

Summary
CD1A is a cell surface protein expressed on Langerhans cells and cortical thymocytes that could potentially be used as an immunotherapeutic target in Langerhans Cell Histiocytosis (LCH), the cortical subtype of T-cell acute lymphocytic leukaemia (T-ALL) and other CD1A-positive tumours. The monoclonal antibody (mAb) CR2113 was selected from a panel of six fully human mAbs isolated from a semi-synthetic phage display library, based on specificity and avidity against cells expressing CD1 antigen variants. CR2113 recognized CD1A in T-ALL cell lines and patient samples. Confocal microscopy revealed that the CR2113-CD1A complex was internalized at 37°C. Furthermore, while CR2113 induced moderate complement-dependent cytotoxicity (CDC), potent antibody-dependent cell cytotoxicity (ADCC) activity was observed against CD1A expressing cell lines as well as T-ALL cell lines and T-ALL patient samples. In vivo experiments showed that CR2113 as a naked antibody has modest but specific anti-tumour activity against CD1A-expressing tumours. CR2113 is a high-affinity human anti-CD1A mAb with significant ADCC activity. These properties make CR2113 a candidate for clinical diagnostic imaging and therapeutic targeting of LCH as well as potential use in other clinical applications.
Keywords: CD1A, monoclonal antibody, immunotherapy, Langerhans cell histiocytosis, T-lineage acute lymphoblastic leukaemia
Introduction
CD1A is a 49,000 Dalton sialoglycoprotein and a member of the CD1 family of non-classical major histocompatibility complex (MHC) class I-like proteins (Porcelli 1995). CD1A is non-covalently associated with β2-microglobulin (β2m) which appears to be necessary for efficient folding and surface expression (Porcelli & Modlin 1999). Similar to MHC I, CD1A molecules are internalized following antibody binding (Ray et al 1989). CD1A can be serologically defined by four different epitopes, which are designated as groups A, B, C, and D. Cross-inhibition studies with different monoclonal antibodies (mAbs) have further revealed that epitopes A, C, and D are distinct while epitope B overlaps with epitopes A and C (Amiot et al 1986, Amiot et al 1987, Kahn-Perles et al 1985, Olive et al 1984).
In normal tissues, CD1A expression is restricted to the surface of Langerhans cells (Fithian et al 1981) and mature cortical thymocytes (Sotzik et al 1993). Both cell populations can be repopulated from CD1A-negative bone marrow precursors. CD1A expression is also used to make a definitive diagnosis of Langerhans cell in Langerhans cell histiocytosis (LCH) when observed on the pathological dendritic cells (DC), which are also characterized by mutations of BRAF1 or activation of BRAF1-related signalling pathways (Badalian-Very et al 2010). In addition, CD1A expression has been reported in a number of haematological malignancies, such as B-cell chronic lymphocytic leukaemia (B-CLL) (Merle-Beral et al 1989), T-cell acute lymphocytic leukaemia (T-ALL) (Salamone et al 1990, Wuchter et al 2002), B-cell acute lymphocytic leukaemia (B-ALL) (Salamone et al 1990), and occasionally in French-American-British classification (FAB) subtypes M4 and M5 acute myeloid leukaemia (AML) (Misery et al 1992).
A radiolabelled version of the murine anti-CD1A antibody, NA1/34, has been tested in patients with refractory LCH. This antibody was able to identify areas of presumed disease activity in patients, but was associated with significant anti-murine antibody host responses (Kelly et al 1994), thus making it less desirable as a therapeutic or diagnostic agent. In an effort to circumvent the adverse immunological effects of repeated administration of a murine-derived mAb, we generated a panel of fully human anti-CD1A mAbs using Mabstract phage antibody technology (Van Ewijk et al 1997). One of these mAbs, CR2113, proved to have specificity for CD1A with a high binding affinity with significant antibody-dependent cell cytotoxicity (ADCC) as well as anti-tumour cell activity.
Methods
Cell lines and patient samples
The NA1/34 hybridoma cell line (kindly supplied by Dr. C. Milstein, Cambridge, United Kingdom) was maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, 2 mM L-glutamine and 10 mM sodium pyruvate. The mouse melanoma cell line (B16) was maintained in Dulbecco’s modified Eagle medium supplemented with 10% FBS, 1% penicillin-streptomycin and 2 mM L-glutamine. The B16 cell line was transfected with either cDNA encoding human CD1A in an expression vector or empty vector only (pCDNA 3.1 (+) neo) and maintained under selection of Geneticin (0.5 mg/ml active). C1R cells transfected with cDNA encoding human CD1A, CD1B, CD1C, CD1D or vector only (kindly supplied by Dr. S.B. Wilson, Boston, MA, USA) were maintained in RPMI 1640 selection media (Geneticin 0.5 mg/ml active) supplemented with 10% FBS, 1% penicillin-streptomycin and 2 mM L-glutamine. T-ALL cell lines were maintained in RPMI 1640 medium containing 10 mM HEPES, 1 mM sodium pyruvate, 4.5 g/l glucose, 1.5 g/l sodium bicarbonate, 10% FBS, 1% penicillin-streptomycin and 2 mM L-glutamine. Cells were cultured at 37° C, 5% CO2. All reagents were purchased from Invitrogen (Carlsbad, CA, USA). Frozen peripheral blood or bone marrow samples from patients with T-ALL were obtained from the Children’s Oncology Group after Institutional Review Board (IRB) approval.
Phage display selection of CD1A specific single-chain variable fragment (scFv)
Immature DCwere generated from cultured monocytes. Monocytes were obtained from pooled normal blood by density gradient separation and cultured for 5 days in the presence of granulocyte-macrophage colony-stimulating factor and interleukin-4 to differentiate them into immature CD1A+/CD83− DC. Phage-selections were performed essentially as described (de Kruif et al 1995, Van Ewijk et al 1997). Briefly, immature DC were mixed with an excess of peripheral blood leucocytes (PBL) used as a subtractor cell population and then combined with a semi-synthetic phage antibody display library (Van Ewijk et al 1997) blocked with phosphate-buffered saline (PBS) containing 5% milk powder (w/v). The cells were incubated overnight at 4° C with continuous rotation and then extensively washed and labelled with CD1A-phycoerythrin (PE) and CD83-fluorescein isothiocyanate (FITC) conjugated antibodies (Becton Dickinson, Erembodegem, Belgium) using standard protocols. Immature CD1A+/CD83− DCs were sorted using a FACStarplus fluorescence-activated cell sorter equipped with an argon laser (Becton Dickinson). Cell-bound phages were eluted with 50 mM glycine-HCl pH 2.2, followed by neutralization with 1 M Tris-HCl pH 7.5, and infection into XL1-Blue competent bacteria (Agilent, Santa Clara, CA, USA). After growth, colonies were scraped from the plates for rescue and a second round of selection was performed. After the completion of three identical selection rounds, individual scFv phages were prepared from single colonies and screened using a fluorescence-activated cell sorting-based phage staining protocol for binding to immature DC, PBL, the cell lines K562, U266, IM9, U937, THP, CEM, Fravel, HL60, Raji, RPMI8226, HepgII, HELA, HT29, Jurkat, a C1R cell line transfected with cDNAs encoding different CD1 antigens, and A431 cell lines transfected with the cDNAs encoding CD80, CD83 and CD86 (de Kruif et al 1995).
Production of Human IgG1 antibodies
Selected scFv-phage clones specifically binding human CD1A were converted into full-length IgG1 molecules as described (Boel et al 2000). Variable heavy (IGHV) and variable light (IGLV) chain genes were amplified by polymerase chain reaction using primers to restore human framework and append restriction sites. The resulting fragments were cloned into eukaryotic expression vectors containing constant antibody domains. IgG1 was expressed by transient transfection of mammalian cells in serum-free medium and purified on protein A affinity columns. The concentration of purified antibodies was measured by optical absorbance at 280 nm and the purity and integrity of the antibodies were analysed by reducing and non-reducing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Purification of mAb NA1/34
Supernatant containing mAb NA1/34 (IgG2a isotype) was collected from cultured NA1/34 hybridoma cells and mAb purified by protein-A affinity chromatography (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. Purity of NA1/34 was assessed using SDS–PAGE under reducing conditions. Protein was dialysed against PBS and the concentration was determined using the Bradford assay. NA1/34 mAb was sterilized using a 0.22-μM low binding protein filter (Millipore, Bedford, MA, USA) and stored at −20°C in phosphate-buffered saline (PBS, pH 7.4).
Commercial antibodies
The following primary antibodies were used: mouse anti-human CD1A-FITC, mouse anti-human CD1A, mouse anti-human CD1B, and mouse anti-human CD1D (BD Biosciences, San Diego, CA, USA). Mouse anti-human CD1C-FITC was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Goat anti-mouse IgG1-FITC, Mouse IgG1, κ anti-human heavy chain-FITC and Streptavidin-FITC (BD Biosciences, San Diego, CA, USA) were used as secondary antibodies in the flow cytometry assays. For apoptosis assays, goat anti-human IgG (Fcγ fragment specific) and goat anti-mouse IgG (Fcγ fragment specific antibodies) secondary antibodies were used (Jackson Immunoresearch Laboratories, Inc., West Grove, PA, USA). Isotype control antibodies included mAb CR2027, IgG1, GBS III-biotin-labelled IgG1 (Crucell Holland BV, Leiden, The Netherlands), purified mouse IgG2a, κ (BD Biosciences, San Diego, CA), and human IgG1 (Lampire Biological Laboratories, Pipersville, PA, USA)
Biotinylation of mAbs
Monoclonal antibodies CR2113, CR2114, CR2115, CR2116, CR2117, CR2118 and NA1/34 were incubated with 1M NaHCO3 and EZ-link Sulfo-NHS-SS-biotin, according to manufacturer’s instruction (Pierce, Rockford, IL, USA). Unincorporated biotin was removed by dialysis against PBS. Biotin-labelled mAb concentration was determined by measurement of the absorbance at a wavelength of 280 nm. The degree of biotinylation was determined by mixing the biotin-labelled mAbs with a HABA/avidin solution according to manufacturer’s instruction (Sigma, St. Louis, MO, USA). The absorbance at a wavelength of 500 nm was measured and compared to a standard curve. The molar ratio of biotin/mAb was then determined.
