

SUMMARY
STING gain-of-function causes autoimmunity and immunodeficiency in mice and STING-associated vasculopathy with onset in infancy (SAVI) in humans. Here, we report that STING gain-of-function in mice prevents development of lymph nodes and Peyer’s patches. We show that the absence of secondary lymphoid organs is associated with diminished numbers of innate lymphoid cells (ILCs), including lymphoid tissue inducer (LTi) cells. Although wild-type (WT) α4β7+ progenitors differentiate efficiently into LTi cells, STING gain-of-function progenitors do not. Furthermore, STING gain-of-function impairs development of all types of ILCs. Patients with STING gain-of-function mutations have fewer ILCs, although they still have lymph nodes. In mice, expression of the STING mutant in RORγT-positive lineages prevents development of lymph nodes and reduces numbers of LTi cells. RORγT lineage-specific expression of STING gain-of-function also causes lung disease. Since RORγT is expressed exclusively in LTi cells during fetal development, our findings suggest that STING gain-of-function prevents lymph node organogenesis by reducing LTi cell numbers in mice.
In Brief
Bennion et al. report that a STING gain-of-function mutation prevents the development of lymph nodes and ILCs in mice. Humans with this mutation also have fewer ILCs. In mice, expression of STING gain-of-function in lymphoid tissue inducer (LTi) cells is sufficient to prevent development of lymph nodes.
Graphical Abstract
INTRODUCTION
Stimulator of interferon genes (STING) is a cytosolic sensor of cyclic dinucleotides that are produced by the host (e.g., cGAMP) or bacteria (e.g., c-di-GMP, c-di-AMP, cGAMP) (Ablasser et al., 2013; Burdette et al., 2011; Sun et al., 2013; Whiteley et al., 2019). Gain-of-function mutations in STING cause a systemic autoinflammatory disease known as STING-associated vasculopathy with onset in infancy (SAVI) (Liu et al., 2014). We previously generated heterozygous STING N153S mice that have a SAVI-associated mutation (Warner et al., 2017). STING N153S mice can only be studied as heterozygous animals since homozygous expression of STING N153S causes early embryonic lethality (Warner et al., 2017). Similar to humans with SAVI, heterozygous STING N153S mice develop systemic inflammation and lung disease as well as T cell cytopenia (Luksch et al., 2019; Warner et al., 2017). However, unlike humans with SAVI, STING N153S mutant mice develop severe combined immunodeficiency (Bennion et al., 2019). The mechanisms of immunodeficiency associated with STING gain-of-function are incompletely understood.
During infection with γ-herpesvirus-68 (γHV68), heterozygous STING N153S mice fail to adequately generate antigen-specific CD8+ T cells and virus-specific immunoglobulin G (IgG) (Bennion et al., 2019). Indeed, STING N153S animals exhibit greater viral burden than Rag1−/− animals, which completely lack B cells and T cells (Bennion et al., 2019). In addition to defects in adaptive immunity, STING N153S causes an innate immunodeficiency (Bennion et al., 2019). Although STING gain-of-function has previously been studied in T cells and myeloid cells, the impact of constitutive STING signaling in innate lymphoid cells is less well defined.
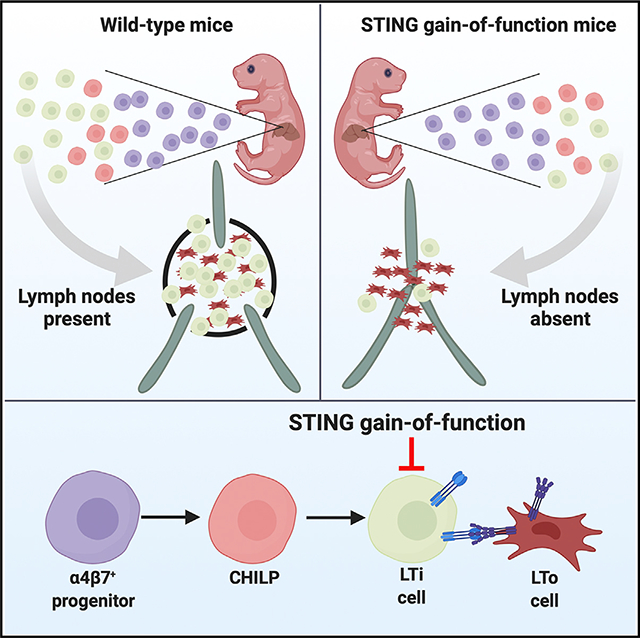
Here, we report that the STING N153S gain-of-function mutation prevents the development of lymph nodes (LNs) and Peyer’s patches in mice. This developmental defect is associated with reduced numbers of all types of ILCs, including lymphoid tissue inducer (LTi) cells. Furthermore, α4β7+ progenitor cells from STING N153S mice lack the capacity to differentiate into LTi cells in an OP9 cell culture system. To define cell-type-specific effects of STING gain-of-function on LN development, we generated mice that express STING N153S in RORγT-positive lineages (e.g., LTi cells in the fetus and in ILC3s and T cells in the adult). Like global STING N153S knock-in mice, these cell-type-specific transgenic mice lack LNs, have reduced numbers of mature LTi cells, and develop autoimmune lung disease. Thus, expression of STING N153S in RORγT-positive lineages prevents lymphoid tissue organogenesis in mice.
RESULTS
Absence of LNs and Peyer’s Patches in STING N153S Mice
We discovered that heterozygous STING N153S mice lack LNs and Peyer’s patches (Figure 1). Independently generated STING N153S mice, produced using a different guide RNA and DNA oligo donor (Luksch et al., 2019), also were found to lack LNs (data not shown). Additionally, mice with a neighboring gain-of-function mutation (STING V154M) were reported to lack LNs, although the severity of the defect and mechanism was not described (Bouis et al., 2019). Therefore, we began to quantitate LNs in heterozygous STING N153S and wild-type (WT) littermate control animals by performing subcutaneous injection of Evans Blue dye, which accumulates in draining lymphatics and LNs (Harrell et al., 2008). This confirmed the absence of visually apparent LNs (Figures 1A and 1B). Serial sectioning of inguinal fat pads (Figure 1C) and the small intestine (Figure 1D), followed by hematoxylin and eosin (H&E) staining, did not reveal histological evidence of even rudimentary LNs or Peyer’s patches. We quantitated the numbers of cervical, inguinal, axillary, brachial, and mesenteric LNs and found a small mesenteric or inguinal LN in only ~10% of animals (Figure 1E). In addition to our histological assessment of the intestine (Figure 1D), there were no visible Peyer’s patches in STING N153S mice (Figure 1F). Since Peyer’s patches are an important site of IgA production (Craig and Cebra, 1971; Reboldi et al., 2016), we measured IgA levels in the serum and the stool of WT and STING N153S animals. We found that STING N153S mice had no detectable IgA in the serum or in stool, in contrast to co-housed WT littermate control mice that had normal levels of IgA (Figure 1G) (Fransen et al., 2015; Klein-Schneegans et al., 1989). Thus, the autosomal dominant STING N153S mutation interferes with LN and Peyer’s patch development in mice, and this likely contributes to STING-N153S-associated immunodeficiency (Bennion et al., 2019).
Figure 1. Absence of Lymph Nodes and Peyer’s Patches in STING N153S Mice.

(A and B) Representative photographs of WT (left panels) and STING N153S (right panels) animals 15 min after unilateral, subcutaneous footpad injection of Evans Blue dye. Inguinal lymph nodes (LNs) are shown in (A), and retroperitoneal LNs are shown in (B). Discernible LNs are marked by black arrows.
(C) Representative H&E staining of serial skin sections and inguinal fat pads of WT and STING N153S mice. Images are representative of 20 sections per mouse from three mice per genotype from two independent experiments. Scale bar: 200 μm.
(D) Representative H&E staining of serial sections from the small intestines of WT and STING N153S mice. n = 3 mice from two independent experiments. Scale bar: 200 μm.
(E and F) Total number of discernible cervical, inguinal, brachial, axillary, and mesenteric LNs (E) and Peyer’s patches (F). Data represent the mean of six STING N153S and six WT littermate control mice.
(G) IgA levels in the serum and stool of STING N153S and WT littermate animals were quantitated by ELISA. Data represent the mean of eight samples per genotype. Dashed line denotes the limit of detection.
(H–L) Flow cytometric analysis of intestinal leukocytes of 6-to-7-week-old STING N153S mice and WT littermate control animals. (H) Representative FACS dot plots of intestinal leukocytes, indicating the gating strategy for ILC2, ILC3, and LTi-like CCR6+ ILC3 populations. Numbers (red text) indicate the percent of CD45+Lin− cells in each gate (lineage markers: CD19, CD5, and CD3). Percent and total number of ILC1 and NK cells (NKp46+GATA3−RORγT−) (I), ILC2s (J), ILC3s (K), and LTi-like CCR6+ ILC3s (L).
Data represent the mean from n = 6 animals per genotype. All data were pooled from at least two independent experiments. Results were analyzed by Mann-Whitney U test. *p < 0.05; **p < 0.01; ***p < 0.001.
Rag1−/− mice have small LNs that can be difficult to visualize (Mombaerts et al., 1992), but Rag1−/− mouse LNs become easier to visualize following adoptive transfer of lymphocytes. Thus, if STING N153S mice had rudimentary LNs, they might become visible after adoptive transfer. After adoptive transfer of splenocytes, we confirmed that Rag1−/− LNs were indeed visible (Figures S1A and S1B). In contrast, we found that Rag1−/− STING N153S recipient mice still had no visually apparent LNs after adoptive transfer (Figures S1A and S1B). To determine whether the cyclic GMP-AMP synthase (cGAS)-STING pathway and subsequent signaling through the type I interferon (IFN) receptor (IFNAR1) is involved in this developmental defect, we crossed STING N153S animals to mice lacking cGAS, the IFN-regulatory factors 3 and 7 (IRF3 and IRF7), or IFNAR1. Similar to our prior observation of IFNAR1-independent lung disease (Luksch et al., 2019), none of these genetic knockouts were sufficient to rescue the LN or Peyer’s patch deficiency (Table S1). Thus, STING N153S disrupts LN and Peyer’s patch formation independently of cGAS and IFNAR1 signaling. This is consistent with what we know about spontaneous autoinflammatory disease pathogenesis in STING N153S animals, which occurs independently of cGAS, IFNAR1, IRF3, and IRF7 (Luksch et al., 2019; Warner et al., 2017). Others also have confirmed that disease in STING gain-of-function mice develops independently of IFNAR1 (Bouis et al., 2019; Motwani et al., 2019).
