

Abstract

Mycoplasma species share a set of features, such as lack of a cell wall, streamlined genomes, simplified metabolism, and the use of a deviant genetic code, that make them attractive approximations of what a chassis strain should ideally be. Among them, Mycoplasma pneumoniae arises as a candidate for synthetic biology projects, as it is one of the most deeply characterized bacteria. However, the historical paucity of tools for editing Mycoplasma genomes has precluded the establishment of M. pneumoniae as a suitable chassis strain. Here, we developed an oligonucleotide recombineering method for this strain based on GP35, a ssDNA recombinase originally encoded by a Bacillus subtilis-associated phage. GP35-mediated oligo recombineering is able to carry out point mutations in the M. pneumoniae genome with an efficiency as high as 2.7 × 10–2, outperforming oligo recombineering protocols developed for other bacteria. Gene deletions of different sizes showed a decreasing power trend between efficiency and the scale of the attempted edition. However, the editing rates for all modifications increased when CRISPR/Cas9 was used to counterselect nonedited cells. This allowed edited clones carrying chromosomal deletions of up to 1.8 kb to be recovered with little to no screening of survivor cells. We envision this technology as a major step toward the use of M. pneumoniae, and possibly other Mycoplasmas, as synthetic biology chassis strains.
One main aim of synthetic biology is to design microorganisms with novel capabilities that could be interesting for a huge variety of applications, such as green chemistry,1,2 bioremediation,3 and microbial therapies.4 As an integrative discipline, synthetic biology aims to bring an engineering perspective to the design of new living forms.5 Following this rationale, designer organisms should be based on a genomic backbone (i.e., chassis), ideally depleted of most of the functions irrelevant to its programmed purpose, with additional modules that can be plugged into this chassis to add novel functionalities. While several interesting modules have been developed,6−10 most synthetic biology projects so far have revolved around a small set of chassis strains, primarily Escherichia coli and Saccharomyces cerevisiae. However, the adoption of these organisms as chassis strains was mainly related to their strong adaptation for laboratory requirements (e.g., fast growth, efficient recombination) rather than their fulfilment of a set of desirable features. For instance, from an engineering point of view, E. coli could represent a reasonable starting structure into which different modules can be introduced to develop a designer strain for gastrointestinal therapies.11,12 In contrast, for other environments or purposes, E. coli might not be the best-suited candidate to use for engineering a designer microorganism. Indeed, it is becoming widely accepted that to move synthetic biology from the laboratory to the field, novel chassis strains should be generated; however, the development of such strains is tightly linked to the establishment of advanced genome editing tools for these less well-studied bacteria.13
Mycoplasma strains share several distinctive features, including the lack of a cell wall, streamlined genomes, limited biosynthetic capabilities, and a variant genetic code in which the UGA codon codes for tryptophan rather than being read as a stop codon.14 All of these features might be of interest for different synthetic biology concerns, such as orthogonality, biosafety, and limited horizontal gene transfer. In other words, the naturally reduced genomes of Mycoplasmas fit perfectly with the chassis concept of synthetic biology. Notably, the human pathogen Mycoplasma pneumoniae arises as candidate for synthetic biology projects because it is one of the most deeply characterized bacteria, as a consequence of being a model organism for systems biology for over a decade.15−19 Thus, by removing the few and well-characterized pathogenicity determinants found in its genome,20M. pneumoniae could become a suitable chassis for plugging in gene platforms to provide the desired functions. Specifically, the natural tropism of M. pneumoniae toward the human respiratory tract might facilitate the development of a designer strain capable to deliver therapeutic molecules into the lung. However, the transition from using M. pneumoniae as a model organism for systems biology to creating a chassis strain for synthetic biology has been hindered so far by the historical paucity of genome editing tools for this bacterium.
There are few reports describing the achievement of targeted gene deletions within the Mycoplasma genus. Initially restricted to Mycoplasma genitalium,21−23 and later expanded with limited success to other strains,24,25 all of these reports rely on the transformation of a nonreplicative plasmid carrying a selectable marker surrounded by regions homologous to the sequences that flank the target gene. In this framework, the appearance of a mutant cell relies on the ability of the Mycoplasma recombination machinery to perform the deletion, making direct editing of Mycoplasma genomes an inefficient and unreliable process. In the case of the M. pneumoniae reference strain M129, only one positively edited clone was obtained, reflecting its poor recombination capability.25 This has been linked to the lack of a functional copy of the RecU Holliday junction resolvase.26,27 Alternatively, it has been proposed that its recombination machinery is tightly controlled by the expression of a sigma factor encoded by the MPN626 gene, whose overexpression is rather toxic, as inferred from data available for its orthologue MG428 in the closely related bacterium M. genitalium.28
The poor performance of the recombination machinery in most Mycoplasma species has forced researchers to develop alternative strategies such as Haystack mutagenesis.29 This technique allows the isolation of clones carrying transposon insertions at the locus of interest, through a comprehensive and iterative PCR screening of an ordered collection of pooled random transposon insertion mutants. Despite being a tedious and time-consuming protocol, it has become the standard method to obtain mutants in most Mycoplasma species. However, it cannot be considered a true genome-editing tool, as it only allows the selection of clones in which a particular gene has been disrupted but not deleted or edited. On the other extreme of technical innovation, we find the chemical synthesis of whole mycoplasma genomes30 and subsequent transplantation into a recipient cell.31 Though strictly this is a genome-writing rather than a genome-editing technique, the possibilities it opens up are fascinating. However, the cost of synthesizing a whole genome is prohibitive for most academic laboratories. Alternatively, a more affordable strategy is to clone naturally existing Mycoplasma genomes as yeast circular centromeric plasmids.32 These genomes can later be comprehensively modified using the state-of-the-art editing tools available for yeast33−35 before their reintroduction into a recipient Mycoplasma cell (i.e., genome transplantation).36 This complete cycle of cloning, in-yeast modification, and genome transplantation, has led to the generation of a fully attenuated M. mycoides subsp. capri strain unable to cause lung lesions in a goat animal model.37 Unfortunately, all genome transplantation experiments so far have used M. capricolum as the recipient cell, and only genomes from species closely related to this strain in terms of phylogeny can be employed as donors. Therefore, genome transplantation continues to be a bottleneck for many synthetic biology projects and does not seem to be broadly applicable to other species, even within the Mycoplasma genus.38
In this work, we developed a reliable genome editing system for M. pneumoniae. Rather than trying to enhance the M. pneumoniae recombination machinery, we developed an oligo recombineering system based on the GP35 recombinase, a protein originally found in the genome of the Bacillus subtilis associated phage SPP1. A recent survey on bacterial recombinases pinpointed GP35 as the most efficient protein to perform recombineering in B. subtilis,39 a species phylogenetically related to the Mycoplasma genus. We conducted a similar survey revealing that, for M. pneumoniae, GP35 also outperforms other putative recombinases in terms of recombineering efficiency. Using GP35-based oligo recombineering, point mutations (i.e., 1-bp deletions) could be obtained with efficiencies as high as 2% of the total amount of cells, whereas larger modifications (i.e., 1800-bp deletions) had efficiencies of ∼10–5. These editing efficiencies were further boosted by using CRISPR/Cas9 technology to counterselect nonedited cells. This allowed edited clones to be recovered with little to no screening of survivor cells. We believe this technology is a great step toward the use of M. pneumoniae as a synthetic biology chassis strain.
Results and Discussion
Establishment of a Recombineering System for M. pneumoniae
To overcome the lack of genome-editing tools available for M. pneumoniae, we aimed to develop an oligo recombineering system functional for this strain.40 This technology relies on two consecutive events. The first step is the homology-driven positioning of oligonucleotides at the lagging strand of the replication fork, a process that in bacteria can be boosted by phage-derived ssDNA recombinases.41 Subsequently, the arranged oligonucleotide is incorporated into the newly synthesized chromosomal copy as an Okazaki fragment, thereby mediating the introduction of the intended modifications in the genome. The apparent simplicity of this process, together with the universal conservation of the replication mechanism, might lead one to assume that oligo recombineering is a broadly portable technology capable of editing genomes independently of the host recombination machinery. However, phage-derived recombinases do not maintain their efficient performance across different bacterial genera, suggesting some sort of dependence on host machinery.42 Indeed, it seems that the recombineering frequency obtained depends on the phylogenetic distance between the native host of the phage and the bacteria being engineered.39
Prompted by these observations, we decided to survey the mycoplasma pan-genome as well as their associated phages for the presence of proteins orthologous to Recβ and RecT. These two proteins from the lambda and Rac phages, respectively, are the best-characterized and most frequently employed phage-derived ssDNA recombinases in oligo recombineering protocols.40 We found three RecT-like proteins coded by the genomes of Spiroplasma melliferum, Spiroplasma citri, and Spiroplasma poulsonii (Figure S1), which we renamed RecTsm, RecTsc, and RecTsp, respectively. Our search for Mycoplasma Recβ orthologues did not produce any relevant candidate. To further complement the screening, we wanted to include a recombinase with a proven capacity to perform recombineering. This led us to choose GP35, a protein recently reported to be the most efficient phage-derived recombinase for performing genome editing in Bacillus subtilis,39 a species phylogenetically related to the Mycoplasma genus.