Surface Plasmon Resonance Analysis
The affinity and kinetic binding properties of human (CR2113) and mouse (NA1/34) anti-hCD1A antibodies binding to hCD1A were measured using a Biacore 3000 spectrometer (Biacore, Piscataway, NJ, USA). Anti-human and anti-mouse Fc antibodies (human and mouse antibody capture kit, Biacore, Pharmacia Biosensor AB, Uppsala, Sweden) were immobilized onto a CM5 sensor chip using the amine coupling method according to the manufacturers instructions. Next, 150–200 response units (RU) of CR2113, NA1/34 or mouse and human isotype control antibodies were captured on the sensor chip at 10 μl/min with running buffer (10 mm HEPES, pH7.4, 150 mm NaCl, 0.005% Tween20, 0.3mM EDTA). Recombinant hCD1A was expressed in Spodoptera frugiperda cells (SF9) using the baculovirus expression system (Clontech, Mountain View, CA, USA) and purified according to published protocols (Zajonc et al 2003). The hCD1A protein was diluted in running buffer at 0.5–32 nM (CR2113) and 10–640 nM (NA1/34). A replicate series of six to seven different concentrations were injected. Binding of hCD1A to the anti-hCD1A antibodies was monitored with a 2- to 3-min injection and 10 min dissociation at 25°C with a flow rate of 30 μl/min (CR2113) or 50 μl/min (NA1/34). The surface was then regenerated with a 20 μl injection of 3 M MgCl2 (CR2113) or 10 mM Glycine, pH1.7 (NA1/34) and subsequent coating with 150–200 RU of the anti-hCD1A antibody. Before analysis, non-specific binding of hCD1A to the isotype control antibody was subtracted for each concentration and instrument noise was removed by subtracting a buffer injection from all curves. Analysis of kinetic rate constants was performed with BIAevaluation 4.1 software using global fitting to a Langmuir 1:1 binding model. The binding experiments were performed either 2 (NA1/34) or 3 times (CR2113) and the statistical mean, including standard deviation, was calculated using Microsoft Excel (Microsoft, Redmond, WA, USA).
Flow cytometry
For all flow cytometry experiments, 5 × 105 cells were stained. Primary mAbs were incubated with live cells on ice for 30 min. Cells were then washed with PBS containing1% BSA and incubated with secondary antibodies for 20 mins on ice. Experiments were done in triplicate. After antibody binding, cells were washed, centrifuged and resuspended in FACS buffer (PBS/1% BSA, 0.5% paraformaldehyde). Cells were analysed using a FACS Calibur flow cytometer and Cell Quest Pro software (BD Biosciences, San Diego, CA, USA) or Flow Jo software (Ashland, OR, USA). Macaque monkey thymocytes and peripheral blood lymphocytes were kindly provided by Dr. Phil Johnson and Mary J. Connell, Childrens Hospital of Philadelphia, Philadelphia, PA, USA).
Fluorescent labelling of primary mAbs
Primary mAbs (CR2113, NA1/34, IgG1) were labelled with Alexa Fluor 488 dye (AF488 Monoclonal Antibody Labeling Kit, Molecular Probes Eugene, OR, USA) according to the manufacturer’s protocol. For each of the mAbs, 100 μl (0.97–1.0 mg/ml) was reacted with Alexa Fluor 488 dye and incubated for 1 h at 25°C. After incubation, the entire reaction volume was transferred to a purification spin column and centrifuged (5 min at 1,100 × g). Protein concentration and degree of labelling were determined by measuring the absorbance of the eluate at both 280 nm and 494 nm.
Confocal Microscopy
B16 CD1A+ and B16 vector only transfected cells were plated (2.5 × 105 cells per well) on coverslips and allowed to attach overnight. Culture medium was changed and cells were incubated with either AF488-labelled CR2113 or AF488-labelled NA1/34 (3 μg/ml) for 1 h at either 4° C or 37° C. Immediately after incubation, plates were placed on ice and transferred to 4° C. All plates were then washed with PBS (3 times × 5 min each wash at 4° C). Cells were fixed with 3% paraformaldehyde in PBS, pH 7.4, 4° C for 30 min, and then washed again with PBS (3 × 5 min for each wash at 4° C). Prepared coverslips were mounted on microscope slides with Slow Fade Antifade reagent (Molecular Probes, Eugene, OR, USA). Cells were analysed by confocal microscopy at 60x magnification (Nikon, Melville, NY, USA) at the Sidney Kimmel Comprehensive Cancer Center Cell Imaging CORE Facility at Johns Hopkins University.
Analysis of Apoptosis
Apoptosis was measured using the Cellular DNA Fragmentation enzyme-linked immunosorbent assay (ELISA) (Roche, Indianapolis, IN, USA). BrdU-labelled target cells (1 × 104 cells per well) were plated in triplicate in U-bottom 96-well plates and allowed to attach over a 5-h period. Cells were incubated for 48 h at 37° C in 5% CO2 with CR2113, NA1/34 or isotype control mAb (4.5 μg/ml) in the presence or absence of a cross-linking secondary antibody (20 μg/ml). Cells treated with mitomycin C (10 μg/ml, Sigma, St. Louis, MO, USA) were used as a positive control. Cell lysates were prepared and measured for BrdU content according to manufacturer’s instruction.
Cytotoxicity assays
Non-radioactive cytotoxicity assays (Cytotox 96, Promega, Madison, WI, USA) were performed to measure complement-dependent cytotoxicity (CDC) and ADCC. For CDC assays, primary mAbs (4.5 μg/ml) alone or in combination with 25 μl of a 1:10 dilution of normal human serum complement (Quidel, San Diego, CA) were added to target cells (5 × 103 cells per sample), followed by a 3-h incubation at 37° C in 5% CO2. For ADCC assays, normal peripheral blood mononuclear cells (PBMC) were isolated from heparinized whole blood from healthy volunteers by Ficoll hypaque density centrifugation. For some ADCC assays, natural killer (NK) cells were isolated using the NK Cell Isolation Kit II (Miltenyi Biotec Inc., Auburn, CA, USA). Target cells were incubated for 5 h at 37° C in 5% CO2 with primary antibodies (4.5 μg/ml) alone or in combination with either PBMC or NK cells using an effector/target ratio of 50:1. Cytotoxicity was determined as a function of lactate dehydrogenase (LDH) enzymatic activity released from the cytosol of damaged target cells into the supernatant. LDH activity was quantitated by monitoring absorbance at a wavelength of 490 nm using an ELISA reader. The percentage of cytolysis was calculated according to the following equation: Cytotoxicity (%) = (experimental effector cell spontaneous release minus target cell spontaneous release/target cell maximum release minus target cell spontaneous release) × 100.
Murine Tumour Growth Inhibition Studies
Non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice (Johns Hopkins University Oncology Department Immune-compromised Animal Facility, Baltimore, MD, USA) were used according to approved institutional protocol. Briefly, 5–7 week-old males were injected subcutaneously with either CD1A+ B16 mouse melanoma cells (1 × 106 in 200 ul PBS per injection) or CD1A- (vector only) B16 cells (same parameters). There were three groups of five mice each. Each mouse received two injections (one per flank) of tumour cells. Two days post-cell injection, the CD1A+ control group received isotype (150 μg /mouse; tail vein injection) while the CD1A+ experimental group received mAb CR2113 (150 μg /mouse; tail vein). The CD1A- group also received mAb CR2113 (150 μg /mouse; tail vein). Injections were repeated every 4 days. At onset of observable tumour burden, tumours were measured every other day (typically starting day seven). Tumours were allowed to grow until they reached maximum size (2 cm3) allowed by protocol. Mice were then sacrificed and tumours were harvested for flow cytometry and immunohistochemical analysis. Tumour xenograft experiments were approved by the Animal Protection Committee under protocol MO10M292 and anonymous patient samples were obtained following IRB approval (05-02-02-01e).
Statistical analysis
Kolmogorov-Smirnov statistics (Cell Quest Pro software, Becton Dickinson) were used to analyse flow cytometry data by determining the maximum shift between the cumulative distributions of peak values (D values). T-test analysis (unpaired, two-tailed) was performed to calculate significance of mAb binding, while two-way analysis of variance (ANOVA) was performed to determine the statistical significance (p <0 0.05) between mAb and isotype treatment of tumours (Prism, Graphpad, La Jolla, CA, USA).
Results
Isolation and specificity of human anti-CD1A mAbs
We employed a semi-synthetic phage display library of 3.6 × 108 human scFv fragments to isolate antibodies binding to CD1A. The scFv were selected for binding to culture-derived CD1A-positive immature DC in the context of a heterogeneous population of CD1A-negative cells that were provided in 10-fold excess. In total, three selection rounds were performed. After both the second and third rounds of screening, phage antibodies were tested for specific binding to a variety of cell-types, including cultured DCs, PBLs, selected myeloid and lymphoid leukaemia cell lines and cell lines transfected with cDNA encoding either CD1A, CD80, CD83 or CD86 (the latter three used as controls).
Twenty scFv phage antibodies were identified that bound exclusively to a C1R cell line transfected with CD1A, as well as to immature DC and mature DC but not to mock transfected C1R line (Supplemental Figure 1). Sequence analysis of the IGHV and IGLV genes of the 20 CD1A-specific phage clones revealed six unique scFv’s, based on V(D)J recombination events, which were reformatted as a complete IgG1 for further characterization.
