

Abstract
While chimeric antigen receptor (CAR) T cell immunotherapy targeting CD19 has shown remarkable success in patients with lymphoid malignancies, the potency of CAR T cells in solid tumors is low so far. To improve the efficacy of CAR T cells targeting prostate carcinoma, we designed a novel CAR that recognizes a new epitope in the prostate-specific membrane antigen (PSMA) and established novel paradigms to apply CAR T cells in a preclinical prostate cancer model. In vitro characterization of the D7 single-chain antibody fragment-derived anti-PSMA CAR confirmed that the choice of the co-stimulatory domain is a major determinant of CAR T cell activation, differentiation, and exhaustion. In vivo, focal injections of the PSMA CAR T cells eradicated established human prostate cancer xenografts in a preclinical mouse model. Moreover, systemic intravenous CAR T cell application significantly inhibited tumor growth in combination with non-ablative low-dose docetaxel chemotherapy, while docetaxel or CAR T cell application alone was not effective. In conclusion, the focal application of D7-derived CAR T cells and their combination with chemotherapy represent promising immunotherapeutic avenues to treat local and advanced prostate cancer in the clinic.
Keywords: CAR T cells, prostate cancer, focal therapy, docetaxel, chemotherapy, combination therapy, prostate specific membrane antigen
Graphical Abstract
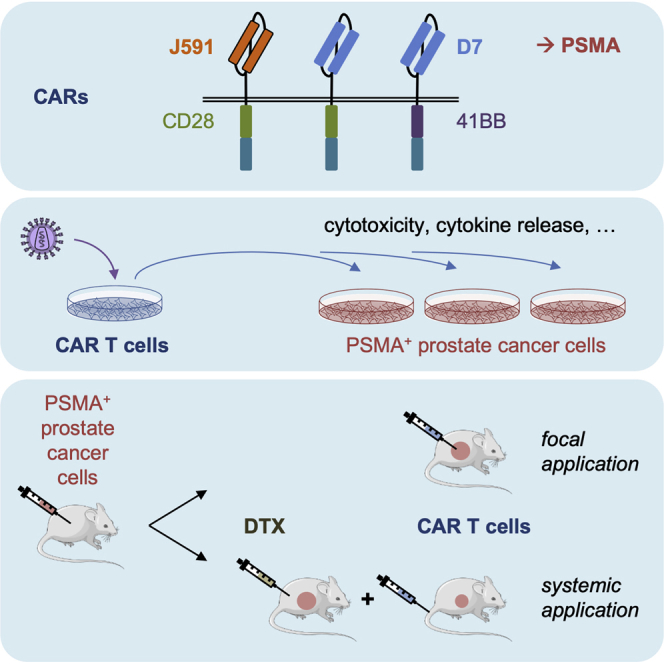
Alzubi and colleagues developed novel PSMA-targeting CARs that enabled the eradication of human prostate cancer xenografts in a mouse tumor model upon focal application of the resulting CAR T cells. Moreover, in combination with a non-ablative dose of docetaxel chemotherapy, systemic PSMA-CAR T cell application significantly inhibited tumor growth.
Introduction
Prostate cancer remains the second most frequent malignancy among men worldwide, with an estimated 1.1 million new cases per year. Moreover, with 307,000 deaths expected, it represents the fifth leading cause of cancer mortality.1,2 Whereas primary tumors can be successfully treated by surgery or local radiation therapy, classical treatment options do not provide a curative treatment for advanced stages.1 In this situation, immunotherapy with redirected T cells is thought to provide an alternative option. A rapidly emerging concept to treat cancer is based on the genetic engineering of T cells with chimeric antigen receptors (CARs), which bind tumor antigens or tumor-associated antigens in a human leukocyte antigen-independent manner. In general, CARs are composed of an extracellular domain harboring the antigen-specific single-chain variable fragment (scFv) antibody, a hinge domain, a transmembrane region, and intracellular signaling domains that activate engineered T cells upon antigen engagement. In clinically explored second-generation CARs, co-stimulatory domains generally derive from CD28 or 4-1BB.3 Targeting hematological malignancies has shown great clinical success, particularly CD19 targeting CAR T cells to treat B cell malignancies.4,5 For solid tumors, however, the potency of CAR T cell therapy is still low due to the immunosuppressive tumor microenvironment (TME) and accessibility of the tumor antigen,6 among others.
Prostate-specific membrane antigen (PSMA) is considered an ideal target for antigen-redirected immunotherapy because it is expressed at the surface of prostate cancer cells,7 present in all tumor stages, and shows an increased expression in androgen-independent and metastatic stages of the disease.8, 9, 10 Several antibodies have been developed to target PSMA both for diagnostic purposes and for the development of antibody-based therapies,11 such as J591,12 3D8,13 D2B,14 and 3/F11.15 Many of these antibodies were the basis for the development of PSMA-targeting CARs,16, 17, 18, 19 of which some have entered clinical trials (e.g., ClinicalTrials.gov: NCT01140373, NCT01929239, and NCT03089203). However, the oncolytic potency of these PSMA CAR T cells is still uncertain. In particular, engineered T cells expressing first-generation CARs derived from 3D818 or J59116,20 scFvs, respectively, showed low potency due to the low persistence of the CAR T cells. CAR T cell potencies improved when second- or third-generation CARs based on either D2B21 or J59117,19 scFvs were used. However, the anti-tumor activity remained moderate in tumor xenograft mouse models, and subjective responses were observed only when high CAR T cell doses or multiple infusions were applied.18,20,21 Nonetheless, PSMA is still considered a good tumor-associated antigen. Although the salivary glands, proximal tubuli of the kidney, and the brush border of the duodenal columnar epithelium express PSMA,22 CAR T cell-mediated “on-target/off-tumor” effects were not reported in any of the clinical trials that used 3D8-based CAR T cells.23 Alternatively, some patients with metastatic castration-resistance prostate cancer developed severe cytokine release syndrome (CRS) in a clinical study with J591-based CAR T cells24 that co-expressed a dominant-negative transforming growth factor β (TGF-β) receptor to render the CAR T cells insensitive to TGF-β.25 While the reason for the reported CRS is not clear yet, the totality of the results indicate that prostate cancer is hard to treat by currently available CAR T cell strategies and that the development of potent anti-PSMA CAR T cells in combination with alternative treatment paradigms is highly warranted.