The ability of the different proteins to introduce changes to the M. pneumoniae genome by catalyzing oligo recombineering was experimentally monitored with a recombineering sensor termed MutCm+1. This sensor is based on a chloramphenicol acetyltransferase gene (cat), whose protein product confers resistance to chloramphenicol (Cm). Nevertheless, the cat coding sequence in the sensor is frame-shifted by the addition of a single nucleotide at position 310, rendering a protein product unable to confer resistance to the antibiotic. To correct the MutCm+1 sensor, we designed two different oligonucleotides termed CmONsense and CmONantisense, following the rules reported in a screening of optimized design criteria for recombineering oligonucleotides.43 Both oligonucleotides have the exact same sequence present in the region surrounding the frame-shift included in the sensor, except for the extra nucleotide. However, whereas CmONsense has the same orientation as the frame-shifted cat gene, the CmONantisense oligonucleotide is antisense to the sequence of the cat gene. In principle, either of the oligonucleotides could mediate the deletion of the frame-shifted nucleotide and the consequent activation of the cat gene. However, as oligonucleotides are incorporated as Okazaki fragments into the newly synthesized chromosome, those targeting the lagging strand produce a substantially higher editing rate than those binding the leading strand of the replication fork.41 Thus, determining the location of the MutCm+1 sensor within the M. pneumoniae genome was essential for our screen, as synthesis of a DNA strand as either leading or lagging depends on its chromosomal location with respect to the origin of replication. To this end, the MutCm+1 sensor was cloned into a transposon vector and transformed into M. pneumoniae WT cells to generate a strain termed M129MutCm+1. After clone isolation, we used an arbitrary PCR (A-PCR) protocol44 to locate the transposon insertion at genome position 60107 (MPN049 locus) of the minus strand. Thus, the CmONsense oligonucleotide would be the one targeting the lagging strand at this location and should yield a higher number of edited cells (Figure 1A).
Figure 1.

Screening of different ssDNA recombinases to perform oligo recombineering in M. pneumoniae. (A) Scheme depicting the chromosome of M129MutCm+1 strain, showing the bidirectional replication fork that starts at the origin of replication (ori) and enlarges until reaching the terminus of replication (ter). The plus and minus strands follow the indicated color code; newly synthesized DNA is either continuous (solid line) or discontinuous (dashed line). The MutCm+1 recombineering sensor located at the minus strand of the MPN049 locus is indicated by a green arrow, as well as the CmONsense or CmONantisense editing oligonucleotides reflected with a color code if their sequence is the same as present on the plus or the minus strand of the chromosome. (B) Bar plot showing in logarithmic scale the colony-forming units (CFU) obtained for M. pneumoniae M129 cells carrying the recombineering sensor (M129MutCm+1 strain) or the recombineering sensor plus a second transposon coding for the different recombinases (M129MutCm+1GP35, M129MutCm+1RecTsm, M129MutCm+1RecTsp, or M129MutCm+1RecTsc). Cells were subjected to a mock transformation (yellow bars) or transformation with the editing oligos CmONantisense (blue bars) or CmONsense (green bars) and then seeded onto nonselective (gray bars) or Cm-selective plates (yellow, blue, and green bars). Total cells (gray bars) are calculated as the mean of CFU counted on nonselective plates for the three transformations for each strain. The editing rate (edited cells/total cells) obtained with the CmONsense oligonucleotide for each strain is shown above the bars. Those differences in terms of editing rate that were found to be statistically significant (P < 0.05) after conducting a paired t test are indicated with an asterisk (*). Error bars represent the mean of the standard deviation (SEM) of three different biological replicas.
Four different strains, all containing the MutCm+1 sensor and the different recombinases found in our orthologue search (i.e., GP35, RecT-sm, RecT-sc, and RecT-sp), were subjected to a mock transformation, or to transformations with the editing oligos CmONsense and CmONantisense. In addition, a control strain not expressing any recombinase was included in this screening (Figure 1B). Two hours post-transformation, serial dilutions of cells were seeded on either nonselective plates to calculate the total amount of cells or on Cm-supplemented plates to determine the amount of edited cells for each condition. These values allowed us to calculate the editing rate (edited cells/total cells) for each recombinase and condition (Table S1).
For all strains assessed, mock transformations yielded a low proportion of cells that were Cm-resistant (Figure 1B). The small amount of cells observed in all the cases (i.e., ∼2 × 102) might represent spontaneous mutants resistant to Cm or might arise as a consequence of poor Cm selective pressure when highly concentrated dilutions are spotted. In any case, this frequency of Cm-resistant cells should be considered as a background signal of our screening, as its occurrence is not mediated by a recombineering phenomenon.
The amount of Cm-resistant cells increased in all the strains when transformed with the CmONantisense oligonucleotide (Figure 1B). However, the amount of edited cells barely overcame the background signal of the screening (i.e., ∼9 × 102 vs ∼2 × 102), except for the strain expressing the GP35 recombinase, for which we detected an increase of almost 2 orders of magnitude in the amount of Cm-resistant cells as compared to the background signal (1.6 × 104 vs ∼2 × 102, respectively). Moreover, the difference between the GP35-expressing strain and all others was further increased when transformed with CmONsense oligonucleotide (Figure 1B). In this scenario, the amount of edited (Cm-resistant) cells was 1.6 × 105 for the strain expressing GP35, but only slightly higher than background signal (i.e., ∼2 × 103 vs ∼2 × 102) for all other strains.
Altogether, these results suggest that none of the RecT-like recombinases are functional in M. pneumoniae, even though the expression levels of at least RecT-sm and RecT-sc were similar to the those in the strain expressing the GP35 recombinase (Figure S2). Surprisingly, RecT-sm and RecT-sc are annotated as a RecT family protein and a putative RecT protein, respectively. Although it cannot be ruled out that these proteins might behave as actual recombinases in their native organisms, it seems that they could be carrying out alternative functions, despite showing a moderate sequence similarity with RecT proteins. In contrast, we found that GP35 is a functional protein that performs oligonucleotide recombineering in M. pneumoniae with an editing efficiency reaching 9.8 × 10–5.
Optimization of GP35-Mediated Oligo Recombineering in M. pneumoniae
Our screening identified GP35 as the first reported recombinase capable of mediating oligonucleotide recombineering in M. pneumoniae. Yet, the editing efficiencies obtained were still far from those reported for other recombinase-microorganism pairs. For instance, in E. coli expressing Recβ protein, oligo recombineering mediates 1 bp gene editing with efficiencies as high as 2 × 10–1,43 whereas the same type of modification is obtained at frequencies of 1.8 × 10–3 for Pseudomonas putida expressing Rec2 protein45 or at 2.5 × 10–3 for Staphylococcus aureus expressing EF2132 protein.46 Consequently, we attempted to increase the efficiency by changing some parameters of our recombineering protocol.
As a starting point, we reasoned that if GP35 protein catalyzes the incorporation of the oligonucleotide at the replication fork, a permissive window of time should be considered to perform its task. For a slow-dividing microorganism such as M. pneumoniae, with a doubling time of approximately 8 h, the 2 h interval between transformation and plate seeding employed in the initial screening might be insufficient to ensure that the replication fork has passed at least once across the desired locus in all the cells of the population. To test this hypothesis, the GP35-expressing strain carrying the MutCm+1 sensor (M129MutCm+1GP35) was transformed with the CmONsense oligonucleotide, but this time the cells were grown under nonselective conditions for either 2, 24, or 48 h before plate seeding (Table S2 and Figure 2A).
Figure 2.

Optimization of GP35-oligo recombineering protocol for M. pneumoniae. (A–C) Bar plots showing in logarithmic scale the CFU obtained for the M129MutCm+1GP35 strain after transformation with the CmONsense oligonucleotide and seeding on either nonselective plates (gray bars) or Cm-selective plates (green bars). The editing rate (edited cells/total cells) is shown above each group of bars. Those differences in terms of editing rate that were found to be statistically significant (P < 0.05) after conducting a paired t test are indicated with an asterisk (*). Error bars represent the SEM of three different biological replicas. (A) Transformed cells were allowed to recover for different time intervals prior to seeding, as indicated on the x-axis. (B) M129MutCm+1GP35 cells were transformed with different amounts of oligo, as indicated on the x-axis, and seeded on plates at 24 h after transformation. (C) M129MutCm+1GP35 cells were transformed with 5 μL of the CmONsense oligo and subjected to 1, 3, 6, or 10 electroporation pulses, as indicated on the x-axis. Cells were allowed to recover for 24 h before seeding. Cell viability for each condition is expressed as a percentage of that observed after one electroporation pulse, as shown below each bar.
