

Abstract
CADASIL is a vascular protein aggregation disorder caused by cysteine-altering NOTCH3 variants, leading to mid-adult-onset stroke and dementia. Here, we report individuals with a cysteine-altering NOTCH3 variant that induces exon 9 skipping, mimicking therapeutic NOTCH3 cysteine correction. The index came to our attention after a coincidental finding on a commercial screening MRI, revealing white matter hyperintensities. A heterozygous NOTCH3 c.1492G>T, p.Gly498Cys variant, was identified using a gene panel, which was also present in four first- and second-degree relatives. Although some degree of white matter hyperintensities was present on MRI in all family members with the NOTCH3 variant, the CADASIL phenotype was mild, as none had lacunes on MRI and there was no disability or cognitive impairment above the age of 60 years. RT-PCR and Sanger sequencing analysis on patient fibroblast RNA revealed that exon 9 was absent from the majority of NOTCH3 transcripts of the mutant allele, effectively excluding the mutation. NOTCH3 aggregation was assessed in skin biopsies using electron microscopy and immunohistochemistry and did not show granular osmiophilic material and only very mild NOTCH3 staining. For purposes of therapeutic translatability, we show that, in cell models, exon 9 exclusion can be obtained using antisense-mediated exon skipping and CRISPR/Cas9-mediated genome editing. In conclusion, this study provides the first in-human evidence that cysteine corrective NOTCH3 exon skipping is associated with less NOTCH3 aggregation and an attenuated phenotype, justifying further therapeutic development of NOTCH3 cysteine correction for CADASIL.
Keywords: CADASIL, small vessel disease, NOTCH3, exon skipping, protein aggregation
Introduction
CADASIL is a hereditary small vessel disease caused by cysteine-altering variants in NOTCH3 (OMIM#125310) (1, 2). More than 250 of such variants have been described in CADASIL families (3), all of which lead to an uneven number of cysteines in one of the epidermal growth factor-like repeat (EGFr) domains of the NOTCH3 ectodomain (NOTCH3ECD). These variants cause mutant NOTCH3ECD to aggregate in the (cerebro)vasculature. Ultrastructurally, CADASIL-causing NOTCH3 variants are associated with granular osmiophilic material (GOM) deposits in the blood vessel wall, including vessels of the brain and skin (4–6). As such, CADASIL is a vascular protein aggregation disorder with secondary central nervous system hypoxia and ischemia, leading to mid-adult-onset strokes and cognitive decline (1). MRI signs include progressive periventricular and deep white matter hyperintensities (WMH), typically but not necessarily including the external capsules and temporal poles, superimposed by lacunes and frequently also microbleeds in later disease stages (7).
We recently described “NOTCH3 cysteine correction” as a therapeutic approach for CADASIL, using antisense-mediated exon skipping to exclude mutant exons from the mRNA (8). Exons were selected in such a way that exclusion from mRNA restores the number of cysteines in the EGFr domains of the NOTCH3ECD, with conservation of signalling function and predicted normal protein folding (8). Many exons harbouring CADASIL-causing variants are eligible for this approach, including exon 9.
Here, we describe a family with a unique cysteine-altering NOTCH3 variant in exon 9, which is predicted to cause natural exon 9 skipping. This effectively mimics the therapeutic NOTCH3 cysteine correction approach, allowing us to study the effect of cysteine corrective exon skipping on NOTCH3 protein aggregation and disease severity in humans. Furthermore, we tested exon 9 skipping in vitro using antisense oligonucleotides (ASOs) and determined the feasibility of NOTCH3 cysteine correction using CRISPR/Cas9-mediated gene editing.
Results
Clinical characteristics of family members with the NOTCH3 c.1492G>T variant
A 63-year-old female (index, #1, Fig. 1A) was referred to the hereditary small vessel disease clinic of our hospital because of confluent WMH seen on a commercial brain scan. This scan was performed at her own initiative when her twin sister was found to have WMH. She had a history of non-migraine headaches, short-lasting attacks of vertigo and tinnitus, but an otherwise unremarkable neurological and neuropsychiatric medical history. Her vascular risk factors included a smoking history of 6 pack-years and hypertension, for which she was treated since age 45, with good response. She was living independently and worked until age 60 years. Since age 62 years, she noticed some short-term memory impairment. Neurological examination revealed a rest tremor and mild dysdiadochokinesia left more than right, which was explained by right-more-than-left nigrostriatal degeneration shown on a DAT-scan. Global neuropsychological examination was normal. MRI performed in our hospital showed bilateral supratentorial confluent WMH in the frontal and parietal lobes, as well as in the anterior temporal lobes, external capsules and in the basal ganglia: a pattern and extension suggestive of CADASIL, although mild for her age. There were no lacunes, which are typically present in CADASIL patients in their sixties, but some microbleeds were observed in the thalamus (Fig. 1B).
Figure 1.