To further refine the analysis of specificity of the mAbs, C1R cells transfected with cDNAs encoding CD1A, CD1B, CD1C or CD1D as well as empty vector (C1R mock) or B16 mouse melanoma cells transfected with cDNA encoding CD1A or empty vector were analysed for binding to each of six candidate mAbs (Figure 1). Cells were tested for binding with CR2113, CR2114, CR2115, CR2116, CR2117 and CR2118 mAbs at 1, 3, and 10 μg/ml. Of these, CR2113 bound specifically to CD1A expressing cells. The low level of binding of CR2113 to CD1D was not statistically significant. CR2114 and CR2117 bound predominantly to CD1A and showed some binding to CD1C. CR2118 reacted to CD1A expressing B16 cells but showed lower binding to CD1A expressing C1R cells. CR2115 and CR2116 showed minimal to no significant binding to any of the CD1 antigens perhaps as a result of the reformatting from scFv, which can alter the antigen binding properties of some antibodies. There was minimal to no binding to both C1R and B16 mock cells for all of the mAbs.
Figure 1.
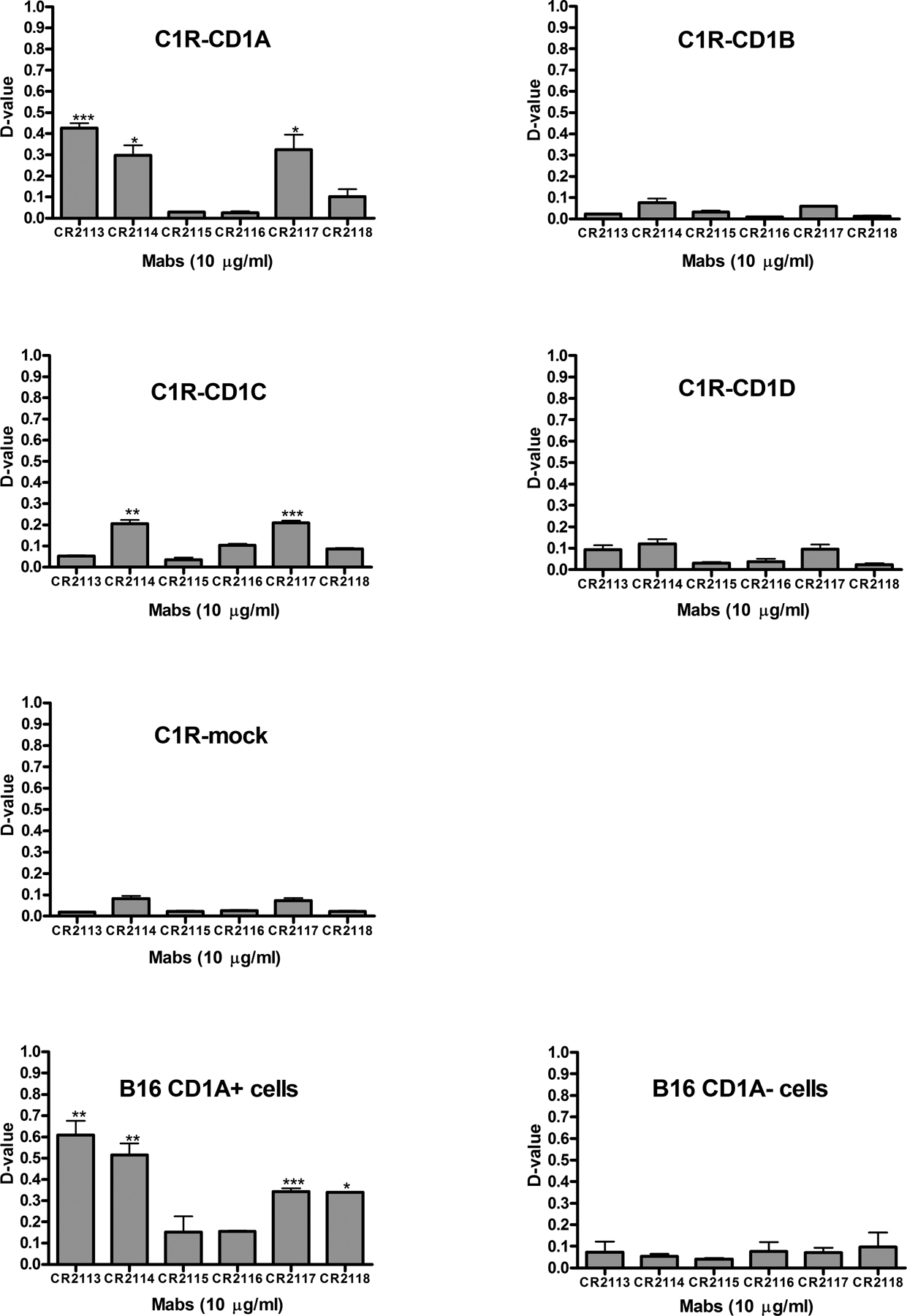
Reactivity and specificity of the human anti-CD1A monoclonal antibodies (mAbs) on different CD1 antigens expressed in the C1R and B16 cell lines. C1R cells expressing either CD1A, CD1B, CD1C or CD1D or no CD1 antigens (termed C1R mock) as well as B16 cells transfected with a viral expression vector containing the cDNA encoding CD1A or empty vector were stained with either CR2113, CR2114, CR2115, CR2116, CR2117 or CR2118 mAbs at 1, 3, and 10 μg/ml. Data for 10 μg mAb/ml are shown and representative of all three doses. Mouse IgG1, κ-FITC was used as a secondary antibody. Cells were analysed by flow cytometry using Kolmogorov-Smirnov statistics to quantitate binding as described in Methods. Specificity was determined by the level of binding to cells expressing specific CD1 isoforms compared to background, isotype control antibody staining. Statistical significance was determined by t-test (unpaired, two-tailed) analysis when comparing the binding of specific monoclonal antibodies to background staining assessed by antibody isotype control staining on cells expressing specific CD1 isoforms. Error bars represent standard deviations. CR2113, CR2114, CR2117 and CR2118 mAbs showed significant binding to CD1A expressing cells with CR2113 having the strongest specific binding characteristics. CR2114 and CR2117 also showed some affinity for CD1C. Legend: ***: p< 0.001, **: p< 0.01, *: p<0.05, ns: p>0.05
Competitive binding experiments of anti-CD1A mAbs
To determine if the different anti-CD1A mAbs recognize the same epitope, competitive binding experiments were done. Competition between CR2113, CR2114, CR2117 and CR2118 was examined by staining B16 CD1A+ cells with the unlabelled form of three of the four mAbs or the isotype control at different concentrations (0.1, 0.3, 1, 3, 10, 30 μg/ml). The cells were then exposed to the competing biotin-labelled fourth mAb. B16 mouse melanoma cells expressing CD1A were incubated with unlabelled CR2113, CR2114, CR2117 or CR2118 at different concentrations (0.1, 0.3, 1, 3, 10, 30, 50, 100 μg/ml) for 30 min at room temperature and then exposed to their respective biotin-labelled mAbs. Isotype control antibody versus each of the biotin-labelled CR2113, CR2114, CR2117 and CR2118 mAbs binding to B16 CD1A+ cells was also tested at different concentrations (0.1, 0.3, 1, 3, 10, 30 μg/ml) under the same conditions. After incubation with primary mAbs, cells were washed and then incubated with biotin-labelled antibody for 5 min at 4° C. Streptavidin-FITC was used as the secondary reagent.
The results showed that CR2113, CR2117 and CR2118 effectively competed with themselves by blocking their own binding (Supplemental Figure 2a). CR2114 displaced itself but only at higher concentrations. CR2113 was not displaced by CR2114, CR2117 or CR2118, but significantly competed with each of the other mAbs. CR2114 competed with CR2117 and CR2118. CR2118 competed with CR2117, but CR2117 did not compete with CR2118 (Supplemental Figure 2b). The isotype control antibody did not compete for binding with any of the mAbs (Supplemental Figure 2c). These data demonstrate that CR2113 probably has the highest binding affinity and binds to at least a completely or partially overlapping epitope of CD1A recognized by the other mAbs. The competitive binding experiments as described above were also performed with mAbs CR2113 and NA1/34 (Supplemental Figure 2d). CR2113 displaced NA1/34, but NA1/34 could only partially compete with CR2113, suggesting that CR2113 has a higher binding affinity than NA1/34 for CD1A. As a control, NA1/34 was shown to effectively compete with itself. These data show that CR2113 and NA1/34 bind to a similar or overlapping epitope(s). Based on these results, the CR2113 mAb was chosen for more extensive analysis.
Affinity and Kinetic Binding Properties of CR2113
The affinity and kinetic binding properties of human anti-human CD1A mAb CR2113 binding to hCD1A were measured by surface plasmon resonance (SPR) and compared with the binding kinetics of mouse mAb NA1/34 (Figure 2a). To measure a monovalent interaction between the Fab arm of the mAb and hCD1A, the anti-hCD1A mAbs were immobilized on the sensor chip in separate experiments and the hCD1A analyte was passed over the bound antibodies of interest. Similar to the competitive binding experiments, human CR2113 mAb had a 6-fold higher binding affinity towards human CD1A than the mouse NA1/34 antibody (KD=0.55 nM vs. 3.39 nM; Figure 2b). The off-rates were almost identical (ka=3.78×10−4/s for NA1/34 and 3.47×10−4/s for CR2113) while the on-rate was five-times faster for CR2113 (kd=6.84 × 105/M/s vs. 1.29×105/M/s). These observed values are in agreement with data from other protective antibodies (Pless et al 2001) and clearly demonstrate that human mAb CR2113 binds efficiently and with high affinity to human CD1A. Furthermore, when hCD1A was immobilized on the sensor chip and the mAb was passed over as the analyte, no dissociation of mAb CR2113 was detectable (data not shown), consistent with the expected gain in avidity through bivalent CD1-mAb interaction.
Figure 2.
