melanoma cells [16], while treatment with antioxidants promotes metastasis [[15], [16], [17]]. In addition, detached cultured tumor cells increase their antioxidant capacity by producing more NADPH [53], which is the main reducing agent used in regenerating oxidized glutathione and thioredoxin, two known ROS scavengers. Intriguingly, our results indicate that NADPH-generating enzymes. ME1 and IDH1, as well as glutathione-producing enzymes, GCLC and GCLC, are targeted by Nrf2 and are induced following ECM detachment. Although NADPH is also required for lipid synthesis [60], AMPK inhibits fatty acid synthesis-mediated NADPH consumption and promotes fatty acid oxidation-related NADPH production instead [31,61]. Thus, activation of AMPK is pivotal for a cancer cell to sense unfavorable conditions, including ECM detachment, and to respond by activating a catabolic switch that augments ATP and NADPH reserves. Although the mechanism of AMPK-mediated NADPH production or its utilization as reducing equivalent to counteract oxidative stress remains unknown, we reveal that AMPK-regulated Nrf2 expression promotes glutamine uptake and glutaminolysis, thereby providing sufficient energy and suppressing oxidative stress following ECM detachment. Importantly, our findings also indicate that AMPK-Nrf2 signaling is involved in induction of xCT expression and is responsible for the proper control of oxidative stress under detached conditions. Based on these results, glutamate produced by AMPK-driven glutamine metabolism is necessary to effectively reduce xCT-mediated oxidative stress, and its induction of xCT expression and downstream glutathione synthesis are required for AMPK-dependent Nrf2 expression. Thus, the AMPK-Nrf2 axis offers a survival strategy based on altered nutrient demand and metabolic rewiring to glutaminolysis. Even though our findings suggest that AMPK and Nrf2 may be effective therapeutic targets in cancer cells undergoing ECM detachment, inhibiting these molecules may have adverse effects as they are critical for survival of non-tumor cells, as well as the regulation of diverse molecules [62,63]. Instead, a combination of CB839 and sulfasalazine, which inhibits both glutamine metabolism and glutaminolysis-associated cystine transporter activity, is expected to be a useful treatment because it can damage cancer cells during ECM detachment.
Both glucose and glutamine are key metabolic substrates in malignant tumor cells and are critical for cancer progression. Although glutamine requirement of invasive and metastatic cells has not been specifically studied, it has been shown that highly invasive ovarian cancer cells exhibit increased glutamine dependence compared with less invasive cells, whereas metastatic prostate tumors show increased glutamate availability and dependence on glutamine uptake [64]. Furthermore, genetic inhibition of glutaminase has been shown to inhibit epithelial-to-mesenchymal transition, a key step in tumor cell invasiveness and eventual metastasis [65,66]. Indeed, we have previously shown that glucose starvation, which enhances glutamine requirement, causes cancer cell migration and invasiveness via induction of MMP-9 expression [20]. In the present study, we reveal that suppression of anoikis relies on elevated glutamine requirement, which is even greater in highly malignant cancer cells, such as A375-MA2 and MDA-MB-231, irrespective of tissue origin and genetic backgrounds. Thus, combinatorial strategies aimed at inhibiting glutamine metabolism and collapsing redox homeostasis could prevent malignant progression and offer important intervention strategies.
Although metabolic reprogramming in cancer cells is now widely recognized to be important for cancer treatment, strategies that target the enhanced glycolytic activity in cancer have, so far, not been very successful clinically [25]. In contrast, several small-molecule glutaminase inhibitors, including BPTES and CB839, are currently being evaluated in pre-clinical and clinical trials for the treatment of various cancers [67]. Sulfasalazine, an FDA-approved therapeutic agent, is already widely utilized because it is applied against rheumatoid arthritis and ulcerative colitis [40,68]. Restriction of glutamine and cystine would not be easy to achieve through dietary intervention in patients. Therefore, the combination therapy shown here represents an option for treating cancer progression during metastasis.
In conclusion, the present study provides new evidence of ECM detachment-induced glutamine metabolism in sustaining cancer cell survival through comprehensive activation of the AMPK-Nrf2 axis. This knowledge broadens our understanding of the mechanisms underlying metastatic processes that may be common to aggressive cancers. We believe that our findings have potential in the development of therapeutic strategies targeting nutrient auxotrophy in various cancer types.
