

SUMMARY
Selective assembly of influenza virus segments into virions is proposed to be mediated through intersegmental RNA-RNA interactions. Here, we developed a method called 2CIMPL that includes proximity ligation under native conditions to identify genome-wide RNA duplexes. Interactions between all eight segments were observed at multiple sites along a given segment and are concentrated at hotspots. Furthermore, synonymous nucleotide changes in a hotspot decreased the formation of RNA-RNA interactions at this site and resulted in a genome-wide rearrangement without a loss in replicative fitness. These results indicate that the viral RNA interaction network is flexible to account for nucleotide evolution. Moreover, comparative analysis of RNA-RNA interaction sites with viral nucleoprotein (NP) binding to the genome revealed that RNA junctions can also occur adjacent to NP peaks, suggesting that NP association does not exclude RNA duplex formation. Overall, 2CIMPL is a versatile technique to map in vivo RNA-RNA interactions.
Graphical Abstract
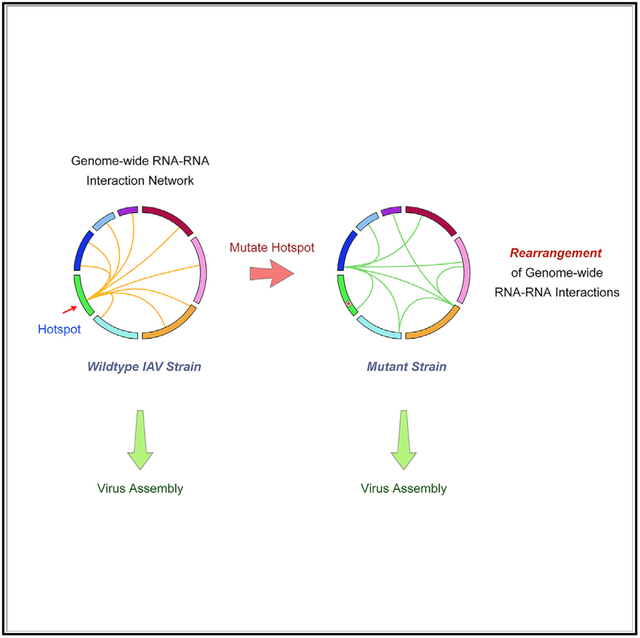
In Brief
Influenza viruses assemble and package all eight viral RNA segments through intersegmental RNA-RNA interactions. Le Sage et al. establish a protocol to capture genome-wide influenza intersegmental RNA-RNA interactions. They show that the viral RNA interaction network is flexible, where hotspots on individual segments coordinate interaction with many other segments.
INTRODUCTION
Many critical questions in the assembly of influenza virions remain open because of the limited availability of tools to study RNA biology. The segmented RNA genome of influenza virus is replicated in the nucleus and transported to the plasma membrane where one copy of each segment is packaged into a progeny virion. Each of the eight segments of the influenza viral genome is bound to the tripartite viral polymerase composed of PB2, PB1, and PA proteins at the panhandle structure formed by the 5′ and 3′ ends. The body of the viral RNA (vRNA) segments is organized into an antiparallel double helix and associates with a scaffold of viral nucleoprotein (NP) molecules to form the viral ribonucleoprotein (vRNP) complex (Figure 1A) (Cros and Palese, 2003; Eisfeld et al., 2015; Palese and Shaw, 2013; Te Velthuis et al., 2016; Whittaker et al., 1996; Wu et al., 2007). The classical architecture of vRNA and NP, depicted as beads on a string, was recently revised by our work, using high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP) assays for NP in influenza A and B virions to reveal that NP binds vRNA in a non-uniform and non-random manner (Le Sage et al., 2018; Lee et al., 2017), and confirmed by other groups (Williams et al., 2018). Furthermore, multiple methodologies have previously been used to examine the mechanism of selective assembly of vRNPs during the packaging process, including electron microscopy and in vitro RNA-binding assays, which cumulatively suggest that RNA-based intersegmental interactions between vRNP pairs promote efficient vRNP packaging (Fournier et al., 2012; Gavazzi et al., 2013; Gilbertson et al., 2016; Noda et al., 2012). Precise identification of these intersegmental RNA-RNA interactions would provide important insight into the assembly process of influenza viruses.
Figure 1. Crosslinking and Proximity Ligation-Based Approach to Identify RNA-RNA Interactions of Influenza Virus.
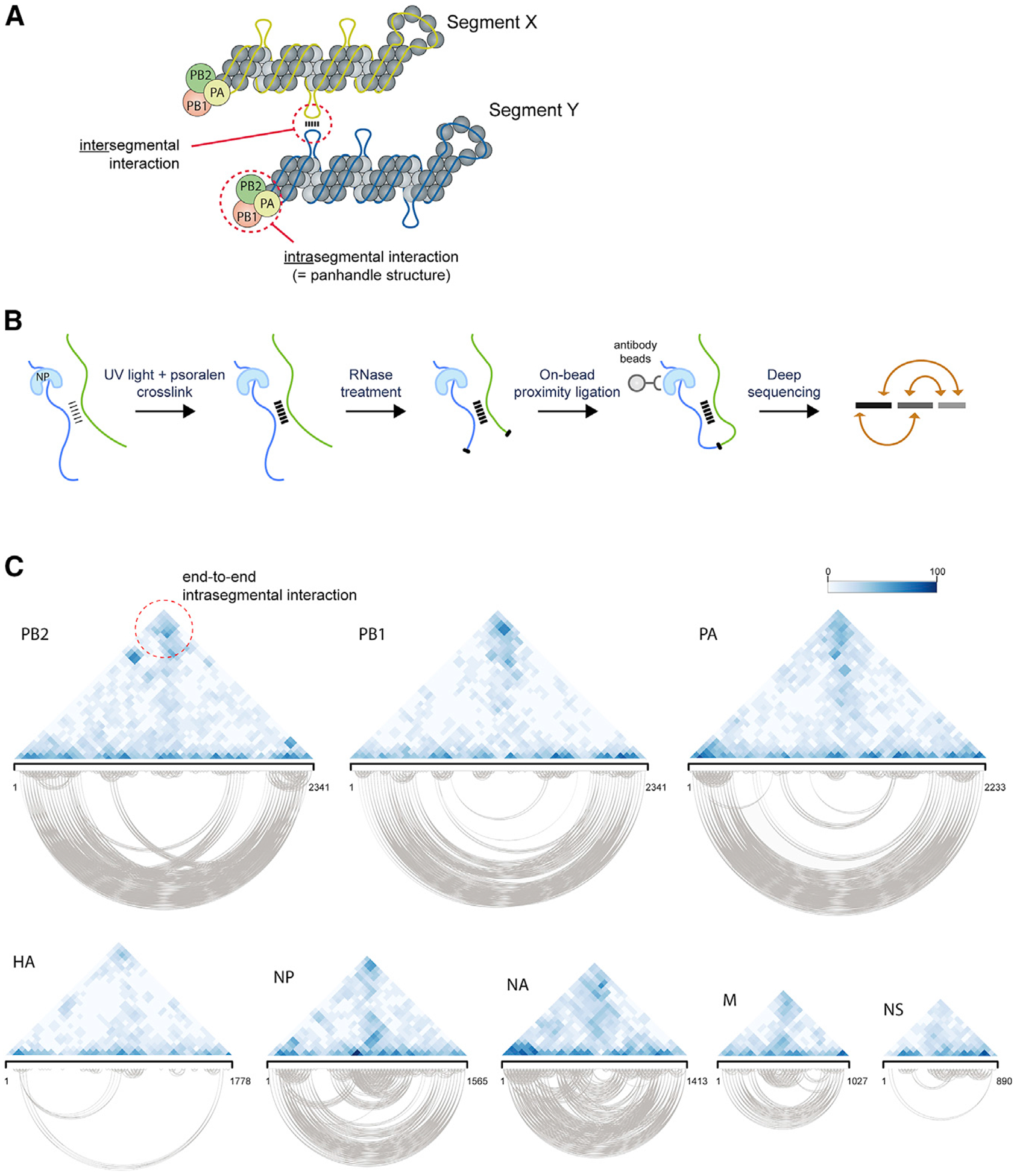
(A) Illustration showing vRNA segments coated with NP molecules and the tripartite viral polymerase complex. Putative intersegmental interaction is indicated as well as the known intrasegmental interaction formed by the segment termini (panhandle structure).
