

Abstract
Alcoholism is associated with brain damage and impaired cognitive functioning. The relative contributions of different etiological factors, such as alcohol, thiamine deficiency and age vulnerability, to the development of alcohol-related neuropathology and cognitive impairment are still poorly understood. One reason for this quandary is that both alcohol toxicity and thiamine deficiency produce brain damage and cognitive problems that can be modulated by age at exposure, aging following alcohol toxicity or thiamine deficiency, and aging during chronic alcohol exposure. Pre-clinical models of alcohol-related brain damage (ARBD) have elucidated some of the contributions of ethanol toxicity and thiamine deficiency to neuroinflammation, neuronal loss and functional deficits. However, the critical variable of age at the time of exposure or long-term aging with ARBD has been relatively ignored. Acute thiamine deficiency created a massive increase in neuroimmune genes and proteins within the thalamus and significant increases within the hippocampus and frontal cortex. Chronic ethanol treatment throughout adulthood produced very minor fluctuations in neuroimmune genes, regardless of brain region. Intermittent “binge–type” ethanol during the adolescent period established an intermediate neuroinflammatory response in the hippocampus and frontal cortex, that can persist into adulthood. Chronic excessive drinking throughout adulthood, adolescent intermittent ethanol exposure, and thiamine deficiency all led to a loss of the cholinergic neuronal phenotype within the basal forebrain, reduced hippocampal neurogenesis, and alterations in the frontal cortex. Only thiamine deficiency results in gross pathological lesions of the thalamus. The behavioral impairment following these types of treatments is hierarchical: Thiamine deficiency produces the greatest impairment of hippocampal- and prefrontal- dependent behaviors, chronic ethanol drinking ensues mild impairments on both types of tasks and adolescent intermittent ethanol exposure leads to impairments on frontocortical tasks, with sparing on most hippocampal-dependent tasks. However, our preliminary data suggest that as rodents age following adolescent intermittent ethanol exposure, hippocampal functional deficits began to emerge. A necessary requirement for the advancement of understanding the neural consequences of alcoholism is a more comprehensive assessment and understanding of how excessive alcohol drinking at different development periods (adolescence, early adulthood, middle-aged and aged) influences the trajectory of the aging process, including pathological aging and disease.
Keywords: Aging, development, alcohol, EtOH, neuroinflammation, frontal cortex, hippocampus, thalamus, basal forebrain, memory, cognition, human, rat, mouse
I. Introduction:
Excessive alcohol use over the lifespan can increase the risk for alcohol-related brain damage (ARBD) and cognitive decline-- including an increased risk for the development of dementia. Over 70% of individuals with chronic alcohol use disorder (AUD) display some degree of brain pathology (Goldstein and Shelley, 1980; Harper, 1998), and most of the brain regions affected by AUDs are similar to the regions that are vulnerable to aging and Alzheimer’s disease. Adults with AUD display reductions in brain volume in several critical regions, including the frontal, temporal, parietal, cingulate, and insular cortices, cerebellum, thalamus and hippocampus, and this loss can be more pronounced in those adults that are 65-years and older (Sullivan et al., 2018; Zahr et al., 2019).
The brain damage associated with chronic AUDs can lead to impaired functioning, even after alcohol abuse ends. For example, 50–75% of detoxified abstinent alcoholics have some type of cognitive or memory disturbance (for reviews, see Dufour, 1993, Parsons and Nixon, 1993, Smith and Atkinson, 1995). Furthermore, middle-aged adults with a history of AUD have a propensity to develop later memory impairment and also have an increased prevalence of dementia later in life (Kuzma et al., 2014; Perreira and Sloan, 2002). The hypothesis that alcoholism interacts or interferes with normal aging was developed because several cross-sectional studies revealed accelerated brain shrinkage by middle-age in patients with AUD, and two recent longitudinal studies provide stronger evidence for age-alcoholism neuropathological interactions in the frontal cortex and hippocampus, even if excessive drinking is initiated later in life (Sullivan et al., 2018; Zahr et al., 2019). Thus, it is hypothesized that heavy alcohol consumption (+5 drinks per day) is associated with accelerated cognitive aging, and aged individuals that are current heavy drinkers exhibited the greatest cognitive deficits (Woods et al., 2016). Furthermore, alcohol consumption has been linked to an increased risk of Alzheimer’s disease in individuals with the ApoE e4 allele (Anttila et al., 2004; Kivipelto et al., 2008). Therefore, as people age with, or after, alcohol addiction the likelihood that they will experience cognitive decline is elevated.
It has been estimated that greater than 10% of alcoholic patients have symptoms of either alcohol-related dementia (ARD) or Wernicke-Korsakoff Syndrome (WKS; see Table 1; Harper and Kril, 1990; Parsons and Nixon, 1993). Traditionally, these two disorders (also called major/mild neurocognitive disorders associated with alcohol use and alcohol-induced persisting amnestic disorder) have been viewed to be on a continuum. Progressive increases in brain and cognitive dysfunction are observed from ARD to WKS (Brion et al., 2014). Patients with WKS demonstrate similar, but more severe, lesions expressed in the form of deficits in regional brain volumes (mammillary bodies, thalamus, cerebellar hemispheres, and vermis) compared to alcoholic patients without a diagnosis of WKS (Sullivan and Pfefferbaum, 2009). However, the continuum hypothesis has been questioned, as the memory and cognitive deficits in alcoholic patients that do not have thiamine deficiency (TD) tend to recover within weeks to months following abstinence (Arts et al., 2017). In contrast, the cognitive recovery in WKS patients is more limited. Thus, it has been suggested that patients with a diagnosis of ARD suffer from residual TD, other types of dementia, or other forms of alcohol-related co-injury (Arts et al., 2017). However, this view has not considered aging as a vulnerability to the emergence of ARD. Thus, it is possible that subclinical brain changes that follow excessive drinking becomes additive, or potentially synergistic, with those that can occur during advanced aging, culminating in the behavioral syndrome of ARD.
Table 1.
Summary of alcohol-related disorders with cognitive impairment and the most common animal models of alcohol-related brain damage.
| Human Condition | Symptoms in the Cognitive Domain | Critical Brain Regions affected |
|---|---|---|
| Alcohol Use Disorder (AUD) | Problem drinking that interferes with daily living/independence | Cortical shrinkage that can recover with abstinence |
| Alcohol Related Dementia (ARD)* | Deficits of working memory, motor speed and executive function | Frontal cortical shrinkage (gray and white matter), hippocampal and cerebellar cell loss, some recovery with abstinence |
| Wernicke Korsakoff Syndrome (WKS)** | Impaired executive function, visuo-perceptual difficulties, and profound memory impairment | Frontal cortical shrinkage (gray & white matter), hippocampal and cerebellar cell loss, lesion within the thalamus and mammillary bodies, limited brain recovery with abstinence |
| Animal Model | Behavioral/Cognitive Symptoms | Critical Brain Regions affected |
| Continuous Intermittent Ethanol Exposure (CIE) | Transient impairment in behavioral (rule-reversal) flexibility, impaired spatial memory | Reduced frontocortical plasticity markers and temporary maladaptive increase in hippocampal excitability |
| Adolescent Intermittent Ethanol Exposure (AIE) | Impaired behavioral (rule-reversal) and cognitive (set-shifting) flexibility, spared spatial memory | Loss of hippocampal neurogenesis, changes in frontocortical connectivity patterns, and loss of cholinergic neurons in basal forebrain |
| Chronic Ethanol Treatment (CET) | Impaired spatial memory, dysfunctional behavioral (rule-reversal) and cognitive (set-shifting) | Frontal cortex cell loss, reduced hippocampal neurogenesis, mature granule and pyramidal cell loss within the hippocampus, loss of forebrain cholinergic neurons |
| Thiamine Deficiency (TD) | Impaired spatial learning and memory, dysfunctional behavioral (rule-reversal) and cognitive (set-shifting), severe perseveration | Frontal cortical shrinkage, reduced hippocampal neurogenesis, loss of forebrain cholinergic neurons, dramatic neuronal loss in the thalamus and mammillary bodies. |
DSM-5: Alcohol-induced neurocognitive disorder (mild/major)
DSM-5: Alcohol-induced persisting amnestic disorder
It has been estimated that 80% of the patients diagnosed with WKS or ARD have executive functioning deficits including impaired planning, higher-order organization, and cognitive flexibility (Van Oort and Kessels, 2009). However, in WKS, the learning and memory deficit is much greater than in ARD, and is irreversible (Arts et al., 2017). It should be noted that even within a diagnosis for WKS there is variability and patients could be classified as “milder” or “more severe”, with the milder form commonly being the non-alcoholic form (see Arts et al., 2017). What is observed in WKS is that the anterograde memory processes are typically more severely affected than retrograde memory processes. A key permanent anterograde memory deficit in WKS patients is contextual memory or source memory that includes both temporal and spatial information, with sparing of semantic memory (facts) and procedural or implicit memory (Kessels and Kopelman, 2012).
Although long-term abstinence (months) can resolve many cognitive deficits associated with chronic AUDs, some patients have deficits that linger (Fein et al., 2006). It has been stated the reduced recovery of cognitive functions that is reported in patients with AUDs may be complicated by factors such as poor eating habits, poly-drug abuse, physical accidents, repeated withdrawal cycles, and advanced age (Fein et al., 1990; Munro et al., 2000; Rourke and Grant, 1999). Thus, the neuropathological profile of persons with chronic AUDs is heterogeneous.
The effects of TD in an animal model are modulated by advanced age and aging. We found that in rats advanced age led to an earlier onset of acute neurological disturbances of TD, relative to young rats, and aged rats developed more extensive neuropathology with a shorter duration of TD. Specifically, we found that aged rats displayed increased brain shrinkage (smaller frontal cortical and callosal thickness) as well as enhanced astrocytic activity within the thalamus and a decrease in cholinergic neurons (ChAT-immunopositive) in the medial septum; the latter two measures of neuropathology were potentiated by TD (Pitkin and Savage, 2001). Aging after experiencing TD as a middle-aged adult rat led to greater loss of thalamic mass (Pitkin and Savage, 2001) than undergoing TD as a young adult rat. This provides evidence that advanced age increases susceptibility to ARBD, as well as aging with ARBD leads to poorer brain outcomes.
Another developmental phenomenon is that an early age of drinking onset (peri-adolescent or adolescent) predicts the later development of AUDs. The prevalence of AUDs is increasing in older adults: heavy drinking ranges from 2.1%−5.6%, whereas binge drinking is even higher (9.1%−23%; SAMHSA, 2014) in older adults. Of those older adults diagnosed with an AUD, about two-thirds started drinking during adolescence or early adulthood (Menninger, 2002). An early onset of excessive drinking (i.e. before 21) is associated with increased neuronal pro-inflammatory mediators and chronic inflammatory reactions associated with neurotoxicity (high-mobility-group box-1 [HMGB1] protein, acting as an activator of Toll-like receptors [TLR] and receptors for advanced glycation end products [RAGE], see Vetreno et al., 2013). Persistent chronic inflammation, even at a low level, is thought to be an accelerator of biological aging (Valero et al., 2017). The concept of a “vulnerable brain” following early onset excessive drinking may explain why cases of alcohol-related dementia generally have a younger age of drinking onset (Ritchie and Villebrun, 2008). Thus, the pathophysiological processes of AUDs may be subchronic for decades, prior to the emergence of symptoms of neurodegeneration in advanced age. Thus, early heavy drinking may be a predictor for the rate or development of neurodegeneration as a function of advanced age.
In this review we examine some of the critical features, in animal models, that modulate the degree, extent and functional consequences of ARBD. Specifically, we will examine how advanced age, both aging with ARBD or developing ARBD when aged, impacts neuroinflammation and neural structure that leads to dysfunctional memory and cognitive abilities. The focus on animal models of ARBD can provide us a means to directly examine how malnourishment, chronic alcohol (ethanol [EtOH]) exposure and aging interact.
2. Neuroinflammation and alcohol-related brain damage
The neuroinflammation response evoked by alcohol exposure has been considered as an important contributor to ARBD (Amor et al., 2014; Crews and Vetreno, 2016), as well as an antecedent to neurodegeneration (Qin and Crews, 2012b). Neuroimmune signaling initiates with the activation of glial cells, microglia and astrocytes , which lead to the production of neuroimmune factors such as cytokines, chemokines, as well as neurotoxic factors such as glutamate and free radical species, which can induce or exacerbate brain damage (Block et al., 2007; Lucas et al., 2006). Studies have shown that alcohol-induced neuroimmune signaling involves changes in brain microglial markers in both animals exposed to EtOH and in the post-mortem brain tissue of humans diagnosed with an alcohol-use disorder (Coleman et al., 2017; Crews et al., 2013; Yang et al., 2014). Studies that expose animals to chronic EtOH have revealed that inflammatory markers can have different responses across brain regions that vary as a function of dose and duration of EtOH exposure, as well as withdrawal and abstinence periods (see review Henriques et al., 2018). Two broad types of rodent models are commonly used to mimic human long-term alcoholism: The forced or semi-voluntary oral intake of EtOH in water or liquid diets for weeks to months (with BEC’s mostly ranging from 60–120 mg/dl), and the “binge-type” experimenter delivered EtOH via intragastric intubation, intraperitoneal injection or vapor inhalation models that last for days or weeks, with continuous or intermittent delivery (achieving BEC’s from 150–400 mg/dl). Furthermore, chronic alcoholism is often associated with a poor diet that can lead to TD, which can also be modeled effectively in rodents. Obviously, these different types of models will evoke different neural and non-neuronal reactions in the brain, but all lead to ARBD and our goal is to understand the role neuroinflammation plays.
There is the hypothesis that alcohol withdrawal leads to hyperexcitability and this response is critical for the induction of ARBD (Butler and Prendergast, 2012; De Witte et al., 2003; Sharrett-Field et al., 2013). However, other data suggests that neurodegeneration associated with chronic binge-type EtOH exposure in the adult brain may be independent of withdrawal, but withdrawal-induced excitotoxicity may be a factor in the developing brain (Collins and Neafsey, 2012; 2016). This appears to be true for shorter-term binge-type of exposure, as NMDA receptor antagonists do not provide neuroprotection (see Collins and Neafsey, 2016 for a review). However, a consistent observation following most chronic intermittent EtOH exposure (CIE) models is the activation of neuroimmune signaling that amplifies innate immune gene induction that triggers neuropathology (Crews et al., 2015). In contrast, TD produces oxidative stress, excitotoxicity and inflammatory responses that contribute to neurodegeneration in distinct brain regions (Abdou and Hazell, 2015). We will review several studies, using different models, to understand how neuroinflammation contributes to ARBD.
A study using chronic EtOH treatment continuous for 5 weeks (free access to aqueous EtOH 5% v/v in water in mice) showed a significant increase in the protein levels of tumor necrosis factor α (TNF-α), monocyte chemotactic protein-1 (MCP-1), and interleukin-1β (IL-1β) in the cerebellum during EtOH exposure (Lippai et al., 2013). Similarly, a longer duration of EtOH treatment (10 week of 10 g/kg, 35% v/v, oral gavage) also significantly elevated TNF-α and IL-1β protein levels in the cerebral cortex and hippocampus of EtOH-treated rats at the end of binge-type exposure (Tiwari and Chopra, 2013). Chronic intermittent EtOH treatment (25%, w/v in a diluted nutritionally complete diet, with i.g. priming dose of 5g/kg EtOH and then 3g/kg every 8h for 4 days, followed by a 3-day EtOH- withdrawal period) for 25 days also elevated TNF-α and IL-1β protein levels (Zhao et al., 2013). However, with that procedure, the production of pro-inflammatory cytokines returned to a normal level after 4 days of abstinence (Zhao et al., 2013). Thus, changes in pro-inflammatory cytokine protein concentration by forced and voluntary EtOH exposure is apparent in the development of ARBD.
Another study using a more prolonged EtOH drinking model (5 months of 5–10% v/v in water), has shown the up-regulation of proinflammatory cytokines gene expression (IL-6, IL-1β and TNF-α) in the cortex and striatum of mice (Alfonso-Loeches et al., 2010; Pascual et al., 2015) and rats (Vallés et al., 2004) during chronic EtOH exposure. Furthermore, Pascual and colleagues showed that IL-1β mRNA in the striatum of mice remained up-regulated at 24-hrs post chronic EtOH drinking (Pascual et al., 2015). The up-regulation of brain pro-inflammatory cytokines (TNFα, IL-6, and MCP-1), down-regulation of IL-10 mRNA and protein, and activation of microglia in the mouse brain also was observed following a short withdrawal period (24- or 27-hrs) from a forced repeated exposure to EtOH protocol (5 g/kg, i.g., 25% EtOH via oral gavage for 10 days; Qin et al., 2008; Qin and Crews, 2012a). These studies examining the effects of repeated EtOH on inflammatory induction have revealed that shortly after the removal EtOH, changes in brain cytokines occur and this response could impact neurodegenerative processes (Qin et al., 2008).
Whitman and colleagues (2013) examined whether intermittent (three 5-day bouts of 7% EtOH diet, separated by 2 days of control diet) or continuous (15 days with 7% v/v in water) EtOH exposure had differential effects on the expression of IL-1β, TNF-α and chemokine ligand 2 (CCL2) genes in rat cortex. They found increases in mRNA expression of these three cytokines (IL-1β, TNF-α and CCL2) that peaked during a 24-hr withdrawal period, regardless of the type of EtOH exposure protocol. However, there were no alterations in these genes during the EtOH consumption period (Whitman et al., 2013). It has also been demonstrated that 29-hrs following 15 days of EtOH consumption (7% v/v in water) there was up-regulation of IL-1β and TNF-α genes in the cortex and IL-1β gene in the hippocampus and hypothalamus of rats (Knapp et al., 2016). Furthermore, following a liquid EtOH diet (36 % of calories as alcohol) for 35 days in rats, withdrawal resulted in an increase of TNF-α in the central nucleus of the amygdala and dorsal vagal complex and this induction was most prominent 48-hrs after cessation of EtOH. These findings suggest that rapid cessation of EtOH exposure may cause acute activation of neuroimmune signaling in regions related to emotional and autonomic dysfunction (Freeman et al., 2012). Thus, acute withdrawal after short-term chronic EtOH exposure seems to contribute to chronic alcohol-induced responses by exacerbation of the proinflammatory state.
However, a recent study from our laboratory using a prolonged life-time chronic EtOH treatment (CET) model (20% v/v EtOH in drinking water for 6–8 months) in adult rats, which produced moderate BECs (40–100 dl/mg), found only small fluctuations in gene expression levels of cytokines in several brain regions (hippocampus, frontal cortex, thalamus; Toledo Nunes et al., 2019). Additionally, these fluctuations varied as a function of active drinking or abstinence phases (see Figure 1). For example, the mRNA of proinflammatory cytokine IL-1β was down-regulated in the hippocampus during CET and up-regulated in thalamus following 3-weeks of abstinence. Also, in the acute withdrawal phase (24-hrs after CET) there was decreased expression of the IL-6 gene in the frontal cortex. In contrast, TD led to rapid burst of neuroimmune genes and proteins during treatment, with profound increase in IL-6, TNF-α and IL-1β within the thalamus. These changes were also observed in the hippocampus and frontal cortex, however, they were moderate relative to the thalamus. Furthermore, the alterations in these neuroimmune markers recovered to normal levels within a 24-hour period. Our results support that TD is a potent driver of neuroinflammation and more broadly alcohol-related brain damage (Toledo Nunes et al., 2019).
Figure 1.
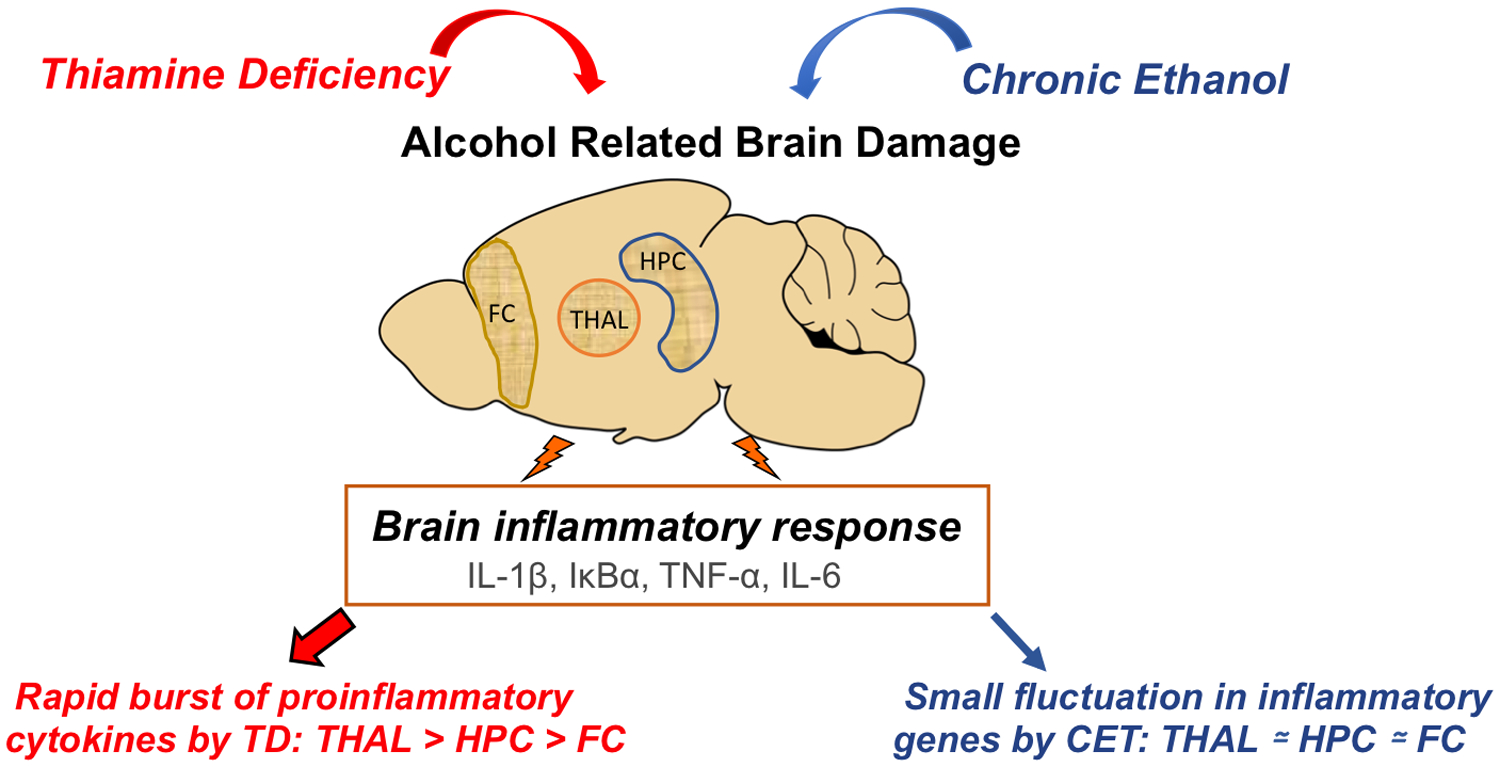
Thiamine deficiency (TD) and chronic ethanol treatment (CET) are critical factors in the induction of alcohol-related brain damage (ARBD) and inflammatory responses in brain. Our findings shown the TD, rather than CET, is a key driver of neuroimmune gene expression and subsequent neuroinflammation across the thalamus (THAL), hippocampus (HPC) and frontal cortex (FC).
Other studies using the long-term EtOH treatment (free access to a 20% aqueous EtOH) model in rats found that exposure to EtOH for 6 months (Cruz et al., 2017) or 12 months (Ehrlich et al., 2012) did not result in an increase of TNF-a, COX-2 and IL-1 β in the hippocampus or cortex. Consistent with those results, Cruz and colleagues (2017) found that chronic EtOH induces partial microglial activation (up to stage 2; see chapter by Perkins et al., this volume), suggesting that induction of these cytokines can be dependent on the stage of microglial activation. Furthermore, the access to EtOH for 6-months significantly increased the expression of IL-15 mRNA, a cytokine that is up-regulated upon activation of microglial and astrocytes. Given that CET did not change other cytokines genes associated with the partial microglial activation, the up-regulation of IL-15 mRNA is likely related to astrocyte activation. This study also found that prolonged EtOH abstinence (2-months) reversed the IL-15 response to CET, but did not reverse the partial microglial activation (Cruz et al., 2017). These results are consistent with our recent work, demonstrating that moderate EtOH consumption for a long period may evoke a neuroimmune response, but the change in pro-inflammatory cytokines genes is relatively modest (Toledo Nunes et al., 2019).
