

Summary
Nerve growth factor elicits signaling outcomes by interacting with both its high-affinity receptor, TrkA, and its low-affinity receptor, p75NTR. Although these two receptors can regulate distinct cellular outcomes, they both activate the extracellular-signal-regulated kinase pathway upon stimulation with ligand. To delineate TrkA subcircuits in PC12 cell differentiation, we developed an optogenetic system where light was used to specifically activate TrkA signaling in the absence of nerve growth factor. By using tyrosine mutants of the optogenetic TrkA in combination with pathway-specific pharmacological inhibition, we find that Y490 and Y785 each contribute to PC12 cell differentiation through the extracellular-signal-regulated kinase pathway in an additive manner. Optogenetic activation of TrkA eliminates the confounding effect of p75NTR and other potential off-target effects of the ligand. This approach can be generalized for the mechanistic study of other receptor-mediated signaling pathways.
Keywords: Optogenetics, Nerve growth factor, TrkA signaling subcircuits, Y490, Y785, ERK, PLCγ, PC12 cell differentiation
Introduction
The dimeric secretory nerve growth factor (NGF), the first discovered neurotrophin, exerts a broad spectrum of neuronal functions including cell survival, growth, differentiation (Chao, 2003), tissue regeneration (Widenfalk, et al., 2001), pain (Hirose, et al., 2016), synaptogenesis, and synaptic plasticity (Poo, 2001). Trophic effects of NGF result from its interaction with the high-affinity receptor tropomyosin receptor kinase A or TrkA (Huang and Reichardt, 2001). Upon binding to NGF, TrkA undergoes auto-phosphorylation of specific tyrosines in its intracellular domain (ICD). Phosphorylated tyrosines in the TrkA ICD serve as docking sites for distinct downstream effectors to activate canonical downstream signaling pathways including the extracellular-signal-regulated kinase (ERK) and phospholipase C-γ (PLCγ) pathways (Segal, 2003).
Outcomes of TrkA signaling have been complicated, however, by the discovery that NGF also binds to its low-affinity receptor, p75NTR (Deshmukh and Johnson, 1997). In fact, p75NTR binds to other neurotrophins including brain-derived neurotrophic factor, neurotrophin-3, and neurotrophin-4/5 with similar affinities (Bothwell, 1995). Evidence suggests a two-fold neuronal function of p75NTR: 1) it can functionally collaborate with TrkA to enhance neurotrophin binding and receptor activation (Barker, 1998; Dechant and Barde, 2002), and 2) it can also induce cell apoptosis via ProNGF-dependent signaling processes (Lee, et al., 2001). Although NGF stimulation leads to ERK phosphorylation primarily through its binding to TrkA, ERK activation can also be mediated by p75NTR (Susen, et al., 1999). Thus, the existence of multiple receptors for NGF makes it challenging to delineate specific roles of TrkA subcircuits in the biological context (Chao, 2003; Lu, et al., 2005).
To delineate sole contributions of TrkA and p75NTR to NGF signaling, in vitro and in vivo models have been developed. Cell lines and transgenic animal models with either gene disrupted have been generated. For instance, the PC12nnr5 cell line expresses only p75NTR (lacking TrkA) (Green, et al., 1986), while PC12-p75- cells express only TrkA (lacking p75NTR) (Bassili, et al., 2010). Additionally, 3T3 cells have been engineered to express TrkA or p75NTR (Huang, et al., 1999). Similarly, p75NTR mutant mice (Lee, et al., 1992) and TrkA knock-out mice (Liebl, et al., 2000) have been developed. However, caution should be warranted with genetic manipulation for at least two reasons: first, complete deletion of a gene may undesirably corrupt its roles in other cellular functions. For instance, p75NTR is widely expressed in non-neuronal tissues (Lomen-Hoerth and Shooter, 1995) and is involved in processes such as liver repair (Passino, et al., 2007) and muscle regeneration (Colombo, et al., 2011). Second, genetic manipulation often causes delayed responses and constitutive alteration of gene expression in cells. Alternatively, pharmacological inhibition can be used to primarily target specific pathways to dissect the NGF signaling network. However, potential off-target effects should be considered (Arcaro and Wymann, 1993; Liu, et al., 2005; Martin, et al., 2011).
Even at the level of TrkA, contradictory evidence exists with respect to the functionality of its downstream subcircuits. Upon phosphorylation, two tyrosine residues, Y490 and Y785, serve as primary docking sites for downstream effectors. Y490 associates with Shc and Src domains (Stephens, et al., 1994), which further activate the Raf/MEK/ERK signaling pathway, whereas Y785 associates with PLCγ (Fig. 1). Work based on a chimeric receptor, a fusion protein of the PDGFR extracellular domain (ECD) and TrkA ICD, shows that the mutation of Y785 in TrkA, which is believed to primarily activate the PLCγ signaling pathway, does not affect PC12 cell differentiation (Obermeier, et al., 1994). However, overexpression of the Src Homology domains of PLCγ1 inhibits NGF-induced PC12 cell differentiation (Bae, et al., 1998), presumably through competition with endogenous PLCγ for receptor interaction. This discrepancy can arise from unknown off-target effects of the ligand or overexpressed protein domains, both of which are challenging to analyze quantitatively. Thus, delineation of TrkA subcircuits calls for the development of new strategies that can evaluate the functionality of specific tyrosines of the TrkA ICD with a clean biological context free from potential off-targets.
Figure 1. Design of the optogenetic TrkA system.
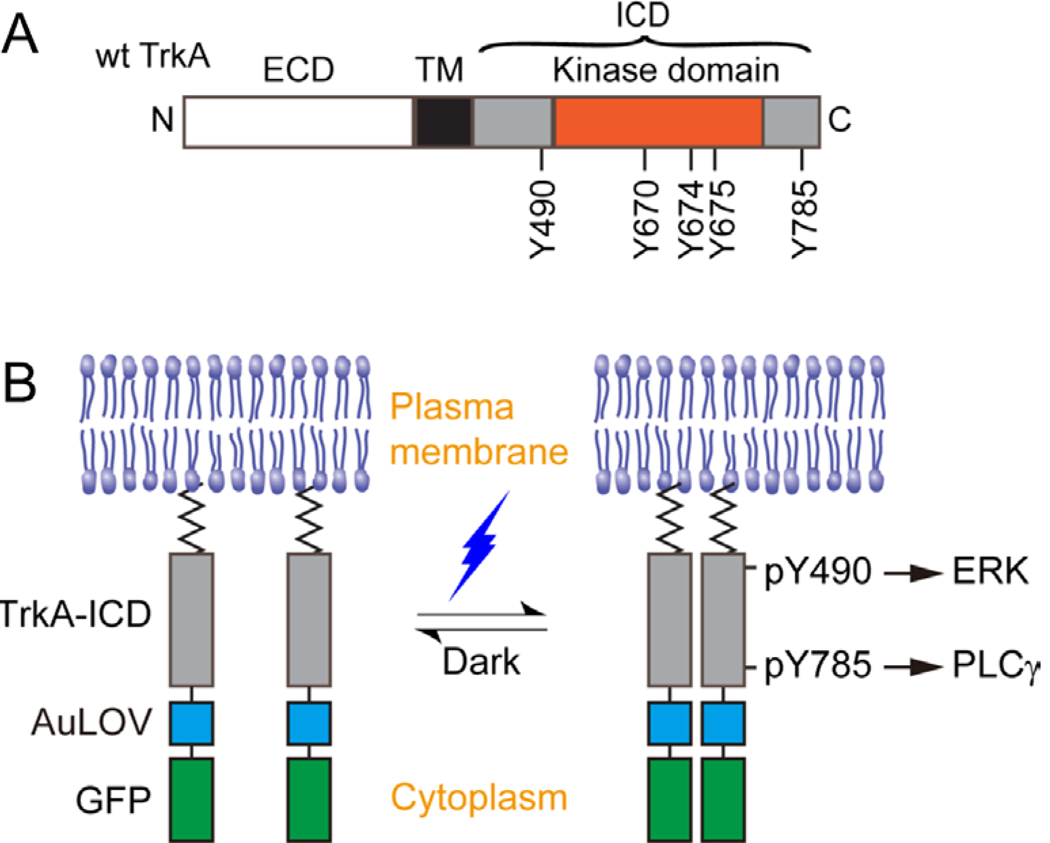
(A) Representation of the wild-type TrkA receptor. TrkA is a Type I transmembrane protein anchored at the plasma membrane by a single-helix transmembrane domain (TM). Nerve growth factor (NGF) associates with the extracellular domain (ECD) to promote receptor dimerization and phosphorylation of key tyrosines within the intracellular domain (ICD). Y670, Y674, and Y675 are critical for kinase activity, while Y490 and Y785 are involved in the initiation of the Raf/MEK/ERK and PLCγ-PKC signaling pathways, respectively. (B) Schematic representation of the optogenetic TrkA receptor. The ECD and TM are replaced by a lipidation motif to abolish ligand sensitivity, while retaining normal orientation and localization at the plasma membrane. Light sensitivity is introduced through fusion with the photosensitive protein, AuLOV. Illumination with blue light should promote dimerization of receptor ICDs and activation of downstream signaling pathways. Green fluorescent protein (GFP) serves as a probe for system expression.