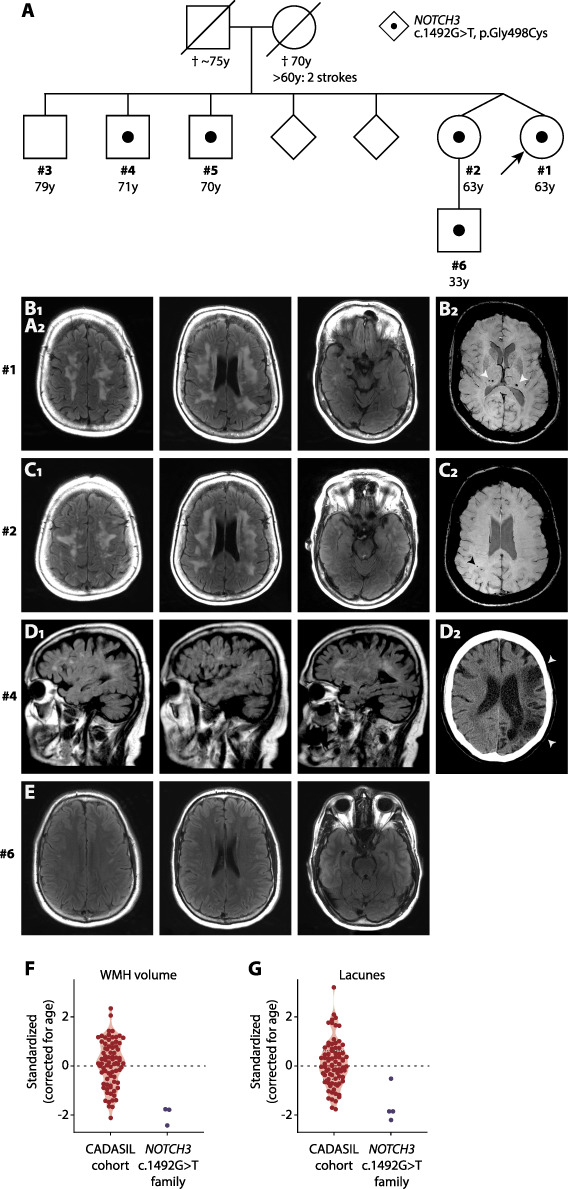
Pedigree and brain imaging of individuals with the NOTCH3 c.1492G>T variant. (A) Pedigree showing the family members with the NOTCH3 c.1492G>T, p.G498C variant. (B–E) FLAIR sequences are shown, unless otherwise stated. (B) MRI of the index patient (#1) shows confluent symmetrical periventricular and subcortical WMH. WMH are also present in the anterior temporal lobes. No lacunes were seen. Microbleeds are present in the thalamus on susceptibility-weighted imaging (SWI) (B2). (C) MRI scan of the twin sister (#2) showing confluent WMH almost identical to those seen in the index, and a parietal subcortical microbleed on the SWI scan, but no lacunes (C2). (D) Brain MRI in the brother (#4) was performed at the age of 63 years, before his large vessel stroke, showing focal and beginning confluent WMH in the semioval centre with some involvement of external capsules, but without lacunes. (D2) A CT scan made after the stroke reveals a large cortico-subcortical infarct in the territory of the left medial cerebral artery (arrowheads). (E) MRI of the nephew (#6) showing minimal focal WMH in the frontal deep white matter. No brain MRI was available for one brother (#5). (F) Violin plot showing age-adjusted standardized WMH scores of the index (#1), her twin sister (#2) and the nephew (#6) compared to a cohort of CADASIL patients. (G) Violin plot showing age-adjusted standardized lacune count scores in the family members #1, #2, #4 and #6. None of the family members had lacunes. The differences the standardized score for lacunes between family members are due to the differences in age (see Supplementary Material, Fig. S1).
Family history was remarkable for a large vessel stroke in a brother with a history of chronic alcohol abuse (#4) and depression in a sibling (#3). No siblings had a history of lacunar stroke or vascular cognitive decline. Except for the brother with large vessel stroke in the left medial cerebral artery with hemiparesis, all were living independently and physically able, with at most complaints of mild short-term memory impairment. Both parents were deceased, and their mother had experienced two strokes and cognitive decline in her seventh decade (Fig. 1A).
A gene panel of 28 genes for small vessel disease and adult-onset leukoencephalopathy was performed in the index patient, revealing a previously unreported cysteine-altering heterozygous missense NOTCH3 variant (NM_000435.2: c.1492G>T, p.(Gly498Cys)). The variant alters the number of cysteines in an EGFr domain of NOTCH3 (EGFr 12), which is typical for a CADASIL-associated variant. However, this variant is remarkable for the fact that it is located adjacent to the exon 9 donor splice site. In silico analyses predict that this variant results in exon 9 skipping, thereby excluding the missense variant from the mature mRNA (Fig. 2). This variant was also found to be present in her twin sister (#2) and in two other siblings (#4 and #5), as well as in a nephew (#6). One tested sibling did not have the variant (#3). Neuroimaging was performed, showing confluent WMH in all siblings, but no lacunes and a few microbleeds in only one individual (Fig. 1C and D). The 33-year-old nephew had a few small focal WMH in the frontal deep white matter (Fig. 1E), but not the more extensive (periventricular and temporal pole) WMH often seen in CADASIL patients in the fourth decade (9). The sibling without the NOTCH3 variant (#3) had a normal MRI scan. Family members with the NOTCH3 variant had a lower age-adjusted WMH volume and lacune lesion load compared to a cohort of CADASIL patients (Fig. 1F and G and Supplementary Material, Fig. S1).
Figure 2.
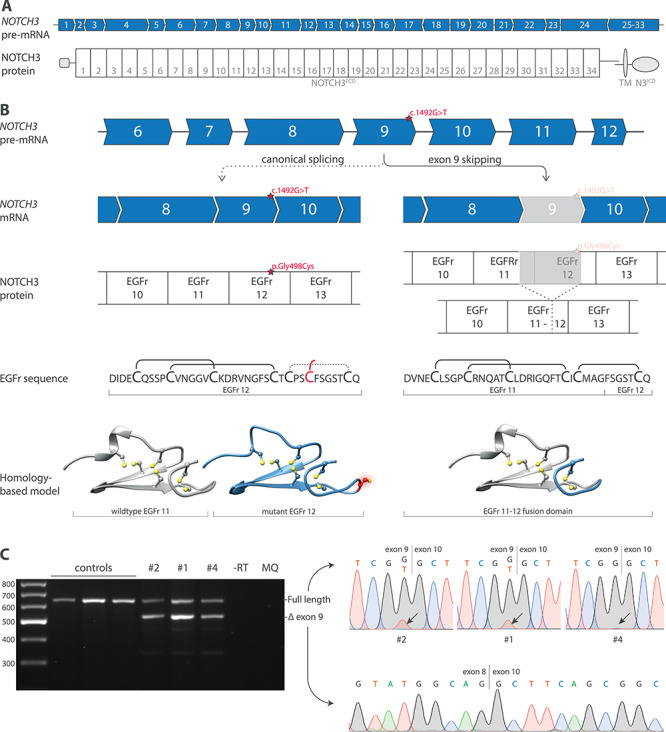
Exon 9 skipping and predicted effect on NOTCH3 protein of NOTCH3 c.1492G>T variant. (A) The NOTCH3 gene contains 33 exons, of which exon 2–24 encode the 34 EGFr domains in the ectodomain of the NOTCH3 protein (NOTCH3ECD). One exon can encode one or more, complete or partial EGFr domains. (B) The NOTCH3 c.1492G>T variant is located adjacent to the splice donor site of exon 9. Upon canonical splicing of the NOTCH3 transcript, the c.1492G>T variant results in an additional cysteine residue in EGFr domain 12. However, because the c.1492G>T variant is located adjacent to the splice donor site, it is predicted to result in exon 9 skipping. Exon 9 encodes part of EGFr 11 and part of EGFr 12; the remaining parts of EGFr 11 and EGFr 12 encoded by exon 8 and exon 10, respectively, are predicted to form an EGFr 11–12 fusion domain, with the correct number of and spacing of cysteine residues. Homology-based models show the predicted structure of EGFr domains. (C) RT-PCR and Sanger sequencing analysis showing exon 9 skipping in fibroblast RNA from individuals with the NOTCH3 c.1492G>T variant. The full-length band predominantly contained the wild-type transcript, but also some mutant transcripts, as indicated by the relatively low mutant T-peak in the sequence. No exon 9 skipping was seen in controls. RT-PCR and Sanger sequencing were performed in two independent experiments. NOTCH3ECD, NOTCH3 extracellular domain. EGFr, Epidermal growth factor-like repeat.