Alterations in cytokine expression induced by CET, intermittent or continuous, or by EtOH withdrawal can be taken as evidence for a role of microglia in EtOH-related neurotoxicity, as well as biomarkers of a neuroimmune response (Alfonso-Loeches and Guerri 2011; Zhao et al., 2013). Beyond withdrawal-induced glutamate excitotoxicity, microglial activation of neuroinflammatory and oxidative stress pathways are other avenues for alcohol-related neurodegeneration (Collins and Neafsey, 2016). A recent study (Tyler et al., 2019) examining the role of EtOH-dependence revealed microglial activation within the hippocampus and thalamus of rats exposed to intermittent EtOH-vapor protocol for 6–10 weeks followed by operant oral EtOH self-administration. They showed that translocator protein (TSPO) ligands, which is considered a biomarker of neuroinflammation, were increased in EtOH-dependent rats compared to nondependent when measured in vitro. Thus, these results support the involvement of neuroinflammation in EtOH dependence.
There is evidence that EtOH exposure sensitizes immune cells in the central nervous system, resulting in the release of innate immune signaling molecules that activate receptors, such as Toll-like receptors (TLR) and cytokine receptors, leading to the transcription of genes that contribute directly to inflammatory brain damage (Alfonso-Loeches and Guerri, 2011; Blanco and Guerri, 2007; Crews and Vetreno, 2016). Specifically, studies suggest that activation of TLR receptors causes the induction of genes that encode inflammation-associated molecules and cytokines through the triggering of the nuclear factor k light-chain-enhancer of activated B cells (NF-κB) (Crews et al., 2017; Song et al., 2014). Among TLRs, TLR2, TLR3, TLR4 and TLR7 have been related to microglia activation and triggered by EtOH treatment in animals, brain slices culture, and in the human post-mortem alcoholic brain (Alfonso-Loeches et al., 2010, 2012, 2016; Coleman et al., 2017; Montesinos et al., 2016; Qin and Crews, 2012a).
The role of TLR4 in EtOH-induced glial activation and brain damage has been demonstrated in studies using long-term EtOH consumption in mice (10% v/v, BECs 87–140 mg/dl; Alfonso-Loeches et al., 2010, 2012). After 5 months of chronic EtOH treatment, TLR4 receptors and the NF-κB pathway are activated, resulting in the expression of cytokines and other genes associated with innate immune responses (IL-6 and TNF-α; Alfonso-Loeches et al., 2010). Importantly, the elimination of TLR4 receptors (TLR4 knockout mice) was able to eliminate the effects of long-term EtOH consumption on neuroinflammation markers, myelin alterations and neural death (Alfonso-Loeches et al., 2010).
The elevation of TLRs in whole brain and cortex of mice was observed during acute withdrawal (24-hrs or 27-hrs) following short-term EtOH treatment using a binge drinking protocol (10 days of i.g. EtOH; Crews et al., 2013; Qin and Crews, 2012a), a continuous EtOH diet (15 days; Knapp et al., 2016; Whitman et al., 2013), forced CIE (three 5-day bouts of EtOH diet separate by 2 days of control diet; Whitman et al., 2013) and voluntary CIE (every-other-day with 15 % v/v EtOH solution for 60 days; McCarthy et al., 2018). Thus, regardless of EtOH exposure model, EtOH exposure and the acute withdrawal period increase the expression of multiple TLR receptors.
TLR signaling leads to activation of the NF-κB pathway through IκB-〈 phosphorylation and degradation, which regulates the expression of pro-inflammatory cytokines that propagate the immune response (Crews et al., 2017; Wu et al., 2009). Studies using a 5-month EtOH drinking model found a decrease in IκB-〈 and an increase NF-κB-p65 subunit protein in the cortex during EtOH exposure (Alfonso-Loeches et al., 2010; Vallés et al., 2004). The increase in protein levels and mRNA expression of NF-κB-p65 were also observed during the drinking phase and following a 24-hr withdrawal period, following 10 EtOH exposures (i.g.) in the hippocampus and cortex of rats (Tiwari and Chopra, 2013) and in the whole brain of mice (Qin and Crews; 2012b). In contrast, the effect of the same period of withdrawal (24-hr), did not change mRNA NF-κB within the cortex of rats following 15 days of continuous or intermittent EtOH drinking (Whitman et al., 2013). We found that following 6–8 months of drinking EtOH (20% v/v) there was no change in IκB-α mRNA expression during the drinking phase; however, 24-hr after the removal of EtOH there was a decrease in gene expression in the thalamus, hippocampus and frontal cortex of rats (Toledo Nunes et al., 2019). These data suggest that although CET did not alter expression of the IκB-α gene, the acute withdrawal period from alcohol triggered the activation of the NF-κB pathway. These conflicting results suggest that immune-related signaling pathways in the brain can vary with different doses and durations of EtOH exposure. However, EtOH also has been shown to induce the transcriptional activity of another factor called the activator protein-1 (AP-1). Both NF-κB and AP-1 transcription factors promote the expression of innate immune markers (Vallés et al., 2004) and may contribute to alcohol-induced neuronal apoptosis (Crews et al., 2015).
The involvement of inflammatory signaling with cytokine release, microglial activation and cognitive dysfunction induced by CET has also been investigated. Using a long-term (12 months) moderate EtOH (20% v/v in drinking water) exposure, Ehrlich and colleagues (2012) showed that CET induced spatial learning impairment was accompanied by activated microglia and partly increased inflammation in the cortex. Moreover, Tiwari and Chopra (2013) found that chronic EtOH exposure for 10 weeks led to elevated levels of neuroimmune markers, such as cytokines and NF-κB, as well as the apoptotic marker Caspase 3 in two brain regions (cerebral cortex and hippocampus) involved in learning and memory. They also revealed learning and spatial memory impairments in EtOH-treated rats, which took more time to find the platform during the learning tasks and spent less time in the target quadrant during the memory test in the Morris Water Maze. These results suggest that activation of inflammatory response as well as apoptotic signaling pathway may contribute to cognitive deficits associated with chronic EtOH exposure (Tiwari and Chopra, 2013). However, abstinence from EtOH resulted in decreased cytokines levels, which was accompanied by change in microglial response, and a recovery in both neuronal damage and cognitive dysfunction (Zhao et al., 2013). These findings suggest that activated microglia have a role in cognitive dysfunctions associated with EtOH exposure and may be time limited and possibly recoverable.
In summary, studies using chronic EtOH exposure, intermittent or continuous, in rodents have shown that EtOH activates the innate immune system, and contributes to brain damage and short-term cognitive dysfunctions. Current studies support the idea that there are multiple mechanisms by which EtOH exposure evokes neuroimmune signaling. One of these mechanisms is the alcohol-induced NF-κB pathway through TLR and cytokines receptors initiating cytokine expression in brain. Nonetheless, changes among inflammatory markers by chronic EtOH exposure are dependent of the dose of EtOH, duration of EtOH exposure, brain regions examined and withdrawal period. The acute burst of neuroimmune genes and proteins (IL-6, TNF-α and IL-1β) that occurs during the apex of TD within the thalamus, and to a lesser degree in the hippocampus and frontal cortex, normalizes within 24-hour period. However, this early neuroinflammatory response by TD could set the stage for tissue regeneration and recovery (barrier maintenance, debris clearance, cytokine and/or neurotrophin production), which could be hampered by concurrent EtOH toxicity and aging.
3. Alcohol Related Brain Damage: Key sites of pathology associated with cognitive dysfunction
As previously stated, a significant percent of alcoholics display cognitive impairments that persist into abstinence. The type, extent and duration of cognitive dysfunction is variable across the spectrum of AUDs. In the next section, we will describe some of the key neuropathology that is seen animal models of AUDs. Figure 2 outlines several critical neural circuits that are damaged by chronic and intermittent EtOH exposure, as well as those damaged by thiamine deficiency. The behavioral dysfunction associated with the pathology is also summarized in the Figure 2 and section 3.5
Figure 2.
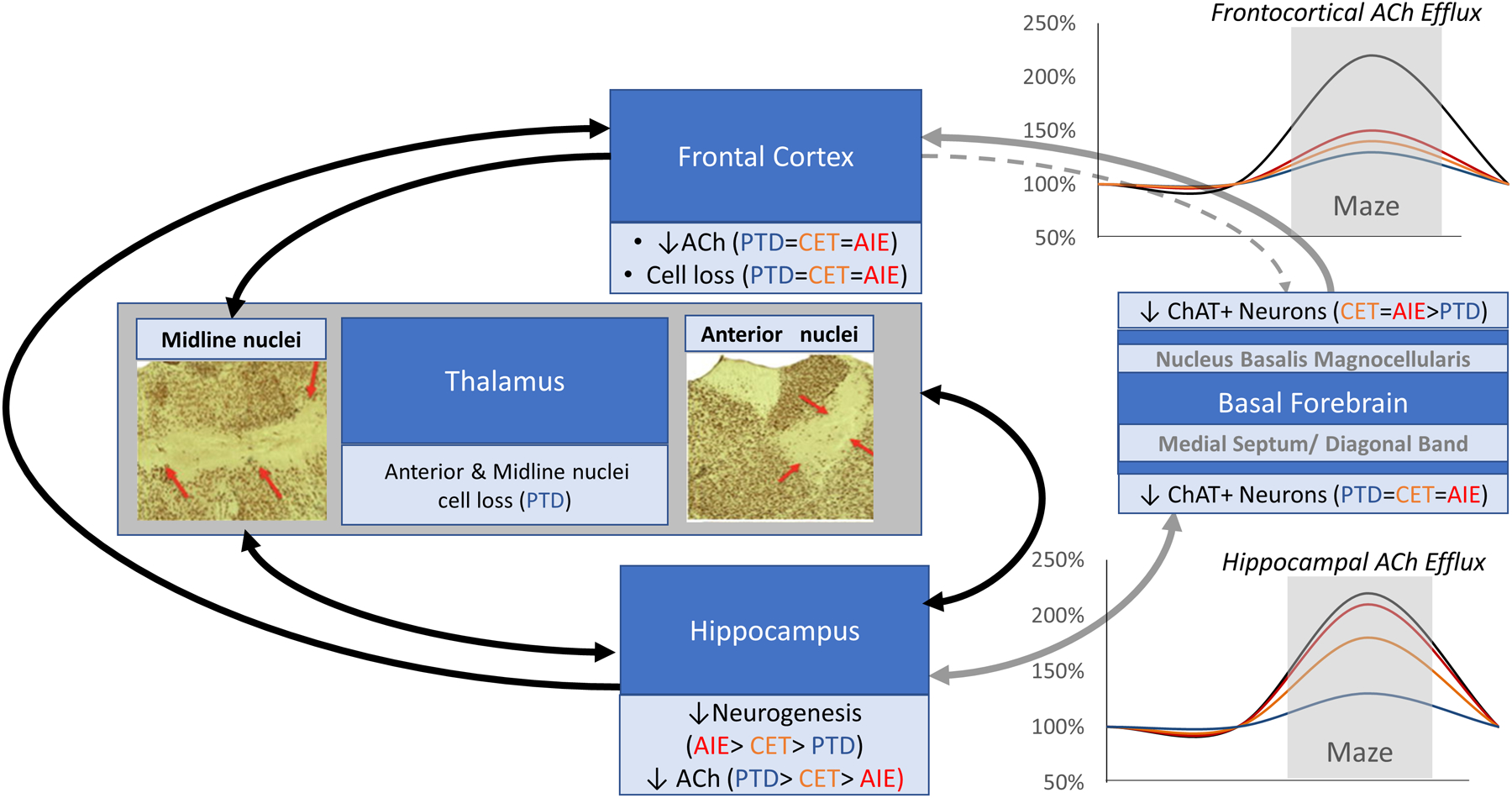
A schematic of the critical brain regions and cognitive circuits damaged by thiamine deficiency (PTD, blue), chronic ethanol treatment (CET; orange) and adolescent intermittent ethanol exposure (AIE, red), and the functional consequences in behaviorally-activated acetylcholine (ACh) release. PTD produces a substantial suppression of both frontocortical and hippocampal ACh efflux, where as CET lead to moderate reduction of frontocortical and hippocampal ACh efflux, AIE only leads to blunting of frontocortical ACh efflux. These functional differences can be explained by the extent of brain damage and critical circuit disruption each model generates. Only TD leads to loss of thalamic neurons involved that are interconnected with the hippocampus and frontal cortex. The anterior thalamus and hippocampus are directly connected, where as the medial and midline thalamus are connected to the frontal cortex. All models of alcohol-related brain damage (PTD, CET, AIE) led to a loss of the cholinergic phenotype (choline acetyltransferase immunopositive neurons [ChAT]) in the basal forebrain. The cholinergic projection from the medial septum/diagonal band is to the hippocampus and is topographically organized. Cholinergic innervation of the cortical mantle in the rodent, including the frontal cortex, is derived from the nucleus basalis magnocellularis (NbM). Thus, cholinergic system likely plays a role in the pathophysiology of alcohol-related cognitive dysfunction that is exacerbated by thalamic pathology when thiamine deficiency co-occurs in alcohol-use disorders.
3.1. Hippocampal dysfunction contributes to memory impairments in ARBD.
Studies examining the effects of EtOH neurotoxicity have identified the hippocampus as a region that experiences cell loss and functional impairment following life-long EtOH exposure that contributes to deficits in learning and memory. Postmortem assessment of hippocampal pathologies in humans with alcoholism has revealed significant reductions in hippocampal volume specifically within the pre-subiculum, fimbria, and subiculum (Lee et al., 2016). Additionally, these findings determined that chronic alcohol consumption produced more significant reductions in hippocampal volume above normal age-related degeneration alone (Lee et al., 2016). Moreover, significant volume reductions were identified in hippocampal CA1 and CA4 subfields alongside additional decreases in the granule cell layer, molecular layer, and dentate gyrus (Zahr et al., 2019). Chronic alcohol consumption appeared to accelerate age-related degeneration in specific hippocampal regions (CA2 and CA3; Zahr et al., 2019).
Preclinical models of alcoholism demonstrate both overall hippocampal atrophy and loss of hippocampal neurogenesis (Cadete-Leite et al., 1990a; Crews et al., 2006; Savage et al., 2000). There is a significant reduction in the overall number of hippocampal neurons during CET extending 6 or more months, which is further exacerbated following withdrawal (Paula-Barbosa et al., 1993; Roberto et al., 2002). Specifically, there is a ~20% reduction in neurons within the CA1 and CA3 regions (Lukoyanov et al., 1999b; Lukoyanov et al., 2000). Both granule cells of the dentate gyrus and hippocampal pyramidal neurons demonstrate significant reduction in total population resulting from chronic EtOH exposure. There is a ~250% increase in granule cell death following 6.4% EtOH treatment for 6 weeks and a 10% loss in the granule cell population following 5 months of 8.1–9.4% EtOH (Walker et al., 1980; Herrera et al., 2003). Higher concentrations and exposure to EtOH for longer periods of time produces more significant cell loss within the hippocampus (Franke et al., 1997; Lukoyanov et al., 1999b). For example, 10% EtOH exposure over the course of 12 weeks produced significant cell loss, however, increasing the duration of exposure to 36 weeks led to more extensive cell loss. CET treatment consisting of 20% EtOH v/v in the drinking water for 36 weeks produced an overall 15% reduction in hippocampal pyramidal neurons, with the CA3 region being the most sensitive (Lukoyanov et al., 1999b). Increased sensitivity of the CA3 subregion is also a consistent finding at lower doses of EtOH (10% v/v in drinking water) identified by the greatest cell loss within the region following 36 weeks of treatment (Franke et al., 1997).
Chronic EtOH treatment in preclinical models identified significant loss of GABAergic neurons within the hippocampus (Lescaudron et al., 1986; Cadete-Leite et al., 1997). Consistent with findings within the cholinergic system, detoxification confounds neuronal cell death of GABAergic neurons and a 24% reduction of GABAergic neurons within the suprapyramidal limb of the hippocampus (Lescaudron et al., 1986; Cadete-Leite et al., 1997). Similarly, glutamatergic pyramidal cells and granule cells were reduced following 5–6 months of 8–20% EtOH v/v in the drinking water and withdrawal (Walker et al., 1980; Herrera et al., 2003; Lukoyanov et al., 2000). EtOH directly inhibited the function of ionotropic glutamate receptors by reduced the magnitude of evoked responses and increasing the rate of desensitization (Lovinger et al., 1989; Gass and Olive, 2008). NMDA receptors are direct targets for EtOH and are inhibited in a dose dependent manner (Lovinger et al., 1989; Gass and Olive, 2008). Following 12 weeks of 5% EtOH treatment, NMDAR1 were elevated by ~65% (Trevisan et al., 1994). EtOH also inhibited AMPA and kainic ionotropic glutamate receptors (Gass and Olive, 2008). Similarly, in human alcoholics, hippocampal expression of AMPA and kainate receptors were upregulated alongside NMDA receptors (Jin et al., 2014). Conversely, glutamate metabotropic receptor mRNA expression was reduced after 8 weeks treatment of 5% EtOH v/v in the drinking water (Simonyi et al., 2004). Specifically, mGlu3 and mGlu5 mRNA expression was reduced in the dentate gyrus, whereas mGlu1, mGlu5, and mGlu7 mRNA were decreased in CA3 following CET. The reduction in mGlu3 and mGu7 mRNA was highlighted as evidence for a state in which glutamate excitotoxicity propagated through impaired presynaptic control of glutamate release (Clapp et al., 2008).
EtOH did not lead to a reduction in GABA-A receptor density in the hippocampus of macaques or rats (Sullivan et al., 2005); however, 5g/kg EtOH CIE every other day for 120 days in rodents reduced the functioning of GABA-A receptor mediated inhibition (Kang et al., 1996; 1998). Reduced GABA-A inhibition may correspond with the downregulation of α1 subunit expression, and the upregulation of α4 subunit expression, was observed following 5g/kg EtOH CIE every other day for 110 days (Cagetti et al., 2003). Taken together these findings highlight one mechanism of action for EtOH-induced glutamate excitotoxicity: EtOH exposure shifts hippocampal GABA/glutamate balance to produce an inhibitory effect by reduced NMDA receptor function and a compensatory increase in NMDA receptor expression (Crews et al., 1996). During detoxification, the GABA/glutamate balance is shifted to an overall excitatory effect and cell death through caspase-3 activation and apoptotic mechanisms (Wang et al., 2018).
Chronic EtOH consumption also results in a significant impairment in hippocampal synaptic plasticity that correlates with functional impairments (Vetreno et al., 2011; Hu et al., 1999). Rats treated with 35–39% of their caloric intake substituted for EtOH over 7 months displayed a significant reduction in the magnitude of evoked LTP. However, rats abstinent for 2 months showed a recovery in hippocampal LTP magnitude (Durand and Carlen, 1984). However, with a longer period of abstinence (5–7 months) the recovery in LTP magnitude was not evident (Tremwel and Hunter, 1994). The lack of LTP recovery after 7 months of abstinence suggests an interaction between CET treatment and aging, where EtOH promotes age-related neurodegeneration. Long-term depression (LTD), on the other hand, was also reduced in the hippocampus and rats exposed 8.1–9.4% EtOH v/v for 40 weeks (Thinschmidt et al., 2003).
Studies examining markers for neurogenesis have revealed that markers for progenitor cells, progenitor cell proliferation, and newly born neurons were all down regulated resulting from alcoholism (Le Maitre et al., 2018). More importantly progenitor cells appear to be especially vulnerable to chronic EtOH exposure and the reduction in immature neurons followed the loss in progenitor cell populations (Le Maitre et al., 2018). Preclinical models confirmed the reduction in hippocampal neurogenesis following CET (Golub et al., 2015). For example, CET treatment in mice consisting of 6.4% EtOH v/v for 4 weeks produced a ~ 66.3% decrease in immature neurons population (Herrera et al., 2003). Additionally, 4 weeks of 5% EtOH v/v led to a reduction in progenitor cell population and immature cell populations within the SGZ of female mice (Golub et al., 2015). Interestingly, markers for proliferation remained unchanged, which may implicate CET in disrupting cell cycle progression (Golub et al., 2015; McClain et al., 2011). Taken together these findings highlight the impaired hippocampal neurogenesis and the vulnerability of immature neurons to EtOH toxicity. However, prolonged abstinence can result in the recovery of neurogenesis (Rice et al., 2004).
Structural remodeling of neurons within the hippocampus is necessary for the maturation of immature neurons and for plasticity within this region (Hoogenraad et al., 2009). Surviving immature neurons also appear to be affected by EtOH exposure as immature neurons have a reduced dendritic growth and dendritic spine density following 4 weeks of 5% EtOH v/v reflecting a disrupted process of forming synaptic connections (Golub et al., 2015). Chronic EtOH consumption results in the downregulation of filamentous actin, polymerized tubulin, MT-associated protein 2, and GTPases, which participate in cytoskeletal reorganization (Romero et al., 2010). These structural proteins are crucial for neuronal development, dendritic spine formation, and intracellular trafficking functions.
However, the hippocampus attempts to recover from EtOH toxicity through synaptic reorganization and expression of neurotrophic factors. Further evidence for hippocampal reorganization during chronic EtOH exposure is supported by increased dendritic length of granule cells following a 35% caloric supplementation of EtOH for 5 months (Durand et al., 1989). Additionally, 6, 12, and 18 months of 20% EtOH exposure resulted in a similar increase of granule cell dendrite length; however, withdrawal obliterated the effect (Paula-Barbosa et al., 1993). An increase in dendritic length may reflect compensatory mechanisms in the recovery of hippocampal synaptic connections following cell loss (Paula-Barbosa et al., 1993). Specifically, the length of dendrites significantly increased in more distal regions, as opposed to proximal regions, with an overall increase in the number of dendrites as well (Durand et al., 1989). During 5% EtOH v/v treatment, NGF in the hippocampus was reported to increase by 21% in the first 2weeks, followed by a further 41% increase at 4 weeks, before normalizing by 12 weeks into CET (Nakano et al., 1996). In a CET model with higher EtOH concentrations (20% EtOH ) NGF expression increased by 210% at the 6 month point, but normalized by the 9th month of treatment (Gericke et al., 2005). However, following 6 months of 10% EtOH the number of NGF+ cells was significantly reduced in the hippocampus (Aloe et al., 1993). It appears that the compensatory change in NGF expression in the presence of EtOH is dependent on the dose and the duration of EtOH exposure, but neurons dependent on NGF eventually enter a pathological state.
Although gross anatomical findings have led to the conclusion that this hippocampus is largely spared from significant TD induced toxicity (Langlais et al., 1992), further examination in preclinical models have identified persistent changes in hippocampal structure and function depending on the model used to produce TD. TD can be induced through the elimination of dietary thiamine alone, however this model of thiamine deficiency does not produce characteristic anterior thalamic lesioning observed in alcoholic TD (Vetreno et al., 2011). In pre- pathological stages of TD, 9 days of dietary thiamine elimination, there is a significant reduction in hippocampal neurogenesis without significant loss of mature hippocampal neurons (Zhao et al., 2008). Only after 22 days, in mice, of diet induced TD is there significant reduction in dopamine, serotonin, and glutamate content in the hippocampus (Makino et al., 2019). Between 25–30 days of dietary TD have identified significant reductions in ChAT activity as well (Nakagawasai et al., 2000; Pires et al., 2001, 2005).
Unlike TD induced by dietary restriction alone, PTD results in diencephalic cell loss and hippocampal functional impairment. Within the hippocampus, PTD results in neuronal fiber degeneration in CA1–3 and the DG (Langlais and Zhang, 1997; Ikarashi et al., 2009, Inaba et al., 2016). Moreover, CA3 and DG regions appear to be more vulnerable to TD neurotoxicity as these regions exhibited severe cell loss (Inaba et al., 2016). Although there are no differences in hippocampal neurogenesis during TD treatment, there is a transient increase in progenitor cell proliferation following the cessation of TD treatment, however long-term sequelae from TD ultimately produces a 20% reduction in hippocampal neurogenesis (Vetreno et al., 2011; Hazell et al., 2014). Concurrently, there is also a reduction in basal forebrain ACh fiber projections to the hippocampus that results in a significant reduction in behaviorally elicited ACh release (Anzalone et al., 2010; Roland and Savage, 2007, Roland et al., 2008, Savage et al., 2003; 2007; Vetreno et al., 2008).
Impaired synaptic plasticity resulting from TD is also evident as treatment led to significant loss of dendritic spine density within the dentate gyrus, driven by the loss of mature dendritic spines opposed to immature spines (Inaba et al., 2016). Loss of dendritic spines correlates with impaired LTP and correspondingly, TD produces deficits in plasticity related markers such as BDNF and a reduction in LTP magnitude at CA3-CA1 synapses with simultaneous reductions in paired pulse facilitations (Vedder and Savage, 2017; Hall and Savage, 2016). Increasing hippocampal BDNF levels, whether through direct administration or indirectly through exercise and environmental enrichment, recovers hippocampal plasticity (Bekinschtein et al., 2011; Chourbaji et al., 2011; Harland et al., 2014). Stimulation of neurotropic activity following TD with bath administration of BDNF also results in recovered LTP magnitude and paired pulse facilitations, while voluntary exercise recovers hippocampal cholinergic deficits (Vedder and Savage, 2017; Hall and Savage, 2016; Hall et al., 2018).
In conclusion, chronic EtOH, thiamine deficiency, and the combination of the two result in global dysregulation of hippocampal structure and function. Significant reductions in cholinergic transmission and neurotrophic activity within the hippocampus accompany dysfunction and we have identified the cholinergic neurons of the MS/DB as a target for therapeutic recovery from ARBD, as neurotrophic stimulation improves hippocampal ACh content which correlates with improved hippocampal dependent cognition (Hall and Savage, 2016; Hall et al., 2018, Vedder and Savage, 2017).