To generate a new PSMA-targeting CAR, we relied on the 3/F11 antibody-derived scFv D7. We previously reported that 3/F11 was less reactive to normal tissues as compared to J591.18 Furthermore, D7 scFv was used to engineer Pseudomonas exotoxin A-based immunotoxins and bispecific anti-PSMA/anti-CD3 diabodies, which showed high and specific cytotoxicity against PSMA-expressing prostate cancer cells in vitro and in vivo in mice bearing human prostate tumors.26, 27, 28, 29, 30, 31 Another critical point in cell-based immunotherapies is that the engineered immune cells reach the tumor. We contemplated that focal injection of PSMA CAR T cells will ensure a high local intratumoral concentration of the engineered cells. Moreover, we speculated that low-dose chemotherapy with docetaxel (DTX), which is commonly used in combination with androgen deprivation therapy for the treatment of prostate cancer in a hormone-sensitive metastatic setting,1 will slow down tumor growth and modify the TME, thereby enabling the CAR T cells to access and fight the cancer cells. In a recent study, the immunomodulatory potential of DTX was demonstrated: pretreatment of non-small-cell lung cancer with DTX elicited an enhanced expression of high-mobility group box 1 (HMGB1) from dying cells, which was followed by a higher secretion of the chemokine CXCL11 and an enhanced tumor infiltration of CD8+ T cells.32 Lastly, the CAR architecture and the CAR expression levels were reported to be major determinants of CAR T cell activity in vivo.6,33, 34, 35
In this study, we show that our new D7-based PSMA-targeting CAR can be expressed at high levels in transduced T cells, correlating with high antigen-specific activation and cytotoxicity of the resulting CAR T cells in vitro. In vivo, D7-based PSMA-targeting CAR T cells completely eradicated human prostate cancer xenografts in a mouse tumor model upon focal application. Furthermore, these CAR T cells significantly inhibited tumor growth in combination with a non-ablative dose of DTX chemotherapy upon intravenous (i.v.) application. These novel treatment paradigms represent promising immunotherapeutic avenues to treat local and disseminated prostate cancer in clinical applications.
Results
Functional Validation of a New Anti-PSMA CAR
To target PSMA, we designed a novel second-generation CAR based on the D7 scFv, a modified immunoglobulin (Ig)G1 domain in the hinge region, which was shown to reduce potential interaction with the Fc receptor of innate cells,36 and a modified CD28 co-stimulatory domain deficient in lymphocyte-specific protein kinase (LcK) binding, which results in an enhanced anti-tumor activity in the presence of regulatory T cells (Tregs).37 For an initial validation of the D7-CAR, the scFvs 3D818 or J59117 were cloned into the same CAR scaffold (Figure 1A; Figure S1A). Although almost 100% of Jurkat T cells were transduced with the corresponding γ-retroviral vectors in all three cases, D7-CAR28 was expressed at higher levels than 3D8- and J591-based CARs, respectively (Figure S1B), suggesting increased stability or improved intracellular transport of the D7-CAR. Antigen-specific activation of these CAR Jurkat cells (Figure S1C) by co-cultivation with PSMA-expressing prostate cancer cell lines LNCaP (programmed cell death ligand 1 [PD-L1]+) or C4-2 (PD-L1−) (Figure S2F) revealed a sound activation of D7-CAR- or J591-CAR-expressing Jurkat cells, respectively, but not of 3D8-CAR-expressing cells (Figure S1C). Next, primary D7-CAR and J591-CAR T cells were generated by retroviral transduction (Figure 1B) and evaluated in terms of cytotoxicity, cytokine release, antigen-specific activation, phenotype, exhaustion, and proliferation (Figures 1C–1E; Figures S1D–S1J). Of note, 3D8-CAR expression could not be detected following retroviral transduction of primary T cells (data not shown), mirroring the low expression levels in Jurkat cells. To assess cytolytic activity, these CAR T cells were co-cultured with C4-2 cells, LNCaP cells, or PSMA− DU145 cells at the indicated effector-to-target (E:T) ratios (Figure 1C; Figures S1D and S1E). D7-CAR T cells eliminated the prostate cancer cells at significantly lower E:T ratios than did the J591-CAR T cells (Figure 1C; Figure S1D), independent of the source of donor T cells or the PD-L1 expression status. As compared to the J591-CAR T cells, the higher cytotoxic activity of D7-CAR T cells correlated with higher interferon γ (IFN-γ) and granzyme A release, respectively (Figure 1D; Figure S1H), as well as higher antigen-specific activation (Figure 1E). Upon co-cultivation of these CAR T cells with C4-2 tumor cells at E:T ratios of 1:1, comparable T cell differentiation pattern (Figure S1I) and exhaustion profiles were detected (Figure S1J). When subjected to a repetitive antigen exposure challenge, D7-CAR T cells were enriched better than J591-CAR T cells (Figure S1F) even though the proliferation rate was comparable (Figure S1G). In conclusion, D7-CAR T cells outperformed J591-CAR T cells in vitro in terms of cytotoxicity, cytokine/granzyme release, and enrichment after repetitive exposure to antigen-positive target cells without having a negative impact on differentiation, exhaustion, and proliferation capacity.
Figure 1.

PSMA-Targeting CARs
(A) Schematic of CAR-expressing γ-retroviral vectors. See Figure S1A for details. (B) Evaluation of CAR expression. Activated T cells were transduced with γ-retroviral vector and stained with anti-human IgG antibody (CAR) and CD3. (C) Cytolytic activity. CAR T cells were co-cultured at the indicated E:T ratios with C4-2 cells (PSMA+/PD-L1−). Cytotoxicity was determined using a cell viability assay (n = 6). (D) Cytokine release. CAR T cells were co-cultured with PSMA+ C4-2 or PSMA− Du145 cells, respectively, and the concentration of IFN-γ was determined in the supernatant (n = 3). (E) PSMA-mediated activation of CAR T cells. Activation of CAR T cells that were co-cultured with PSMA+ C4-2 tumor cells was assessed by evaluating CD25 expression (n = 3). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. UT, untransduced cells; PSMA, prostate-specific membrane antigen; PD-L1, programmed cell death ligand 1; MFI, mean fluorescent intensity.
Impact of Co-stimulatory Domains on Activity of D7-CAR T Cells In Vitro
To assess the impact of the co-stimulatory signaling domain on D7-CAR T cell activity (Figure 2A), a CAR harboring the 4-1BB costimulatory domain (CAR41) was compared to CD28-based CAR T cells (CAR28) with regard to CAR expression levels, PSMA-specific activation, cytotoxicity, cytokine release, and antigen-specific differentiation. Both CARs were stably expressed upon γ-retroviral transduction (Figures S2A and S2B), and the resulting CAR T cells preserved a high percentage of early undifferentiated T cell subtypes at the end of the expansion phase (Figures S2C and S2D). Interestingly, CAR28 was expressed at higher levels at the cell surface (Figure S2A), although total CAR expression was greater for CAR41 (Figure S2B). This might be due to a more efficient intracellular transport of CAR28 to the cell surface.