Skin punch biopsies were taken to determine whether CADASIL-associated vessel wall pathology was present. NOTCH3 immunohistochemistry using two NOTCH3ECD antibodies showed only very slight positive, but typically granular, NOTCH3 staining compared to positive controls. There was no granular staining present in negative controls nor in the sibling lacking the NOTCH3 variant (Fig. 3). Exhaustive electron microscopic analysis did not reveal CADASIL-associated GOM deposits in the vessel walls of any of the family members with the NOTCH3 c.1492G>T variant, while many GOM deposits were observed in vessel walls of CADASIL patients (Supplementary Material, Table S5).
Figure 3.
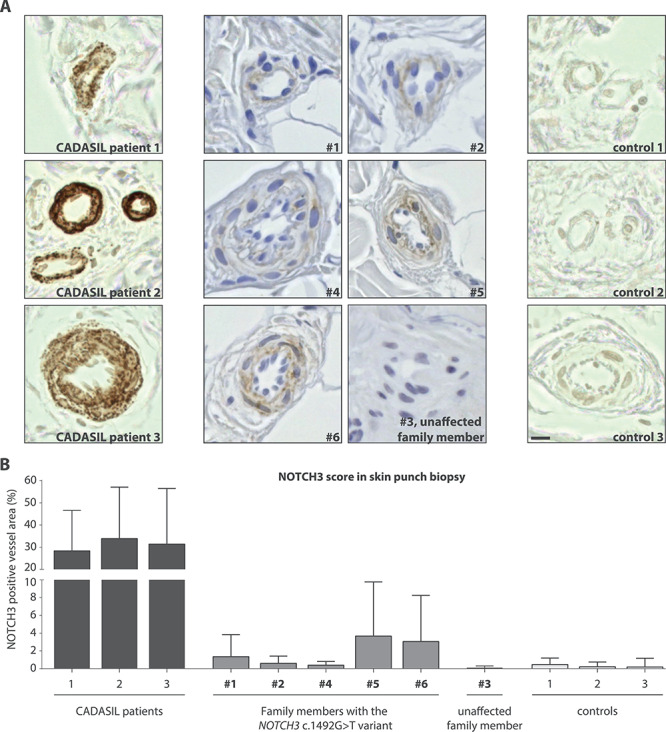
Individuals with the NOTCH3 c.1492G>T variant show much less granular NOTCH3ECD staining in skin vasculature than CADASIL patients. (A) Representative images of NOTCH3ECD staining in blood vessels of skin punch biopsies: CADASIL patients, individuals with the NOTCH3 c.1492G>T variant and controls. All family members with the NOTCH3 c.1492G>T variant had minimal granular NOTCH3ECD staining, which was much less compared to NOTCH3ECD staining typically seen in CADASIL patients. No NOTCH3ECD staining was seen in the brother without the NOTCH3 variant (#3) or in controls. (B) Quantification of the area of NOTCH3ECD immunostaining within the vessel wall showed that individuals with the NOTCH3 c.1492G>T variant have significantly less NOTCH3ECD staining compared to CADASIL patients (1.81% ± 1.48 versus 31.2% ± 2.75 NOTCH3ECD positive area within vessel wall boundaries, ANOVA, P < 0.001). Bar represents 10 μm. Mean ± standard deviation.
Characterization of the NOTCH3 cysteine-altering splice variant
To confirm that the NOTCH3 c.1492G>T variant impairs exon 9 inclusion during the splicing process, RT-PCR analysis was performed on RNA from skin fibroblasts from three family members, which confirmed exon 9 skipping in all three individuals. Exon 9 skipping was highly efficient, with only a minority of mutant transcripts escaping exon skipping and therefore still harbouring the cysteine-altering missense variant (Fig. 2C). Skipping of exon 9 leaves the open reading frame intact and is predicted to result in the production of a slightly shorter internally deleted NOTCH3 protein. Exon 9 encodes part of EGFr 11 and part of EGFr 12, and these parts are therefore excluded from the exon 9-skipped NOTCH3 protein; the remaining parts of EGFr 11 and EGFr 12 encoded by exon 8 and exon 10, respectively, are predicted to form an EGFr 11–12 fusion domain. This EGFr fusion domain resembles a wild-type EGFr domain, with six canonically spaced cysteine residues (Fig. 2B), mimicking NOTCH3 cysteine correction (8). There is one notable difference with other NOTCH3 exons eligible for cysteine corrective exon skipping, such as exon 4–5 skipping, namely that the exon 9-skipped protein lacks a small part of EGFr 11 which is part of the putative NOTCH3 ligand-binding domain (i.e. EGFr 10 and 11) (LBD).
To assess translation, processing and signalling of the exon 9-skipped NOTCH3 protein, we transfected control fibroblasts and NIH 3T3 cells with cDNA constructs lacking exon 9 (NOTCH3Δexon9). NOTCH3Δexon9 was expressed at the cell surface of fibroblasts, comparable to wild-type NOTCH3 (Fig. 4A). However, ligand-dependent signalling was impaired, comparable to the protein lacking the entire NOTCH3 ligand-binding domain (NOTCH3ΔLBD) (Fig. 4B and Supplementary Material, Table S4).
Figure 4.
