

Abstract
Acute myeloid leukemia (AML) is a heterogeneous hematologic malignancy in terms of clinical features, underlying pathogenesis and treatment outcomes. Recent advances in genomic techniques have unraveled the molecular complexity of AML leukemogenesis, which in turn have led to refinement of risk stratification and personalized therapeutic strategies for patients with AML. Incorporation of prognostic and druggable genetic biomarkers into clinical practice to guide patient-specific treatment is going to be the mainstay in AML therapeutics. Since 2017 there has been an explosion of novel treatment options to tailor personalized therapy for AML patients. In the past 3 years, the U.S. Food and Drug Administration approved a total of eight drugs for the treatment of AML; most specifically target certain gene mutations, biological pathways, or surface antigen. These novel agents are especially beneficial for older patients or those with comorbidities, in whom the treatment choice is limited and the clinical outcome is very poor. How to balance efficacy and toxicity to further improve patient outcome is clinically relevant. In this review article, we give an overview of the most relevant genetic markers in AML with special focus on the therapeutic implications of these aberrations.
Keywords: Acute myeloid leukemia, Genetic markers, Risk stratification, Therapeutics
Introduction
Acute myeloid leukemia (AML) is a clonal hematologic malignancy with great variability in the clinical features, pathogenesis and treatment outcomes [1, 2]. The incidence of AML is increasing over time and males are more prone to develop AML than female [3]. It is the most common form of acute leukemia in adults and accounts for the highest percentage of leukemia death [4].
Although the majority of fit patients initially achieve complete remission (CR) after induction chemotherapy, a significant number of patients eventually experience disease relapse or refractoriness [5, 6], which underscores the unmet need for novel therapies. Until recently, the treatment options for AML have been limited to cytotoxic chemotherapy and allogeneic hematopoietic stem cell transplantation (HSCT). The combination of an anthracycline and cytarabine, widely known as the “7 + 3” regimen, has been the cornerstone of induction therapy for AML for decades. Efforts to improve the response rate and overall survival (OS) had previously focused on dose intensification of cytarabine and the addition of pharmaceutically distinct agents to induction and the following consolidation chemotherapy [7–10]. Nevertheless, the clinical outcome of AML patients treated with these cytotoxic drugs, even in combination with HSCT, is not satisfactory. The long-term survival in de novo patients younger than 60 years is approximately 30–50%, and that in older patients and those with secondary AML is less than 10% [2, 11, 12], highlighting the urgent need for novel treatment to improve the survival. Herein we give an overview of the most relevant genetic markers in AML and their clinical implications in risk-stratification and targeted therapy.
Changes of AML classification over time with the advance of genomics
AML was initially classified by the French-American-British (FAB) Cooperative Group in 1976 according to the cell lineage of leukemic cells and the extent of their differentiation based on the cell morphology and cytochemical staining of bone marrow (BM) cells [13]. However, it was far from perfect to precisely stratify this heterogeneous disease and predict outcome. The identification of recurrent cytogenetic abnormalities advance our understanding of the AML biology and drive decision-making in clinical practice [14–16]. In 2001, the World Health Organization (WHO) introduced a new classification system by including recurrent cytogenetic abnormalities as criteria [17], which was followed by a revised version in 2008 [18].
Advances in genomic techniques and research have greatly shed light on our understanding of cancer biology. It is found that more than 95% of AML patients have driving and co-concurring mutations regardless of the presence of cytogenetic abnormalities [19–21]. Because of the importance of genetic aberrations, AML with recurrent genetic abnormalities (either cytogenetic or molecular genetic) is classified as the first subtype of AML, together with five other subtypes, in the latest 2016 WHO Classification (Table 1) [1].
Table 1.
The 2016 WHO classification of acute myeloid leukemia (AML) and related neoplasms
| AML with recurrent genetic abnormalities | |
| AML with t(8;21)(q22;q22.1);RUNX1-RUNX1T1 | |
| AML with inv. (16)(p13.1q22) or t(16;16)(p13.1;q22);CBFB-MYH11 | |
| APL with PML-RARA | |
| AML with t(9;11)(p21.3;q23.3);MLLT3-KMT2A | |
| AML with t(6;9)(p23;q34.1);DEK-NUP214 | |
| AML with inv. (3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2, MECOM | |
| AML (megakaryoblastic) with t(1;22)(p13.3;q13.3);RBM15-MKL1 | |
| Provisional entity: AML with BCR-ABL1 | |
| AML with mutated NPM1 | |
| AML with biallelic mutations of CEBPA | |
| Provisional entity: AML with mutated RUNX1 | |
| AML with myelodysplasia-related changes | |
| Therapy-related myeloid neoplasms | |
| AML, NOS | |
| AML with minimal differentiation | |
| AML without maturation | |
| AML with maturation | |
| Acute myelomonocytic leukemia | |
| Acute monoblastic/monocytic leukemia | |
| Pure erythroid leukemia | |
| Acute megakaryoblastic leukemia | |
| Acute basophilic leukemia | |
| Acute panmyelosis with myelofibrosis | |
| Myeloid sarcoma | |
| Myeloid proliferations related to Down syndrome | |
| Transient abnormal myelopoiesis (TAM) | |
| Myeloid leukemia associated with Down syndrome |
APL, acute promyelocytic leukemia; NOS, not otherwise specified
Genomic landscape in AML and its implication in risk classification
The mutations that have a putative role in AML pathogenesis are classified into eight categories according to their biological function, including those involving myeloid transcription-factor genes, NPM1, tumor suppressors, signaling genes, DNA methylation, chromatin modifier, cohesin complex and splicing factors (Table 2) [2, 19, 21]. The incidences of common molecular mutations in our AML cohort is shown in Fig. 1. It’s common that more than one mutations occur concurrently in the same patient (Fig. 2) indicating a role of concerted interaction of mutations in the pathogenesis of AML [22, 23]. The discovery of molecular genetic alterations has led to the refinement of prognostication in AML. The 2017 European LeukemiaNet (ELN) recommendation for risk-stratification of AML [24] (Table 3) is the most widely used model in current clinical practice since it incorporates cytogenetic changes and gene mutation status, including FLT3-ITD allelic ratio, into the risk classification which largely enhances the stratification power compared with the 2010 version of the ELN recommendations [25]. Based on integrated analysis of clinical features, survivals and patterns of mutual cooperativeness or exclusivity among cytogenetic and molecular genetics in large cohorts of patients, it’s clearly shown that the majority of AML cases can be classified into a number of biologically and prognostically distinct subgroups [23]. The 2017 ELN risk classification also works well in AML patients in Taiwan. (Fig. 3) It is suggested that patients with adverse-risk AML should be treated more aggressively to improve their survival.