(B) Schematic outline of 2CIMPL. After UV light irradiation and psoralen crosslinking, intact virions are lysed and subjected to partial RNase treatment under native buffer conditions. Viral RNA is tethered to magnetic beads through their widespread interaction with NP, thus allowing for cleanup of RNase and buffer exchange for subsequent enzymatic reactions before proximity ligation. The hybrid RNAs are converted into an Illumina-compatible sequencing library, and the junctions are identified computationally.
(C) Triangular heatmaps of all eight WSN segments illustrating the location and relative abundance of intrasegmental RNA-RNA interactions. The coordinates of the two RNA hybrid junctions are displayed by the diagonal projections on a given segment axis, such that the top of the triangle (dashed circle) depicts interactions between the 5′ and 3′ termini (panhandle structure). Robust interactions are expected at this site for all segments. Relative abundance of each interaction is indicated by color intensity shown in the legend. All of the intrasegmental interactions captured in the triangle heatmap are also displayed below in the base-pairing plots to provide a visual representation of the RNA hybrid junctions. See also Figure S1.
Effective protocols for high-resolution mapping of three-dimensional nucleic acid organization have been developed for DNA, but equivalent techniques for RNA are only beginning to emerge. Chromosome conformation capture (3C) and its derivatives employ formaldehyde crosslinking to preserve intracellular DNA interactions, restriction enzyme digestion to fragment the genome, and subsequent ligation of nearby ends to produce hybrids of DNA fragments in spatial proximity (Dekker et al., 2002, 2013). As molecular tools to manipulate RNA are not as refined as for DNA, an exact RNA equivalent of 3C has not yet been reported. However, three approximating methods based on similar concepts have been developed to interrogate transcriptome-wide RNA-RNA interactions, which are termed LIGR (ligation of interacting RNA), PARIS (psoralen analysis of RNA interactions and structures), or SPLASH (sequencing of psoralen cross-linked, ligated, and selected hybrids) (Aw et al., 2016; Lu et al., 2016; Sharma et al., 2016). Of these, LIGR and PARIS use the psoralen-derivative AMT (aminomethyltrioxsalen), a cell-permeable and reversible crosslinking agent, to covalently link RNA duplexes in intact cells, followed by partial RNase digest and subsequent proximity ligation. The resulting RNA hybrids generated by ligation are subjected to next-generation sequencing, and the transcriptome-wide RNA junctions are mapped computationally to the reference genome. These unbiased approaches can provide a glimpse into the putative global RNA-RNA interactions, but because naked RNA is used as a substrate for proximity ligation, essential information regarding pre-existing RNA-protein interaction is lost, and artifactual RNA hybrids may be generated that consist of in vivo unavailable regions because of protein interactions.
In this study, we set out to capture genome-wide influenza intersegmental RNA-RNA interactions while considering in vivo vRNA-protein association. Our approach, referred to as 2CIMPL (dual crosslinking, immunoprecipitation, and proximity ligation) includes (1) UV light irradiation in addition to AMT crosslinking, and (2) on-bead enzymatic reactions to perform proximity ligation under native conditions, thus taking into account in vivo constraints from protein association and bypassing the need for RNA purification between each enzymatic reaction. Using this refined methodology, we reproducibly generated intersegmental RNA-RNA interaction maps for an influenza A virus (IAV) H1N1 strain (A/WSN/1933, referred to as “WSN”), which, compared with LIGR/PARIS applications on influenza virions, yielded higher frequencies of intersegmental RNA hybrids. Our analysis revealed multiple interactions between all segments, suggesting a complex and redundant vRNA interaction network. Notably, some RNA-RNA junctions are concentrated at specific regions of vRNA, referred to as hotspots, which suggests that a single re gion can coordinate multiple interactions with other segments. Synonymous codon mutations over the most pronounced hotspot observed in the NP gene segment abrogated the formation of RNA-RNA interactions at that site and resulted in a genome-wide rearrangement of the RNA network. Collectively, our 2CIMPL methodology demonstrates that the genome-wide network is not rigid but adjustable to sequence variations.
RESULTS
Developing a Method to Interrogate RNA-RNA Interactions of Influenza Virus Genome
Selective assembly of influenza vRNA segments into a progeny virion is thought to be assisted by RNA-RNA interactions. To identify those interactions within the IAV genome, we first adapted the recently reported LIGR/PARIS approach (Lu et al., 2016; Sharma et al., 2016) to virions (Figures 1A and S1A). Both methods are based on the same concept and employ the psoralen-derivative AMT to crosslink RNA duplexes, followed by partial RNA digest and subsequent RNA purification. Finally, a proximity ligation step is performed with naked RNA in the absence of bound protein (non-native conditions), which would not exclude regions of vRNA that are inaccessible in vivo because of protein association. We conducted several experiments of LIGR/PARIS for the laboratory-adapted WSN H1N1 strain and, to validate our results, first searched for intrasegmental RNA-RNA interactions between the 5′ and 3′ ends of each segment, which are known to form the panhandle structure of vRNP segments to which the tripartite viral polymerase binds. Although our LIGR/PARIS experiments indicated that this protocol can indeed be applied to identify RNA-RNA interactions, the recovery frequency of these known end-to-end interactions was low and mostly short-range intrasegmental RNA hybrids were detected with LIGR/PARIS (Figure S1B), indicative of ligation between proximal RNA ends and not necessarily stemming from an RNA duplex. The overall percentage of RNA hybrids containing sequence reads covering two distinct loci of the viral genome was less than 0.004% of the total mapped reads to the reference IAV genome. Intersegmental interactions were also observed, but at an even lower frequency of less than 0.002% of the total mapped reads. Moreover, replicate experiments yielded only a low degree of reproducibility because RNA-RNA interactions observed in one experiment were not detected in the other and vice versa, suggesting that modifications to the LIGR/PARIS protocol may be necessary to improve the detection of intersegmental RNA-RNA interactions within the influenza virus genome.
To overcome those issues, we added a second crosslinking step before AMT crosslinking to preserve the spatial information of the vRNP segments by crosslinking the vRNA to nearby proteins, as performed in the HITS-CLIP or 3C protocol. To that end, UV light irradiation was used because crosslinking with formaldehyde, even at a final concentration as low as 0.2%, yielded no RNA hybrids in the deep-sequencing library. RNA sequencing (RNA-seq) experiments with virions (deep sequencing of vRNA after RNase treatment of native crosslinked vRNPs) suggested that the absence of RNA hybrids is likely due to formaldehyde crosslinking rendering the vRNPs greatly inaccessible to the RNase enzyme because sequencing coverage did not indicate any RNA digestion (Figure S1D). Moreover, we sought to conduct the proximity ligation step with vRNPs instead of using naked RNA as substrate. Therefore, after virion lysis, we tethered the crosslinked vRNPs to magnetic beads through anti-NP antibody, given the binding of NP to extended stretches of the viral genome. This immunoprecipitation step allows for all necessary enzymatic reactions before library preparation, such as phosphatase treatment, polynucleotide kinase reaction, and RNA ligation, to be performed “on-bead” and for clean-up of these reactions in the absence of RNA precipitation between each step. After Proteinase K treatment to release RNA bound to beads and reversal of AMT-crosslinking with short-wave UV light irradiation, the resulting RNA products were then reverse transcribed and converted into an Illumina-compatible deep-sequencing library; the junctions of the RNA hybrids were determined computationally from the sequencing library (Figure 1B). To validate the ability of this methodology, which we refer to as 2CIMPL, to reproducibly capture genome-wide RNA-RNA interactions, we again examined whether the end-to-end interactions (panhandle structure) of the eight viral segments were present in the sequencing library. Indeed, 2CIMPL retrieved those intrasegmental interactions between the 5′ and 3′ termini and at a higher frequency than the LIGR/PARIS approach (Figures 1C, S1B, and S1C). With 2CIMPL, RNA hybrids occurred at a frequency of 1.14%–1.21% of the total mapped reads for replicate experiments, being markedly more abundant than with LIGR/PARIS approaches.