The emerging optogenetic techniques provide a new way to study signaling mechanisms by integrating photoactivatable proteins with signaling molecules (Khamo, et al., 2017; Kim and Lin, 2013; Muller, et al., 2014; Tischer and Weiner, 2014; Toettcher, et al., 2011; Tucker, 2012; Zhang and Cui, 2015; Zoltowski and Gardner, 2011). As light can be easily manipulated with high spatial and temporal resolution, optogenetic control of signaling can provide insights into the kinetic and spatial features of signal transduction. In addition, light can bypass ligand-binding to specifically interrogate optogenetically engineered receptors, whose ligand can interact with and signal through multiple known and potentially unidentified receptors.
Here, we use an optogenetic approach to specifically elucidate the signaling outcomes of TrkA subcircuits in the absence of NGF. By constructing wild-type and tyrosine mutants of the optogenetic TrkA receptor, we control canonical TrkA signaling without introducing a ligand-based chimeric receptor or disturbing the endogenous TrkA expression and function. We find that both Y490 and Y785 of TrkA regulate PC12 cell differentiation, with Y490 primarily activating the ERK pathway and Y785 contributing to the ERK signaling cascade in a PLCγ- and PKC-dependent manner.
Results
System construction
Natural signal transduction mediated by TrkA sequentially involves NGF interaction with the receptor ECD, receptor dimerization, and a series of autophosphorylation at key tyrosines throughout the receptor ICD (Fig. 1A). Several tyrosines located within the receptor kinase domain are critical for optimal kinase activity, and some tyrosines located outside of the kinase domain serve as docking sites for initiating factors of intracellular signaling pathways. To bypass the ligand requirement for receptor activity, we constructed an optogenetically controllable TrkA (Lyn-TrkAICD-AuLOV-GFP) by fusing a homo-associating domain, the light-oxygen-voltage domain of aureochrome1 from V. frigida (AuLOV) (Grusch, et al., 2014), to the ICD of the wild-type TrkA receptor (Fig. 1B). This construct will be referred to as “WT ICD” throughout the remainder of the text. Lyn is the lipidation motif of the Src family Lyn kinase, which serves as a membrane-targeting peptide that replaces the transmembrane and extracellular domain of TrkA. This construct is different from a previously reported optogenetic system for TrkA, which used the full-length TrkA fused to the photoactivatable protein, cryptochrome 2 (Chang, et al., 2014). We chose to control TrkA ICD alone to avoid the potential interaction of TrkA ECD with other receptors such as p75NTR (Covaceuszach, et al., 2015), although this interaction is a subject of debate (Wehrman, et al., 2007). For practical convenience, we chose AuLOV because of its smaller size (145 a.a.) compared to cryptochrome 2 (498 a.a.). In our system, we expected that light-induced homo-association of AuLOV would bring TrkA kinase domains within proximity of each other to initiate the cross- and autophosphorylation of specific tyrosines throughout the ICDs, including Y490 and Y785. To delineate the signaling outcomes of these two docking sites, we also constructed single tyrosine-to-phenylalanine mutants (Y490F, Y785F) and a double mutant (Y490/785F) of WT ICD. Note that light-induced activation of the optogenetic TrkA system should not activate the endogenous wild-type TrkA.
Blue light illumination induces homo-association of optogenetic TrkA in live cells
When overexpressed in PC12 cells, WT ICD and its mutant variants localized primarily on the plasma membrane (Fig. 2A). Compared to a cytosolic GFP, optogenetic TrkA variants showed a higher fluorescence intensity at the plasma membrane relative to the cytoplasm (Fig. 2B). In the absence of ligand binding, the plasma membrane is the main subcellular localization of wild-type TrkA in PC12 cells, which was revealed in previous work by crosslinking NGF with wild-type TrkA (Hartman, et al., 1992) as well as immunostaining of TrkA (Grimes, et al., 1996). In addition, overexpression of fluorescently labeled TrkA also results in primary targeting to the plasma membrane (Wang, et al., 2011; Zhang, et al., 2013). Here, we reproduced the results of TrkA overexpression and found that TrkA-mCherry overlapped with WT ICD in PC12 cells (Fig. 2C), suggesting that the Lyn sequence allows for subcellular localization of TrkA ICD to the natural membrane compartment of full-length TrkA.
Figure 2. Optogenetic TrkA localizes to the plasma membrane and homo-associates in response to blue light.
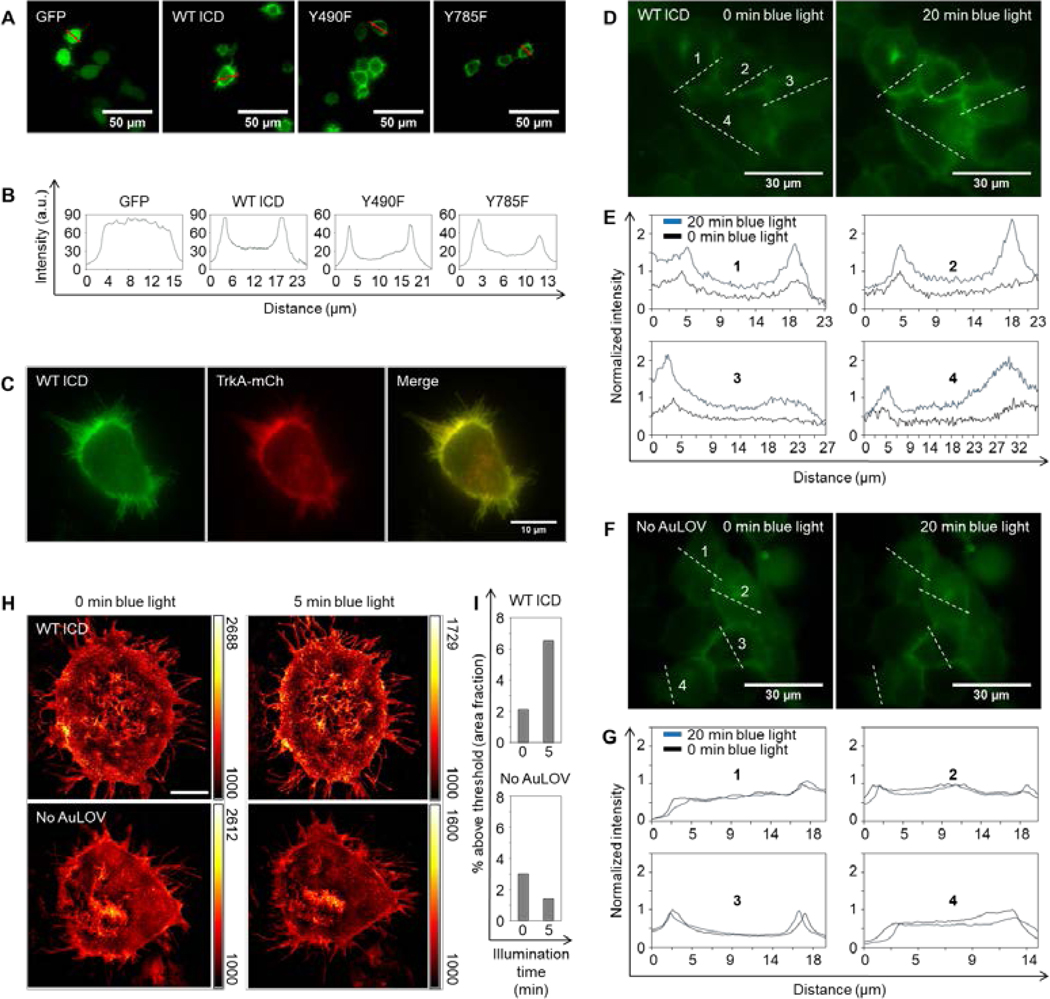
(A) Fluorescence microscopy images of PC12 cells transiently expressing a cytosolic GFP or variants of the optogenetic TrkA system. (B) Red-line profile analysis of fluorescence images in (A) reveals a strong membrane localization of the optogenetic constructs. (C) Overexpression of WT ICD and TrkA-mCherry shows that both fusion proteins primarily localize to the plasma membrane. (D) Bimolecular fluorescence complementation assay based on split Venus fragments. A 20-minute blue light illumination (5 mW/cm2) increased the fluorescence intensity in cells co-transfected with Lyn-TrkAICD-AuLOV-VN and Lyn-TrkAICD-AuLOV-VC. Images of the same cells were acquired before and after blue light treatment. (E) Intensity quantification along four dash-line profiles outlined in (D). (F-G) Same as (D-E) except that a No AuLOV control, Lyn-TrkAICD-VN and Lyn-TrkAICD-VC, is used. The same blue light illumination does not enhance the fluorescence intensity. (H) Structured illumination microscopy (SIM) images of MDA-MB-231 cells expressing WT ICD (top panels) or No AuLOV control (bottom panels) before and after blue light (405 nm) irradiation. (I) Quantification of the fraction of images whose intensity is above a threshold set at 50% of the maximum intensity of the background-subtracted images. Scale bar = 10 μm.