Declaration of competing interest
The authors declare no conflict of interest.
Acknowledgements
We would like to thank Editage (www.editage.com) for English language editing.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101643.
Contributor Information
Hitoshi Endo, Email: h-endo@tokai-u.jp.
Satoshi Owada, Email: sowada@tsc.u-tokai.ac.jp.
Yutaka Inagaki, Email: yutakai@is.icc.u-tokai.ac.jp.
Yukari Shida, Email: 8bmud013@mail.u-tokai.ac.jp.
Masayuki Tatemichi, Email: tatemichi@tokai-u.jp.
Funding
This work was supported in part by 2017 Tokai University School of Medicine Research Aid (to H.E.), Research and Study Program of Tokai University Educational System General Research Organization (to H.E.), 2019 Tokai University Supporters Association Research and Study Grant (to H.E.), and Grant-in-Aid for Scientific Research (26870600 to H.E.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Author contributions
H.E., S.O., Y.I., Y.S., and M.T. conceived and designed the experiments. H.E. and S.O. performed the experiments. H.E., S.O., and Y.I. analyzed the data. H.E., S.O., and Y.S. contributed reagents/materials/analysis tools. H.E. wrote the paper and all authors reviewed and approved the final manuscript.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
figs1.

figs2.

figs3.
figs4.

figs5.

References
- 1.Steeg P.S. Tumor metastasis: mechanistic insights and clinical challenges. Nat. Med. 2006;12:895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D., Weinberg R.A. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Sethi N., Kang Y. Unravelling the complexity of metastasis - molecular understanding and targeted therapies. Nat. Rev. Canc. 2011;11:735–748. doi: 10.1038/nrc3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wirtz D., Konstantopoulos K., Searson P.C. The physics of cancer: the role of physical interactions and mechanical forces in metastasis. Nat. Rev. Canc. 2011;11:512–522. doi: 10.1038/nrc3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guadamillas M.C., Cerezo A., Del Pozo M.A. Overcoming anoikis--pathways to anchorage-independent growth in cancer. J. Cell Sci. 2011;124:3189–3197. doi: 10.1242/jcs.072165. [DOI] [PubMed] [Google Scholar]
- 6.Paoli P., Giannoni E., Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta. 2013;1833:3481–3498. doi: 10.1016/j.bbamcr.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 7.Hawk M.A., Schafer Z.T. Mechanisms of redox metabolism and cancer cell survival during extracellular matrix detachment. J. Biol. Chem. 2018;293:7531–7537. doi: 10.1074/jbc.TM117.000260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buchheit C.L., Weigel K.J., Schafer Z.T. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat. Rev. Canc. 2014;14:632–641. doi: 10.1038/nrc3789. [DOI] [PubMed] [Google Scholar]
- 9.Mason J.A., Hagel K.R., Hawk M.A., Schafer Z.T. Metabolism during ECM detachment: achilles heel of cancer cells? Trends Canc. 2017;3:475–481. doi: 10.1016/j.trecan.2017.04.009. [DOI] [PubMed] [Google Scholar]
- 10.DeBerardinis R.J., Chandel N.S. Fundamentals of cancer metabolism. Sci Adv. 2016;2 doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ward P.S., Thompson C.B. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Canc. Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cairns R.A., Harris I.S., Mak T.W. Regulation of cancer cell metabolism. Nat. Rev. Canc. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J., Pavlova N.N., Thompson C.B. Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. EMBO J. 2017;36:1302–1315. doi: 10.15252/embj.201696151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeBerardinis R.J., Cheng T. Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sayin V.I., Ibrahim M.X., Larsson E., Nilsson J.A., Lindahl P., Bergo M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014;6 doi: 10.1126/scitranslmed.3007653. 221ra215. [DOI] [PubMed] [Google Scholar]