3.2. Cholinergic basal forebrain dysfunction is seen following both chronic EtOH exposure and thiamine deficiency.
The basal forebrain cholinergic system consists of several groups of nuclei that are implicated in modulating learning, memory, and attentional processes (Boskovic et al., 2019; Blake and Boccia, 2016). This region is also a key site of ARBD, which is persistent and contributes to deficits in cognitive functioning (Pepeu and Grazia, 2017; Vetreno et al., 2011; Savage et al., 2012). Four anatomically distinct subregions comprise this system which innervates cortical and hippocampal regions, and to a lesser extent thalamic and amygdalar areas. The medial septal nucleus (MS) and the vertical limb of the diagonal band (DB) innervate the hippocampus, whereas the horizontal limb of the diagonal band innervates the olfactory bulb, and the nucleus basalis magnocellularis (NbM) innervates much of the neocortex with lesser projections to the amygdala (Mesulam et al., 1983). Moreover, these projections are not exclusively cholinergic either, but a heterogeneous composition of cholinergic, GABAergic, and glutamatergic fibers that modulate learning, memory, and attention (Mesulam et al., 1983; Wainer et al., 1985; Dutar et al., 1995; Solari and Hangya, 2018).
Chronic EtOH consumption models of alcoholism consistently find a reduction in choline acetyltransferase (ChAT) and acetylcholine esterase (AChE) expressing cells within the MS/DB and NbM (Aloe and Tirassa, 1992; Arendt et al., 1988, 1989, 1995a, 1995b). The loss of ChAT expressing neurons is accompanied by a significant decrease in acetylcholine synthesis by 60–80% in surviving neurons (Arendt et al., 1995a) leading to a corresponding reduction in ACh activity (Arendt et al., 1988, Floyd et al., 1997) and terminal release in the hippocampus (Inagawa et al., 2004) and cortex (Arendt et al., 1988, 1989; Ehrlich et al., 2012). There is also a significant reduction AChE activity (Arendt et al., 1995a; Hodges et al., 1991). Not only is there an impairment in ACh and AChE activity, CET leads to impaired vesicular storage of ACh and choline transport functioning (Floyd et al., 1997; Arendt et al., 1989) in the basal forebrain. The cholinergic neurons that survive CET display morphological changes that suggest there is some extent of compensatory remodeling taking place (Arendt et al., 1995a, 1995b).
There is also evidence of altered neurotrophic activity within the basal forebrain following CET that is dose and duration dependent. Following 8.1–9.4% EtOH treatment for 28 weeks, there is a reduction in TrkA mRNA receptor expression as well as a downregulation of TrkA proteins within the MS/DB (Miller et al., 2002), with a corresponding reduction in NGF (Miller and Mooney, 2004). Six months of 10% EtOH fortified diet produced a reduction in hippocampal NGF positive cells, with a concurrent reduction in ChAT activity (Aloe and Tirassa, 1992). A reduction in NGF, NT-3, as well as the p75NTR, have been reported following CET (Cadete-Leite et al., 2003; Ehrlich et al., 2012). In rats treated with 20% EtOH v/v in drinking water, NGF mRNA expression was examined in the basal forebrain alongside ChAT, ACh synthesis, and ACh content at 4, 8, 18, and 28- week time points (Arendt et al., 1995a). ACh, ChAT, and ACh synthesis were reduced as a function of the duration of CET treatment and NGF mRNA expression was higher in the basal forebrain compared to controls. Follow up studies that examined NGF protein expression in the basal forebrain following the same CET treatment identified an 850% increase in expression, whereas 9 months of CET did not result in significant differences from controls (Gericke et al., 2005). Basal forebrain neurons that survive CET express p75NTR, but there is an overall reduction by 20–30%, suggesting that neurons expressing this receptor may have increased resilience to EtOH toxicity (Arendt et al., 1995a).
Target derived neurotrophins regulate cholinergic forebrain populations and reductions in neurotrophic activity lead to cholinergic dysfunction (Sofroniew et al., 1993). NGF and its precursor, proNGF, act upon p75NTR and TrkA receptors in the brain but induce very different biological effects driven by varying affinities between these receptors. For example, NGF preferentially binds with TrkA receptor with reduced affinity to p75NTR, while proNGF demonstrates high affinity for p75NTR, but TrkA to a lesser extent (Boskovic et al., 2019). While TrkA-NGF interactions produce a neuroprotective effect through various signal transduction mechanisms, p75-proNGF binding induces apoptotic mechanisms through JNK signaling (Fahnestock et al., 2019). Although NGF and proNGF produce contrasting biological effects, most antibody-mediated methods of detecting NGF through ELISA and IHC methods fail to differentiate the two signaling molecules and findings generally represent the combination of the two (Malerba et al., 2016). Upregulation of detected NGF in frontal cortex and hippocampus using ELISA assays following 6 months of 20% v/v CET treatment (Gericke et al., 2005), likely reflects increases in both proNGF and NGF. While impaired retrograde transport of mature NGF may contribute to basal forebrain pathology in ARBD, alterations in proNGF to NGF ratios appear to be more likely as administration of intracerebral injections of NGF following 16 weeks of CET treatment led to recovery of ChAT and neurotrophin receptor expression of forebrain cholinergic neurons (Aloe and Tirassa, 1992).
As previously stated, TD co-occurs in alcoholics. Thiamine is a necessary cofactor in several metabolic reactions, as well as the production of acetylcholine in cholinergic neurons (Todd and Butterworth, 1999b). Since thiamine is essential for normal energy production and neurotransmitter production in cholinergic neurons, these cells are especially vulnerable to complications associated with TD (Szutowicz et al., 2006). Additionally, cholinergic neurons are more sensitive to the effects of glucose deprivation as they display greater energy demands (Szutowicz et al., 2006). Dietary restriction of thiamine produces basal forebrain cholinergic cell loss in as early as 9 days (Zhao et al., 2008), while continued TD leads to more pronounced lesioning at day 30 with protraction of cholinergic projections to the hippocampus (Nakagawasai et al., 2000; Roland and Savage, 2009). Dietary TD-induced BF cell loss reduced AChE activity in projection regions as measured in the cortex and hippocampus (Pires et al., 2001; 2005). Differential sensitivity of BF cholinergic regions to combined CET and TD can be inferred with the use of rodent models. It appears that the presence of p75-NGF receptor confers additional sensitivity to EtOH and TD related toxicities (Nardone et al., 2013). We have observed a sparing of NBM ChAT+ neuron in the NbM, but not MS/DB following TD (Roland and Savage, 2009). The increased sensitivity of the MS/DB compared to the NbM results from the neurotrophic regulation in maintenance of MS/DB neurons by GABAergic hippocampal neurons (Lauterborn et al., 1993; Acsady et al., 2000). The p75 NGF receptor is expressed by ChAT+ cells in the MS/DB that is activated upon the retrograde transport of NGF from hippocampal projections (Acsady et al., 2000). Conversely, NbM cholinergic neurons projecting to the amygdala do not express p75 NGF receptors (Berger-Sweeney et al., 1994; Heckers et al., 1994). Studies on the effects of PTD in rodent models have revealed that subclinical TD does not produce significant cell loss of ChAT cells within the MS/DB, nor do they result in significant reduction in ChAT activity; while severe TD consistently produces ~30% reduction of ChAT+ neurons within this region (Thompson and McGeer, 1985; Pitkin and Savage, 2001; 2004; Roland and Savage, 2009). The effects of TD appear to be ChAT specific as GABAergic neurons within the BF are largely unaffected by mild and severe TD (Roland and Savage, 2009). Severe TD leads to significant blunting of ACh efflux in the hippocampus and cortex, as measured through in vivo microdialysis (Savage et al., 2003; Roland and Savage, 2007; Savage et al., 2007; Vetreno et al., 2008). As observed in human alcoholic WKS cases (Schliebs and Arendt, 2011), the extent of BF ChAT loss correlates with the extent of behavioral impairment on BF target-dependent tasks such as hippocampal dependent spatial working memory.
There is evidence that basal forebrain neurons can be rescued following EtOH and TD. For example, grafting fetal neurons rich in ACh into the cortex or hippocampus results in a recovery of phenotypic expression of ChAT in neurons, improved ACh synthesis, release, and reuptake within the MS/DB (Arendt et al., 1989). The grafting of purified astrocytes also recovered the ChAT expression in neurons to levels indistinguishable from controls (Brückner and Arendt, 1992). Forebrain administration of NGF can recover cholinergic ARBD deficits through the restoration of the ChAT phenotype (Aloe and Tirassa, 1992). Following PTD, exercise upregulated neurotrophic activity in the form of NGF led to a recovery of ChAT+ neurons in the MS/DB following PTD (Hall et al., 2018). It is apparent that a subset of cholinergic neurons expressing nestin has increased sensitivity to the effects of PTD. However, voluntary exercise produced a more robust recovery of this subtype of cholinergic neurons, suggesting that these neurons are in a quiescent state with undetectable ChAT expression (Hall and Savage, 2016). More recently, evidence from our lab demonstrated that NGF activity specifically drives this recovery (Hall et al., 2018). Only inhibition of TrkA activity prevented the recovery of cholinergic activity through the reemergence of the ChAT/Nestin expressing cells within the MS/DB, while TrkB inhibition still allowed for exercise mediated recovery from cholinergic dysfunction.
TrkA expression in basal forebrain cholinergic neurons acts as a metaphorical functional telomere in the preservation of the ChAT phenotype of basal forebrain neurons in lieu of age-related or disease state induced upregulation of proNGF. Taken together with findings of CET induced reductions in cholinergic basal forebrain expression of TrkA, but not p75NTR following 28 weeks of 8.1–9.4% EtOH v/v (Miller et al., 2002), EtOH may accelerate the ability of cholinergic neurons to function and express phenotypical markers. However, we have shown that exercise, via NGF upregulation, can revive forebrain cholinergic neurons (Hall and Savage, 2016, Hall et al., 2018). Inadequate conversion of proNGF to mature NGF could create a concentration-dependent competitive antagonist effect on TrkA receptor binding sites, thereby limiting the biological response of TrkA and the extent to which mature NGF provides trophic support, contributing to ARBD.
To date, no studies have examined the relative differences in the expression of proNGF to NGF in response to chronic EtOH exposure or TD, and interventions directed at NGF metabolism may provide increased resilience to the neurotoxic effects of severe TD or CET. Specifically, blocking MMP9, which is responsible for rapid degradation of mature NGF (Bruno and Cuello, 2006), may be an effective avenue in increasing the abundance of mature NGF to act upon TrkA receptors and suppress ARBD in the basal forebrain.
3.3. The frontal cortex is a key site of ARBD
Neuropathological studies in humans have shown reductions of cortical gray and white matter with enlarging of the sulci, as well as loss of cortical neurons after chronic EtOH consumption (see review: Harper, 1998; Fadda and Rossetti, 1998). The significant white matter loss (the prefrontal cortex, corpus callosum, and cerebellum) and gray volume loss, as well as, astrocytic loss and death was observed in conjunction with glial dysfunction in alcoholic patients (Harper, 1998; 2009; Kril et al., 1997; Miguel-Hidalgo et al., 2002). In uncomplicated alcoholics and alcoholics with WKS, the prefrontal cortex is more vulnerable to chronic EtOH exposure than other cortical regions (Fadda and Rossetti, 1998), as there are decreases in neuron density (15–23%; Harper and Matsumoto 2005), shrinkage of frontal lobe volume (Pfefferbaum et al., 1997), and decreased metabolic rate (Adams et al., 1993; Gilman et al., 1990). Reduction of regional cerebral blood flow was also observed in the frontal lobe of alcoholics (Chung et al., 2009; Hunter et al., 1989), which can recover with prolonged abstinence (Gansler et al., 2000).
Neuropathological changes in frontal cortex associated with chronic EtOH also have been demonstrated in a rodent model of alcoholism. Cadete-Leite and collaborators (1990a) revealed that CET for 6, 12 and 18 months (20% aqueous EtOH v/v) and withdrawal periods resulted in reduction of cell density, as well as decreased cortical thickness in layers I-III in the prelimbic cortex of rats. Similar to these findings, our research group also showed alteration in the frontal cortex after CET (20% v/v for 20 weeks) and subsequent withdrawal: CET resulted in shrinkage of white and gray matter in the frontal cortex, as well as memory deficits (Savage et al., 2000). These findings suggest that neuropathological alterations in the frontal cortex associated with prolonged EtOH exposure can contribute to learning and memory impairments.
The mechanisms underlying these cortical pathological changes caused by CET remain somewhat elusive. The neuroinflammation process engaged by prolonged CET and withdrawal is one proposed mechanism (Crews and Vetreno, 2016; Yang et al., 2014). Changes in neuroimmune markers were observed in cortex of rodents after long-term CET (5 months of 5 to 10% v/v in drinking water; Alfonso- Loeches et al., 2010; Vallés et al., 2004). However, our recent study showed that CET (20% v/v for 6 months) resulted in only small fluctuations in neuroimmune response in frontal cortex, as well as in hippocampus and thalamus. Also, the combination of CET and PTD model did not exacerbate the neuroinflammation response in frontal cortex, but decreased the IκB-α gene during acute withdrawal (24-hrs post EtOH), which suggests activation of immune-related signaling pathway through NF-κB during EtOH withdrawal (Toledo Nunes et al., 2019).
Decreased neurotrophic activity is another mechanism that may be involved in EtOH-induced neurodegeneration in the adult brain (Davis, 2008). Boyadjieva and Sarkar (2013) state that BDNF and cAMP are potential inhibitors of microglia activation, which may inhibit neuroinflammation, and protect against neurodegeneration induced by EtOH. Furthermore, our previous study showed a decrease in BDNF and NGF protein levels in the frontal cortex of rats treated with CET (20% v/v) for 6 months (Fernandez et al., 2016; Vedder et al., 2015). This down-regulation of BDNF was also observed during acute and protracted abstinence from CET (Fernandez et al., 2016), and when CET was combined with PTD (Vedder et al., 2015). These findings suggest that long-term EtOH exposure can influence plasticity within the frontal cortex, which can modulate functional and structural process associated with alcoholism (Koob and Volkow, 2010).
In contrast, Hansen and colleagues (2017) showed that EtOH exposure and withdrawal did not change BDNF mRNA in the frontal cortex of rats treated with EtOH 2 g/kg twice daily by oral gavage for 28 days. This contrast also was found in other studies that have shown that CET alters BDNF levels in different directions, with increase, decrease or no change (see review by Davis, 2008). Some different results in BDNF levels can be explained by the periods analyzed, such as during EtOH treatment, acute withdrawal or recovery (see Crews and Nixon, 2009). Also, the duration of treatment and concentration of EtOH exposure can affect the expression of neurotrophin genes and protein levels.
Studies using a CIE paradigm in mice have shown that intermittent EtOH exposure and withdrawal alters cortical glutamatergic synaptic function by increasing the NMDA/AMPA current ratio at glutamatergic synapses in medial prefrontal cortex (mPFC; Kroener et al., 2012). Also increases in expression of NMDA receptor subunits (NR1, NR2A and NR2B) were found after cessation of CIE (Nelson et al., 2005; Qiang et al., 2007; Rani and Ticku, 2006). However, this increase was not observed after 1 week of abstinence (Kroener et al., 2012). These changes in glutamatergic transmission in mPFC pyramidal cell described by Kroener and collaborators (2012) were suggested to contribute with deficits in behavioral control seen in alcohol dependent mice.
Impairment on GABAA receptor-mediated neurotransmission in deep-layer pyramidal cells of the mPFC has also been observed in rats treated with 5.0 g/kg; intragastric gavage; for 14 to 15 consecutive days (Hughes et al., 2019). This finding indicated that inhibitory neurotransmission was impaired by EtOH exposure and are consistent with clinical reports of elevated PFC activity observed in human alcoholics (Kamarajan et al., 2004; Seo et al., 2013). Other studies have shown that CET alters the expression of GABAA receptor subunit in the cerebral cortex of rodents (Fadda and Rossetti, 1998; Roberto and Varodayan, 2017), which resulted in a decrease in mRNA and protein expression of the α1, α2 and α3 subunits and increased expression of the α4, β1, β2, β3, γ1 and γ2 subunits (Devaud et al., 1995, Devaud et al., 1997). The dysregulation of neurotransmitters in the PFC by chronic EtOH might be associated with neural functions important to the loss of control of drinking behavior (Roberto and Varodayan, 2017; Moselhy et al., 2001).
Studies using a PTD protocol in rodents have revealed that TD, but not CET, significantly decreased in vitro glutamate release in the frontal cortex and resulted in learning deficits observed in the Morris water maze (Carvalho et al., 2006). Glutamate excitotoxicity is considered one mechanism of cell death in TD (Savage et al., 2012), which can be associated with cortical tissue loss in prefrontal cortex of TD rats (Langlais and Savage, 1995). Furthermore, TD in rodents produces frontal cortical shrinkage (Langlais and Savage, 1995), neural degeneration in layers III through VI of cerebral cortex (Le Roch et al., 1987; Langlais and Zhang 1997), and have shown a strong association between cognitive deficits and the degree of cortical and diencephalic damage (Pires et al., 2005; Anzalone et al., 2010).
Reduced acetylcholinesterase (AChE) activity and ACh release in cerebral cortex was verified after 30 days of recovery from PTD treatment in rats, and AChE activity was correlated with impairment on spatial memory performance (Pires et al., 2001, 2005). Our lab has shown that PTD treatment led to 25% reduction in density of AChE-positive nerve fiber in prefrontal frontal cortex and a 19% reduction in retrosplenial cortex (Anzalone et al., 2010). Furthermore, ACh efflux was blunted in prefrontal frontal cortex, which was positively correlated with spontaneous alternation performance of PTD rats.
Overall, the mechanisms involved in neuropathology induced by EtOH exposure or TD in the frontal cortex contribute to behavioral dysfunction such as impaired impulsivity, cognitive and behavioral flexibility and working memory. It appears that the adult CIE paradigms do not produce protracted frontal cortical dysfunction, whereas CET that progresses across months does, but not to the same degree as TD. One key difference between the CIE and CET paradigm is duration: CIE paradigms last 1–2 months, whereas CET paradigms often last 6–18 months. The longer duration of CET paradigm engages the aging process and this likely contributes to the more persistent frontocortical pathology. For example, only 12 and 18 months of CET led to a decrease in neural density in the prelimbic cortex, as well as a reduction in pyramidal cell dendritic length (Cadete-Leite et al., 1990a, 1990b).
3.4. Thalamus as a critical region of ARBD associated with thiamine deficiency:
Chronic and excessive EtOH consumption results in thalamic damage and affects two brain networks associated with cognitive functions: the frontocerebellar circuit and the Papez circuit. Both networks share the thalamus, a diencephalic region that plays an important role as a node of these networks and appears to be vulnerable in alcoholics (Fadda and Rossetti, 1998; Pitel et al., 2011; 2015). Clinicopathological studies in alcoholics report shrinkage of the thalamus as a consequence of chronic and heavy alcohol consumption, and the resultant TD. In addition, the consequence of chronic alcohol consumption seems to be more severe in alcoholics with WKS than in uncomplicated alcoholics (Pitel et al., 2012; 2015; Sullivan and Pfefferbaum, 2009). A recent study in humans has suggested that atrophy of anterior nuclei observed in patients with WKS can be considered a potential neuroimaging marker of the pathophysiology of WKS, and the disconnection between the anterior nuclei and hippocampus may be the leading factor in the persisting amnestic syndrome (Segobin et al., 2019). In alcoholics with or without a contaminant diagnosis of WKS, reductions in the medial dorsal nucleus and its connections to the prefrontal cortex were found. Although loss of thalamic mass was reported in alcoholics without WKS, it was not as severe as alcoholic WKS patients. Thus, pathology in the thalamus, in particular the anterior thalamic group, is a reaction to TD associated with chronic alcohol abuse.
Rodent models have shown that TD clearly causes brain lesions and degenerative changes in several nuclei of the thalamus (anterior thalamus, midline thalamus, posterior thalamus), which are affected progressively with rapid neuronal loss as a consequence of the severity of TD (Ke et al., 2003; Langlais and Savage 1995; Zhang et al., 1995; Savage et al., 2012; Vetreno et al., 2012). TD-induced glutamate excitotoxicity also occurs through glutamate transporter disruption, which is thought to be the key mediator of thalamic pathology (Langlais and Mair, 1990; Langlais and Zang, 1993).
Our laboratory has also examined whether chronic ethanol potentiates the thalamic pathology associated with TD. Using a prolonged EtOH drinking model (a progression to 20% v/v EtOH in drinking water across 8 months) alone or combined with PTD treatment in rats we investigated the thalamic damage observed as a consequence of chronic EtOH, TD and the interaction of both experimental conditions (Vedder et al., 2015). Lesions in the midline thalamic region were seen in rats treated with PTD, and lesions ranged in magnitude from modest to large lesions with the severity of TD. Modest lesions were also observed in rats treated with PTD and CET, but the lesions were not amplified relative to the PTD alone conditions. Furthermore, CET alone did not produce thalamic lesions. It was found that even though CET alone produced a significant suppression of thiamine diphosphate, the active form of thiamine, there was no loss of thalamic volume in the CET alone group. This follows the expression of neuroinflammation genes outlined in section 2 and suggests that activation of neuroinflammatory markers contribute to thalamus degeneration (Qin and Crews, 2014; Wang and Hazell, 2010; Yang et al., 2011).
Astrocyte dysfunction with loss of astrocyte markers and glutamate transporters also contribute to the TD metabolic dysfunction (Todd and Butterworth, 1998; 1999a; Hazell and Butterworth, 2009; Hazell et al., 2010). Furthermore, glutamate-mediated excitotoxicity is considered an important mechanism of cell death in the TD model which suggests that glutamate plays a key role in the development of thalamic lesions (Hazell et al., 1993; Langlais and Zhang, 1993; Todd and Butterworth, 1998). The increase in the levels of extracellular glutamate were observed in the midline and lateral posterior nuclei of the thalamus after 4–5 hours post seizure (Hazell et al., 1993; Langlais and Zhang, 1993).
Investigations of long-term EtOH exposure in animal models have shown that prolonged EtOH exposure can produce some neuronal loss in the thalamus, as well as other brain structures involved in memory processes (Beracochea et al., 1987a; 1987b). An experimental study using 7 months of forced EtOH consumption alone (12% v/v EtOH in drinking water) showed a decrease in the volume of the medial dorsal and anterior nuclei of the thalamus in mice (Beracochea et al., 1987a). However, the loss of cells in the dorsomedial and anterior thalamus was small (11–16%), compared to what is observed following TD, which is an almost complete loss of neurons in the anterior ventroventral lateral and intralaminar midline nuclei (see Savage et al., 2012). However, heavy chronic drinking during adolescence in monkeys attenuated the developmental growth rate of the thalamus (Shnitko et al., 2019), suggesting that developmental EtOH exposure is more damaging to the thalamus than adult exposure.
Although thalamic pathology does exist in WKS patients with and without an alcoholic history, animal models have demonstrated that significant cell loss and lesions only occur with TD. Chronic EtOH does alter some properties of the thalamus, but whether that translates into functional abnormalities, in particular cognitive dysfunction is unknown. It has been stated that EtOH-induced thalamic alterations cannot account for the memory dysfunction associated with chronic EtOH treatment (Célérier et al., 2000). In contrast, with TD the extent of thalamic lesions has been associated with the degree of thalamic pathology (Langlais and Savage, 1995; Mair, 1994).
3.5. Behavioral impairment following chronic EtOH exposure and/or thiamine deficiency is both hippocampal and frontocortical dependent.
Alcohol use disorders are characterized by dynamic periods of binge level consumption, withdrawal, abstinence and relapse across a significant period of time (Le Berre et al., 2014). Correspondingly, there is significant variance in the degree of cognitive impairment associated with AUD’s (Fein et al., 1990; Oscar-Berman et al., 2014; Sullivan et al., 2010). Animal models have helped determine the sources of variability in behavioral dysfunction associated with AUDs.
The relative contributions of different etiological factors are key in the development of ARBD and its functional consequences. Both EtOH toxicity and TD result in brain damage and cognitive problems (Vetreno et al., 2011). CET can lead to impairments in hippocampal-dependent spatial learning, working and reference memory (Abadi et al., 2013; Lukoyanov et al., 1999b, 2003; Santucci et al., 2004; Vedder et al., 2015; Wang et al., 2018). It appears that the duration of EtOH dictates the severity of spatial working memory impairment as 10% EtOH v/v CET, and 20% EtOH v/v CET produced spatial working deficits on the radial arm maze in mice only after 180 days of treatment (Wang et al., 2018). Similarly, spatial reference memory impairments on the Morris water maze task only became evident following 90 days of CET treatment. Moreover, 20% EtOH v/v CET for 196 days led to a reduction in the rate of spontaneous alterations compared to control conditions (Vedder et al., 2015). The rate of alternation was found to be negatively correlated with BECs and hippocampal BDNF levels. Spatial learning in the Morris water maze was also affected by daily EtOH administered (4g/kg 20%) by gavage for 4 weeks, which was recovered by exercise following EtOH exposure (Abadi et al., 2013). The grafting of ACh rich fetal neurons into the hippocampus or cortex following CET (20% v/v for 28 weeks) also reversed spatial working memory impairments on the radial arm maze task (Arendt et al., 1989).