Figure 2.

In Vitro Analysis
(A) Schematic of γ-retroviral vectors. See Figure S1A for details. Costimulatory domains were derived either from CD28 (CAR28) or 4-1BB (CAR41). (B) Cytolytic activity. CAR T cells were co-cultured at the indicated E:T ratios with PSMA+ C4-2 tumor cells. Cytotoxicity was determined using a cell viability assay (n = 3). (C) PSMA-mediated activation of CAR T cells. CAR T cells were co-cultured with PSMA+ (C4-2) or PSMA− (Du145) tumor cells. T cell activation was assessed by evaluating expression of CD25. Shown is mean fluorescent intensity (MFI, n = 6). (D) Cytokine release. CAR T cells were co-cultured with PSMA+ (C4-2) or PSMA− (Du145) cells and IFN-γ in supernatant was measured (n = 3). (E) CAR T cell phenotype. CAR T cells were co-cultured with PSMA+ tumor cells before the phenotype was assessed based on CD62L and CD45RA expression. Shown are the average percentages of the different T cell subsets (n = 3 or 4). (F) Exhaustion. CAR T cells were co-cultured with PSMA+ tumor cells and the extent of T cell exhaustion was assessed by measuring expression of CD223 (LAG-3). Shown are the average percentages of LAG-3+ cells (n = 3 or 4). ∗p < 0.05, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. UT, untransduced T cells; Tn/Tscm, T cell naive or T stem cell memory; Tcm, T cell central memory; Tem, T cell effector memory; Teff, T cell effector; LAG-3, lymphocyte activation gene 3.
While both CARs mediated killing in a PSMA-dependent manner (Figure S2E), CAR28 T cells eliminated antigen-positive prostate cancer cells at a lower E:T ratio in a short-term in vitro assay than did CAR41 T cells (Figure 2B). As compared to CAR41 T cells, co-cultivation of CAR28 T cells with PSMA+ tumor cells induced a higher upregulation of activation markers CD25 (Figure 2C) and CD69 (Figure S3A). Analysis of the supernatants revealed that CAR28 T cells secreted significantly higher amounts of IFN-γ (Figure 2D) and granzyme B (Figure S3B) than did CAR41 T cells when co-incubated with PSMA+ tumor cells. The percentages of differentiated T cell subsets, such as effector memory and terminally effector T cells, was significantly higher for CAR28 T cells in comparison to CAR41 T cells (Figure 2E; Figure S3C), and CAR28 T cells were found to be more exhausted, as indicated by upregulated LAG3 (Figure 2F; Figure S3D). In summary, and in agreement with published data,38 the co-stimulatory domain had a major impact on activity and phenotype of D7-CAR T cells.
Elimination of Prostate Cancer Xenografts upon Focal Application of CAR T Cells
In order to explore the in vivo potency of the CAR T cells, a xenograft mouse model was used. To this end, C4-2luc+ cells were subcutaneously applied into the right flank of the animals. When tumors reached a mean volume of 70 mm3, one dose of 5 × 106 CAR28 T cells, CAR41 T cells, or non-transduced T cells, respectively, were intratumorally (i.t.) injected and changes in tumor volume were monitored by in vivo bioluminescence imaging (BLI) until day 22 (Figure 3A). In line with the in vitro data, CAR28 T cell treatment was more efficacious than CAR41 T cell-based therapy. Treatment with CAR28 T cells eliminated the tumors in five out of five mice between day 3 and day 8 (Figures 3B and 3C), with four out of five mice maintaining complete remission (CR) until the end of the experiment (Figure 3D). Treatment with CAR41 T cells resulted in one case each of CR and partial remission (PR), whereas mice injected with non-transduced T cells did not respond to treatment. The calculation of the area under the curve (AUC), which considers the temporal course of tumor growth, confirmed the increased antitumor activity of CAR28 T cells compared to CAR41 T cells (Figure S4A). In conclusion, focal injection of CAR T cells may be a promising approach to treat local prostate cancer.
Figure 3.

Focal CAR T Cell Therapy
(A) Schematic of experimental setup. 5- to 6-week-old SCID mice were injected subcutaneously (s.c.) with 1.5 × 106 PSMA+ C4-2luc+ tumor cells. When tumors reached ~70 mm3, mice were injected intratumorally (i.t.) with a single dose of 5 × 106 CAR28 (n = 5) or CAR41 (n = 5) T cells (day 1 of treatment). As controls, mice were left untreated (control, n = 6) or injected with untransduced T cells (UT, n = 7). (B) In vivo imaging. Shown are representative in vivo bioluminescence images (BLIs) of mice in different treatment groups. (C) Quantification of tumor size. Tumor volumes were determined on days 1, 3, 8, 15, and 22 of treatment. (D) Summary. Shown is antitumor response in different treatment groups. CR, complete response; PR, partial response. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, by unpaired t test.
Combination Therapy with DTX Controls Prostate Cancer Xenograft Growth upon i.v. CAR T Cell Application
Using the same experimental conditions as above, one i.v. injection of 5 × 106 CAR T cells did not affect tumor growth during the course of the experiment (Figure S4B), although the CAR T cells reached the tumor site as detected by in vivo imaging (Figure S4C). We conjectured that treatment with the chemotherapeutic agent DTX would render the tumors more susceptible to CAR T cell therapy by reducing tumor growth and/or altering the TME. For combination treatment, mice bearing C4-2luc+-derived large tumors (mean tumor volume of 150–200 mm3) were injected intraperitoneally (i.p.) with two cycles of DTX at days 1 and 2 of treatment, followed by i.v. injection of a single dose of 5 × 106 CAR T cells on day 8. Control groups were treated with DTX alone or left untreated (Figure 4A). Concurring with our approved animal protocol, tumor growth was monitored by BLI until day 22. For ethical reasons, some mice bearing large and aggressive tumors had to be euthanized before the end of the experiment. Treatment with DTX alone did not considerably affect tumor growth (Figures 4B and 4C; Figure S4D). While the combination of DTX with CAR41 T cell therapy had some effect on tumor growth, the combination of DTX with CAR28 T cells led to a significant reduction in tumor growth from day 17 on (Figures 4B and 4C). While a TME hardly fully develops in the immunodeficient mouse background, our histological analysis of the tumors indicated the formation of some features of a primitive TME, as indicated by the presence of connective tissue and tumor stroma with low cellular density and some infiltrated macrophages (Figures 5A and 5B). The application of DTX chemotherapy induced tumor damage marked by vacuolization and nuclear condensation of the tumor cells. Moreover, a high proportion of tumor cells with metaphase arrest was found, which can be attributed to the cell cycle inhibitory effect of DTX. Compared to control, the TME showed a higher cellularity with an infiltration of innate immune cells, accompanied by some stromal edema (Figures 5C and 5D). In tumors from mice treated with combination therapy, extensive damage of the tumor marked by necrotic tumor cells on the tumor stroma border was found. The TME was marked by stromal edema and infiltration of CD3+ CAR T cells. These characteristics were less pronounced in tumors treated with DTX and CAR41 T cells (Figures 5E and 5F) as compared to tumors treated with DTX and CAR28 T cells (Figures 5G and 5H), which was in line with a higher number of infiltrating CD3+ cells. In summary, we engineered CAR T cells that completely eradicated prostate cancer xenografts after a single focal application. Moreover, in combination with low-dose, non-ablative DTX chemotherapy, significant inhibition of tumor growth was achieved after systemic application of PSMA-targeting CAR28 T cells.