Table 2.
Functional categories of genes that are commonly mutated in acute myeloid leukemia (AML)
| Functional category | Gene members | Role in AML Leukemogenesis |
|---|---|---|
| Myeloid transcription-factor genes |
Transcription factor fusions by chromosomal rearrangements, such as t(8;21)(q22;q22); RUNX1-RUNX1T1 and inv(16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11 GATA2, RUNX1 and CEBPA |
Transcriptional deregulation and impaired hematopoietic differentiation. |
| Nucleophosmin (NPM1) gene | NPM1 | Aberrant cytoplasmic localization of NPM1 and its interacting proteins |
| Tumor suppressor genes | TP53, WT1, PHF6 | Transcriptional deregulation and impaired degradation via the negative regulator (MDM2 and PTEN oncogenes) |
| Signaling genes | FLT3, KIT, PTPN11, RAS | Proliferative advantage through the RAS-RAF, JAK-STAT, and PI3K-AKT signaling pathways |
| DNA methylation | DNMT3A, TET2, IDH1, IDH2 | Deregulation of DNA methylation and oncometabolite production |
| Chromatin modifier | ASXL1, EZH2 and KMT2A | Deregulation of chromatin modification and impairment of methyltransferases function |
| Cohesin complex | STAG1, STAG2, RAD21, SMC1A, SMC3, | Impairment of accurate chromosome segregation and transcriptional regulation |
| Splicing factors | SRSF2, SF3B1, U2AF1, ZRSR2 | Deregulated RNA processing and aberrant splicing patterns |
Fig. 1.

Common molecular gene mutations and their incidences in 763 AML patients in Taiwan. The data are derived from the mutation analyses of 763 patients diagnosed and treated at the National Taiwan University Hospital
Fig. 2.
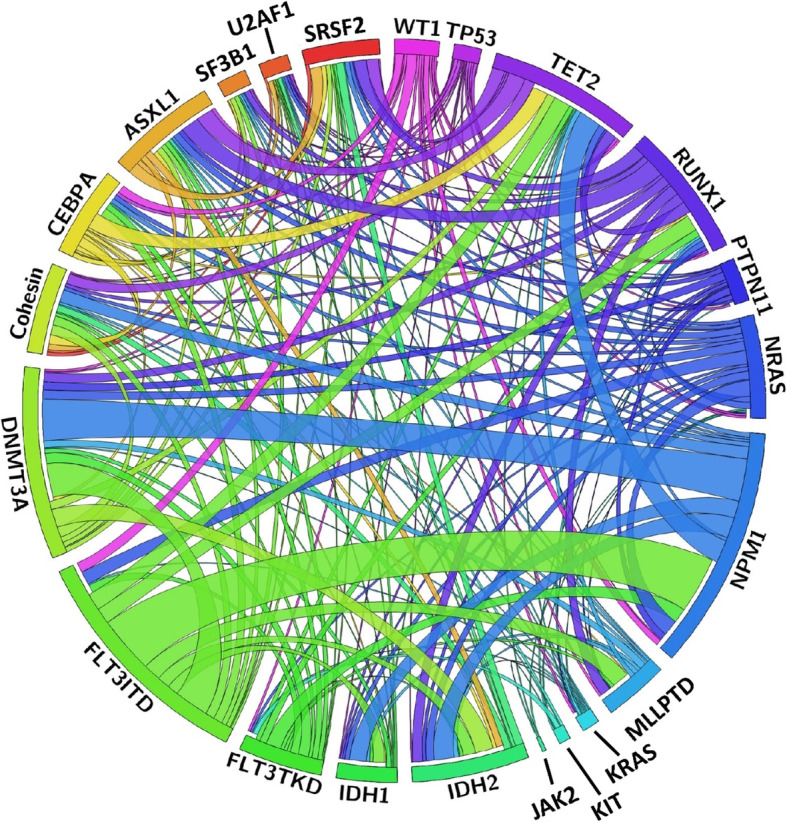
The Circos plots depicting the relative frequency and pairwise co-occurrence of genetic alterations in 500 AML patients in Taiwan. The length of the arc corresponds to the frequency of the first gene mutation, and the width of the ribbon corresponds to the proportion of the second gene mutation. The data are derived from the mutation analyses of 500 patients diagnosed and treated at the National Taiwan University Hospital
Table 3.
Risk stratification of AML according To 2017 ELN recommendations [24]
| Risk profiles | Subgroups |
|---|---|
| Favorable | t(8;21)(q22;q22.1); RUNX1-RUNX1T1 |
| inv (16)(p13.1q22) or t(16;16)(p13.1;q22); CBFB-MYH11 | |
| Mutated NPM1 without FLT3-ITD | |
| Mutated NPM1 with FLT3-ITDlow | |
| Biallelic mutated CEBPA | |
| Intermediate | Mutated NPM1 and FLT3-ITDhigh |
| Wild-type NPM1 without FLT3-ITD | |
| Wild-type NPM1 with FLT3-ITDlow | |
| t(9;11)(p21.3;q23.3); MLLT3-KMT2A | |
| Cytogenetic abnormalities not classified | |
| Adverse | t(6;9)(p23;q34.1); DEK-NUP214 |
| t(v;11q23.3); KMT2A rearranged | |
| t(9;22)(q34.1;q11.2); BCR-ABL1 | |
| inv (3)(q21.3q26.2) or t(3;3)(q21.3;q26.2); GATA2,MECOM(EVI1) | |
| Complex karyotype, monosomal karyotype | |
| -5 or del(5q); −7; −17/abn(17p) | |
| Wild-type NPM1 and FLT3-ITDhigh | |
| Mutated RUNX1 | |
| Mutated ASXL1 | |
| Mutated TP53 |
Low, low allelic ratio (< 0.5); high, high allelic ratio (≥0.5)
Fig. 3.