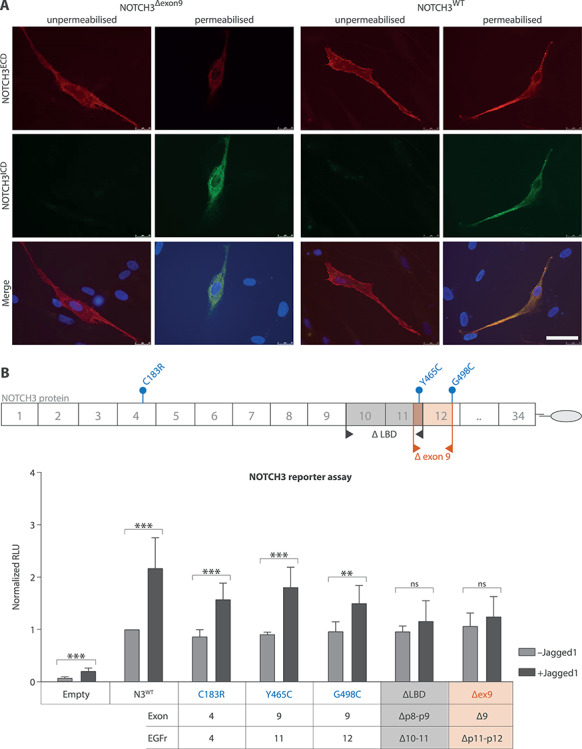
NOTCH3Δexon9 protein has normal cellular localisation but has reduced signalling properties in vitro. (A) Immunocytochemistry of fibroblasts transfected with NOTCH3 cDNA constructs shows that the NOTCH3Δexon9 protein is translated and expressed on the cell surface similar to NOTCH3WT, as indicated by NOTCH3ECD staining of the cell membrane in unpermeabilized cells. (B) NOTCH3 signalling in a CBF1 reporter assay with and without JAGGED1 stimulation. JAGGED1 significantly activated NOTCH3 signalling in NOTCH3WT, NOTCH3Y465C and NOTCH3G498C, but not in NOTCH3Δexon9 and NOTCH3ΔLBD (independent samples t-test; see Supplementary Material, Table S4 for details). RLU, relative luciferase units; N3WT, NOTCH3 wild-type protein; NOTCH3Δexon9, NOTCH3 exon 9 skip protein; scale bar represents 50 μm. Mean ± standard deviation of seven independent experiments. **P < 0.01, ***P < 0.001.
Targeted NOTCH3 exon exclusion in vitro using ASOs and CRISPR/Cas9
For purposes of translatability, we assessed whether it was possible to induce exon 9 antisense-mediated skipping or an exon 9 genomic deletion in control cell cultures. Exon 9 skipping was induced in VSMCs transfected with ASOs targeting exon 9 (Fig. 5A and B). Using CRISPR/Cas9-mediated gene editing with guide RNAs targeting introns 8 and 9, exon 9 was deleted from genomic DNA in HEK293 cells (Fig. 5C and D), with mRNA transcripts showing a correct exon 8–10 boundary similar to the mRNA transcript expressed after ASO-mediated exon 9 skipping. We also tested whether we could delete NOTCH3 exons 4 and 5, as this cysteine corrective exon skipping would ensure a single treatment approach for most CADASIL patients (8). Genome editing with guide RNAs targeting introns 3 and 5 resulted in a correct genomic exon 4–5 deletion in VSMCs and mRNA transcripts with a correct exon 3–6 boundary (Fig. 5E and F).
Figure 5.
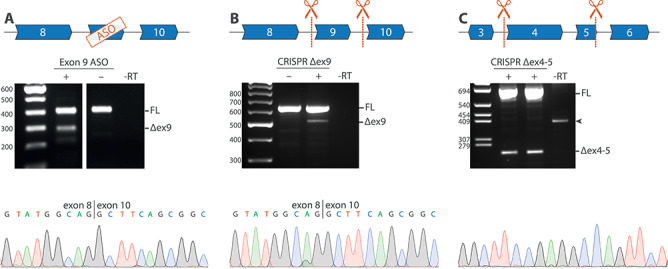
Targeted NOTCH3 exon exclusion using ASOs and CRISPR/Cas9 in vitro. (A) RT-PCR analysis of VSMCs after transfection with an ASO targeting exon 9, showing a band at ~ 300 bp, corresponding to the expected fragment size after exon 9 skipping, which was confirmed to be a correct exon 9 skip with Sanger sequencing. (B) Guide RNAs targeting NOTCH3 introns 8 and 9 were used to delete exon 9 from the genomic DNA of HEK293 cells. RT-PCR analysis confirmed exon 9 exclusion at the RNA level. (C) Guide RNAs targeting intron 3 and intron 5 were used to delete exon 4 and exon 5 from the genomic DNA from control (left lane) and CADASIL patient-derived VSMCs (right lane), which was confirmed with Sanger sequencing. FL, full length; −RT, condition without reverse transcriptase; Δex9, PCR product lacking exon 9; Δex4–5, PCR product lacking exon 4–5; arrowhead, aspecific genomic PCR product.
Discussion
Here, we present a family with a unique cysteine-altering NOTCH3 variant that leads to exon 9 skipping, effectively excluding the mutant exon and correcting the number of cysteines in the EGFr domains of the NOTCH3ECD. This mimics the “NOTCH3 cysteine correction” therapeutic strategy for CADASIL we previously described (8), offering the unique opportunity to study the effect of cysteine correction on NOTCH3 protein aggregation and disease severity in humans. We show that exon 9 skipping is associated with strongly reduced levels of vascular NOTCH3ECD aggregation and that individuals with exon 9 skipping have a milder small vessel disease phenotype compared to most CADASIL patients, although CADASIL can also be variable (10). Furthermore, we show that NOTCH3 cysteine correction can be accomplished at the RNA level using ASOs (8) and at the DNA level using CRISPR/Cas9-mediated gene editing.
All family members with naturally occurring exon 9 skipping had only minimal levels of NOTCH3ECD protein aggregation in their skin vasculature, suggesting that the exon 9-skipped NOTCH3 protein does not aggregate. The minimal aggregation that was seen is likely due to the low levels of mutant NOTCH3 proteins containing exon 9, translated from the low levels of unskipped mutant transcripts. In line with this, the individual with the most efficient exon 9 skipping had the least vascular NOTCH3ECD positive staining in skin biopsy. Notably, none of the individuals with the NOTCH3 c.1492G>T variant had CADASIL-associated GOM deposits. In CADASIL mouse models, it has been shown that progressive NOTCH3 aggregation precedes GOM formation, showing that GOM are likely only formed once a certain threshold of NOTCH3 aggregation has been reached (11, 12). Thus, we cautiously pose that the natural cysteine correction that occurs in these patients strongly reduces progressive NOTCH3 aggregation, such that the GOM stage is not reached, vessel wall integrity is better preserved, and therefore there is a milder later-onset disease course. Cysteine-altering NOTCH3 variants are known to be associated with a variable disease severity, part of which is explained by mutation position (10) and environmental factors (13, 14), but other genetic factors are widely held to play a role (14–16), of which this naturally occurring exon skipping may be a rare example.