Detection of Intersegmental Interactions between Viral RNA Segments
We next searched for intersegmental RNA-RNA interactions within our deep sequencing libraries. Most RNA hybrids comprised short-range intrasegmental interactions, and intersegmental RNA hybrids were found at 0.08%–0.09% of total mapped reads. Notably, the junctions of those RNA duplexes were not evenly distributed throughout all eight WSN segments, and each segment had multiple intersegmental interactions (Figure 2A). Interestingly, we observed intersegmental interactions to be concentrated around hotspots, the most predominant one being found in the central region of the NP gene segment (Figure 2A, arrow), where one region of the genome interacts with multiple other segments and also with multiple sites within those segments. Importantly, the 2CIMPL protocol was highly reproducible because an independent biological replicate resulted in an almost identical genome-wide RNA-RNA interaction map with an overall Pearson correlation coefficient of 0.908 (Figures S2A and S2B). Further quantification of the intersegmental junctions, normalized to segment length, showed that the NP segment generated the greatest number of RNA hybrids among the eight segments (Figure 2B). Most intersegmental interactions of the NP segment were detected with the NA segment, which primarily occurred between the hotspot region in NP and the 5′ region of the NA segment (Figure 2C, boxed regions; Table S1). To identify the nucleotide sequences of this particular RNA duplex, we surveyed the deep-sequencing reads that corresponded to RNA hybrids and spanned both loci to narrow down the regions that could form potential base pairs (Figure S3A). Prediction of RNA duplexes among those overlapping read regions was performed with the program RNAhybrid (Krüger and Rehmsmeier, 2006). One of the predicted RNA-RNA interactions with the greatest thermodynamic stability is shown in Figure 2D. Taken together, our 2CIMPL approach reproducibly identifies genome-wide RNA-RNA interactions that form at multiple sites along a given segment and cluster at specific regions (hotspots) within the influenza virus genome.
Figure 2. Intersegmental RNA-RNA Interactions Can Be Concentrated at Hotspots.
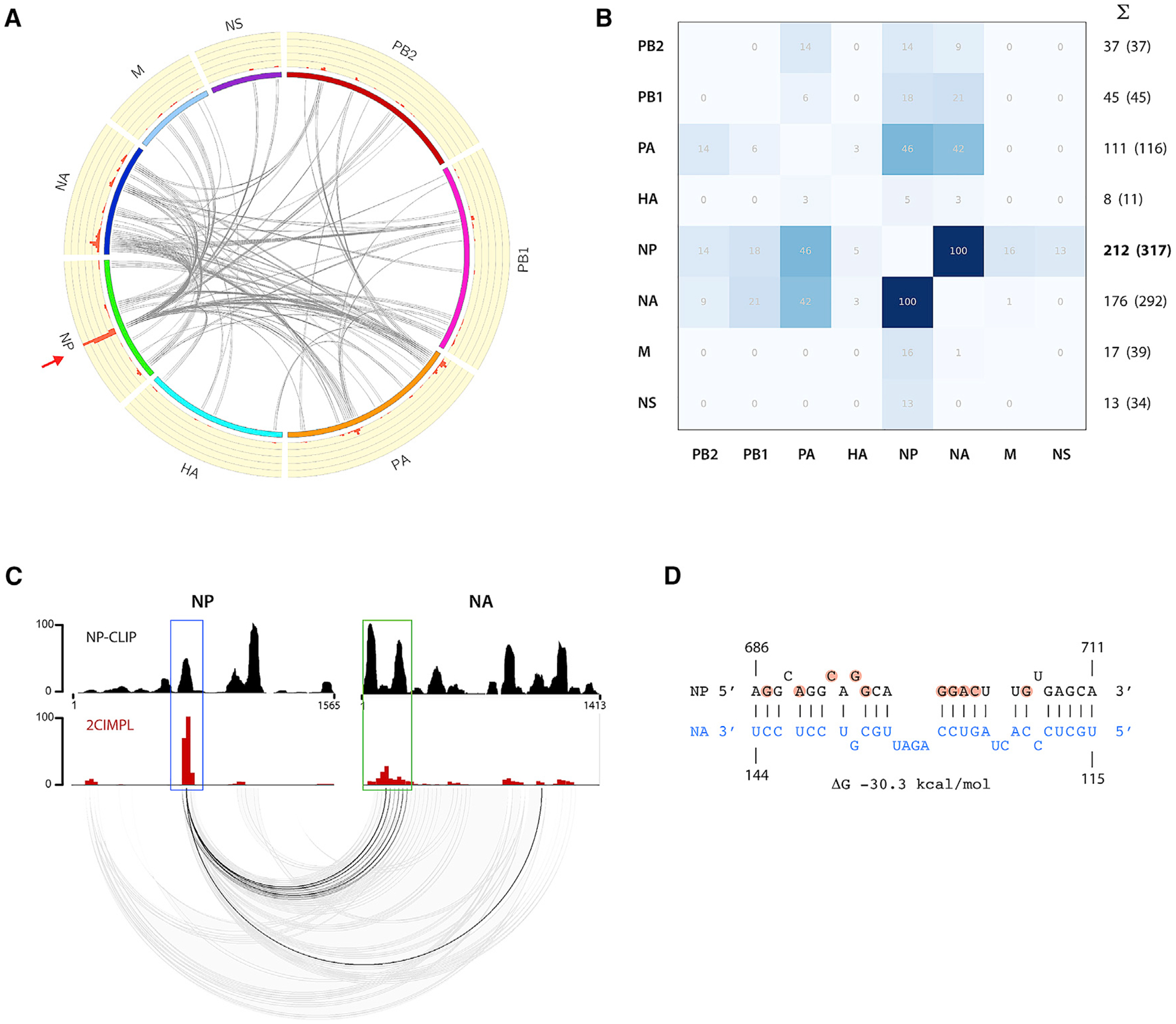
(A) Circos plot showing intersegmental RNA-RNA interactions between all segment pairs that are observed in two independent biological replicates of the wild-type WSN strain. The top 25% of interactions binned into windows of 25 nucleotides are shown. Please see Figure S2A for individual results of each replicate. Each line indicates the hybrid junctions of an RNA duplex, which result from proximity ligation. The histogram on the outer circle (red bars on yellow background) indicates the relative frequency of intersegmental junctions at a given site. A major hotspot is observed in the center region of the NP segment (arrow).
(B) Heatmap showing the relative abundance of intersegmental hybrid junctions for each segment. The highest absolute number was normalized to 100. The sum of all intersegmental interactions for each segment is indicated. Numbers in parentheses are normalized to segment length. The NP segment forms the most intersegmental interactions with other segments (numbers in bold).
(C) Comparison of the NP binding profile and intersegmental interactions between the NP and NA segments. The NP CLIP profile (top; data previously described [Lee et al., 2017]) and 2CIMPL histogram, as shown in (A), are also shown on top of each other for comparison.
(D) A predicted RNA duplex between the hotspot region in the NP and the 5′ region of the NA segment is shown. Highlighted nucleotides indicate mutations in the WSN [NP-HSMUT] strain. See also Figures S2 and S3.