In line with our hypothesis, when cells that received the oligonucleotide were grown for 24 h before plating, the editing efficiency increased to 1.3 × 10–3. Indeed, during the time window between 2 and 24 h post-transformation, the total number of cells rose almost six times (5.3 × 108 vs 3.1 × 109, respectively), whereas the numbers of edited cells increased ∼43 times (9.2 × 104 vs 4 × 106, respectively). This result further suggests that the GP35-oligo editing mechanism is linked to the replication machinery. A similar editing efficiency was observed when comparing 24- and 48-h post transformation time points (1.3 × 10–3 vs 8.9 × 10–4, respectively).
To further increase the transformation efficiency, we fixed 24 h as the optimal time frame before plating and examined the contribution of the amount of recombination substrate (Table S2, and Figure 2B). Increasing the oligonucleotide amount from 1 to 5 μL enhanced the editing rate almost 7 times (9.4 × 10–4 vs 6.3 × 10–3, respectively), whereas using 10 μL did not significantly improve this rate (giving 6.8 × 10–3). Thus, we concluded that using 5 μL of the editing oligonucleotide is sufficient to saturate the recombineering process.
Finally, we explored whether multiple electroporation pulses could improve editing efficiency. A similar strategy was reported to increase the number of plasmid-transformed cells by 2–5 times in Agrobacterium tumefaciens.47 Thus, M129MutCm+1GP35 cells were transformed with 5 μL of CmONsense oligonucleotide, subjected to a variable number of pulses (i.e., 1, 3, 6 or 10), and seeded on plates 24 h post-transformation (Table S2 and Figure 2C). We observed a general trend of compromised viability with increasing number of pulses: of the cells that survived a single pulse, only ∼30% remained viable after 6 or 10 pulses. However, the number of edited cells increased with the number of pulses, at least until the viability of the total population started to be severely compromised. Altogether, this screening identified 6 electroporation pulses as the trade-off point between an increased number of edited cells and total cell viability, resulting in an editing rate 2.3 times higher than that obtained with one pulse (2.7 × 10–2 vs 1.2 × 10–2, respectively).
In sum, after a limited screening of a series of parameters that could affect the recombineering process, we increased the editing rate 165 times (from 1.6 × 10–4 to 2.7 × 10–2). Indeed, after optimization, the frequency of incorporation of a 1-bp deletion in M. pneumoniae outperformed that reported for other bacteria such as S. aureus(46) or P. putida(45) by 1 order of magnitude.
The Scale of Genetic Modifications Affects Recombineering Efficiency
The future adoption of M. pneumoniae as a suitable synthetic biology chassis strain would unarguably require more than point mutations to its genome. Therefore, we determined which efficiencies could be obtained when using GP35-mediated oligo recombineering to carry out gene edits larger than 1 bp. To this end, we created three new reporters in which a 50-bp, 750-bp, or 1800-bp frame-shifting sequence was placed in the cat coding sequence. Next, M. pneumoniae WT cells were subjected to a first transformation with transposon vectors carrying the above-mentioned sensors, and a second transformation with the GP35-coding transposon. The resulting clonal strains termed M129MutCm+50GP35, M129MutCm+750GP35, and M129MutCm+1800GP35 were found to harbor the sensors in the positive strand of the MPN493, MPN582, and MPN034 loci, respectively. In line with these locations, the CmONsense oligonucleotide was used as a recombineering substrate for both M129MutCm+50GP35 and M129MutCm+750GP35 strains, whereas the CmONantisense oligonucleotide was used for the M129MutCm+1800 strain (Figure S3). The three reporter strains were then subjected to 6 electroporation pulses with 5 μL of their corresponding oligos, and seeded on plates 24 h post-transformation. (Table S3 and Figure 3A).
Figure 3.

Efficiency of GP35-oligo recombineering for large chromosomal modifications. (A) Bar plot showing in logarithmic scale the CFU obtained for different recombineering sensor strains (x-axis) after transformation with their respective editing oligos following the conditions established in the optimization screening and seeding on nonselective medium (gray bars) or on Cm-selective medium (green bars). All strains expressed GP35 recombinase and different recombineering sensors whose activation required the deletion of 50 bp, 750 bp, or 1800 bp, depending on the strain. The editing rate (edited cells/total cells) obtained for each strain is shown on top of each group of bars. Those differences in terms of editing rate that were found to be statistically significant (P < 0.05) after conducting a paired t test are indicated with an asterisk (*). Error bars represent the SEM of three different biological replicas. (B) Plot comparing the size of the attempted chromosomal deletion and the editing rate obtained for that modification. Each green rectangle represents the mean editing rate of three independent biological replicas performed with M129MutCm+1GP35, M129MutCm+50GP35, M129MutCm+750GP35, or M129MutCm+1800GP35 strains. Error bars represent the SEM of three different biological replicas. Dotted line represents the decreasing power trend observed between deletion size and efficiency. The equation describing this trend as well as the coefficient of determination (R2) is shown inside the square.
For all three reporter strains, there was a clear anticorrelation between recombineering efficiency and length of the attempted deletion (Figure 3A). Specifically, the editing rate obtained for a 50-bp deletion was 8.1 × 10–3, which is only 3-fold lower than that observed for a 1-bp deletion (2.7 × 10–2). In contrast, deletions of 750 bp and 1800 bp occurred at much lower frequencies, with editing rates of 3.4 × 10–4 and 9.5 × 10–5, respectively. Collectively, these results showed that GP35-mediated oligo recombineering can perform targeted chromosomal deletions of various sizes, though the efficiency is affected by the length of the deletion. Indeed, when the editing rates obtained for each reporter strain are plotted against the deletion size, the results adjust quite well to a decreasing power trend (Figure 3B). These results are in line with those previously reported for recombineering-mediated chromosomal deletions in E. coli.43 Of note, the different recombineering sensors were incorporated at chromosomal locations quite distant from each other (i.e., MPN049, MPN493, MPN582, and MPN034), suggesting that the whole chromosome is capable of being edited by GP35-mediated oligo recombineering.
Adaptation of CRISPR/Cas9 Technology to M. pneumoniae as a Tool to Counterselect Nonedited Cells
The main limitation of oligo recombineering lies in its inability to select for those cells carrying the intended modification, as the limited oligonucleotide length precludes the inclusion of a selection marker into the chromosome of edited cells to facilitate their identification. To solve this, spCas9, a Streptococcus pyogenes-derived protein forming part of the widely known CRISPR/Cas system,48 has been recently repurposed as counterselection tool for recombineering protocols49 given its ability to specifically cleave a target DNA sequence in an easily reprogrammable manner. This ability relies on short guide RNAs (sgRNAs) that guide the endonuclease Cas9 to their complementary strand on the target DNA, and also on the presence of a 5′-NGG-3′ consensus sequence immediately downstream of the target site, which is called the protospacer adjacent motif (PAM).48 Cas9 chromosomal cleavage is highly lethal in bacteria, presumably due to the lack of NHEJ systems in most of the genera.50 This toxicity has been exploited to counterselect nonedited cells in oligo recombineering protocols developed for different strains.46,51−53
For this reason, the transposon vector employed to introduce the GP35 recombinase into the three reporter strains also contained a Cas9-based counterselection platform. Specifically, this platform was composed of: (i) an inducible system responding to anhydrotetracycline (aTc), based on the tet repressor (TetR) and a promoter termed Pxyl/tetO2mod,54 (ii) a copy of the enhanced-Cas9 (eCas9) coding sequence,55 and (iii) a sgRNA termed eNT2 that targets the nontemplate strand of the gene coding for the Venus fluorescent protein.56 Note that the sequence recognized by eNT2 sgRNA is present in the three different recombineering sensors, as part of the frame-shifting sequences. Thus, in principle only edited cells—that is, those that have incorporated a recombineering oligo and consequently deleted the Cm frame-shifting sequence—would survive once eCas9 expression is induced. Nevertheless, nonedited “escapee” cells carrying mutations that somehow affect Cas9 activity or expression would also survive and still carry the sequence recognized by eNT2 sgRNA in their chromosomes (also termed Cas9 evaders). An outline of the recombineering and Cas9-mediated counter-selection strategy can be found in Figure 4.
Figure 4.

Outline of the recombineering and Cas9-mediated counterselection strategy. Top: scheme depicting a bacterial population along the different steps of the protocol. The initial population is composed of unedited cells (gray bacteria) carrying a Cas9 system that is either functional (indicated by orange chromosomes) or nonfunctional (Cas9 evaders, indicated by red chromosomes). Upon oligo transformation, some cells become edited (green cells), and subsequent Cas9 induction (represented by orange or red outer shades) results in the selective killing of unedited cells (gray bacteria with broken chromosomes) and the survival of edited cells and Cas9 evaders. Bottom: scheme depicting the molecular mechanisms of this selection. In counterselected cells the functional Cas9 protein (orange molecule) forms a complex with eNT2 sgRNA (purple molecule) that specifically cuts the frameshifting sequence (black box) found in the MutCm sensor. The same complex is formed in edited cells, but the oligo-mediated deletion of the frameshifting sequence precludes Cas9-mediated cleavage of the chromosome. In Cas9 evaders, a nonfunctional (NF) copy of Cas9 is expressed (red molecule), resulting in the survival of cells still carrying the frameshifting sequence on their chromosomes.