We have previously shown that NOTCH3 cysteine corrected proteins with EGFr fusion domains retain signalling function comparable to wild type (8). Here, we show that the exon 9-skipped NOTCH3 protein has expression and localisation properties comparable to wild type but that ligand-induced signalling properties are strongly reduced, comparable to complete lack of the LBD (EGFr domains 10 and 11). Whether reduced NOTCH3 signalling may contribute to the phenotype in this family is unclear, as the role of heterozygous loss-of-function variants in CADASIL or CADASIL-like small vessel disease is still subject of debate (10, 17, 18). Of note, we found that NOTCH3Y465C, located in EGFr domain 11, has similar JAGGED1-induced signalling capacity as variants located outside the LBD, as opposed to the NOTCH3C455R variant in EGFr 11 which has been shown to impair JAGGED1-induced signalling (19). As the NOTCH3Y465C variant is located in the second-to-the-last amino acid of EGFr domain 11, this suggests that the distal part of EGFr domain 11 is not involved in binding to JAGGED1.
We previously showed that cysteine-altering variants associated with a severe CADASIL phenotype, i.e. those located EGFr domains 1–6, are also eligible for cysteine correction by skipping exons 4 and 5 simultaneously using ASOs (8). Here, we showed that CRISPR/Cas9-mediated genome editing to exclude exons 4 and 5 or exon 9 from genomic DNA is feasible and thereby may be a potential future alternative for ASO-based cysteine correction. Therapeutic genome editing has the potential of a single treatment approach, instead of the potentially lifelong repeated ASO administration, which would be necessary for an RNA-based exon skipping approach. However, before in vivo genome editing of VSMCs can be applied in CADASIL patients, major hurdles need to be taken, including delivery, immunogenicity and off-target effects (20), whereas ASO-mediated RNA modifications are already FDA-approved for the treatment of a number of neurodegenerative disorders (21), for both intrathecal and systemic administration (22, 23).
In conclusion, we provide the first in-human evidence that cysteine corrective NOTCH3 exon skipping is associated with only minimal vascular NOTCH3ECD aggregation and a relatively mild later-onset phenotype. These findings support continuing efforts in developing NOTCH3 cysteine correction for treatment of CADASIL patients.
Materials and Methods
This study was approved by the Medical Ethics Committee of the Leiden University Medical Center (B19.057). All participants gave written informed consent for publication.
Phenotype description
Family members were evaluated at the multidisciplinary outpatient clinic for cerebral small vessel disease of our hospital. A neurologist (G.M.T.) and clinical geneticist (S.A.J.L.O.) with longstanding expertise in CADASIL took their history and family history. Neuroimaging was performed as part of diagnostic care. MRI scans were re-evaluated by a neuroradiologist with broad expertise in MRI scans of CADASIL patients (M.C.K.). WMH volume was quantified relative to the brain parenchyme volume as described previously (24). Age-adjusted z-score of lacune count (for family member #1, #2, #4, #6) and WMH volume (for family member #1, #2, #6) were calculated using simple linear regression, with a previously published CADASIL cohort as reference (24). WMH volume in family member #4 could not be quantified because only sagittal brain MRI images were available.
Genetic variant analysis
In the index patient, a panel of 26 cerebral small vessel disease-associated genes (Supplementary Material, Table S1) was sequenced by the Leiden Laboratory for Diagnostic Genome Analysis using the Illumina HiSeq platform with the Agilent SureSelectXT Clearseq inherited disease panel target enrichment kit. The complete coding sequence was analysed, including 20 nucleotides in the flanking introns. In the other family members, the presence of the NOTCH3 variant was determined using Sanger sequencing. NOTCH3 transcript (NM_000435.2) was used as reference. The identified NOTCH3 variant was submitted to the Leiden Open Variant Database (lovd.nl/notch3) (25).
Qualitative and quantitative analysis of vascular NOTCH3 aggregation in skin biopsies
Skin punch biopsies of all six family members, as well as of three CADASIL patients (positive controls with NOTCH3 variants p.Arg182Cys and p.Cys144Phe located in exon 4, and with NOTCH3 variant p.Cys446Phe located in exon 8) and three unaffected individuals (negative controls), were taken from the lateral upper arm, fixed in formalin and embedded in paraffin. Two 5-μm sections were pretreated with proteinase K and washed three times for 5 min with PBS. The primary antibody (rabbit-anti-NOTCH3ECD, #25070002, Novus, dilution 1:500), was incubated for 2 h at room temperature. The secondary antibody (swine-anti-rabbit/biotin, Dako, dilution 1:400) was incubated for 30 min and developed with the Vectastain Elite ABC HRP Kit (PK-6100, Vectorlabs). Finally, slices were stained with 0.05% 3,3′-diaminobenzidine (DAB, Sigma-Aldrich) supplemented with 30% H2O2 for 10 min. To validate immunohistochemistry results, sections were also stained with another primary mouse anti-NOTCH3ECD antibody (clone 1E4, Millipore, dilution 1:1000; data not shown).
For quantification of vascular NOTCH3 aggregation, full-colour images were obtained with the Keyence BZ-X710 microscope (Keyence, Itasca, USA) at 1-ms capture time using 20× magnification. Ten images in the z-axis (pitch 1 μm) per location were made and stitched into one high-resolution, full-focus image by the Keyence BZ-X Analyzer software version 1.3.0.3. Regions of interest were drawn manually around vessel wall inner and outer boundaries. A median of 40 vessels per individual was analysed (minimally 13 vessels per family member). The NOTCH3ECD positive area was determined using a colour threshold (hue 0–50, saturation 0–255, brightness 0–175) in ImageJ and expressed as percentage of vessel wall area.