- 16.Piskounova E., Agathocleous M., Murphy M.M., Hu Z., Huddlestun S.E., Zhao Z., Leitch A.M., Johnson T.M., DeBerardinis R.J., Morrison S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–191. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H., Liu X., Long M., Huang Y., Zhang L., Zhang R., Zheng Y., Liao X., Wang Y., Liao Q., Li W., Tang Z., Tong Q., Wang X., Fang F., Rojo de la Vega M., Ouyang Q., Zhang D.D., Yu S., Zheng H. NRF2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci. Transl. Med. 2016;8 doi: 10.1126/scitranslmed.aad6095. 334ra351. [DOI] [PubMed] [Google Scholar]
- 18.Xu L.H., Yang X., Bradham C.A., Brenner D.A., Baldwin A.S., Jr., Craven R.J., Cance W.G. The focal adhesion kinase suppresses transformation-associated, anchorage-independent apoptosis in human breast cancer cells. Involvement of death receptor-related signaling pathways. J. Biol. Chem. 2000;275:30597–30604. doi: 10.1074/jbc.M910027199. [DOI] [PubMed] [Google Scholar]
- 19.Owada S., Ito K., Endo H., Shida Y., Okada C., Nezu T., Tatemichi M. An adaptation system to avoid apoptosis via autophagy under hypoxic conditions in pancreatic cancer cells. Anticancer Res. 2017;37:4927–4934. doi: 10.21873/anticanres.11902. [DOI] [PubMed] [Google Scholar]
- 20.Endo H., Owada S., Inagaki Y., Shida Y., Tatemichi M. Glucose starvation induces LKB1-AMPK-mediated MMP-9 expression in cancer cells. Sci. Rep. 2018;8 doi: 10.1038/s41598-018-28074-w. 10122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Endo H., Niioka M., Kobayashi N., Tanaka M., Watanabe T. Butyrate-producing probiotics reduce nonalcoholic fatty liver disease progression in rats: new insight into the probiotics for the gut-liver axis. PloS One. 2013;8 doi: 10.1371/journal.pone.0063388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Endo H., Niioka M., Sugioka Y., Itoh J., Kameyama K., Okazaki I., Ala-Aho R., Kahari V.M., Watanabe T. Matrix metalloproteinase-13 promotes recovery from experimental liver cirrhosis in rats. Pathobiology. 2011;78:239–252. doi: 10.1159/000328841. [DOI] [PubMed] [Google Scholar]
- 23.Hildyard J.C., Ammala C., Dukes I.D., Thomson S.A., Halestrap A.P. Identification and characterisation of a new class of highly specific and potent inhibitors of the mitochondrial pyruvate carrier. Biochim. Biophys. Acta. 2005;1707:221–230. doi: 10.1016/j.bbabio.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 24.Vacanti N.M., Divakaruni A.S., Green C.R., Parker S.J., Henry R.R., Ciaraldi T.P., Murphy A.N., Metallo C.M. Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol. Cell. 2014;56:425–435. doi: 10.1016/j.molcel.2014.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin X., Xiao Z., Chen T., Liang S.H., Guo H. Glucose metabolism on tumor plasticity, diagnosis, and treatment. Front Oncol. 2020;10 doi: 10.3389/fonc.2020.00317. 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Klarer A.C., O'Neal J., Imbert-Fernandez Y., Clem A., Ellis S.R., Clark J., Clem B., Chesney J., Telang S. Inhibition of 6-phosphofructo-2-kinase (PFKFB3) induces autophagy as a survival mechanism. Canc. Metabol. 2014;2 doi: 10.1186/2049-3002-2-2. 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin L., Alesi G.N., Kang S. Glutaminolysis as a target for cancer therapy. Oncogene. 2016;35:3619–3625. doi: 10.1038/onc.2015.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hensley C.T., Wasti A.T., DeBerardinis R.J. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J. Clin. Invest. 2013;123:3678–3684. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Timmerman L.A., Holton T., Yuneva M., Louie R.J., Padro M., Daemen A., Hu M., Chan D.A., Ethier S.P., van 't Veer L.J., Polyak K., McCormick F., Gray J.W. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Canc. Cell. 2013;24:450–465. doi: 10.1016/j.ccr.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Combs J.A., DeNicola G.M. The non-essential amino acid cysteine becomes essential for tumor proliferation and survival. Cancers. 2019;11 doi: 10.3390/cancers11050678. 678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hardie D.G., Ross F.A., Hawley S.A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones R.G., Plas D.R., Kubek S., Buzzai M., Mu J., Xu Y., Birnbaum M.J., Thompson C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 33.Ng T.L., Leprivier G., Robertson M.D., Chow C., Martin M.J., Laderoute K.R., Davicioni E., Triche T.J., Sorensen P.H. The AMPK stress response pathway mediates anoikis resistance through inhibition of mTOR and suppression of protein synthesis. Cell Death Differ. 