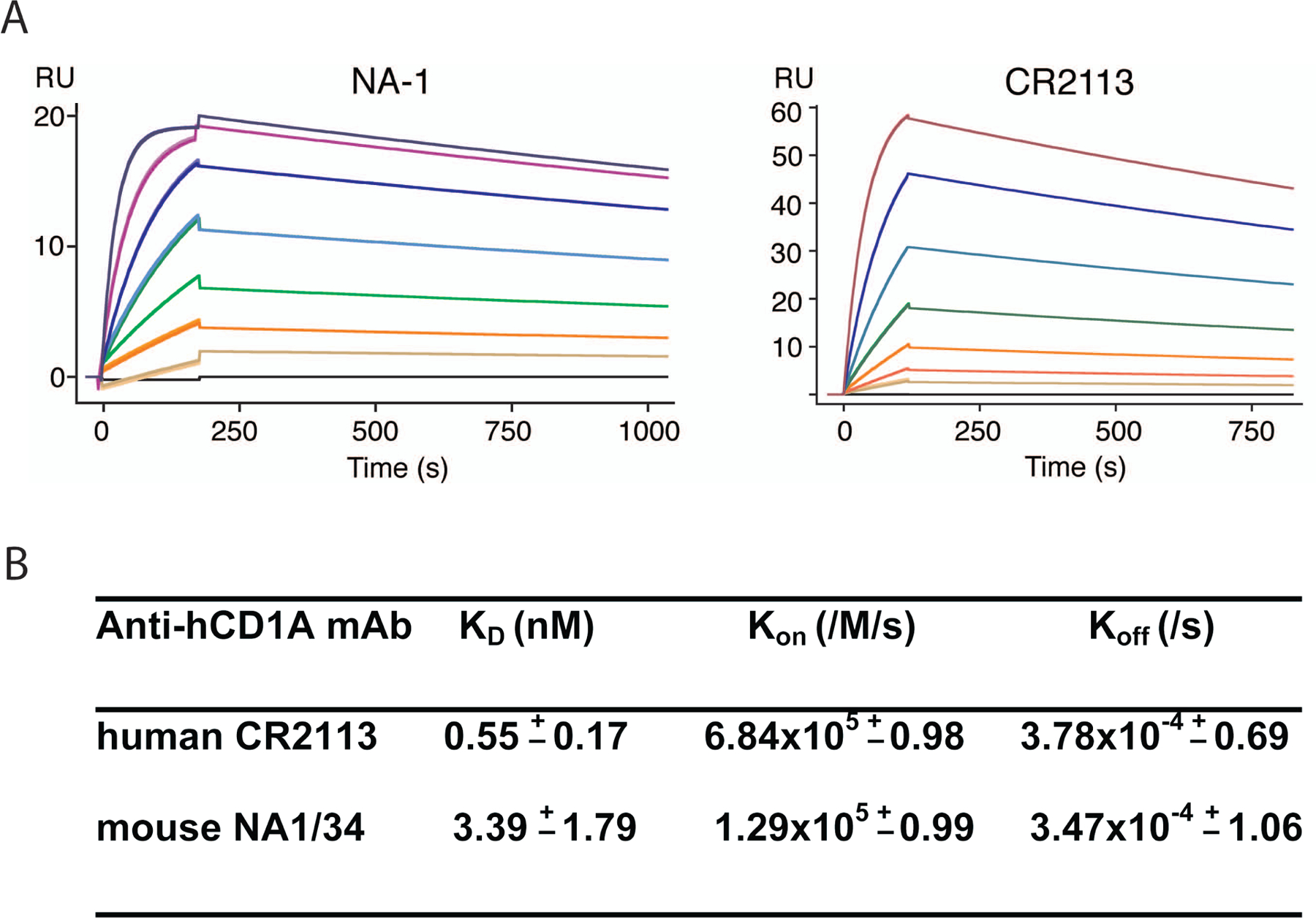
Affinity and kinetic binding measurements by surface plasmon resonance. Binding kinetics of mouse and human anti-human CD1A mAbs NA1/34 and CR2113, respectively, were determined by surface plasmon resonance. Sensograms of mouse mAb NA1/34 (left panel) and human CR2113 (right panel) are depicted (A). Increasing concentrations of hCD1A from 0.5–32nM (CR2113) and 10–640 nM (NA1/34) in two-fold dilutions were injected and the binding responses were superimposed. A representative experiment is shown. Affinity and kinetic parameters are summarized in B. Numbers are mean (± s.e.m) of two (NA1/34) and three (CR2113) separate experiments.
Internalization of CR2113 visualized by confocal microscopy
A potentially important characteristic of therapeutic mAbs is their ability to internalize following binding to their cell surface epitope. In order to determine whether there is internalization of CR2113 after binding to CD1A, B16 mouse melanoma cells transfected with CD1A or vector only were incubated with CR2113-AF488 at either 37° C or 4° C for 1 h. Subsequently all cells were kept at 4° C and analysed by confocal microscopy. As a positive control B16 CD1A+ cells were exposed to NA1/34-AF488. CR2113-AF488/CD1A complex internalization was visible in cells incubated at 37° C, but not at 4° C (Figure 3). NA1/34-AF488 staining of the cells showed similar results. B16 vector only cells incubated with either CR2113-AF488 or NA1/34-AF488 did not show any internal staining (data not shown).
Figure 3.
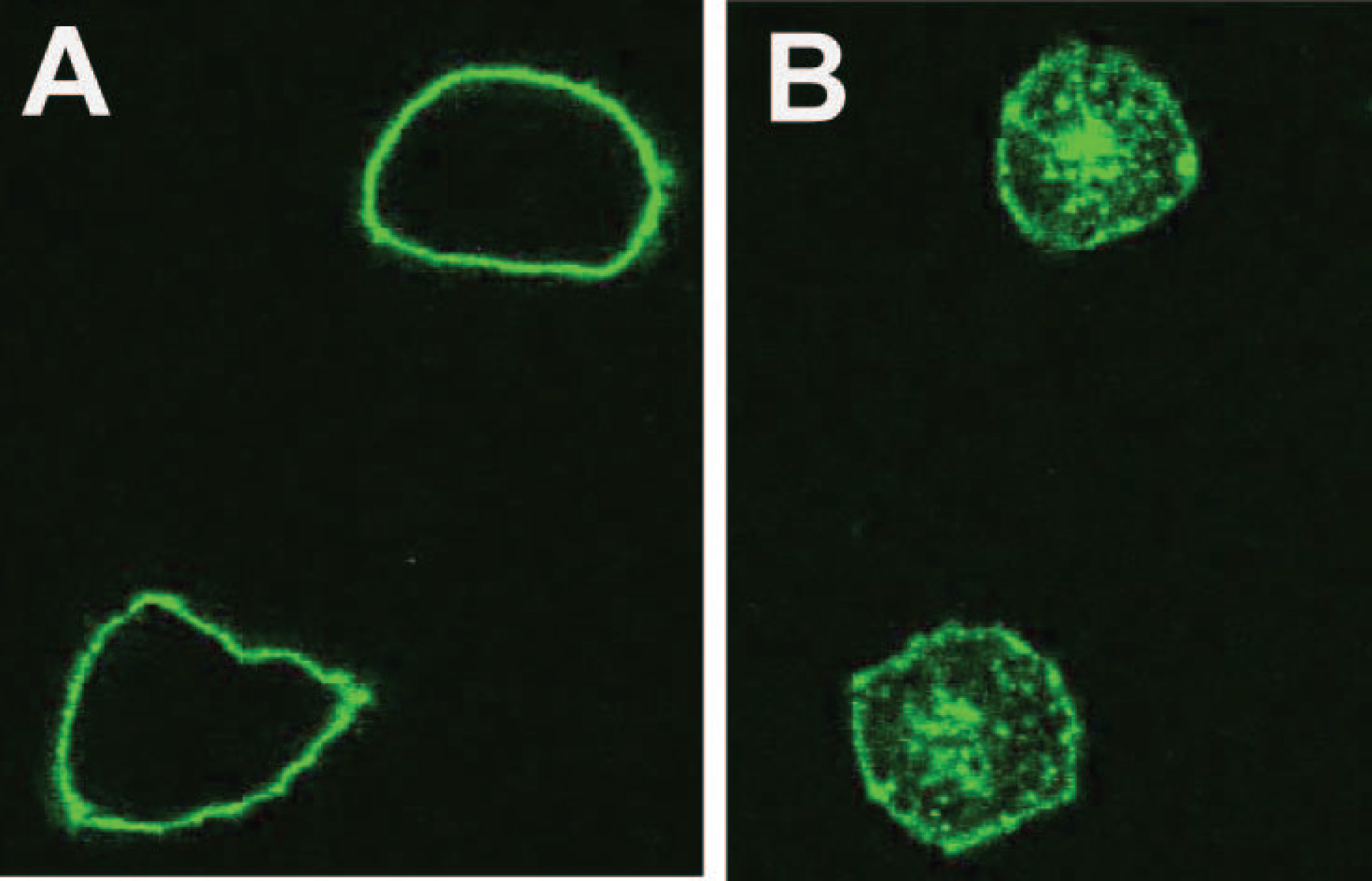
Internalization of AF488-CR2113. B16 CD1A+ cells were incubated at either 4°C (left) or 37°C (right) with CR2113-AF488for 1 h. Cells kept at 4°C during incubation showed only surface binding of CR2113-AF488. Internalization of the CD1A-CR2113-AF488 complex at 37°C was evident from capping and punctate staining of endocytic vessels. AF488 labelled isotype control antibody did not show binding or internalization on CD1A-positive or -negative cells and CR2113-AF488 did not show binding or internalization on CD1A-negative cells (data not shown).
CR2113 detects CD1A in leukaemia cell lines and patient samples
Given that the cortical subtype of T-ALL is known to express CD1A, flow cytometric analysis of CD1A antigen expression levels was determined on several T-ALL cell lines as well as T-ALL patient samples. CD1A expression was detected by CR2113 in 3 of the 5 T-ALL cell lines (Figure 4a) as well as on T-ALL patient samples (Figure 4b). In both assays, NA1/34 mAb showed equivalent results (data not shown). Flow cytometric analyses of binding to a panel of myeloid and lymphoid leukaemia samples are shown in Supplementary Figure 3. Of note, CR2113 demonstrated specific binding to macaque but not mouse thymocytes (Supplementary Figure 4), demonstrating that CR2113 was specific for human and non-human primate CD1A expressing cell. CR2113 did not bind to mouse, macaque or human peripheral blood lymphocytes (data not shown).