For electron microscopy, skin tissue blocks of ≤ 1 mm3 were postfixed for 90 min in 2% osmium tetroxide and 2% potassium ferrocyanide. After dehydration, tissue blocks were polymerized in Epon LX-112 for 48 h at 70°C. 80-nm sections were stained with uranyl acetate and lead citrate. Images were acquired on a digital camera (One View, Gatan Inc., Pleasanton, USA) mounted on a 120 kV transmission electron microscope (Tecnai T12 with a twin objective lens, Fei, Eindhoven, The Netherlands). Overlapping images were collected and stitched together into one image, as previously described (26). The presence of GOM in the walls of skin vessels was evaluated by independent observers (G.G., A.A.M., C.R.J.) in 5–14 vessels per individual, in all family members with the NOTCH3 variant, in two positive control CADASIL patients and in one healthy control.
Analysis of exon 9 skipping in fibroblast RNA
To obtain patient-derived fibroblasts, part of the skin biopsy was washed in PBS and incubated for 30 min at room temperature in collagenase A (0.42 units/ml, Roche). Primary fibroblast culture was performed in DMEM/F12 GlutaMAX supplement medium (Gibco Life Technologies, The Netherlands), supplemented with 10% heat-inactivated foetal calf serum (FCS) (Gibco), 2 μM MEM sodium pyruvate (Gibco), 0.5 U/ml penicillin and 0.5 μg/ml streptomycin (Gibco). Fibroblast RNA was isolated from ~ 1 × 106 cells according to the manufacturer’s instruction using the High Pure RNA Isolation Kit (Roche Diagnostics, Almere, The Netherlands). cDNA was synthesized from 1 μg RNA with the transcriptor first-strand cDNA synthesis kit according to the manufacturer’s protocol (Roche). RT-PCR was performed using a 10× PCR buffer with 1.5 mm MgCl2 (Roche) supplemented with 0.2 mm dNTP, 0.2 pmol/μL NOTCH3 exon 7 forward primer and exon 11 reverse primer with an annealing temperature of 60°C (Supplementary Material, Table S2) and 1 U FastStart Taq DNA polymerase (Roche). PCR products were then excised from gel and DNA was extracted using the Zymoclean Gel DNA Recovery Kit (Zymo Research, Irvine, USA) and analysed using Sanger sequencing (Macrogen Europe, Amsterdam, The Netherlands).
3D modelling of NOTCH3Δexon9
3D modelling of EGFr domains was performed with the online Swiss-Model tool (https://swissmodel.expasy.org/), using the sequence of NOTCH3 EGFr domains and a model template 5fma.1.A from the NOTCH1 protein. EGFr domains were visualized using Chimera software version 1.11.2.
Generation of NOTCH3 cDNA constructs
cDNA constructs (NOTCH3G498C, NOTCH3Y465C, NOTCH3Δexon9) were generated by adapting previously generated cDNA construct pTThN3FL and pSEhN3 (8). pSEhN3 was trimmed by removing exons 20–33 by HindIII digestion. Inverse PCR was performed on this clone using specific primer sets (Supplementary Material, Table S2) and Q5 High-Fidelity DNA Polymerase (New England Biolabs) to introduce the pathogenic variants in exon 9 or to delete exon 9. Following the PCR, the mixture was digested using DpnI to remove the methylated input DNA and purified using the GenElute™ PCR Clean-Up Kit (Sigma-Aldrich, Saint Louis, USA). The products were ligated and subsequently NEB5α cells were transformed with the ligation mixes. Clones were verified using Sanger sequencing (Macrogen). The KpnI-HindIII fragment of the clones carrying the modification was transferred to the pTThN3FL KpnI-HindIII backbone. Plasmids were isolated using NucleoSpin Plasmid QuickPure Kit according to the manufacturer’s protocol (Macherey-Nagel, Düren, Germany).
Protein expression, processing and signalling function of NOTCH3 Δexon9
Control fibroblasts were transfected with wild-type NOTCH3Δexon9 and empty vector constructs and stained for NOTCH3ECD to assess cellular localisation as described previously (8). In short, cells were either fixed and permeabilized with 4% paraformaldehyde and 0.1% Triton X-100 or only fixed without permeabilization using 4% paraformaldehyde and stained with a mouse anti-NOTCH3ECD antibody (clone 1E4, Millipore, dilution 1:10 000). To determine the signalling activity of NOTCH3Δexon9, a CBF1-responsive luciferase assay was performed as described previously (8). In short, NIH 3T3 cells were transfected with NOTCH3 cDNA constructs together with the Renilla luciferase expression vector (19, 27). One day after transfection, cells were co-cultured with 3T3 cells expressing the NOTCH3 ligand human JAGGED1 or with mock transfected 3T3 cells. Luciferase activity of NOTCH3Δexon9 was compared to NOTCH3 with a deletion of the LBD, NOTCH3C183R, NOTCH3Y465C, NOTCH3G498C and NOTCH3WT. Seven independent luciferase experiments were performed, with four technical replicates per experiment. Signalling values were normalized to unstimulated wild-type NOTCH3 signalling activity.
ASO-mediated exon 9 skipping
ASOs targeting NOTCH3 exon 9 were designed according to guidelines for ASO development using Human Splicing Finder (www.umd.be/HSF) (Supplementary Material, Table S3) (28, 29). ASOs consisted of 2′-O-methyl riboses and contained a full-length phosphorothioate backbone (2′-O-MePS). ASOs were tested on primary control arachnoid vascular smooth muscle cells (VSMCs) (8). Cultures of 1 × 105 cells were transfected with a final concentration of 100 nM ASO complexed with Lipofectamine2000 (ratio 2.67:1, Life Technologies) in serum-free medium. Exon 9 skipping was analysed 24 h after transfection using RT-PCR analysis with an exon 7 forward and exon 11 reverse primer. Results of the most efficient ASO are shown.