To confirm that blue light could induce the homo-association of WT ICD, we performed a bimolecular fluorescence complementation (BiFC) assay based on split Venus fragments composed of VN (amino acid 1–154) and VC (amino acids 155–238) (Saka, et al., 2007; Shyu, et al., 2006). Two C-terminal fusion proteins were constructed: Lyn-TrkAICD-AuLOV-VN and Lyn-TrkAICD-AuLOV-VC. As expected, after 20 minutes of blue light illumination, fluorescence intensity from cells co-transfected with both plasmids significantly increased (Fig. 2D–E). In cells co-transfected with the No AuLOV control (Lyn-TrkAICD-VN and Lyn-TrkAICD-VC) the same dose of blue light did not increase the fluorescence intensity (Fig. 2F–G).
We also used structured illumination microscopy (SIM) to probe blue-light-induced receptor homo-association. MDA-MB-231 cells were transiently transfected with WT ICD and were exposed to blue light. To quantify the light-induced homo-association of WT ICD, we analyzed the SIM images using the thresholding technique. By setting the threshold at the midpoint value between the minimal and maximal intensity of the background-subtracted cell images, we calculated the percentage of above-threshold regions over the whole cell area using ImageJ based on a published protocol (Jensen, 2013). Following 5 minutes of blue light (405 nm) illumination, the above-threshold percentage increased in cells transfected with WT ICD, in stark contrast to cells transfected with the No AuLOV control (Lyn-TrkAICD-GFP), which showed no increase in the above-threshold percentage after blue light illumination (Fig. 2H–I).
In addition to fluorescence microscopy, we were inspired by a previous optogenetic study (Zhou, et al., 2012) to use a native PAGE assay to resolve the light response of AuLOV. Cells were transiently transfected with WT ICD and lysates were harvested in 0.5% Triton X-100 in PBS. Lysates were treated with blue light (5 mW/cm2) for 5 minutes or kept in the dark. Equal amounts of each lysate were loaded on a native gel. The lane containing the illuminated sample underwent continued exposure to blue light throughout the electrophoresis. Samples were then analyzed by western blot using an anti-GFP antibody. As expected, blue light resulted in a reduced-mobility shift of the most prominent band in the dark control, indicative of light-mediated homo-association (Supplementary Figure 1).
Light-induced activation of optogenetic TrkA results in PC12 cell differentiation
PC12 cells project neurites in response to NGF stimulation, a process referred to as differentiation (Fig. 3A). We proceeded to determine whether WT ICD activation could achieve a similar cellular outcome. PC12 cells transiently transfected with WT ICD were exposed to low-power blue light (300 μW/cm2) for two days in a humidified 37 °C CO2 incubator before their fluorescence images were acquired. Light stimulation of these cells resulted in significant differentiation (45%) compared to cells kept in the dark (12%) (Fig. 3B). Cells expressing the optogenetic construct without the TrkA ICD (No ICD) showed little differentiation in both light and dark (<5%), indicating that light-induced differentiation was ICD dependent. Cells expressing No ICD showed robust differentiation (>68%) when treated with 50 ng/mL NGF, indicating that endogenous TrkA retained ligand sensitivity and signaling activity.
Figure 3. Optogenetic TrkA promotes PC12 cell differentiation in response to blue-light stimulation.
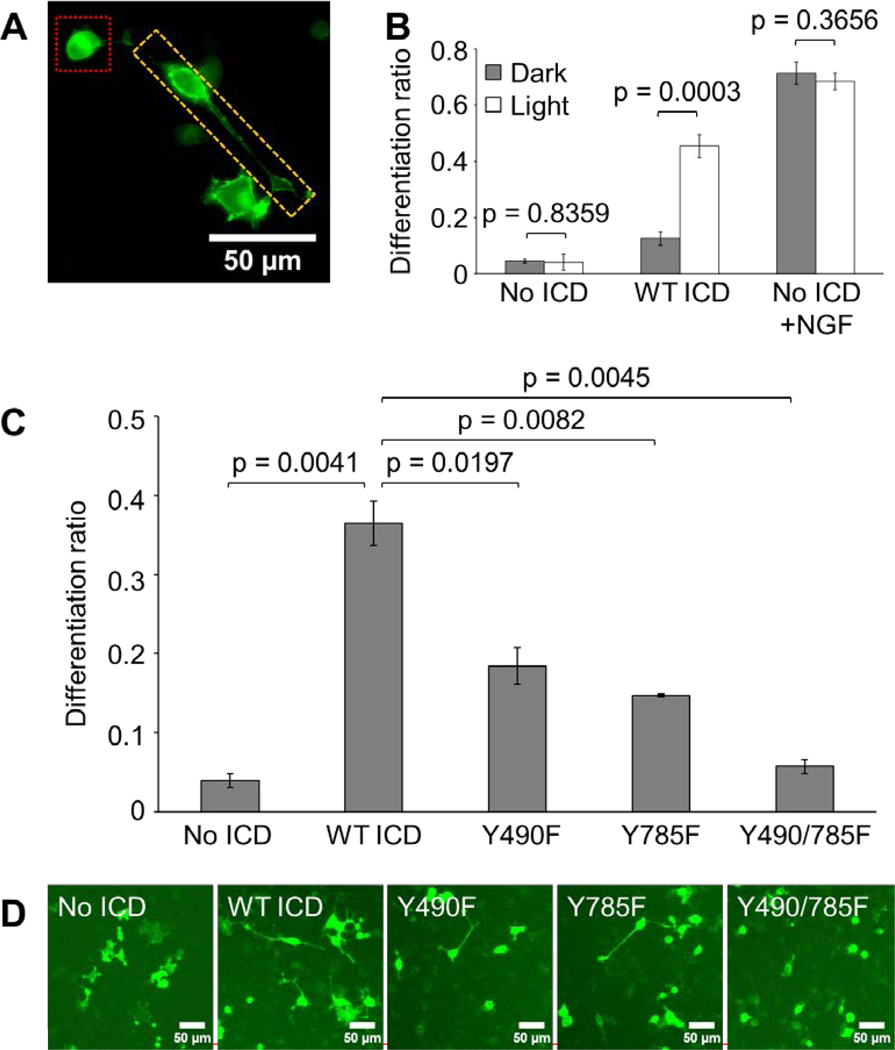
(A) Image for reference depicting differentiated (yellow box) and undifferentiated (red box) PC12 cells transfected with No ICD and treated with 50 ng/mL NGF for 24 hours. (B) Differentiation ratios calculated for PC12 cells transiently expressing WT ICD or No ICD. Transfected cells were illuminated with 300 μW/cm2 blue light or kept in the dark for 44 hours prior to imaging. WT ICD mediated significant light-induced differentiation compared to No ICD. Cells expressing No ICD in the presence of 50 ng/mL NGF underwent robust differentiation in a light-independent manner. Values represent the mean ± standard deviation of three biological replicates (n=3) with >70 cells counted per replicate. (C) Differentiation ratios calculated for PC12 cells transiently expressing variants of the optogenetic TrkA system. Transfected cells were illuminated with 300 μW/cm2 blue light for 24 hours prior to imaging. Y490F and Y785F showed a significant reduction in light-induced differentiation compared to WT ICD. Y490/785F resulted in a differentiation ratio similar to that of No ICD. Values represent the mean ± standard deviation of two biological replicates (n=2) with >900 cells counted per replicate. (D) Representative fluorescence images of the conditions reported in (C). Scale bar = 50 μm.
Light-induced activation of optogenetic TrkA mutants results in diminished PC12 cell differentiation
To determine the functional role of Y490 and Y785 in the differentiation phenotype, we transiently transfected PC12 cells with mutant constructs and illuminated for 24 hours as previously described. Cells expressing Y490F or Y785F exhibited significantly reduced levels of light-induced differentiation (18% or 15%, respectively) compared to cells expressing WT ICD (36%) (Fig. 3C–D). Furthermore, the degree of differentiation undergone by cells expressing Y490/785F (6%) was similar to that displayed by cells expressing No ICD (4%), suggesting that Y490 and Y785 each contributed to TrkA-mediated PC12 cell differentiation.