Figure 4.
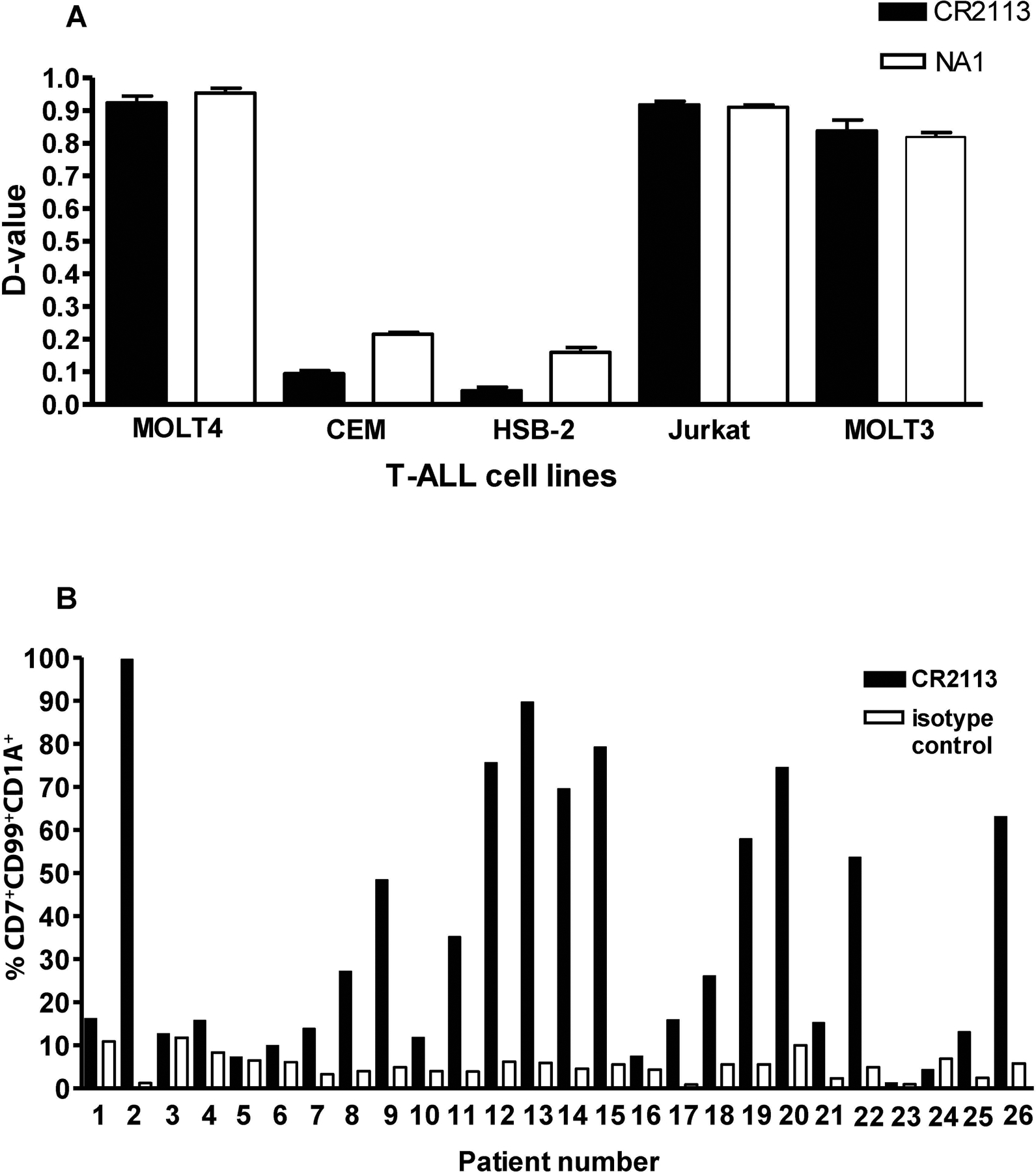
CD1A expression in leukaemia cell lines and patient samples. Five T-ALL cell lines and 26 T-ALL patient samples were stained with CR2113 and screened for CD1A expression by flow cytometry. CR2113 detected CD1A expression in 3 of 5 T-ALL cell lines (A) and 9 of 26 T-ALL patient samples (B). Panel B also shows the percentage of CD7+/CD99+/CD1A+ cells detected for each of the leukaemia samples compared to isotype control mAb.
CR2113-mediated Cytotoxicity
CR2113 does not directly induce apoptosis
Induction of apoptosis was evaluated by an ELISA-based DNA fragmentation assay. B16 cells expressing CD1A or CD1D were labelled with BrdU and then cultured for 24 h with CR2113, NA1/34 or isotype control alone or in the presence of a secondary antibody. Mitomycin C was used to induce apoptosis and served as a positive control. After incubation, cell lysates were prepared and DNA fragmentation was monitored by absorbance at a wavelength of 450 nm. Neither CR2113 nor NA1/34 induced apoptosis alone or in the presence of a cross-linking secondary antibody (Supplementary Figure 5).
CR2113 induces Complement Dependent Cytotoxicity (CDC)
The ability of CR2113 to induce CDC in B16 CD1A or CD1D expressing cell lines was determined by incubating the cell lines with CR2113 (4.5 μg/ml achieved optimal activity) either alone or in the presence of human complement (25 μl of 1:10 dilution) for 3 h. Both CR2113 and NA1/34 induced a moderate level of CDC in CD1A expressing B16 cells, although NA1/34 consistently showed a higher level of CDC activity. B16 CD1D and vector only cells were not affected in this assay (Figure 5). Neither CR2113 nor NA1/34 were able to induce significant CDC activity in the CD1A expressing T-ALL cell lines (data not shown). CDC resistance to mAbs has been described to be due to expression of membrane-bound complement binding and regulatory proteins (Geis et al 2010).
Figure 5.
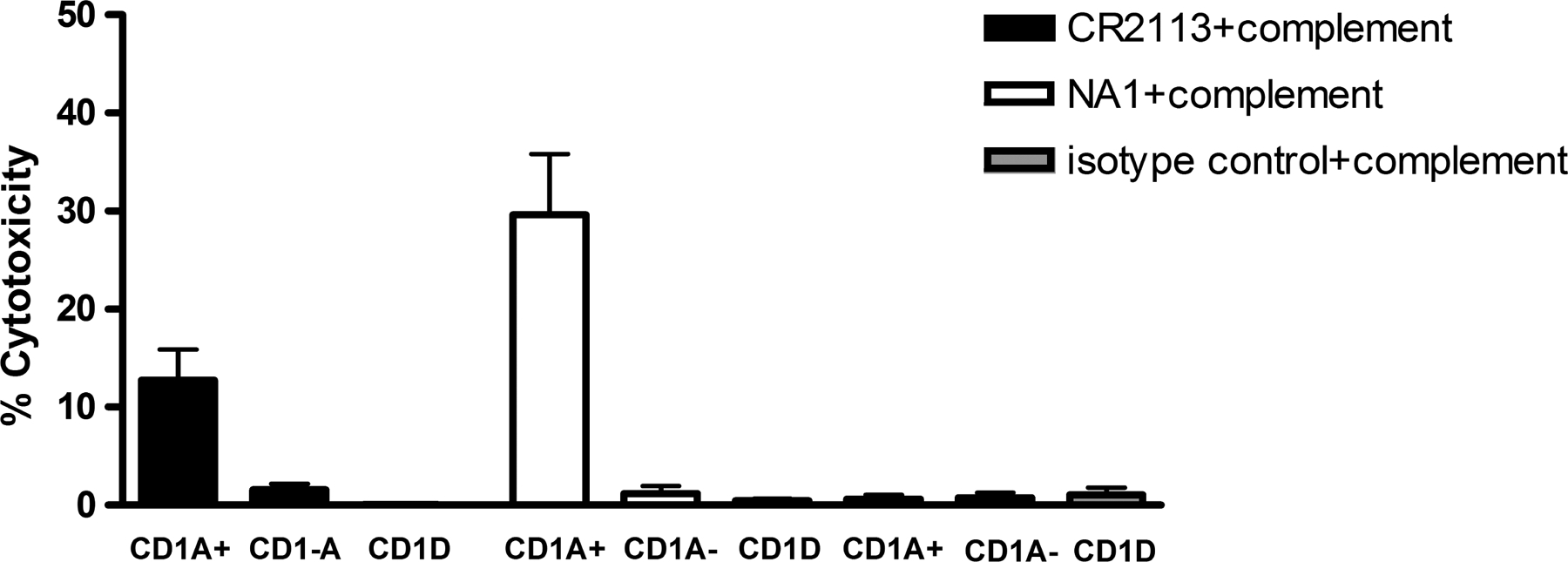
Complement-Dependent Cytotoxicty. B16 cells expressing CD1A, CD1D or B16 cells transfected with vector only were incubated with CR2113, NA1/34 or isotype control alone or with human serum for 5 h at 37°C. CR2113 induced a moderate level while NA1/34 produced a higher cytotoxicity level of CDC in CD1A+ cells.
CR2113 strongly induces antibody-dependent cell cytotoxicity (ADCC)
To determine if CR2113 can induce ADCC, B16 cells expressing CD1A or CD1D as well as CD1A expressing T-ALL cell lines and patient samples were tested in ADCC assays. Cells were incubated with CR2113 (4.5 μg/ml) alone or in the presence of PBMC or NK cells (50:1 Effector (E) to Target (T) cell ratio) for 5 h. Lower E:T ratios showed titratable killing but the greatest cytotoxicity was observed at the ET ratio of 50:1. Of note, spontaneous lysis from target cells or from effector cells incubated alone was low (data not shown). Visualization of the reactions showed intact effector cells but damaged target cells, further supporting the specificity of the cytotoxic effects. CR2113 induced a high level of ADCC with B16 CD1A+ cells. B16 cells transfected with vector only were unaffected. NA1/34 showed a lower ADCC effect than CR2113 (Figure 6a), possibly relating to different Fc receptor interactions of the murine and human mAbs. CR2113 also showed significant ADCC activity against all the tested CD1A expressing T-ALL cell lines (Figure 6b) and against 7 of 9 tested T-ALL patient samples (Figure 6c).