STING N153S Reduces Numbers of All Types of ILCs, Including LTi Cells
LN development requires LTi cells, which are a type of ILC3 (Sawa et al., 2010). To test whether STING N153S has an effect on ILCs, we quantitated intestinal ILCs in 6-to-7-week-old STING N153S mice and WT littermate control animals (Figure 1H). In the adult small intestine, we observed a reduced frequency of ILC1s and natural killer (NK) cells, as well as ILC3s, and an increased frequency of ILC2s (Figures 1I–1K). Upon quantitation of absolute numbers of ILCs, all types of ILCs were reduced in the STING N153S small intestine (~38-fold fewer ILC1s and NK cells, ~2.9-fold fewer ILC2s, and ~28-fold fewer ILC3s than in WT; p < 0.005 for ILC2s and p < 0.0001 for ILC1s and ILC3s) (Figures 1I–1K). Thus, STING N153S has a large effect on development and/or survival of all three groups of ILCs in the adult small intestine. LTi-like ILC3s, defined by expression of CCR6 (Klose et al., 2013; Rankin et al., 2013; Sawa et al., 2010; Vonarbourg et al., 2010), exhibited a ~41-fold reduction in STING N153S mice compared to WT littermates (Figure 1L). In the STING N153S spleen, ILC numbers also were globally reduced. To distinguish between NK cells and ILC1s, we performed EOMES staining, which confirmed that there were fewer ILC1s (1.7-fold reduction, p < 0.05) and NK cells in STING N153S mice (30.2-fold reduction, p < 0.0001) (Figures S1C–S1E).
We hypothesized that STING N153S may result in a deficiency of committed ILC progenitors, leading to reduced numbers of mature ILCs. However, STING N153S had no effect on the number of alpha-lymphoid progenitor (αLP) cells, which are precursors to all types of ILCs. Furthermore, there was no difference in the numbers of common helper-like innate lymphoid cell progenitors (CHILPs) 1 or CHILP2 cells (Figures S1F–S1J), which are precursors to specific subsets of ILCs (Constantinides et al., 2014; Klose et al., 2014). NK cells have distinct progenitors known as refined NK-cell progenitors (Fathman et al., 2011). STING N153S diminished the numbers of refined NK-cell progenitors (3.7-fold reduction, p < 0.001) in the bone marrow of STING N153S animals (Figures S1K–S1M). Thus, STING N153S reduces the number of refined NK-cell progenitors, but not other ILC progenitor cells in the bone marrow. Collectively, these results suggest that STING N153S preferentially impacts mature ILCs.
STING N153S Mouse Spleens Have B Cell Zones and Express Chemokines Known to Regulate Splenic Organization
STING N153S mice have splenomegaly and exhibit histological abnormalities of the spleen (Luksch et al., 2019; Warner et al., 2017). To more carefully evaluate the spleens of STING S153S animals, we performed histological staining and flow cytometry. Consistent with prior reports (Luksch et al., 2019; Warner et al., 2017), histological analysis of STING N153S and WT littermate spleens revealed disorganized architecture (Figure S2A). However, unlike IκBα mutant mice that lack LNs (Mooster et al., 2015), we found that the mucosal addressin cell adhesion molecule (MADCAM-1)-expressing marginal sinus remained intact in the STING N153S spleen (Figure S2B). Immunofluorescent staining of CD3 revealed that T cell zones were absent in STING N153S spleens (Figure S2C), a finding that may reflect a large reduction in T cell numbers (Warner et al., 2017). Flow cytometric analysis of STING N153S and WT littermate splenocytes revealed no difference in the number of follicular dendritic cells (FDCs), a 5-fold increase in fibroblastic reticular cells (FRCs) in STING N153S spleens, and no difference in the numbers of WT and STING N153S endothelial cells (Figures S2D–S2G). Furthermore, we confirmed expression of the chemokines CCL19 and CCL21, which organize T cell zones (Dieu et al., 1998; Gunn et al., 1998; Luther et al., 2000), as well as the B-cell-attracting chemokine CXCL13, which is produced by FDCs (Figure S2H) (Cyster et al., 2000). Thus, STING N153S does not create a deficiency of splenic FDCs or FRCs, which regulate lymphoid tissue organization.
STING N153S Mice Have Fewer LTi Cells in Fetal Tissues
Because adult STING N153S mice have fewer ILCs and LTi-like cells, we hypothesized that STING N153S fetuses would also have fewer LTi cells, which are required for LN organogenesis. Indeed, in STING N153S fetuses, we observed a reduction in the percent and number of gut LTi cells (~4.5-fold reduction, p < 0.0001) (Figures 2A and 2B). CD4 is used as a surrogate marker for LTi cells in embryonic LN anlagen, since most CD4+ cells in developing LNs represent CD3−RORγT+ LTi cells (Eberl et al., 2004; Kelly and Scollay, 1992). Histological analysis of cervical LN anlagen from embryonic day 18.5 (E18.5) STING N153S and WT littermate fetuses revealed diminished CD4 staining intensity, suggesting a reduced accumulation of LTi cells (Figures 2C and 2D). There was no difference in the intensity of VCAM-1 staining, a marker of lymphoid tissue organizer (LTo) cells, or in the two-dimensional size of the LN anlagen (Figures 2E and 2F). Thus, the STING N153S mutation reduces the accumulation of LTi cells in the fetal gut and LN anlagen.
Figure 2. STING N153S Reduces Numbers of LTi Cells in Fetal Tissues as Well as Their Accumulation in Developing LN Anlagen.

In (A) and (B), leukocytes were harvested from the fetal gut on E16.5–E18.5 and analyzed by flow cytometry. In (C)–(F), frozen sections of cervical LN anlagen of E18.5 fetuses were analyzed by widefield fluorescence microscopy.
(A) Representative FACS dot plots of Lin−CD45+CD127+cKIT+ cells isolated from the fetal gut. Numbers indicate the percent of events in each gate.
(B) Total number α4β7+RORγT+ LTi cells in the fetal gut on E16.5–E18.5. Data represent the mean of n = 20–23 fetuses per genotype pooled from three independent experiments.
(C) Cervical LN anlagen sections were stained with DAPI and with antibodies against VCAM-1 (left panel) and CD4 (middle panel). Scale bar: 40 μm.
(D) Quantitation of CD4 staining intensity relative to the size of LN anlagen, defined as the total area of VCAM-1 and CD4 staining.
(E) Quantitation of VCAM-1 staining intensity relative to the size of LN anlagen.
(F) Quantitation of the size of LN anlagen, based on merged VCAM-1 and CD4 staining of WT and STING N153S cervical LN anlagen on E18.5.
Quantitation is from n = 7–9 cervical LN anlagen per genotype from two independent experiments. FACS data were analyzed by unpaired t test, and immunofluorescence data were analyzed by Mann-Whitney U test. *p < 0.05; ****p < 0.0001.
LTi cells activate noncanonical NF-κB signaling in LTo cells by engaging the lymphotoxin-β receptor (LTβR), leading to signaling events required for LN development (Mooster et al., 2015; Onder et al., 2013; Rennert et al., 1998). Therefore, we set out to test whether non-canonical NF-κB signaling downstream of the LTβR is intact in STING N153S cells. Mouse embryonic fibroblasts (MEFs) have previously been used to study LTβR signaling (Dejardin et al., 2002; Mooster et al., 2015), so we generated MEFs and treated them with a monoclonal antibody (mAb) agonist of LTβR or isotype control mAb, as described previously (Dejardin et al., 2002). SDS-PAGE and western blot of IκBα and RelB revealed no differences in NF-κB activation downstream of LTβR Figure S2I). Treatment of WT and STING N153S MEFs with the LTβR agonist antibody caused a similar upregulation of genes that contribute to lymphoid tissue organogenesis, including Vcam1, Icam1, Madcam1, and Cxcl10 (Figures S2J–S2M), which are upregulated downstream of LTβR signaling (Caposio et al., 2007; Dejardin et al., 2002; Hoffmann et al., 2003; Mooster et al., 2015). Thus, LTβR-mediated noncanonical NF-κB signaling remains intact in STING N153S MEFs, suggesting that LTβR signaling is not dysregulated by the mutation.
Single-Cell RNA Sequencing of α4β7+ Cells from the Fetal Liver Suggests Fewer Mature LTi Cells in STING N153S Fetuses
To confirm that α4β7+ cells from WT and STING N153S fetal livers (Figure 3A) express STING as well as transcription factors known to impact ILC differentiation, we performed fluorescence-activated cell sorting sequencing (FACS-seq), a type of single-cell RNA sequencing. We detected STING (Tmem173) gene expression in at least a subset of α4β7+ cells (Figure 3B) and additionally confirmed that there was no difference in expression of Id2 and Tox, transcription factors required for LN development (Figures 3C and 3D) (Aliahmad et al., 2010; Cupedo and Mebius, 2005). However, α4β7+ cells from STING N153S fetuses expressed higher levels of Tcf7, a gene highly expressed in early lymphoid progenitors (Figure 3E) (Yang et al., 2015). This may suggest that there are fewer mature α4β7+ cells in STING N153S fetal livers than in the WT control livers. Using flow cytometry, we confirmed similar expression levels of ID2, TOX, and NFIL3, all of which are involved in α4β7+ progenitor cell differentiation into mature LTi cells (Figures 3F–3I) (Aliahmad et al., 2010; Cherrier et al., 2012; Geiger et al., 2014; Yokota et al., 1999). Additionally, FACS-seq did not reveal differences in the expression of type-I-IFN-stimulated genes (Figure S3A). Finally, no other genes known to be involved in LTi cell differentiation and function were appreciably affected (Figure S3B) (Chea et al., 2016). Thus, our FACS-seq and flow cytometric analyses confirmed that STING is expressed in at least a subset of α4β7+ cells, but without impacting the expression of most key transcription factors involved in LN and ILC development.
Figure 3. Gene Expression and Transcription Factor Analysis of STING N153S α4β7+ Fetal Liver Progenitor Cells.

In (A)–(H), fetal livers were harvested on E14.5 from WT and heterozygous STING N153S animals.
(A) Illustration depicting α4β7+ fetal liver cells and their key transcription factors.
(B–E) α4β7+ cells were single-cell sorted by FACS into 96 well plates, followed by RNA sequencing (FACS-seq). Violin plots of FACS-seq results demonstrating expression levels of Tmem173 (STING) (B), Id2 (C), Tox (D), and Tcf7 (E) in α4β7+ progenitor cells. Data represent the mean number of counts per gene from 48 cells per genotype performed as a screening experiment.
(F) Total number of α4β7+ cells (Lin−CD45+cKITintCD127+α4β7+) in WT and STING N153S fetal livers.
(G–I) Mean fluorescence intensity (MFI) of the transcription factors ID2 (G), TOX (H), and NFIL3 (I) in α4β7+ cells from the fetal livers of WT and STING N153S mice.
Data in (E)–(H) represent the mean of n = 20–24 (F), n = 13 (G), n = 6–9 (H), and n = 5–11 (I). Data in (F)–(I) were pooled from at least two independent experiments and analyzed by unpaired t test. *p < 0.05; **p < 0.01.