Light-induced optogenetic TrkA mutants differentially activate the ERK and PLCγ pathways
To interrogate the mechanism behind the reduced differentiation mediated by the optogenetic TrkA mutants, we performed western blot analysis to probe the activity of downstream signaling pathways. To minimize baseline signaling activity and capture subtle differences between conditions, PC12 cells were transiently transfected with mixtures of the variant ICD constructs and the No ICD construct. Cells were illuminated for 10 minutes with 5 mW/cm2 blue light prior to lysis. Both Y490F and Y785F showed a significant reduction in ERK signaling compared to WT ICD, with the reduction for Y490F being more pronounced (Fig. 4A, Lanes 4, 6, and 8). Cells expressing Y490F showed no change in light-induced PLCγ activity compared to WT ICD, while Y785F showed a complete loss of PLCγ activity (Fig. 4B, Lanes 4, 6, and 8). These results indicate that Y490 primarily contributed to ERK activity, while Y785 primarily contributed to PLCγ activity. Additionally, Y785 seemed to partially contribute to ERK signaling as evidenced by the persistent, residual ERK activity observed for Y490F (Fig. 4A, Lane 6) and the reduction in ERK activity observed for Y785F (Fig. 4A, Lane 8) compared to WT ICD. To confirm that the observed variations in signaling were a result of specific mutations and not due to significant variability in system expression, lysates were probed with an antibody against GFP and all constructs were expressed at a comparable level (Fig. 4C).
Figure 4. Light-induced optogenetic TrkA mutants differentially activate the ERK and PLCγ pathways.

Western blot analysis of light-induced (A) ERK and (B) PLCγ activity exhibited by PC12 cells transiently expressing variants of the optogenetic TrkA system. Transfected cells were serum-starved overnight following transfection, and were illuminated with 5 mW/cm2 blue light or kept in the dark for 10 minutes prior to lysis. Compared to WT ICD, Y490F displayed a dramatic reduction in ERK activity with no change in PLCγ activity. Y785F showed a relatively modest reduction in ERK activity with nearly abrogated PLCγ activity. Y490/785F showed no light-induced activity for either pathway, similar to No ICD. Values represent the mean ± standard deviation of three separate experiments (n=3) for ERK and two separate experiments (n=2) for PLCγ. (C) Western blot analysis of system expression by probing GFP. Expression level across samples fluctuates within 16%.
Y785 contributes to ERK signaling and PC12 cell differentiation in a PLCγ-PKC dependent manner
Given that Y785 is essential for PLCγ activity, we speculated that the contribution of Y785 to ERK signaling was mediated by pathway crosstalk. Indeed, findings from previous studies support this notion (Mauro, et al., 2002; Ueda, et al., 1996). To validate this, we transiently transfected PC12 cells as previously described with a combination of Y490F and No ICD. Transfected cells were then treated with a PLCγ inhibitor (U73122, 1 μM) or a PKC inhibitor (GF 109203X, 1 μM) 10 minutes prior to illumination. With the inhibitors still present, cells were then illuminated for 10 minutes with 5 mW/cm2 blue light before lysis and subsequent immunoblotting (Fig. 5A). Compared to untreated cultures, all cells treated with inhibitors displayed an abrogation of the light-induced residual ERK activity mediated by Y490F (Fig. 5A, Lanes 2, 4, and 6), suggesting that the role of Y785 in ERK signaling is dependent on the PLCγ-PKC pathway. To confirm that the observed variations in signaling were not due to significant variability in system expression, lysates were probed with an antibody against GFP and all constructs were expressed at a comparable level (Fig. 5B). In addition to interrogating the short-term effect of inhibitors on light-induced ERK signaling mediated by Y490F, we set out to determine the effect of long-term inhibition on PC12 cell differentiation. PC12 cells overexpressing Y490F were illuminated with blue light (300 μW/cm2) for 24 hours in the presence or absence of U73122 (1 μM) or GF 109203X (1 μM). Inhibitors were added 1 hour prior to illumination. Cell cultures treated with either inhibitor showed significantly reduced differentiation, suggesting that Y785 also contributes to PC12 cell differentiation in a PLCγ-PKC dependent manner (Fig. 5C–D).
Figure 5. Y490F-mediated ERK signaling is abolished by PLCγ-PKC inhibitors.
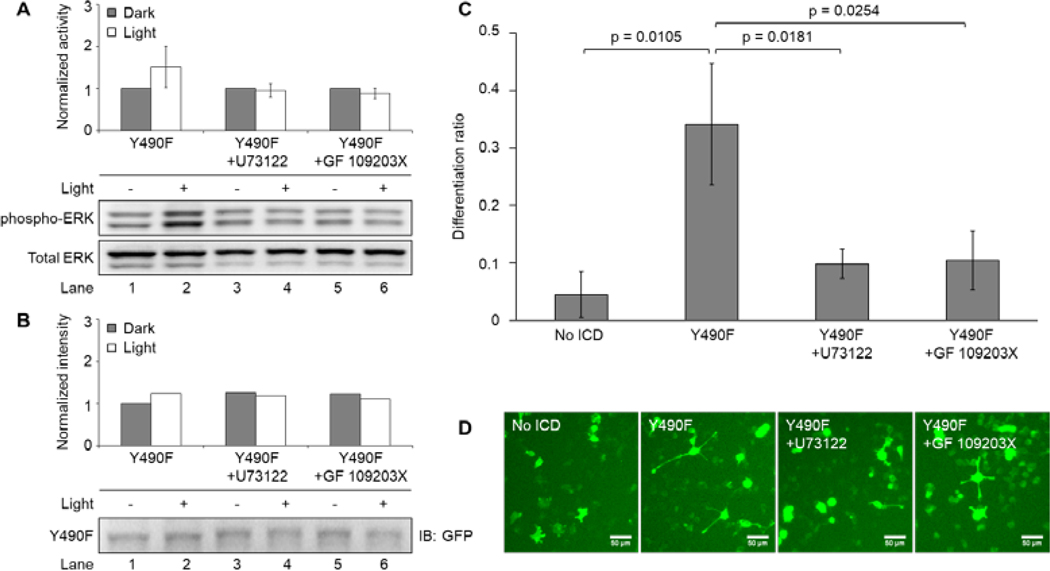
(A) Western blot analysis of light-induced ERK activity exhibited by PC12 cells transiently expressing Y490F. Transfected cells were serum-starved overnight following transfection and were treated with a PLCγ inhibitor (U73122, 1 μM) or a PKC inhibitor (GF 109203X, 1 μM) for 10 minutes prior to illumination. Cells were subsequently illuminated with 5 mW/cm2 blue light or kept in the dark for 10 minutes prior to lysis. Cells treated with U73122 or GF 109203X had abolished light-induced ERK activity compared to the untreated control. Values represent the mean ± standard deviation of two separate experiments (n=2) for untreated, and four separate experiments (n=4) for U73122 and GF 109203X. (B) Western blot analysis of system expression by probing GFP. Expression level for illuminated conditions fluctuates within 14%. (C) Differentiation ratios calculated for PC12 cells transiently expressing Y490F in the presence of U73122 (1 μM) or GF 109203X (1 μM). Inhibitors were added to cells 1 hour prior to illumination. Cells were illuminated with 300 μW/cm2 blue light for 24 hours prior to imaging. Cells treated with U73122 or GF 109203X had decreased light-induced differentiation compared to the untreated control, which received inhibitor vehicle (DMSO). Values represent the mean ± standard deviation of three biological replicates (n=3) with >40 cells counted per replicate (D) Representative fluorescence images of the conditions reported in (C). Scale bar = 50 μm.
Discussion
We developed an optogenetic TrkA system to study differentiation in PC12 cells by photo-activation of the TrkA ICD and its downstream signaling pathways. Light-mediated homo-association of TrkA resulted in elevated ERK and PLCγ activity. Light stimulation specifically activated the optogenetic TrkA, efficiently decoupling its signaling from endogenous TrkA and p75NTR. By generating mutants of the signal pathway-specific tyrosines, Y490 and Y785, we used this system to determine the mechanisms by which TrkA regulates PC12 cell differentiation.
In a previous study, we demonstrated that optogenetic activation of Raf/MEK/ERK signaling is sufficient to induce PC12 cell differentiation, suggesting that this pathway is a strong correlating factor for the phenotype (Krishnamurthy, et al., 2016). From our experiments employing optogenetic TrkA mutants in combination with pathway-specific inhibitors, we propose a model of the TrkA signaling network in the context of PC12 cell differentiation (Fig. 6). We find that Y490 regulates ERK signaling and Y785 primarily regulates PLCγ signaling. Y785 also promotes ERK signaling in a PLCγ-PKC dependent manner. Indeed, previous studies showed that PKC could feed into the Raf/MEK/ERK signaling cascade (Mauro, et al., 2002; Ueda, et al., 1996). The reduced differentiation caused by the Y490F and Y785F single mutants, and the abolished differentiation observed for the Y490/785F double mutant, suggest that TrkA uses both Y490 and Y785 to activate the Raf/MEK/ERK signaling pathway in promoting PC12 cell differentiation.
Figure 6. Proposed model for the role of Y490 and Y785 in TrkA-mediated PC12 cell differentiation.