2012;19:501–510. doi: 10.1038/cdd.2011.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scalise M., Pochini L., Console L., Losso M.A., Indiveri C. The human SLC1A5 (ASCT2) amino acid transporter: from function to structure and role in cell biology. Front Cell Dev Biol. 2018;6 doi: 10.3389/fcell.2018.00096. 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Taguchi K., Yamamoto M. The KEAP1-NRF2 system in cancer. Front Oncol. 2017;7 doi: 10.3389/fonc.2017.00085. 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okazaki K., Papagiannakopoulos T., Motohashi H. Metabolic features of cancer cells in NRF2 addiction status. Biophys Rev. 2020;12:435–441. doi: 10.1007/s12551-020-00659-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lacher S.E., Slattery M. Gene regulatory effects of disease-associated variation in the NRF2 network. Curr Opin Toxicol. 2016;1:71–79. doi: 10.1016/j.cotox.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitsuishi Y., Motohashi H., Yamamoto M. The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front Oncol. 2012;2 doi: 10.3389/fonc.2012.00200. 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mitsuishi Y., Taguchi K., Kawatani Y., Shibata T., Nukiwa T., Aburatani H., Yamamoto M., Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Canc. Cell. 2012;22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 40.Gorrini C., Harris I.S., Mak T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 41.Stupack D.G., Puente X.S., Boutsaboualoy S., Storgard C.M., Cheresh D.A. Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J. Cell Biol. 2001;155:459–470. doi: 10.1083/jcb.200106070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stupack D.G., Cheresh D.A. Get a ligand, get a life: integrins, signaling and cell survival. J. Cell Sci. 2002;115:3729–3738. doi: 10.1242/jcs.00071. [DOI] [PubMed] [Google Scholar]
- 43.Reddig P.J., Juliano R.L. Clinging to life: cell to matrix adhesion and cell survival. Canc. Metastasis Rev. 2005;24:425–439. doi: 10.1007/s10555-005-5134-3. [DOI] [PubMed] [Google Scholar]
- 44.Hatley R.J.D., Macdonald S.J.F., Slack R.J., Le J., Ludbrook S.B., Lukey P.T. An alphav-RGD integrin inhibitor toolbox: drug discovery insight, challenges and opportunities. Angew Chem. Int. Ed. Engl. 2018;57:3298–3321. doi: 10.1002/anie.201707948. [DOI] [PubMed] [Google Scholar]
- 45.Yoshida G.J. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J. Exp. Clin. Canc. Res. 2015;34 doi: 10.1186/s13046-015-0221-y. 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Clark E.A., Golub T.R., Lander E.S., Hynes R.O. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 47.Holliday D.L., Speirs V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011;13 doi: 10.1186/bcr2889. 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cantor J.R., Sabatini D.M. Cancer cell metabolism: one hallmark, many faces. Canc. Discov. 2012;2:881–898. doi: 10.1158/2159-8290.CD-12-0345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boroughs L.K., DeBerardinis R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015;17:351–359. doi: 10.1038/ncb3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pavlova N.N., Thompson C.B. The emerging hallmarks of cancer metabolism. Cell Metabol. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Saha M., Kumar S., Bukhari S., Balaji S.A., Kumar P., Hindupur S.K., Rangarajan A. AMPK-Akt double-negative feedback loop in breast cancer cells regulates their adaptation to matrix deprivation. Canc Res. 2018;78:1497–1510. doi: 10.1158/0008-5472.CAN-17-2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pereira P.M.R., Berisha N., Bhupathiraju N., Fernandes R., Tome J.P.C., Drain C.M. Cancer cell spheroids are a better screen for the photodynamic efficiency of glycosylated photosensitizers. PloS One. 2017;12 doi: 10.1371/journal.pone.0177737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schafer Z.T., Grassian A.R., Song L., Jiang Z., Gerhart-Hines Z., Irie H.Y., Gao S., Puigserver P., Brugge J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature. 