Nucleotide Changes Cause Genome-wide Rearrangements of Intersegmental RNA-RNA Interactions
To determine the importance of the NP segment hotspot for establishing the genome-wide intersegmental RNA-RNA interaction network, we mutated nucleotides 656–705 of the NP segment with synonymous codons to disrupt putative RNA duplex formation and maintain the amino acid sequence of the encoded viral protein (Figure 3A). We verified that the change in codon usage did not affect the translation efficiency of NP (Figure 3B). A mutant WSN strain, referred to as WSN [NP-HSMUT], was generated by reverse genetics, which differs from the wild-type WSN strain by the mutations in the NP hotspot only. To examine how the genome-wide RNA-RNA interaction network would change upon disrupting a region enriched for intersegmental connections, we performed 2CIMPL with the WSN [NP-HSMUT] strain, which unexpectedly replicated similarly to the wild type (Figure S4A). The 2CIMPL data of the mutant revealed that the genome-wide RNA-RNA interactions were rearranged, as evidenced by the disappearance of the original hotspot in the NP gene segment and the appearance of new junction hotspots in the PA, HA, and NA segments (Figures 3C, arrows). An independent biological replicate of this mutant strain produced a similar RNA-RNA interactome with a Pearson correlation coefficient of 0.901 (Figure S2D). Analysis of the rear ranged 2CIMPL interaction network demonstrated a shift from the NP segment to the NA segment as the one with the most connections and the PA-NA pair generating the most intersegmental RNA hybrids (Figure 3D). Consistent with a rearranged genome-wide RNA-RNA interaction network in WSN [NP-HSMUT], the new hotspot in the 5′ region of the NA segment, which, in the wild-type strain, interacts with the NP hotspot (Figures 2C and 2D), shifted to form interactions predominantly with the 5′ region of the mutant NP segment (Figure 3E, green boxes). RNA-RNA structure predictions indicated that the 5′ region of the NA segment forms a less-favorable RNA duplex with the mutated nucleotides of the NP hotspot and a thermodynamically more stable duplex with the 5′ region of the NP segment (Figures 3F and S3B). A similar observation was made for the 5′ region of the PA segment, which forms one of the most abundant RNA hybrids with the 5′ region of the NA segment in WSN [NP-HSMUT] (Figures 4A and 4B; Table S1), whereas this region interacts preferentially with the NP hotspot in the wild-type strain (Figures 2A and 4C). RNA duplex predictions indicated that interactions between the mutated sequence of the NP hotspot and the 5′ region of the PA segment are thermodynamically less favorable (Figure 4D) than the observed interactions with the NA segment (Figure 4B). Taken together, our data suggest that genome-wide intersegmental interaction networks are flexible and can reorganize as a consequence of nucleotide mutations.
Figure 3. Altering the Nucleotide Sequence of the RNA Junction Hotspot Results in Rearrangement of the Genome-wide RNA-RNA Interaction Network.
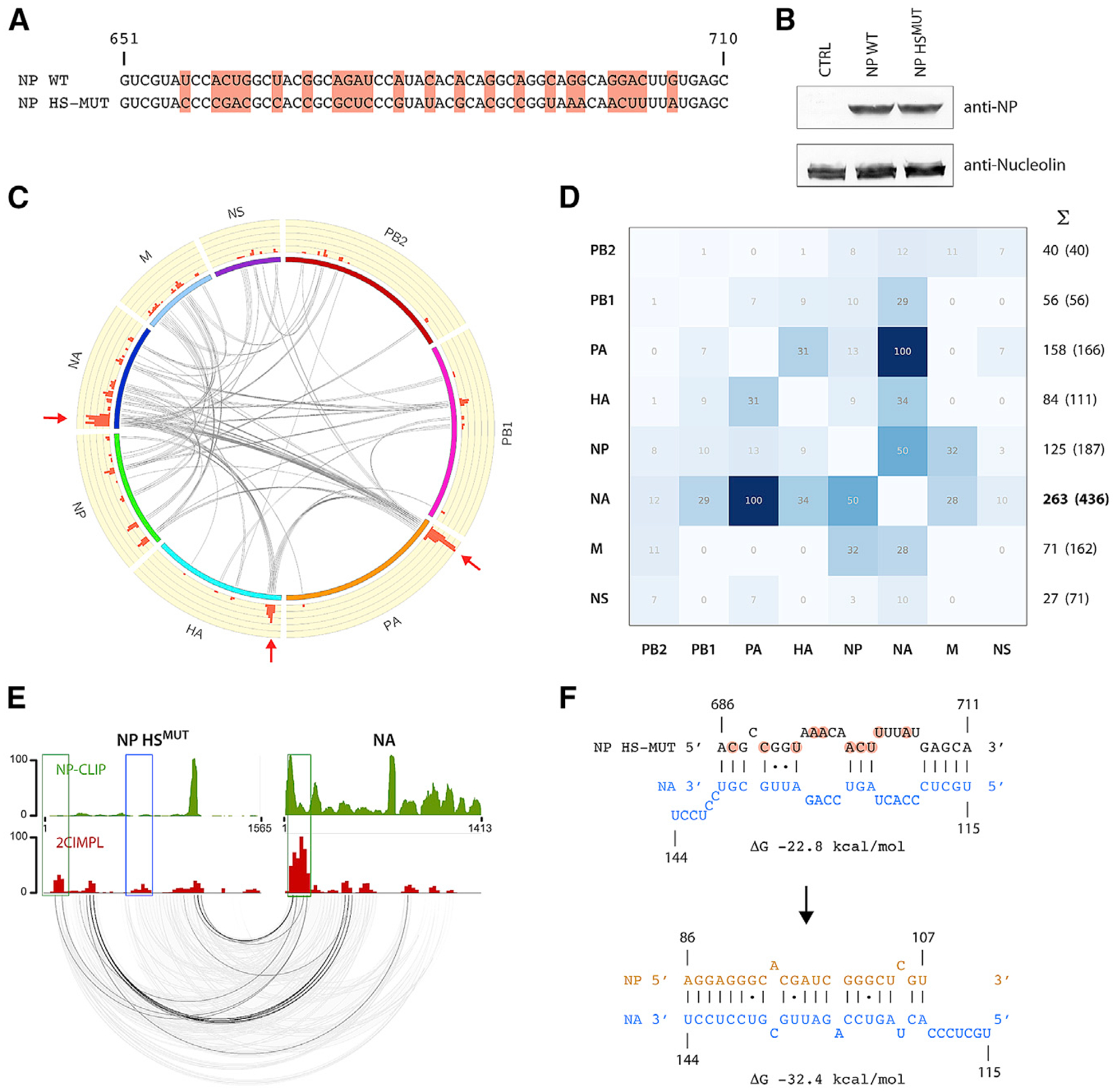
(A) Sequence alignment of the NP gene segment comparing WSN wild type and the hotspot mutant. Altered nucleotides are highlighted and result in synonymous codons and an unaffected amino acid sequence. The nucleotide positions of the negative-sense vRNA sequence are indicated.
(B) Synonymous codons do not affect translation efficiency of NP. Either the NP wild-type or hotspot mutant cDNA was expressed in HEK293T cells; lysates were subjected to western blot analysis using an anti-NP antibody. Nucleolin was probed as a loading control.
(C) Circos plot of the WSN [NP-HSMUT] strain showing intersegmental interactions observed in twoindependent biological replicates. The top25% of interactions binned into windows of 25 nucleotides are shown. Please see Figure S2D for individual results of each replicate. The original hotspot in the NP segment is lost, but other hotspots appear in the PA, HA, and NA segments (arrows).
(D) Heatmap showing the relative abundance of intersegmental junctions for each segment. In contrast to the wild-type interaction map, the NA segment forms the most intersegmental interactions with the other segments in the mutant.
(E) Intersegmental interactions between the NP hotspot mutant and NA segments are shown. The NP CLIP profile of the WSN [HSMUT] strain is shown on top. Please see Figure S2E for the CLIP profile of all segments. The blue box indicates the original hotspot region. Absence of the NP peak at the original NP hotspot location was observed in the WSN [HSMUT] strain.
(F) Predicted RNA duplex between the mutated region in NP and the same region of NA as in Figure 2D (top). The predicted duplex between the same region of the NA segment and the 5′ region of NP, which are indicated by green boxes in (E), are shown at the bottom. The latter interaction occurs frequently, whereas RNA hybrids encompassing the mutated hotspot region are not observed. See also Figures S2 and S4.
Figure 4. Intersegmental Interactions between PA and NA Segments in the WSN [HSMUT] Strain.