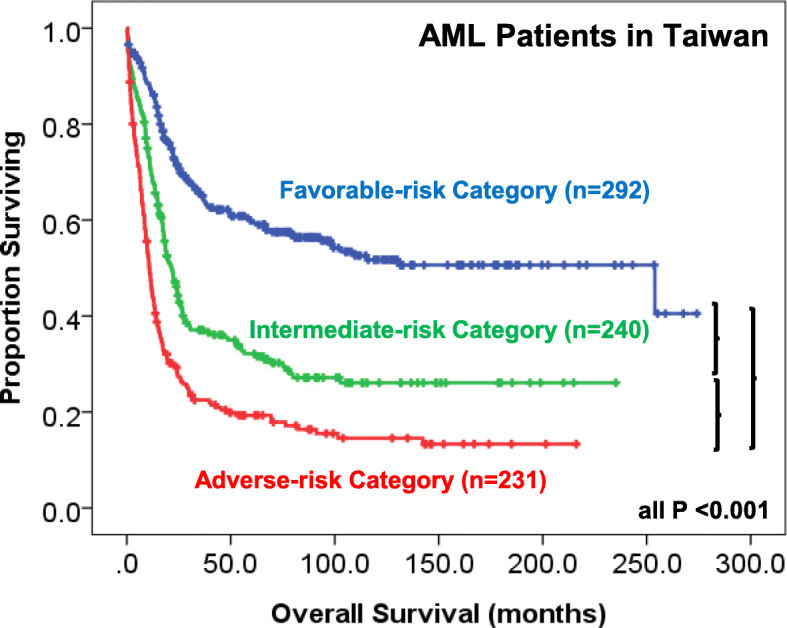
Survival curves of 763 AML patients in Taiwan stratified according to the 2017 ELN risk stratification. The data are derived from the mutation analyses of 763 patients diagnosed and treated at the National Taiwan University Hospital
Relevant genetic markers in AML
FLT3 mutations
FMS-like tyrosine kinase 3 gene (FLT3), located on chromosome 13q12, encodes a receptor tyrosine kinase that plays a major role in the regulation of hematopoiesis [26, 27]. There are two types of FLT3 mutations; internal tandem duplication (ITD) of the FLT3 juxtamembrane domain, which are gain-of- function mutations [28, 29], and tyrosine kinase domain (TKD) point mutations, which occur mainly at codon 835 or 836 within the activation loop of the second kinase domain [30, 31]. The FLT3 mutant protein constitutively activates the cascade of FLT3 signaling in the absence of FLT3 ligand promoting cell proliferation and decreased apoptosis [32–34].
Mutations of FLT3 are detected in approximately 25–30% of newly diagnosed AML patients as either ITD (20%) or point mutations in TKD (5–10%) [35–37]. Of note, the patients with FLT3-ITD have shorter disease-free survival (DFS), higher relapse rate and poorer OS [37–39], particularly among patients with high FLT3-ITD allelic ratio [40] or absence of NPM1 mutation [39, 41, 42]. Besides, the insertion site and ITD length of FLT3 as well as concomitant mutations also appear to influence the prognosis [40, 43, 44]. For these reason, patients with FLT3/ITD are frequently referred for early allogeneic HSCT in first CR. Accordingly, the ELN and the National Comprehensive Cancer Center Consensus panels designated FLT3/ITD with high allelic ratio as an unfavorable prognostic subgroup [24, 45]. On the other hand, the prognostic impact of FLT3-TKD is not well defined [46, 47]. Up to one third of AML patients with FLT3-ITD or FLT3-TKD would lose the mutation at relapse whereas acquisition of novel FLT3 mutations was detected in 20% patients at disease progression [48–50]. It is clinically significant to recheck FLT3-ITD or FLT3-TKD status at all subsequent treatment decision points in every patient regardless the FLT3 status at diagnosis [51].
KIT mutations
KIT, known as stem cell factor receptor (cluster of differentiation 117, CD117), belongs to type III receptor tyrosine kinase family and is involved in the regulation of survival and proliferation of hematopoietic progenitor cells [52, 53]. KIT is highly expressed in the majority of leukemic blasts [54, 55], and KIT mutations, most commonly affecting exons 8 and 17 (especially D816 mutations), are identified in approximately 25% of AML with core binding factor (CBF) rearrangements [56–59], but infrequently found in other AML types [20]. The prognostic impact of KIT mutations in AML is controversial. Recently, the targeted high-throughput sequencing in 331 patients with t(8;21), which leads to CBF rearrangement, showed that KIT mutation, especially with higher allele burden, was independently associated with increased relapse rate and reduced OS.
TP53 mutations
Somatic mutation of the tumor suppressor gene TP53, located in 17p13, is frequently detected in patients with therapy-related AML [60, 61] or AML with complex karyotype or monosomal karyotype (53–73%) [62–65], in contrast to 7–8% in de novo AML patients [19, 65]. In general, TP53 mutations independently predict lower CR rate, higher relapse rate, shorter event-free survival (EFS) and OS.
RAS mutations
RAS proteins (HRAS, KRAS and NRAS), which are activated by cytokine receptors in response to ligand stimulation, control proliferation and survival of hematopoietic progenitors [66–69]. Activated RAS mutations are mostly single nucleotide substitutions in codon 12, 13 or 61 [70–73]. NRAS and KRAS mutations are found in 8–12% and 2–5% of AML patients, respectively. The prognostic relevance of RAS mutations in AML remains controversial. Higher dose of cytarabine was reported to decrease the relapse rate in RAS-mutated AML patients [74].
Mutations of genes involving in epigenetic modifications
Deregulation of epigenetic modifiers, including alterations in DNA methylation, DNA hydroxymethylation and histone modifications, such as methylation, acetylation, phosphorylation, sumoylation and ubiquitination etc., is now recognized as an important mechanism in the pathogenesis of AML [75]. Somatic mutations in genes regulating epigenetic modifications, such as IDH, TET2, DNMT3A, ASXL1, EZH2 and KMT2A are frequently detected in patients with AML, especially those with intermediate-risk cytogenetics [19, 76–80]. The epigenetic alterations usually present as the early pre-leukemic events [81–83] which cooperate with other genetic alterations in the development of AML. Mutations in ASXL1, TET2, and DNMT3A as well as MLL rearrangements confer poor prognosis, whereas IDH and EZH2 mutations may bear clinical significance [75]. The treatment of choice for patients with epigenetic mutations is still under exploration. Induction chemotherapy with high-dose daunorubicin, as compared with standard-dose daunorubicin, improves OS among patients with DNMT3A mutations or MLL translocations [41]. Further, retrospective studies suggested that allogeneic HSCT in first CR can overcome the poor prognosis of patients with MLL rearrangements [84–86], DNMT3A mutations [87, 88], ASXL1 mutations [89], and IDH mutations [90]. Nevertheless, prospective studies are warrant to clarify the point. Here we will specifically focus on IDH, MLL, and EZH2 mutations in which targeted agents are either available in clinical use or under investigation.