Thus, the three different reporter strains expressing GP35 recombinase and the enhanced and inducible Cas9 (eiCas9) system were transformed with either CmONsense or CmONantisense oligos and, after 24 h, seeded on Cm-selective or nonselective plates supplemented with different aTc concentrations (Table S4). This screening of inducer concentrations allowed us to determine two important parameters for the counter-selection protocol: the optimal inducer concentration and the Cas9 evader rate. The optimal inducer concentration is defined as the aTc dose at which the viability of the total cells is affected without having an impact on the survival of edited cells. For instance, in the case of M129MutCm+50GP35 strain, this optimal inducer concentration was found at 0.66 ng/mL, a condition that led to a 50-fold reduction in the viability of total cells (1.2 × 109 vs 2.3 × 107) without affecting the edited population (1.5 × 107 vs 2.2 × 107). However, in the cases of the M129MutCm+750GP35 and M129MutCm+1800GP35 strains, the optimal Cas9 induction was found to take place at 1.25 ng/mL aTc (Table S4, Figure S4). Note that these three sensor strains differ in the chromosomal location of the eiCas9 gene cassette, and this probably accounts for the variable optimal inducer concentrations found. On the other hand, the Cas9 evader rate is defined as the proportion of cells that survive to eiCas9-mediated counterselection, regardless of the inducer concentration. Thus, the ratio between survivor cells at the highest aTc concentration and total cells when no induction is applied allowed us to calculate the evader rate for each strain (Table S4). Strikingly, this parameter also showed variations depending on the strain. This suggests that the tightness in the regulation of the eiCas9 inducible system is also affected by the chromosomal location of the cassette, resulting in higher evader rates for those locations favoring leaky transcription of eiCas9 gene. In any case, is it clear that the evader rate will influence the outcome of the recombineering–counterselection protocol. Specifically, if the proportion of evaders is higher than the proportion of edited cells, the selection of the latter would require numerous clones to be screened. In contrast, if the rate of evaders is lower than the rate of editing, virtually all cells surviving eiCas9 expression should carry the intended modification.
Taking advantage of all the data generated in the screening of eiCas9 inducer concentrations (Table S4 and Figure S4), we compared the editing rates obtained for each strain in the absence of eiCas9-mediated counterselection or under optimal eiCas9 induction conditions (Figure 5). For the M129MutCm+50GP35 strain, optimal induction of eiCas9 expression boosted the editing rate from the initial value 1.3 × 10–2 to 9.9 × 10 –1. This implies that virtually all cells surviving eiCas9 induction are edited cells. To further confirm this, we randomly picked 20 colonies from nonselective plates supplemented with the optimal aTc dose for this strain, and inoculated them into a 96-well plate containing either nonselective or Cm-selective medium. Of note, 19 out the 20 clones analyzed were found to be chloramphenicol resistant (Figure S5). This efficient selection of edited cells relies on the fact that, for this strain, the initial editing rate is higher than the frequency of evaders (1.3 × 10–2 and 3.1 × 10–3, respectively). In contrast, the initial editing rates obtained for M129MutCm+750GP35 and M129MutCm+1800GP35 strains (i.e., 4.9 × 10–4 and 1.1 × 10–4, respectively) were lower than the Cas9 evader rate (3.9 × 10–3 and 8.3 × 10–4, respectively) (Table S4 and Figure S4). In other words, in these two strains the number of cells that do not respond to eiCas9 induction is higher than the amount cells that were positively edited by oligo recombineering. As a consequence, optimal eiCas9 induction boosted the editing rates to 1.2 × 10–1 in the M129MutCm+750GP35 strain, and 6.4 × 10–2 in the M129MutCm+1800GP35 strain, both values being lower than the editing rate observed in M129MutCm+50GP35 strain after eiCas9-mediated counterselection (9.9 × 10 –1) (Figure 5). Nevertheless, these values should enable the isolation of positively edited cells by analyzing a reasonable number of clones. In the case of the M129MutCm+750GP35 strain, the analysis of 20 colonies randomly picked from nonselective plates supplemented with 1.25 ng/mL aTc allowed the identification of two positively edited clones, as inferred from their ability to grow on Cm-selective medium. This ratio of edited clones (2/20) is substantially lower that the one obtained for 50 bp deletions (19/20), and therefore the identification of clones carrying deletions of 750 bp would inarguably require a PCR screening. Thus, as a proof of concept, the same 20 clones assessed in the antibiotic selection screening were analyzed by PCR, confirming the deletion of 750 bp in the same clones that were found to be resistant to chloramphenicol (Figure S6). Likewise, the same screenings conducted in 20 clones of M129MutCm+1800GP35 strain led to the identification of one positively edited clone that carried a targeted 1.8 kb deletion (Figure S7).
Figure 5.
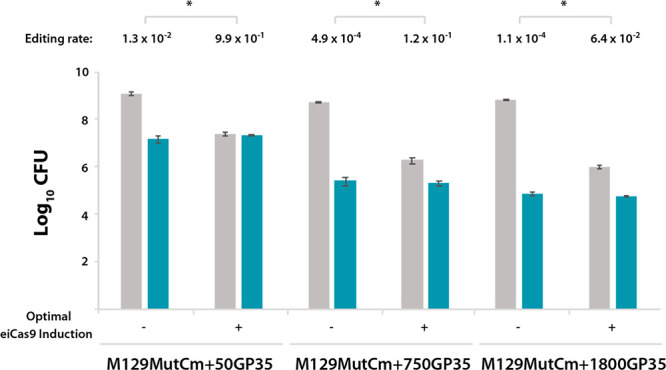
Improvement of editing rates mediated by Cas9-based counterselection. Bar plot showing in logarithmic scale the CFU values obtained for the indicated recombineering sensor strains after transformation with their corresponding editing oligos and seeding on nonselective medium (gray bars) or selective medium (green bars), nonsupplemented with aTc (−) or supplemented with the optimal aTc dose for each strain (+) as indicated on the x-axis. The editing rate (edited cells/total cells) obtained for each strain and experimental condition is shown on top of each group of bars. Those differences in terms of editing rate that were found to be statistically significant (P < 0.05) after conducting a paired t test are indicated with an asterisk (*). Error bars represent the SEM of three different biological replicas.
Altogether, these results indicate that GP35 recombineering coupled to eiCas9-mediated counterselection enables cells that have undergone a 50-bp chromosome editing to be isolated with virtually no screening, and allows simple and affordable screening experiments for deletions as large as 1.8 kb. Paradoxically, small scale modifications such as point mutations might be the hardest to obtain with this technology, as PAM sequences required to be recognized by Cas9 might not be always present in the vicinity of the nucleotide to be modified. However, Cas-9 mediated counterselection is not an absolute requirement to select for small scale modifications. Indeed, GP35 oligo recombineering generates clones carrying point mutations with a frequency high enough (i.e., 2.7 × 10–2) to allow their identification through a screening based on allele-specific PCR protocols.57
Conclusion
Research in the Mycoplasma field has traditionally been hampered by the lack of reliable genome editing tools. The cloning of natural Mycoplasma genomes as yeast centromeric plasmids was a major breakthrough that seemed to overcome the scarcity of tools for this genus.32,58 Indeed, complete genomes of several Mycoplasma species have been cloned and modified in yeast with a variety of genome-editing techniques such as TREC,33 TREC-IN,34 and CreasPy-cloning35 that take advantage of the proficient recombination machinery found in S. cerevisiae. Perhaps the best example to illustrate the potential of this approach is the recent report of massive genome engineering in M. mycoides using the TREC technique that involved the multistep targeted deletion of up to 10% of the original genome. Once the in-yeast edited genome was transplanted into a Mycoplasma acceptor cell, a fully attenuated M. mycoides strain was generated.37 Given that this strain shows a host preference in principle restricted to ruminants, it might become a standard for synthetic biology projects focused on the veterinary field. However, synthetic biology projects focused on human health still miss a member of the Mycoplasma genus as a suitable chassis strain. That strain could be M. pneumoniae, as it is a bacterium that naturally infects the human respiratory tract and is extensively characterized. Still, the few pathogenicity determinants encoded in its genome should be removed before it can be used therapeutically.