Figure 6.
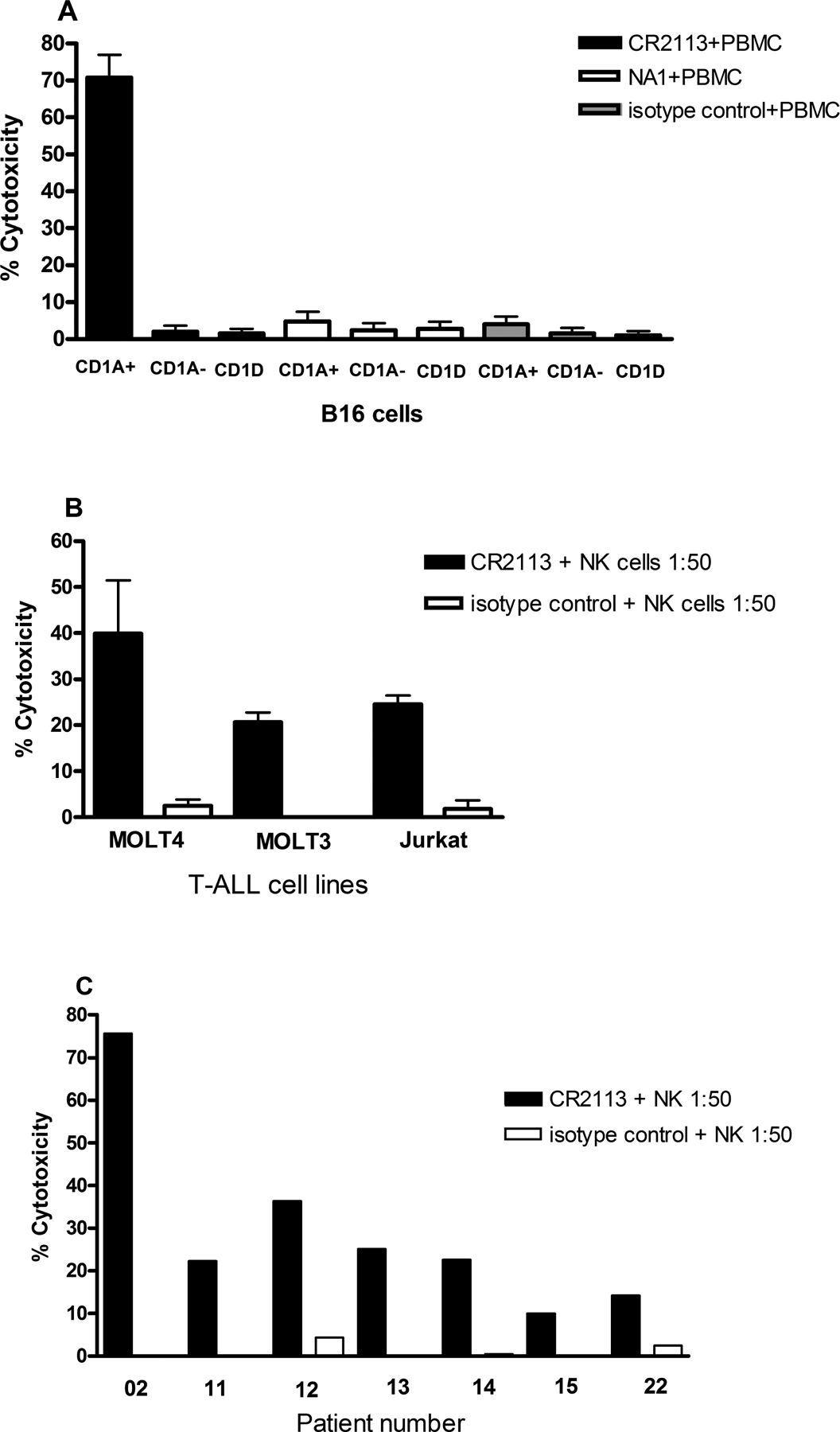
Antibody-Dependent Cell Cytotoxicity. B16 cells expressing CD1A, CD1D or B16 cells transfected with vector only were incubated with CR2113, NA1/34 or isotype control alone or with either peripheral blood mononuclear cells (PBMC) or natural killer (NK) cells for 5 hs at 37°C. CR2113 induced a high level of ADCC in the CD1A+ cells (A). NA1/34 was not effective in inducing cytotoxicity by ADCC. ADCC experiments with CR2113 in combination with NK cells (1:50 ratio) were also performed on the CD1A positive cell lines and patient samples. CR2113 induced ADCC in 3 of 3 CD1A+ T-ALL cell lines (B) and in 7 of 9 CD1A+ T-ALL patient samples (C).
In vivo activity of CR2113 in a CD1A positive melanoma model
We next tested the activity of the CR2113 against a murine B16 melanoma cell line transfected with the human cDNA encoding CD1A or vector only (CD1A−) as a model only for whether CR2113 could target selectively CD1 expressing cells in vitro. NOD/SCID mice were injected subcutaneously with 1×106 CD1A positive or negative B16 mouse melanoma cells and antibody injections were started 2 days later and continued every four days. Subcutaneous injection of the melanoma cells gave rise to tumours that had stable expression of surface CD1A on tumour cells determined by flow cytometry of dissociated cells and by immunohistochemistry (Figure 7a - d). A modest but statistically significant effect on tumour cell growth was observed with treatment using mAb CR2113 compared to isotype control (Figure 7e).
Figure 7.
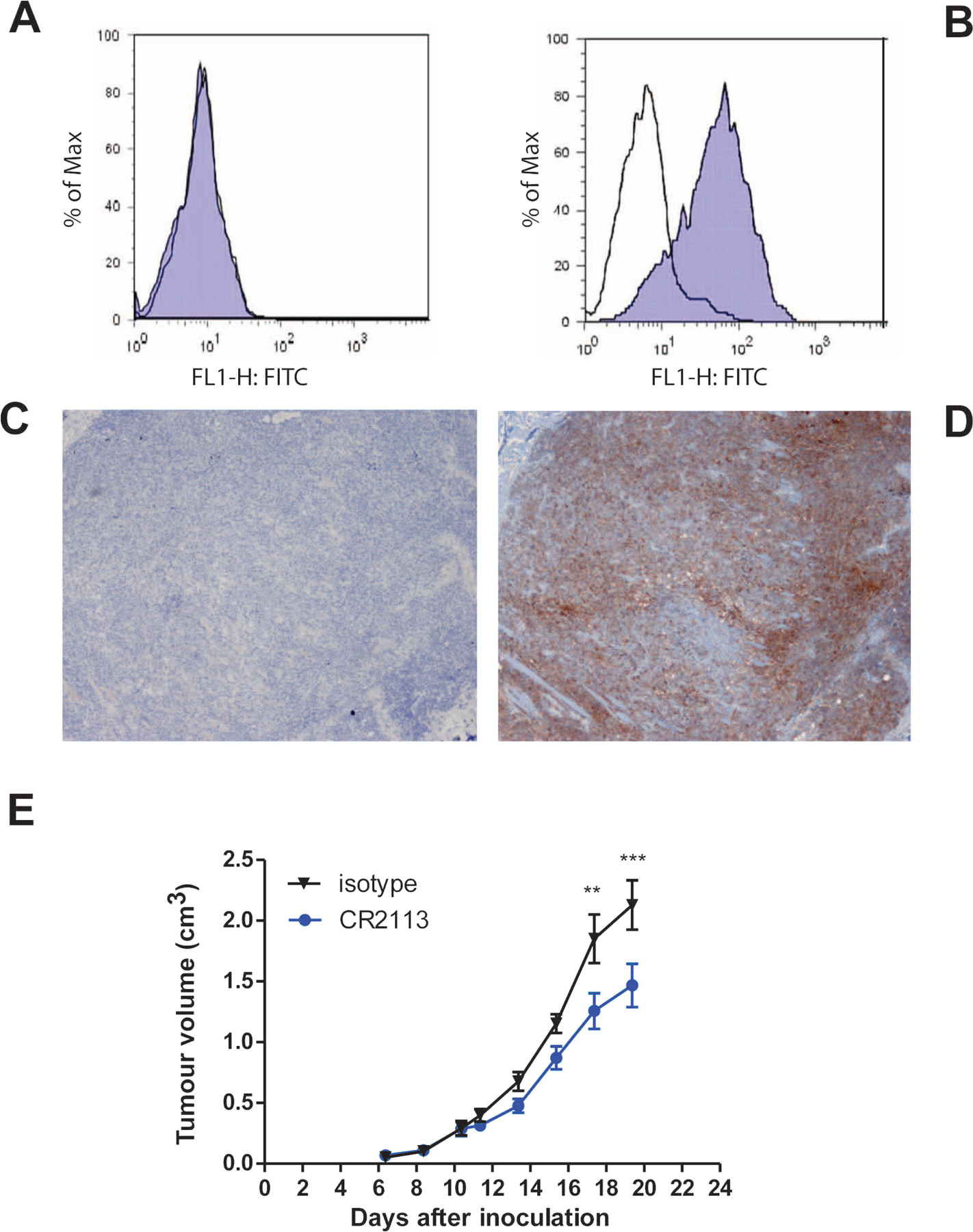
Engrafted B16 mouse melanoma tumours retain CD1A expression by flow cytometry and immunohistochemistry. (A, B) By flow cytometry, harvested B16 melanoma tumours derived from CD1A-B16 melanoma cells do not stain with mAb CR2113 anti-human CD1A antibody (A) while CD1A+ B16 melanoma cells stain positive with mAb CR2113 anti-human CD1A antibody (B). The transparent curve represents isotype control mAb (superimposed over blue curve in panel A) and the blue curve is staining with mAb CR2113. (C, D) B16 melanoma tumours derived from CD1A+ B16 melanoma cells did not show positive staining with isotype control antibody followed by staining with Haematoxylin (C) while CD1A+ B16 melanoma tumours stain showed strong staining with an anti-CD1A antibody (Becton Dickinson, Franklin Lakes, NJ) (D). (E) Efficacy of mAb CR2113 antibody against B16 mouse melanoma tumours expressing CD1A antigen. NOD/SCID mice were subcutaneously injected with 1×106 tumour cells in each flank while they were under anesthesia to assure consistent site and injection characteristics. Tumours grew at a uniform rate. Animals were treated with mAb CR2113 (Blue) or isotype control antibody (Black) on days 2, 6, 10, 14 and 18 as described in the text. Graphs represent the average tumour volume over time for all animals in a particular arm of the experiment. Error bars represent the standard error of the mean. Two–way ANOVA identifies statistical significance of p<0.001 (***) as indicated in the figure. Of note, anti-CD1A CR2113 did not have an inhibitory effect on non-CD1A expressing B16 derived tumours (data not shown).