IDH mutations
IDH1 and IDH2 genes encode two isoforms of isocitrate dehydrogenase, which catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG) [91]. Mutant IDH proteins convert α-KG to 2-hydroxyglutarate (2-HG), an onco-metabolite that contributes to tumor growth or malignant transformation [92, 93]. IDH mutations impair TET2-mediated hydroxymethylation of cytosine through reduced production of α-KG, a cofactor of TET2, thus result in global DNA hypermethylation [94]. Mutations in IDH1 occur in 7.8–16% of patients with cytogenetically normal (CN) AML (CN-AML), and IDH2 mutations, in 10–19% of those with CN-AML [75, 76, 78]. Both are less frequently detected in cytogenetically abnormal AML. Occasionally, IDH mutations can be detected in healthy older individuals with age-related clonal hematopoiesis, suggesting IDH mutations occur early in leukemogenesis [95]. The impact of IDH mutations on prognosis remains to be elucidated.
MLL rearrangements
The mixed lineage leukemia gene (MLL), also called Histone-lysine N-methyltransferase 2A (KMT2A), is located on chromosome 11q23 which encodes a DNA-binding protein that methylates histone H3 lysine 4 position (H3K4) and positively regulates Hox gene expression [96]. The incidence of MLL fusion and partial tandem duplication (MLL/PTD) in AML is approximately 5–12% and 5–6%, respectively [75] and the presence of MLL rearrangements usually predict aggressive course and poor outcome.
EZH2 mutations
Enhancer of Zest Homologue 2 gene (EZH2), located at chromosome 7q, is a H3K27 methyltransferase that catalyzes the Polycomb Repressive Complex 2 (PRC2) [97]. Mutations in EZH2 exert context-specific and sometimes opposing effects on tumorigenesis. Oncogenic gain-of-function mutations are found in patients with lymphoid malignancies [98]; in contrast, loss-of-function EZH2 mutations at diverse sites were detected in myeloid neoplasms [99, 100], including AML (0–2%) [75]. Further, derepression of stage-specific gene profiles induces polymorphic and contradictory phenotypes of EZH2 protein in different phases of AML [101, 102]. During AML maintenance, wild-type EZH2 exerts an oncogenic function as a facilitator of disease that may be therapeutically targeted. In contrast, EZH2 acts as a tumor suppressor during leukemia induction. The findings offer a potentially attractive therapeutic approach in AML with EZH2 mutations and the EZH2 inhibitor is currently under development or early phase trials.
Splicing factor mutations
RNA splicing, a crucial post-transcription process, plays an important role in gene regulation and increases genomic diversity [103]. However, aberrant splicing pathologically drives the initiation and progression of cancers, including hematologic malignancies. Mutations of the splicing factor (SF) genes occur most frequently in SRSF2, U2AF1, SF3B1 and ZRSR2 [104]. The reported incidence of SF mutations in AML varied from 4.5 to 12.5% [19, 105–107]. Presence of SF mutations predict lower CR rate and shorter DFS and OS [107]. The discovery of somatic mutations in the spliceosome and/or aberrant splicing in cancers has prompted interest to novel therapeutic approaches by targeting splicing catalysis, splicing regulatory proteins, and individual key altered splicing events [108, 109].
Targeted agents
Annotation of the mutational landscape in AML has greatly facilitated the development of targeted therapy. The breakthrough discovery of all-trans retinoic acid and arsenic trioxide in the treatment of acute promyelocytic leukemia [110], a specific subtype of AML, and tyrosine kinase inhibitor in chromic myeloid leukemia [111] has encouraged the searching for more novel targeted therapy in AML. In the past 3 years, the U.S. Food and Drug Administration (FDA) approved a total of eight drugs for AML; some specifically target certain gene mutations, biological pathways, or surface antigen. Based on the patients recruited in the clinical trials, most agents are approved at this moment for older patients or those with comorbidities, in whom the treatment choice is limited and the clinical outcome is very poor. It is expected that some of them may also be beneficial for younger and fit patients, but further clinical trials targeting this group of patients are needed to clarify it.
FLT3 inhibitor
Given the adverse prognostic impact of FLT3-ITD and high frequency of FLT3 mutations in de novo AML, FLT3 mutations are attractive targets for therapy. Several FLT3 inhibitors are now available for clinical use, while more are under development in preclinical and clinical studies. FLT3 inhibitors can be classified into first and second generation inhibitors based on the potency and target specificity of the drugs. First generation FLT3 inhibitors, such as sunitinib, sorafenib, lestaurtinib and midostaurin, are relatively nonspecific multi-kinase inhibitors and generally have higher toxicities, while second generation inhibitors, such as quizartinib, crenolanib, and gilteritinib, are more selective and potent FLT3 inhibitors and have fewer toxicities. FLT3 inhibitors can be further classified into type I and II inhibitors based on the mechanism of interaction with the receptor. Type I inhibitors are effective for both FLT3-ITD and FLT3-TKD, while type II inhibitors, for FLT3-ITD only [35].
Midostaurin (PKC412) is the first FLT3 inhibitor approved by the U.S. as well as Taiwan FDA for the treatment of newly-diagnosed FLT3-mutated AML patients based on its effect on improving OS when combined with traditional chemotherapy [112, 113]. It is Type I inhibitor and effective regardless of types of FLT3 mutations (ITD or TKD) or the allelic ratio of FLT3-ITD. Recently, maintenance of sorafenib, another first generation inhibitor, following allogeneic HSCT has shown encouraging results in FLT3-mutated AML by reducing the post-transplant relapse rate [114, 115].