Strikingly, the genome of M. pneumoniae has also been cloned as a yeast centromeric plasmid and successfully edited using the CreasPy-cloning technique.35 In spite of this, the reintroduction of this in-yeast engineered M. pneumoniae genome into a Mycoplasma acceptor cell (i.e., genome transplantation) has never been reported. Therefore, although the M. pneumoniae genome can be edited in yeast, the generation of M. pneumoniae edited cells remains unsolved. The same applies for M. hominis, whose genome has been cloned and edited in yeast but never transplanted to generate M. hominis mutant cells.59 This is not surprising, as it was recently reported that the phylogenetic distance between the donor genome and the recipient cell constrains the efficiency of genome transplantation, requiring around 90% of identity of the core proteome between donor and acceptor organisms to obtain a successful outcome.38 As the only available acceptor cell for genome transplantation to date is M. capricolum, in-yeast engineering of Mycoplasma genomes and subsequent transplantation is a feasible approach for only a few species. Thus, there is a need for strategies enabling direct editing of Mycoplasma genomes without requiring the use of yeast as the engineering platform.
In this work, we report the development of a recombineering technology coupled to CRISPR/Cas9-based counterselection for M. pneumoniae. Our work may represent the foundation for the adoption of this bacterium as a synthetic biology chassis strain by providing a technology that should enable the removal of the few and well-characterized pathogenicity determinants found in the M. pneumoniae genome. Remarkably, gene editing with the GP35-CRISPR/Cas9 system leaves no marks behind, so that iterative rounds can be performed without requiring selection marker recycling.
Once the undesired elements of the naturally streamlined genome of M. pneumoniae have been erased, it is likely that novel functions can be plugged into this genomic backbone. However, while oligo recombineering can easily mediate gene deletions, the small size of oligonucleotides precludes the use of this technology to mediate targeted insertion of gene cassettes. To solve this, we envision that lox sites are small enough (i.e., 34 bp) to be included into oligonucleotide molecules, which would enable GP35-mediated generation of targeted landing platforms that can later be loaded with the gene cassettes of interest. Notably, the functionality of Cre/lox technology has been already demonstrated in M. genitalium,54 a species closely related with M. pneumoniae.
Finally, it should be noted that GP35 is a ssDNA recombinase from a phage that uses B. subtilis as a host. Despite this phylogenetic distance, GP35 performs quite efficiently in M. pneumoniae, so it would not be surprising if its efficacy is maintained across the whole Mycoplasma genus.
Methods
Bacterial Strains and Culture Conditions
All the M. pneumoniae strains generated in this work are described in Table S5 and derive from the wild-type strain M129-B7 (ATTC 29342, subtype 1, broth passage no. 35). All strains were grown in Hayflick modified medium16 at 37 °C under 5% CO2 in tissue culture flasks (Corning). Hayflick broth was supplemented with tetracycline (2 μg mL–1), puromycin (3 μg mL–1), or chloramphenicol (20 μg mL–1) for selection of cells as needed or with anhydrotetracycline at the indicated concentrations for inducing Cas9 expression. When growth on the plate was required, Hayflick broth was supplemented with 0.8% bacto agar (Difco). All strains generated in this work are available upon request.
For cloning purposes, the E. coli NEB 5-alpha High Efficiency strain (New England Biolabs) was grown at 37 °C in LB broth or on LB agar plates supplemented with ampicillin (100 μg mL–1)
Plasmids and Oligonucleotides
All of the plasmids generated in this work were assembled following the Gibson method60 unless otherwise indicated. When required, IDT Incorporation performed gene synthesis. Oligonucleotides were synthesized by Sigma-Aldrich. Gene amplifications were carried out with Phusion DNA polymerase (Thermo Fisher Scientific) A detailed list of plasmids and the sequences of the different modules included in them is available in Table S6. Note that M. pneumoniae does not require a Shine–Dalgarno region at the 5′ end of the mRNA to efficiently translate the transcripts, so the promoter sequences indicated in Table S6 contain the region placed immediately upstream of each of the coding sequences controlled by them. Thus, researchers working with other Mycoplasma species may need to define their own regulatory regions or use a recently reported regulatory region that seems to be functional in all Mycoplasma species.61 All plasmids generated in this work are available upon request. All oligonucleotides employed for plasmid assembly as well as details for vector construction are available in Table S7. The correct assembly of all plasmids was verified by Sanger sequencing (GATC biotech). The sequences of the editing oligonucleotides employed as substrates for recombineering are available in Table S8. The sequences of the oligonucleotides employed for the arbitrary PCR protocol and for the screening of edited clones are available in Table S9 and Table S10, respectively.
BLAST Search of Orthologues, CLUSTALW Multiple Sequence Alignment, and Accession Numbers of Recombinases
Mycoplasma orthologues of E. coli-derived proteins RecT (P33228) and Recβ (P03698) were sought using BLASTp (protein–protein BLAST). Parameters of the search were restricted to Mycoplasmas and walled relatives (taxid: 31969), Mycoplasma phage phiMFV1 (taxid: 280702), Mycoplasma phage MAV1 (taxid: 75590), and Mycoplasma phage P1 (taxid: 35238). RecT-associated positive hits of this search, here renamed as RecTsm (WP_004028097.1), RecTsp (WP_127093247.1), and RecTsc (CAK99285.1), were later aligned using CLUSTALW software taking the native E. coli-derived RecT protein as a reference. A search for Recβ-like orthologues did not produce any positive hits. As our search did not produce any candidate with a proven capacity to perform recombineering, GP35 protein (CAA66543.1) was included in the screening of recombinases.
M. pneumoniae Transformations
Transformations were performed as described previously62 with few modifications. Briefly, M. pneumoniae cultures were grown to late-exponential phase in 75 cm2 tissue culture flasks. The adherent layer of M. pneumoniae cells was washed three times with chilled electroporation buffer,62 scraped off, and resuspended in 500 μL of this buffer at a concentration of approximately 1010 cells mL–1. Next, this cell suspension was passed 10 times through a 25-gauge (G25) syringe needle, and 50 μL aliquots were mixed with the desired DNA molecules for transformation. For transposon vector transformations, 2 μg of DNA was added to the mix, whereas for oligo transformations, the volumes employed were 1, 5, or 10 μL of a 100 μM stock, corresponding to 0.1, 0.5, or 1 nmol, respectively. The mixture of DNA and cells was adjusted to a final volume of 80 μL and transferred into 0.1 cm electrocuvettes, letting it sit for 15 min on ice before being electroporated in a BIO-RAD Gene Pulser Xcell apparatus. The settings employed were 1250 V/25 μF/100 Ω. After the pulse, cells were incubated on ice for 15 min and subsequently harvested by adding 420 μL of Hayflick into the cuvette. In the case of transposon vector transformations, cells were allowed to recover at 37 °C for 2 h before inoculating one-fifth of the transformation volume into a 25 cm2 flask filled with 5 mL of Hayflick supplemented with the appropriate antibiotic. In those cases in which individual clones of the transformation were required, after the 2 h of recovering time, serial dilutions were seeded on plates, and an individual clone was picked and expanded. In the case of oligo transformations where several pulses were performed, cells were allowed to recover 3 min on ice between the pulses. Later, the total volume of the transformation was directly inoculated into T75 flasks containing 25 mL of Hayflick medium.
Editing Rate Determination
M. pneumoniae cells carrying one of the different recombineering sensors generated in this work, a second transposon harboring one of the recombinases screened, and in some cases also the eiCas9 system were transformed with an editing oligo. The sequences of the editing oligos can be found in Table S8. At the indicated post-transformation time, transformed cells were scraped from the flask in 500 μL of Hayflick medium. Subsequently, 10-fold serial dilutions were performed (from −1 to −8). Dilutions were made in a total volume of 100 μL, and 10 μL of each dilution was spotted onto Hayflick 0.8% bacto agar plates supplemented with chloramphenicol and/or anhydrotetracycline where required. Thus, the detection limit of these experiments was 500 CFU. When the number of cells obtained for a given condition was below this detection limit, the maximum possible number of cells (i.e., 499 CFU) was considered for statistical analyses. The editing rate is defined as the number of cells resistant to chloramphenicol divided by the total number of cells obtained for each condition. Paired t test analysis of the editing rates obtained in the three biological replicates conducted for each condition was performed using GraphPad QuickCalcs software. An asterisk (*) was included in the figures when the difference in the editing rate for two given conditions was found to be statistically significant (p < 0.05). The actual value of all p-values can be found in the Supplementary Tables.
Western Blot Analysis
Plasmids employed in this work to assess the functionality of different proteins were free of protein tags to avoid the possibility that their inclusion could affect the activity. Therefore, to confirm the expression of the different recombinases, a set of dedicated plasmids containing FLAG tags was constructed (Table S6).