STING N153S Impairs Generation of α4β7+ Progenitors into LTi Cells and ILCs
Increased expression of Tcf7 may reflect the differentiation status of α4β7+ cells in the fetal liver (Yang et al., 2015), consistent with what we observed in the fetal gut (Figure 2B). To begin to determine whether STING N153S impacts the differentiation of ILCs, we assessed the capacity of WT and STING N153S α4β7+ cells to differentiate into mature ILCs cells for 6 or 14 days in an OP9 stromal cell culture system in the presence of interleukin-7 (IL-7) and stem cell factor (SCF) (Cherrier et al., 2012). On day 6, only ~5% of Lin− (CD19−CD3−) STING N153S cells were LTi cells, compared with ~50% in the WT cell culture (Figures 4A, top panels, and 4B). We also detected a higher percentage of NK cells and ILC2s in STING N153S cell cultures compared with WT controls (Figures 4A, bottom panels, and 4B). Cell-intrinsic nucleic acid sensing may direct T cells into a T helper 2-type lineage (Imanishi et al., 2014), and an increase in ILC2 and NK cell frequency may suggest that STING gain-of-function biases ILC progenitors into helper ILC lineages. On day 6, we observed ~6.5-fold fewer STING N153S ILC1s and ~2.6-fold fewer ILC2s compared with WT controls. However, there was a much larger effect on the number of ILC3s (~21-fold) and LTi cells (~145-fold) in STING N153S cultures (Figures 4C–4G). Since STING N153S impairs the differentiation of multiple ILC subsets after 6 days in the OP9 stromal cell system, we reasoned that a longer experiment (14 days) may elucidate whether this reflects a delay in differentiation. In contrast to what was observed on day 6, we found no difference in the numbers of WT and STING N153S ILC1s and ILC2s on day 14. However, the numbers of ILC3s and LTi cells were still reduced in STING N153S cultures compared with WT controls (~10-fold and ~8.6-fold, p < 0.0001 and p < 0.001) (Figure S4). Thus, STING N153S delays the differentiation of ILC1s, ILC2s, ILC3s, and LTi cells in a cell culture system but has its largest effect on ILC3s and LTi cells (Figure 4H). This result is consistent with our in vivo findings demonstrating a preferential effect of STING N153S on ILC3s and LTi cells in mice (Figures 1H–1L and 4H).
Figure 4. STING N153S Fetal Liver α4β7+ Progenitor Cells Do Not Differentiate into LTi Cells after 6 Days in an OP9 Cell Culture System.

α4β7+ progenitor cells from the fetal liver were co-cultured with OP9 stromal cells, SCF, and IL-7. Cells were allowed to differentiate for 6 days and analyzed by FACS.
(A) Representative FACS plots of adult WT (left panels) and STING N153S (right panels) CD45+CD3—CD19− cells. Cell frequencies within each gate are denoted in red, and cell population names are labeled in blue.
(B) Average frequencies of ILC and α4β7+ cell populations as a fraction of total CD45+CD3−CD19− cells.
(C–G) Percent and number of ILC1 (C), NK cells (D), ILC2 (E), ILC3 (F), and LTi-like cells (G).
(H) Graphical summary of the fold reduction in numbers of ILCs by STING N153S compared to WT.
Data represent the mean of 6–10 replicates per group pooled from at least two independent experiments. Results were analyzed by unpaired t test. *p < 0.05; **p < 0.01; ****p < 0.0001.
STING gain-of-function mutations cause apoptosis of T cells (Gulen et al., 2017; Wu et al., 2019), so we hypothesized that STING N153S may similarly cause apoptosis of ILCs. However, we observed no difference in the percent or number of annexin V-positive or annexin V-negative α4β7+CD127+ cells, which represent a mixture of mature LTi and LTi progenitor cells (Figures S5A–S5C). STING gain-of-function mutations also reduce the proliferative capacity of T cells (Cerboni et al., 2017), but bromodeoxyuridine (BrdU) labeling in vivo did not reveal any differences in the percent or number of proliferating LTi cells (Figure S5D). We reasoned that dead or dying cells may be difficult to detect in vivo, so we also tested for apoptosis of ILCs in the OP9 co-culture system. We observed a small increase in the frequency of total dead cells in the STING N153S culture when compared to WT samples (Figures S5E–S5G). Thus, STING N153S may produce subtle effects on apoptosis of ILCs, although we only detected this effect in cell culture and not in cells freshly isolated from mice.
Human SAVI Patients Have Diminished Frequencies of ILCs in Peripheral Blood
To test whether STING gain-of-function affects ILCs in humans with SAVI, we performed flow cytometric analysis of peripheral blood mononuclear cells (PBMCs). In healthy control subjects, circulating ILCs represent approximately 0.1% of total CD45+ cells; are myeloid-, T-, B-, and NK-cell lineage negative; and express CD7 and CD127 (IL-7R) (Hazenberg and Spits, 2014; Lim et al., 2017). The two main peripheral blood ILC subsets include CRTH2+ ILC2s, some of which express CD117, and ILC precursors (ILCPs) that express CD117 but not CRTH2 (Lim et al., 2017; Mjösberg et al., 2011). A minor subset of CD117−CRTH2− ILCs (also referred to as ILC1) has been reported (Roan et al., 2016; Spits et al., 2016), although the identity of this rare subset is the subject of ongoing study (Bernink et al., 2017; Simoni et al., 2017). To determine whether humans with STING gain-of-function mutations have an ILC deficiency, we performed FACS analysis on healthy donor and SAVI patient PBMCs. Five unique SAVI patient samples with either the N154S, V155M, or R281Q mutations were analyzed. Four patients were being treated with a JAK1/2 inhibitor, and one was being treated with mycophenolate mofetil (MMF) (Figure 5A). Obtaining samples from patients with untreated disease was not possible for ethical reasons. Frequencies of Lin−CD7+ cells were similar in healthy control and JAK-inhibitor-treated SAVI patients (Figure 5B). However, patients with STING gain-of-function exhibited a ~3-fold reduction in the frequency of circulating total ILCs and ILC2s (Figures 5C–5E). Thus, STING gain-of-function reduces the numbers of circulating ILCs, consistent with a prior report that numbers of circulating NK cells are reduced in patients with STING gain-of-function mutations (Liu et al., 2014). Since SAVI patients have LNs, which we confirmed histologically (Figures S6A and S6B), our results indicate that the effects on lymphoid organogenesis only occur in mice. This may suggest species-specific differences in the effects of STING gain-of-function in LTi cells. Studies of LTi cells in this rare patient population were not possible.
Figure 5. Quantitation of ILC and ILC Progenitor Cells in the Blood of Human SAVI Patients.

(A) Gating strategy and representative FACS plots of peripheral blood ILCP and ILC2 populations gated on the CD45+ live lymphocytes.
(B–E) Percent of total ILC & NK cells (Lin−CD7+) (B), total ILCs (CD56−CD127+CD94−NKG2A−CD16−CD2−) (C), ILCP (CD117+CRTH2−) (D), and ILC2 (CD117intCRTH2+) (E) as a frequency of CD45 live lymphocytes. Black dots (12 donors, left columns) denote healthy donors, purple dots (4 patients, middle columns) denote SAVI patients on JAK inhibitors (ruxolitinib), and yellow dots (1 patient, right columns) denote a SAVI patient on mycophenolate mofetil (MMF). Results were analyzed using unpaired t test. **p < 0.01.
Cell-Type-Specific Expression of STING Gain-of-Function in RORγT+ Lineages Causes T Cell Cytopenia and Lung Disease
Next, we set out to examine whether the expression of STING N153S in mature LTi cells might be sufficient to reduce the number of LTi cells and prevent LN development in mice. We generated transgenic LoxP-STOP-LoxP STING N153S (floxed-STOP STING N153S) mice where Cre-mediated recombination leads to expression of the STING N153S mutant (Figure 6A). RORγT is expressed specifically in LTi cells in the developing fetus (Sun et al., 2000). In adult mice, RORγT is expressed in LTi-like cells, ILC3s, Th17 cells, and T cells at the double-positive (DP) stage of development (Sawa et al., 2010; Sun et al., 2000; Takatori et al., 2009). We crossed the floxed-stop STING N153S mice to transgenic RORγT-Cre animals and confirmed excision of the STOP cassette in DP but not double-negative (DN) thymocytes (Figure 6B). We previously found that αβ T cells drive STING-N153S-associated lung disease (Luksch et al., 2019), but we did not examine whether the expression of the mutant in T cells was sufficient to cause disease. Here, we found that STING N153S expression induced by RORγT-Cre was sufficient to cause perivascular lung inflammation in 13-week-old mice (Figure 6C), suggesting that the expression of STING N153S in T cells may be necessary and sufficient for certain features of disease in STING N153S mice. Flow cytometric analysis of thymocytes from 3-to-4-week-old RORγT-Cre+ floxed-stop STING N153S mice and Cre-negative littermate control animals revealed a reduced frequency of single-positive T cells, but not DN or DP T cells, in RORγT-Cre-positive mice (Figure 6D). STING N153S expression resulted in an increase in the percent of DP thymocytes but without affecting DN thymocytes (Figures 6E, 6F, and S7A–S7D). This likely reflects a block in T cell differentiation caused by the expression of STING N153S at the DP stage (Figure 6F). Indeed, there was a corresponding reduction in the percent and number of both CD4+ and CD8+ single-positive T cells expressing the STING N153S mutant (~7-fold reduction in CD4+ T cells, p < 0.001; ~2-fold reduction in CD8+ T cells, p < 0.01) (Figures 6G and 6H). Thus, STING N153S expression at the DP stage causes a reduction in the number of single-positive thymocytes without impacting the numbers of DN thymocytes. Flow cytometric analysis of splenocytes confirmed an effect of cell-type-specific expression of STING N153S on numbers of T cells but not B cells or myeloid cells (Figures 6I–6L and S7E–S7J).
Figure 6. RORγT-Cre-Mediated Cell-Type-Specific Expression of STING N153S Diminishes Numbers of Single-Positive T Cells in the Thymus and Spleen.

(A) Schematic of the floxed-STOP STING N153S construct. Mice with Cre-dependent expression of the STING N153S gene in the ROSA locus (floxed-stop STING N153S mice) were generated using transcription activator-like effector nucleases (TALENs).
(B) PCR amplification of transcriptional stop containing ROSA homology arms from double-negative (DN; CD8−CD4−) and double-positive (DP; CD8+CD4+) thymocyte DNA from an adult RORγT-Cre+ floxed-STOP STING N153S mouse.
(C) Images of H&E staining on lungs of 13-week-old RORγT-Cre+ floxed-STOP STING N153S mice (right panel) and floxed-STOP STING N153S control animals (left panel). n = 3 mice per genotype.