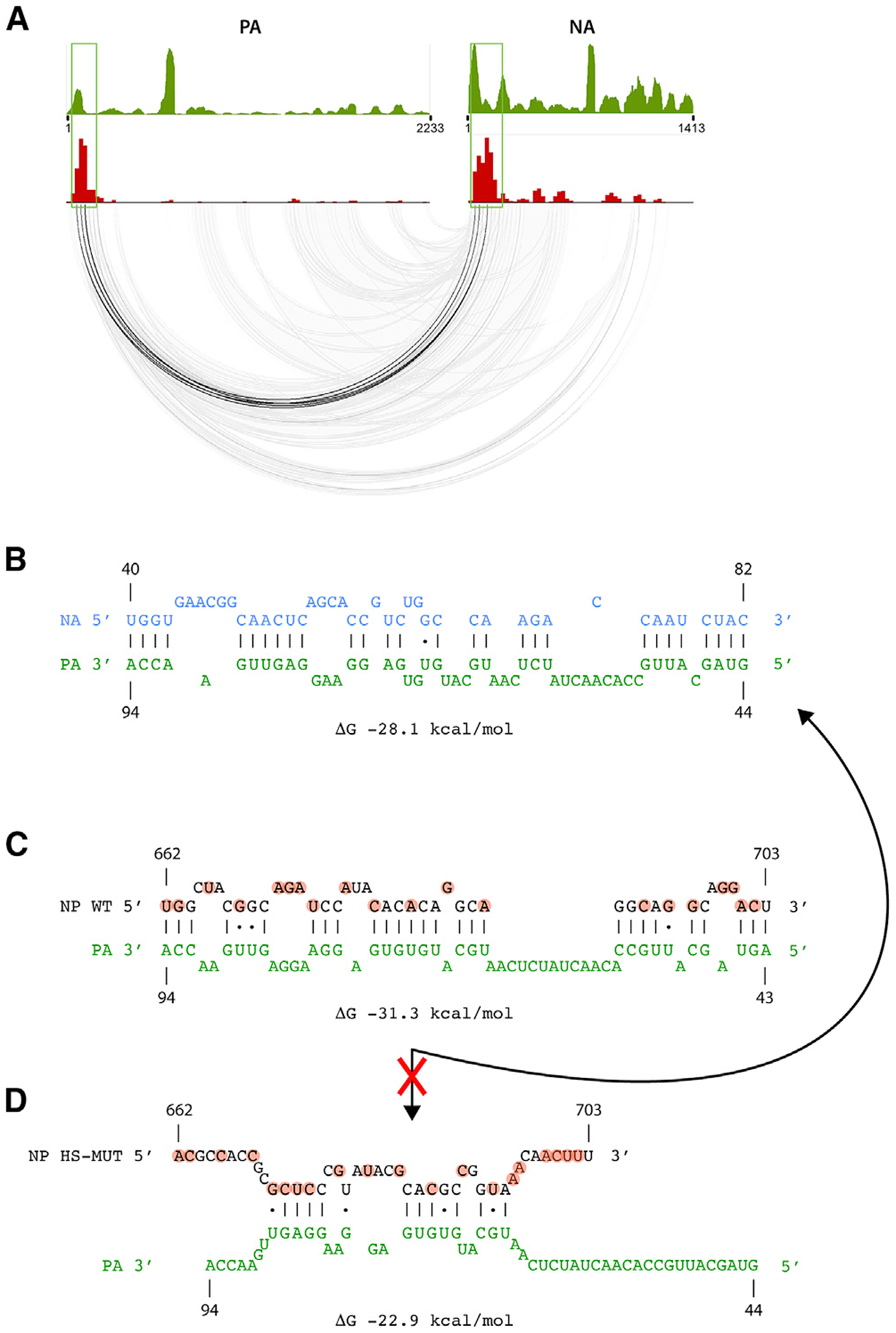
(A) Base-pairing plot and NP-CLIP profile of PA and NA segments.
(B) Predicted RNA-RNA interaction between regions in PA and NA segments based on frequently observed RNA hybrids in the hotspot mutant, indicated by boxed regions in (A).
(C and D) Predicted RNA-RNA interactions between wild-type (C) or the mutated hotspot (D) region in the NP and the same region of the PA segment as in (A). See also Figure S3 and Table S1.
Comparison of 2CIMPL Data with RNA-RNA Interaction Network Derived from SPLASH
We compared our 2CIMPL result with a recently published RNA-RNA interaction map of the WSN strain, which was obtained using SPLASH (Dadonaite et al., 2019). This approach uses a biotinylated psoralen derivative, both as a crosslinking agent and as a means to enrich for RNA duplexes, and performs the proximity ligation step under non-native conditions in the absence of bound proteins (Aw et al., 2016). To compare the identified networks from 2CIMPL and SPLASH, we applied the same analysis pipeline on both datasets to locate intersegmental interactions. While the 2CIMPL method retrieved 312 unique RNA hybrids, 529 unique junctions were observed with SPLASH and 10.3% of the RNA hybrids were found in both datasets (Figure S5C), indicating that both approaches identify an overlapping cohort of RNA-RNA interactions. Importantly, the 2CIMPL hotspot located in the central region of the NP segment was also one of the top RNA junctions in SPLASH (Figures S5A and S5B, arrows), which solidifies the significance of the NP hotspot to coordinate RNA-RNA interactions with other viral RNA segments.
Relationship between RNA-RNA Interactions and NP Binding to vRNA
Use of CLIP-based techniques has recently provided evidence that NP is distributed along the viral genome in a non-uniform and non-random manner (Lee et al., 2017; Williams et al., 2018). This observation led us and others to speculate that regions of the genome with low NP binding coordinate RNA-RNA interactions. Indeed, we observed hotspots at regions with low NP binding, such as in the 5′ region of the HA segment in the WSN [HSMUT] strain (Figure S2E). However, our analysis also revealed that, in wild-type WSN, the hotspot in the NP gene segment was adjacent to a strong NP protein binding site (Figures 2C and S2C), suggesting that NP-bound regions of vRNA may still participate in RNA-RNA interactions. To quantitatively assess the relationship between NP-binding and intersegmental RNA-RNA interaction, we recorded the number of mapped RNA hybrid reads for each nucleotide in the genome and plotted them based on the classification of whether the nucleotide position overlapped with a called NP peak or a non-peak region. Nucleotides belonging to NP peaks showed a small but significant overrepresentation of aligned RNA hybrids in both the wild-type and the mutant WSN strain (Figures 5A and 5B), suggesting that NP binding does not preclude RNA duplex formation and that regions of both NP peaks and valleys contribute to RNA-RNA interactions.
Figure 5. NP Peak as well as Non-peak Regions Participate in RNA-RNA Interactions.
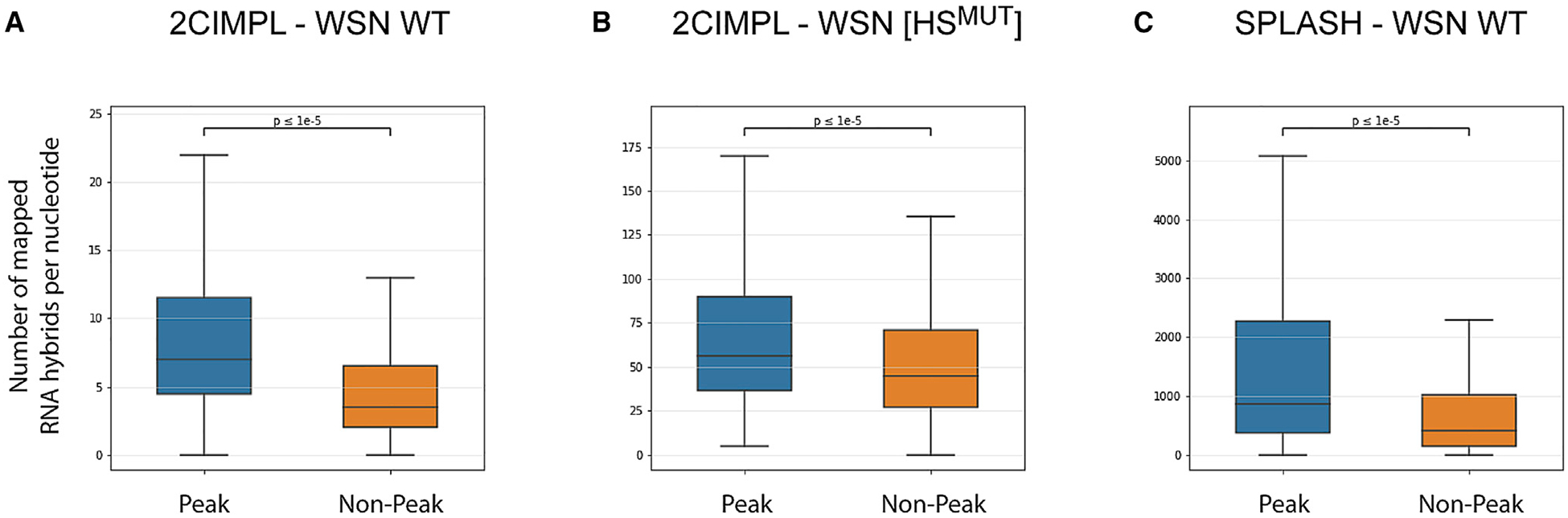
(A–C) Quantity of RNA-RNA interactions occuring in peak and non-peak regions is shown for wild-type WSN (A), the hotspot mutant (B), and wild-type WSN
derived from SPLASH (C). The bottom and top of the boxes represent the 25th and 75th percentiles; the bar in the middle represents the median value, and the whiskers represent 1.53 the interquartile range extended out from the 25th and 75th percentiles. The values on the y axis represent the absolute number of mapped RNA hybrids at each nucleotide, which were plotted based on their classification of overlapping with either a called NP peak or a non-peak (Lee et al., 2017); 3,958 and 9,623 nucleotides of the WSN IAV genome were called as peaks and non-peaks, respectively. Please note that values are dependent on sequencing coverage (SPLASH data contained more sequencing reads than the 2CIMPL data, hence the higher values); p values were determined using the Wilcoxon signed-rank test. See also Figures S5.