Discussion
Monoclonal antibody-directed therapy has proven to be a successful treatment in a variety of cancers, such as B cell malignancies (anti-CD20), breast cancer (anti-HER2/neu) and colon cancer (anti-epidermal growth factor receptor) (Adams & Weiner 2005). We have generated and characterized the binding specificity and functional properties of a novel, fully human anti-CD1A mAb, CR2113. The CR2113 mAb is a high affinity antibody with specificity for CD1A and demonstrates potent ADCC activity. While the in vivo anti-tumour activity in the CD1A positive and negative B16 melanoma xenograft studies was modest, these studies were done to primarily demonstrate that in vivo targeting could occur. Additional studies are planned using immunotoxins and/or radiolabelled mAb conjugates. Unfortunately, there are currently no human LCH xenograft models available to study.
The murine anti-CD1A mAb NA1/34 has served as a starting point for development of antibody-mediated therapy for LCH. In an earlier study (Kelly et al. 1994), 111Indium-labelled NA1/34 was administered intravenously to 5 patients. Comparison of skeletal radiographs with radiolabelled NA1/34 scans demonstrated that the mAb was able to localize to areas of disease activity. However, toxic effects due to the murine origin of NA1/34 were observed and made further clinical evaluation less attractive (Kelly et al 1994). Our results show that CR2113 binds to a similar or overlapping epitope as NA1/34, but with increased affinity and greater ADCC activity. These data suggest that CR2113 should exhibit similar in vivo properties as NA1/34 regarding localization of CD1A expressing cells. However, being a purely human antibody, CR2113 would probably show fewer or less intense adverse immunogenic-associated toxicities as those seen in patients treated with NA1/34 and could be used for repeated courses of therapy or imaging (Kelly et al 1994).
The specificity, high binding capacity, high ADCC activity and internalization of the mAb/antigen complex, along with in vivo targeting provide the basis upon which the completely human CR2113 could be further developed as a clinical imaging and/or treatment modality. Further, improved anti-tumour activity might be achieved by conjugation with immunotoxins and/or by radiolabelling. Such an application would be particularly relevant for CD1A+ LCH, in which the radiolabelled murine mAb NA1/34 conjugate has been shown to localize to LCH lesions.
Treatment of patients with LCH is currently based on the use of combination chemotherapy. The outcome for patients with multisystem disease causing dysfunction of established risk organs and who do not respond to initial chemotherapy remains poor, with survival rates under 60% (Minkov et al 2003). In addition, adults with LCH have decreased responses and more toxicity to standard chemotherapy regimens that are used for children (Arico et al 2003, Nquyen & Tazi 2006). As the CR2113 mAb would target pathological cells of LCH, along with potentially some normal Langerhans cells, some of the adverse side effects associated with chemotherapy may be avoided. Further, no depletion of mature T lymphocytes along with expected regeneration of T lymphocytes from precursor cells would be expected following anti-CD1A directed antibody therapy. Another potential immunological target in LCH is CD52, although this antigen is also expressed on mature T lymphocytes (Jordan et al 2005). Whether CD1A is present on the LCH initiating cell and, thus a potential target for curative therapy, is unknown. Further experimental and potentially clinical studies with anti-CD1A mAbs may contribute to resolving this issue (Allen et al 2010). Thus, monoclonal antibody-directed therapy against CD1A would provide a potential alternative treatment alone or in combination with other agents for patients with LCH. Further, the identification of BRAF1 activating mutations in over half of the patients with LCH is now leading to specific pathway inhibition clinical trials (Badalian-Very et al 2010). The use of a mAb, such as CR2113, in combination with targeted therapies or as a vehicle to selectively target inhibitors to lesional cells may also represent a novel treatment strategy.
Using CR2113 as a radiolabelled conjugate for in vivo diagnostic and disease evaluation imaging could be an important application. For example, the specificity of CR2113 for CD1A might provide an approach for reducing the number and different types of non-specific radiographic imaging currently being used to evaluate patients with LCH. In addition, for patients in whom the biopsy of an anatomic site may not be safe or in patients with a question of disease progression or recurrence, specific imaging with such an anti-CD1A mAb could be particularly informative.
Expression of CD1A on has also been reported on several types of haematological malignancies. Here we showed that CR2113 was able to detect CD1A expression in T-ALL and other leukaemia samples, although there was heterogeneity in the percentage of cells expressing CD1A, based in part on the arbitrary D value cut-off point of 0.25. Furthermore, we demonstrated that CR2113 induced ADCC in some CD1A expressing T-ALL cell lines and CD1A expressing-leukaemia patient samples, suggesting that CR2113 could potentially prove useful as a component of therapy for CD1A-positive lymphoid malignancies, particularly coupled with a toxin or as a radiolabelled antibody. Furthermore, the ADCC activity of the antibody could be further enhanced by Fc modifications, such as a-fucosylation (Davies et al 2001). An additional potential application for an anti-CD1A human mAb such as CR2113 might include in vivo targeting of specific infectious or cancer-associated antigens to DC to enhance immunostimulatory vaccines (Bonifaz et al 2004, Cheong et al 2007, Idoyaga et al 2008). There may be a potential role for CR2113 in the treatment of refractory eczema in which CD1A-positive Langerhans cells are increased and probably play an important pathophysiological role (Allam & Novak 2006, Hussein 2008). Another potential application might also be in graft-versus-host disease (GVHD), a major complication of allogeneic bone marrow transplantation (BMT). A murine allogeneic BMT model has shown that host DC are key cellular elements for the initiation of GVHD (Shlomchik et al 1999). In addition, Merad et al reported that host Langerhans cells are responsible for initiating cutaneous GVHD (Merad 2005, Merad et al 2004), although this remains an area of controversy (Fujita et al 2007, Hemmerling et al 2011, Klangsinsirikul et al 2002, Li et al 2011, Wilson et al 2009). A mAb with the characteristics of CR2113 might therefore be worth exploring in terms of depletion of host DC pre-transplant as a strategy to prevent or abrogate GVHD.
In summary, CR2113 is a fully human mAb specific for an external epitope of the non-classical MHC antigen, CD1A. CR2113 displays potent ADCC activity against CD1A expressing cell lines and T-ALL samples as well as modest anti-tumour activity used as a naked antibody in a CD1A expressing xenograft model. The initial studies using the radiolabelled murine mAb, NA1/34, in patients with LCH stimulated the generation of CR2113. While many questions remain to be tested in terms of the potential clinical application of CR2113 or similar mAbs and their conjugated forms, such questions can now be further experimentally explored. Thus, future work is being directed toward assessing the activity of radiolabelled or toxin-conjugates of CR2113 for in vivo imaging and treatment using additional animal models, such as those involving nonhuman primates, with the goal of clinical testing in patients with LCH.
Supplementary Material
Acknowledgements:
Support for this work was came from the Teresa Bogstad Foundation for Histiocytosis, the Histiocytosis Association of America, the Histiocytosis Association of Canada, the Fighting Children’s Cancer Foundation and The Children’s Cancer Foundation, Baltimore, Maryland. DMZ was supported by an investigator award from the Cancer Research Institute and by National Institute of Health Grant AI074952. Dr. Arceci is partly supported by the endowed King Fahd Chair in Pediatric Oncology. We also thank the late Drs. Cesar Milstein and Jon Pritchard for their encouragement to initially pursue these studies.
Footnotes
Conflicts of Interest. Authors do not have any conflicts of interest. Marja van Meijer, and A.M. Kruisbeek are employees of Crucell, NV, The Netherlands. While some of the work involved in this manuscript was being done, Mark Throsby and Wilfred. Germeraad, were employees of Crucell, NV.