2009;461:109–113. doi: 10.1038/nature08268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rayavarapu R.R., Heiden B., Pagani N., Shaw M.M., Shuff S., Zhang S., Schafer Z.T. The role of multicellular aggregation in the survival of ErbB2-positive breast cancer cells during extracellular matrix detachment. J. Biol. Chem. 2015;290:8722–8733. doi: 10.1074/jbc.M114.612754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khan I.A., Yoo B.H., Masson O., Baron S., Corkery D., Dellaire G., Attardi L.D., Rosen K.V. ErbB2-dependent downregulation of a pro-apoptotic protein Perp is required for oncogenic transformation of breast epithelial cells. Oncogene. 2016;35:5759–5769. doi: 10.1038/onc.2016.109. [DOI] [PubMed] [Google Scholar]
- 56.Blagih J., Coulombe F., Vincent E.E., Dupuy F., Galicia-Vazquez G., Yurchenko E., Raissi T.C., van der Windt G.J., Viollet B., Pearce E.L., Pelletier J., Piccirillo C.A., Krawczyk C.M., Divangahi M., Jones R.G. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. 2015;42:41–54. doi: 10.1016/j.immuni.2014.12.030. [DOI] [PubMed] [Google Scholar]
- 57.Yang C., Sudderth J., Dang T., Bachoo R.M., McDonald J.G., DeBerardinis R.J. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Canc Res. 2009;69:7986–7993. doi: 10.1158/0008-5472.CAN-09-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guan X. Cancer metastases: challenges and opportunities. Acta Pharm. Sin. B. 2015;5:402–418. doi: 10.1016/j.apsb.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang J., Gao Q., Zhou Y., Dier U., Hempel N., Hochwald S.N. Focal adhesion kinase-promoted tumor glucose metabolism is associated with a shift of mitochondrial respiration to glycolysis. Oncogene. 2016;35:1926–1942. doi: 10.1038/onc.2015.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carracedo A., Cantley L.C., Pandolfi P.P. Cancer metabolism: fatty acid oxidation in the limelight. Nat. Rev. Canc. 2013;13:227–232. doi: 10.1038/nrc3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jeon S.M., Chandel N.S., Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485:661–665. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mihaylova M.M., Shaw R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Qin J.J., Cheng X.D., Zhang J., Zhang W.D. Dual roles and therapeutic potential of Keap1-Nrf2 pathway in pancreatic cancer: a systematic review. Cell Commun. Signal. 2019;17 doi: 10.1186/s12964-019-0435-2. 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang L., Moss T., Mangala L.S., Marini J., Zhao H., Wahlig S., Armaiz-Pena G., Jiang D., Achreja A., Win J., Roopaimoole R., Rodriguez-Aguayo C., Mercado-Uribe I., Lopez-Berestein G., Liu J., Tsukamoto T., Sood A.K., Ram P.T., Nagrath D. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014;10 doi: 10.1002/msb.20134892. 728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiang L., Mou J., Shao B., Wei Y., Liang H., Takano N., Semenza G.L., Xie G. Glutaminase 1 expression in colorectal cancer cells is induced by hypoxia and required for tumor growth, invasion, and metastatic colonization. Cell Death Dis. 2019;10 doi: 10.1038/s41419-018-1291-5. 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee S.Y., Jeon H.M., Ju M.K., Jeong E.K., Kim C.H., Park H.G., Han S.I., Kang H.S. Dlx-2 and glutaminase upregulate epithelial-mesenchymal transition and glycolytic switch. Oncotarget. 2016;7:7925–7939. doi: 10.18632/oncotarget.6879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu X., Meng Y., Li L., Xu P., Wang J., Li Z., Bian J. Overview of the development of glutaminase inhibitors: achievements and future directions. J. Med. Chem. 2019;62:1096–1115. doi: 10.1021/acs.jmedchem.8b00961. [DOI] [PubMed] [Google Scholar]
- 68.Altman B.J., Stine Z.E., Dang C.V. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat. Rev. Canc. 2016;16:619–634. doi: 10.1038/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.