CRISPR/Cas9-induced exon 9 and exon 4–5 genomic deletions
To obtain a genomic exon 9 or exon 4–5 deletion, using CRISPR/Cas9 genome editing, guide RNAs were designed targeting NOTCH3 introns 8 and 9, and introns 3 and 5 with the online tool of the Zhang lab (https://zlab.bio/guide-design-resources). Guide RNAs were cloned into a modified pSQT1313 vector (Addgene #53370), with both guide RNAs flanked by Csy4 recognition sites. HEK293 cells and VSMCs were transfected during 4 h with 0.5 μg DNA encoding the guide RNAs and 3.5 μg DNA of a modified eSpCas9(1.1) (Addgene #71814), expressing both the SpCas9 protein and the Csy4 RNAse, together with Lipofectamine2000 (DNA/Lipofectamine ratio 1:2.5) in serum-free medium. Two days after transfection, DNA and RNA were isolated, and the deletion of exon 9 was assessed by PCR using primers in introns 8 and 9, and by RT-PCR using primers in exon 7 and 11, respectively. The deletion of exon 4–5 was assessed by RT-PCR using primers in exon 3 and 6.
Statistical analysis
Differences between groups were compared using one-way ANOVA analysis with Bonferroni’s post hoc correction in the immunohistochemistry analysis and with Dunnett’s post hoc correction relative to the wild-type condition in the NOTCH3 signalling assay. Ligand-dependent increase in luciferase activity was tested per condition using independent samples t-tests. Normality of the data was assessed using histograms and could be assumed. All statistical analyses were two-sided tests with threshold for statistical significance of 0.05, using the IBM SPSS Statistics version 23.0.0.2 software.
Supplementary Material
Acknowledgements
We acknowledge H.M. van der Klift and N. Lamzira-Arichi for their assistance in the variant analysis. We thank A.M. van Opstal for the quantification of the WMH.
Conflict of Interest Statement
M.O., G.B., I.H., A.A.M., S.G.v.D., M.C.K., G.M.T. and C.R.J. report no conflict of interest. G.G., J.W.R. and S.A.J.L.O. were financially supported by the Netherlands Brain Foundation (HA2016-02-03; BG2015-2). NOTCH3 antisense therapies have been patented by the Leiden University Medical Center. As co-inventors on this patent J.G.D., A.A.R. and S.A.J.L.O. are entitled to a share of potential royalties. F.B. is employed by LUMC and named inventor on patents related to neuroregeneration and is founder of Complement Pharma BV. He receives funding for contract research from WAVE technologies and is a member of scientific advisory board of CMTA USA. Renumeration is paid to LUMC. A.A.R. discloses being employed by LUMC which has patents on exon skipping technology, including NOTCH3. As co-inventor of these patents, A.A.R. is entitled to a share of royalties. For full transparency (not related to this work), A.A.R. discloses being ad hoc consultant for PTC Therapeutics, Sarepta Therapeutics, CRISPR Therapeutics, Summit PLC, Alpha Anomeric, BioMarin Pharmaceuticals Inc., Eisai, Astra Zeneca, Santhera, Audentes, Guidepoint Global and GLG consultancy, Grunenthal, Wave and BioClinica, having been a member of the Duchenne Network Steering Committee (BioMarin) and being a member of the scientific advisory boards of ProQR and Philae Pharmaceuticals. Remuneration for these activities is paid to LUMC. LUMC also received speaker honoraria from PTC Therapeutics and BioMarin Pharmaceuticals and funding for contract research from Italfarmaco and Alpha Anomeric.
Funding
The Netherlands Brain Foundation (HA2016-02-03; BG2015-2).
References
- 1. Chabriat H., Joutel A., Dichgans M., Tournier-Lasserve E. and Bousser M.-G. (2009) Cadasil. Lancet Neurol., 8, 643–653. [DOI] [PubMed] [Google Scholar]
- 2. Joutel A., Corpechot C., Ducros A., Vahedi K., Chabriat H., Mouton P., Alamowitch S., Domenga V., Cécillion M., Marechal E. et al. (1996) Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature, 383, 707–710. [DOI] [PubMed] [Google Scholar]
- 3. Rutten J.W., Haan J., Terwindt G.M., van Duinen S.G., Boon E.M.J. and Lesnik Oberstein S.A.J. (2014) Interpretation of NOTCH3 mutations in the diagnosis of CADASIL. Expert. Rev. Mol. Diagn., 14, 593–603. [DOI] [PubMed] [Google Scholar]
- 4. Brulin P., Godfraind C., Leteurtre E. and Ruchoux M.-M. (2002) Morphometric analysis of ultrastructural vascular changes in CADASIL: analysis of 50 skin biopsy specimens and pathogenic implications. Acta Neuropathol., 104, 241–248. [DOI] [PubMed] [Google Scholar]
- 5. Joutel A., Favrole P., Labauge P., Chabriat H., Lescoat C., Andreux F., Domenga V., Cécillon M., Vahedi K., Ducros A. et al. (2001) Skin biopsy immunostaining with a Notch3 monoclonal antibody for CADASIL diagnosis. Lancet, 358, 2049–2051. [DOI] [PubMed] [Google Scholar]
- 6. Lesnik Oberstein S.A.J., van Duinen S.G., van den Boom R., Maat-Schieman M.L.C., van Buchem M.A., van Houwelingen H.C., Hegeman-Kleinn I.M., Ferrari M.D., Breuning M.H. and Haan J. (2003) Evaluation of diagnostic NOTCH3 immunostaining in CADASIL. Acta Neuropathol., 106, 107–111. [DOI] [PubMed] [Google Scholar]
- 7. Schoemaker D., Quiroz Y.T., Torrico-Teave H. and Arboleda-Velasquez J.F. (2019. Available from:) Clinical and research applications of magnetic resonance imaging in the study of CADASIL. Neurosci. Lett. [Internet]. https://linkinghub.elsevier.com/retrieve/pii/S0304394019300205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rutten J.W., Dauwerse H.G., Peters D.J.M., Goldfarb A., Venselaar H., Haffner C., Ommen G.J., Aartsma-Rus A.M. and Lesnik Oberstein S.A. (2016) Therapeutic NOTCH3 cysteine correction in CADASIL using exon skipping: in vitro proof of concept. Brain, 139, 1123–1135. [DOI] [PubMed] [Google Scholar]
- 9. van den Boom R., Lesnik Oberstein S.a.J., Ferrari M.D., Haan J. and van Buchem M.A. (2003) Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy: MR imaging findings at different ages—3rd–6th decades. Radiology, 229, 683–690. [DOI] [PubMed] [Google Scholar]