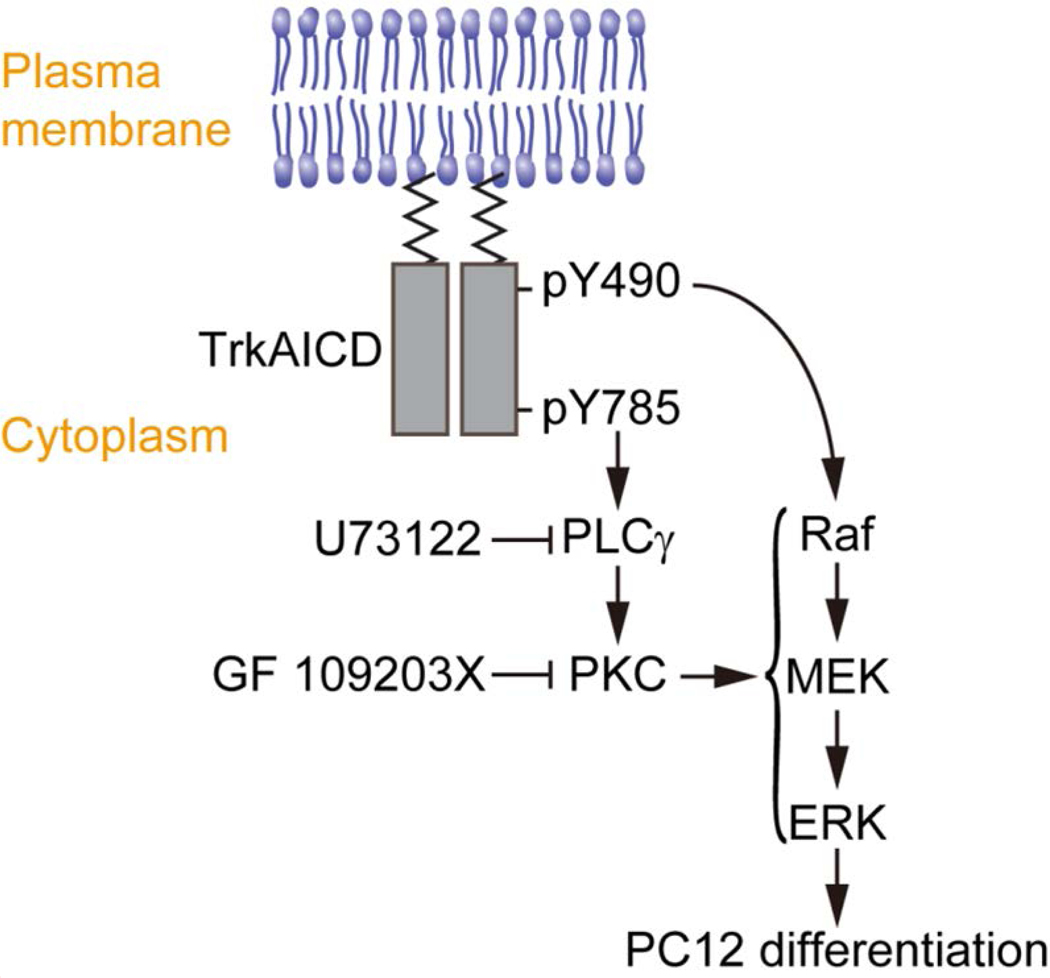
The Raf/MEK/ERK signaling cascade, primarily instigated by Y490 of TrkA, is essential for PC12 cell differentiation. Y785 instigates the PLCγ-PKC pathway, which feeds into the Raf/MEK/ERK signaling cascade. Mutating Y785 or inhibiting the PLCγ-PKC pathway results in diminished receptor-mediated ERK signaling and differentiation in PC12 cells.
Based on previous studies (Obermeier, et al., 1994; Stephens, et al., 1994), PC12 cells expressing the Y490/785F double mutant did not differentiate in the presence of NGF, in agreement with our optogenetic result. However, the individual contribution of Y490 and Y785 to PC12 cell differentiation has been elusive. In one case, expression of single mutants Y490F and Y785F did not affect NGF-induced PC12 cell differentiation, suggesting a redundancy in their phenotypical role (Stephens, et al., 1994). In the other case, Y490F resulted in a dramatic loss of differentiation while Y785F had no effect (Obermeier, et al., 1994). This discrepancy may be due to the nature of the ligands (i.e., NGF versus PDGF) used in these studies.
Using our optogenetic TrkA system, we show that Y490 and Y785 make significant and additive contributions to PC12 cell differentiation. We found that Y490F and Y785F each diminished PC12 cell differentiation to approximately half of that promoted by WT ICD (Fig. 3C). A similar additive trend was observed for their capacity to activate ERK (Fig. 4A). Stephens and colleagues (Stephens, et al., 1994) observed a similar reduction of ERK activity for both Y490F and Y785F compared to wild-type TrkA. At the phenotypic level, however, Y490F and Y785F promoted a degree of PC12 cell differentiation comparable to that promoted by wild-type TrkA. These results indicate that an ERK signaling threshold exists for establishing maximal differentiation. It is possible that this threshold was exceeded by both Y490F and Y785F when stimulated with NGF.
Here, we note that PC12 cell differentiation mediated by WT ICD was less than that of NGF treatment (Fig. 3B). We speculated that this was a result of a difference in signaling intensity between ligand- and light-simulated receptors. To test this idea, we compared the ERK activity induced by WT ICD (Fig. 7A) to NGF treatment (Fig. 7B). Given that not all cells are light-responsive in transfected cultures, we defined the signaling response of optogenetic TrkA as We defined the NGF signaling response of untransfected cells as R is the signal ratio of phosphorylated ERK over total ERK probed by western blot. Figure 7A–B shows that light elicited a milder signaling response () compared to NGF treatment (). This result suggests that NGF elicits higher ERK activity than WT ICD, at least on a short time scale (5–10 minutes). To determine if a similar signaling difference exists at a longer time scale, we compared the ERK activity generated by NGF and light-activated WT ICD after 4 hours and 24 hours of treatment. As expected, NGF promoted higher ERK activity than WT ICD at both time points (Fig. 7C).
Figure 7. NGF elicits higher ERK activity compared to optogenetic TrkA and NGF-mediated PC12 cell differentiation is reduced by pharmacological inhibition of PLCγ and PKC in a dose-dependent manner.
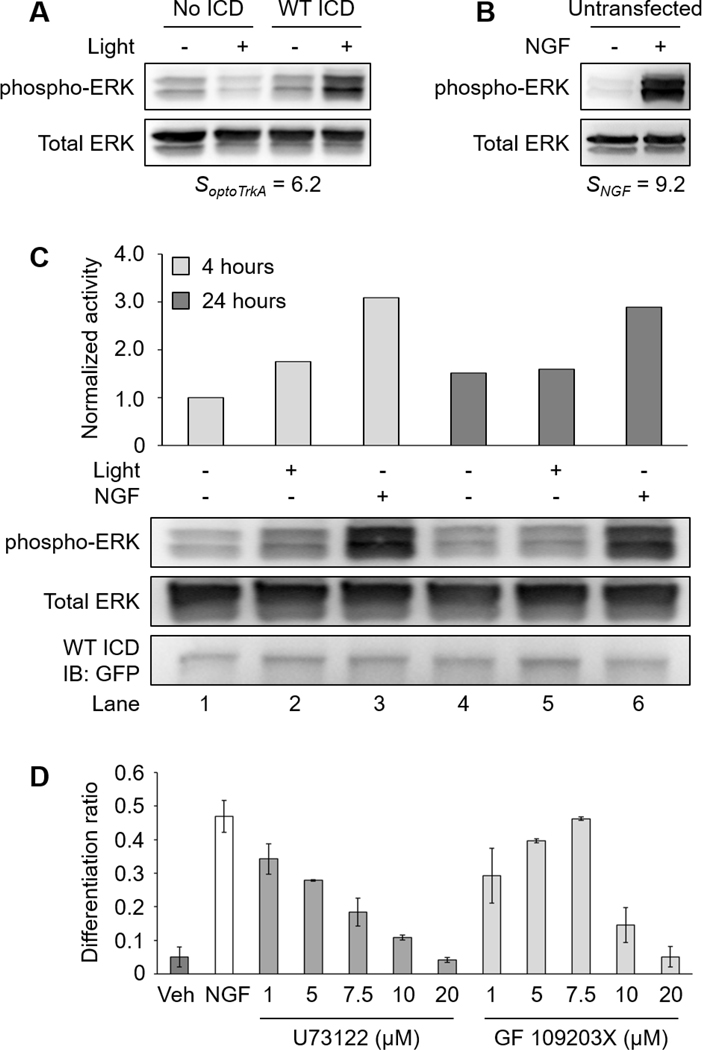
(A) Western blot analysis of two separate experiments. Transfected cells (No ICD and WT ICD) were serum-starved overnight following transfection. Cells were illuminated with 5 mW/cm2 blue light or kept in the dark for 10 minutes prior to lysis. (B) Untransfected cells were serum-starved overnight prior to treatment. Cells were untreated or treated with NGF (100 ng/mL) for 5 minutes prior to lysis. The ERK activity mediated by optogenetic TrkA was lower than that of NGF. (C) Western blot analysis of ERK signaling mediated by long-term NGF or WT ICD activity. PC12 cells were transfected with WT ICD and serum-starved overnight following transfection. Cells were then kept in the dark, illuminated with 300 μW/cm2 blue light, or treated with NGF (50 ng/mL) for 4 or 24 hours prior to lysis. Untreated cells received NGF vehicle (sodium acetate). NGF stimulation elicited higher ERK activity than WT ICD for both durations. All samples are normalized to Lane 1. (D) Differentiation ratios for PC12 cells treated with NGF in the presence of U73122 or GF 109203X. Serum-starved cells were treated with inhibitors (1, 5, 7.5, 10, and 20 μM) 1 hour prior to NGF treatment. Cells were treated with NGF (50 ng/mL) for 24 hours. Cells were stained with 1 μM Calcein AM 10 minutes before imaging. NGF-induced PC12 cell differentiation was reduced by U73122 or GF 109203X in a dose-dependent manner. Untreated cells received NGF vehicle (sodium acetate) and inhibitor vehicle (DMSO). Values represent the mean ± standard deviation of two biological replicates (n=2) with >400 cells counted per replicate.