Figure 4.

Combination of Chemotherapy with Systemic Application of CAR T Cells
(A) Schematic of combination therapy. 5- to 6-week-old SCID mice were injected subcutaneously (s.c.) with 1.5 × 106 C4-2luc+ tumor cells. When tumors reached a mean tumor volume of 150–200 mm3, mice were treated intraperitoneally (i.p.) with two cycles of DTX (6 mg/kg body weight [bw]) at days 1 and 2 of treatment. At day 8, mice received one i.v. dose of 5 × 106 CAR28 (n = 9) or CAR41 T cells (n = 8). As controls, mice were injected with DTX alone (DTX, n = 9) or left untreated (control, n = 8). (B) In vivo monitoring. Shown are representative in vivo bioluminescence images (BLIs) of mice in different treatment groups. (C) Quantification of tumor size. Tumor volumes were determined on days 1, 8, 15, 17, and 22 of treatment. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, by unpaired t test.
Figure 5.

Histology
At the end of the combined docetaxel (DTX) and CAR T cell therapy, tumor slices of indicated groups were subjected to histopathological analysis by hematoxylin and eosin (H&E) staining (A, C, E, and G) or immunohistochemical analysis using anti-human CD3 that highlights human CD3+ T cells in red (B, D, F, and H). (A and B) Untreated group (control) with few nuclear condensations (arrowhead, exemplary) of tumor cells near the tumor margin, low cellular density in the tumor microenvironment (TME), consisting of connective tissue with few macrophages (asterisk), and absence of CD3+ T cell infiltrates. (C and D) DTX-treated group with nuclear condensations and cellular vacuolization but higher cellularity in the TME due to infiltration of monocytes and neutrophil granulocytes (asterisk), accompanied by stromal edema, and absence of CD3+ T cells. (E and F) Combination therapy group (DTX and CAR41 T cells) with presence of TME with high stromal edema and stroma cellularity due to increased monocytes, macrophages, and lymphocytes infiltrating the connective tissue (asterisk in E), accompanied by tumor necrosis at the tumor-stroma border (arrowhead in E), and few infiltrating CD3+ T cells in the TME (arrowheads in F). (G and H) Combination therapy group (DTX and CAR28 T cells) with presence of TME with high stromal edema and stroma cellularity (asterisk in G), accompanied by tumor necrosis zone (arrowhead in G), and high amount of infiltrating CD3+ T cells (arrowheads in H) and macrophage with CD3+ cytoplasm (asterisk in H). The average numbers ± standard error of the mean of infiltrating CD3 cells per tumor slice is indicated below. p values are indicated for comparison between the DTX group versus the DTX+CAR41 or DTX+CAR28 groups.
Discussion
The great success of CAR T cells to treat certain hematological malignances could not be transferred to solid tumors thus far. A main reason for this failure might be the TME that includes various kinds of immune cells, fibroblasts, and an extracellular matrix. Any of these factors could either intrinsically or physically inhibit the migration and/or activity of CAR T cells,6 suggesting that current CAR T cell therapies may not be successful without additive therapeutic interventions, such as checkpoint blockade or chemotherapy. We hypothesized that the focal administration of CAR T cells or their application in combination with chemotherapy may pave the way to a more successful treatment of solid tumors. To this end, we designed novel D7 scFv-based CARs to target PSMA and validated these CAR T cells in a xenograft mouse model. We demonstrate that local application of anti-PSMA CAR T cells led to complete eradication of prostate cancer, while systemic application of CD28-based CAR T cells significantly inhibited tumor growth when combined with low-dose DTX chemotherapy.
Our combined in vitro and in vivo data confirmed that D7-based CARs mediated high antigen-specific activation and cytotoxicity of the engineered T cells. The observed difference in comparison to previously reported PSMA-CARs in terms of efficacy may be found in the binding properties of the used scFvs and/or the heightened CAR expression, as previously shown for CARs targeting mesothelin or CD22.33,39 We previously demonstrated that the D7 scFv binds PSMA with high affinity and specificity.15 Moreover, we showed that its parental 3/F11 antibody recognizes a different extracellular epitope in PSMA than J591.15,40 It is therefore interesting to speculate that the high in vitro and in vivo efficacy of D7-based PSMA CAR T cells is based on optimal positioning of the CAR to recognize the target epitope.
Both CD28- and 4-1BB-based CAR T cells showed promising clinical results against B cell malignancies, albeit with different tumor-killing kinetics: inclusion of CD28 appears to mediate faster tumor reduction, whereas 4-1BB promotes T cell persistence.4,5,38 Our results are in line with these studies for lymphoid malignancies and showed that 4-1BB-based PSMA-CARs mediated an attenuated activation of the CAR T cells, resulting in less differentiation and less exhaustion upon antigen-specific stimulation.6 Although our animal protocol did not allow us to assess long-term effects, CD28-bearing PSMA-CAR T cells induced complete tumor eradication upon local application. PSMA-targeting CAR T cells were previously applied i.t. with ∼50% of treated mice showing CR upon injection of two doses of J591-based CAR T cells.20 This underlines the excellent performance of the new D7-based CAR for focal therapy. A local therapy could be highly attractive to treat early stage prostate cancer as it will avoid, or at least reduce, systemic-associated toxicities related to CAR T cell activation at “off-tumor” tissues that express the PSMA antigen at lower levels. It will also enable CAR T cells to bypass the TME, so overcoming the challenge of CAR T cell trafficking to and infiltration of the tumor.41
In agreement with previous publications,21,42 the D7-CAR T cells were not able to eradicate large solid tumors upon systemic application. However, two cycles of non-ablative low-dose chemotherapy were sufficient to modify the TME, thereby allowing the PSMA-targeting CAR T cells to infiltrate the tumor and control tumor growth upon a single dose of i.v. administration. It will be interesting to assess whether DTX treatment induces the secretion of CD8+ T cell-attracting cytokines from prostate cancer cells, as previously reported for non-small-cell lung cancer.32 Furthermore, it will be attractive to combine DTX treatment with “enhanced” CAR T cells43,44 by knocking out, for example, genes coding for inhibitory receptors, such as PD-1,45 CTLA4,46 LAG3,47 or the T cell receptor,48 or to introduce simple modifications to the CAR scaffold that have proven to increase safety and efficacy of CAR T cells in preclinical models of leukemia and lymphoma.34,35
In conclusion, we developed a novel D7-based anti-PSMA CAR that demonstrated excellent in vitro and in vivo activity. Notably, the CAR T cells were able to completely eradicate prostate cancer upon focal application and control tumor growth in combination with non-ablative low-dose chemotherapy upon systemic application. These pre-clinical data are encouraging and are enough to move forward to the clinic.