Parallel to spatial learning and memory deficits, CET treatment has been shown to also produce impairments in nonspatial discrimination as well (Fernandez et al., 2016). Following 20 % EtOH v/v exposure for 6–8 months, rats required significantly greater number of trials to reach criterion on compound discrimination tasks (Fernandez et al., 2016). Behavioral flexibility, or the propensity to change behavior in response to changes in environmental contingencies was also notably reduced following CET (Fernandez et al., 2016). Parallel to this, CET treatment of the same concentration and duration led to significantly greater trials required to reach criterion on an attentional set shifting paradigm (Birrell and Brown, 2000) on extradimensional set shift, and reversal tasks (Vedder et al., 2015). Generally, impairments in behavioral (reversal learning) or cognitive flexibility (extradimensional set shift) are reflective of perseverative behavior, as animals will continue to engage in previously learned behavior.
CIE exposure models of episodic drinking produce cognitive vulnerabilities, although not as global as those induced by CET. We found that rats treated with 5g/kg EtOH via gavage in accordance to a 2 day on, 2 day off cycle for 3 weeks, required significantly more trials in order to reach criteria on a simple discrimination task, and this treatment also led to impairments in behavioral flexibility, as the animals performed worse than water controls on reversal tasks (Fernandez et al., 2016). Kuzmin and colleagues (2012) identified similar deficits in behavioral flexibility assessed by reversal learning on the Morris water maze, suggesting CIE induced frontocortical dysfunction. However, the impairment was transient on spatial memory, persisting only for the first 3–6 days following EtOH withdrawal, but recovered with protracted abstinence. Thus, CET with its longer duration appears to produce spatial memory impairments that are more protracted than CIE.
PTD models of thiamine deficiency lead to more robust decrements in cognitive functioning and more closely resemble the pattern of neuropathology observed in human WKS. Performance of PTD treated animals on delayed matching-to-place tasks and non-matching to place tasks demonstrate working memory and spatial reference memory impairments (Mair et al., 1985; Langlais and Savage, 1995; Mumby et al., 1999). This dysfunction is also evident on auditory- and odor-based matching-to-position and matching-to-sample tasks (Mair et al., 1991a, 1991b). Similarly, our lab as consistently identified spatial working memory dysfunction on the spontaneous alternation task as well (Roland et al., 2008; Savage et al., 2003; 2007; Vetreno et al., 2008; Resende et al., 2012; Vedder et al., 2015; Hall et al., 2016), and PTD associated pathology of the intramedullary lamina confers additional deficits in performance on this task (Langlais and Savage, 1995). Additionally, when placed in the Morris water maze PTD treated animals maintained a thigmotaxic search strategy (Langlais et al., 1992). The aforementioned incidents of perseverative behavior most likely reflect behavioral inflexibility, as PTD treated animals require significantly more trials to master tasks during reversal learning and attentional set shifting (Vedder et al., 2015).
Combined CET-PTD treatment does not always reveal synergistic behavioral deficits. beyond those observed with thiamine deficiency alone (Ba et al., 1999; Ciccia and Langlais, 2000; Homewood et al., 1997; Pires et al., 2001). For example, rats undergoing 3 months of 20% EtOH v/v CET treatment combined with PTD did not produce additional deficits on acquisition of the Morris water maze task beyond rats treated only with PTD (Pires et al., 2005). Similarly, when examining the effect of pre-training on the Morris water maze prior to CET and or TD treatments, only rat exposed to TD (with or without EtOH) did not demonstrate improved performance, when compared to their non-pretrained counterparts (Pires et al., 2005). The memory impairment observed in TD treated rats was found to be correlated with reductions in cortical and hippocampal AChE activity. Additionally, rats treated on a 20% EtOH v/v CET treatment with PTD demonstrated significant behavioral inflexibility on the attentional set shifting task, congruent with the neurocognitive dysfunction observed in CET and PTD treated rats (Vedder et al., 2015). However, our lab has also identified potential synergistic effects of combined CET and PTD treatment in the form of attentional deficits in an operant attentional set shifting paradigm.
Thus, data across several studies confirm that the most severe and persistent spatial or working memory deficits, as well as cognitive/behavioral flexibility impairments are seen following severe TD. Although EtOH exposure may cause deficits in spatial memory, the effects are transient in the case of CIE or less severe in the case of CET. Such data supports the hypothesis that is emerging from the human clinical data that the majority of ARBD and behavioral sequelae of alcoholism is driven by TD (see Arts et al., 2017; Pitel et al., 2011).
4. Early alcohol abuse may prime the brain for accelerated aging
Aging is often characterized by maladaptive alterations at the molecular, cellular and behavioral levels. Although several neural circuits are affected by advanced age, we will focus on those involved in learning and memory. The hippocampus is the hub of two critical circuits involved in learning and memory: The septohippocampal pathway and the prefrontal- hippocampal pathway. Dysfunction in these pathways contributes to age- and alcohol- related declines in spatial and episodic memory (Fernandez and Savage, 2017; Hall and Savage, 2016; Mufson et al., 2015; Ohara, 2013; Vetreno and Crews, 2015). The interaction between alcohol abuse and aging has led to the “premature aging hypothesis” of alcohol dependence (Guggenmos et al., 2017; Pfefferbaum et al., 1992, Zahr et a., 2019; Zhao et al., 2019). In this section, we will summarize the critical neuronal dysfunction associated with adolescent intermittent EtOH exposure (AIE) alone, aging alone, and the combination of the two. Specifically, we will consider how early alcohol abuse may prime the brain for accelerated aging. In addition, we will examine the neurocognitive consequences of EtOH exposure and aging to determine critical intersections.
4.1. Critical brain regions affected by adolescent EtOH exposure
The hippocampus and its associated pathways are critical to learning and memory (Bird and Burgess, 2008). Furthermore, the hippocampus and the septohippocampal pathway are targets of both acute and chronic EtOH actions (Geil et al., 2014; Givens et al., 2000; Zahr et al., 2019). Interestingly, adolescents are more sensitive than adults to many of the effects of both acute and chronic alcohol, including hippocampal ARBD (Nixon and Crews, 2004; Broadwater et al., 2014). A reliable neuropathological finding associated with adolescent intermittent EtOH exposure (AIE) is an enduring reduction in neurogenesis across the hippocampal axis (Broadwater et al., 2014; Spear, 2018; Vetreno and Crews 2015; Vetreno et al., 2018; 2019). This is interesting as the adolescent period is normally a period of heightened neurogenesis (Curlik et al., 2014). However, adult rodents exposed to binge type EtOH exposure display a transient loss of hippocampal neurogenesis that recovers following a prolonged (30-day) abstinence period (Nixon and Crews, 2004; Broadwater et al., 2014). In contrast, the reduction in neurogenesis that follows AIE has been detected at least 7-months post-AIE, and as the rodent begins to enter middle-age there is an accelerated loss of hippocampal neurogenesis (Vetreno and Crews, 2015).
Vetreno and Crews (2015) also found a long-term upregulation of proinflammatory cytokines and proteases, which could explain the increased neuronal death and loss of neurogenesis in the hippocampus. A reduced expression of brain-derived neurotrophic factor (BDNF) protein and BDNF exon IV (but not exon I) mRNA has also been shown following AIE (Sakharkar et al., 2016). This significant and persistent decrease in BDNF expression could explain the loss of neurogenesis into adulthood (and likely into aged adulthood) following AIE. Briones and Woods (2013) found that rats who self-administer EtOH to intoxication levels show decreased levels of BDNF, followed by the development of despair behaviors, and anhedonia. When the EtOH treated animals were then treated with a BDNF-like agonist, neurogenesis was restored and despair behaviors and anhedonia were abolished (Briones and Woods, 2013). Thus, the recovery of neurotrophins restore neurogenesis following ARBD (Hall and Savage 2016; Vetreno et al., 2019).
Several other hippocampal abnormalities are found after AIE. There is an increased propensity for LTP induction in the adult hippocampus following AIE, which is likely due to an AIE-induced hyperplastic hippocampal circuit (Brun et al., 2001; Moser et al., 1998; Risher et al., 2015). In addition, AIE results in a greater proportion of CA1 immature to mature dendritic spines, suggesting impairment in experience-dependent pruning. Additionally, AIE produces an increase in length of both mature and immature spine types, but had no effect on overall spine density (Risher et al., 2015). Specifically, AIE resulted in more filopodia and long/thin spines (immature) and less stubby and mushroom spines (mature). Risher and colleagues (2015) also noted a decrease in PSD-95 and SAP102, two postsynaptic density proteins, within the CA1 region of the hippocampus. Dentate gyrus granule neurons also express decreased dendritic diameters as well as a reduction in total spine density following AIE. However, treatment of AIE-exposed adult rats with donepezil, an acetylcholinesterase inhibitor that increases acetylcholine levels, reversed hippocampal dendritic spine adaptations (Mulholland et al., 2018).
Cholinergic deficits have been observed following AIE (Fernandez and Savage, 2017). The septohippocampal circuit is critical for memory, and although spatial memory deficits may be absent or transient soon after AIE, the reduction in cholinergic medial septal/diagonal band (MS/DB) neurons is persistent into advanced adulthood (Fernandez and Savage, 2017; Swartzwelder et al., 2015; Vetreno et al., 2014, 2019). This effect is seen following adolescent, but not adult, alcohol exposure (Vetreno et al., 2014). This loss of cholinergic neurons may contribute to reductions in neurogenesis seen following AIE. A main input into the hippocampus is the fornix, which carries the axons from the MS/DB. The hippocampus, including the dentate gyrus, receives cholinergic, GABAergic and glutamatergic projections from the MS/DB (Bao et al., 2017; Cooper-Kuhn et al., 2004). Cholinergic neurons within the MS/DB contact new neurons and established granular cells of the dentate gyrus. Drugs that enhance acetylcholine levels increase hippocampal adult neurogenesis (Kotani et al., 2006) and the survival of newly born neurons is regulated in part by nicotinic α7 receptor (Kita et al., 2014). However, the neurogenic effect of AChRα7 may be limited to males (Otto and Yakel, 2019). In reciprocal fashion, neurogenesis appears to be critical for maintaining and stabilizing the cholinergic septohippocampal circuit, which is critical to successful spatial memory during normal aging.
Although several cholinergic nuclei throughout the brain are sensitive to the effects of AIE (Boutros et al., 2014; Ehlers et al., 2011; Fernandez and Savage, 2017; Galaj et al., 2019; Swartzwelder et al., 2015; Vetreno et al., 2014), unique phenotypic populations of cholinergic neurons (nestin+, nestin-) are differentially affected (Fernandez and Savage 2017). About 35% of the neurons that express choline acetyltransferase (ChAT), the enzyme responsible for the biosynthesis of acetylcholine (ACh), also co-express nestin, a filament protein implicated in the structure, plasticity, and remodeling of cells (Gilyarov, 2008; Hall and Savage, 2016; Hendrickson et al., 2011; Michalczyk and Ziman, 2005). Due to the high excitability and high expression of nerve growth factor receptors in ChAT+/nestin+ populations, as well as their role in cell support, it is likely that these neurons serve a neuroprotective role (Guo et al., 2010; Zhu et al., 2011). Within MS/DB, there was a decrease in the number of neurons that express the ChAT+/nestin- in rats exposed to AIE, but no change in the number of ChAT+/nestin+ expressing neurons (Fernandez and Savage, 2017). In contrast, within the nucleus basalis magnocellularis, which projects to the cortical mantle, there was a loss of ChAT+/nestin+ neurons, with sparing of the ChAT+/nestin- neurons. We did find, however, a decrease in soma size of the ChAT+/nestin+ neurons in the MS/DB following AIE, suggesting that this population displays some vulnerability. Like other studies (Fernandez et al., 2017; Swartzwelder et al., 2015; Vetreno and Crews, 2012), we did not find dorsal hippocampal-dependent learning impairments following AIE on a novel object recognition task or spontaneous alternation, nor was there blunted behaviorally-activated ACh efflux within the hippocampus (see Fernandez and Savage, 2017; Fernandez et al., 2017). However, AIE treated subjects displayed impairments on a social recognition task, which is partially mediated by the ventral hippocampus (Galaj et al., 2019). Thus, despite loss of hippocampal neurogenesis and reduced ChAT expression in cholinergic neurons that project to the hippocampus, AIE fails to induce hippocampal-dependent spatial learning and memory impairments in young adulthood.
However, there is evidence that when animals treated with AIE face an additional acute EtOH challenge in adulthood, they display greater hippocampal behavioral disruption relative to control rats (Risher et al., 2013). Specifically, AIE rats make more working memory errors on the radial arm maze following acute EtOH exposure compared to control. Such results suggest that there may be a sub-threshold septohippocampal dysfunction that may emerge as the circuit is strained by other neurobiological perturbations.
The hippocampus (from MS/DB) and the prefrontal cortex (from nucleus basalis) are two major areas of afferent cholinergic projections, strongly implicating them in the learning and memory deficits following EtOH exposure. AIE results in decreased connectivity between subregions of the prefrontal cortex and between PFC-striatal regions (Broadwater et al., 2018). These critical brain regions are likely affected the most by AIE due to their continued development throughout adolescence and that development and deleterious effects of EtOH toxicity. Interestingly, these pathways (septohippocampal and prefrontal-hippocampal) are also vulnerable to aging.
4.2. Aging affects critical pathways involved in learning and memory:
Normal aging leads to changes in neurotransmitter systems, neuronal organization and survival, and neuroimmune reactivity. The inability of a brain to adapt to normal aging dysfunctions contributes to the development of neurodegenerative and cognitive diseases. However, several compensatory mechanisms exist in the brain to deal with age-related neurobiological perturbations (Gray and Barnes, 2015). For example, areas of the aged brain exhibit a reduction in total number of synapses, yet the size of the synapses in that same area can increase (Morrison and Baxter, 2012; Terry et al., 1991). Age-associated synaptic deficits are likely due to alterations in the expression of several proteins involved in synaptogenesis and synaptic stabilization (postsynaptic density protein 95 (PSD-95), synapsin I, synaptophysin) and decreases in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and N-methyl-D-aspartate (NMDA) receptor subunits (Bettio et al., 2017). In addition, aged brains exhibit increased phosphorylation of the microtubule associated protein (MAP) tau in the entorhinal cortex and the hippocampus (Mattson et al., 1997). This increased phosphorylation of MAP tau results in the inhibition of microtubule assembly, which is noteworthy as aggregation of MAP tau is one of the main characterizations of Alzheimer’s disease (Jameson et al., 1980; Lindwall and Cole, 1984).
Several neurotrophins are altered in the senescent brain. Although BDNF and NGF protein levels do not change in the hippocampus of aged rats (male), there is reduced BDNF and NGF transcription, as well as a decrease in the cleavage of pro-BDNF and pro-NGF, leading to the accumulation of pro-BDNF and pro-NGF precursor proteins (Perovic et al., 2013). However, others have found that aged rats express less NGF in the hippocampus than young rats do, which could explain the decreased function of forebrain cholinergic neurons following aging (Lärkfors et al., 1987). However, other studies have found no change or even an increase in neurotrophins as rodents age (Bimonte-Nelson et al., 2008). It appears that factors such as strain, sex, and exact age all influence the levels of neurotrophins during the aging process. However, whether assays are sensitive to both the mature and pro- forms of the neurotrophins is critical. The neurotrophic imbalance between mature and pro-NGF is sufficient to induce a loss of cholinergic synapses (Allard et al., 2018). In addition, neurotrophins can increase in response to neuropathology: when rats undergo electrolytic lesioning within the medial septum, young rats exhibit an increase in NGF-like activity, where aged rats do not (Scott et al., 1994). Similarly, after total cholinergic immunolesion (192 IgG-saporin), NGF protein levels significantly increase in the hippocampus, cortex, and olfactory bulb of young rats, but not in aged rats (Gu et al., 1998). The lack of neurotrophic response to injury in the aged brain may be due to failed neurotrophic transport and signal transduction pathways between the hippocampus and the basal forebrain (Gu et al., 1998).
Neurogenesis exponentially declines as rodents age. Collectively, there is not substantial hippocampal neuronal loss due to aging (Rapp and Gallagher, 1996; Rasmussen et al., 1996), but there is a decrease in neurogenesis and deterioration of hippocampal formation processing (Morrison and Hof, 1997). Specifically, there is an 80% reduction in the rate of neural stem/progenitor cell (NPC) proliferation in the dentate gyrus of aged brains (Jin et al., 2003). Several rodent studies have found that by middle age (9–12 months) the drop in hippocampal neurogenesis is between 85–90% (Bondolfi et al., 2004; Ben Abdallah et al., 2010) and by the time rodents age to over 20 months, the loss of neurogenesis can be greater than 94% (Gil-Mohapel et al., 2013; Morel et al., 2015). Further, NPC migration rate and distance traveled decreases as rats age (Shuboni-Mulligan et al., 2019). As rodents age, the decline in hippocampal neurogenesis occurs more sharply in the ventral dentate gyrus than in the dorsal dentate gyrus (Jinno, 2011).
The aged brain is accompanied by a loss of hippocampal neurogenesis. However, increasing BDNF levels along with increasing hippocampal neurogenesis, by exercise or gene therapy, can recover age-related behavioral impairments in the 5xFAD mouse model of Alzheimer’s disease (Choi et al., 2018). Use of a peptidergic neurotrophin compound in aged rats leads to enhanced hippocampal neurogenesis, increased BDNF, and improve learning Bolognin et al., 2014). In contrast, increasing pro-BDNF in age mice reduced hippocampal neurogenesis and induced greater deficits in the Morris water maze (Chen et al., 2016). BDNF appears to be a critical factor involved in the regulation of cellular aging within the hippocampus (von Bohlen und Halbach, 2010).
Additionally, decreased levels of nerve growth factor (NGF) mRNA and protein have been reported in aged rats, notably in the basal forebrain of aged rats and humans (Hefti and Mash, 1989; Koh, et al., 1989; Lärkfors et al., 1987). Aged animals considered cognitively-impaired have exhibited improved cognitive function after administration of NGF, supporting the importance of neurogenesis in healthy learning and memory (Fischer, Gage, and Björklund, 1989; Gage et al., 1989). Implantation of a cholinergic septal graft, which is rich in NGF, into the hippocampus improves Morris water maze performance in aged, cognitively impaired rats, and the graft also prevents further behavioral deterioration (Gage and Bjorklund, 1986). When given atropine, a muscarinic receptor antagonist, aged and grafted rats are unable to utilize a successful spatial cue-search behavior (Gage and Bjorklund, 1986). The single dose of atropine resulted in the aged and grafted rats engaging in a thigmotaxis response, instead of exploring within the maze where they could use spatial cues. This could be indicative of the graft repairing presynaptic and axonal functioning but failing to mediate the postsynaptic shortcomings.
Aged rats that are cognitively-impaired also show a significant loss of cholinergic neurons in the MS/DB compared to young controls, but show no change in the pontine reticular cholinergic neurons, implicating the basal forebrain as a key structure in age-associated memory impairment (Baskerville et al., 2006). Overall, aging primate brains show a 17% reduction in basal forebrain neurons and a 16% decrease in the size of ChAT+ neurons, however, administration of NGF almost completely recovers the loss and shrinkage of the neurons (Nagahara et al., 2009). Retrograde labeling has shown a decrease in MS/DB projection neurons in aged rat brains (De Lacalle et al., 1996). There is an age-associated reduction of the density of cholinergic fibers within the dentate gyrus that is more profound in aged male rats relative to aged female rats (Lukoyanov et al., 1999a). In addition, cortical cholinergic innervation becomes less dense as rats age, and this is further augmented when the expression of tropomyosin-related kinase A (trkA) receptors are suppressed by viral vector-based RNA interference (Parikh et al., 2013). This suppression also decreases the ability of cholinergic neurons to release ACh, more so in aged-suppressed rats compared to aged or young controls (Parikh et al., 2013), suggesting that cholinergic transmission declines slowly over the lifespan (Shen and Barnes, 1996).
However, Xie and colleagues (2019) showed a preservation of basal forebrain cholinergic neuron area in aged rats following a three-month administration a small molecule p75NTR modulator, LM11A-31, which down regulates degenerative and upregulates trophic receptor-associated signaling. When treated later in life (21 to 25 months), administration of LM11A-31 resulted in increased ChAT cell size, and led to longer MS/VDB neurites, greater density of cholinergic fibers, and higher levels of synaptophysin presynaptic markers (Xie et al., 2019). Aged brains also exhibit deficits in choline transport, a decrease in acetylcholine release and synthesis, and the inability for muscarinic receptors to couple with their GTP-binding effector proteins. Repeated exposure to galantamine or donepezil (acetylcholinesterase inhibitors) improves spatial learning and increases some cholinergic receptors in the aged rat brain (Hernandez et al., 2006). A dysfunctional cholinergic system can make an organism more vulnerable to the aging process, and whether adaptive aging responses are able to compensate under such disease states is unknown (Gray and Barnes, 2015).
Beyond cellular and structural brain changes associated with aging, there are also functional impairments. Aging results in the impairment of LTP within the hippocampus (Barnes, 1979; Lynch, 1998), potentially due to the lack of calcium influx related to the NMDA receptors (Kumar and Foster, 2019). Aging can lead to impairment in both the induction and maintenance of LTP. Furthermore, there is an increase in susceptibility to LTD that was demonstrated to contribute to the decay of LTP, and the induction or magnitude of LTD is correlated with increased forgetting in older rodents (Kumar and Foster, 2019). Aged rats exhibit rates of LTP decay that are twice as fast as young rats (Barnes and McNaughton, 1985). Although most properties of pyramidal or granule cells are unchanged by aging (Barnes, 1994), there is a reduction in the total number of perforant-path synaptic contacts on granule cells (Geinisman et al., 1992). An interesting age-related compensatory LTP response has been observed. Although as rodents age there is a substantial reduction in NMDAR-dependent LTP, there is an increased NMDA-independent LTP that occurs distinctively in cognitively spared aged rats (Boric et al., 2008). Furthermore, an upregulation of synaptic inhibition may be needed for successful cognitive aging. Hippocampal extracellular recordings revealed that aged mice have more frequent spikes in the CA3, relative to young mice, suggesting a state of hyper-excitability within the aged hippocampus (El-Hayek et al., 2013). In aged rats that do not display cognitive impairment, miniature excitatory and inhibitory currents in dentate granule cells display larger amplitudes, which is not seen in pyramidal cells, and extrasynaptic tonic inhibition is selectively enhanced in the CA1 region (Tran et al., 2018). Thus, successful cognitive aging involves unique functional adaptations, distinct from those used in the younger brain.
4.3. Behavioral Deficits of Aging
The losses and dysfunctions of critical neuronal populations, pathways and connections due to aging can have significant behavioral consequences, if successful neural adaptations do not occur. A subset of aged animals and humans experience deficits in episodic and working memory, spatial learning, and attention (Gallagher et al., 1993; Geinisman et al., 1995; Kausler, 1994). Populations of aged rats are typically divided into cognitive-impaired and cognitive-spared in order to study the learning and memory consequences of aging. Presence or absence of cognitive impairment in aged rats is commonly determined by their performance on the Morris water maze (Albeck et al., 2003; Smith, et al., 2000; Aubert, et al., 1995; Fischer, et al., 1992; Rowe et al., 2007; Vaucher et al., 2001). When tested on the W-track, a measure of both working and spatial memory, aged animals make more working memory errors and take significantly longer to meet criterion (Kapellusch et al., 2018). Aged-impaired rats display deficits in recollection, but intact item recognition based on familiarity, on an odor nonmatching-to sample-task (Fortin et al., 2004, Robitsek et al., 2008). Cognitive-impaired aged rats exhibit increased rates of forgetting, greater susceptibility to interference, and worse long-term recall of specific experiences, compared to young controls (Winocur, 1988). Aged rats impaired on the Barnes maze, another hippocampal-dependent learning task, show impairments not in just learning and retention of the location of the escape box, but also displayed impaired flexibility to adapt to a change in the escape location (Bach et al., 1999; Barnes and McNaughton, 1980; Barnes and McNaughton, 1985; Markowska et al., 1989). This may be related to the aged rats reduced CA3/CA1 place field adaptation with alteration of the environment (Tanila et al., 1997; Wilson et al., 2003). Johnson et al., (2016) found that aged animals perform not just significantly worse on the Morris water maze, but are also much more susceptible to cumulative interference. Aged rats show a particular type of error on maze tasks: There is more likely to be an error when the non-reward arm is adjacent to the correct reward arm on a radial 8-arm maze (Gracian et al., 2013). This suggests that when spatial interference is high, aged rats are less efficient, potentially due to a pattern separation deficit.