Because the 2CIMPL approach uses immunoprecipitation of NP to capture vRNA, RNA junctions adjacent to strong NP peaks may be overrepresented in the sequencing libraries. To rule out that potential bias within our dataset, we correlated the RNA-RNA interactions generated by SPLASH (Dadonaite et al., 2019), which does not use NP immunoprecipitation, to the genome-wide NP binding in WSN (Figure 5C). That analysis confirmed that NP-peak regions were slightly overrepresented in intersegmental RNA-RNA interactions as observed with 2CIMPL. In addition, we compared the NP binding profile of WSN generated with a different anti-NP antibody to the RNA-RNA interaction sites identified by 2CIMPL and again found that more interactions occur at NP peaks than in NP-low regions (Figure S5D). Finally, NP peak heights do not correlate with the frequencies of RNA-RNA interactions at a given location, as exemplified in the 5′ regions of the PA and HA segments in the WSN [HSMUT] strain (Figure S2E, arrows). Consistent with that notion, the genome-wide NP binding profile determined by HITS-CLIP and the RNA-RNA interactome identified by 2CIMPL do not show any correlation (Pearson correlation coefficient r = 0.159). Taken together, these data support the notion that 2CIMPL is not biased toward NP bound regions of vRNA and that NP-bound regions also participate in intersegmental RNA-RNA interactions.
DISCUSSION
A lack of tools to identify intersegmental RNA-RNA interactions within the influenza virus genome has limited our understanding of virus assembly. In this study, we refined the existing methodology for uncovering in vivo RNA duplexes and developed a protocol termed 2CIMPL to study these interactions in influenza virions in the context of vRNPs (Figure 1). A highly reproducible RNA-RNA interaction map was identified using this approach (Figure 2A), which indicated that intersegmental RNA-RNA interactions occur throughout the viral genome and are not restricted to the proposed 5′ and 3′ packaging regions of the vRNA segments. In addition, interactions can cluster at select regions within the genome, creating hotspots (Figure 2C). Our data indicate that hotspots interact with multiple regions in other segments, creating flexibility in the system, rather than functioning as a static binary interaction network. This flexibility in RNA-RNA interaction networks can accommodate for changes in the nucleotide sequence through viral evolution, providing an advantage to the virus. For example, mutations within the NP hotspot region resulted in a large-scale reorganization of intersegmental RNA-RNA interactions (Figure 3) but did not affect viral replication kinetics (Figure S4A), suggesting that genome-wide RNA-RNA interactions can adjust to compensate for changes to the RNA sequence. Notably, the observation that genome-wide RNA-RNA interactions of IAV are flexible may be an essential characteristic, which allows the maintenance of efficient virus replication by tolerating the high mutation rate of its own polymerase and the propagation as quasispecies.
Based on our previous report on genome-wide NP-binding sites (Lee et al., 2017), we proposed that regions not bound by NP would preferentially engage in intersegmental RNA-RNA interactions. However, our observation that the NP segment hotspot is located in the vicinity of a NP-binding peak as well as our genome-wide comparative analysis between NP binding and RNA duplex sites (Figure 5) suggests that both NP peak and valley regions contribute to RNA-RNA interactions. This observation was confirmed by comparative analysis of a previously published RNA-RNA interaction network derived using SPLASH (Dadonaite et al., 2019). Both approaches yielded an overlapping cohort of RNA hybrids found in both datasets (Figure S5). Differences between the networks derived from both techniques could be due to variations in experimental approaches and introduction of distinct biases. For example, not all RNA duplexes are covalently crosslinked by psoralen and different psoralen derivatives, as used in SPLASH, may create distinct biases. Analogous to how variations in CLIP protocols, such as HITS-CLIP and PAR-CLIP, which use distinct crosslinking methods, can produce variable outcomes because of RNA geometry, 2CIMPL and SPLASH may also be complementary methods to delineate the IAV RNA interaction network. The differences may also represent many of the flexibilities within the interaction networks that are revealed by examining a large population of viruses.
Given the striking observation that local mutations can affect RNA-RNA interaction networks, other IAV strains with divergent nucleotide sequences and of another subtype may form distinct RNA-RNA interaction hotspots. The distinct RNA-RNA interactions between IAV strains will also likely affect reassortment potential between strains. Therefore, the development of this highly reproducible protocol will enable us to examine other strains and mutants to uncover which parameters control intersegmental RNA-RNA interactions and provide insight into how reassortment bias is established. Overall, the 2CIMPL methodology can be applied to define RNA-RNA interactions of any RNA type bound by known proteins to address a broad array of biological questions and to provide an alternative to current methodologies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Nara Lee (nara.lee@pitt.edu).
Materials Availability
Reagents (plasmid for virus rescue) generated in this study (see KEY RESOURCES TABLE) are available upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-NP (IAV) | Millipore | Cat# MAB8251; RRID: AB_95293 |
| Bacterial and Virus Strains | ||
| IAV H1N1 strain A/WSN/1933 | Dr. Richard Webby, St. Jude Children’s Research Hospital | N/A |
| WSN [NP-HSMUT] | This study | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Aminomethyltrioxsalen (AMI) | Sigma-Aldrich | A4330 |
| Critical Commercial Assays | ||
| NEBNext Ultra II RNA Library Prep Kit | NEB | E7770 |
| NEBNext Ultra II DNA Library Prep Kit | NEB | E7645 |
| Deposited Data | ||
| 2CIMPL datasets | This study | SRA BioProject accession number PRJNA546584 (SRR9204625, SRR9204626, SRR9204628, and SRR9204629) |
| HITS-CLIP dataset | This study | SRA BioProject accession number PRJNA546584 (SRR9204627) |
| Experimental Models: Cell Lines | ||
| MDCK cells | ATCC | Cat# PTA-6500; RRID: CVCL-IQ72 |
| HEK293T cells | ATCC | Cat# CRL-3216; RRID: CVCL_0063 |
| Recombinant DNA | ||
| Plasmid pHW2000-(NP-HSMUT) | This study | N/A |
| Software and Algorithms | ||
| 2CIMPL analysis pipeline | This study | https://github.com/NaraLee-Lab/2CIMPL |
Data and Code Availability
The accession numbers for the 2CIMPL datasets generated during this study are Sequence Read Archive: PRJNA546584 (SRR9204625, SRR9204626, SRR9204628, and SRR9204629). The accession number for the HITS-CLIP dataset is Sequence Read Archive: PRJNA546584 (SRR9204627). A detailed data analysis pipeline can be found in the Github repository at https://github.com/NaraLee-Lab/2CIMPL.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines
MDCK cells (ATCC) were cultured in Eagle’s Minimum Essential Medium (EMEM) supplemented with 10% FBS, L-glutamine and penicillin/streptomycin at 37°C in a 5% CO2 atmosphere. HEK293T cells (ATCC) were cultured in Dulbecco’s Modified Eagle Medium (DMEM) with 10% FBS, L-glutamine and penicillin/streptomycin at 37°C in a 5% CO2 atmosphere.