(D–H) Thymocytes from 3-to-4-week-old RORγT-Cre+ floxed-STOP STING N153S mice and floxed-STOP STING N153S control animals were harvested, and single-cell suspensions were prepared for FACS analysis. (D) Representative FACS plots of CD4 and CD8 expression gated on CD45+CD19—NK1.1− cells. Percent and number of DN (E), DP (F), CD4+ (G), and CD8+ (H) cells. Data represent the mean of five mice per group pooled from two independent experiments. (I–L) Splenocytes from 3-to-4-week-old RORγT-Cre-positive floxed-stop STING N153S mice and floxed-stop STING N153S control animals were harvested, and single-cell suspensions were prepared for FACS analysis. Percent and number of CD3+ (I), CD4+ (J), CD8+ (K), and NK1.1+ (L) cells.
Data represent the mean of 10 mice per group pooled from at least two independent experiments. Results were analyzed by unpaired t test. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
STING N153S Expression in RORγT+ Fetal LTi Cells Is Sufficient to Interfere with LN and Peyer’s Patch Development
RORγT expression during fetal development is restricted to LTi cells (Sun et al., 2000), which act in concert with LTo cells to initiate LN and Peyer’s patch organogenesis (van de Pavert and Mebius, 2010). We found that expression of STING N153S, specifically in RORγT+ LTi cells, was sufficient to prevent LN and Peyer’s patch development based on visual assessment and Evans blue staining of LNs (Figures 7A and 7B). However, we still observed very small mesenteric LNs and occasionally a single unilateral inguinal, brachial, or axillary LN in some animals that express the STING N153S mutant in LTi cells (Figure 7B). The sporadic presence of a residual LN despite the expression of STING N153S in LTi cells might suggest additional effects of STING N153S on nonhematopoietic cells. Alternatively, sporadic residual LNs might result from the incomplete excision of the floxed-STOP by Cre, which would be expected to occur in a small subset of LTi cells.
Figure 7. Expression of STING N153S in LTi Cells Disrupts LN Development.

(A) Representative photographs of inguinal LNs after Evans Blue dye injection into the foot pad of Cre-negative floxed-STOP STING N153S (left panel) or RORγT-Cre-positive floxed-stop STING N153S (right panel) animals.
(B) Quantitation of LNs and Peyer’s patches from floxed-stop STING N153S and RORγT-Cre+ floxed-STOP STING N153S mice. n = 13–14.
(C–M) Leukocytes were harvested from the small intestines of 3-to-4-week-old RORγT-Cre+ and floxed-stop STING N153S animals and then analyzed by flow cytometry. Percent (C) and total number (D) of Lin−CD45+ cell (lineage markers: CD3−CD5−CD19−). Representative FACS plots (E) of GATA3+ (ILC2), RORγT+ (ILC3), and GATA3−RORγT− populations gated on Lin−CD45+ cells from floxed-STOP STING N153S (left panel) and RORγT-Cre+ floxed-STOP STING N153S (right panel) animals. Red text denotes percent of cells in each gate, and blue text denotes the cell population. Percent and total number (respectively) of ILC3s (F and G), LTi-like cells (H and I), NKp46+ ILC1 and NK cells (J and K), and ILC2 cells (L and M).
Data represent the mean from 8–9 mice per genotype. All data were pooled from at least two independent experiments. Results were analyzed by unpaired t test. ***p < 0.001, ****p < 0.0001.
Postnatally, RORγT is expressed in mature ILC3s, including LTi-like cells (Sawa et al., 2010; Sun et al., 2000; Takatori et al., 2009). In the adult gut, we found that there was a similar percentage and number of CD45+ cells in WT and STING N153S animals. Cell-type-specific expression of STING N153S in RORγT-positive lineages caused a ~95-fold reduction in the number of ILC3s and a ~91-fold reduction in the number of LTi-like cells (Figures 7C–7I). There also was a corresponding increase in the percent of ILC1s and ILC2s, but no difference in the total number of these cell types (Figures 7J–7M). Collectively, these results demonstrate that the expression of STING N153S in DP T cells, ILC3s, and LTi-like cells is sufficient to reduce their numbers. Furthermore, our results suggest that STING gain-of-function signaling can impact the differentiation of progenitor cells as well as the lifespan of mature T cells and ILCs. Finally, expression of STING N153S in fetal LTi cells is sufficient to prevent the development of LNs and Peyer’s patches, revealing a deleterious role of STING gain-of-function during lymphoid tissue organogenesis in mice.
DISCUSSION
We discovered that the STING N153S gain-of-function mutation disrupts LN and Peyer’s patch organogenesis and interferes with LTi cell differentiation. Furthermore, we demonstrated that expression of STING N153S in RORγT-positive lineages is sufficient to interfere with the development of LNs and Peyer’s patches. Thus, STING gain-of-function dysregulates lymphoid tissue organogenesis in mice by interfering with the development of LTi cells and by reducing the numbers of mature LTi cells.
An effect of STING gain-of-function in lymphoid tissue organogenesis was unexpected, especially since other pattern recognition receptors have not been implicated in lymphoid tissue organogenesis. Furthermore, the impact of STING signaling in ILCs is incompletely understood (Canesso et al., 2018; Marcus et al., 2018). Since CRISPR/Cas9 can sometimes produce off-target effects (Cradick et al., 2013; Frock et al., 2015; Fu et al., 2013; Hsu et al., 2013; Pattanayak et al., 2013; Wang et al., 2015), one potential question regarding the LN deficiency phenotype is whether a second mutation due to an off-target effect may be responsible for this particular defect. However, we corroborated the LN deficiency phenotype in independently generated STING N153S mice, and a similar defect was observed but not studied in another STING gain-of-function mouse that has a mutation in the neighboring amino acid (STING V154M mice) (Bouis et al., 2019). LN deficiency also occurred in our heterozygous transgenic mice that express the STING N153S cDNA from the ROSA locus. It is unlikely that an off-target mutation elsewhere in the genome could explain the universal absence of LNs and Peyer’s patches in four independently generated heterozygous mouse models.
We previously found that STING N153S is sufficient to cause an immunodeficiency during viral infection that is more severe than that of STING goldenticket mice, which lack functional STING signaling. The immunodeficiency of STING N153S mice is also more severe than that of Rag1−/− animals, which lack adaptive immunity (Bennion et al., 2019). Severe immunodeficiency in STING N153S mice distinguishes the animal model from the disease associated with the analogous STING N154S mutation in humans. In contrast to what we observed in mice, patients with the STING N154S mutation have LNs (Liu et al., 2014). In mice, the deficiency of ILCs, LNs, and Peyer’s patches likely contributes to the severe immunodeficiency phenotype, including IgA deficiency and a failure to adequately produce virus-specific IgG. We previously found that STING N153S dysregulates virus-specific CD8+ T cell responses after intranasal inoculation with γHV68, leading to reduced gHV68-specific CD8+ T cell responses in the lung. This might result from diminished antigen presentation as a consequence of LN deficiency (Banchereau and Steinman, 1998). Finally, since CD4+ T cells in the spleen help B cells to undergo antibody class switching (Victora and Nussenzweig, 2012), the absence of T cell zones in the spleen may explain the impaired ability of STING N153S animals to produce virus-specific IgG (Bennion et al., 2019).
Development of LNs requires LTi cells, which represent a subset of ILC3s (Artis and Spits, 2015; Eberl et al., 2004). We found that there were fewer CD4+ LTi cells in STING N153S fetuses compared to WT littermate fetuses, and this defect corresponded with reduced CD4 staining in LN anlagen on E18.5. Although RORγT lineage-restricted expression of STING N153S blocked LN development, we cannot exclude contributions of STING N153S expression in other cell types. For example, STING is also highly expressed in lymphatic endothelial cells and may be expressed in LTo stromal cells (Heng et al., 2008).
An open question is whether alterations in the quantity or quality of LTi cells may explain the LN deficiency in STING N153S mice. LN anlagen develops as LTα on LTi cells ligates the LTβR on stromal LTo cells (Fütterer et al., 1998; Rennert et al., 1996; van de Pavert and Mebius, 2010). LTβR stimulation induces noncanonical NF-κB signaling, which upregulates adhesion molecules and chemokines to recruit more LTi cells as well as T and B cells to the developing LN (Cupedo and Mebius, 2005; Dejardin et al., 2002; Ngo et al., 1999). Studies of LN development in mice deficient in noncanonical NF-κB signaling have suggested that LTi cell accumulation must exceed a threshold level of cells for LN anlagen to develop and persist postnatally (Kim et al., 2000; Onder et al., 2013). Our results are consistent with the hypothesis that diminished numbers of LTi cells, below a threshold, can cause LN deficiency. However, mice deficient for the transcription factor Nfil3 also have reduced numbers of LTi cells in the fetus, although Nfil3−/− mice still develop LNs (Xu et al., 2015). This may indicate that LTi cells are a heterogenous population with only a subset of cells contributing to LN development. In STING N153S mice, an alternative explanation might be that the LTi cells, although reduced in number, also are somehow unfit in their capacity to promote LN development. An answer to this question will likely require more extensive mechanistic phenotyping and subset analysis of LTi cells.
We found that SAVI patients have lower frequencies of circulating ILCs in the blood. However, humans with STING gain-of functions mutations have LNs (Liu et al., 2014). These species-specific differences may reflect STING expression levels in ILC subsets or, alternatively, differential effects of the STING mutant in human and mouse cells. To better explain species-specific effects of STING gain-of-function on LN development, we would have liked to assess LTi-like cells in SAVI patient samples. However, this was not possible, in part because ILC3s and LTi cells are not readily detectable in circulation (Shikhagaie et al., 2017). Future studies examining LTi and ILC3 cells from tonsils or intestinal tissues from SAVI patients may help to further define species-specific effects on these cell types.
Although we uncovered an immunological mechanism of LN deficiency caused by STING gain-of-function, the molecular mechanisms that underlie STING-N153S-mediated impairment of ILCs remain elusive. STING gain-of-function mutations cause pro-apoptotic and anti-proliferative effects in T cells (Cerboni et al., 2017; Gui et al., 2019; Wu et al., 2019), and we observed subtle effects on apoptosis in a cell culture differentiation system of ILCs, but not in freshly isolated ILCs. If STING gain-of-function induces apoptosis in ILCs, the effect appears to be subtle. Alternatively, STING N153S may cause ILC cytopenia via an alternative mechanism that remains to be identified.
Our group and others continue to pursue the molecular and cellular effects of STING gain-of-function mutations (Cerboni et al., 2017; Gui et al., 2019; Wu et al., 2019). Definitive reversal of the physiological and immunological effects of STING gain-of-function, which are established as type I IFN independent in mice (Bouis et al., 2019; Luksch et al., 2019), will likely require characterization of pathways that are less well understood, as well as genetic approaches that rigorously confirm mechanism under physiological conditions. This ongoing work may lead to a greater understanding of WT and mutant STING biology, in addition to insights regarding fundamental mechanisms of LN development and ILC differentiation and survival in humans and in mice.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jonathan J. Miner (jonathan.miner@wustl.edu).