The strains carrying these tagged constructs were grown on 25 cm2 flasks until reaching confluence. The adherent layer of cells was washed twice with PBS and scraped off in 500 μL of this buffer. Cell solution was centrifuged (12 000g, 5 min), and the resulting pellet was lysed in 150 of lysis buffer (SDS 4%, Hepes 100 mM). Subsequently, mycoplasma cell lysates were quantified using the Pierce BCA Protein Assay Kit, and 10 μg of cell extracts was subjected to electrophoresis on NuPAGE 4–12% Bis-Tris precast polyacrylamide gels (Invitrogen). Next, proteins were transferred onto nitrocellulose membranes using an iBlot dry blotting system (Invitrogen). Novex Sharp Prestained Proteins Standards allowed cutting the membrane into two pieces to process individually. Both membrane pieces were blocked with 5% skim milk (Sigma) in Tris-buffered saline (TBS) solution, containing 0.1% Tween 20 (TBST). The upper membrane piece (containing proteins above 20 kDa) was probed with monoclonal anti-FLAG M2 (Sigma) as primary antibody (1:5000) and antimouse IgG (1:10 000) conjugated to horseradish peroxidase (Sigma) as secondary antibody. The lower membrane piece (containing proteins below 20 kDa) was probed with anti-RL7 polyclonal serum (kind gift of Dr. Herrmann, Heidelberg University) as primary antibody (1:1000) and antirabbit IgG (1:10 000) coupled to horseradish peroxidase (Sigma) as secondary antibody. Blots were developed with the Supersignal West Femto Chemiluminescent Substrate Detection Kit (ThermoScientific), and signals were detected in a LAS-3000 Imaging System (Fujifilm).
Transposon Insertion Localization by A-PCR
Cultures of the clones of interest were grown in 25 cm2 flasks until reaching confluence. The adherent layer of cells was washed three times with PBS and scraped off in 300 μL of this buffer. This cell solution was treated with MasterPure DNA Purification Kit (Epicenter) following the manufacturer’s instructions to isolate genomic DNA.
The A-PCR protocol followed is a variant of the one previously described44 to adapt it to M. pneumoniae genome composition. Specifically, we modified the 3′ end of the arbitrary oligo to mimic the most frequent pentanucleotide sequence in the M. pneumoniae genome that ends in a “GC clump”. The script for finding the most frequent pentanucleotides for a given genome and sort them by the number of hits is available at: https://github.com/jdelgadoblanco/pentanucleotides.git. The sequences of the four primers employed for A-PCR in this study are detailed in Table S9.
Screening of Edited Clones
96 well plates were prepared as follows: all the perimeter wells were filled with 200 μL of Hayflick medium as color reference. Then, the “inoculation wells” were filled with 200 μL of Hayflick medium, the “non-selective wells” with 150 μL of Hayflick medium, and the “Cm-selective wells” with 150 μL of Hayflick medium supplemented with Cm at 1.25× concentration. Colonies were picked from the plates of interest and transferred into the inoculation wells by pipetting up and down several times. Subsequently, separate aliquots of 50 μL were transferred from the inoculation well to both the “non-selective well” and the “Cm-selective well”. Plates were incubated at 37 °C under 5% CO2 for 7 days before taking pictures of them with ImageScannerIII (Epson).
For the PCR screening of edited clones, a fast genomic DNA extraction of Mycoplasma cells was performed. Briefly, cells grown in the multiwell plates were scraped off and 100 μL of the resulting cell suspensions were transferred to Eppendorf tubes and boiled for 10 min. Next, the inactivated cell suspension was mixed with 20 μL of StrataClean resin (Agilent). This mixture was incubated for 10 min at RT with gentle mixing every 2 min before being centrifuged (10 000g, 1 min). Finally, 3 μL of the resulting supernatants was used as a template for the PCR with the screening oligos detailed in Table S10.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.0c00022.
Seven supplementary figures (Figure S1: multiple sequence alignment of RecT proteins; Figure S2: Western blot analysis of the expression of the different recombinases used in the screening; Figure S3: chromosome locations of the different recombineering sensors; Figure S4: screening of optimal inducer concentrations for Cas9-based counterselection on each recombineering sensor strain; Figures S5–S7: screening of edited clones of the M129MutCm+50GP35, M129MutCm+750GP35, and M129MutCm+1800GP35 strains, respectively upon eiCas9-mediated counterselection) (PDF)
Ten supplementary tables (Tables S1–S3: crude data from the transformations performed for the screening of recombinases, for the protocol optimization, and for the different recombineering sensor strains, respectively; Table S4: screening of optimal Cas9 inducer concentration and determination of Cas9 evader rate in the different recombineering sensor strains; Table S5: strains generated in this work; Table S6: plasmids generated in this work with detailed information on the sequences present in them; Table S7: oligonucleotides employed for plasmid assembly and details on vector assembly; Table S8: editing oligonucleotides employed in this work; Table S9: oligonucleotides employed for A-PCR protocol; Table S10: oligonucleotides employed for the screening of edited clones) (XLSX)
This project has received funding from the European Union’s Horizon 2020 research and innovation program under Grant 634942 (MycoSynVac) and was also financed by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program under Grant 670216 (MYCOCHASSIS) and the FEDER project from Instituto Carlos III (ISCIII, Acción Estratégica en Salud 2016) (reference CP16/00094). We also acknowledge support of the Spanish Ministry of Science and Innovation, to the EMBL partnership, the Centro de Excelencia Severo Ochoa, and the CERCA Programme/Generalitat de Catalunya. Finally, we would like to thank Dr. Sarah A. Head for critical manuscript revision.
The authors declare the following competing financial interest(s): C.P.-L., L.S., and M.L.-S. are inventors of the patent application EP20382051.9, filed based partially on the technique and results described in this manuscript.
Supplementary Material
References
- Anderson L. A.; Islam M. A.; Prather K. L. J. (2018) Synthetic Biology Strategies for Improving Microbial Synthesis of “Green” Biopolymers. J. Biol. Chem. 293 (14), 5053–5061. 10.1074/jbc.TM117.000368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K. Y.; Araki M.; Okai N.; Wakai S.; Hasunuma T.; Kondo A. (2014) Development of Bio-Based Fine Chemical Production through Synthetic Bioengineering. Microb. Cell Fact. 13, 173. 10.1186/s12934-014-0173-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvořák P.; Nikel P. I.; Damborský J.; de Lorenzo V. (2017) Bioremediation 3.0: Engineering Pollutant-Removing Bacteria in the Times of Systemic Biology. Biotechnol. Adv. 35 (7), 845–866. 10.1016/j.biotechadv.2017.08.001. [DOI] [PubMed] [Google Scholar]
- Piñero-Lambea C.; Ruano-Gallego D.; Fernández L. Á. (2015) Engineered Bacteria as Therapeutic Agents. Curr. Opin. Biotechnol. 35, 94–102. 10.1016/j.copbio.2015.05.004. [DOI] [PubMed] [Google Scholar]