Leal and Yassa (2015) define pattern separation as the process of reducing interference among similar inputs by using non-overlapping representations, and is believed to require the hippocampus, specifically the dentate and its connection to the CA3. A more recently developed theory is that aged cognitive-impaired rodents have deficits in the balance between pattern separation and pattern completion, with diminished pattern separation, but not pattern completion (Leal and Yassa, 2015). It has been proposed that the age-related cognitive deficits are caused by the failure to quickly develop distinctive pattern separations of spatial or non-spatial representations, and this leads to interference among episodic associations and representations (Haberman et al., 2017). Such pattern separation deficits have been linked to the age-related disruption of neurogenesis in the dentate gyrus and the faulty integration and functions of adult-born dentate granule cells (McAvoy and Sahay, 2017). A second, potentially related theory of successful aging is the need for the age-dependent recruitment of inhibitory processes within the hippocampus, as previously discussed. Behavioral support for this theory is that following a cue mismatch experience, where local cues (within maze) are placed in maximum conflict with the global cues (extra maze cues) on a circular track task, the mRNA expression of the inhibitory gene GAD1 was selectively enhanced in the hippocampus of aged cognitively-spared rats, suggesting that recruitment of inhibitory processes is a compensatory mechanism to control for age-related increased hippocampal neural activity (Branch et al., 2019). A lack of compensation is a significant feature of many neurodegenerative disorders (Dekosky et al., 1996; Terry et al., 1991).
While late aging-related neural vulnerabilities exist and can lead to cognitive impairment, there is plasticity and adaptations in aged brains that serve to compensate for some dysfunctional age-related changes (Gray and Barnes, 2015). How developmental perturbations alter age-related neural adaptations is virtually unknown. Greater insight to the distinctions between adaptive and pathological age-related alterations is crucial for our understanding to why some rodents, and humans, do not display age-related cognitive decline. Such knowledge would also help with the development of therapeutics for age-related neurodegeneration and developmental brain disruption.
4.4. Developmental ethanol exposure and aging interactions
While the literature on EtOH consumption and aging on brain pathology and behavior are abundant when investigated individually, the study of the two in combination is unfortunately scarce. There have been a few studies examining the effects of chronic EtOH exposure as an effect of age, with a focus on the resulting behavioral deficits. Aged animals that undergo chronic EtOH exposure show more sensitivity to the effects of EtOH by a loss of righting reflex, compared to adolescent and adult treated rats (Matthews et al., 2019, and this volume). This same study reported that aged rats undergoing CIE lost significantly more weight than adolescent or adult CIE-treated animals, and that they slept 79% less than aged saline-treated animals. Previous work from our lab has shown effects of chronic EtOH treatment, over a 6–8 months period, at different ages of onset (Fernandez et al., 2016). Animals who begin forced EtOH drinking (20% continuous access) at the onset of adolescence drink significantly more throughout an extended 6–8 month period than animals who begin drinking in adulthood. Regardless of age of EtOH drinking onset, a life-long pattern of EtOH drinking (rats were 250 and 290 days old at the end of treatment) results in suppressed mature forms of BDNF and NGF levels in the frontal cortex during intoxication that persist into long-term abstinence. However, in the hippocampus, only adult onset drinking leads to a selective suppression of BDNF. The behavioral performance on the spatial working memory task of spontaneous alternation has a negative correlation with high BECs. Non-spatial discrimination learning was impaired in rats that begin long-term drinking as adolescents, and when multiple cues or reversal of a cue rule are included, rats that drink EtOH for up to 8 months are impaired—regardless of age at drinking onset. Thus, exposure to EtOH over the life span does lead to disruption in neurotrophins and cognition, especially when high BECs are reached.
It is still unknown how AIE will modulate the aging process. Vetreno and colleagues (2015, 2019) showed a persistent reduction in DCX-immunopositive neurons in the hippocampus as well as 20–28% reduction in ChAT-immunopositive neurons within the MS/DB from AIE treatment throughout adulthood and into middle age (up to 220 days of age). The reduction in neurogenesis is directly related to the discrimination ratio of a novel object recognition task (hippocampal-dependent memory) and latency to enter the center of an open field (measure of anxiety; see Vetreno and Crews, 2015). The AIE-induced loss of the cholinergic phenotype within the MS/DB was associated with epigenetic changes in DNA methylation at the promoter regions of the ChAT gene (Vetreno et al., 2019). This phenotypic loss of cholinergic neuronal markers (ChAT, TrkA, p75) following AIE is recoverable with post-treatment voluntary exercise (Vetreno et al., 2019), presumably through exercise-induced increases in NGF (see Hall et al., 2018).
The development of hippocampal deficits when animals are aged after AIE may be due to an amplified loss of neurogenesis, dysfunctional acetylcholine release, or receptor dysregulation. It is likely that the compensatory mechanisms that are utilized by the young brain following AIE begin to fail as animals begin to age. Preliminary data from our lab suggests an emergence of hippocampal cholinergic deficits in aged adulthood following AIE. As shown in Figure 3, young adult rats exposed to AIE show no significant impairment in behaviorally-activated hippocampal ACh efflux, but at 12 months of age a reduction in behaviorally-evoked ACh emerges. However, a complete mapping of the behavioral and brain changes that emerge following developmental EtOH exposure has yet to be determined. Understanding how compromises in brain developmental trajectory ensued by developmental EtOH exposure (prenatal or adolescent) interacts with the aging process is crucial.
Figure 3.
Peri-adolescent rats (PD 25) were exposed to intermittent (2 day on/ 2 day off) ethanol exposure (AIE) until the end of adolescence (PD 55) or where gavaged with water (Con). Two-months post-treatment there was no consequences of AIE on behaviorally-activated acetylcholine (ACh) efflux in the hippocampus. However, as the AIE rats aged (12-months post AIE), a blunting of behaviorally-activated ACh efflux within the hippocampus emerged. Also, there was an overall effect of age on ACh efflux, as 14-month old rats had lower level of behaviorally-activated hippocampal ACh efflux than 3-month old rats.
5. Conclusions
Animal models have revealed that TD, life-long chronic ethanol drinking, and intermittent EtOH exposure during adolescence adversely affect several brain regions and produce a range of behavioral impairment (see Figure 2). The neuroimmune response to TD and/or ethanol exposure varies (see Figure 1) as a function of active state (intoxication or neurological sequelae of TD), recovery (after thiamine normalization) or abstinence (acute withdrawal or long-term EtOH free). During an active thiamine deficient state there is a massive increase in neuroimmune genes and proteins within the thalamus, and significant increases within the hippocampus and frontal cortex. In contrast, CET throughout adulthood produces more minor fluctuations in neuroimmune genes, regardless of brain region. Intermittent “binge–type” ethanol during the adolescent period produces an intermediate neuroinflammatory response in the hippocampus and frontal cortex, that can persist into adulthood. The induction of neuroimmune genes by excess exposure to EtOH or TD likely leads to neuronal excitability and oxidative stress, which may prime neurodegeneration (Crews et al., 2015; Toledo Nunes et al., 2019; Qin and Crews, 2014). There is some overlap in ARBD produced by these manifestations: loss of cholinergic cells in the basal forebrain, hypocholinergic functioning, reduction of hippocampal neurogenesis, as well as loss of frontal cortical grey and white matter (Wolstenholme et al., 2017; Pitkin and Savage, 2001; Vetreno et al., 2011). TD selectively induces lesions and significant cell loss within the thalamus.
Hippocampal and frontal cortical cholinergic dysfunctions are emerging as critical variables in the cognitive dysfunction associated with ARBD (Anzalone et al., 2010; Fernandez and Savage, 2017; Schliebs and Arendt, 2011; Vetreno and Crews, 2018), as well as important for the recovery of both structure and function. If neurotrophin and/or acetylcholine levels within the basal forebrain system, hippocampus, or frontal cortex are enhanced, then cognitive performance can be recovered (Hall and Savage, 2016; Hall et al., 2018), the cholinergic phenotype re-emergences within the MS/DB (Hall et al., 2018; Vetreno et al., 2019), and there is a recovery of hippocampal spine density (Mulholland et al., 2019). Although the use of cholinergic modulators is not new for the treatment of cognitive dysfunction, new therapies that still rely on the cholinergic system are still a viable treatment approach (Douchamps and Mathis, 2017).
Studies that have directly compared the EtOH or TD treatment protocols are few—as are the number of studies that have examined the interactive effects of ethanol toxicity and TD. To date, EtOH appears to hasten the progression of TD, and recently our data suggest that when EtOH exposure and TD is combined there are additive deficits, compared to moderate or mild TD. However, a variable that has been ignored in the development of ARBD and cognitive dysfunction is aging. Subclinical neuropathology from AUDs may make an organism more vulnerable to the aging process, and normal compensatory adaptive aging responses may fail leading to the behavioral sequelae of alcohol-related dementia. Future research is needed to examine the functional adaptations and neural changes that occur across the lifespan following heavy alcohol consumption or exposure. Including aged at time of EtOH exposure, as well as advanced aging following EtOH exposure will result in a more valid model system for use in exploring potential therapeutic interventions for disorders of cognition and memory associated with ARBD.
Acknowledgements:
Supported by NIH grant numbers P50AA017823 and R01AA021775 to LMS and the Center for Development and Behavioral Neuroscience at Binghamton University. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the above stated funding agencies. The authors have no conflicts of interest to declare.
References
- Abadi THN, Vaghef L, Babri S, Mahmood-Alilo M, & Beirami M (2013). Effects of different exercise protocols on ethanol-induced spatial memory impairment in adult male rats. Alcohol, 47(4), 309–316. [DOI] [PubMed] [Google Scholar]
- Abdou E, & Hazell AS (2015). Thiamine deficiency: An update of pathophysiologic mechanisms and future therapeutic considerations. Neurochemical Research, 40(2), 353–361. [DOI] [PubMed] [Google Scholar]
- Acsady L, Pascual M, Rocamora N, Soriano E, & Freund TF (2000). Nerve growth factor but not neurotrophin-3 is synthesized by hippocampal GABAergic neurons that project to the medial septum. Neuroscience, 98, 23–31. [DOI] [PubMed] [Google Scholar]
- Adams KM, Gilman S, Koeppe RA, Kluin KJ, Brunberg JA, Dede D, Berent S, & Kroll PD (1993). Neuropsychological deficits are correlated with frontal hypometabolism in positron emission tomography studies of older alcoholic patients. Alcoholism: Clinical and Experimental Research, 17(2), 205–210. [DOI] [PubMed] [Google Scholar]
- Albeck D, Mesches MH, Juthberg S, Browning M, Bickford PC, Rose GM, & Granholm AC (2003). Exogenous NGF restores endogenous NGF distribution in the brain of the cognitively impaired aged rat. Brain Research, 967(1–2), 306–310. doi: 10.1016/S0006-8993(03)02272-8 [DOI] [PubMed] [Google Scholar]
- Alfonso-Loeches S, & Guerri C (2011). Molecular and behavioral aspects of the actions of alcohol on the adult and developing brain. Critical Reviews in Clinical Laboratory Sciences, 48(1), 19–47. [DOI] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Pascual M, Gómez-Pinedo U, Pascual-Lucas M, Renau-Piqueras J, & Guerri C (2012). Toll-like receptor 4 participates in the myelin disruptions associated with chronic alcohol abuse. Glia, 60(6), 948–964. [DOI] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Pascual-Lucas M, Blanco AM, Sanchez-Vera I, & Guerri C (2010). Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. Journal of Neuroscience, 30(24), 8285–8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Ureña-Peralta J, Morillo-Bargues MJ, Gómez-Pinedo U, & Guerri C (2016). EtOH-induced TLR4/NLRP3 neuroinflammatory response in microglial cells promotes leukocyte infiltration across the BBB. Neurochemical Research, 41(1–2), 193–209. [DOI] [PubMed] [Google Scholar]
- Allard S, Jacobs ML, Do Carmo S, & Cuello AC (2018). Compromise of cortical proNGF maturation causes selective retrograde atrophy in cholinergic nucleus basalis neurons. Neurobiology of Aging, 67, 10–20. doi: 10.1016/j.neurobiolaging.2018.03.002 [DOI] [PubMed] [Google Scholar]
- Aloe L & Tirassa P (1992). The effect of long-term alcohol intake on brain NGF-target cells of aged rats. Alcohol, 9(4), 299–304. [DOI] [PubMed] [Google Scholar]
- Aloe L, Bracci-Laudiero L, & Tirassa P (1993). The effect of chronic EtOH intake on brain NGF level and on NGF-target tissues of adult mice. Drug and Alcohol Dependence, 31(2), 159–167. [DOI] [PubMed] [Google Scholar]
- Amor S, Peferoen LA, Vogel DY, Breur M, van der Valk P, Baker D, & van Noort JM (2014). Inflammation in neurodegenerative diseases--An update. Immunology, 142(2), 151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone S, Vetreno RP, Ramos RL, & Savage LM (2010), Cortical cholinergic abnormalities contribute to the amnesic state induced by pyrithiamine-induced thiamine deficiency in the rat. European Journal of Neuroscience, 32(5), 847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anttila T, Helkala EL, Viitanen M, Kåreholt I, Fratiglioni L, Winblad B, Soininen H, Tuomilehto J, Nissinen A, & Kivipelto M (2004). Alcohol drinking in middle age and subsequent risk of mild cognitive impairment and dementia in old age: A prospective population based study. BMJ (Clinical Research ed.), 329(7465), 539. doi: 10.1136/bmj.38181.418958.BE [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Allen Y, Marchbanks RM, Schugens MM, Sinden J, Lantos PL, & Gray JA (1989). Cholinergic system and memory in the rat: Effects of chronic EtOH, embryonic basal forebrain brain transplants and excitotoxic lesions of cholinergic basal forebrain projection system. Neuroscience, 33, 435–62. [DOI] [PubMed] [Google Scholar]
- Arendt T, Brückner MK, Krell T, Pagliusi S, Kruska L, & Heumann R (1995a). Degeneration of rat cholinergic basal forebrain neurons and reactive changes in nerve growth factor expression after chronic neurotoxic injury—II. Reactive expression of the nerve growth factor gene in astrocytes. Neuroscience, 65(3), 647–659. [DOI] [PubMed] [Google Scholar]
- Arendt T, Brückner MK, Pagliusi S, & Krell T (1995b). Degeneration of rat cholinergic basal forebrain neurons and reactive changes in nerve growth factor expression after chronic neurotoxic injury—I. Degeneration and plastic response of basal forebrain neurons. Neuroscience, 65(3), 633–645. [DOI] [PubMed] [Google Scholar]
- Arendt T, Hennig D, Gray JA, & Marchbanks R (1988). Loss of neurons in the rat basal forebrain cholinergic projection system after prolonged intake of EtOH. Brain Research Bulletin, 21(4), 563–569. [DOI] [PubMed] [Google Scholar]
- Arts NJ, Walvoort SJ, & Kessels RP (2017). Korsakoff’s syndrome: A critical review. Neuropsychiatric Disease and Treatment, 13, 2875–2890. doi: 10.2147/NDT.S130078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aubert I, Rowe W, Meaney MJ, Gauthier S, & Quirion R (1995). Cholinergic markers in aged cognitively impaired long-evans rats. Neuroscience, 67(2), 277–292. doi: 10.1016/0306-4522(95)00056-O [DOI] [PubMed] [Google Scholar]
- Ba A, Seri BV, Aka KJ, Glin L & Tako A (1999). Comparative effects of developmental thiamine deficiencies and ethanol exposure on the morphometry of the CA3 pyramidal cells. Neurotoxicology and Teratology, 21, 579–586 [DOI] [PubMed] [Google Scholar]
- Bach ME, Barad M, Son H, Zhuo M, Lu YF, Shih R, Mansuy I, Hawkins RD & Kandel ER (1999). Age related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proceeding of the National Academy of Science. USA, 96, 5280– 5285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H, Asrican B, Li W, Gu B, Wen Z, Lim SA, Haniff I, Ramakrishnan C, Deisseroth K, Philpot B, & Song J (2017). Long-range GABAergic inputs regulate neural stem cell quiescence and control adult hippocampal neurogenesis. Cell Stem Cell, 21(5), 604–617. 10.1016/j.stem.2017.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes CA (1979). Memory deficits associated with senescence: A neurophysiological and behavioral study in the rat. Journal of Comparative and Physiological Psychology, 93, 74–104. [DOI] [PubMed] [Google Scholar]
- Barnes CA (1994). Normal aging: Regionally specific changes in hippocampal synaptic transmission. Trends in Neuroscience, 17, 13–18. [DOI] [PubMed] [Google Scholar]
- Barnes CA & McNaughton BL (1980). Spatial memory and hippocampal synaptic plasticity in middle-aged and senescent rats In Psychobiology of Aging: Problems and Perspectives (ed. Stein D), pp. 253–272. New York: Elsevier. [Google Scholar]
- Barnes CA & McNaughton BL (1985). An age comparison of the rates of acquisition and forgetting of spatial information in relation to long-term enhancement of hippocampal synapses. Behavioral Neuroscience, 99, 1040–1048. [DOI] [PubMed] [Google Scholar]
- Baskerville KA, Kent C, Nicolle MM, Gallagher M, & McKinney M (2006). Aging causes partial loss of basal forebrain but no loss of pontine reticular cholinergic neurons. Neuroreport, 17(17), 1819–1823. [DOI] [PubMed] [Google Scholar]
- Bekinschtein P, Oomen CA, Saksida LM, & Bussey TJ (2011). Effects of environmental enrichment and voluntary exercise on neurogenesis, learning and memory, and pattern separation: BDNF as a critical variable? Seminars in Cell & Developmental Biology, 22(5), 536–542. [DOI] [PubMed] [Google Scholar]
- Ben Abdallah NM, Slomianka L, Vyssotski AL, & Lipp HP (2010). Early age-related changes in adult hippocampal neurogenesis in C57 mice. Neurobiology of Aging, 31, 151–161. doi: 10.1016/j.neurobiolaging.2008.03.002 [DOI] [PubMed] [Google Scholar]
- Beracochea D, Lescaudron L, Tako A, Verna A, & Jaffard R (1987a). Build-up and release from proactive interference during chronic ethanol consumption in mice: A behavioral and neuroanatomical study. Behavioural Brain Research, 25(1), 63–74. [DOI] [PubMed] [Google Scholar]
- Beracochea D, Lescaudron L, Verna A, & Jaffard R (1987b). Neuroanatomical effects of chronic ethanol consumption on dorsomedial and anterior thalamic nuclei and on substantia innominata in mice. Neurosci Letters, 73(1), 81–84. [DOI] [PubMed] [Google Scholar]
- Berger-Sweeney J, Heckers S, Mesulam MM, Wiley RG, Lappi DA, & Sharma M (1994). Differential effects on spatial navigation of immunotoxin-induced cholinergic lesions of the medial septal area and nucleus basalis magnocellularis. Journal of Neuroscience, 14, 4507–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettio LEB, Rajendran L, & Gil-Mohapel J (2017). The effects of aging in the hippocampus and cognitive decline. Neuroscience and Biobehavioral Review, 79, 66–86. doi: 10.1016/j.neubiorev.2017.04.030 [DOI] [PubMed] [Google Scholar]
- Bimonte-Nelson HA, Granholm AC, Nelson ME, & Moore AB (2008). Patterns of neurotrophin protein levels in male and female Fischer 344 rats from adulthood to senescence: How young is “young” and how old is “old”?. Experimental Aging Research, 34(1), 13–26. doi: 10.1080/03610730701761908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird CM & Burgess N (2008). The hippocampus and memory: Insights from spatial processing. Nature Reviews Neuroscience, 9(3), 182–194. doi: 10.1038/nrn2335. [DOI] [PubMed] [Google Scholar]
- Birrell JM, & Brown VJ (2000). Medial frontal cortex mediates perceptual attentional set shifting in the rat. Journal of Neuroscience, 20, 4320–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake M, & Boccia MM (2016). Basal forebrain cholinergic system and memory. Current Topics in Behavioral Neuroscience, 37. [DOI] [PubMed] [Google Scholar]
- Blanco AM, & Guerri C (2007). EtOH intake enhances inflammatory mediators in brain: role of glial cells and TLR4/IL-1RI receptors. Frontiers in Bioscience, 12, 2616–2630. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, & Hong JS (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nature Reviews Neuroscience, 8(1), 57–69. [DOI] [PubMed] [Google Scholar]
- Bolognin S, Buffelli M, Puoliväli J, & Iqbal K (2014). Rescue of cognitive-aging by administration of a neurogenic and/or neurotrophic compound. Neurobiology of Aging, 35(9), 2134–2146. doi: 10.1016/j.neurobiolaging.2014.02.017 [DOI] [PubMed] [Google Scholar]
- Bondolfi L, Ermini F, Long JM, Ingram DK, & Jucker M (2004). Impact of age and caloric restriction on neurogenesis in the dentate gyrus of C57BL/6 mice. Neurobiology of Aging, 25(3), 333–340. doi: 10.1016/S0197-4580(03)00083-6 [DOI] [PubMed] [Google Scholar]
- Boric K, Muñoz P, Gallagher M, & Kirkwood A (2008). Potential adaptive function for altered long-term potentiation mechanisms in aging hippocampus. Journal of Neuroscience, 28(32), 8034–8039. doi: 10.1523/JNEUROSCI.2036-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boskovic Z, Meier S, Wang Y, Milne MR, Onraet T, Tedoldi A, & Coulson EJ (2019). Regulation of cholinergic basal forebrain development, connectivity and function by neurotrophin receptors. Neuronal Signaling, 3(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros N, Semenova S, Liu W, Crews FT, & Markou A (2014). Adolescent intermittent EtOH exposure is associated with increased risky choice and decreased dopaminergic and cholinergic neuron markers in adult rats. International Journal of Neuropsychopharmacology, 18(2). doi: 10.1093/ijnp/pyu003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyadjieva NI, & Sarkar DK (2013). Cyclic adenosine monophosphate and brain-derived neurotrophic factor decreased oxidative stress and apoptosis in developing hypothalamic neuronal cells: role of microglia. Alcoholism: Clinical and Experimental Research, 37(8), 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branch A, Monasterio A, Blair G, Knierim JJ, Gallagher M, & Haberman RP (2019). Aged rats with preserved memory dynamically recruit hippocampal inhibition in a local/global cue mismatch environment. Neurobiology of Aging, 76, 151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brion M, Pitel AL, Beaunieux H, & Maurage P (2014). Revisiting the continuum hypothesis: toward an in-depth exploration of executive functions in korsakoff syndrome. Frontiers in Human Neuroscience, 8, 498. doi: 10.3389/fnhum.2014.00498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briones TL & Woods J (2013). Chronic binge-like alcohol consumption in adolescence causes depression-like symptoms possibly mediated by the effects of BDNF on neurogenesis. Neuroscience, 254, 324–334. doi: 10.1016/j.neuroscience.2013.09.03 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadwater MA, Lee SH, Yu Y, Zhu H, Crews FT, Robinson DL, & Shih YI (2018). Adolescent alcohol exposure decreases frontostriatal resting-state functional connectivity in adulthood. Addiction Biology, 23(2), 810–823. doi: 10.1111/adb.12530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadwater MA, Liu W, Crews FT, & Spear LP (2014). Persistent loss of hippocampal neurogenesis and increased cell death following adolescent, but not adult, chronic EtOH exposure. Developmental Neuroscience, 36, 297–305. doi: 10.1159/000362874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brückner MK, & Arendt T (1992). Intracortical grafts of purified astrocytes ameliorate memory deficits in rat induced by chronic treatment with EtOH. Neuroscience Letters, 141(2), 251–254. [DOI] [PubMed] [Google Scholar]