As for second generation FLT3 inhibitors, Crenolanib and gilteritinib are type I inhibitors, whereas quizartinib is type II inhibitor [116]. Gilteritinib has single-agent activity in FLT3-mutated AML and was approved by the U.S. FDA in November 2018 for treating adult patients who have relapsed or refractory (R/R) FLT3-mutated AML based on safety data and an interim analysis of the response rate in the ADMIRAL trial (NCT02421939) [117]. The final results showed that the median OS and event-free survival (EFS) in the gilteritinib group was significantly longer than that in the chemotherapy group. The clinical trials to investigate its use as frontline treatment or maintenance in AML patients with FLT3 mutations are undergoing. Quizartinib (AC220) was shown effective as single agent in R/R FLT3/ITD patients with improving OS compared to chemotherapy. However, Quizartinib is only approved in this setting in Japan, but not in the USA and European Union (European Medicines Agency, EMA) due to marginal survival benefits and safety concerns. A number of other novel FLT3 inhibitors, such as tandutinib, crenolanib, cabozantinib, etc., are currently under development or in clinical trials [118].
Collectively, FLT3 inhibitors has emerged as an important part of therapy for FLT3-mutated patients in both frontline and R/R status. Much is still to be learned about how to advance the use of FLT3 inhibitors in fit or frail patients (such as novel combinations), overcome the primary and secondary acquired resistance, and manage the adverse effects, especially in maintenance therapy.
KIT inhibitor
KIT mutations occur frequently in CBF AML and may confer poorer prognosis in this group of patients. Since dasatinib is a potent oral multi-kinase inhibitor with strong activity on KIT oncoprotein, it has the potential to target this molecular aberration in AML patients. A phase 2, open-label, multicenter trial (CALGB10801) showed that chemotherapy plus dasatinib was well tolerated without any unexpected or dose-limiting toxicities [119]. It provided excellent outcomes (90% CR rate and 77% OS at 3-year) to both younger and older patients with KIT mutations, supporting further large-scale, prospective randomized phase 3 trials to evaluate KIT inhibitors in combination with cytotoxic chemotherapy in the treatment of KIT-mutated CBF AML.
TP53 inhibitor
A growing number of small low-molecular-weight compounds including PRIMA-1 and the PRIMA-1 analog APR-246 have been developed to restore tumor suppressor function to mutant p53 [120–123]. In a preliminary analysis of 45 TP53-mutated patients with myelodysplastic syndrome (MDS), a pre-leukemia state, and oligoblastic AML in a phase Ib/II combination study (APR-246 and azacitidine; NCT03072043) [124], the overall response rate (ORR) was 87% with 53% CR. The median time to response was 2.1 months (range, 0.1–5.4) and the median duration of response was 6.5 months with a median follow-up of 10.5 months. The randomized phase 3 study of APR-246 and AZA versus AZA alone in TP53-mutated MDS is ongoing (NCT03745716).
RAS inhibitor
Since farnesylation is the primary translational modification essential for the transforming activity of RAS oncoprotein, attempts to target RAS with farnesylation inhibitors have been developed since 2000 [69, 125]. Although preclinical activity was observed in RAS-mutant cell lines and animal models, clinical activity of farnesylation inhibitors in AML patients has been largely unsuccessful and disappointing [126–128]. Clinical trials targeting mitogen-activated protein kinase (MAPK) signaling in NRAS-mutated leukemia with MAP-ERK kinase (MEK) inhibitors are ongoing; the response is only minimal though MEK inhibitors are generally well tolerated [129, 130]. Further studies are required to explore other small-molecular inhibitors, select suitable patient cohort and investigate synergistic combination therapies.
IDH inhibitor
Enasidenib (AG-221), first IDH mutation-specific inhibitor, suppresses 2-HG production and induces cellular differentiation in primary human IDH2-mutated AML cells and xenograft mouse models [131]. In the interim analysis of the landmark first-in-human phase I/II trial for enasidenib (NCT01915498) [132], the ORR was 38.5%, including 20.2% CR in 109 adult R/R patients with IDH2-mutated AML receiving 100 mg daily. The median time to CR was 3.7 months and the duration of response in patients who attained CR was 8.8 months (range, 6.4-not reached). Accordingly, the U.S. FDA approved enasidenib in August 2017 for the treatment of IDH2-mutated R/R AML. The final analysis of this trial (n = 345) showed 46% attained their best response by cycle 4 and 80%, by cycle 6 among responding patients, implying failure to obtain early response with enasidenib does not necessarily indicate treatment failure and the importance of continuing enasidenib therapy for at least 5–6 cycles [133]. The clearance of IDH2-mutant clones was associated with achievement of CR. Clinical trials including enasidenib for R/R AML (phase 3, NCT02577406), newly diagnosed AML (NCT02632708) and post allogeneic HSCT maintenance (NCT03515512), and in combination with azacitidine for R/R AML (NCT03683433), etc. are ongoing.
Ivosidenib (AG-120) is a highly selective inhibitor for IDH1 mutants. It lowers 2-HG in tumor models and enhances differentiation of primary AML samples [134]. Ivosidenib monotherapy was associated with durable remissions in 179 patients with IDH1-mutated, R/R AML in a phase 1 dose-escalation and dose-expansion study (NCT02074839) [135]. The ORR was 41.6%, including 30.4% CR/CR with partial hematologic recovery (CRh); 21% of patients who had a CR or CRh had no residual detectable IDH1 mutations on digital polymerase-chain-reaction assay. The median time to CR/CRh was 2.7 months (range, 0.9–5.6) and the duration of response in these patients was 8.2 months (range, 5.5–12). In a trial of ivosidenib for patients with newly-diagnosed IDH1-mutated AML, the CR/CRh rate was 42% and the median duration of response was not reached in 34 patients who received ivosidenib 500 mg once daily [136]. Based on these findings, the U.S FDA approved ivosidenib in both the frontline and salvage treatment of IDH1-mutated AML.