Thus, the previously observed reduction of ERK activity for NGF-treated PC12 cells expressing either Y490F or Y785F may still surpass the signaling threshold required for saturated differentiation, thereby obscuring the contribution of individual tyrosines (Stephens, et al., 1994). On the other hand, the optogenetic Y490F and Y785F likely produce a sub-threshold ERK activity when stimulated with light, providing advantages in interpreting the phenotypic role of each tyrosine. These results suggest that the difference between light- and ligand-induced PC12 cell differentiation resulted from their distinct potency in the induction of ERK signaling, the reason for which has yet to be determined, although it can be speculated that p75NTR plays a role in the presence of the ligand. In the physiological context, while Y490 primarily mediates the ERK-dependent trophic effects of NGF, we speculate that the role of Y785 in ERK signaling becomes more critical where NGF concentrations are low.
Finally, we asked if pharmacological inhibition of NGF-induced PLCγ-PKC activity would support our proposed model of the TrkA signaling network in the context of PC12 cell differentiation. We performed a PC12 cell differentiation assay in the presence of U73122 or GF 109203X. As expected, NGF-induced PC12 cell differentiation was reduced by U73122 or GF 109203X in a dose-dependent manner, supporting our previous finding that the PLCγ-PKC axis contributes to PC12 cell differentiation (Fig. 7D).
Optogenetic activation of TrkA provides an attractive approach to dissect NGF signaling processes by leaving the endogenous TrkA expression unperturbed and by bypassing p75NTR signaling. As we have demonstrated, optogenetics can also be applied together with conventional inhibitor-based assays to further delineate signaling mechanisms. Although this work has primarily focused on delineation of TrkA signaling in mammalian cells, utilization of this optogenetic TrkA system in multicellular organisms should be applicable. Indeed, recent work from multiple laboratories including our own has demonstrated optogenetic control of intracellular signaling pathways in drosophila (Guglielmi, et al., 2015; Johnson, et al., 2017), zebrafish (Buckley, et al., 2016; Reade, et al., 2016), Xenopus (Krishnamurthy, et al., 2016), and mice (Konermann, et al., 2013; Kyung, et al., 2015; Lee, et al., 2017; Wang, et al., 2017). Notably, in vivo application of optogenetics has been significantly improved by the utilization of novel nanometer materials, such as upconversion nanoparticles (Chen, et al., 2018; He, et al., 2015; Huang, et al., 2016; Zhang, et al., 2016), to convert near-infrared light to visible light and facilitate deep-tissue light delivery. As receptor tyrosine kinase activation is a common function for a variety of growth factors and cytokines, we expect that this strategy can be generalized to study other receptor-mediated signal transduction pathways.
Significance
Cell fate is largely determined by integrating and processing a variety of signaling inputs from the environment. To effectively and precisely transmit extracellular signals into the cell, delicate signaling machinery is required. Transmembrane receptors serve as the major machinery for transducing membrane-impermeant extracellular signals. In some cases, one type of ligand may bind to a variety of receptors to elicit distinct signaling outcomes. The signaling mechanisms of these receptors are often confounded particularly where common downstream signaling pathways are activated. In these cases, mechanistic delineation of the signaling mediated by each type of receptor calls for a strategy that can successfully decouple receptor activity. Nerve growth factor is one such ligand that interacts with both its high-affinity and low-affinity receptors, TrkA and p75NTR, both of which activate the extracellular-signal-regulated kinase pathway. To delineate TrkA subcircuits from those of p75NTR, we developed an optogenetic system where light was used to specifically activate TrkA signaling in the absence of nerve growth factor. By combining optogenetics with pharmacological assays, we demonstrated that the tyrosine residues Y490 and Y785 of the TrkA intracellular domain each contributes to PC12 cell differentiation through the extracellular-signal-regulated kinase pathway in an additive manner. Because receptor tyrosine kinase signaling is involved in neurological disorders, development, and cancer, we believe that delineating the signaling outcomes of TrkA subcircuits could lead to new insights into pathological conditions.
Materials and Methods
Materials
Phusion DNA polymerase master mix was purchased from NEB (Cat. #M0531). Oligonucleotides and gBlock Gene Fragments for cloning were purchased from IDT. In-Fusion HD Cloning Plus kit was purchased from Clontech (Cat. #638909). BamHI, NheI, Turbofect transfection reagent, protease/phosphatase inhibitors, Bradford reagent, and Calcein AM were purchased from Thermo Fisher Scientific (Cat. #FD0054, #FD0973, #R0533, #A32959, #23238, #C3100MP). F12K cell media and horse serum were purchased from Gibco (Cat. #21127–022, #26050–088). Fetal bovine serum (FBS) was purchased from Sigma-Aldrich (Cat. #12303C). Penicillin-Streptomycin solution and DPBS were purchased from Corning (Cat. #30–002-CI, #21–031-CV). RIPA lysis buffer was purchased from Millipore (Cat. #20–188). LDS sample buffer was purchased from Invitrogen (Cat. #NP0007). Polyacrylamide gels, PVDF membrane, and protein standards were purchased from Bio-Rad (Cat. # 4561046, # 1620177, # 1610374). NGF and antibodies were purchased from Cell Signaling Technology (Cat. #5221, #9101, #9102, #2821, #2822, #2956, #7074). Erlotinib was purchased from Selleck Chemicals (Cat. #S7786). U73122 and GF 109203X were purchased from Tocris (Cat. #1268, #0741).
Plasmids construction
Lyn-TrkAICD-AuLOV-GFP was constructed by inserting a gBlock fragment coding the Lyn lipidation tag fused to the rat TrkA ICD and AuLOV (Lyn-TrkAICD-AuLOV) into a pEGFP-N1 vector (Clontech, discontinued; www.addgene.org/vector-database/2491/; linearized by NheI and BamHI digestion) using In-Fusion cloning. ICD mutants were generated using overlap extension PCR.
Cell culture and transfection
PC12 cells were cultured in F12K medium supplemented with 15% horse serum, 2.5% FBS, and 1× Penicillin-Streptomycin solution (complete medium). Cultures were maintained in a standard humidified incubator at 37 °C with 5% CO2. For differentiation assays, 2400 ng of DNA were combined with 7.2 μL of Turbofect in 240 μL of serum-free F12K. For western blots, 1200 ng of DNA (1200 ng No ICD construct alone or 700 ng No ICD construct + 500 ng ICD-containing construct) were combined with 3.6 μL of Turbofect in 120 μL of serum-free F12K. The transfection mixtures were incubated at room temperature for 20 minutes prior to adding to cells cultured in 35 mm dishes with 500 μL complete medium. For differentiation assays, the transfection medium was replaced with 2 mL complete medium after 3 hours of transfection to recover cells overnight. For western blots, the transfection medium was replaced with 1 mL serum-free F12K supplemented with 1× Penicillin-Streptomycin solution after 3 hours of transfection to serum-starved cells overnight.
PC12 cell differentiation assay
Transfected and recovered PC12 cells were switched to F12K supplemented with 0.15% horse serum, 0.025% FBS, and 1× Penicillin-Streptomycin solution (starvation medium) immediately prior to incubating cells on a homemade blue LED light box emitting at 300 μW/cm2. Untransfected cells were similarly switched to starvation medium immediately prior to NGF treatment. Any inhibitors were added prior to NGF or light treatment. Cells were incubated with light for 24 or 44 hours before imaging GFP fluorescence at 10× magnification using a Leica DMI8 microscope. Untransfected cells were treated with 1 μM Calcein AM for 10 minutes before imaging. Differentiation ratios were calculated as follows:
Epi-illumination fluorescence live-cell microscopy
An epi-illumination inverted fluorescence microscope (Leica DMI8) equipped with a 10×, 100× objective (HCX PL FLUOTAR 100×/1.30 oil) and a light-emitting diode illuminator (SOLA SE II 365) transfected cells. Green fluorescence was detected using the GFP filter cube (Leica, excitation filter 472/30, dichroic mirror 495, and emission filter 520/35). Exposure time for both fluorescence channel was 200 ms.