Materials and Methods
Culturing of Prostate Cancer Cells
Prostate cancer cell lines were grown in RPMI 1640 medium (Gibco) supplemented with penicillin (100 U/mL), streptomycin (100 mg/L), and 10% fetal calf serum (FCS, Biochrom) at 37°C in a humidified atmosphere of 5% CO2. For in vivo tumor establishment and bioluminescence imaging, C4-2 cells were transduced with a lentiviral vector encoding firefly luciferase and a neomycin resistance gene as previously described.49
CAR Design and Preparation of γ-Retroviral Vectors
Second-generation anti-PSMA CARs were cloned into the backbone of a self-inactivating γ-retroviral vector50 by conventional cloning. All CARs are under control of the EFS promotor and contain an optimized hinge domain36 and a CD3ζ intracellular signaling domain. The various CARs harbor scFv domains derived from monoclonal antibodies 3D8 (US2017/0232070 A1; SEQ ID 21), J591 (WO 2009/017823 A2), or D7.15 The CAR28 scaffolds harbor the transmembrane domain of CD28 and the CD28 co-stimulatory domain with a modified LcK binding moiety.37 The CAR41 scaffold contains a CD8α transmembrane domain and the 4-1BB co-stimulatory domain. Retroviral vectors were generated as previously described.51 Biological titers were determined by transducing Jurkat T cells, followed by staining of the transduced cells with anti-human IgG to determine CAR+ cells.
Generation and Culturing of CAR T Cells
CAR-transduced Jurkat cells were expanded in RPMI complete medium (RPMI 1640 medium [Gibco] supplemented with 10% FCS [Biochrom], penicillin [100 U/mL], streptomycin [100 mg/L], and 10 mM HEPES buffer [Sigma-Aldrich]). Primary CAR T cells were generated from peripheral blood mononuclear cells (PBMCs) as described before52 with modifications. In short, PBMCs were isolated from leukocyte reduction system (LRS) chambers obtained from the Blood Donation Center (informed consent of donors) of the Medical Center using phase separation (Ficoll, Sigma-Aldrich) and frozen in liquid nitrogen until used. For generation of CAR T cells, PBMCs were thawed in RPMI complete medium, activated with anti-CD2/CD3/CD28 antibodies (ImmunoCult, STEMCELL Technologies), and cultured with RPMI complete medium supplemented with cytokine cocktail (100 U/mL of IL-2, 25 U/mL of IL-7, and 50 U/mL of IL-15; all Miltenyi Biotec) for 3 days before transduction with γ-retroviral constructs with a dose of 50–300 transducing units/cell. Transduced cells were cultured in RPMI complete medium supplemented with 5 μg/mL of protamine sulfate (Sigma-Aldrich) and 1,000 U/mL of IL-2, 25 U/mL of IL-7, and 50 U/mL of IL-15 on poly-d-lysine (PDL, Sigma-Aldrich)-coated wells. After 1 day, medium was changed and cells were expanded for 9–12 days in RPMI complete medium supplemented with cytokine cocktail before being frozen in liquid nitrogen until further use.
Phenotyping of CAR T Cells
To determined transduction efficiency, surface CAR expression was evaluated by flow cytometry (FACSCanto II or Accuri, BD Biosciences) by staining transduced cells with anti-human IgG-phycoerythrin (PE) (SouthernBiotech) and CD3-allophycocyanin (APC) (Miltenyi Biotec), respectively. For phenotyping, CAR-transduced cells were stimulated with PSMA+ tumor cells (C4-2 or LNCaP) for 24 h at an E:T ratio of 1:1, before cells were harvested and evaluated. Activation was evaluated based on the expression of CD69 or CD25 (CD69-APC, clone CH4; CD25-PE, clone 3G10; both Thermo Fisher Scientific). T cell subsets were determined by staining cells with anti-human CD62L-Brilliant Violet 421 (BV421) (BD Biosciences), anti-human CD45RA-fluorescein isothiocyanate (FITC) (BioLegend), anti-human CD3-APC/H7 (BD Biosciences), and anti-human IgG-PE (SouthernBiotech). To determine exhaustion, cells were stained with anti-human CD279-FITC (PD-1, BD Biosciences), anti-human CD223-eFluor 710 (LAG-3, BD Biosciences), anti-human CD3-APC/H7, and anti-human IgG-PE.
Cytokine Release
CAR T cells were co-cultured with PSMA+ C4-2 tumor cells for 48 h at an E:T ratio of 1:1 in RPMI complete medium without cytokines. Supernatants were collected and evaluated by a multiplexed cytometric bead array (CBA, BD Biosciences) according to the manufacturer’s recommendations.
Cytolytic Activity of CAR T Cells
To determine the cytotoxic potential of CAR T cells, the viability of target cells was determined using the XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)2H-tetrazolium-5-carboxanilide) assay as described previously.53 In short, to determine cell viability as a function of metabolic activity, 100 μL of medium was removed and replaced with 100 μL/well of XTT solution (Sigma-Aldrich) and cells were incubated at 37°C. Colorimetric changes were quantified using an ELISA reader (Infinite F50, Tecan) at 450 nm. Cytotoxicity is indicated as 100% minus the percentage of viable cells, which was calculated according to the equation (ODE+T – ODE only)/(ODT only – ODmedium only), where OD indicates optical density, E represents effector cells (CAR T cells), and T represents target cells (tumor cells).