Epigenetic therapies
Hypomethylating agents (HMA)
The hypomethylating agents (HMA), azacytidine and decitabine, have long been known for their effects in AML patients with low blast percentages (20–30%) [137, 138]. However, their effects in AML patients with higher blast percentages are not impressive, and efforts now are focused on the combination of the drug with other novel agents. Guadecitabine (SGI-110), a next-generation HMA, is a dinucleotide of decitabine and deoxyguanosine resulting in slow release of decitabine and prolonged in vivo half-life; therefore, guadecitabine is potentially more effective and less toxic than its parent drug [139]. Unfortunately, phase III ASTRAL-1 study failed to meet the primary endpoint of a statistical difference in CR and OS between guadecitabine and control arm [140]. Nevertheless, a benefit was observed in subgroup of patients who received 4 or more cycles, indicating that treatment duration is crucial to response. The phase III QUAZAR AML-001 study (NCT01757535) demonstrated that maintenance with CC-486, an oral formulation of 5-azacitidine, resulted in significant improvements in OS and RFS, compared with placebo [141]. CC-486 is the first HMA used in the maintenance setting to improve clinical outcome in patients with AML after achieving remission following induction chemotherapy, with or without consolidation.
DOT1L inhibitor
The histone 3 lysine 79 (H3K79) methyltransferase disruptor of telomeric silencing-1 like (DOT1L) is proposed to play a role in the development of leukemia in patients with MLL translocations [142, 143]. Pharmacological inhibition of DOT1L enzymatic activity has been of interest for the treatment of MLL-rearranged leukemias [144]. The DOT1L inhibitor Pinometostat (EPZ − 5676) exhibited modest clinical activity in a phase I study, which paves a road for further exploration of combination therapies in leukemia [145].
Bromodomain and extra-terminal (BET) protein inhibitors
Bromodomain and extra-terminal (BET) proteins bind acetylated lysine residues on histone tail to facilitate transcriptional activation [146]. BET proteins are involved in aberrant chromatin states in AML through MYC upregulation [147]. BET inhibitors, such as JQ1 and OTX015 (MK-8628) showed efficacy in cell lines, mouse models and ex vivo patient samples of MLL-fused, NPM1-, FLT3-, or IDH2-mutated leukemias [148]. So far single-agent activity is modest in R/R AML [149] and the early phase studies of several BET inhibitors are ongoing.
Lysine-specific demethylase 1 (LSD1) inhibitors
Lysine-specific demethylase 1 (LSD1), an enzyme responsible for the demethylation of H3K4 and H3K9, is an essential regulator to sustain the oncogenic potential of leukemic stem cells [150]. Several LSD1 inhibitors have shown in vitro anti-leukemic activity but also striking hematologic toxicity in mouse models [150, 151]. Ladademstat (ORY-1001), a highly potent and selective LSD1 inhibitor, induced blast differentiation and reduction of leukemic stem cell capacity in AML [152], and exhibited potent synergy with standard-of-care drugs and selective epigenetic inhibitors [152].
Histone deacetylase inhibitors
Several inhibitors of histone deacetylases (HDAC), such as panobinostat and vorinostat, have been developed [153, 154]. However, monotherapy with HDAC inhibitors only showed modest effect in AML [155].
BCL2 inhibition
The B-cell leukemia/lymphoma-2 (BCL-2), a key regulator of the mitochondrial apoptotic pathway, supports cell survival by suppressing programmed cell death [156, 157]. BCL2 is aberrantly overexpressed in AML blasts, specifically in leukemic stem cells [158], and enhanced BCL-2 expression mediates chemotherapy resistance [159, 160]. Venetoclax is a highly selective BH3 mimetic agent showing potent BCL-2 inhibition. Venetoclax-based therapy for heavily pretreated patients with R/R AML showed fair activity with 19–22.5% of patients achieving CR or CR with incomplete hematologic recovery (CRi) [161, 162]. Impressively, in phase Ib/II clinical trials, venetoclax in combination with low-dose cytarabine (LDAC) or HMAs in treatment-naive patients showed very exciting results; rapid and deep response could be seen in 54–67% of patients aged 75 years or older or those with comorbidities that precluded intensive chemotherapy [163, 164]. The response to venetoclax-based therapy is mostly observed within 1–2 cycles and the median survival is not reached for patients obtaining CR/CRi. The results served as the basis for accelerated approval by the U.S. FDA in Nov 2018 and herald a new era of AML therapy that largely avoids traditional cytotoxic agents in unfit patients.
The phase III trials comparing venetoclax and azacitidine to azacitidine alone (NCT02993523) and venetoclax and LDAC to LDAC alone (NCT03069352) are ongoing to confirm the clinical benefits. Besides, venetoclax is also tested to combine with other targeted agents, such as IDH inhibitors in IDH-mutated patients or FLT3 inhibitors in FLT3-mutated patients, to evaluate if such combinations can enhance anti-leukemic efficacy.
Other agents
In addition to the targeted therapies mentioned above, the most promising agents for non-mutation-targeted novel agents approved by the U.S. FDA include CPX-351 (Vyxeos), gemtuzumab ozogamicin (Mylotarg), and glasdegib (Daurismo). CPX-351 is a dual drug liposomal encapsulation of daunorubicin and cytarabine which is approved for the treatment of therapy-related AML and AML with myelodysplasia-related change [165]. Gemtuzumab ozogamicin (Mylotarg) is a humanized anti-CD33 monoclonal antibody linked to calicheamicin which is approved for newly-diagnosed and R/R CD33+ AML [166]. Glasdegib (Daurismo) is the first Hedgehog pathway inhibitor. It is approved in combination with low-dose cytarabine for newly diagnosed AML aged 75 or more or those who have comorbidities that preclude use of intensive induction chemotherapy [167].
Conclusion
Recent advances in genomics techniques have unraveled the molecular heterogeneity of AML leukemogenesis and further help refine risk stratification and prognostication. Patients with adverse-risk AML require more aggressive treatment including allogeneic HSCT in first CR and possibly novel targeted agents, to improve the prognosis. However, the complex pattern of cooperativity and mutual exclusivity among different mutations remain a clinical challenging. Since 2017 there has been an explosion of newly approved treatment options to tailor personalized treatment for AML. Each of these targeted therapies has unique treatment timing, dosing, efficacy, and adverse effects and appropriate management is crucial to the success of treatment. Further combinations of molecularly targeted therapies and standard cytotoxic chemotherapy or other novel agents to enhance efficacy are still under investigation. We believe it is clinically relevant to comprehensively elucidate the molecular signatures to better characterize the AML biology, precisely predict prognosis and tailor treatment strategies with targeted agents.
Acknowledgements
Nil
Authors’ contributions
H-AH and H-FT reviewed the literature and wrote the manuscript. The author(s) read and approved the final manuscript.