Materials Availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement. Commercially available reagents are indicated in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-CD4 | Biolegend | RRID:AB_312686 |
| Mouse anti-CD4 | ThermoFisher | RRID:AB_467063 |
| Mouse anti-CD45 | ThermoFisher | RRID:AB_467251 |
| Mouse anti-NKp46 | ThermoFisher | RRID:AB_1210743 |
| Mouse anti-CCR6 | BD Biosciences | RRID:AB_2738926 |
| Mouse anti-CD3 (biotin) | Biolegend | RRID:AB_2563946 |
| Mouse anti-CD3 | Biolegend | RRID:AB_893320 |
| Mouse anti-CD5 | Biolegend | RRID:AB_2563432 |
| Mouse anti-CD19 | Biolegend | RRID:AB_2259869 |
| Mouse anti-RORγt | ThermoFisher | RRID:AB_1834470 |
| Mouse anti-GATA3 | BD Biosciences | RRID:AB_1645302 |
| Mouse anti-CD127 (IL-7Rα) | Biolegend | RRID:AB_1937216 |
| Mouse anti-CD135 (FLT3) | Biolegend | RRID:AB_2562338 |
| Mouse anti-CD117 (cKit) | Biolegend | RRID:AB_1877215 |
| Mouse anti-CD25 | Biolegend | RRID:AB_961212 |
| Mouse anti-α4β7 | Biolegend | RRID:AB_10730607 |
| Mouse anti-α4β7 | Biolegend | RRID:AB_493267 |
| Mouse anti-Eomes | ThermoFisher | RRID:AB_2574062 |
| Mouse anti-Madcam1 | R&D Biosystems | RRID:AB_355772 |
| Mouse anti- CD11c | Biolegend | RRID:AB_2129642 |
| Mouse anti-Cd11b | Biolegend | RRID:AB_893233 |
| Mouse anti-TCRg/d | Biolegend | RRID:AB_10612572 |
| Mouse anti-CD244.2 | ThermoFisher | RRID:AB_657875 |
| Mouse anti-CD122 | ThermoFisher | RRID:AB_465836 |
| Mouse anti-PLZF | ThermoFisher | RRID:AB_2574445 |
| Mouse anti-ID2 | ThermoFisher | RRID:AB_2735056 |
| Mouse anti-CD27 | ThermoFisher | RRID:AB_2722949 |
| Mouse anti-CD90.2 | ThermoFisher | RRID:AB_2717157 |
| Mouse anti-Tox | ThermoFisher | RRID:AB_10853657 |
| Mouse anti-E4BP4 (NFIL3) | ThermoFisher | RRID:AB_11150965 |
| Mouse anti-B220 | Biolegend | RRID:AB_492875 |
| Mouse anti-VCAM1 | R&D Biosystems | RRID:AB_355499 |
| Mouse anti-CD31 | Biolegend | RRID:AB_2629682 |
| Mouse anti-CD21/35 | Biolegend | RRID:AB_10965544 |
| Mouse anti-CD54 | BD Biosciences | RRID:AB_394735 |
| Mouse anti-PDPN | ThermoFisher | RRID:AB_1106990 |
| Mouse anti-B220 (biotin) | Biolegend | RRID:AB_312989 |
| Mouse anti-Ter119 (biotin) | Biolegend | RRID:AB_313704 |
| Mouse anti-GR1 (biotin) | Biolegend | RRID:AB_313368 |
| Lymphotoxin beta receptor monoclonal antibody | ThermoFisher | RRID:AB_763451 |
| Mouse anti-RelB | Cell Signaling Technologies | RRID:AB_2179173 |
| Mouse anti-IκBα | Cell Signaling Technologies | RRID:AB_390781 |
| Goat anti-rabbit IgG (H+L) secondary antibody, HRP | ThermoFisher | RRID:AB_228341 |
| CD159a (NKG2A)-VioBright FITC, human antibody | Miltenyi Biotec | RRID:AB_2726450 |
| CD94-APC-Vio770, human antibody | Miltenyi Biotec | RRID:AB_2811494 |
| CD127 Monoclonal Antibody (eBioRDR5), PE-Cyanine7 | ThermoFisher | RRID:AB_1659672 |
| PE/Dazzle 594 anti-human CD294 (CRTH2) | BioLegend | RRID:AB_2572052 |
| Brilliant Violet 605 anti-human CD117 (c-kit) | BioLegend | RRID:AB_2562024 |
| Brilliant Violet 650 anti-human CD16 | BioLegend | RRID:AB_11125578 |
| Brilliant Violet 785 anti-human CD56 (NCAM) | BioLegend | RRID:AB_2566058 |
| CD7 antibody | BD Biosciences | RRID:AB_10898348 |
| CD2 antibody | BD Biosciences | RRID:AB_2744356 |
| CD3 antibody | BD Biosciences | RRID:AB_2744390 |
| Mouse Anti-Human CD5 Clone UCHT2 | BD Biosciences | RRID:AB_2714177 |
| CD14 antibody | BD Biosciences | RRID:AB_2744285 |
| BUV737 Mouse Anti-Human CD19 | BD Biosciences | RRID:AB_2716867 |
| CD45 antibody | BD Biosciences | RRID:AB_2744401 |
| Biological Samples | ||
| Human peripheral blood mononuclear cells | Bénédicte Neven, Necker-Enfants Malades Hospital | N/A |
| Human lymph nodes | Bénédicte Neven, Necker-Enfants Malades Hospital | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Evans Blue | Millipore Sigma | Cat# E2129 |
| Prolong Diamond Antifade Mountant with Dapi | Fischer | Cat# P36966 |
| RIPA buffer | Cell Signaling Technologies | Cat# 9806S |
| Halt Protease Inhibitor | ThermoFisher | Cat# 78429 |
| PierceTM ECL Substrate | ThermoFisher | Cat# 32106 |
| Collagenase IV | Sigma | Cat# C5138 |
| Intracellular Fixation and Permeabilization kit | ThermoFisher | Cat# 88-8824-00 |
| Recombinant murine stem cell factor (SCF) | Peprotech | Cat# 250-03 |
| Recombinant murine IL-7 | Peprotech | Cat# 217-17 |
| Recombinant murine FLT3-ligand | Peprotech | Cat# 250-31L |
| Promega RNasin Plus Rnase Inhibitor | Promega | Cat# N2611 |
| Annexin V | Biolegend | Cat# 640917 |
| Annexin V | BD Biosciences | Cat# 556421 |
| Critical Commercial Assays | ||
| Mouse IgA ELISA Kit | Immunology Consultants Laboratory, Inc | Cat# E90-A |
| FITC BrdU Flow Kit | BD Biosciences | Cat# 559619 |
| Deposited Data | ||
| Single-cell RNA FACS-seq on WT and STING N153S α4β7+ fetal liver (E14.5) cells | This paper | Mendeley DOI: https://dx.doi.org/10.17632/9nck2z26tf.1 |
| Experimental Models: Cell Lines | ||
| OP9 cells | ATCC | Cat# CRL-2749 |
| Experimental Models: Organisms/Strains | ||
| Mouse: CD45.1 mice (B6.SJL-Ptprca Pepcb/BoyJ) | Jackson Laboratory | Stock# 002014 |
| Mouse: STING N153S mice | Jackson Laboratory | Stock# 033543 |
| Mouse: Floxed-STOP STING N153S mice | This paper | N/A |
| Mouse: RORγt-Cre (B6.FVB-Tg(Rorc-cre)1Litt/J | Jackson Laboratory | Stock# 022791 |
| Mouse: Rag1−/− (B6.129S7-Rag1tm1Mom/J) | Jackson Laboratory | Stock# 002216 |
| Mouse: Ifnar1−/− | Hwang et al., 1995 | N/A |
| Mouse: cGas−/− (B6(C)-Cgastm1d(EUCOMM)Hmgu/J | Jackson Laboratory | Stock# 026554 |
| Mouse: Irf3−/− | Sato et al., 2000 | N/A |
| Mouse: Irf7−/− | Honda et al., 2005 | N/A |
| Oligonucleotides | ||
| CCL19 primers: 5′CAGACAGGCAGCAGTCTT-3′ & 5′-GTGGCCTGCCTCAGATTAT-3′ | Integrated DNA Technologies | N/A |
| CCL21 primers: 5′TTCTTCTGGCTGTACTTAAGGC-3′ & 5′-TGATGACTCTGAGCCTCCTTAG-3′ | Integrated DNA Technologies | N/A |
| Cxcl13 PrimeTime qPCR Assay | Integrated DNA Technologies | Assay ID: Mm.PT.58.31389616 |
| Vcam1 PrimeTime qPCR Assay | Integrated DNA Technologies | Assay ID: Mm.PT.58.9687546 |
| Icam1 PrimeTime qPCR Assay | Integrated DNA Technologies | Assay ID: Mm.PT.58.43714327 |
| Madcam1 PrimeTime qPCR Assay | Integrated DNA Technologies | Assay ID: Mm.PT.58.28611018 |
| Cxcl10 PrimeTime qPCR Assay | Integrated DNA Technologies | Assay ID: Mm.PT.58.43575827 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| GraphPad Prism 8 | GraphPad | https://www.graphpad.com |
| Other | ||
| EasySep Mouse Streptavidin RapidSpheres Isolation Kit | STEMCELL Technologies | Cat# 19860 |
Data and Code Availability
The dataset generated by the single-cell FACs-Sequencing of α4β7+ fetal liver (E14.5) progenitor cells during this study is available at Mendeley DOI: https://dx.doi.org/10.17632/9nck2z26tf.1
EXPERIMENTAL MODEL AND SUBJECT DETAILS
In vivo animal models
STING N153S mice were generated by our lab and published previously (Warner et al., 2017). All animals were housed in specific pathogen free facilities at Washington University in St. Louis. All STING N153S expressing animals were heterozygous and aged matched, co-housed littermate control animals were used for all experiments. Both sexes were used in all experiments and animals were randomly assigned to experimental groups. Floxed-STOP STING N153S mice were generated by and obtained from the Hope Center Transgenic Vectors Core at Washington University in Saint Louis. TALENs genome editing for the creation of transgenic mice has been described previously (Meyer et al., 2010). Briefly, to generate mice that conditionally expressed the STING N153S protein a targeting vector specific to the Rosa26 locus was assembled. This vector included a Rosa26 homology arm, a CAG promoter region, a transcriptional stop sequence flanked by loxp sequences, the STING N153S cDNA sequence, and a second Rosa26 homology arm. The targeting vector and TALENs were injected into C57BL/6J single-cell embryos obtained from superovulated C57BL/6J female mice mated to male C57BL/6J animals. Modified embryos were transferred into pseudo-pregnant female recipient mice. PCR assays and DNA sequencing confirmed targeted insertion of the vector into the Rosa26 locus. Expression of the mutant STING N153S protein was obtained by crossing heterozygous floxed-STOP STING N153S mice to RORγT-Cre-expressing animals. All other mouse strains were obtained as indicated in the Key Resources Table. Power analysis was conducted for Institutional Animal Care and Use Committee-approved in vivo studies in order to determine the number of animals needed per experimental group. At least two independent experiments were conducted to replicate findings. No outliers were excluded from analyses. The age and number of animals used for each experiment is listed in the figure legends.