In Vivo Evaluation of CAR T Cells
Male severe combined immunodeficiency (SCID) CB17/lcr-Prkdcscid/Crl mice (5–6 weeks old, 20-25 g) were purchased from Janvier Labs (Saint-Berthevin, France) and kept under sterile and standardized environmental conditions. All experiments were carried out according to the animal protection law and ARRIVE guidelines with permission from the responsible local authorities. For tumor establishment, 1.5 × 106 C4-2luc+ cells diluted in 50% Matrigel/PBS (Collaborative Biomedical Products) were injected subcutaneously into the right flank of the animals. Tumor growth was monitored by palpation and BLI. To this end, 150 μg/kg of luciferin (BioSynth AG) were injected i.p. into the animals and BLI was done 10–30 min after injection under anesthesia using the in vivo imaging system (IVIS) 200 (Xenogen VivoVision). Tumor volumes were calculated from BLI using the software Living Image 3.0 and the formula V = (d2 × D)/2, where d was the smaller diameter and D the larger diameter of the tumor. When tumors reached a mean volume of about 70 mm3, mice were injected either i.t. or i.v. with one dose of 5 × 106 CAR28 T cells (n = 5) or CAR41 T cells (n = 5), untransduced T cells with a dose matching the final total cells in the CAR-treated groups (UT, n = 5–7), or left untreated (control, n = 6). For combinatorial treatment, mice with mean tumor volumes of 150–200 mm3 were treated i.p. with two doses of 6 mg/kg DTX on days 1 and 2 of treatment, followed by an i.v. injection of CAR28 T cells (n = 9) or CAR41 T cells (n = 8), respectively, on day 8 of treatment. Control mice were injected with DTX alone (n = 9) or left untreated (control, n = 8). Mice were euthanized at day 22 or earlier, when one of the following termination criteria were met: tumor diameter >15 mm, weight loss >20%, body condition score (BCS) >3, spasms, paralysis, body curvature respiratory disorders, apathy, or aggressiveness.
Histological Analysis
2-μm sections were prepared from formalin-fixed paraffin-embedded tumors and stained with hematoxylin and eosin. Immunohistochemistry was carried out with mouse anti-human CD3 antibody (clone F7.2.38; Dako-Agilent flex kit, Denmark), followed by rabbit anti-mouse secondary antibody mix, and visualized by an alkaline-based red chromogen reaction (K5005 kit, Dako-Agilent). Images were taken with an Olympus BX 51 microscope (Olympus, Germany) with the AxioCam MRc microscope camera (Carl Zeiss, Germany). For semiquantitative analysis, CD3+ lymphocytes were counted on three ×60 high-power field (HPF) slides (Olympus BX51; ×60/0.9/FN26.5).
Statistical Analysis
Statistical significance in all experiments was determined with the help of GraphPad Prism software by using the unpaired Student’s t test.
Author Contributions
T.C., P.W., and H.A. conceived and supervised the study; J.A., J.K., and N.T. carried out the in vitro experiments; V.D.-M. and S.T. performed the in vivo experiments and the statistical analysis of the data; M.S. performed the histological analyses; S.T., R.Z., W.S., and H.A. provided protocols and advice for the project; J.A., T.C., P.W., and H.A. planned the experiments and interpreted the data; J.A., P.W., and T.C. wrote the manuscript.
Conflicts of Interest
Research in the laboratory of T.C. is supported by Cellectis S.A. and Miltenyi Biotec. The remaining authors declare no competing interests.
Acknowledgments
This work was supported by grants from the Horizon 2020 Programme of the European Commission (CARAT-667980 to T.C.), the German Federal Ministry of Education and Research (IFB-01EO0803 to T.C.), and the Research Commission of the Faculty of Medicine of the Albert-Ludwigs-University of Freiburg (no. WOL1111/16 to P.W. and T.C.). The authors thank Beate vom Hövel for viral vector preparation and Susanne Schultze-Seemann, Irina Kuckuck, and Justin Mastroianni for technical assistance with the experiments, the Lighthouse Core Facility (Medical Center - University of Freiburg) for flow cytometry support, and the Blood Donation Center (Medical Center - University of Freiburg) for providing leukocyte reduction system (LRS) chambers.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.06.014.
Contributor Information
Toni Cathomen, Email: toni.cathomen@uniklinik-freiburg.de.
Philipp Wolf, Email: philipp.wolf@uniklinik-freiburg.de.
Supplemental Information
References
- 1.Litwin M.S., Tan H.J. The diagnosis and treatment of prostate cancer: a review. JAMA. 2017;317:2532–2542. doi: 10.1001/jama.2017.7248. [DOI] [PubMed] [Google Scholar]
- 2.Zhou C.K., Check D.P., Lortet-Tieulent J., Laversanne M., Jemal A., Ferlay J., Bray F., Cook M.B., Devesa S.S. Prostate cancer incidence in 43 populations worldwide: an analysis of time trends overall and by age group. Int. J. Cancer. 2016;138:1388–1400. doi: 10.1002/ijc.29894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zabel M., Tauber P.A., Pickl W.F. The making and function of CAR cells. Immunol. Lett. 2019;212:53–69. doi: 10.1016/j.imlet.2019.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park J.H., Rivière I., Gonen M., Wang X., Sénéchal B., Curran K.J., Sauter C., Wang Y., Santomasso B., Mead E. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl. J. Med. 2018;378:449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maude S.L., Laetsch T.W., Buechner J., Rives S., Boyer M., Bittencourt H., Bader P., Verneris M.R., Stefanski H.E., Myers G.D. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018;378:439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez M., Moon E.K. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019;10:128. doi: 10.3389/fimmu.2019.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mesters J.R., Barinka C., Li W., Tsukamoto T., Majer P., Slusher B.S., Konvalinka J., Hilgenfeld R. Structure of glutamate carboxypeptidase II, a drug target in neuronal damage and prostate cancer. EMBO J. 2006;25:1375–1384. doi: 10.1038/sj.emboj.7600969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wright G.L., Jr., Grob B.M., Haley C., Grossman K., Newhall K., Petrylak D., Troyer J., Konchuba A., Schellhammer P.F., Moriarty R. Upregulation of prostate-specific membrane antigen after androgen-deprivation therapy. Urology. 1996;48:326–334. doi: 10.1016/s0090-4295(96)00184-7. [DOI] [PubMed] [Google Scholar]