- Brun VH, Ytterbo K, Morris RG, Moser MB, & Moser EI (2001). Retrograde amnesia for spatial memory induced by NMDA receptor-mediated long-term potentiation. Journal of Neuroscience, 21(1), 356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno MA & Cuello AC (2006). Acitivity-dependent release of precursor nerve growth factor, conversion to mature nerve growth factor, and it degradation by protease cascade, Proceeding of the National Academy of Sciences of the United States of America, 103 (17), 67-35-6740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler TR, & Prendergast MA (2012). Neuroadaptations in adenosine receptor signaling following long-term EtOH exposure and withdrawal. Alcoholism: Clinical and Experimental Research, 36(1), 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadete-Leite A, Alves MC, Paula-Barbosa MM, Uylings HB, & Tavares MA (1990a). Quantitative analysis of basal dendrites of prefrontal pyramidal cells after chronic alcohol consumption and withdrawal in the adult rat. Alcohol and Alcoholism, 25(5), 467–475. [PubMed] [Google Scholar]
- Cadete-Leite A, Alves MC, Tavares MA, & Paula-Barbosa MM (1990b). Effects of chronic alcohol intake and withdrawal on the prefrontal neurons and synapses. Alcohol, 7(2), 145–152. [DOI] [PubMed] [Google Scholar]
- Cadete-Leite A, Brandao F, Andrade JP, Ribeiro-Da-Silva A, & Paula-Barbosa MM (1997). The gabaergic system of the dentate gyrus after withdrawal from chronic alcohol consumption: Effects of intracerebral grafting and putative neuroprotective agents. Alcohol and Alcoholism, 32(4), 471–484. [DOI] [PubMed] [Google Scholar]
- Cadete-Leite A, Pereira PA, Madeira MD, & Paula-Barbosa MM (2003). Nerve growth factor prevents cell death and induces hypertrophy of basal forebrain cholinergic neurons in rats withdrawn from prolonged ethanol intake. Neuroscience, 119(4), 1055–1069. [DOI] [PubMed] [Google Scholar]
- Cagetti E (2003). Withdrawal from chronic intermittent ethanol treatment changes subunit composition, reduces synaptic function, and decreases behavioral responses to positive allosteric modulators of GABAA receptors. Molecular Pharmacology, 63(1), 53–64. [DOI] [PubMed] [Google Scholar]
- Carvalho FM, Pereira SR, Pires RG, Ferraz VP, Romano-Silva MA, Oliveira-Silva IF, & Ribeiro AM (2006). Thiamine deficiency decreases glutamate uptake in the prefrontal cortex and impairs spatial memory performance in a water maze test. Pharmacology, Biochemistry & Behavior, 83(4), 481–489. [DOI] [PubMed] [Google Scholar]
- Célérier A, Ognard R, Decorte L, & Beracochea D. (2000). Deficits of spatial and non-spatial memory and of auditory fear conditioning following anterior thalamic lesions in mice: comparison with chronic alcohol consumption. European Journal of Neuroscience, 12(7), 2575–2584. [DOI] [PubMed] [Google Scholar]
- Chen J, Li CR, Yang H, Liu J, Zhang T, Jiao SS, Wang YJ, & Xu ZQ (2016). proBDNF attenuates hippocampal neurogenesis and induces learning and memory deficits in aged mice. Neurotoxicity Research, 29(1), 47–53. [DOI] [PubMed] [Google Scholar]
- Choi SH, Bylykbashi E, Chatila ZK, Lee SW, Pulli B, Clemenson GD, Kim E, Rompala A, Oram MK, Asselin C, Aronson J, Zhang C, Miller SJ, Lesinski A, Chen JW, Kim DY, van Praag H, Spiegelman BM, Gage FH, & Tanzi RE (2018). Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science, 361(6406), eaan8821. doi: 10.1126/science.aan8821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourbaji S, Brandwein C, & Gass P (2011). Altering BDNF expression by genetics and/or environment: impact for emotional and depression-like behaviour in laboratory mice. Neuroscience & Biobehavioral Reviews, 35(3), 599–611. [DOI] [PubMed] [Google Scholar]
- Chung YA, Choi SW, Joe KH, Jeong J, Cheon Y, & Kim DJ (2009). Regional cerebral blood flow in patients with alcohol-related dementia: a SPECT study. International Journal of Neuroscience, 119(11), 2100–2111. [DOI] [PubMed] [Google Scholar]
- Ciccia RM, & Langlais PJ (2000). An examination of the synergistic interaction of ethanol and thiamine deficiency in the development of neurological signs and long-term cognitive and memory impairments. Alcoholism: Clinical and Experimental Research, 24(5), 622–634. [PubMed] [Google Scholar]
- Clapp P, Bhave SV, & Hoffman PL (2008). How adaptation of the brain to alcohol leads to dependence: A pharmacological perspective. Alcohol Research & Health, 31, 310–39 [PMC free article] [PubMed] [Google Scholar]
- Coleman LG, Zou J, & Crews FT (2017). Microglial-derived miRNA let-7 and HMGB1 contribute to EtOH-induced neurotoxicity via TLR7. Journal of Neuroinflammation, 14(1), 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, & Neafsey EJ (2012). EtOH and adult CNS neurodamage: oxidative stress, but possibly not excitotoxicity. Frontiers in Bioscience (Elite Ed), 4, 1358–1367. [DOI] [PubMed] [Google Scholar]
- Collins MA, & Neafsey EJ (2016). Alcohol, excitotoxicity and adult brain damage: An experimentally unproven chain-of-events. Frontiers in Molecular Neuroscience, 9, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Kuhn CM, Winkler J, & Kuhn HG (2004). Decreased neurogenesis after cholinergic forebrain lesion in the adult rat. Journal of Neuroscience Research, 77(2), 155–165. [DOI] [PubMed] [Google Scholar]
- Crews FT, Lawrimore CJ, Walter TJ, & Coleman LG (2017). The role of neuroimmune signaling in alcoholism. Neuropharmacology, 122, 56–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Morrow AL, Criswell H, & Breese G (1996). Effects of ethanol on ion channels. International Review Neurobiology, 39, 283–367. [DOI] [PubMed] [Google Scholar]
- Crews FT, Mdzinarishvili A, Kim D, He J, & Nixon K (2006). Neurogenesis in adolescent brain is potently inhibited by ethanol. Neuroscience, 137(2), 437–445. [DOI] [PubMed] [Google Scholar]
- Crews FT, & Nixon K (2009). Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol and Alcoholism, 44(2), 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Qin L, Sheedy D, Vetreno RP, & Zou J (2013). High mobility group box 1/Toll-like receptor danger signaling increases brain neuroimmune activation in alcohol dependence. Biological Psychiatry, 73(7), 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Sarkar DK, Qin L, Zou J, Boyadjieva N, & Vetreno RP (2015). Neuroimmune Function and the Consequences of Alcohol Exposure. Alcohol Research, 37(2), 331–341. [PMC free article] [PubMed] [Google Scholar]
- Crews FT, & Vetreno RP (2016). Mechanisms of neuroimmune gene induction in alcoholism. Psychopharmacology (Berl), 233(9), 1543–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz C, Meireles M, & Silva SM (2017). Chronic EtOH intake induces partial microglial activation that is not reversed by long-term EtOH withdrawal in the rat hippocampal formation. Neurotoxicology, 60, 107–115. [DOI] [PubMed] [Google Scholar]
- Curlik II DM, DiFeo G, & Shors TJ (2014). Preparing for adulthood: Thousands upon thousands of new cells are born in the hippocampus during puberty, and most survive with effortful learning. Frontiers in Neuroscience, 8, 70 10.3389/fnins.2014.00070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI (2008). Ethanol-BDNF interactions: still more questions than answers. Pharmacology & Therapeutics, 118(1), 36–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lacalle S, Cooper JD, Svendsen CN, Dunnett SB, & Sofroniew MV (1996). Reduced retrograde labelling with fluorescent tracer accompanies neuronal atrophy of basal forebrain cholinergic neurons in aged rats. Neuroscience, 1(6), 19–27. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW, & Styren SD (1996). Structural correlates of cognition in dementia: Quantification and assessment of synapse change. Neurodegeneration, 5(4), 417–421. [DOI] [PubMed] [Google Scholar]
- Devaud LL, Fritschy JM, Sieghart W, & Morrow AL (1997). Bidirectional alterations of GABA(A) receptor subunit peptide levels in rat cortex during chronic ethanol consumption and withdrawal. Journal of Neurochemistry, 69(1), 126–130. [DOI] [PubMed] [Google Scholar]
- Devaud LL, Smith FD, Grayson DR, & Morrow AL (1995). Chronic ethanol consumption differentially alters the expression of gamma-aminobutyric acidA receptor subunit mRNAs in rat cerebral cortex: competitive, quantitative reverse transcriptase-polymerase chain reaction analysis. Molecular Pharmacology, 48(5), 861–868. [PubMed] [Google Scholar]
- de Witte P, Pinto E, Ansseau M, & Verbanck P (2003). Alcohol and withdrawal: from animal research to clinical issues. Neuroscience and Biobehavioral Reviews, 27(3), 189–197. [DOI] [PubMed] [Google Scholar]
- Douchamps V, & Mathis C (2017). A second wind for the cholinergic system in Alzheimer’s therapy. Behavioral Pharmacology, 28, 112–123. [DOI] [PubMed] [Google Scholar]
- Dufour MC (1993). The epidemiology of alcohol-induced brain damage In: Hunt WA, and Nixon SJ (Ed.), Alcohol-induced Brain Damage. The National Institute on Alcohol Abuse and Alcoholism, Rockville, MD, pp. 39–69. [Google Scholar]
- Durand D, & Carlen PL (1984). Impairment of long-term potentiation in rat hippocampus following chronic EtOH treatment. Brain Research, 308(2), 325–332 [DOI] [PubMed] [Google Scholar]
- Durand D, Saint-Cyr JA, Gurevich N, & Carlen PL (1989). EtOH-induced dendritic alterations in hippocampal granule cells. Brain Research, 477(1–2), 373–377. [DOI] [PubMed] [Google Scholar]
- Dutar P, Bassant MH, Senut MC, & Lamour Y (1995). The septohippocampal pathway: Structure and function of a central cholinergic system. Physiological Reviews, 75(2), 393–427. [DOI] [PubMed] [Google Scholar]
- Ehlers CL, Criado JR, Wills DN, Liu W, & Crews FT (2011). Periadolescent EtOH exposure reduces adult forebrain ChAT+IR neurons: Correlation with behavioral pathology. Neuroscience, 29, 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich D, Pirchl M, & Humpel C (2012). Effects of long-term moderate EtOH and cholesterol on cognition, cholinergic neurons, inflammation, and vascular impairment in rats. Neuroscience, 205, 154–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hayek YH, Wu C, Ye H, Wang J, Carlen PL, & Zhang L (2013). Hippocampal excitability is increased in aged mice. Experimental Neurology, 247, 710–719. [DOI] [PubMed] [Google Scholar]
- Fadda F, & Rossetti ZL (1998). Chronic ethanol consumption: From neuroadaptation to neurodegeneration. Progress in Neurobiology, 56(4), 385–431. [DOI] [PubMed] [Google Scholar]
- Fahnestock M, & Shekari A (2019). ProNGF and neurodegeneration in Alzheimer’s Disease. Frontiers in Neuroscience, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fein G, Bachman L, Fisher S, & Davenport L (1990). Cognitive impairments in abstinent alcoholics. The Western Journal of Medicine, 152(5), 531–537. [PMC free article] [PubMed] [Google Scholar]
- Fein G, Torres J, Price LJ, & Di Sclafani V (2006). Cognitive performance in long-term abstinent alcoholic individuals. Alcoholism: Clinical and Experimental Research, 30(9), 1538–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez GM, & Savage LM (2017). Adolescent binge EtOH exposure alters specific forebrain cholinergic cell populations and leads to selective functional deficits in the prefrontal cortex. Neuroscience, 361, 129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez GM, Stewart WN, & Savage LM (2016). Chronic drinking during adolescence predisposes the adult rat for continued heavy drinking: Neurotrophin and behavioral adaptation after long-term, continuous EtOH exposure. PloS one, 11(3), e0149987. doi: 10.1371/journal.pone.0149987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez GM, Lew BJ, Vedder LC, & Savage LM (2017). Chronic intermittent EtOH exposure leads to alterations in brain-derived neurotrophic factor within the frontal cortex and impaired behavioral flexibility in both adolescent and adult rats. Neuroscience, 348, 324–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer W, Chen KS, Gage FH, & Bjorklund A (1992). Progressive decline in spatial learning and integrity of forebrain cholinergic neurons in rats during aging. Neurobiology of Aging, 13(1), 9–23. [DOI] [PubMed] [Google Scholar]
- Fischer W, Gage F, & Bjorklund A (1989). Degenerative changes in forebrain cholinergic nuclei correlate with cognitive impairments in aged rats. European Journal of Neuroscience, 1(1), 34–45. [DOI] [PubMed] [Google Scholar]
- Floyd EA, Young-Seigler AC, Ford BD, Reasor JD, Moore EL, Townsel JG, & Rucker HK (1997). Chronic EtOH ingestion produces cholinergic hypofunction in rat brain. Alcohol, 14(1), 93–98. [DOI] [PubMed] [Google Scholar]
- Fortin NJ, Wright SP, & Eichenbaum H (2004). Recollection-like memory retrieval in rats is dependent on the hippocampus. Nature, 431, 188–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke H, Kittner H, Berger P, Wirkner K, & Schramek J (1997). The reaction of astrocytes and neurons in the hippocampus of adult rats during chronic EtOH treatment and correlations to behavioral impairments. Alcohol, 14(5):445–54. [DOI] [PubMed] [Google Scholar]
- Freeman K, Brureau A, Vadigepalli R, Staehle MM, Brureau MM, Gonye GE, Hoek JB, Hooper DC, &Schwaber JS (2012). Temporal changes in innate immune signals in a rat model of alcohol withdrawal in emotional and cardiorespiratory homeostatic nuclei. Journal of Neuroinflammation, 9, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH, Batchelor P, Chen KS, Chin D, Higgins GA, Koh S, Deputy S, Rosenberg MB, Fischer W, & Bjorklund A (1989). NGF receptor reexpression and NGF-mediated cholinergic neuronal hypertrophy in the damaged adult neostriatum. Neuron, 2(2), 1177–1184. [DOI] [PubMed] [Google Scholar]
- Gage FH, & Bjorklund A (1986). Cholinergic septal grafts into the hippocampal formation improve spatial learning and memory in aged rats by an atropine-sensitive mechanism. Journal of Neuroscience, 6(10), 2837–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaj E, Kipp BT, Floresco SB, & Savage LM (2019). Persistent alterations of accumbal cholinergic interneurons and cognitive dysfunction after adolescent intermittent EtOH exposure. Neuroscience, 404, 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher M, Burwell R, & Burchinal M (1993). Severity of spatial learning impairment in aging: Development of a learning index for performance in the Morris water maze. Behavioral Neuroscience, 107(4), 618–626. [DOI] [PubMed] [Google Scholar]
- Gansler DA, Harris GJ, Oscar-Berman M, Streeter C, Lewis RF, Ahmed I, & Achong D (2000). Hypoperfusion of inferior frontal brain regions in abstinent alcoholics: A pilot SPECT study. Journal of Studies on Alcohol and Drugs, 61(1), 32–37. [DOI] [PubMed] [Google Scholar]
- Gass JT, & Olive MF (2008). Glutamatergic substrates of drug addiction and alcoholism. Biochemistry Pharmacology, 75, 218–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geil CR, Hayes DM, McClain JA, Liput DJ, Marshall SA, Chen KY, & Nixon K (2014). Alcohol and adult hippocampal neurogenesis: Promiscuous drug, wanton effects. Progress in Neuropsychopharmacology and Biological Psychiatry, 54, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geinisman Y, de Toledo-Morrell L, Morrell F, & Heller RE (1995). Hippocampal markers of age-related memory function: Behavioral electrophysiological and morphological perspectives. Progress in Neurobiology, 45, 223–252. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, de Toledo-Morrell L, Morrell F, Persina IS & Rossi M (1992). Age- related loss of axospinous synapses formed by two afferent systems in the rat dentate gyrus as revealed by the unbiased stereological dissector technique. Hippocampus, 2, 437–444. [DOI] [PubMed] [Google Scholar]
- Gericke CA, Schulte-Herbrüggen O, Arendt T, & Hellweg R (2005). Chronic alcohol intoxication in rats leads to a strong but transient increase in NGF levels in distinct brain regions. Journal of Neural Transmission, 113(7), 813–820. [DOI] [PubMed] [Google Scholar]
- Gilman S, Adams K, Koeppe RA, Berent S, Kluin KJ, Modell JG, Kroll P, & Brunberg JA (1990). Cerebellar and frontal hypometabolism in alcoholic cerebellar degeneration studied with positron emission tomography. Annals of Neurology, 28(6), 775–785. [DOI] [PubMed] [Google Scholar]
- Gil-Mohapel J, Brocardo PS, Choquette W, Gothard R, Simpson JM, & Christie BR (2013). Hippocampal neurogenesis levels predict water maze search strategies in the aging brain. PLoS One, 8(9). doi: 10.1371/journal.pone.0075125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilyarov AV (2008). Nestin in central nervous system cells. Neuroscience and Behavioral Physiology, 38, 165–169. [DOI] [PubMed] [Google Scholar]
- Givens B, Williams JM, & Gill TM (2000). Septohippocampal pathway as a site for the memory-impairing effects of EtOH. Hippocampus, 10(1), 111–121. [DOI] [PubMed] [Google Scholar]
- Goldstein G, & Shelly C (1980). Neuropsychological investigation of brain lesion localization in alcoholism. In: Begleiter H (Ed). Biological Effect of Alcohol, Plenum Press, New York, NY, pp. 731–743. [DOI] [PubMed] [Google Scholar]
- Golub HM, Zhou Q-G, Zucker H, McMullen MR, Kokiko-Cochran ON, Ro EJ, Nagy LE, & Suh H (2015). Chronic alcohol exposure is associated with decreased neurogenesis, aberrant integration of newborn neurons, and cognitive dysfunction in female mice. Alcoholism: Clinical and Experimental Research, 39(10), 1967–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracian EI, Shelley LE, Morris AM, & Gilbert PE (2013). Age-related changes in place learning for adjacent and separate locations. Neurobiology of Aging, 34(10), 2304–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray DT, & Barnes CA (2015). Distinguishing adaptive plasticity from vulnerability in the aging hippocampus. Neuroscience, 309, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guggenmos M, Schmack K, Sekutowicz M, Garbusow M, Sebold M, Sommer C, Smolka MN, Wittchen HU, Zimmerman US, Heinz A, & Sterzer P (2017). Quantitative neurobiological evidence for accelerated brain aging in alcohol dependence. Translational Psychiatry, 7, 1279. doi: 10.1038/s41398-017-0037-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Yu J, & Perez-Polo JR (1998). Responses in the aged rat brain after total immunolesion. Journal of Neuroscience Research, 54, 7–16. [DOI] [PubMed] [Google Scholar]
- Guo KH, Zhu JH, Yao ZB, Gu HY, Zou JT, & Li DP (2010). Chemical identification of nestin- immunoreactive neurons in the rat basal forebrain: a re-examination. Neurochemistry International, 56, 694–702. [DOI] [PubMed] [Google Scholar]
- Haberman RP, Branch A, & Gallagher M (2017). Targeting neural hyperactivity as a treatment to stem progression of late-onset Alzheimer’s Disease. Neurotherapeutics, 14(3), 662–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JM, & Savage LM (2016). Exercise leads to the re-emergence of the cholinergic/nestin neuronal phenotype within the medial septum/diagonal band and subsequent rescue of both hippocampal ACh efflux and spatial behavior. Experimental Neurology, 278, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JM, Gomez-Pinilla F, & Savage LM (2018). Nerve growth factor is responsible for exercise-induced recovery of septohippocampal cholinergic structure and function. Frontiers in Neuroscience, 12, 773. doi: 10.3389/fnins.2018.00773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen AW, Almeida FB, Bandiera S, Pulcinelli RR, Fragoso ALR, Schneider R Jr., Barros HMT, & Gomez R (2017). Taurine restores the exploratory behavior following alcohol withdrawal and decreases BDNF mRNA expression in the frontal cortex of chronic alcohol-treated rats. Pharmacology, Biochemistry & Behavior, 161, 6–12. [DOI] [PubMed] [Google Scholar]
- Harland BC, Collings DA, McNaughton N, Abraham WC, & Dalrymple-Alford JC (2014). Anterior thalamic lesions reduce spine density in both hippocampal CA1 and retrosplenial cortex, but enrichment rescues CA1 spines only. Hippocampus, 24(10), 1232–1247. [DOI] [PubMed] [Google Scholar]
- Harper CG (1998). The neuropathology of alcohol-specific brain damage, or does alcohol damage the brain? Journal of Neuropathology and Experimental Neurology, 57, 101–110. [DOI] [PubMed] [Google Scholar]
- Harper C (2009). The neuropathology of alcohol-related brain damage. Alcohol and Alcoholism, 44(2), 136–140. [DOI] [PubMed] [Google Scholar]
- Harper CG, & Kril JJ (1990). Neuropathology of alcoholism. Alcohol, 25, 207–216. [DOI] [PubMed] [Google Scholar]
- Harper C, & Matsumoto I (2005). Ethanol and brain damage. Current Opinion in Pharmacology, 5(1), 73–78. [DOI] [PubMed] [Google Scholar]
- Hazell AS, & Butterworth RF (2009). Update of cell damage mechanisms in thiamine deficiency: focus on oxidative stress, excitotoxicity and inflammation. Alcohol and Alcoholism, 44(2), 141–147. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Butterworth RF, & Hakim AM (1993). Cerebral vulnerability is associated with selective increase in extracellular glutamate concentration in experimental thiamine deficiency. Journal of Neurochemistry, 61(3), 1155–1158. [DOI] [PubMed] [Google Scholar]
- Hazell AS, Sheedy D, Oanea R, Aghourian M, Sun S, Jung JY, Wang D, & Wang C (2010). Loss of astrocytic glutamate transporters in Wernicke encephalopathy. Glia, 58(2), 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazell AS, Wang D, Oanea R, Sun S, Aghourian M, & Yong JJ (2014). Pyrithiamine- induced thiamine deficiency alters proliferation and neurogenesis in both neurogenic and vulnerable areas of the rat brain. Metabolic Brain Disease, 29(1), 145–152. [DOI] [PubMed] [Google Scholar]
- Heckers S, Ohtake T, Wiley RG, Lappi DA, Geula C, & Mesulam MM (1994). Complete and selective cholinergic denervation of rat neocortex and hippocampus but not amygdala by an immunotoxin against the p75 NGF receptor. Journal of Neuroscience, 14, 1271–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefti F, & Mash DC (1989). Localization of nerve growth factor receptors in the normal human brain and in Alzheimer’s disease. Neurobiology of Aging, 10, 75–87. [DOI] [PubMed] [Google Scholar]
- Hendrickson ML, Rao AJ, Demerdash ONA, & Kalil RE (2011). Expression of nestin by neural cells in the adult rat and human brain. PLoS ONE, 6(4). doi: 10.1371/journal.pone.0018535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques JF, Portugal CC, Canedo T, Relvas JB, Summavielle T, & Socodato R (2018). Microglia and alcohol meet at the crossroads: Microglia as critical modulators of alcohol neurotoxicity. Toxicology Letters, 283, 21–31. [DOI] [PubMed] [Google Scholar]
- Herrera DG, Yague AG, Johnsen-Soriano S, Bosch-Morell F, Collado-Morente L, Muriach M, Romero FJ, & Garcia-Verdugo JM (2003). Selective impairment of hippocampal neurogenesis by chronic alcoholism: protective effects of an antioxidant. PNAS, 100(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez CM, Gearhart DA, Parikh V, Hohnadel EJ, Davis LW, Middlemore ML, Warsi SP, Waller JL, & Terry AV Jr. (2006). Comparison of galantamine and donepezil for effects on nerve growth factor, cholinergic markers, and memory performance in aged rats. Journal of Pharmacology and Experimental Therapeutics. 316(2), 679–694. [DOI] [PubMed] [Google Scholar]
- Hodges H, Allen Y, Sinden J, Mitchell SN, Arendt T, Lantos PL, & Gray JA (1991). The effects of cholinergic drugs and cholinergic-rich foetal neural transplants on alcohol-induced deficits in radial maze performance in rats. Behavioural Brain Research, 43(1), 7–28 [DOI] [PubMed] [Google Scholar]
- Homewood JM, Bond NW, & Mackenzie A (1997). The effects of single and repeated episodes of thiamin deficiency on memory in alcohol-consuming rat. Alcohol, 14, 81–91. [DOI] [PubMed] [Google Scholar]
- Hoogenraad CC, & Bradke F (2009). Control of neuronal polarity and plasticity – A renaissance for microtubules? Trends in Cell Biology, 19(12), 669–676. [DOI] [PubMed] [Google Scholar]
- Hu M, Walker DW, Vickroy TW, & Peris J (1999) Chronic ethanol exposure increases 3H–GABA release in rat hippocampus by presynaptic muscarinic receptor modulation. Alcoholism: Clinical and Experimental Research, 23, 1587–1595. [PubMed] [Google Scholar]
- Hughes BA, Bohnsack JP, O’Buckley TK, Herman MA, & Morrow AL (2019). Chronic Ethanol Exposure and Withdrawal Impair Synaptic GABA. Alcoholism: Clinical and Experimental Research, 43(5), 822–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter R, McLuskie R, Wyper D, Patterson J, Christie JE, Brooks DN, McCulloch J, Fink G, & Goodwin GM (1989). The pattern of function-related regional cerebral blood flow investigated by single photon emission tomography with 99mTc-HMPAO in patients with presenile Alzheimer’s disease and Korsakoff’s psychosis. Psychological Medicine, 19(4), 847–855. [DOI] [PubMed] [Google Scholar]