SAVI patient samples
ILCs were analyzed from blood samples drawn from a total of 5 patients with STING gain-of-function mutations (V155M, N154S, or R281Q). There were 2 males (2 years-old, V155M and 10 years-old, N154S) and 3 females (8 years-old, V155M, 24 years-old, V155M, and 10 years old, R281Q). Four patients were on a JAK1/2 inhibitor, and one patient was on mycophenolate mofetil. For ethical reasons, SAVI patients were not removed from medical treatments. Hilar lymph nodes were obtained from SAVI patients (1 male, 1 female, both age 14) who underwent lung transplantation. Written informed consent (parental consent, in case of minors) was obtained from all participants of the study. The study and protocols conform to the 1975 Declaration of Helsinki and were approved by the comité de protection des personnes Ile de France II and the French advisory committee on data processing in medical research.
Cell lines
OP9 cell lines were used in the co-culture of mouse primary bone marrow cells. OP9 cell lines were cultured in MEM-α medium supplemented with 20% FCS and 1% penicillin/streptomycin (OP9 medium) at 37°C in a humidified atmosphere at 5% CO2.
METHOD DETAILS
Quantitation of lymph nodes
Bilateral cervical, inguinal, brachial, and axillary lymph nodes as well as mesenteric lymph nodes from WT and STING N153S mice were dissected and the number of discernible nodes counted. A string of mesenteric lymph nodes was counted as one lymph node. For Evans Blue staining of lymphatics and lymph nodes, mice were anesthetized and 25 μL of 5% Evans Blue dye in PBS was injected into 1 forefoot and 1 hindfoot. Fifteen minutes after Evans Blue injection, mice were euthanized and dissected for lymph node visualization and quantitation (Harrell et al., 2008). Any sign of a lymph node, regardless of size, was counted. To quantitate Peyer’s patches, the small intestine was divided into proximal, middle, and distal segments, and quantitated visually. For adoptive transfer studies, splenocytes were isolated from adult WT mice and single cell suspensions were obtained after disruption of tissue through a 70-μm filter. After erythrocyte lysis in ACK buffer, splenocytes were washed and counted. 5 million bulk splenocytes were transferred into Rag1−/− STING N153Sor Rag1−/− animals via intravenous injection. Mice were euthanized 3.5 weeks later, and Evans Blue staining was used to aid in quantitation of lymph nodes and Peyer’s patches.
Inguinal fat pad and intestine histology
Inguinal fat pads (and attached abdominal skin) were harvested at the bifurcation of the superficial epigastric vein (approximately 1 cm diameter). Small intestines were removed and flushed with PBS to remove any fecal matter. Tissues were fix in 4% paraformaldehyde for 24 hours and then embedded in paraffin. Inguinal fat pads were sequentially sectioned with 20 sectioned analyzed per mouse. Small intestines were serially sectioned with at least 48 sections analyzed per mouse.
Quantitation of IgA
IgA levels in the serum and stool were determined using a commercial mouse IgA ELISA kit (Immunology Consultants Laboratory catalog no. E-90A) according to the manufacturer’s protocol. All fecal samples were weighed and then suspended in sterile PBS at 100 μL per 10 mg, vortexed, and then spun down and supernatant transferred to new tube. Samples were diluted 1:300 and then analyzed.
Spleen and fetal tissue immunofluorescence
Fetuses were harvested on E18.5, and spleens were harvested from adult mice (7–16 weeks old). Tissues were immediately embedded in OCT, frozen on dry ice and stored at −80°C. Sections (8-μm) were cut at −20°C and then stored at −80°C until time of staining. At the time of staining, sections were removed from freezer and fixed in ice-cold acetone for 15 minutes, washed with PBS, and blocked for 10 minutes. Primary antibodies were applied overnight at 4°C and secondary antibodies were applied for 1 hour at room temperature. Coverslips were mounted using Prolong Diamond Antifade Mountant with Dapi (Fisher Cat# P36966). Images were taken using a Zeiss Axio Imager M2 Plus Wide Field Fluorescence Microscope. Antibodies used: CD4 (Biolegend catalog no. 100401), Madcam1 (R&D Biosystems catalog no. AF993), CD3 (Biolegend Cat# 100243), B220 (Biolegend catalog no. 103229), VCAM-1 (R&D Biosystems catalog no. AF643). Quantitation of staining intensity was performed using ImageJ and normalized to the area analyzed.
Splenic stromal cell analysis
Isolation of follicular dendric cells (FDC), fibroblastic reticular cells (FRC), and endothelial cells was performed as previously described (Sato et al., 2016). Briefly, spleens were harvested and digested using a cocktail of Collagenase D, DNase I, and Dispase I. Digested spleens were filtered, washed, and then enriched for FDCs and FRCs by negatively selecting for CD45− B220− Ter119− cells. Cells were washed and then stained with antibodies against CD45, CD19, CD31 (clone 390), PDPN (clone eBio8.1.1), CD54 (clone 3e2), and CD21/35 (clone 7e9).
Gene expression analysis
Total RNA from spleen homogenates or MEFs was isolated using the RNeasy kit (QIAGEN) per the manufacturer’s protocol. TaqMan RNA-to-Ct 1-Step kit (Applied Biosystems) was used to measure mRNA expression. Primer and probe assays were obtained from Integrated DNA Technologies. ΔΔCt values were calculated and then normalized to the mock WT samples. All samples analyzed were normalized to the house keeping gene (GAPDH). Samples where the target gene did not amplify were assigned a CT value of 38 as the limit of detection.
Noncanonical NF-κB signaling in MEFs
For studies of noncanonical NF-κB signaling, primary MEFS were stimulated with 2 μg/ml of anti-LTβR antibody (clone eBio3C8) for 24 hours. Cells were then harvested and analyzed by western blot or qRT-PCR.
SDS-PAGE and western blot
Primary MEFs were lysed in RIPA buffer (CST, catalog no. 9806S) supplemented with a protease inhibitor (Thermo Fisher, catalog no. 78430) and phosphatase inhibitor (Thermo Fisher, catalog no. 88667). An equal amount of protein was loaded and separated on 10% SDS-PAGE gels (Bio-Rad), then transferred to polyvinylidene fluoride membranes (EMD Millipore). Primary antibody against RelB (clone C1E4), IκBα (clone L34A5), and GAPDH were stained and detected by the use of horseradish-peroxidase-conjugated secondary anti-rabbit antibody (Invitrogen, catalog no. 31460). All the blots were performed using PierceTM ECL Substrate (Thermo Fisher Scientific) and scanned with a ChemiDocTM Touch Imaging System (Bio-Rad).
Flow cytometric analysis of leukocytes
For analysis of intestinal leukocytes, small intestines were harvested, flushed, and opened longitudinally, and Peyer’s patches were removed. Epithelial cells were removed via two rounds of gentle agitation with HBSS containing 10% FBS, 150 mM HEPES, and 5 mM EDTA. Tissues were washed with HBSS and digested with Collagenase IV (Sigma C5138) by shaking in a 37°C incubator for 40 minutes. Digested tissues were filtered through 100-μm filters and cellular debris removed via Percoll gradient for flow cytometric analysis. Single-cell suspensions were stained for CD45 (Invitrogen: clone 30-F11), NKp46 (Invitrogen: clone 29A1.4), CD4 (eBioscience: clone GK1.5), CCR6 (BD Biosciences: clone 140706), and lineage (Lin) markers, which consist of CD3ε (Biolegend: clone 145–2C11), CD5 (Biolegend: clone 53–7.3), CD19 (Biolegend: clone 6D5). Intracellular staining for RORγT (eBioscience: clone AFKJS-9) and GATA3 (BD Biosciences: clone L50–823) was performed according to eBioscience’s Intracellular Fixation and Permeabilization kit (Thermo Fischer Scientific: catalog no. 88–8824-00) before analysis on a Fortessa X-20. For analysis of bone marrow leukocytes, femurs and tibias of adult mice were collected and flushed with PBS for collection of bone marrow. Bone marrow was filtered, red blood cells lysed, and remaining cells were washed and resuspended for FACS analysis. Single-cell suspensions were stained with antibodies against lineage markers CD19, CD5, TER119 (Biolegend: clone TER119), B220 (Biolegend: clone RA3–6B2), CD11b (Biolegend: clone M1/70), CD11c (Biolegend: clone N418), NK1.1 (Biolegend: clone PK136), CD4 (Biolegend: clone Gk1.5), CD3ε, CD8α (Biolegend: clone 53–6.7), GR1 (Biolegend: clone RB6–8C5), Ly6G (Biolegend: clone 1A8), TCRγ/δ (Biolegend: clone GL3), CD45, CD127 (Biolegend: clone A7R34), FLT3 (Biolegend: clone A2F10), cKIT (Biolegend: clone ACK2), CD25 (Biolegend: clone 3C7) α4β7 (Biolegend: clone DATK32), PLZF (Invitrogen: clone Mags.21F7), ID2 (Invitrogen: clone ILCID2), CD27 (Invitrogen: clone LG.7F9), CD244 (Invitrogen: clone eBio244F4), CD122 (Invitrogen: clone TM-b1), and fixable live dead stain (Biolegend catalog no. 423105 or Thermo Fisher catalog no. L34965).
Isolation of fetal gut and fetal liver cells
Fetal livers were harvested at E13.5–14.5 and mechanically dissociated using a 1 mL pipette before being passed through a 70-μm filter. Cells were washed and resuspended for flow cytometric analysis. Fetal gut cells were isolated as described previously (Bando et al., 2015). Briefly, mouse fetuses were harvested at E16.5–18.5, and the small intestine was removed. Intestinal tissue was minced and then digested in complete RPMI media and collagenase at 37°C for 25 minutes while shaking at 200 rpm. Following digestion, the tissue was dissociated in Gentlemacs tubes (Milteny Biotec catalog no. 130–096-334). Dissociated tissue was filtered through a 70-μm filter into a 50 mL conical tube and cell suspension was washed with cold FACS buffer (4% FBS in PBS). For in vivo BrdU labeling of fetal gut cells, 0.1 mg/gram of body weight BrdU was injected into a pregnant dam. Two-hours post BrdU injection, fetal livers were harvested as described above.
LTi cell differentiation assays
In vitro LTi cell differentiation was described previously (Cherrier et al., 2012). Briefly, one day prior to sorting, OP9 cells were plated into 24-well plates at a density of 3.0 × 104 cells per well. Fetal livers were harvested on E13.5-E14.5, and single-cell suspensions were obtained. Negative selection was performed to remove CD3+, Ter119+, Gr-1+, and CD11c+ cells, and then CD3−CD19−B220−Gr1−CD45+cKITintCD127+α4β7+ cells were sorted and ~1000 fetal liver progenitor cells were plated onto OP9 stromal cells along with 10 ng/μl of rSCF (Peprotech catalog no. 250–03) and rIL-7 (Peprotech catalog no. 217–17). A half-media change was performed on days 4 and 11. Cells were passaged onto new OP9 stromal cells on day 7. Analysis by flow cytometry was performed 6 or 14 days after sorting and co-culture.