- 9.Silver D.A., Pellicer I., Fair W.R., Heston W.D., Cordon-Cardo C. Prostate-specific membrane antigen expression in normal and malignant human tissues. Clin. Cancer Res. 1997;3:81–85. [PubMed] [Google Scholar]
- 10.Kawakami M., Nakayama J. Enhanced expression of prostate-specific membrane antigen gene in prostate cancer as revealed by in situ hybridization. Cancer Res. 1997;57:2321–2324. [PubMed] [Google Scholar]
- 11.Wüstemann T., Haberkorn U., Babich J., Mier W. Targeting prostate cancer: prostate-specific membrane antigen based diagnosis and therapy. Med. Res. Rev. 2019;39:40–69. doi: 10.1002/med.21508. [DOI] [PubMed] [Google Scholar]
- 12.Chang S.S., Reuter V.E., Heston W.D., Bander N.H., Grauer L.S., Gaudin P.B. Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm PSMA expression in tumor-associated neovasculature. Cancer Res. 1999;59:3192–3198. [PubMed] [Google Scholar]
- 13.Ma Q., Safar M., Holmes E., Wang Y., Boynton A.L., Junghans R.P. Anti-prostate specific membrane antigen designer T cells for prostate cancer therapy. Prostate. 2004;61:12–25. doi: 10.1002/pros.20073. [DOI] [PubMed] [Google Scholar]
- 14.Frigerio B., Fracasso G., Luison E., Cingarlini S., Mortarino M., Coliva A., Seregni E., Bombardieri E., Zuccolotto G., Rosato A. A single-chain fragment against prostate specific membrane antigen as a tool to build theranostic reagents for prostate cancer. Eur. J. Cancer. 2013;49:2223–2232. doi: 10.1016/j.ejca.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 15.Wolf P., Freudenberg N., Bühler P., Alt K., Schultze-Seemann W., Wetterauer U., Elsässer-Beile U. Three conformational antibodies specific for different PSMA epitopes are promising diagnostic and therapeutic tools for prostate cancer. Prostate. 2010;70:562–569. doi: 10.1002/pros.21090. [DOI] [PubMed] [Google Scholar]
- 16.Stephan M.T., Ponomarev V., Brentjens R.J., Chang A.H., Dobrenkov K.V., Heller G., Sadelain M. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat. Med. 2007;13:1440–1449. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 17.Zhong X.S., Matsushita M., Plotkin J., Riviere I., Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol. Ther. 2010;18:413–420. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma Q., Gomes E.M., Lo A.S., Junghans R.P. Advanced generation anti-prostate specific membrane antigen designer T cells for prostate cancer immunotherapy. Prostate. 2014;74:286–296. doi: 10.1002/pros.22749. [DOI] [PubMed] [Google Scholar]
- 19.Santoro S.P., Kim S., Motz G.T., Alatzoglou D., Li C., Irving M., Powell D.J., Jr., Coukos G. T cells bearing a chimeric antigen receptor against prostate-specific membrane antigen mediate vascular disruption and result in tumor regression. Cancer Immunol. Res. 2015;3:68–84. doi: 10.1158/2326-6066.CIR-14-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gade T.P., Hassen W., Santos E., Gunset G., Saudemont A., Gong M.C., Brentjens R., Zhong X.S., Stephan M., Stefanski J. Targeted elimination of prostate cancer by genetically directed human T lymphocytes. Cancer Res. 2005;65:9080–9088. doi: 10.1158/0008-5472.CAN-05-0436. [DOI] [PubMed] [Google Scholar]
- 21.Zuccolotto G., Fracasso G., Merlo A., Montagner I.M., Rondina M., Bobisse S., Figini M., Cingarlini S., Colombatti M., Zanovello P., Rosato A. PSMA-specific CAR-engineered T cells eradicate disseminated prostate cancer in preclinical models. PLoS ONE. 2014;9:e109427. doi: 10.1371/journal.pone.0109427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ristau B.T., O’Keefe D.S., Bacich D.J. The prostate-specific membrane antigen: lessons and current clinical implications from 20 years of research. Urol. Oncol. 2014;32:272–279. doi: 10.1016/j.urolonc.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Junghans R.P., Ma Q., Rathore R., Gomes E.M., Bais A.J., Lo A.S., Abedi M., Davies R.A., Cabral H.J., Al-Homsi A.S., Cohen S.I. Phase I trial of anti-PSMA designer CAR-T cells in prostate cancer: possible role for interacting interleukin 2-T cell pharmacodynamics as a determinant of clinical response. Prostate. 2016;76:1257–1270. doi: 10.1002/pros.23214. [DOI] [PubMed] [Google Scholar]
- 24.Narayan V., Gladney W., Plesa G., Vapiwala N., Carpenter E., Maude S.L., Lal P., Lacey S.F., Melenhorst J.J., Sebro R. A phase I clinical trial of PSMA-directed/TGFβ-insensitive CAR-T cells in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2019;37(Suppl):TPS347. [Google Scholar]
- 25.Kloss C.C., Lee J., Zhang A., Chen F., Melenhorst J.J., Lacey S.F., Maus M.V., Fraietta J.A., Zhao Y., June C.H. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol. Ther. 2018;26:1855–1866. doi: 10.1016/j.ymthe.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michalska M., Schultze-Seemann S., Bogatyreva L., Hauschke D., Wetterauer U., Wolf P. In vitro and in vivo effects of a recombinant anti-PSMA immunotoxin in combination with docetaxel against prostate cancer. Oncotarget. 2016;7:22531–22542. doi: 10.18632/oncotarget.8001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michalska M., Schultze-Seemann S., Kuckuck I., Wolf P. In vitro evaluation of humanized/de-immunized anti-PSMA immunotoxins for the treatment of prostate cancer. Anticancer Res. 2018;38:61–69. doi: 10.21873/anticanres.12192. [DOI] [PubMed] [Google Scholar]
- 28.Baum V., Bühler P., Gierschner D., Herchenbach D., Fiala G.J., Schamel W.W., Wolf P., Elsässer-Beile U. Antitumor activities of PSMA×CD3 diabodies by redirected T-cell lysis of prostate cancer cells. Immunotherapy. 2013;5:27–38. doi: 10.2217/imt.12.136. [DOI] [PubMed] [Google Scholar]
- 29.Fortmüller K., Alt K., Gierschner D., Wolf P., Baum V., Freudenberg N., Wetterauer U., Elsässer-Beile U., Bühler P. Effective targeting of prostate cancer by lymphocytes redirected by a PSMA × CD3 bispecific single-chain diabody. Prostate. 2011;71:588–596. doi: 10.1002/pros.21274. [DOI] [PubMed] [Google Scholar]
- 30.Wolf P., Alt K., Wetterauer D., Bühler P., Gierschner D., Katzenwadel A., Wetterauer U., Elsässer-Beile U. Preclinical evaluation of a recombinant anti-prostate specific membrane antigen single-chain immunotoxin against prostate cancer. J. Immunother. 2010;33:262–271. doi: 10.1097/CJI.0b013e3181c5495c. [DOI] [PubMed] [Google Scholar]