METHODS DETAILS
Virus Rescue
Rescue of recombinant wild-type and mutant A/WSN/1933 strains were previously described (Bhagwat et al., 2018; Lakdawala et al., 2013, 2014). Briefly, eight bidirectional plasmids (pHW2000) encoding WSN gene segments (provided by Dr. Richard Webby, St. Jude Children’s Research Hospital) were transfected into HEK293T cells and the media was collected at 24, 48, and 72 hpi and placed onto a 10 cm-dish of MDCK cells. Viral stocks were collected at the onset of visual CPE, titered on MDCK and passaged a second time in MDCK for these studies. Mutant NP segment was generated by chemical synthesis and cloned into the reverse genetics plasmid for virus rescue.
Viral Growth Curves and Titration
Multicycle growth curves were performed by infecting with a multiplicity of infection (MOI) of 0.01. Confluent MDCK cells were inoculated in triplicate with each virus and incubated for 1 h at room temperature with shaking, after which the inoculum was replaced with 500 μL of serum-free medium. Samples were titered by tissue culture infectious dose 50 (TCID50) on MDCK cells as described (Reed and Muench, 1938). All growth curve measurements were performed in at least three independent biological replicates.
Dual crosslinking, immunoprecipitation, and proximity ligation (2CIMPL)
Two confluent T225 flasks of MDCK cells were washed twice with phosphate-buffered saline (PBS) and infected at a dilution of 1:100,000 with the indicated virus (cell passage 1) in serum-free EMEM. At 48 hours post-infection, 40 mL of the culture medium containing ~106 infectious virus particle per ml was harvested and cellular debris was pelleted by centrifugation at 2,000 × g for 20 min. UV light irradiation at 254 nm (400 mJ/cm2 followed by 200 mJ/cm2) was performed on clarified culture medium. Crosslinked virus supernatant was layered onto a 30% sucrose-NTE (100 mM NaCl, 10 mM Tris pH 7.4, 1 mM EDTA) cushion and centrifuged at 200,000 × g for 2 hours at 4°C. Pelleted virions were resuspended in 100 μL PBS including 50 mg/ml AMT (Sigma-Aldrich) and irradiated for 30 min from 15 cm distance with a handheld 365 nm UV lamp on ice while covered with a 2 mm-thick glass plate. Equal volume of 2× PXL buffer (1× PBS, 2% NP40, 1% deoxycholate, 0.2% SDS) was added to lyse the virions followed by DNase treatment with 2 μL RQ1 DNase (Promega) for 5 min at 37°C. Partial RNase digestions were carried out for 5 min at 37°C by adding either 0.25 μg or 0.025 μg RNase A. Reaction was stopped by adding 5 μL RNase inhibitor (RNasin Plus, Promega) and spun at full speed for 10 min. 40 μL of anti-NP antibody-beads (mouse monoclonal antibody MAB8251 (Millipore) coupled to Protein G Dynabeads to full capacity) were added to the supernatant and incubated for 4 h at 4°C in a rotator. Beads were washed three times with PXL buffer and twice with PNK buffer (50 mM Tris pH 7.5, 10 mM MgCl2, 0.5% NP40). Dephosphorylation reaction was performed using Calf Intestinal Phosphate (NEB) for 20 min at 37°C with shaking, and after washing the beads once with PXL, once with 50 mM Tris pH 7.5, 20 mM EGTA, 0.5% NP40, and twice with PNK buffer, beads were PNK (NEB)-treated for 20 min at 37°C with shaking. Reaction was cleaned up by washing beads once with PXL and twice with PNK buffer followed by an overnight RNA ligation reaction (10 μL 10× T4 RNA ligase 1 buffer, 10 μL 10 mM ATP, 5 μL T4 RNA ligase 1 (NEB), 2 μL RNasin, 73 μL H2O). Beads were washed twice with PXL and twice with PNK buffer, and Proteinase K-treated for 1 h at 50°C. Phenol-chloroform was added to the beads; the supernatant was collected and irradiated for 8 min with 254 nm UV light on ice to reverse AMT-crosslinks (Nanni and Lee, 2018). RNA was isolated by ethanol precipitation in the presence of 5 μg glycogen and resuspended in 10 μL H2O. Illumina-compatible deep sequencing library was constructed using the NEBNext Ultra II RNA Library Prep Kit (NEB). For the LIGR/PARIS application on influenza virions, the same starting material was used as for 2CIMPL and the PARIS protocol was followed as described (Lu et al., 2016) to generate a deep sequencing library. Data analysis was performed by adapting the LIGR pipeline (Sharma et al., 2016) as outlined below.
2CIMPL data analysis
Raw sequencing reads were processed via a data analysis pipeline that can be found at https://github.com/NaraLee-Lab/2CIMPL. In brief, sequence reads from a paired-end 110-cycle run were paired using PEAR (Zhang et al., 2014) and identical results were removed using a Perl script from the CLIP Toolkit (Shah et al., 2017). Chimeric read alignment and junction discovery were performed using a modified version of Aligater (Sharma et al., 2016). To create upper triangle plots, intrasegmental interactions were isolated and assigned to bins of size 50, then plotted on a rotated heatmap using Matplotlib. Circos plots depicting intersegmental interactions were generated with Circos (Krzywinski et al., 2009) and using BEDTools to create the surrounding histograms (Quinlan and Hall, 2010). For circos plots showing the top 25% of interactions occurring in two independent biological replicates (Figures 2 and 3), interactions were binned into windows of 25 nucleotides. Pearson correlations were determined by pairwise comparison of the intersegmental interaction histograms with pandas. Intersegmental links shared between replicates were found by generating an interaction matrix between bins of genomic loci and checking for the presence of a read from both replicates in every bin. This was achieved using in-house python code that can be found in the Github repository.
HITS-CLIP and deep sequencing data analysis
HITS-CLIP and subsequent data analysis for the WSN [NP-HSMUT] mutant strain was performed as previously described (Lee et al., 2017). In brief, two confluent T175 flasks of MDCK cells were washed twice with phosphate-buffered saline (PBS) and infected at a dilution of 1:100,000 with the WSN [NP-HSMUT] mutant virus in serum-free EMEM containing TCPK-trypsin (Worthington Biochemicals). At 96 hours post-infection, 40 mL of the culture medium was harvested and cellular debris was pelleted by centrifugation at 2,000 × g for 20 min. UV light irradiation at 254 nm (400 mJ/cm2 followed by 200 mJ/cm2) was performed on clarified culture medium. Crosslinked virus supernatant was layered onto a 30% sucrose-NTE (100 mM NaCl, 10 mM Tris pH 7.4, 1 mM EDTA) cushion and centrifuged at 200,000 × g for 2 hours at 4°C. Virus particles concentrated from 25 mL of culture supernatant were resuspended in 300 μL PXL buffer (1× PBS, 1% NP40, 0.5% deoxycholate, 0.1% SDS), followed by DNase and RNase treatment. For each viral strain sample, partial RNase digestions were carried out for 5 min at 37°C with three aliquots of 100 μL viral lysate and a tenfold dilution series of RNase A (0.25 mg, 0.025 μg, and 0.0025 mg total amount of enzyme, respectively). An anti-NP mouse monoclonal antibody (Millipore cat. no. MAB8251) was used for immunoprecipitating influenza NP. For each IP reaction, 25 μL of antibody-Dynabeads Protein G complexes were used. Ligation of 5′ and 3′ adapters, RT reaction and first-round PCR amplification step were carried out as described (Moore et al., 2014). The first-round PCR products were then converted into an Illumina-compatible deep sequencing library using the NEBNext Ultra DNA Library Prep Kit (NEB), followed by Illumina deep sequencing. Data analysis was performed as described (Moore et al., 2014) using the NovoAlign alignment program and mapping the reads to reference genomes available from the NCBI database.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical methods are described in the Methods Details section and figure legends. Pearson’s r was used to obtain correlations between independent samples of HITS-CLIP and/or 2CIMPL data, with all displayed correlation values having p < 0.001. HITS-CLIP data corresponded to total number of NP-bound reads observed at each nucleotide position in the WSN Influenza genome, while 2CIMPL data corresponded to number of intersegmental interactions that overlapped each nucleotide position. The same 2CIMPL data was used to quantify interaction overlap with NP peaks or valleys, and Wilcoxon signed-rank test was used to determine significance with p < 0.001. All statistical tests were performed in python using pandas or SciPy.