- Ikarashi Y, Iizuka S, Imamura S, Yamaguchi T, Sekigucki T, Kanno H, Kawakami Z, Yuzurihara M, Kase Y, & Takeda S (2009). Effects of yokukansan, a traditional Japanese medicine, on memory disturbance and behavioral and psychological symptoms of dementia in thiamine-deficient rats. Biological and Pharmaceutical Bulletin, 32(10), 1701–1709. [DOI] [PubMed] [Google Scholar]
- Inaba H, Kishimoto T, Oishi S, Nagata K, Hasegawa S, Watanabe T, & Kida S (2016). Vitamin B1-deficient mice show impairment of hippocampus-dependent memory formation and loss of hippocampal neurons and dendritic spines: Potential microendophenotypes of Wernicke–Korsakoff syndrome. Bioscience, Biotechnology, and Biochemistry, 80(12), 2425–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagawa G, Sato K, Kikuchi T, Nishihama M, Shioda M, Koyama Y, Yamada Y, & Andoh T (2004). Chronic EtOH consumption does not affect action of propofol on rat hippocampal acetylcholine release in vivo. British Journal of Anaesthesia, 93(5), 737–739. [DOI] [PubMed] [Google Scholar]
- Jameson L, Frey T, Zeeberg B, Dalldorf F & Caplow W (1980). Inhibition of microtubule assembly by phosphorylation of microtubule-associated proteins. Biochemistry, 19 , 2472–2479. [DOI] [PubMed] [Google Scholar]
- Jin Z, Bhandage AK, Bazov I, Kononenko O, Bakalkin G, Korpi ER, & Birnir B (2014). Selective increases of AMPA, NMDA, and kainate receptor subunit mRNAs in the hippocampus and orbitofrontal cortex but not in prefrontal cortex of human alcoholics. Frontiers in Cellular Neuroscience, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Sun Y, Xie L, Batteur S, Mao XO, Smelick C, Logvinova A, & Greenberg DA (2003). Neurogenesis and aging: FGF-2 and HB-EGF restore neurogenesis in hippocampus and subventricular zone of aged mice. Aging Cell, 2(3), 175–183. [DOI] [PubMed] [Google Scholar]
- Jinno S (2011). Decline in adult neurogenesis during aging follows a topographic pattern in the mouse hippocampus. Journal of Comparative Neurology, 519(3), 451–466. [DOI] [PubMed] [Google Scholar]
- Johnson SA, Sacks PK, Turner SM, Gaynor LS, Ormerod BK, Maurer AP, Bizon JL, & Burke SN (2016). Discrimination performance in aging is vulnerable to interference and dissociable from spatial memory. Learning and Memory, 23, 339–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamarajan C, Porjesz B, Jones KA, Choi K, Chorlian DB, Padmanabhapillai A, Rangaswamy M, Stimus AT, & Begleiter H (2004). The role of brain oscillations as functional correlates of cognitive systems: a study of frontal inhibitory control in alcoholism. International Journal of Psychophysiology, 51(2), 155–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, Spigelman I, Sapp DW, & Olsen RW (1996). Persistent reduction of GABA(A) receptor-mediated inhibition in rat hippocampus after chronic intermittent EtOH treatment. Brain Research, 709(2), 221–8. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, & Nedergaard M (1998). Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nature Neuroscience, 1, 683–692. [DOI] [PubMed] [Google Scholar]
- Kapellusch AJ, Lester AW, Schwartz BA, Smith AC, & Barnes CA (2018). Analysis of learning deficits in aged rats on the W-track continuous spatial alternation task. Behavioral Neuroscience, 132(6), 512–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kausler DH (1994). Learning and memory in normal aging. San Diego, CA, US: Academic Press. [Google Scholar]
- Ke ZJ, DeGiorgio LA, Volpe BT, & Gibson GE (2003). Reversal of thiamine deficiency-induced neurodegeneration. Journal of Neuropathology & Experimental Neurology, 62(2), 195–207. [DOI] [PubMed] [Google Scholar]
- Kessels RP, & Kopelman MD (2012). Context memory in Korsakoff’s Syndrome. Neuropsychology Review, 22(2), 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita Y, Ago Y, Higashino K, Asada K, Takano E, Takuma K, & Matsuda T Galantamine promotes adult hippocampal neurogenesis via M1 muscarinic and a7 nicotinic receptors in mice. International Journal of Neuropsychopharmacology, 17(12), 1957–1968. [DOI] [PubMed] [Google Scholar]
- Kivipelto M, Rovio S, Ngandu T, Kåreholt I, Eskelinen M, Winblad B, Hachinski V, Cedazo-Minguez A, Soininen H, Tuomilehto J, & Nissinen A (2008). Apolipoprotein E epsilon4 magnifies lifestyle risks for dementia: a population-based study. Journal of Cellular and Molecular Medicine, 12(6B), 2762–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp DJ, Harper KM, Whitman BA, Zimomra Z, & Breese GR (2016). Stress and withdrawal from chronic EtOH induces selective changes in neuroimmune mRNAs in differing brain sites. Brain Sciences, 6(3), 25. doi: 10.3390/brainsci6030025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh S, Chang P, Collier TJ, & Loy R (1989). Loss of NGF receptor immunoreactivity in basal forebrain neurons of aged rats: correlation with spatial memory impairment. Brain Research, 498, 397–404. [DOI] [PubMed] [Google Scholar]
- Koob GF, & Volkow ND (2010). Neurocircuitry of addiction. Neuropsychopharmacology, 35(1), 217–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopelman MD, Thomson AD, Guerrini I, & Marshall EJ (2009). The Korsakoff syndrome: Clinical aspects, psychology and treatment. Alcohol and Alcoholism, 44, 148–154. [DOI] [PubMed] [Google Scholar]
- Kotani S, Yamauchi T, Teramoto T, & Ogura H (2006). Pharmacological evidence of cholinergic involvement in adult hippocampal neurogenesis in rats. Neuroscience, 142(2), 505–514. [DOI] [PubMed] [Google Scholar]
- Kril JJ, Halliday GM, Svoboda MD, & Cartwright H (1997). The cerebral cortex is damaged in chronic alcoholics. Neuroscience, 79(4), 983–998. [DOI] [PubMed] [Google Scholar]
- Kroener S, Mulholland PJ, New NN, Gass JT, Becker HC, & Chandler LJ (2012). Chronic alcohol exposure alters behavioral and synaptic plasticity of the rodent prefrontal cortex. PLoS One, 7(5), e37541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A,& Foster TC (2019). Alteration in NMDA receptor mediated glutamatergic neurotransmission in the hippocampus during senescence. Neurochemistry Research, 44(1), 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuźma E, Llewellyn DJ, Langa KM, Wallace RB, & Lang IA (2014). History of alcohol use disorders and risk of severe cognitive impairment: A 19-year prospective cohort study. The American Journal of Geriatric Psychiatry, 22(10), 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmin A, Liljequist S, Meis J, Chefer V, Shippenberg T, & Bakalkin G (2012) Repeated moderate-dose ethanol bouts impair cognitive function in Wistar rats. Addiction Biology, 17, 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais PJ, & Mair RG (1990). Protective effects of the glutamate antagonist MK-801 on pyrithiamine-induced lesions and amino acid changes in rat brain. Journal of Neuroscience, 10(5), 1664–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais PJ, Mandel RJ, & Mair RG (1992). Diencephalic lesions, learning impairments and intact retrograde memory following acute thiamine deficiency in the rat. Behavioural Brain Research, 48, 177–185. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, & Savage LM (1995). Thiamine deficiency in rats produces cognitive and memory deficits on spatial tasks that correlate with tissue loss in diencephalon, cortex and white matter. Behavioural Brain Research, 68(1), 75–89. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, & Zhang SX (1993). Extracellular glutamate is increased in thalamus during thiamine deficiency-induced lesions and is blocked by MK-801. Journal of Neurochemistry, 61(6), 2175–2182. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, & Zhang SX (1997). Cortical and subcortical white matter damage without Wernicke’s encephalopathy after recovery from thiamine deficiency in the rat. Alcoholism: Clinical and Experimental Research, 21(3), 434–443. [DOI] [PubMed] [Google Scholar]
- Lärkfors L, Ebendal T, Whittemore SR, Persson H, Hoffer B, & Olson L (1987). Decreased level of nerve growth factor (NGF) and its messenger RNA in the aged brain. Brain Research, 427(1), 55–60. [DOI] [PubMed] [Google Scholar]
- Lauterborn JC, Tran TM, Isackson PJ, & Gall CM (1993). Nerve growth factor mRNA is expressed by GABAergic neurons in rat hippocampus. Neuroreport, 5(3), 273–276. [DOI] [PubMed] [Google Scholar]
- Leal SL, & Yassa MA (2015). Neurocognitive aging and the hippocampus across species. Trends in Neurosciences, 38(12), 800–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Berre AP, Pitel AL, Chanraud S, Beaunieux H, Eustache F, Martinot JL, Reynaud M, Martelli C, Rohlfing T, Sullivan EV, & Pfefferbaum A (2014). Chronic alcohol consumption and its effect on nodes of frontocerebellar and limbic circuitry: Comparison of effects in France and the United States. Human Brain Mapping, 35(9), 4635–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Im SJ, Lee SG, Stadlin A, Son JW, Shin CJ Ju G, Lee SI, & Kim S (2016). Volume of hippocampal subfields in patients with alcohol dependence. Psychiatry Research: Neuroimaging, 258, 16–22. [DOI] [PubMed] [Google Scholar]
- Le Maître TW, Dhanabalan G, Bogdanovic N, Alkass K, & Druid H (2018). Effects of alcohol abuse on proliferating cells, stem/progenitor cells, and immature neurons in the adult human hippocampus. Neuropsychopharmacology, 43(4), 690–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roch K, Riche D, & Sara SJ (1987). Persistence of habituation deficits after neurological recovery from severe thiamine deprivation. Behavioural Brain Research, 26(1), 37–46. [DOI] [PubMed] [Google Scholar]
- Lescaudron L, Seguela P, Geffard M, & Verna A (1986). Effects of long-term EtOH consumption on gabaergic neurons in the mouse hippocampus: A quantitative immunocytochemical study. Drug and Alcohol Dependence, 18(4), 377–384. [DOI] [PubMed] [Google Scholar]
- Lindwall G, & Cole RD (1984). Phosphorylation affects the ability of tau protein to promote microtubule assembly. Journal of Biological Chemistry, 259, 5301–5305. [PubMed] [Google Scholar]
- Lippai D, Bala S, Petrasek J, Csak T, Levin I, Kurt-Jones EA, & Szabo G (2013). Alcohol-induced IL-1β in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. Journal of Leukocytes Biology, 94(1), 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, White G, & Weight FF (1989). Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science, 243(4899), 1721–1724. [DOI] [PubMed] [Google Scholar]
- Lucas SM, Rothwell NJ, & Gibson RM (2006). The role of inflammation in CNS injury and disease. British Journal of Pharmacology, 147 Suppl 1, S232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukoyanov NV, Andrade JP, Dulce Madeira M, & Paula-Barbosa MM (1999a). Effects of age and sex on the water maze performance and hippocampal cholinergic fibers in rats. Neuroscience Letters, 269(3), 141–144. [DOI] [PubMed] [Google Scholar]
- Lukoyanov NV, Brandão F, Cadete-Leite A, Madeira MD, & Paula-Barbosa MM (2000). Synaptic reorganization in the hippocampal formation of alcohol-fed rats may compensate for functional deficits related to neuronal loss. Alcohol, 20(2),139–48. [DOI] [PubMed] [Google Scholar]
- Lukoyanov NV, Madeira MD, & Paula–Barbosa MM (1999b). Behavioral and neuroanatomical consequences of chronic EtOH intake and withdrawal. Physiology & Behavior, 66(2), 337–346. [DOI] [PubMed] [Google Scholar]
- Lukoyanov NV, Pereira PA, Paula-Barbosa MM, & Cadete-Leite A (2003). Nerve growth factor improves spatial learning and restores hippocampal cholinergic fibers in rats withdrawn from chronic treatment with ethanol. Experimental Brain Research, 148, 88–94. [DOI] [PubMed] [Google Scholar]
- Lynch MA (1998). Age-related impairment in long-term potentiation in hippocampus: A role for the cytokine, interleukin-1 beta? Progress in Neurobiology, 56, 571–589. [DOI] [PubMed] [Google Scholar]
- Mair RG (1994). On the role of thalamic pathology in diencephalic amnesia. Reviews in Neuroscience, 5(2), 105–140. [DOI] [PubMed] [Google Scholar]
- Mair RG, Anderson CD, Langlais PJ, McEntee WJ (1985). Thiamine deficiency depletes cortical norepinephrine and impairs learning processes in the rat. Brain Research, 360, 273–284. [DOI] [PubMed] [Google Scholar]
- Mair RG, Knoth RL, Rabchenuk SA, & Langlais PJ (1991a). Impairment of olfactory, auditory, and spatial serial reversal learning in rats recovered from pyrithiamine-induced thiamine deficiency. Behavioral Neuroscience, 105, 360–374 [PubMed] [Google Scholar]
- Mair RG, Otto TA, Knoth RL, Rabchenuk SA, & Langlais PJ (1991b). Analysis of aversively conditioned learning and memory in rats recovered from pyrithiamine-induced thiamine deficiency. Behavioral Neuroscience, 105, 351–359. [PubMed] [Google Scholar]
- Makino M, Takahashi-Ito K, Murasawa H, Pawlak A, Kashimoto Y, & Kitano Y (2019). Memantine ameliorates learning and memory disturbance and the behavioral and psychological symptoms of dementia in thiamine-deficient mice. Pharmacology Biochemistry and Behavior, 183, 6–13. [DOI] [PubMed] [Google Scholar]
- Malerba F, Paoletti F, & Cattaneo A (2016). NGF and proNGF reciprocal interference in immunoassays: Open questions, criticalities, and ways forward. Frontiers in Molecular Neuroscience, 9, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowska AL, Stone WS, Ingram DK, Reynolds J, Gold PE, Conti LH, Pontecorvo MJ, Wenk GL & Olton DS (1989). Individual differences in aging: behavioral and neurobiological correlates. Neurobiology of Aging, 10, 31–43. [DOI] [PubMed] [Google Scholar]
- Matthews DB, Watson MR, James K, Kastner A, Schneider A, & Mittleman G (2019). The impact of low to moderate chronic intermittent EtOH exposure on behavioral endpoints in aged, adult, and adolescent rats. Alcohol, 78, 33–42. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Fu W, Waeg G, & Uchida K (1997). 4-hydroxynonenal, a product of lipid peroxidation, inhibits dephosphorylation of the microtubule-associated protein tau. NeuroReport, 8, 2275–2281. [DOI] [PubMed] [Google Scholar]
- McAvoy KM, & Sahay A (2017). Targeting adult neurogenesis to optimize hippocampal circuits in aging. Neurotherapeutics, 14(3), 630–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy GM, Warden AS, Bridges CR, Blednov YA, & Harris RA (2018). Chronic EtOH consumption: Role of TLR3/TRIF-dependent signaling. Addiction Biology, 23(3), 889–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClain JA, Hayes DM, Morris SA, & Nixon K (2011). Adolescent binge alcohol exposure alters hippocampal progenitor cell proliferation in rats: Effects on cell cycle kinetics. The Journal of Comparative Neurology, 519(13), 2697–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menninger JA (2002). Assessment and treatment of alcoholism and substance-related disorders in the elderly. Bulletin of the Menninger Clinic, 66(2), 166–183. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Levey AI, & Wainer BH (1983). Cholinergic innervation of cortex by the basal forebrain: Cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. The Journal of Comparative Neurology, 214(2), 170–197. [DOI] [PubMed] [Google Scholar]
- Michalczyk K, & Ziman M (2005). Nestin structure and predicted function in cellular cytoskeletal organisation. Histology Histopathology, 20, 665–671. [DOI] [PubMed] [Google Scholar]
- Miguel-Hidalgo JJ, Wei J, Andrew M, Overholser JC, Jurjus G, Stockmeier CA, & Rajkowska G (2002). Glia pathology in the prefrontal cortex in alcohol dependence with and without depressive symptoms. Biological Psychiatry, 52(12), 1121–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MW, & Mooney SM (2004). Chronic exposure to EtOH alters neurotrophin content in the basal forebrain-cortex system in the mature rat: Effects on autocrine-paracrine mechanisms. Journal of Neurobiology, 60(4), 490–498. [DOI] [PubMed] [Google Scholar]
- Miller R, King M, Heaton M, & Walker D (2002). The effects of chronic EtOH consumption on neurotrophins and their receptors in the rat hippocampus and basal forebrain. Brain Research, 950(1–2), 137–147. [DOI] [PubMed] [Google Scholar]
- Montesinos J, Alfonso-Loeches S, & Guerri C (2016). Impact of the Innate Immune Response in the Actions of EtOH on the Central Nervous System. Alcoholism: Clinical and Experimental Research, 40(11), 2260–2270. [DOI] [PubMed] [Google Scholar]
- Morel GR, Andersen T, Pardo J, Zuccolilli GO, Cambiaggi VL, Hereñú CB, & Goya RG (2015). Cognitive impairment and morphological changes in the dorsal hippocampus of very old female rats. Neuroscience, 303, 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, & Baxter MG (2012). The aging cortical synapse: Hallmarks and implications or cognitive decline. Nature Neuroscience, 13(4), 240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, & Hof PR (1997). Life and death of neurons in the aging brain. Science, 278, 412–419. [DOI] [PubMed] [Google Scholar]
- Moselhy HF, Georgiou G, & Kahn A (2001). Frontal lobe changes in alcoholism: a review of the literature. Alcohol and Alcoholism, 36(5), 357–368. [DOI] [PubMed] [Google Scholar]
- Moser EI, Krobert KA, Moser MB, & Morris RG (1998). Impaired spatial learning after saturation of long-term potentiation. Science, 281(5385), 2038–2042. [DOI] [PubMed] [Google Scholar]
- Mufson EJ, Mahady L, Waters D, Counts SE, Perez SE, DeKosky ST, Ginsberg SD, Ikonomovic MD, Scheff SW, & Binder LI (2015). Hippocampal plasticity during the progression of Alzheimer’s disease. Neuroscience, 309, 51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulholland PJ, Teppen TL, Miller KM, Sexton HG, Pandey SC, & Swartzwelder HS (2018). Donepezil reverses dendritic spine morphology adaptations and Fmr1 epigenetic modifications in the hippocampus of adult rats after adolescent alcohol e18 exposure. Alcoholism: Clinical and Experimental Research, 42(4), 706–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumby DG, Cameli L, & Glenn MJ (1999). Impaired allocentric spatial working memory and intact retrograde memory after thalamic damage cause by thiamine deficiency in rats. Behavioral Neuroscience, 113, 42–50. [PubMed] [Google Scholar]
- Munro CA, Saxton J, & Butters MA (2000). The neuropsychological consequences of abstinence among older alcoholics: a cross-sectional study. Alcoholism: Clinical and Experimental Research, 24(10), 1510–1516. doi: 10.1111/j.1530-0277.2000.tb04569.x [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Bernot T, Moseanko R, Brignolo L, Blesch A, Conner JM, Ramirez A, Gasmi M, & Tuszynski MH (2009). Long-term reversal of cholinergic neuronal decline in aged non-human primates by lentiviral NGF gene delivery. Experimental Neurology, 215(1), 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawasai O, Tadano T, Hozumi S, Tan-No K, Niijima F, & Kisara K (2000). Immunohistochemical estimation of brain choline acetyltransferase and somatostatin related to the impairment of avoidance learning induced by thiamine deficiency. Brain Research Bulletin, 52(3), 189–196. [DOI] [PubMed] [Google Scholar]
- Nakano T, Fujimoto T, Shimooki S, Fukudome T, Uchida T, Tsuji T, Mitsuyama Y, Akimoto H, & Furukawa S (1996). Transient elevation of nerve growth factor content in the rat hippocampus and frontal cortex by chronic EtOH treatment. Psychiatry and Clinical Neuroscience, 50(3), 157–60. [DOI] [PubMed] [Google Scholar]
- Nardone R, Holler Y, Storti M, Christova M, Tezzon F, Golaszewski S, Trinka E, & Brigo F (2013). Thiamine deficiency induced neurochemical, neuroanatomical, and neurophysical alterations: A reappraisal. Science World Journal, 309143. doi: 10.1155/2013/309143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson TE, Ur CL, & Gruol DL (2005). Chronic intermittent ethanol exposure enhances NMDA-receptor-mediated synaptic responses and NMDA receptor expression in hippocampal CA1 region. Brain Res, 1048(1–2), 69–79. [DOI] [PubMed] [Google Scholar]
- Nixon K, & Crews FT (2004). Temporally specific burst in cell proliferation increases hippocampal neurogenesis in protracted abstinence from alcohol. Journal of Neuroscience, 24, 9714–9722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara S, Tsutsui K, Witter MP, & Iijima T (2013) Organization of multisynaptic inputs to the dorsal and ventral dentate gyrus: Retrograde trans-synaptic tracing with rabies virus vector in the rat. PLoS ONE, 8(11). doi: 10.1371/journal.pone.0078928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oscar-Berman M, Valmas MM, Sawyer KS, Ruiz SM, Luhar RB, & Gravitz ZR (2014). Profiles of impaired, spared, and recovered neuropsychologic processes in alcoholism. Handbook of Clinical Neurology, 125, 183–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SL, & Yakel JL (2019). The α7 nicotinic acetylcholine receptors regulate hippocampal adult-neurogenesis in a sexually dimorphic fashion. Brain Structure and Function, 224(2), 829–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh V, Howe WM, Welchko RM, Naughton SX, D’Amore DE, Han DH, & Sarter M (2013). Diminished trkA receptor signaling reveals cholinergic-attentional vulnerability of aging. The European Journal of Neuroscience, 37(2), 278–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons OA, & Nixon SJ (1993). Neurobehavioral sequelae of alcoholism. Neurologic Clinics, 11, 205–218. [PubMed] [Google Scholar]
- Pascual M, Baliño P, Aragón CM, & Guerri C (2015). Cytokines and chemokines as biomarkers of EtOH-induced neuroinflammation and anxiety-related behavior: role of TLR4 and TLR2. Neuropharmacology, 89, 352–359. [DOI] [PubMed] [Google Scholar]
- Paula-Barbosa MM, Brandao F, Madeira MD, & Cadete-Leite A (1993). Structural changes in the hippocampal formation after long-term alcohol consumption and withdrawal in the rat. Addiction, 88(2), 237–247. [DOI] [PubMed] [Google Scholar]
- Pepeu G, & Grazia GM (2017). The fate of the brain cholinergic neurons in neurodegenerative diseases. Brain Research, 1670, 173–184. [DOI] [PubMed] [Google Scholar]
- Perreira KM, & Sloan FA (2002). Excess alcohol consumption and health outcomes: A 6- year follow-up of men over age 50 from the health and retirement study. Addiction, 97(3), 301–310. [DOI] [PubMed] [Google Scholar]
- Perovic M, Tesic V, Djordjevic AM, Smiljanic K, Loncarevic-Vasiljkovic N, Ruzdijic S, & Kanazir S (2013). BDNF transcripts, proBDNF and proNGF, in the cortex and hippocampus throughout the life span of the rat. Age, 35(6), 2057–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfefferbaum A, Lim KO, Zipursky RB, Mathalon DH, & Rosenbloom MJ (1992). Brain gray and white matter volume loss accelerates with aging in chronic alcoholics: A quantitative MRI study. Alcoholism: Clinical and Experimental Research, 16(6), 1078–1089. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Sullivan EV, Mathalon DH, & Lim KO (1997). Frontal lobe volume loss observed with magnetic resonance imaging in older chronic alcoholics. Alcoholism: Clinical and Experimental Research, 21(3), 521–529. [DOI] [PubMed] [Google Scholar]
- Pires R, Pereira S, Pittella J, Franco GC, Ferreira C, Fernandes PA, & Ribeiro AM (2001). The contribution of mild thiamine deficiency and ethanol consumption to central cholinergic parameter dysfunction and rats’ open-field performance impairment. Pharmacology, Biochemistry and Behavior, 70, 227–235. [DOI] [PubMed] [Google Scholar]
- Pires RGW, Pereira SRC, Oliveira-Silva IF, Franco GC, & Ribeiro AM (2005). Cholinergic parameters and the retrieval of learned and re-learned spatial information: A study using a model of Wernicke–Korsakoff Syndrome. Behavioural Brain Research, 162(1), 11–21. [DOI] [PubMed] [Google Scholar]
- Pitel AL, Chételat G, Le Berre AP, Desgranges B, Eustache F, & Beaunieux H (2012). Macrostructural abnormalities in Korsakoff syndrome compared with uncomplicated alcoholism. Neurology, 78(17), 1330–1333. [DOI] [PubMed] [Google Scholar]
- Pitel AL, Segobin SH, Ritz L, Eustache F, & Beaunieux H (2015). Thalamic abnormalities are a cardinal feature of alcohol-related brain dysfunction. Neuroscience & Biobehavioral Reviews, 54, 38–45. [DOI] [PubMed] [Google Scholar]
- Pitel AL, Zahr NM, Jackson K, Sassoon S, Rosenbloom M, Pfefferbaum A, & Sullivan E (2011). Signs of preclinical Wernicke’s encephalopathy and thiamine levels as predictors of neuropsychological deficits in alcoholism without Korsakoff syndrome. Neuropsychopharmacology, 36, 580–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkin SR, & Savage LM (2001). Aging potentiates the acute and chronic neurological symptoms of pyrithiamine-induced thiamine deficiency in the rodent. Behavioural Brain Research, 119, 167–177. [DOI] [PubMed] [Google Scholar]
- Pitkin SR, & Savage LM (2004). Age-related vulnerability to diencephalic amnesia produced by thiamine deficiency: The role of time of insult. Behavioral Brain Research, 148, 93–105. [DOI] [PubMed] [Google Scholar]