Funding
His work was partially sponsored by grants MOST 104–2314-B-002-128-MY4, 106–2314-B-002-226-MY3 and MOST 108–2628-B-002 -015 from the Ministry of Science and Technology (Taiwan) and MOHW 107-TDU-B-211-114009 from the Ministry of Health and Welfare (Taiwan.
Availability of data and materials
Non applicable.
Ethics approval and consent to participate
Non applicable.
Consent for publication
Non applicable.
Competing interests
The authors declare no competing financial interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 2.Dohner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373:1136–1152. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- 3.Surveillance, epidemiology, and end results (SEER) program cancer stat facts: Leukemia - acute myeloid leukemia (AML) https://seer.cancer.gov/statfacts/html/amyl.html; 2018, Accessed 18 Apr 2019.
- 4.Shallis RM, Wang R, Davidoff A, Ma X, Zeidan AM. Epidemiology of acute myeloid leukemia: recent progress and enduring challenges. Blood Rev. 2019;36:70–87. doi: 10.1016/j.blre.2019.04.005. [DOI] [PubMed] [Google Scholar]
- 5.Tang JL, Hou HA, Chen CY, et al. AML1/RUNX1 mutations in 470 adult patients with de novo acute myeloid leukemia: prognostic implication and interaction with other gene alterations. Blood. 2009;114:5352–5361. doi: 10.1182/blood-2009-05-223784. [DOI] [PubMed] [Google Scholar]
- 6.Hou HA, Huang TC, Lin LI, et al. WT1 mutation in 470 adult patients with acute myeloid leukemia: stability during disease evolution and implication of its incorporation into a survival scoring system. Blood. 2010;115:5222–5231. doi: 10.1182/blood-2009-12-259390. [DOI] [PubMed] [Google Scholar]
- 7.Weick JK, Kopecky KJ, Appelbaum FR, et al. A randomized investigation of high-dose versus standard-dose cytosine arabinoside with daunorubicin in patients with previously untreated acute myeloid leukemia: a southwest oncology group study. Blood. 1996;88:2841–2851. [PubMed] [Google Scholar]
- 8.Geller RB, Burke PJ, Karp JE, et al. A two-step timed sequential treatment for acute myelocytic leukemia. Blood. 1989;74:1499–1506. [PubMed] [Google Scholar]
- 9.Lowenberg B, Pabst T, Vellenga E, et al. Cytarabine dose for acute myeloid leukemia. N Engl J Med. 2011;364:1027–1036. doi: 10.1056/NEJMoa1010222. [DOI] [PubMed] [Google Scholar]
- 10.Burnett AK, Russell NH, Hills RK, et al. A randomized comparison of daunorubicin 90 mg/m2 vs 60 mg/m2 in AML induction: results from the UK NCRI AML17 trial in 1206 patients. Blood. 2015;125:3878–3885. doi: 10.1182/blood-2015-01-623447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thol F, Schlenk RF, Heuser M, Ganser A. How I treat refractory and early relapsed acute myeloid leukemia. Blood. 2015;126:319–327. doi: 10.1182/blood-2014-10-551911. [DOI] [PubMed] [Google Scholar]
- 12.Tsai CH, Hou HA, Tang JL, et al. Genetic alterations and their clinical implications in older patients with acute myeloid leukemia. Leukemia. 2016;30:1485–1492. doi: 10.1038/leu.2016.65. [DOI] [PubMed] [Google Scholar]
- 13.Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33:451–458. doi: 10.1111/j.1365-2141.1976.tb03563.x. [DOI] [PubMed] [Google Scholar]
- 14.Grimwade D, Walker H, Harrison G, et al. The predictive value of hierarchical cytogenetic classification in older adults with acute myeloid leukemia (AML): analysis of 1065 patients entered into the United Kingdom Medical Research Council AML11 trial. Blood. 2001;98:1312–1320. doi: 10.1182/blood.v98.5.1312. [DOI] [PubMed] [Google Scholar]
- 15.Slovak ML, Kopecky KJ, Cassileth PA, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a southwest oncology group/eastern cooperative oncology group study. Blood. 2000;96:4075–4083. [PubMed] [Google Scholar]
- 16.Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116:354–365. doi: 10.1182/blood-2009-11-254441. [DOI] [PubMed] [Google Scholar]
- 17.Vardiman JW, Harris NL, Brunning RD. The World Health Organization (WHO) classification of the myeloid neoplasms. Blood. 2002;100:2292–2302. doi: 10.1182/blood-2002-04-1199. [DOI] [PubMed] [Google Scholar]
- 18.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114:937–951. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas Research N. Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bullinger L, Dohner K, Dohner H. Genomics of acute myeloid leukemia diagnosis and pathways. J Clin Oncol. 2017;35:934–946. doi: 10.1200/JCO.2016.71.2208. [DOI] [PubMed] [Google Scholar]
- 22.Hou HA, Lin CC, Chou WC, et al. Integration of cytogenetic and molecular alterations in risk stratification of 318 patients with de novo non-M3 acute myeloid leukemia. Leukemia. 2014;28:50–58. doi: 10.1038/leu.2013.236. [DOI] [PubMed] [Google Scholar]
- 23.Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127:29–41. doi: 10.1182/blood-2015-07-604496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dohner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453–474. doi: 10.1182/blood-2009-07-235358. [DOI] [PubMed] [Google Scholar]
- 26.Kiyoi H, Towatari M, Yokota S, et al. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia. 1998;12:1333–1337. doi: 10.1038/sj.leu.2401130. [DOI] [PubMed] [Google Scholar]
- 27.Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532–1542. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 28.Nakao M, Yokota S, Iwai T, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–1918. [PubMed] [Google Scholar]
- 29.Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene. 2002;21:2555–2563. doi: 10.1038/sj.onc.1205332. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97:2434–2439. doi: 10.1182/blood.v97.8.2434. [DOI] [PubMed] [Google Scholar]
- 31.Spiekermann K, Bagrintseva K, Schoch C, Haferlach T, Hiddemann W, Schnittger S. A new and recurrent activating length mutation in exon 20 of the FLT3 gene in acute myeloid leukemia. Blood. 2002;100:3423–3425. doi: 10.1182/blood-2002-03-0953. [DOI] [PubMed] [Google Scholar]