Supplementary Material
Highlights.
RNA-RNA interactions between influenza virus genome segments are redundant
One region on a segment coordinates interaction with multiple other segments
Hotspot interaction sites are rearranged with synonymous mutations
Binding to nucleoprotein does not prevent RNA-RNA interactions
ACKNOWLEDGMENTS
We thank members of the Lakdawala laboratory for thoughtful discussions. V.L.S., S.S.L., J.P.K., and N.L. are supported by a New Initiative Award from the Charles E. Kaufman Foundation, a supporting organization of the Pittsburgh Foundation. S.S.L. and V.L.S. are supported by the NIH (grant number 1R01AI139063-01A1). S.S.L. is also supported by the American Lung Association biomedical research grant. D.J.S. and V.S.C. are supported by the NIH (grant number U01AI124303).
Footnotes
DECLARATION OF INTERESTS
S.S.L. and N.L. are named inventors on a patent application describing the use of antisense oligonucleotides against specific NP binding sites as therapeutics.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107823.
REFERENCES
- Aw JG, Shen Y, Wilm A, Sun M, Lim XN, Boon KL, Tapsin S, Chan YS, Tan CP, Sim AY, et al. (2016). In Vivo Mapping of Eukaryotic RNA Interactomes Reveals Principles of Higher-Order Organization and Regulation. Mol. Cell 62, 603–617. [DOI] [PubMed] [Google Scholar]
- Bhagwat AR, Le Sage V, and Lakdawala SS (2018). Live Imaging of Influenza Viral Ribonucleoproteins Using Light-Sheet Microscopy. Methods Mol. Biol 1836, 303–327. [DOI] [PubMed] [Google Scholar]
- Cros JF, and Palese P (2003). Trafficking of viral genomic RNA into and out of the nucleus: influenza, Thogoto and Borna disease viruses. Virus Res. 95, 3–12. [DOI] [PubMed] [Google Scholar]
- Dadonaite B, Gilbertson B, Knight ML, Trifkovic S, Rockman S, Laederach A, Brown LE, Fodor E, and Bauer DLV (2019). The structure of the influenza A virus genome. Nat. Microbiol 4, 1781–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, Rippe K, Dekker M, and Kleckner N (2002). Capturing chromo-some conformation. Science 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Dekker J, Marti-Renom MA, and Mirny LA (2013). Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat. Rev. Genet 14, 390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisfeld AJ, Neumann G, and Kawaoka Y (2015). At the centre: influenza A virus ribonucleoproteins. Nat. Rev. Microbiol 13, 28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier E, Moules V, Essere B, Paillart JC, Sirbat JD, Isel C, Cavalier A, Rolland JP, Thomas D, Lina B, and Marquet R (2012). A supramolecular assembly formed by influenza A virus genomic RNA segments. Nucleic Acids Res. 40, 2197–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavazzi C, Isel C, Fournier E, Moules V, Cavalier A, Thomas D, Lina B, and Marquet R (2013). An in vitro network of intermolecular interactions between viral RNA segments of an avian H5N2 influenza A virus: comparison with a human H3N2 virus. Nucleic Acids Res. 41, 1241–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson B, Zheng T, Gerber M, Printz-Schweigert A, Ong C, Marquet R, Isel C, Rockman S, and Brown L (2016). Influenza NA and PB1 Gene Segments Interact during the Formation of Viral Progeny: Localization of the Binding Region within the PB1 Gene. Viruses 8, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger J, and Rehmsmeier M (2006). RNAhybrid: microRNA target prediction easy, fast and flexible. Nucleic Acids Res. 34, W451–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, and Marra MA (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakdawala SS, Shih AR, Jayaraman A, Lamirande EW, Moore I, Paskel M, Kenney H, Sasisekharan R, and Subbarao K (2013). Receptor specificity does not affect replication or virulence of the 2009 pandemic H1N1 influenza virus in mice and ferrets. Virology 446, 349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakdawala SS, Wu Y, Wawrzusin P, Kabat J, Broadbent AJ, Lamirande EW, Fodor E, Altan-Bonnet N, Shroff H, and Subbarao K (2014). Influenza a virus assembly intermediates fuse in the cytoplasm. PLoS Pathog. 10, e1003971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Sage V, Nanni AV, Bhagwat AR, Snyder DJ, Cooper VS, Lakdawala SS, and Lee N (2018). Non-uniform and non-random binding of nucleoprotein to influenza A and B viral RNA. Viruses 10, 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Le Sage V, Nanni AV, Snyder DJ, Cooper VS, and Lakdawala SS (2017). Genome-wide analysis of influenza viral RNA and nucleoprotein association. Nucleic Acids Res. 45, 8968–8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Zhang QC, Lee B, Flynn RA, Smith MA, Robinson JT, Davidovich C, Gooding AR, Goodrich KJ, Mattick JS, et al. (2016). RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell 165, 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Zhang C, Gantman EC, Mele A, Darnell JC, and Darnell RB (2014). Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat. Protoc 9, 263–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanni AV, and Lee N (2018). Identification of host RNAs that interact with EBV noncoding RNA EBER2. RNA Biol. 15, 1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, Sugita Y, Aoyama K, Hirase A, Kawakami E, Miyazawa A, Sagara H, and Kawaoka Y (2012). Three-dimensional analysis of ribonucleoprotein complexes in influenza A virus. Nat. Commun 3, 639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palese P, and Shaw ML (2013). Orthomyxoviridae: The Viruses and Their Replication. (Lippincott Williams & Wilkins). [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LJ, and Muench H (1938). A simple method of estimating fifty per cent Endpoints12. Am. J. Epidemiol 27, 493–497. [Google Scholar]
- Shah A, Qian Y, Weyn-Vanhentenryck SM, and Zhang C (2017). CLIP Tool Kit (CTK): a flexible and robust pipeline to analyze CLIP sequencing data. Bioinformatics 33, 566–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma E, Sterne-Weiler T, O’Hanlon D, and Blencowe BJ (2016). Global mapping of human RNA-RNA interactions. Mol. Cell 62, 618–626. [DOI] [PubMed] [Google Scholar]
- Te Velthuis AJ, Robb NC, Kapanidis AN, and Fodor E (2016). The role of the priming loop in influenza A virus RNA synthesis. Nat. Microbiol 1, 16029. [DOI] [PubMed] [Google Scholar]
- Whittaker G, Bui M, and Helenius A (1996). The role of nuclear import and export in influenza virus infection. Trends Cell Biol. 6, 67–71. [DOI] [PubMed] [Google Scholar]
- Williams GD, Townsend D, Wylie KM, Kim PJ, Amarasinghe GK, Kutluay SB, and Boon ACM (2018). Nucleotide resolution mapping of influenza A virus nucleoprotein-RNA interactions reveals RNA features required for replication. Nat. Commun 9, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WW, Weaver LL, and Panté N (2007). Ultrastructural analysis of the nuclear localization sequences on influenza A ribonucleoprotein complexes. J. Mol. Biol 374, 910–916. [DOI] [PubMed] [Google Scholar]
- Zhang J, Kobert K, Flouri T, and Stamatakis A (2014). PEAR: a fast and accurate Illumina paired-end read merger. Bioinformatics 30, 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the 2CIMPL datasets generated during this study are Sequence Read Archive: PRJNA546584 (SRR9204625, SRR9204626, SRR9204628, and SRR9204629). The accession number for the HITS-CLIP dataset is Sequence Read Archive: PRJNA546584 (SRR9204627). A detailed data analysis pipeline can be found in the Github repository at https://github.com/NaraLee-Lab/2CIMPL.