Single-cell RNA-seq
E14.5 fetal liver cells were prepared according to the established protocols that have been previously described for LTi cell isolation. Single Lin−CD45+cKITintCD127+α4β7+ cells were sorted directly into a 96-well plate (one cell per well) containing 2 μL of 10x lysis buffer (Takara catalog no. 635013) and 5% RNase inhibitor (Promega catalog no. PRN2611). After sorting, plates were immediately frozen at −80°C. Single-cell RNA sequencing of each well was performed at the Genome Technology Access Center (GTAC) at Washington University in St. Louis. Data are available at Mendeley DOI: https://dx.doi.org/10.17632/9nck2z26tf.1
Flow cytometric analysis of human PBMCs
Isolation of human PBMCs was performed using a Ficoll-Paque gradient. Cells were stained for the following markers: CD159a (NKG2A) (Miltenyi Biotec: VioBright FITC, clone REA110), CD294 (CRTH2) (BioLegend: PE-Dazzle594, clone BM16), CD127 (IL-7R) (Thermo Fisher: PE-Cy7, clone eBioRDR5), CD7 (BD Biosciences: Alexa Fluor 700, clone M-T701), CD94 (Miltenyi Biotec: APC-Vio770 clone REA113), CD117 (c-Kit) (BioLegend: BV605, clone 104D2), CD16 (BioLegend: BV605, clone 3G8), CD56 (BioLegend: BV785, clone 5.1H11), CD2 (BD Biosciences: BUV395, clone RPA-2.10), CD45 (BD Biosciences: BUV805, clone HI30), and lineage markers consisting of CD3 (BD Biosciences: BUV737, clone UCHT1), CD5 (BD Biosciences: BUV737, clone UCHT2), CD14 (BD Biosciences: BUV737, clone M5E2) and CD19 (BD Biosciences: BUV737, SJ25C1). Human serum IgG was used to block Fc receptors (Sigma-Aldrich). Surface membrane staining was performed using Brilliant Stain Buffer (BD Biosciences). Dead cells were excluded using the fixable viability dye eFluor506 (Thermo Fisher). Cells were fixed with 2% PFA prior to acquisition on a BD LSRFortessa (BD Biosciences) and subsequent analysis with FlowJo 10 (BD Biosciences)
Histopathology of SAVI patient lymph nodes
During lung transplantation, mediastinal lymph nodes from explanted lungs were retrieved and analyzed. Biopsies were fixed in 10% neutral buffered formalin, embedded in paraffin and stained with hematoxylin-eosin. Immunohistochemical staining was performed on an automated stainer (Bond Max; Leica Biosystems).
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise specified, all data were analyzed using GraphPad Prism software by Mann-Whitney or unpaired t test as specified in the figure legends. Flow cytometry data were analyzed using Cytobank or FlowJo v10.4.1.
Supplementary Material
Highlights.
STING gain-of-function in LTi cells prevents lymph node development in mice
STING gain-of-function impacts all types of ILCs, but especially ILC3s
Humans with STING gain-of-function mutations have fewer ILCs
ACKNOWLEDGMENTS
We thank the Hope Center Transgenic Vectors Core for assistance with generation of transgenic mice and the Washington University Morphology and Imaging Core for assistance with tissue processing and staining. D.J.P. is supported by the Washington University Chancellors Graduate Fellowship Program and the Initiative to Maximize Student Development. The Miner laboratory is supported by grants from the NIH (K08 AR070918 and R01 AI143982). RNA sequencing data analysis was supported by the WUSTL Rheumatic Diseases Research Resource-based Core (E.D.O.R., P30-AR073752). Single-cell RNA-seq data were generated at the Washington University GTAC MGI sequencing core, which is partially supported by NCI grant P30 CA91842 (Siteman Cancer Center) and ICTS/CTSA grant UL1 TR002345. C.A.C. is part of the Pasteur-Paris University (PPU) International PhD Program that received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie grant agreement 665807 and from the Labex Revive, Institut Pasteur. The study was also supported by the Institut National de la Santé et de la Recherche Médicale (INSERM), by a government grant managed by the Agence National de la Recherche as part of the “Investment for the Future” program (ANR-10-IAHU-01), and by an AAPG grant from the Agence National de la Recherche (ANR-14-CE14-0026-01 “Lumugene” to F.R.-L.). The Rösen-Wolff laboratory is supported by the German Research Foundation (TRR237,B18).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107771.
REFERENCES
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner KP, Ludwig J, and Hornung V (2013). cGAS produces a 2′−5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aliahmad P, de la Torre B, and Kaye J (2010). Shared dependence on the DNA-binding factor TOX for the development of lymphoid tissue-inducer cell and NK cell lineages. Nat. Immunol 11, 945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artis D, and Spits H (2015). The biology of innate lymphoid cells. Nature 517, 293–301. [DOI] [PubMed] [Google Scholar]
- Banchereau J, and Steinman RM (1998). Dendritic cells and the control of immunity. Nature 392, 245–252. [DOI] [PubMed] [Google Scholar]
- Bando JK, Liang HE, and Locksley RM (2015). Identification and distribution of developing innate lymphoid cells in the fetal mouse intestine. Nat. Immunol 16, 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennion BG, Ingle H, Ai TL, Miner CA, Platt DJ, Smith AM, Baldridge MT, and Miner JJ (2019). A Human Gain-of-Function STING Mutation Causes Immunodeficiency and Gammaherpesvirus-Induced Pulmonary Fibrosis in Mice. J. Virol 93, e01806–e01818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernink JH, Mjösberg J, and Spits H (2017). Human ILC1: To Be or Not to Be. Immunity 46, 756–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouis D, Kirstetter P, Arbogast F, Lamon D, Delgado V, Jung S, Ebel C, Jacobs H, Knapp AM, Jeremiah N, et al. (2019). Severe combined immunodeficiency in stimulator of interferon genes (STING) V154M/wild-type mice. J. Allergy Clin. Immunol 143, 712–725.e715. [DOI] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, and Vance RE (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canesso MCC, Lemos L, Neves TC, Marim FM, Castro TBR, Veloso ES, Queiroz CP, Ahn J, Santiago HC, Martins FS, et al. (2018). The cytosolic sensor STING is required for intestinal homeostasis and control of inflammation. Mucosal Immunol. 11, 820–834. [DOI] [PubMed] [Google Scholar]
- Caposio P, Musso T, Luganini A, Inoue H, Gariglio M, Landolfo S, and Gribaudo G (2007). Targeting the NF-kappaB pathway through pharmacological inhibition of IKK2 prevents human cytomegalovirus replication and virus-induced inflammatory response in infected endothelial cells. Antiviral Res. 73, 175–184. [DOI] [PubMed] [Google Scholar]
- Cerboni S, Jeremiah N, Gentili M, Gehrmann U, Conrad C, Stolzenberg MC, Picard C, Neven B, Fischer A, Amigorena S, et al. (2017). Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytes. J. Exp. Med 214, 1769–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chea S, Schmutz S, Berthault C, Perchet T, Petit M, Burlen-Defranoux O, Goldrath AW, Rodewald HR, Cumano A, and Golub R (2016). Single-Cell Gene Expression Analyses Reveal Heterogeneous Responsiveness of Fetal Innate Lymphoid Progenitors to Notch Signaling. Cell Rep. 14, 1500–1516. [DOI] [PubMed] [Google Scholar]
- Cherrier M, Sawa S, and Eberl G (2012). Notch, Id2, and RORγt sequentially orchestrate the fetal development of lymphoid tissue inducer cells. J. Exp. Med 209, 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinides MG, McDonald BD, Verhoef PA, and Bendelac A (2014). A committed precursor to innate lymphoid cells. Nature 508, 397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cradick TJ, Fine EJ, Antico CJ, and Bao G (2013). CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 41, 9584–9592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig SW, and Cebra JJ (1971). Peyer’s patches: an enriched source of precursors for IgA-producing immunocytes in the rabbit. J. Exp. Med 134, 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupedo T, and Mebius RE (2005). Cellular interactions in lymph node development. J. Immunol 174, 21–25. [DOI] [PubMed] [Google Scholar]
- Cyster JG, Ansel KM, Reif K, Ekland EH, Hyman PL, Tang HL, Luther SA, and Ngo VN (2000). Follicular stromal cells and lymphocyte homing to follicles. Immunol. Rev 176, 181–193. [DOI] [PubMed] [Google Scholar]
- Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li ZW, Karin M, Ware CF, and Green DR (2002). The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity 17, 525–535. [DOI] [PubMed] [Google Scholar]
- Dieu MC, Vanbervliet B, Vicari A, Bridon JM, Oldham E, Aït-Yahia S, Briè re F, Zlotnik A, Lebecque S, and Caux C (1998). Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med 188, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, and Littman DR (2004). An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat. Immunol 5, 64–73. [DOI] [PubMed] [Google Scholar]
- Fathman JW, Bhattacharya D, Inlay MA, Seita J, Karsunky H, and Weissman IL (2011). Identification of the earliest natural killer cell-committed progenitor in murine bone marrow. Blood 118, 5439–5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen F, Zagato E, Mazzini E, Fosso B, Manzari C, El Aidy S, Chiavelli A, D’Erchia AM, Sethi MK, Pabst O, et al. (2015). BALB/c and C57BL/6 Mice Differ in Polyreactive IgA Abundance, which Impacts the Generation of Antigen-Specific IgA and Microbiota Diversity. Immunity 43, 527–540. [DOI] [PubMed] [Google Scholar]
- Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, and Alt FW (2015). Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol 33, 179–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, and Sander JD (2013). High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol 31, 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fütterer A, Mink K, Luz A, Kosco-Vilbois MH, and Pfeffer K (1998). The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity 9, 59–70. [DOI] [PubMed] [Google Scholar]
- Geiger TL, Abt MC, Gasteiger G, Firth MA, O’Connor MH, Geary CD, O’Sullivan TE, van den Brink MR, Pamer EG, Hanash AM, and Sun JC (2014). Nfil3 is crucial for development of innate lymphoid cells and host protection against intestinal pathogens. J. Exp. Med 211, 1723–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui X, Yang H, Li T, Tan X, Shi P, Li M, Du F, and Chen ZJ (2019). Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567, 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, Radtke F, and Ablasser A (2017). Signalling strength determines pro-apoptotic functions of STING. Nat. Commun 8, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn MD, Tangemann K, Tam C, Cyster JG, Rosen SD, and Williams LT (1998). A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc. Natl. Acad. Sci. USA 95, 258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrell MI, Iritani BM, and Ruddell A (2008). Lymph node mapping in the mouse. J. Immunol. Methods 332, 170–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazenberg MD, and Spits H (2014). Human innate lymphoid cells. Blood 124, 700–709. [DOI] [PubMed] [Google Scholar]