- 31.Bühler P., Molnar E., Dopfer E.P., Wolf P., Gierschner D., Wetterauer U., Schamel W.W., Elsässer-Beile U. Target-dependent T-cell activation by coligation with a PSMA×CD3 diabody induces lysis of prostate cancer cells. J. Immunother. 2009;32:565–573. doi: 10.1097/CJI.0b013e3181a697eb. [DOI] [PubMed] [Google Scholar]
- 32.Gao Q., Wang S., Chen X., Cheng S., Zhang Z., Li F., Huang L., Yang Y., Zhou B., Yue D. Cancer-cell-secreted CXCL11 promoted CD8+ T cells infiltration through docetaxel-induced-release of HMGB1 in NSCLC. J. Immunother. Cancer. 2019;7:42. doi: 10.1186/s40425-019-0511-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haso W., Lee D.W., Shah N.N., Stetler-Stevenson M., Yuan C.M., Pastan I.H., Dimitrov D.S., Morgan R.A., FitzGerald D.J., Barrett D.M. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ying Z., Huang X.F., Xiang X., Liu Y., Kang X., Song Y., Guo X., Liu H., Ding N., Zhang T. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019;25:947–953. doi: 10.1038/s41591-019-0421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feucht J., Sun J., Eyquem J., Ho Y.J., Zhao Z., Leibold J., Dobrin A., Cabriolu A., Hamieh M., Sadelain M. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019;25:82–88. doi: 10.1038/s41591-018-0290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hombach A., Hombach A.A., Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc “spacer” domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010;17:1206–1213. doi: 10.1038/gt.2010.91. [DOI] [PubMed] [Google Scholar]
- 37.Kofler D.M., Chmielewski M., Rappl G., Hombach A., Riet T., Schmidt A., Hombach A.A., Wendtner C.M., Abken H. CD28 costimulation Impairs the efficacy of a redirected T-cell antitumor attack in the presence of regulatory T cells which can be overcome by preventing Lck activation. Mol. Ther. 2011;19:760–767. doi: 10.1038/mt.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Stegen S.J., Hamieh M., Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 2015;14:499–509. doi: 10.1038/nrd4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Z., Jiang D., Yang H., He Z., Liu X., Qin W., Li L., Wang C., Li Y., Li H. Modified CAR T cells targeting membrane-proximal epitope of mesothelin enhances the antitumor function against large solid tumor. Cell Death Dis. 2019;10:476. doi: 10.1038/s41419-019-1711-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bander N.H., Nanus D.M., Milowsky M.I., Kostakoglu L., Vallabahajosula S., Goldsmith S.J. Targeted systemic therapy of prostate cancer with a monoclonal antibody to prostate-specific membrane antigen. Semin. Oncol. 2003;30:667–676. doi: 10.1016/s0093-7754(03)00358-0. [DOI] [PubMed] [Google Scholar]
- 41.Schmidts A., Maus M.V. Making CAR T cells a solid option for solid tumors. Front. Immunol. 2018;9:2593. doi: 10.3389/fimmu.2018.02593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bobisse S., Rondina M., Merlo A., Tisato V., Mandruzzato S., Amendola M., Naldini L., Willemsen R.A., Debets R., Zanovello P., Rosato A. Reprogramming T lymphocytes for melanoma adoptive immunotherapy by T-cell receptor gene transfer with lentiviral vectors. Cancer Res. 2009;69:9385–9394. doi: 10.1158/0008-5472.CAN-09-0494. [DOI] [PubMed] [Google Scholar]
- 43.Cornu T.I., Mussolino C., Cathomen T. Refining strategies to translate genome editing to the clinic. Nat. Med. 2017;23:415–423. doi: 10.1038/nm.4313. [DOI] [PubMed] [Google Scholar]
- 44.Mussolino C., Alzubi J., Pennucci V., Turchiano G., Cathomen T. Genome and epigenome editing to treat disorders of the hematopoietic system. Hum. Gene Ther. 2017;28:1105–1115. doi: 10.1089/hum.2017.149. [DOI] [PubMed] [Google Scholar]
- 45.Menger L., Sledzinska A., Bergerhoff K., Vargas F.A., Smith J., Poirot L., Pule M., Hererro J., Peggs K.S., Quezada S.A. TALEN-mediated inactivation of PD-1 in tumor-reactive lymphocytes promotes intratumoral T-cell persistence and rejection of established tumors. Cancer Res. 2016;76:2087–2093. doi: 10.1158/0008-5472.CAN-15-3352. [DOI] [PubMed] [Google Scholar]
- 46.Ren J., Liu X., Fang C., Jiang S., June C.H., Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res. 2017;23:2255–2266. doi: 10.1158/1078-0432.CCR-16-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y., Zhang X., Cheng C., Mu W., Liu X., Li N., Wei X., Liu X., Xia C., Wang H. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front. Med. 2017;11:554–562. doi: 10.1007/s11684-017-0543-6. [DOI] [PubMed] [Google Scholar]
- 48.Poirot L., Philip B., Schiffer-Mannioui C., Le Clerre D., Chion-Sotinel I., Derniame S., Potrel P., Bas C., Lemaire L., Galetto R. Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res. 2015;75:3853–3864. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 49.Dürr C., Pfeifer D., Claus R., Schmitt-Graeff A., Gerlach U.V., Graeser R., Krüger S., Gerbitz A., Negrin R.S., Finke J., Zeiser R. CXCL12 mediates immunosuppression in the lymphoma microenvironment after allogeneic transplantation of hematopoietic cells. Cancer Res. 2010;70:10170–10181. doi: 10.1158/0008-5472.CAN-10-1943. [DOI] [PubMed] [Google Scholar]
- 50.Dettmer V., Bloom K., Gross M., Weissert K., Aichele P., Ehl S., Cathomen T. Retroviral UNC13D gene transfer restores cytotoxic activity of T cells derived from familial hemophagocytic lymphohistiocytosis type 3 patients in vitro. Hum. Gene Ther. 2019;30:975–984. doi: 10.1089/hum.2019.025. [DOI] [PubMed] [Google Scholar]
- 51.Bobis-Wozowicz S., Galla M., Alzubi J., Kuehle J., Baum C., Schambach A., Cathomen T. Non-integrating gamma-retroviral vectors as a versatile tool for transient zinc-finger nuclease delivery. Sci. Rep. 2014;4:4656. doi: 10.1038/srep04656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Golumba-Nagy V., Kuehle J., Abken H. Genetic modification of T cells with chimeric antigen receptors: a laboratory manual. Hum. Gene Ther. Methods. 2017;28:302–309. doi: 10.1089/hgtb.2017.083. [DOI] [PubMed] [Google Scholar]
- 53.Koehler H., Kofler D., Hombach A., Abken H. CD28 costimulation overcomes transforming growth factor-β-mediated repression of proliferation of redirected human CD4+ and CD8+ T cells in an antitumor cell attack. Cancer Res. 2007;67:2265–2273. doi: 10.1158/0008-5472.CAN-06-2098. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.