Three-dimensional structured illumination microscopy (SIM)
A total of 2 × 105 MDA-MB-231 cells were seeded into a 35 mm dish containing a 14 mm coverslip 24 hours prior to transfection with WT ICD. Super-resolution images were acquired on an N-SIM Microscope (Nikon, Tokyo, Japan) equipped with solid-state lasers (405 nm and 488 nm). Light-induced WT ICD homo-association was stimulated by 405 nm, and SIM images were acquired under 488 nm excitation. The cells were exposed to continuous blue light (405 nm) for 300 seconds. SIM images were captured at the beginning and the end of illumination using an electron-multiplying charge coupled device (EMCCD) camera (iXon 897, Andor, USA). To reduce photobleaching during SIM image acquisition, laser power (488 nm) was reduced to <20% with a minimum exposure time of 200 ms for each image. Images were obtained at 512 × 512 using Z-stacks with a step size of 0.125 μm. SIM frames were deliberately spaced at 2-s intervals. SIM images were analyzed with Nikon Elements and ImageJ.
Western blot
All transfected and serum-starved PC12 cells were treated with 10 μM Erlotinib (an EGFR inhibitor) for 5 minutes prior to illumination to further minimize baseline signaling. Cell were then illuminated for 10 minutes using a homemade blue LED light box emitting at 5 mW/cm2. For PLCγ and PKC inhibitor experiments, cells were treated with inhibitors for 10 minutes prior to light treatment. Following illumination, cells were washed once with 1 mL cold DBPS and lysed with 100 μL cold lysis buffer (RIPA + protease/phosphatase inhibitor cocktail). Lysates were centrifuged at 17,000 RCF, 4 °C for 10 minutes to pellet cell debris. Purified lysates were normalized using Bradford reagent. Normalized samples were mixed with LDS buffer and loaded onto 12% polyacrylamide gels. SDS-PAGE was performed at room temperature. Samples were transferred to PVDF membranes overnight at 30 V, 4 °C. Membranes were blocked in 5% BSA/TBST for 1 hour at room temperature and probed with the primary and secondary antibodies according to company guidelines. Membranes were incubated with ECL substrate and imaged using a Bio-Rad ChemiDoc XRS chemiluminescence detector. Signal analysis was performed using ImageJ. Activity is defined as the signal ratio of phospho-target/total target. All reported activity is normalized to the “dark” activity of each tested condition.
Statistical analysis
The p-values were determined by performing two-tailed, unpaired t-test using the GraphPad Prism software. The p-values were determined by performing two-tailed, unpaired t-test using the GraphPad Prism software.
Supplementary Material
ACKNOWLEDGMENTS
We thank Prof. Tobias Meyer at Stanford University for providing the PC12 cell line and Dr. Jun-Lin Guan at University of Cincinnati for MDA-MB-231 cells. We thank Prof. Erik Procko at the University of Illinois at Urbana-Champaign for providing split Venus plasmids. We thank Dr. Sandra McMasters from the cell media facility at the University of Illinois at Urbana-Champaign for providing DH5α competent cells. K. Z. thanks the funding support from University of Illinois at Urbana-Champaign. J.D. thanks the support from National Institutes of Health (R35GM128837).
Footnotes
COMPETING INTERESTS STATEMENT
The authors declare no competing interests.
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Kai Zhang (kaizkaiz@illinois.edu)
REFERENCES
- Arcaro A, and Wymann MP (1993). Wortmannin Is a Potent Phosphatidylinositol 3-Kinase Inhibitor - the Role of Phosphatidylinositol 3,4,5-Trisphosphate in Neutrophil Responses. Biochem J 296, 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae SS, Lee YH, Chang JS, Galadari SH, Kim YS, Ryu SH, and Suh PG (1998). Src homology domains of phospholipase C gamma 1 inhibit nerve growth factor-induced differentiation of PC12 cells. Journal of neurochemistry 71, 178–185. [DOI] [PubMed] [Google Scholar]
- Barker PA (1998). p75NTR: A study in contrasts. Cell death and differentiation 5, 346–356. [DOI] [PubMed] [Google Scholar]
- Bassili M, Birman E, Schor NF, and Saragovi HU (2010). Differential roles of Trk and p75 neurotrophin receptors in tumorigenesis and chemoresistance ex vivo and in vivo. Cancer Chemoth Pharm 65, 1047–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothwell M (1995). Functional interactions of neurotrophins and neurotrophin receptors. Annual review of neuroscience 18, 223–253. [DOI] [PubMed] [Google Scholar]
- Buckley CE, Moore RE, Reade A, Goldberg AR, Weiner OD, and Clarke JD (2016). Reversible Optogenetic Control of Subcellular Protein Localization in a Live Vertebrate Embryo. Dev Cell 36, 117–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KY, Woo D, Jung H, Lee S, Kim S, Won J, Kyung T, Park H, Kim N, Yang HW, et al. (2014). Light-inducible receptor tyrosine kinases that regulate neurotrophin signalling. Nature communications 5, 4057. [DOI] [PubMed] [Google Scholar]
- Chao MV (2003). Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat Rev Neurosci 4, 299–309. [DOI] [PubMed] [Google Scholar]
- Chen S, Weitemier AZ, Zeng X, He LM, Wang XY, Tao YQ, Huang AJY, Hashimotodani Y, Kano M, Iwasaki H, et al. (2018). Near-infrared deep brain stimulation via upconversion nanoparticle-mediated optogenetics. Science 359, 679–683. [DOI] [PubMed] [Google Scholar]
- Colombo E, Romaggi S, Medico E, Menon R, Mora M, Falcone C, Lochmuller H, Confalonieri P, Mantegazza R, Morandi L, et al. (2011). Human neurotrophin receptor p75NTR defines differentiation-oriented skeletal muscle precursor cells: implications for muscle regeneration. Journal of neuropathology and experimental neurology 70, 133–142. [DOI] [PubMed] [Google Scholar]
- Covaceuszach S, Konarev PV, Cassetta A, Paoletti F, Svergun DI, Lamba D, and Cattaneo A (2015). The conundrum of the high-affinity NGF binding site formation unveiled? Biophys J 108, 687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dechant G, and Barde YA (2002). The neurotrophin receptor p75(NTR): novel functions and implications for diseases of the nervous system. Nat Neurosci 5, 1131–1136. [DOI] [PubMed] [Google Scholar]
- Deshmukh M, and Johnson EM Jr. (1997). Programmed cell death in neurons: focus on the pathway of nerve growth factor deprivation-induced death of sympathetic neurons. Molecular pharmacology 51, 897–906. [DOI] [PubMed] [Google Scholar]
- Green SH, Rydel RE, Connolly JL, and Greene LA (1986). Pc12-Cell Mutants That Possess Low-Affinity but Not High-Affinity Nerve Growth-Factor Receptors Neither Respond to nor Internalize Nerve Growth-Factor. Journal of Cell Biology 102, 830–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes ML, Zhou J, Beattie EC, Yuen EC, Hall DE, Valletta JS, Topp KS, LaVail JH, Bunnett NW, and Mobley WC (1996). Endocytosis of activated TrkA: Evidence that nerve growth factor induces formation of signaling endosomes. Journal of Neuroscience 16, 7950–7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grusch M, Schelch K, Riedler R, Reichhart E, Differ C, Berger W, Ingles-Prieto A, and Janovjak H (2014). Spatio-temporally precise activation of engineered receptor tyrosine kinases by light. Embo J 33, 1713–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guglielmi G, Barry JD, Huber W, and De Renzis S (2015). An Optogenetic Method to Modulate Cell Contractility during Tissue Morphogenesis. Dev Cell 35, 646–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman DS, McCormack M, Schubenel R, and Hertel C (1992). Multiple trkA proteins in PC12 cells bind NGF with a slow association rate. The Journal of biological chemistry 267, 24516–24522. [PubMed] [Google Scholar]
- He L, Zhang Y, Ma G, Tan P, Li Z, Zang S, Wu X, Jing J, Fang S, Zhou L, et al. (2015). Near-infrared photoactivatable control of Ca(2+) signaling and optogenetic immunomodulation. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose M, Kuroda Y, and Murata E (2016). NGF/TrkA Signaling as a Therapeutic Target for Pain. Pain Pract 16, 175–182. [DOI] [PubMed] [Google Scholar]
- Huang C, Zhou J, Feng AK, Lynch CC, Klumperman J, DeArmond SJ, and Mobley WC (1999). Nerve growth factor signaling in caveolae-like domains at the plasma membrane. Journal of Biological Chemistry 274, 36707–36714. [DOI] [PubMed] [Google Scholar]