- 10. Rutten J.W., Van Eijsden B.J., Duering M., Jouvent E., Opherk C., Pantoni L., Federico M., Dichgans H.S. and Markus H. (2018) The effect of NOTCH3 pathogenic variant position on CADASIL disease severity: NOTCH3 EGFr 1-6 pathogenic variant are associated with a more severe phenotype and lower survival compared with EGFr 7-34 pathogenic variant. Genet. Med., 21, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rutten J.W., Klever R.R., Hegeman I.M., Poole D.S., Dauwerse H.G., Broos L.A.M., Breukel C., Aartsma-Rus A.M., Verbeek J.S., Weerd L. et al. (2015) The NOTCH3 score: a pre-clinical CADASIL biomarker in a novel human genomic NOTCH3 transgenic mouse model with early progressive vascular NOTCH3 accumulation. Acta Neuropathol. Commun., 3, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Joutel A., Monet-Leprêtre M., Gosele C., Baron-menguy C., Hammes A., Schmidt S., Lemaire-Carrette B., Domenga V., Schedl A., Lacombe P. et al. (2010) Cerebrovascular dysfunction and microcirculation rarefaction precede white matter lesions in a mouse genetic model of cerebral ischemic small vessel disease. J. Clin. Invest., 120, 433–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mykkänen K., Junna M., Amberla K., Bronge L., Kääriäinen H., Pöyhönen M., Kalimo H. and Viitanen M. (2009) Different clinical phenotypes in monozygotic cadasil twins with a novel notch3 mutation. Stroke, 40, 2215–2218. [DOI] [PubMed] [Google Scholar]
- 14. Singhal S., Bevan S., Barrick T., Rich P. and Markus H.S. (2004) The influence of genetic and cardiovascular risk factors on the CADASIL phenotype. Brain, 127, 2031–2038. [DOI] [PubMed] [Google Scholar]
- 15. Opherk C., Gonik M., Duering M., Malik R., Jouvent E., Hervé D., Adib-Samii P., Bevan S., Pianese L., Silvestri S., Dotti M.T., De Stefano N. et al. (2014) Genome-wide genotyping demonstrates a polygenic risk score associated with white matter hyperintensity volume in CADASIL. Stroke, 45, 968–972. [DOI] [PubMed] [Google Scholar]
- 16. Opherk C., Peters N., Holtmannspötter M., Gschwendtner A., Müller-Myhsok B. and Dichgans M. (2006) Heritability of MRI lesion volume in CADASIL: evidence for genetic modifiers. Stroke, 37, 2684–2689. [DOI] [PubMed] [Google Scholar]
- 17. Arboleda-Velasquez J.F., Manent J., Lee J.H., Tikka S., Ospina C., Vanderburg C., Frosch M.P., Rodriguez-Falcon M., Villen J., Gygi S., Lopera F., Kalimo H., et al. (2011) Hypomorphic notch 3 alleles link notch signaling to ischemic cerebral small-vessel disease. Proc. Natl. Acad. Sci. USA, 108, E128–E135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moccia M., Mosca L., Erro R., Cervasio M., Allocca R., Vitale C., Leonardi A., Caranci F., Del Basso-De Caro M.L., Barone P. and Penco A. (2015) Hypomorphic NOTCH3 mutation in an Italian family with CADASIL features. Neurobiol. Aging., 36, 547.e5–547.e547.e11. [DOI] [PubMed] [Google Scholar]
- 19. Peters N., Opherk C., Zacherle S., Capell A., Gempel P. and Dichgans M. (2004) CADASIL-associated Notch3 mutations have differential effects both on ligand binding and ligand-induced Notch3 receptor signaling through RBP-Jk. Exp. Cell Res., 299, 454–464. [DOI] [PubMed] [Google Scholar]
- 20. Rahman S., Datta M., Kim J. and Jan A.T. (2019) CRISPR/Cas: an intriguing genomic editing tool with prospects in treating neurodegenerative diseases. Semin. Cell Dev. Biol. [Internet], 96, 22–31. Available from:http://www.ncbi.nlm.nih.gov/pubmed/31102655. [DOI] [PubMed] [Google Scholar]
- 21. Wurster C.D. and Ludolph A.C. (2018) Antisense oligonucleotides in neurological disorders. Ther. Adv. Neurol. Disord., 11, 175628641877693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Luu K.T., Norris D.A., Gunawan R., Henry S., Geary R. and Wang Y. (2017) Population pharmacokinetics of Nusinersen in the cerebral spinal fluid and plasma of Pediatric patients with spinal muscular atrophy following Intrathecal administrations. J. Clin. Pharmacol., 57, 1031–1041. [DOI] [PubMed] [Google Scholar]
- 23. Geary R.S., Norris D., Yu R. and Bennett C.F. (2015) Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev., 87, 46–51. [DOI] [PubMed] [Google Scholar]
- 24. Liem M.K., Haan J., Van Der Neut I.L., Ferrari M.D., Van Buchem M.A. and Van Der Grond J. (2009) MRI correlates of cognitive decline in CADASIL. Neurology, 72, 143–148. [DOI] [PubMed] [Google Scholar]
- 25. Fokkema I.F.A.C., Taschner P.E.M., Schaafsma G.C.P., Celli J., Laros J.F.J. and den Dunnen J.T. (2011) LOVD v.2.0: the next generation in gene variant databases. Hum. Mutat., 32, 557–563. [DOI] [PubMed] [Google Scholar]
- 26. Faas F.G.A., Cristina Avramut M., van den B.M., Mieke Mommaas A., Koster A.J. and Ravelli R.B.G. (2012) Virtual nanoscopy: generation of ultra-large high resolution electron microscopy maps. J. Cell Biol., 198, 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karlström H., Beatus P., Dannaeus K., Chapman G., Lendahl U. and Lundkvist J. (2002) A CADASIL-mutated notch 3 receptor exhibits impaired intracellular trafficking and maturation but normal ligand-induced signaling. Proc. Natl. Acad. Sci. USA., 99, 17119–17124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aartsma-Rus A., De Winter C.L., Janson A.A.M., Kaman W.E., Van Ommen G.-J.B., Den Dunnen J.T. and Van Deutekom J.C.T. (2005) Functional analysis of 114 exon-internal AONs for targeted DMD exon skipping: indication for steric hindrance of SR protein binding sites. Oligonucleotides, 15, 284–297. [DOI] [PubMed] [Google Scholar]
- 29. Aartsma-Rus A., Fokkema I., Verschuuren J., Ginjaar I., Van Deutekom J., Van Ommen G.J. and Den Dunnen J.T. (2009) Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat., 30, 293–299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.