- Serrano L. (2007) Synthetic Biology: Promises and Challenges. Mol. Syst. Biol. 3, 158. 10.1038/msb4100202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzadfard F.; Lu T. K. (2014) Synthetic Biology. Genomically Encoded Analog Memory with Precise in Vivo DNA Writing in Living Cell Populations. Science 346 (6211), 1256272. 10.1126/science.1256272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedland A. E.; Lu T. K.; Wang X.; Shi D.; Church G.; Collins J. J. (2009) Synthetic Gene Networks That Count. Science 324 (5931), 1199–1202. 10.1126/science.1172005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner T. S.; Cantor C. R.; Collins J. J. (2000) Construction of a Genetic Toggle Switch in Escherichia Coli. Nature 403 (6767), 339–342. 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- Piñero-Lambea C.; Bodelón G.; Fernández-Periáñez R.; Cuesta A. M.; Álvarez-Vallina L.; Fernández L. Á. (2015) Programming Controlled Adhesion of E. Coli to Target Surfaces, Cells, and Tumors with Synthetic Adhesins. ACS Synth. Biol. 4 (4), 463–473. 10.1021/sb500252a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeidi N.; Wong C. K.; Lo T.-M.; Nguyen H. X.; Ling H.; Leong S. S. J.; Poh C. L.; Chang M. W. (2011) Engineering Microbes to Sense and Eradicate Pseudomonas Aeruginosa, a Human Pathogen. Mol. Syst. Biol. 7, 521. 10.1038/msb.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isabella V. M.; Ha B. N.; Castillo M. J.; Lubkowicz D. J.; Rowe S. E.; Millet Y. A.; Anderson C. L.; Li N.; Fisher A. B.; West K. A.; Reeder P. J.; Momin M. M.; Bergeron C. G.; Guilmain S. E.; Miller P. F.; Kurtz C. B.; Falb D. (2018) Development of a Synthetic Live Bacterial Therapeutic for the Human Metabolic Disease Phenylketonuria. Nat. Biotechnol. 36 (9), 857–864. 10.1038/nbt.4222. [DOI] [PubMed] [Google Scholar]
- Kurtz C. B.; Millet Y. A.; Puurunen M. K.; Perreault M.; Charbonneau M. R.; Isabella V. M.; Kotula J. W.; Antipov E.; Dagon Y.; Denney W. S.; Wagner D. A.; West K. A.; Degar A. J.; Brennan A. M.; Miller P. F. (2019) An Engineered E. Coli Nissle Improves Hyperammonemia and Survival in Mice and Shows Dose-Dependent Exposure in Healthy Humans. Sci. Transl. Med. 11 (475), eaau7975. 10.1126/scitranslmed.aau7975. [DOI] [PubMed] [Google Scholar]
- Adams B. L. (2016) The Next Generation of Synthetic Biology Chassis: Moving Synthetic Biology from the Laboratory to the Field. ACS Synth. Biol. 5 (12), 1328–1330. 10.1021/acssynbio.6b00256. [DOI] [PubMed] [Google Scholar]
- Razin S.; Yogev D.; Naot Y. (1998) Molecular Biology and Pathogenicity of Mycoplasmas. Microbiol. Mol. Biol. Rev. 62 (4), 1094–1156. 10.1128/MMBR.62.4.1094-1156.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Güell M.; van Noort V.; Yus E.; Chen W.-H.; Leigh-Bell J.; Michalodimitrakis K.; Yamada T.; Arumugam M.; Doerks T.; Kühner S.; Rode M.; Suyama M.; Schmidt S.; Gavin A.-C.; Bork P.; Serrano L. (2009) Transcriptome Complexity in a Genome-Reduced Bacterium. Science 326 (5957), 1268–1271. 10.1126/science.1176951. [DOI] [PubMed] [Google Scholar]
- Yus E.; Maier T.; Michalodimitrakis K.; van Noort V.; Yamada T.; Chen W.-H.; Wodke J. A. H.; Güell M.; Martínez S.; Bourgeois R.; Kühner S.; Raineri E.; Letunic I.; Kalinina O. V.; Rode M.; Herrmann R.; Gutiérrez-Gallego R.; Russell R. B.; Gavin A.-C.; Bork P.; Serrano L. (2009) Impact of Genome Reduction on Bacterial Metabolism and Its Regulation. Science 326 (5957), 1263–1268. 10.1126/science.1177263. [DOI] [PubMed] [Google Scholar]
- Maier T.; Schmidt A.; Güell M.; Kühner S.; Gavin A.; Aebersold R.; Serrano L. (2011) Quantification of MRNA and Protein and Integration with Protein Turnover in a Bacterium. Mol. Syst. Biol. 7 (1), 511. 10.1038/msb.2011.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yus E.; Güell M.; Vivancos A. P.; Chen W.-H.; Lluch-Senar M.; Delgado J.; Gavin A.-C.; Bork P.; Serrano L. (2012) Transcription Start Site Associated RNAs in Bacteria. Mol. Syst. Biol. 8, 585. 10.1038/msb.2012.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lluch-Senar M.; Luong K.; Lloréns-Rico V.; Delgado J.; Fang G.; Spittle K.; Clark T. A.; Schadt E.; Turner S. W.; Korlach J.; Serrano L. (2013) Comprehensive Methylome Characterization of Mycoplasma Genitalium and Mycoplasma Pneumoniae at Single-Base Resolution. PLoS Genet. 9 (1), e1003191 10.1371/journal.pgen.1003191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J.; Liu M.; Ye Z.; Tan T.; Liu X.; You X.; Zeng Y.; Wu Y. (2016) Insights into the Pathogenesis of Mycoplasma Pneumoniae. Mol. Med. Rep. 14 (5), 4030–4036. 10.3892/mmr.2016.5765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhandayuthapani S.; Rasmussen W. G.; Baseman J. B. (1999) Disruption of Gene Mg218 of Mycoplasma Genitalium through Homologous Recombination Leads to an Adherence-Deficient Phenotype. Proc. Natl. Acad. Sci. U. S. A. 96 (9), 5227–5232. 10.1073/pnas.96.9.5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhandayuthapani S.; Blaylock M. W.; Bebear C. M.; Rasmussen W. G.; Baseman J. B. (2001) Peptide Methionine Sulfoxide Reductase (MsrA) Is a Virulence Determinant in Mycoplasma Genitalium. J. Bacteriol. 183 (19), 5645–5650. 10.1128/JB.183.19.5645-5650.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgos R.; Pich O. Q.; Querol E.; Piñol J. (2008) Deletion of the Mycoplasma Genitalium MG_217 Gene Modifies Cell Gliding Behaviour by Altering Terminal Organelle Curvature. Mol. Microbiol. 69 (4), 1029–1040. 10.1111/j.1365-2958.2008.06343.x. [DOI] [PubMed] [Google Scholar]
- Allam A. B.; Reyes L.; Assad-Garcia N.; Glass J. I.; Brown M. B. (2010) Enhancement of Targeted Homologous Recombination in Mycoplasma Mycoides Subsp. Capri by Inclusion of Heterologous RecA. Appl. Environ. Microbiol. 76 (20), 6951–6954. 10.1128/AEM.00056-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar R.; Assad-Garcia N.; Benders G. A.; Phan Q.; Montague M. G.; Glass J. I. (2010) Targeted Chromosomal Knockouts in Mycoplasma Pneumoniae. Appl. Environ. Microbiol. 76 (15), 5297–5299. 10.1128/AEM.00024-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluijter M.; Kaptein E.; Spuesens E. B. M.; Hoogenboezem T.; Hartwig N. G.; Van Rossum A. M. C.; Vink C. (2010) The Mycoplasma Genitalium MG352-Encoded Protein Is a Holliday Junction Resolvase That Has a Non-Functional Orthologue in Mycoplasma Pneumoniae. Mol. Microbiol. 77 (5), 1261–1277. 10.1111/j.1365-2958.2010.07288.x. [DOI] [PubMed] [Google Scholar]
- Sluijter M.; Estevão S.; Hoogenboezem T.; Hartwig N. G.; van Rossum A. M. C.; Vink C. (2012) The RuvA Homologues from Mycoplasma Genitalium and Mycoplasma Pneumoniae Exhibit Unique Functional Characteristics. PLoS One 7 (5), e38301 10.1371/journal.pone.0038301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Puig S.; Broto A.; Querol E.; Piñol J.; Pich O. Q. (2015) A Novel Sigma Factor Reveals a Unique Regulon Controlling Cell-Specific Recombination in Mycoplasma Genitalium. Nucleic Acids Res. 43 (10), 4923–4936. 10.1093/nar/gkv422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbedel S.; Stulke J. (2007) Tools for the Genetic Analysis of Mycoplasma. Int. J. Med. Microbiol. 297 (1), 37–44. 10.1016/j.ijmm.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Gibson D. G.; Benders G. A.; Andrews-Pfannkoch C.; Denisova E. A.; Baden-Tillson H.; Zaveri J.; Stockwell T. B.; Brownley A.; Thomas D. W.; Algire M. A.; Merryman C.; Young L.; Noskov V. N.; Glass J. I.; Venter J. C.; Hutchison C. A.; Smith H. O. (2008) Complete Chemical Synthesis, Assembly, and Cloning of a Mycoplasma Genitalium Genome. Science 319 (5867), 1215–1220. 10.1126/science.1151721. [DOI] [PubMed] [Google Scholar]
- Gibson D. G.; Glass J. I.; Lartigue C.; Noskov V. N.; Chuang R.-Y.; Algire M. A.; Benders G. A.; Montague M. G.; Ma L.; Moodie M. M.; Merryman C.; Vashee S.; Krishnakumar R.; Assad-Garcia N.; Andrews-Pfannkoch C.; Denisova E. A.; Young L.; Qi Z.-Q.; Segall-Shapiro T. H.; Calvey C. H.; Parmar P. P.; Hutchison C. A.; Smith H. O.; Venter J. C. (2010) Creation of a Bacterial Cell Controlled by a Chemically Synthesized Genome. Science 329 (5987), 52–56. 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- Benders G. A.; Noskov V. N.; Denisova E. A.; Lartigue C.; Gibson D. G.; Assad-Garcia N.; Chuang R.-Y.; Carrera W.; Moodie M.; Algire M. A.; Phan Q.; Alperovich N.; Vashee S.; Merryman C.; Venter J. C.; Smith H. O.; Glass J. I.; Hutchison C. A. (2010) Cloning Whole Bacterial Genomes in Yeast. Nucleic Acids Res. 38 (8), 2558–2569. 10.1093/nar/gkq119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noskov V. N.; Segall-Shapiro T. H.; Chuang R.-Y. (2010) Tandem Repeat Coupled with Endonuclease Cleavage (TREC): A Seamless Modification Tool for Genome Engineering in Yeast. Nucleic Acids Res. 38 (8), 2570–2576. 10.1093/nar/gkq099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandran S.; Noskov V. N.; Segall-Shapiro T. H.; Ma L.; Whiteis C.; Lartigue C.; Jores J.; Vashee S.; Chuang R.-Y. (2014) TREC-IN: Gene Knock-in Genetic Tool for Genomes Cloned in Yeast. BMC Genomics 15, 1180. 10.1186/1471-2164-15-1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz E.; Talenton V.; Dubrana M.-P.; Guesdon G.; Lluch-Senar M.; Salin F.; Sirand-Pugnet P.; Arfi Y.; Lartigue C. (2019) CReasPy-Cloning: A Method for Simultaneous Cloning and Engineering of Megabase-Sized Genomes in Yeast Using the CRISPR-Cas9 System. ACS Synth. Biol. 8 (11), 2547–2557. 10.1021/acssynbio.9b00224. [DOI] [PubMed] [Google Scholar]