- Heng TS, and Painter MW; Immunological Genome Project Consortium (2008). The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol 9, 1091–1094. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Leung TH, and Baltimore D (2003). Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. EMBO J. 22, 5530–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, and Taniguchi T (2005). IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. (2013). DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol 31, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SY, Hertzog PJ, Holland KA, Sumarsono SH, Tymms MJ, Hamilton JA, Whitty G, Bertoncello I, and Kola I (1995). A null mutation in the gene encoding a type I interferon receptor component eliminates anti-proliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc Natl Acad Sci USA 92, 11284–11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imanishi T, Ishihara C, Badr Mel.S., Hashimoto-Tane A, Kimura Y, Kawai T, Takeuchi O, Ishii KJ, Taniguchi S, Noda T, et al. (2014). Nucleic acid sensing by T cells initiates Th2 cell differentiation. Nat. Commun 5, 3566. [DOI] [PubMed] [Google Scholar]
- Kelly KA, and Scollay R (1992). Seeding of neonatal lymph nodes by T cells and identification of a novel population of CD3-CD4+ cells. Eur. J. Immunol 22, 329–334. [DOI] [PubMed] [Google Scholar]
- Kim D, Mebius RE, MacMicking JD, Jung S, Cupedo T, Castellanos Y, Rho J, Wong BR, Josien R, Kim N, et al. (2000). Regulation of peripheral lymph node genesis by the tumor necrosis factor family member TRANCE. J. Exp. Med 192, 1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein-Schneegans AS, Kuntz L, Fonteneau P, and Loor F (1989). Serum concentrations of IgM, IgG1, IgG2b, IgG3 and IgA in C57BL/6 mice and their congenics at the lpr (lymphoproliferation) locus. J. Autoimmun 2, 869–875. [DOI] [PubMed] [Google Scholar]
- Klose CS, Kiss EA, Schwierzeck V, Ebert K, Hoyler T, d’Hargues Y, Göppert N, Croxford AL, Waisman A, Tanriver Y, and Diefenbach A (2013). A T-bet gradient controls the fate and function of CCR6-RORγt+ innate lymphoid cells. Nature 494, 261–265. [DOI] [PubMed] [Google Scholar]
- Klose CSN, Flach M, Möhle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, et al. (2014). Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157, 340–356. [DOI] [PubMed] [Google Scholar]
- Lim AI, Li Y, Lopez-Lastra S, Stadhouders R, Paul F, Casrouge A, Serafini N, Puel A, Bustamante J, Surace L, et al. (2017). Systemic Human ILC Precursors Provide a Substrate for Tissue ILC Differentiation. Cell 168, 1086–1100.e1010. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, et al. (2014). Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med 371, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luksch H, Stinson WA, Platt DJ, Qian W, Kalugotla G, Miner CA, Bennion BG, Gerbaulet A, Rosen-Wolff A, and Miner JJ (2019). STING-associated lung disease in mice relies on T cells but not type I interferon. J. Allergy Clin. Immunol 144, 254–266.e258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther SA, Tang HL, Hyman PL, Farr AG, and Cyster JG (2000). Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc. Natl. Acad. Sci. USA 97, 12694–12699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus A, Mao AJ, Lensink-Vasan M, Wang L, Vance RE, and Raulet DH (2018). Tumor-Derived cGAMP Triggers a STING-Mediated Interferon Response in Non-tumor Cells to Activate the NK Cell Response. Immunity 49, 754–763.e754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer M, de Angelis MH, Wurst W, and Kühn R (2010). Gene targeting by homologous recombination in mouse zygotes mediated by zinc-finger nucleases. Proc. Natl. Acad. Sci. USA 107, 15022–15026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mjösberg JM, Trifari S, Crellin NK, Peters CP, van Drunen CM, Piet B, Fokkens WJ, Cupedo T, and Spits H (2011). Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol 12, 1055–1062. [DOI] [PubMed] [Google Scholar]
- Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, and Papaioannou VE (1992). RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877. [DOI] [PubMed] [Google Scholar]
- Mooster JL, Le Bras S, Massaad MJ, Jabara H, Yoon J, Galand C, Heesters BA, Burton OT, Mattoo H, Manis J, and Geha RS (2015). Defective lymphoid organogenesis underlies the immune deficiency caused by a heterozygous S32I mutation in IκBα. J. Exp. Med 212, 185–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motwani M, Pawaria S, Bernier J, Moses S, Henry K, Fang T, Burkly L, Marshak-Rothstein A, and Fitzgerald KA (2019). Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models. Proc. Natl. Acad. Sci. USA 116, 7941–7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo VN, Korner H, Gunn MD, Schmidt KN, Riminton DS, Cooper MD, Browning JL, Sedgwick JD, and Cyster JG (1999). Lymphotoxin alpha/beta and tumor necrosis factor are required for stromal cell expression of homing chemokines in B and T cell areas of the spleen. J. Exp. Med 189, 403–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onder L, Danuser R, Scandella E, Firner S, Chai Q, Hehlgans T, Stein JV, and Ludewig B (2013). Endothelial cell-specific lymphotoxin-β receptor signaling is critical for lymph node and high endothelial venule formation. J. Exp. Med 210, 465–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, and Liu DR (2013). High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol 31, 839–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankin LC, Groom JR, Chopin M, Herold MJ, Walker JA, Mielke LA, McKenzie AN, Carotta S, Nutt SL, and Belz GT (2013). The transcription factor T-bet is essential for the development of NKp46+ innate lymphocytes via the Notch pathway. Nat. Immunol 14, 389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboldi A, Arnon TI, Rodda LB, Atakilit A, Sheppard D, and Cyster JG (2016). IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science 352, aaf4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennert PD, Browning JL, Mebius R, Mackay F, and Hochman PS (1996). Surface lymphotoxin alpha/beta complex is required for the development of peripheral lymphoid organs. J. Exp. Med 184, 1999–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennert PD, James D, Mackay F, Browning JL, and Hochman PS (1998). Lymph node genesis is induced by signaling through the lymphotoxin beta receptor. Immunity 9, 71–79. [DOI] [PubMed] [Google Scholar]
- Roan F, Stoklasek TA, Whalen E, Molitor JA, Bluestone JA, Buckner JH, and Ziegler SF (2016). CD4+ Group 1 Innate Lymphoid Cells (ILC) Form a Functionally Distinct ILC Subset That Is Increased in Systemic Sclerosis. J. Immunol 196, 2051–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, and Taniguchi T (2000). Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13, 539–548. [DOI] [PubMed] [Google Scholar]
- Sato K, Honda SI, Shibuya A, and Shibuya K (2016). Improved protocol for the isolation of naïve follicular dendritic cells. Mol. Immunol 78, 140–145. [DOI] [PubMed] [Google Scholar]
- Sawa S, Cherrier M, Lochner M, Satoh-Takayama N, Fehling HJ, Langa F, Di Santo JP, and Eberl G (2010). Lineage relationship analysis of RORgammat+ innate lymphoid cells. Science 330, 665–669. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikhagaie MM, Björklund AK, Mjösberg J, Erjefält JS, Cornelissen AS, Ros XR, Bal SM, Koning JJ, Mebius RE, Mori M, et al. (2017). Neuropilin-1 Is Expressed on Lymphoid Tissue Residing LTi-like Group 3 Innate Lymphoid Cells and Associated with Ectopic Lymphoid Aggregates. Cell Rep. 18, 1761–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoni Y, Fehlings M, Kløverpris HN, McGovern N, Koo SL, Loh CY, Lim S, Kurioka A, Fergusson JR, Tang CL, et al. (2017). Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity 46, 148–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits H, Bernink JH, and Lanier L (2016). NK cells and type 1 innate lymphoid cells: partners in host defense. Nat. Immunol 17, 758–764. [DOI] [PubMed] [Google Scholar]
- Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, Mebius RE, and Littman DR (2000). Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science 288, 2369–2373. [DOI] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatori H, Kanno Y, Watford WT, Tato CM, Weiss G, Ivanov II, Littman DR, and O’Shea JJ (2009). Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med 206, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Pavert SA, and Mebius RE (2010). New insights into the development of lymphoid tissues. Nat. Rev. Immunol 10, 664–674. [DOI] [PubMed] [Google Scholar]
- Victora GD, and Nussenzweig MC (2012). Germinal centers. Annu. Rev. Immunol 30, 429–457. [DOI] [PubMed] [Google Scholar]
- Vonarbourg C, Mortha A, Bui VL, Hernandez PP, Kiss EA, Hoyler T, Flach M, Bengsch B, Thimme R, Hölscher C, et al. (2010). Regulated expression of nuclear receptor RORγt confers distinct functional fates to NK cell receptor-expressing RORγt(+) innate lymphocytes. Immunity 33, 736–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang Y, Wu X, Wang J, Wang Y, Qiu Z, Chang T, Huang H, Lin RJ, and Yee JK (2015). Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat. Biotechnol 33, 175–178. [DOI] [PubMed] [Google Scholar]
- Warner JD, Irizarry-Caro RA, Bennion BG, Ai TL, Smith AM, Miner CA, Sakai T, Gonugunta VK, Wu J, Platt DJ, et al. (2017). STING-associated vasculopathy develops independently of IRF3 in mice. J. Exp. Med 214, 3279–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteley AT, Eaglesham JB, de Oliveira Mann CC, Morehouse BR, Lowey B, Nieminen EA, Danilchanka O, King DS, Lee ASY, Mekalanos JJ, and Kranzusch PJ (2019). Bacterial cGAS-like enzymes synthesize diverse nucleotide signals. Nature 567, 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ, and Yan N (2019). STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J. Exp. Med 216, 867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Domingues RG, Fonseca-Pereira D, Ferreira M, Ribeiro H, Lopez-Lastra S, Motomura Y, Moreira-Santos L, Bihl F, Braud V, et al. (2015). NFIL3 orchestrates the emergence of common helper innate lymphoid cell precursors. Cell Rep. 10, 2043–2054. [DOI] [PubMed] [Google Scholar]
- Yang Q, Li F, Harly C, Xing S, Ye L, Xia X, Wang H, Wang X, Yu S, Zhou X, et al. (2015). TCF-1 upregulation identifies early innate lymphoid progenitors in the bone marrow. Nat. Immunol 16, 1044–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota Y, Mansouri A, Mori S, Sugawara S, Adachi S, Nishikawa S, and Gruss P (1999). Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature 397, 702–706. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The dataset generated by the single-cell FACs-Sequencing of α4β7+ fetal liver (E14.5) progenitor cells during this study is available at Mendeley DOI: https://dx.doi.org/10.17632/9nck2z26tf.1