- Qiang M, Denny AD, & Ticku MK (2007). Chronic intermittent ethanol treatment selectively alters N-methyl-D-aspartate receptor subunit surface expression in cultured cortical neurons. Molecular Pharmacology, 72(1), 95–102. [DOI] [PubMed] [Google Scholar]
- Qin L, & Crews FT (2012a). Chronic EtOH increases systemic TLR3 agonist-induced neuroinflammation and neurodegeneration. Journal of Neuroinflammation, 9, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, & Crews FT (2012b). NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. Journal of Neuroinflammation, 9(1), 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, & Crews FT (2014). Focal thalamic degeneration from ethanol and thiamine deficiency is associated with neuroimmune gene induction, microglial activation and lack of moncarboxylic acid transporters. Alcoholism: Clinical and Experimental Research, 38(3), 657–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, He J, Hanes RN, Pluzarev O, Hong JS, & Crews FT (2008). Increased systemic and brain cytokine production and neuroinflammation by endotoxin following EtOH treatment. Journal of Neuroinflammation, 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani CSS, & Ticku MK Comparison of chronic ethanol and chronic intermittent ethanol treatments on the expression of GABAA and NMDA receptor subunits. Alcohol, 38(2), 89–97. [DOI] [PubMed] [Google Scholar]
- Rapp PR, & Gallagher M (1996). Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. PNAS, 93(18), 9926–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen T, Schliemann T, Sørensen JC, Zimmer J, & West MJ (1996). Memory impaired aged rats: No loss of principal hippocampal and subicular neurons. Neurobiology of Aging, 17(1), 143–7. [DOI] [PubMed] [Google Scholar]
- Resende LS, Ribeiro AM, Werner D, Hall JM, & Savage LM (2012). Thiamine deficiency degrades the link between spatial behavior and hippocampal synapsin I and phosphorylated synapsin I protein levels. Behavioural Brain Research, 232(2), 421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice AC, Bullock MR, & Shelton KL (2004). Chronic EtOH consumption transiently reduces adult neural progenitor cell proliferation. Brain Research, 1011(1), 94–98. [DOI] [PubMed] [Google Scholar]
- Risher ML, Fleming RL, Boutros N, Semenova S, Wilson WA, Levin ED, Markou A, Swartzwelder HS, & Acheson SK (2013). Long-term effects of chronic intermittent EtOH exposure in adolescent and adult rats: Radial-arm maze performance and operant food reinforced responding. PloS One, 8(5). doi: 10.1371/journal.pone.0062940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risher ML, Fleming RL, Risher WC, Miller KM, Klein RC, Wills T, Acheson SK, Moore SD, Wilson WA, Eroglu C, & Swartzwelder HS (2015). Adolescent intermittent alcohol exposure: Persistence of structural and functional hippocampal abnormalities into adulthood. Alcoholism: Clinical and Experimental Research, 39(6), 989–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie K, & Villebrun D (2008). Epidemiology of alcohol related dementia. Handbook of Clinical Neurology, 89, 845–850. [DOI] [PubMed] [Google Scholar]
- Roberto M, Nelson TE, Ur CL, & Gruol DL (2002). Long-term potentiation in the rat hippocampus is reversibly depressed by chronic intermittent EtOH exposure. Journal of Neurophysiology, 87(5), 2385–2397. [DOI] [PubMed] [Google Scholar]
- Roberto M, & Varodayan FP (2017). Synaptic targets: Chronic alcohol actions. Neuropharmacology, 122, 85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitsek J, Fortin NJ, Koh MT, Gallagher M, & Eichenbaum H (2008). Cognitive aging: A common decline of episodic recollection and spatial memory in rats. Journal of Neuroscience, 28(36), 8945–8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Mark K, Vetreno RP, & Savage LM (2008). Increasing hippocampal acetylcholine levels enhance behavioral performance in an animal model of diencephalic amnesia. Brain Research, 1234, 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, & Savage LM (2007). Blunted hippocampal, but not striatal, acetylcholine efflux parallels learning impairment in diencephalic-lesioned rats. Neurobiology of Learning and Memory, 87, 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, & Savage LM (2009). The role of cholinergic and GABAergic medial septal/diagonal band cell populations in the emergence of diencephalic amnesia. Neuroscience, 160, 32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero AM, Esteban-Pretel G, Marín MP, Ponsoda X, Ballestín R, Canales JJ, & Renau-Piqueras J (2010). Chronic EtOH exposure alters the levels, assembly, and cellular organization of the actin cytoskeleton and microtubules in hippocampal neurons in primary culture. Toxicological Sciences, 118(2), 602–612. [DOI] [PubMed] [Google Scholar]
- Rourke SB, & Grant I (1999). The interactive effects of age and length of abstinence on the recovery of neuropsychological functioning in chronic male alcoholics: A 2-year follow-up study. Journal of the International Neuropsychological Society, 5(3), 234–246. [DOI] [PubMed] [Google Scholar]
- Rowe WB, Blalock EM, Chen K, Kadish I, Wang D, Barrett JE, Thibault O, Porter NM, Rose GM, & Landfield PW (2007). Hippocampal expression analyses reveal selective association of immediate-early, neuroenergetic, and myelinogenic pathways with cognitive impairment in aged rats. Journal of Neuroscience, 27(12), 3098–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakharkar AJ, Vetreno RP, Zhang H, Kokare DM, Crews FT, & Pandey SC (2016). A role for histone acetylation mechanisms in adolescent alcohol exposure-induced deficits in hippocampal brain-derived neurotrophic factor expression and neurogenesis markers in adulthood. Brain Structure & Function, 221(9), 4691–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santucci AC, Mercado M, Bettica A, Cortes C, York D, & Moody E (2004). Residual behavioral and neuroanatomical effects of short-term chronic ethanol consumption in rats. Cognitive Brain Research, 20(3), 449–461. [DOI] [PubMed] [Google Scholar]
- Savage LM, Candon PM, & Hohmann HL (2000). Alcohol-induced brain pathology and behavioral dysfunction: Using an animal model to examine sex differences. Alcoholism: Clinical and Experimental Research, 24, 465–475. [PubMed] [Google Scholar]
- Savage LM, Chang Q, & Gold PE (2003) Diencephalic damage decreases hippocampal acetylcholine release during spontaneous alternation testing. Learning & Memory, 10, 242–246. [DOI] [PubMed] [Google Scholar]
- Savage LM, Hall JM, & Resende LS (2012). Translational rodent models of Korsakoff syndrome reveal the critical neuroanatomical substrates of memory dysfunction and recovery. Neuropsychology review, 22(2), 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage LM, Roland JJ, & Klintsova A (2007). Selective septohippocampal-but not forebrain amygdalar–cholinergic dysfunction in diencephalic amnesia. Brain Research, 1139, 210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliebs R, & Arendt T (2011). The cholinergic system in aging and neuronal degeneration. Behavioural Brain Research, 221(2), 555–563. [DOI] [PubMed] [Google Scholar]
- Scott SA, Liang S, Weingartner JA, & Crutcher KA (1994). Increased NGF-like activity in young but not aged rat hippocampus after septal lesions. Neurobiology of Aging, 15(3), 337–346. [DOI] [PubMed] [Google Scholar]
- Segobin S, Laniepce A, Ritz L, Lannuzel C, Boudehent C, Cabé N, Urso L, Vabret F, Eustache F, Beaunieux H, & Pitel AL (2019). Dissociating thalamic alterations in alcohol use disorder defines specificity of Korsakoff’s syndrome. Brain, 142(5), 1458–1470. [DOI] [PubMed] [Google Scholar]
- Seo D, Lacadie CM, Tuit K, Hong KI, Constable RT, & Sinha R (2013). Disrupted ventromedial prefrontal function, alcohol craving, and subsequent relapse risk. JAMA Psychiatry, 70(7), 727–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharrett-Field L, Butler TR, Reynolds AR, Berry JN, & Prendergast MA (2013). Sex differences in neuroadaptation to alcohol and withdrawal neurotoxicity. Pflugers Archive, 465(5), 643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, & Barnes CA (1996). Age-related decrease in cholinergic synaptic transmission in three hippocampal subfields. Neurobiology of Aging, 17(3), 439–451. [DOI] [PubMed] [Google Scholar]
- Shnitko TA, Gonzales SW, & Grant KA (2019). Low cognitive flexibility as a risk for heavy alcohol drinking in non-human primates. Alcohol, 74, 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuboni-Mulligan DD, Chakravarty S, Mallett CL, Wolf AM, Dmitriev PM, Forton SM, & Shapiro EM (2019). In vivo serial MRI of age-dependent neural progenitor cell migration in the rat brain. Neuroimaging, 199, 153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonyi A, Christian MR, Sun AY, & Sun GY (2004). Chronic EtOH-induced subtype- and subregion-specific decrease in the mRNA expression of metabotropic glutamate receptors in rat hippocampus. Alcoholism: Clinical & Experimental Research, 28(9), 1419–1423. [DOI] [PubMed] [Google Scholar]
- Smith DM, & Atkinson RM (1995). Alcoholism and dementia. The International Journal of the Addictions, 30, 1843–1869. [DOI] [PubMed] [Google Scholar]
- Smith TD, Adams MM, Gallagher M, Morrison JH, & Rapp PR (2000). Circuit- specific alterations in hippocampal synaptophysin immunoreactivity predict spatial learning impairment in aged rats. Journal of Neuroscience, 20(17), 6587–6593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Cooper JD, Svendsen CN, Crossman P, Ip NY, Lindsay RM, Zafra F, & Lindholm D (1993). Atrophy but not death of adult septal cholinergic neurons after ablation of target capacity to produce mRNAs for NGF, BDNF, and NT3. Journal of Neuroscience, 13, 5263–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solari N, & Hangya B (2018). Cholinergic modulation of spatial learning, memory and navigation. European Journal of Neuroscience, 48(5), 2199–2230. doi: 10.1111/ejn.14089. Epub 2018 Aug 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song BJ, Akbar M, Abdelmegeed MA, Byun K, Lee B, Yoon SK, & Hardwick JP (2014). Mitochondrial dysfunction and tissue injury by alcohol, high fat, nonalcoholic substances and pathological conditions through post-translational protein modifications. Redox Biology, 3, 109–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP (2018). Effects of adolescent alcohol consumption on the brain and behavior. Nature Reviews Neuroscience, 19, 197–214. [DOI] [PubMed] [Google Scholar]
- Swartzwelder HS, Acheson SK, Miller KM, Sexton HG, Liu W, Crews FT, & Risher ML (2015). Adolescent intermittent alcohol exposure: Deficits in object recognition memory and forebrain cholinergic markers. PloS one, 10(11), e0140042. doi: 10.1371/journal.pone.0140042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EV, Harris RA, & Pfefferbaum A (2010). Alcohol’s effects on brain and behavior. Alcohol Research & Health, 33(1–2), 127. [PMC free article] [PubMed] [Google Scholar]
- Sullivan EV, & Pfefferbaum A (2009). Neuroimaging of the Wernicke-Korsakoff syndrome. Alcohol and Alcoholism, 44(2), 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan EV, Sable HJK, Strother WN, Friedman DP, Davenport A, Tillman-Smith H, Kraft RA, Wyatt C, Szeliga KT, Buccheimer NC, Daunais JB, Adalsteinsson E, Pfefferbaum A, & Grant KA (2005). Neuroimaging of rodent and primate models of alcoholism: Initial reports from the integrative neuroscience initiative on alcoholism. Alcoholism: Clinical & Experimental Research, 29(2), 287–294. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Zahr NM, Sassoon SA, Thompson WK, Kwon D, Pohl KM, & Pfefferbaum A (2018). The role of aging, drug dependence, and Hepatitis C comorbidity in alcoholism cortical compromise. JAMA Psychiatry, 75(5), 474–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szutowicz A, Bielarczyk H, Gul S, Ronowska A & Pawełczyk T (2006) Phenotype-dependent susceptibility of cholinergic neuroblastoma cells to neurotoxic inputs. Met. Brain Dis, 21, 149–161. [DOI] [PubMed] [Google Scholar]
- Tanila H, Shapiro M, Gallagher M, & Eichenbaum H (1997). Brain aging: Changes in the nature of information coding by the hippocampus. Journal of Neuroscience, 71, 5155–5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, & Katzman R (1991). Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Annals of Neurology, 30(4), 572–580. [DOI] [PubMed] [Google Scholar]
- Thinschmidt JS, Walker DW, & King MA (2003). Chronic EtOH treatment reduces the magnitude of hippocampal LTD in the adult rat. Synapse, 48(4), 189–197. [DOI] [PubMed] [Google Scholar]
- Thompson SG, & McGeer EG (1985). GABA-transaminase and glutamic acid decarboxylase changes in the brain of rats treated with pyrithiamine. Neurochemical Research, 10(12), 1653–60. [DOI] [PubMed] [Google Scholar]
- Tiwari V, & Chopra K (2013). Protective effect of curcumin against chronic alcohol-induced cognitive deficits and neuroinflammation in the adult rat brain. Neuroscience, 244, 147–158. [DOI] [PubMed] [Google Scholar]
- Todd KG, & Butterworth RF (1998). Evaluation of the role of NMDA-mediated excitotoxicity in the selective neuronal loss in experimental Wernicke encephalopathy. Experimental Neurology, 149(1), 130–138.9454622 [Google Scholar]
- Todd KG, & Butterworth RF (1999a). Early microglial response in experimental thiamine deficiency: an immunohistochemical analysis. Glia, 25(2), 190–198. [DOI] [PubMed] [Google Scholar]
- Todd KG, & Butterworth RF (1999b). Mechanisms of selective neuronal cell death due to thiamine deficiency. Annals of the New York Academy of Sciences, 893(1), 404–411. [DOI] [PubMed] [Google Scholar]
- Toledo Nunes P, Vedder LC, Deak T, & Savage LM (2019). A Pivotal Role for Thiamine Deficiency in the Expression of Neuroinflammation Markers in Models of Alcohol-Related Brain Damage. Alcoholism: Clinical and Experimental Research, 43(3), 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran T, Gallagher M, & Kirkwood A (2018). Enhanced postsynaptic inhibitory strength in hippocampal principal cells in high-performing aged rats. Neurobiology of Aging, 70, 92–101. 08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremwel MF, & Hunter BE (1994). Effects of chronic EtOH ingestion on long-term potentiation remain even after a prolonged recovery from EtOH exposure. Synapse, 17(2), 141–148 [DOI] [PubMed] [Google Scholar]
- Trevisan L, Fitzgerald LW, Brose N, Gasic GP, Heinemann SF, Duman RS, & Nestler EJ (1994). Chronic ingestion of EtOH up-regulates NMDAR1 receptor subunit immunoreactivity in rat hippocampus. Journal of Neurochemistry, 62(4), 1635–1638. [DOI] [PubMed] [Google Scholar]
- Tyler RE, Kim SW, Guo M, Jang YJ, Damadzic R, Stodden T, Vendruscolo LF, Koob GF, Wang GJ, Wiers CE, & Volkow ND (2019). Detecting neuroinflammation in the brain following chronic alcohol exposure in rats: A comparison between in vivo and in vitro TSPO radioligand binding. European Journal of Neuroscience, Epub ahead of print. [DOI] [PMC free article] [PubMed]
- U.S. Department of Health and Human Services, Substance Abuse and Mental Health Services Administration, Center for Behavioral Health Statistics and Quality. (2014). The National Survey on Drug Use and Health 2013 (NSDUH-2016-DS0001). Retrieved from https://datafiles.samhsa.gov/ [PubMed]
- Valero J, Bernardino L, Cardoso FL, Silva AP, Fontes-Ribeiro C, Ambrósio AF, & Malva JO (2017). Impact of Neuroinflammation on Hippocampal Neurogenesis: Relevance to Aging and Alzheimer’s Disease. Journal of Alzheimer’s Disease. 60(s1), S161–S168. [DOI] [PubMed] [Google Scholar]
- Vallés SL, Blanco AM, Pascual M, & Guerri C (2004). Chronic EtOH treatment enhances inflammatory mediators and cell death in the brain and in astrocytes. Brain Pathology, 14(4), 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Oort R & Kessels RP (2009). Executive dysfunction in Korsakoff’s syndrome: Time to revise the DSM criteria for alcohol-induced persisting amnestic disorder? International Journal of Psychiatry in Clinical Practice, 13(1), 78–81. [DOI] [PubMed] [Google Scholar]
- Vaucher E, Aumont N, Pearson D, Rowe W, Poirier J, & Kar S (2001). Amyloid Beta peptide levels and its effects on hippocampal acetylcholine release in aged, cognitively-impaired and -unimpaired rats. Journal of Chemical Neuroanatomy, 21(4), 323–329. [DOI] [PubMed] [Google Scholar]
- Vedder LC, Hall JM, Jabrouin KR, & Savage LM (2015). Interactions between chronic ethanol consumption and thiamine deficiency on neural plasticity, spatial memory, and cognitive flexibility. Alcoholism: Clinical and Experimental Research, 39(11), 2143–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedder LC, & Savage LM (2017). BDNF regains function in hippocampal long-term potentiation deficits caused by diencephalic damage. Learning & Memory, 24(2), 81–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Anzalone SJ, & Savage LM (2008). Impaired, spared, and enhanced ACh efflux across the hippocampus and striatum in diencephalic amnesia is dependent on task demands. Neurobiology of Learning and Memory, 90(1), 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Bohnsack JP, Kusumo H, Liu W, Pandey SC, & Crews FT (2019). Neuroimmune and epigenetic involvement in adolescent binge EtOH-induced loss of basal forebrain cholinergic neurons: Restoration with voluntary exercise. Addiction Biology, Early View. doi: 10.1111/adb.12731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Broadwater M, Liu W, Spear LP, & Crews FT (2014). Adolescent, but not adult, binge EtOH exposure leads to persistent global reductions of choline acetyltransferase expressing neurons in brain. PLOS One, 9(11). doi: 10.1371/journal.pone.0113421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, & Crews FT (2012). Adolescent binge drinking increases expression of the danger signal receptor agonist HMGB1 and Toll-like receptors in the adult prefrontal cortex. Neuroscience, 226, 475–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, & Crews FT (2015). Binge EtOH exposure during adolescence leads to a persistent loss of neurogenesis in the dorsal and ventral hippocampus that is associated with impaired adult cognitive functioning. Frontiers in Neuroscience, 9, 35. doi: 10.3389/fnins.2015.00035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, & Crews FT (2018). Adolescent binge EtOH-induced loss of basal forebrain cholinergic neurons and neuroimmune activation are prevented by exercise and indomethacin. PLoS One, 13(10). e0204500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Hall JM, & Savage LM (2011). Alcohol-related amnesia and dementia: animal models have revealed the contributions of different etiological factors on neuropathology, neurochemical dysfunction and cognitive impairment. Neurobiology of Learning and Memory, 96(4), 596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Lawrimore CJ, Rowsey PJ, & Crews FT (2018). Persistent adult neuroimmune activation and loss of hippocampal neurogenesis following adolescent EtOH exposure: Blockade by exercise and the anti-inflammatory drug indomethacin. Frontiers in Neuroscience, 12, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Qin L, & Crews FT (2013). Increased receptor for advanced glycation end product expression in the human alcoholic prefrontal cortex is linked to adolescent drinking. Neurobiology of Disease, 59, 52–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Ramos RL, Anzalone S, & Savage LM (2012). Brain and behavioral pathology in an animal model of Wernicke’s encephalopathy and Wernicke–Korsakoff Syndrome. Brain Research, 1436, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bohlen und Halbach O (2010). Involvement of BDNF in age-dependent alterations in the hippocampus. Frontiers in Aging Neuroscience, 2(36). doi: 10.3389/fnagi.2010.00036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainer BH, Levey AI, Rye DB, Mesulam MM, & Mufson EJ (1985). Cholinergic and non-cholinergic septohippocampal pathways. Neuroscience Letters, 54(1), 45–52. [DOI] [PubMed] [Google Scholar]
- Walker DW, Barnes DE, Zornetzer SF, Hunter BE, & Kubanis P (1980). Neuronal loss in hippocampus induced by prolonged EtOH consumption in rats. Science, 209(4457), 711–713. [DOI] [PubMed] [Google Scholar]
- Wang D, & Hazell AS (2010). Microglial activation is a major contributor to neurologic dysfunction in thiamine deficiency. Biochemical and Biophysical Research Communications, 402(1), 123–128. [DOI] [PubMed] [Google Scholar]
- Wang X, Yu H, You J, Wang C, Feng C, Liu Z, Li Y, Wie R, Xu S, Zhao R, Wu X, & Zhang G (2018). Memantine can improve chronic EtOH exposure-induced spatial memory impairment in male C57BL/6 mice by reducing hippocampal apoptosis. Toxicology, 406–407, 21–32. [DOI] [PubMed] [Google Scholar]
- Whitman BA, Knapp DJ, Werner DF, Crews FT, & Breese GR (2013). The cytokine mRNA increase induced by withdrawal from chronic EtOH in the sterile environment of brain is mediated by CRF and HMGB1 release. Alcoholism: Clinical and Experimental Research, 37(12), 2086–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson IA, Ikonen S, McMahan RW, Gallagher M, Eichenbaum H, & Tanila H (2003). Place cell rigidity correlates with impaired spatial learning in aged rats. Neurobiology of Aging, 24(2), 297–305. [DOI] [PubMed] [Google Scholar]
- Winocur G (1988). A neuropsychological analysis of memory loss with age. Neurobiology of Aging, 9, 487–494. [DOI] [PubMed] [Google Scholar]
- Wolstenholme JT, Mahmood T, Harris GM, Abbas S, & Miles MF (2017). Intermittent ethanol during adolescence leads to lasting behavioral changes in adulthood and alters gene expression and histone methylation in the pfc. Frontiers in Molecular Neuroscience, 10, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods AJ, Porges EC, Bryant VE, Seider T, Gongvatana A, Kahler CW, de la Monte S, Monti PM, & Cohen RA (2016). Current heavy alcohol consumption is associated with greater cognitive impairment in older adults. Alcoholism: Clinical and Experimental Research, 40(11), 2435–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Siow YL, & O, K. (2009). Induction of hepatic cyclooxygenase-2 by hyperhomocysteinemia via nuclear factor-kappaB activation. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 297(4), 1086–1094. [DOI] [PubMed] [Google Scholar]
- Xie Y, Meeker RB, Massa SM, & Longo FM (2019). Modulation of the p75 neurotrophin receptor suppresses age-related basal forebrain cholinergic neuron degeneration. Scientific Reports, 9, 5273. doi: 10.1038/s41598-019-41654-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Meng Y, Li W, Yong Y, Fan Z, Ding H, Wie Y, Luo J, & Ke ZJ (2011). Neuronal MCP-1 mediates microglia recruitment and neurodegeneration induced by the mild impairment of oxidative metabolism. Brain Pathology, 21(3), 279–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Xue X, Tian H, Wang XX, Dong YX, Wang F, Zhao YN, Yao XC, Cui W, & Wu CF (2014). Role of microglia in EtOH-induced neurodegenerative disease: Pathological and behavioral dysfunction at different developmental stages. Pharmacology and Therapeutics, 144(3), 321–337. [DOI] [PubMed] [Google Scholar]
- Zahr NM, Kilian MP, Manojkumar S, Sullivan EV, & Pfefferbaum A (2019). Hippocampal subfield CA2+3 exhibits accelerated aging in Alcohol use Disorder: A preliminary study. NeuroImage: Clinical, 22. doi:10/1016/j.nicl.2019.101764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SX, Weilersbacher GS, Henderson SW, Corso T, Olney JW, & Langlais PJ (1995). Excitotoxic cytopathology, progression, and reversibility of thiamine deficiency-induced diencephalic lesions. Journal of Neuropathology & Experimental Neurology, 54(2), 255–267. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Pfefferbaum A, Podhajsky S, Pohl KM & Sullivan EV (2019). Accelerated aging and motor control deficits are related to regional deformation of central cerebellar white matter in alcohol use disorder. Addiction Biology, Early Access. doi: 10.1111/adb.12746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YN, Wang F, Fan YX, Ping GF, Yang JY, & Wu CF (2013). Activated microglia are implicated in cognitive deficits, neuronal death, and successful recovery following intermittent EtOH exposure. Behavioural Brain Research, 236(1), 270–282. [DOI] [PubMed] [Google Scholar]
- Zhao N, Zhong C, Wang Y, Zhao Y, Gong N, Zhou C, Xu T, & Hong Z (2008). Impaired hippocampal neurogenesis is involved in cognitive dysfunction induced by thiamine deficiency at early prepathological lesion stage. Neurobiology of Disease, 29, 176–185. [DOI] [PubMed] [Google Scholar]
- Zhu J, Gu H, Yao Z, Zou J, Guo K, Li D, & Gao T (2011). The nestin-expressing and non-expressing neurons in rat basal forebrain display different electrophysiological properties and project to hippocampus. BMC Neuroscience, 12, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]