- Lartigue C.; Glass J. I.; Alperovich N.; Pieper R.; Parmar P. P.; Hutchison C. A.; Smith H. O.; Venter J. C. (2007) Genome Transplantation in Bacteria: Changing One Species to Another. Science 317 (5838), 632–638. 10.1126/science.1144622. [DOI] [PubMed] [Google Scholar]
- Jores J.; Ma L.; Ssajjakambwe P.; Schieck E.; Liljander A.; Chandran S.; Stoffel M. H.; Cippa V.; Arfi Y.; Assad-Garcia N.; Falquet L.; Sirand-Pugnet P.; Blanchard A.; Lartigue C.; Posthaus H.; Labroussaa F.; Vashee S. (2019) Removal of a Subset of Non-Essential Genes Fully Attenuates a Highly Virulent Mycoplasma Strain. Front. Microbiol. 10, 664. 10.3389/fmicb.2019.00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labroussaa F.; Lebaudy A.; Baby V.; Gourgues G.; Matteau D.; Vashee S.; Sirand-Pugnet P.; Rodrigue S.; Lartigue C. (2016) Impact of Donor-Recipient Phylogenetic Distance on Bacterial Genome Transplantation. Nucleic Acids Res. 44 (17), 8501–8511. 10.1093/nar/gkw688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z.; Deng A.; Hu T.; Wu J.; Sun Q.; Bai H.; Zhang G.; Wen T. (2015) A High-Efficiency Recombineering System with PCR-Based SsDNA in Bacillus Subtilis Mediated by the Native Phage Recombinase GP35. Appl. Microbiol. Biotechnol. 99 (12), 5151–5162. 10.1007/s00253-015-6485-5. [DOI] [PubMed] [Google Scholar]
- Thomason L. C.; Sawitzke J. A.; Li X.; Costantino N.; Court D. L. (2014) Recombineering: Genetic Engineering in Bacteria Using Homologous Recombination. Curr. Protoc. Mol. Biol. 106, 1.16.1–39. 10.1002/0471142727.mb0116s106. [DOI] [PubMed] [Google Scholar]
- Ellis H. M.; Yu D.; DiTizio T.; Court D. L. (2001) High Efficiency Mutagenesis, Repair, and Engineering of Chromosomal DNA Using Single-Stranded Oligonucleotides. Proc. Natl. Acad. Sci. U. S. A. 98 (12), 6742–6746. 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S.; Costantino N.; Zhou X.; Court D. L. (2008) Identification and Analysis of Recombineering Functions from Gram-Negative and Gram-Positive Bacteria and Their Phages. Proc. Natl. Acad. Sci. U. S. A. 105 (5), 1626–1631. 10.1073/pnas.0709089105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. H.; Isaacs F. J.; Carr P. A.; Sun Z. Z.; Xu G.; Forest C. R.; Church G. M. (2009) Programming Cells by Multiplex Genome Engineering and Accelerated Evolution. Nature 460 (7257), 894–898. 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S.; Noe J. C.; Paik S.; Kitten T. (2005) An Improved Arbitrary Primed PCR Method for Rapid Characterization of Transposon Insertion Sites. J. Microbiol. Methods 63 (1), 89–94. 10.1016/j.mimet.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Ricaurte D. E.; Martínez-García E.; Nyerges Á.; Pál C.; de Lorenzo V.; Aparicio T. (2018) A Standardized Workflow for Surveying Recombinases Expands Bacterial Genome-Editing Capabilities. Microb. Biotechnol. 11 (1), 176–188. 10.1111/1751-7915.12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penewit K.; Holmes E. A.; McLean K.; Ren M.; Waalkes A.; Salipante S. J. (2018) Efficient and Scalable Precision Genome Editing in Staphylococcus Aureus through Conditional Recombineering and CRISPR/Cas9-Mediated Counterselection. mBio 9 (1), 1. 10.1128/mBio.00067-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmood T.; Zar T.; Naqvi S. M. S. (2008) Multiple Pulses Improve Electroporation Efficiency in Agrobacterium Tumefaciens. Electron. J. Biotechnol. 11 (1), 0–0. 10.2225/vol11-issue1-fulltext-1. [DOI] [Google Scholar]
- Doudna J. A.; Charpentier E. (2014) Genome Editing. The New Frontier of Genome Engineering with CRISPR-Cas9. Science 346 (6213), 1258096. 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Jiang W.; Bikard D.; Cox D.; Zhang F.; Marraffini L. A. (2013) RNA-Guided Editing of Bacterial Genomes Using CRISPR-Cas Systems. Nat. Biotechnol. 31 (3), 233–239. 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L.; Bikard D. (2016) Consequences of Cas9 Cleavage in the Chromosome of Escherichia Coli. Nucleic Acids Res. 44 (9), 4243–4251. 10.1093/nar/gkw223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio T.; de Lorenzo V.; Martínez-García E. (2019) CRISPR/Cas9-Enhanced SsDNA Recombineering for Pseudomonas Putida. Microb. Biotechnol. 12 (5), 1076–1089. 10.1111/1751-7915.13453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisch C. R.; Prather K. L. J. (2015) The No-SCAR (Scarless Cas9 Assisted Recombineering) System for Genome Editing in Escherichia Coli. Sci. Rep. 5, 15096. 10.1038/srep15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J.-H.; van Pijkeren J.-P. (2014) CRISPR-Cas9-Assisted Recombineering in Lactobacillus Reuteri. Nucleic Acids Res. 42 (17), e131 10.1093/nar/gku623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariscal A. M.; González-González L.; Querol E.; Piñol J. (2016) All-in-One Construct for Genome Engineering Using Cre-Lox Technology. DNA Res. 23 (3), 263–270. 10.1093/dnares/dsw015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaymaker I. M.; Gao L.; Zetsche B.; Scott D. A.; Yan W. X.; Zhang F. (2016) Rationally Engineered Cas9 Nucleases with Improved Specificity. Science 351 (6268), 84–88. 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi L. S.; Larson M. H.; Gilbert L. A.; Doudna J. A.; Weissman J. S.; Arkin A. P.; Lim W. A. (2013) Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 152 (5), 1173–1183. 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X.; Morris S. L.; Langone J. J.; Bockstahler L. E. (2005) Microarray and Allele Specific PCR Detection of Point Mutations in Mycobacterium Tuberculosis Genes Associated with Drug Resistance. J. Microbiol. Methods 63 (3), 318–330. 10.1016/j.mimet.2005.04.026. [DOI] [PubMed] [Google Scholar]
- Lartigue C.; Vashee S.; Algire M. A.; Chuang R.-Y.; Benders G. A.; Ma L.; Noskov V. N.; Denisova E. A.; Gibson D. G.; Assad-Garcia N.; Alperovich N.; Thomas D. W.; Merryman C.; Hutchison C. A.; Smith H. O.; Venter J. C.; Glass J. I. (2009) Creating Bacterial Strains from Genomes That Have Been Cloned and Engineered in Yeast. Science 325 (5948), 1693–1696. 10.1126/science.1173759. [DOI] [PubMed] [Google Scholar]
- Rideau F.; Le Roy C.; Descamps E. C. T.; Renaudin H.; Lartigue C.; Bébéar C. (2017) Cloning, Stability, and Modification of Mycoplasma Hominis Genome in Yeast. ACS Synth. Biol. 6 (5), 891–901. 10.1021/acssynbio.6b00379. [DOI] [PubMed] [Google Scholar]
- Gibson D. G.; Young L.; Chuang R.-Y.; Venter J. C.; Hutchison C. A.; Smith H. O. (2009) Enzymatic Assembly of DNA Molecules up to Several Hundred Kilobases. Nat. Methods 6 (5), 343–345. 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Montero-Blay A.; Miravet-Verde S.; Lluch-Senar M.; Piñero-Lambea C.; Serrano L. (2019) SynMyco Transposon: Engineering Transposon Vectors for Efficient Transformation of Minimal Genomes. DNA Res. 26 (4), 327–339. 10.1093/dnares/dsz012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedreyda C. T.; Lee K. K.; Krause D. C. (1993) Transformation of Mycoplasma Pneumoniae with Tn4001 by Electroporation. Plasmid 30 (2), 170–175. 10.1006/plas.1993.1047. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.