- Huang EJ, and Reichardt LF (2001). Neurotrophins: Roles in neuronal development and function. Annual review of neuroscience 24, 677–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K, Dou QQ, and Loh XJ (2016). Nanomaterial mediated optogenetics: opportunities and challenges. Rsc Adv 6, 60896–60906. [Google Scholar]
- Jensen EC (2013). Quantitative Analysis of Histological Staining and Fluorescence Using ImageJ. Anatomical Record-Advances in Integrative Anatomy and Evolutionary Biology 296, 378–381. [DOI] [PubMed] [Google Scholar]
- Johnson HE, Goyal Y, Pannucci NL, Schupbach T, Shvartsman SY, and Toettcher JE (2017). The Spatiotemporal Limits of Developmental Erk Signaling. Dev Cell 40, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khamo JS, Krishnamurthy VV, Sharum SR, Mondal P, and Zhang K (2017). Applications of Optobiology in Intact Cells and Multicellular Organisms. J Mol Biol 429, 2999–3017. [DOI] [PubMed] [Google Scholar]
- Kim B, and Lin MZ (2013). Optobiology: optical control of biological processes via protein engineering. Biochemical Society Transactions 41, 1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino A, Hsu PD, Heidenreich M, Cong L, Platt RJ, Scott DA, Church GM, and Zhang F (2013). Optical control of mammalian endogenous transcription and epigenetic states. Nature 500, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy VV, Khamo JS, Mei W, Turgeon AJ, Ashraf HM, Mondal P, Patel DB, Risner N, Cho EE, Yang J, et al. (2016). Reversible optogenetic control of kinase activity during differentiation and embryonic development. Development 143, 4085–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyung T, Lee S, Kim JE, Cho T, Park H, Jeong YM, Kim D, Shin A, Kim S, Baek J, et al. (2015). Optogenetic control of endogenous Ca(2+) channels in vivo. Nat Biotechnol 33, 1092–1096. [DOI] [PubMed] [Google Scholar]
- Lee D, Creed M, Jung K, Stefanelli T, Wendler DJ, Oh WC, Mignocchi NL, Luscher C, and Kwon HB (2017). Temporally precise labeling and control of neuromodulatory circuits in the mammalian brain. Nat Methods 14, 495–503. [DOI] [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, and Jaenisch R (1992). Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell 69, 737–749. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, and Hempstead BL (2001). Regulation of cell survival by secreted proneurotrophins. Science 294, 1945–1948. [DOI] [PubMed] [Google Scholar]
- Liebl DJ, Klesse LJ, Tessarollo L, Wohlman T, and Parada LF (2000). Loss of brain-derived neurotrophic factor-dependent neural crest-derived sensory neurons in neurotrophin-4 mutant mice. Proceedings of the National Academy of Sciences of the United States of America 97, 2297–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YS, Shreder KR, Gai WZ, Corral S, Ferris DK, and Rosenblum JS (2005). Wortmannin, a widely used phosphoinositide 3-kinase inhibitor, also potently inhibits mammalian polo-like kinase. Chemistry & biology 12, 99–107. [DOI] [PubMed] [Google Scholar]
- Lomen-Hoerth C, and Shooter EM (1995). Widespread neurotrophin receptor expression in the immune system and other nonneuronal rat tissues. Journal of neurochemistry 64, 1780–1789. [DOI] [PubMed] [Google Scholar]
- Lu B, Pang PT, and Woo NH (2005). The yin and yang of neurotrophin action. Nat Rev Neurosci 6, 603–614. [DOI] [PubMed] [Google Scholar]
- Martin KJ, Shpiro N, Traynor R, Elliott M, and Arthur JS (2011). Comparison of the specificity of Trk inhibitors in recombinant and neuronal assays. Neuropharmacology 61, 148–155. [DOI] [PubMed] [Google Scholar]
- Mauro A, Ciccarelli C, De Cesaris P, Scoglio A, Bouche M, Molinaro M, Aquino A, and Zani BM (2002). PKC alpha-mediated ERK, JNK and p38 activation regulates the myogenic program in human rhabdomyosarcoma cells. J Cell Sci 115, 3587–3599. [DOI] [PubMed] [Google Scholar]
- Muller K, Naumann S, Weber W, and Zurbriggen MD (2014). Optogenetics for gene expression in mammalian cells. Biological chemistry. [DOI] [PubMed] [Google Scholar]
- Obermeier A, Bradshaw RA, Seedorf K, Choidas A, Schlessinger J, and Ullrich A (1994). Neuronal differentiation signals are controlled by nerve growth factor receptor/Trk binding sites for SHC and PLC gamma. Embo J 13, 1585–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passino MA, Adams RA, Sikorski SL, and Akassoglou K (2007). Regulation of hepatic stellate cell differentiation by the neurotrophin receptor p75NTR. Science 315, 1853–1856. [DOI] [PubMed] [Google Scholar]
- Poo MM (2001). Neurotrophins as synaptic modulators. Nat Rev Neurosci 2, 24–32. [DOI] [PubMed] [Google Scholar]
- Reade A, Motta-Mena LB, Gardner KH, Stainier DY, Weiner OD, and Woo S (2016). TAEL: A zebrafish-optimized optogenetic gene expression system with fine spatial and temporal control. Development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka Y, Hagemann AI, Piepenburg O, and Smith JC (2007). Nuclear accumulation of Smad complexes occurs only after the midblastula transition in Xenopus. Development 134, 4209–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal RA (2003). Selectivity in neurotrophin signaling: theme and variations. Annu Rev Neurosci 26, 299–330. [DOI] [PubMed] [Google Scholar]
- Shyu YJ, Liu H, Deng X, and Hu CD (2006). Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions. Biotechniques 40, 61–66. [DOI] [PubMed] [Google Scholar]
- Stephens RM, Loeb DM, Copeland TD, Pawson T, Greene LA, and Kaplan DR (1994). Trk Receptors Use Redundant Signal-Transduction Pathways Involving Shc and Plc-Gamma-1 to Mediate Ngf Responses. Neuron 12, 691–705. [DOI] [PubMed] [Google Scholar]
- Susen K, Heumann R, and Blochl A (1999). Nerve growth factor stimulates MAPK via the low affinity receptor p75(LNTR). Febs Lett 463, 231–234. [DOI] [PubMed] [Google Scholar]
- Tischer D, and Weiner OD (2014). Illuminating cell signalling with optogenetic tools. Nat Rev Mol Cell Bio 15, 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toettcher JE, Gong DQ, Lim WA, and Weiner OD (2011). Light Control of Plasma Membrane Recruitment Using the Phy-Pif System. Method Enzymol 497, 409–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker CL (2012). Manipulating cellular processes using optical control of protein-protein interactions. Prog Brain Res 196, 95–117. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Hirai S, Osada S, Suzuki A, Mizuno K, and Ohno S (1996). Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. The Journal of biological chemistry 271, 23512–23519. [DOI] [PubMed] [Google Scholar]
- Wang L, Liang Z, and Li G (2011). Rab22 controls NGF signaling and neurite outgrowth in PC12 cells. Mol Biol Cell 22, 3853–3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wildes CP, Pattarabanjird T, Sanchez MI, Glober GF, Matthews GA, Tye KM, and Ting AY (2017). A light- and calcium-gated transcription factor for imaging and manipulating activated neurons. Nat Biotechnol 35, 864–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrman T, He XL, Raab B, Dukipatti A, Blau H, and Garcia KC (2007). Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron 53, 25–38. [DOI] [PubMed] [Google Scholar]
- Widenfalk J, Lundstromer K, Jubran M, Brene S, and Olson L (2001). Neurotrophic factors and receptors in the immature and adult spinal cord after mechanical injury or kainic acid. The Journal of neuroscience : the official journal of the Society for Neuroscience 21, 3457–3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, and Cui B (2015). Optogenetic control of intracellular signaling pathways. Trends in biotechnology 33, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Fishel Ben Kenan R, Osakada Y, Xu W, Sinit RS, Chen L, Zhao X, Chen JY, Cui B, and Wu C (2013). Defective axonal transport of Rab7 GTPase results in dysregulated trophic signaling. The Journal of neuroscience : the official journal of the Society for Neuroscience 33, 7451–7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Huang L, Li Z, Ma G, Zhou Y, and Han G (2016). Illuminating Cell Signaling with Near-Infrared Light-Responsive Nanomaterials. ACS nano 10, 3881–3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou XX, Chung HK, Lam AJ, and Lin MZ (2012). Optical control of protein activity by fluorescent protein domains. Science 338, 810–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoltowski BD, and Gardner KH (2011). Tripping the light fantastic: blue-light photoreceptors as examples of environmentally modulated protein-protein interactions. Biochemistry 50, 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.