References
- Adams GP & Weiner LM (2005) Monoclonal antibody therapy of cancer. Nat Biotechnol, 23, 1147–1157. [DOI] [PubMed] [Google Scholar]
- Allam JP & Novak N (2006) The pathophysiology of atopic eczema. Clinical and Experimental Dermatology, 31, 89–93. [DOI] [PubMed] [Google Scholar]
- Allen CE, Li L, Peters TL, Leung HC, Yu A, Man TK, Gurusiddappa S, Phillips MT, Hicks MJ, Gaikwad A, Merad M & McClain KL (2010) Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. Journal of Immunology, 184, 4557–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amiot M, Bernard A, Raynal B, Knapp W, Deschildre C & Boumsell L (1986) Heterogeneity of the first cluster of differentiation: characterization and epitopic mapping of three CD1 molecules on normal human thymus cells. Journal of Immunology, 136, 1752–1758. [PubMed] [Google Scholar]
- Amiot M, Dastot H, Degos L, Schmid M & Boumsell L (1987) ‘Leukocyte Typing III’. Oxford Univ Press, Oxford. [Google Scholar]
- Arico M, Girschikofsky M, Genereau T, Klersy C, McClain K, Grois N, Emile JF, Lukina E, De Juli E & Danesino C (2003) Langerhans cell histiocytosis in adults. Report from the International Registry of the Histiocyte Society. Eur J Cancer, 39, 2341–2348. [DOI] [PubMed] [Google Scholar]
- Badalian-Very G, Vergilio JA, Degar BA, MacConaill LE, Brandner B, Calicchio ML, Kuo FC, Ligon AH, Stevenson KE, Kehoe SM, Garraway LA, Hahn WC, Meyerson M, Fleming MD & Rollins BJ (2010) Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood, 116, 1919–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boel E, Verlaan S, Poppelier MJ, Westerdaal NA, Van Strijp JA & Logtenberg T (2000) Functional human monoclonal antibodies of all isotypes constructed from phage display library-derived single-chain Fv antibody fragments. J Immunol Methods, 239, 153–166. [DOI] [PubMed] [Google Scholar]
- Bonifaz LC, Bonnyay DP, Charalambous A, Darguste DI, Fujii S, Soares H, Brimnes MK, Moltedo B, Moran TM & Steinman RM (2004) In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. The Journal of Experimental Medicine, 199, 815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong C, Idoyaga J, Do Y, Pack M, Park SH, Lee H, Kang YS, Choi JH, Kim JY, Bonito A, Inaba K, Yamazaki S, Steinman RM & Park CG (2007) Production of monoclonal antibodies that recognize the extracellular domain of mouse langerin/CD207. Journal of Immunological Methods, 324, 48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J, Jiang L, Pan LZ, LaBarre MJ, Anderson D & Reff M (2001) Expression of GnTIII in a recombinant anti-CD20 CHO production cell line: Expression of antibodies with altered glycoforms leads to an increase in ADCC through higher affinity for FC gamma RIII. Biotechnology and Bioengineering, 74, 288–294. [PubMed] [Google Scholar]
- de Kruif J, Terstappen L, Boel E & Logtenberg T (1995) Rapid selection of cell subpopulation-specific human monoclonal antibodies from a synthetic phage antibody library. Proceedings of the National Academy of Sciences of the United States of America, 92, 3938–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fithian E, Kung P, Goldstein G, Rubenfeld M, Fenoglio C & Edelson R (1981) Reactivity of Langerhans cells with hybridoma antibody. Proc Natl Acad Sci U S A, 78, 2541–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita S, Sato Y, Sato K, Eizumi K, Fukaya T, Kubo M & Yamashita N (2007) Regulatory dendritic cells protect against cutaneous chronic graft-versus-host disease mediated through CD4+CD25+Foxp3+ regulatory T cells. Blood, 110, 3793–3803. [DOI] [PubMed] [Google Scholar]
- Geis N, Zell S, Rutz R, Li W, Giese T, Mamidi S, Schultz S & Kirschfink M (2010) Inhibition of membrane complement inhibitor expression (CD46, CD55, CD59) by siRNA sensitizes tumor cells to complement attack in vitro. Current Cancer Drug Targets, 10, 922–931. [DOI] [PubMed] [Google Scholar]
- Hemmerling J, Wegner-Kops J, von Stebut E, Wolff D, Wagner EM, Hartwig UF, Andre MC, Theobald M, Schopf RE, Herr W & Meyer RG (2011) Human Epidermal Langerhans Cells Replenish Skin Xenografts and Are Depleted by Alloreactive T Cells In Vivo. Journal of Immunology, 187, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Hussein MR (2008) Evaluation of Langerhans’ cells in normal and eczematous dermatitis skin by CD1a protein immunohistochemistry: preliminary findings. Journal of Cutaneous Pathology, 35, 554–558. [DOI] [PubMed] [Google Scholar]
- Idoyaga J, Cheong C, Suda K, Suda N, Kim JY, Lee H, Park CG & Steinman RM (2008) Cutting edge: langerin/CD207 receptor on dendritic cells mediates efficient antigen presentation on MHC I and II products in vivo. Journal of Immunology, 180, 3647–3650. [DOI] [PubMed] [Google Scholar]
- Jordan MB, McClain KL, Yan X, Hicks J & Jaffe R (2005) Anti-CD52 antibody, alemtuzumab, binds to Langerhans cells in Langerhans cell histiocytosis. Pediatric Blood & Cancer, 44, 251–254. [DOI] [PubMed] [Google Scholar]
- Kahn-Perles B, Wietzerbin J, Caillol DH & Lemonnier F (1985) Delineation of three subsets of class I human T antigens (HTA) on Molt-4 cells: serologic and regulatory relationship to HLA class I antigens. Journal of Immunology, 134, 1759–1765. [PubMed] [Google Scholar]
- Kelly KM, Beverley PC, Chu AC, Davenport V, Gordon I, Smith M & Pritchard J (1994) Successful in vivo immunolocalization of Langerhans cell histiocytosis with use of a monoclonal antibody, NA1/34. J Pediatr, 125, 717–722. [DOI] [PubMed] [Google Scholar]
- Klangsinsirikul P, Carter GI, Byrne JL, Hale G & Russell NH (2002) Campath-1G causes rapid depletion of circulating host dendritic cells (DCs) before allogeneic transplantation but does not delay donor DC reconstitution. Blood, 99, 2586–2591. [DOI] [PubMed] [Google Scholar]
- Li H, Kaplan DH, Matte-Martone C, Tan HS, Venkatesan S, Johnson K, Demetris AJ, McNiff J, Shlomchik MJ & Shlomchik WD (2011) Langerhans cells are not required for graft-versus-host disease. Blood, 117, 697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M (2005) Ontogeny of Lagerhans cells and graft versus host disease. Advances in Experimental Medicine and Biology, 560, 115–123. [DOI] [PubMed] [Google Scholar]
- Merad M, Hoffmann P, Ranheim E, Slaymaker S, Manz MG, Lira SA, Charo I, Cook DN, Weissman IL, Strober S & Engleman EG (2004) Depletion of host Langerhans cells before transplantation of donor alloreactive T cells prevents skin graft-versus-host disease. Nat Med, 10, 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merle-Beral H, Boumsell L, Michel A & Debre P (1989) CD1 expression on B-CLL lymphocytes. Br J Haematol, 71, 209–212. [DOI] [PubMed] [Google Scholar]
- Minkov M, Grois N, Braier J, Rosso D, Arico M, Broadbent V, Gadner H & Ladisch S (2003) Immunosuppressive treatment for chemotherapy-resistant multisystem Langerhans cell histiocytosis. Med Pediatr Oncol, 40, 253–256. [DOI] [PubMed] [Google Scholar]
- Misery L, Campos L, Dezutter-Dambuyant C, Guyotat D, Treille D, Schmitt D & Thivolet J (1992) CD1-reactive leukemic cells in bone marrow: presence of Langerhans cell marker on leukemic monocytic cells. Eur J Haematol, 48, 27–32. [DOI] [PubMed] [Google Scholar]
- Nquyen K & Tazi A (2006) [Langerhans cell histiocytosis in adults]. La Revue du praticien, 56, 1863–1871. [PubMed] [Google Scholar]
- Olive D, Dubreuil P & Mawas C (1984) Two distinct TL-like molecular subsets defined by monoclonal antibodies on the surface of human thymocytes with different expression on leukemia lines. Immunogenetics, 20, 253–264. [DOI] [PubMed] [Google Scholar]
- Pless DD, Torres ER, Reinke EK & Bavari S (2001) High-affinity, protective antibodies to the binding domain of botulinum neurotoxin type A. Infect Immun, 69, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcelli SA (1995) The CD1 Family: A third lineage of antigen-presenting molecules. Academic Press Inc.,U.S. [DOI] [PubMed] [Google Scholar]
- Porcelli SA & Modlin RL (1999) The CD1 system: antigen-presenting molecules for T cell recognition of lipids and glycolipids. Annu Rev Immunol, 17, 297–329. [DOI] [PubMed] [Google Scholar]
- Ray A, Schmitt D, Dezutter-Dambuyant C, Fargier MC & Thivolet J (1989) Reappearance of CD1a antigenic sites after endocytosis on human Langerhans cells evidenced by immunogoldrelabeling. The Journal of Investigative Dermatology, 92, 217–224. [DOI] [PubMed] [Google Scholar]
- Salamone MC, Roisman FR, Santiago J, Satz ML & Fainboim L (1990) Analysis of CD1 molecules on haematological malignancies of myeloid and lymphoid origin. I. Cell surface antigen expression. Dis Markers, 8, 265–274. [PubMed] [Google Scholar]
- Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, Shlomchik MJ & Emerson SG (1999) Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science, 285, 412–415. [DOI] [PubMed] [Google Scholar]
- Sotzik F, Boyd A & Shortman K (1993) Surface antigens of human thymocyte populations defined by CD3, CD4 and CD8 expression: CD1a is expressed by mature thymocytes but not peripheral T cells. Immunol Lett, 36, 101–106. [DOI] [PubMed] [Google Scholar]
- Van Ewijk W, de Kruif J, Germeraad WT, Berendes P, Ropke C, Platenburg PP & Logtenberg T (1997) Subtractive isolation of phage-displayed single-chain antibodies to thymic stromal cells by using intact thymic fragments. Proc Natl Acad Sci U S A, 94, 3903–3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson J, Cullup H, Lourie R, Sheng Y, Palkova A, Radford KJ, Dickinson AM, Rice AM, Hart DN & Munster DJ (2009) Antibody to the dendritic cell surface activation antigen CD83 prevents acute graft-versus-host disease. The Journal of Experimental Medicine, 206, 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuchter C, Ruppert V, Schrappe M, Dorken B, Ludwig WD & Karawajew L (2002) In vitro susceptibility to dexamethasone- and doxorubicin-induced apoptotic cell death in context of maturation stage, responsiveness to interleukin 7, and early cytoreduction in vivo in childhood T-cell acute lymphoblastic leukemia. Blood, 99, 4109–4115. [DOI] [PubMed] [Google Scholar]
- Zajonc DM, Elsliger MA, Teyton L & Wilson IA (2003) Crystal structure of CD1a in complex with a sulfatide self antigen at a resolution of 2.15 Å. Nat Immunol, 4, 808–815. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.