- 32.Mizuki M, Fenski R, Halfter H, et al. Flt3 mutations from patients with acute myeloid leukemia induce transformation of 32D cells mediated by the Ras and STAT5 pathways. Blood. 2000;96:3907–3914. [PubMed] [Google Scholar]
- 33.Hayakawa F, Towatari M, Kiyoi H, et al. Tandem-duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL-3-dependent cell lines. Oncogene. 2000;19:624–631. doi: 10.1038/sj.onc.1203354. [DOI] [PubMed] [Google Scholar]
- 34.Grafone T, Palmisano M, Nicci C, Storti S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: biology and treatment. Oncol Rev. 2012;6:e8. doi: 10.4081/oncol.2012.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33:299–312. doi: 10.1038/s41375-018-0357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kottaridis PD, Gale RE, Linch DC. Flt3 mutations and leukaemia. Br J Haematol. 2003;122:523–538. doi: 10.1046/j.1365-2141.2003.04500.x. [DOI] [PubMed] [Google Scholar]
- 37.Kottaridis PD, Gale RE, Frew ME, et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98:1752–1759. doi: 10.1182/blood.v98.6.1752. [DOI] [PubMed] [Google Scholar]
- 38.Kottaridis PD, Gale RE, Langabeer SE, Frew ME, Bowen DT, Linch DC. Studies of FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood. 2002;100:2393–2398. doi: 10.1182/blood-2002-02-0420. [DOI] [PubMed] [Google Scholar]
- 39.Gale RE, Green C, Allen C, et al. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood. 2008;111:2776–2784. doi: 10.1182/blood-2007-08-109090. [DOI] [PubMed] [Google Scholar]
- 40.Schlenk RF, Kayser S, Bullinger L, et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood. 2014;124:3441–3449. doi: 10.1182/blood-2014-05-578070. [DOI] [PubMed] [Google Scholar]
- 41.Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 43.Schnittger S, Schoch C, Dugas M, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002;100:59–66. doi: 10.1182/blood.v100.1.59. [DOI] [PubMed] [Google Scholar]
- 44.Garg M, Nagata Y, Kanojia D, et al. Profiling of somatic mutations in acute myeloid leukemia with FLT3-ITD at diagnosis and relapse. Blood. 2015;126:2491–2501. doi: 10.1182/blood-2015-05-646240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.NCCN clinical oncology guideline acute meyloid leukemia. 2020 Available from: www.nccn.org.
- 46.Bacher U, Haferlach C, Kern W, Haferlach T, Schnittger S. Prognostic relevance of FLT3-TKD mutations in AML: the combination matters--an analysis of 3082 patients. Blood. 2008;111:2527–2537. doi: 10.1182/blood-2007-05-091215. [DOI] [PubMed] [Google Scholar]
- 47.Whitman SP, Ruppert AS, Radmacher MD, et al. FLT3 D835/I836 mutations are associated with poor disease-free survival and a distinct gene-expression signature among younger adults with de novo cytogenetically normal acute myeloid leukemia lacking FLT3 internal tandem duplications. Blood. 2008;111:1552–1559. doi: 10.1182/blood-2007-08-107946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kronke J, Bullinger L, Teleanu V, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122:100–108. doi: 10.1182/blood-2013-01-479188. [DOI] [PubMed] [Google Scholar]
- 49.Shih LY, Huang CF, Wu JH, et al. Internal tandem duplication of FLT3 in relapsed acute myeloid leukemia: a comparative analysis of bone marrow samples from 108 adult patients at diagnosis and relapse. Blood. 2002;100:2387–2392. doi: 10.1182/blood-2002-01-0195. [DOI] [PubMed] [Google Scholar]
- 50.McCormick SR, McCormick MJ, Grutkoski PS, et al. FLT3 mutations at diagnosis and relapse in acute myeloid leukemia: cytogenetic and pathologic correlations, including cuplike blast morphology. Arch Pathol Lab Med. 2010;134:1143–1151. doi: 10.5858/2009-0292-OA.1. [DOI] [PubMed] [Google Scholar]
- 51.Elshoury A, Przespolewski A, Baron J, Wang ES. Advancing treatment of acute myeloid leukemia: the future of FLT3 inhibitors. Expert Rev Anticancer Ther. 2019;19:273–286. doi: 10.1080/14737140.2019.1573679. [DOI] [PubMed] [Google Scholar]
- 52.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 53.Ogawa M, Matsuzaki Y, Nishikawa S, et al. Expression and function of c-kit in hemopoietic progenitor cells. J Exp Med. 1991;174:63–71. doi: 10.1084/jem.174.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ikeda H, Kanakura Y, Tamaki T, et al. Expression and functional role of the proto-oncogene c-kit in acute myeloblastic leukemia cells. Blood. 1991;78:2962–2968. [PubMed] [Google Scholar]
- 55.Reuss-Borst MA, Buhring HJ, Schmidt H, Muller CA. AML: immunophenotypic heterogeneity and prognostic significance of c-kit expression. Leukemia. 1994;8:258–263. [PubMed] [Google Scholar]
- 56.Beghini A, Peterlongo P, Ripamonti CB, et al. C-kit mutations in core binding factor leukemias. Blood. 2000;95:726–727. [PubMed] [Google Scholar]
- 57.Beghini A, Ripamonti CB, Cairoli R, et al. KIT activating mutations: incidence in adult and pediatric acute myeloid leukemia, and identification of an internal tandem duplication. Haematologica. 2004;89:920–925. [PubMed] [Google Scholar]
- 58.Christen F, Hoyer K, Yoshida K, et al. Genomic landscape and clonal evolution of acute myeloid leukemia with t(8;21): an international study on 331 patients. Blood. 2019;133:1140–1151. doi: 10.1182/blood-2018-05-852822. [DOI] [PubMed] [Google Scholar]
- 59.Itzykson R, Duployez N, Fasan A, et al. Clonal interference of signaling mutations worsens prognosis in core-binding factor acute myeloid leukemia. Blood. 2018;132:187–196. doi: 10.1182/blood-2018-03-837781. [DOI] [PubMed] [Google Scholar]
- 60.Pedersen-Bjergaard J, Christiansen DH, Desta F, Andersen MK. Alternative genetic pathways and cooperating genetic abnormalities in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2006;20:1943–1949. doi: 10.1038/sj.leu.2404381. [DOI] [PubMed] [Google Scholar]
- 61.Ok CY, Patel KP, Garcia-Manero G, et al. TP53 mutation characteristics in therapy-related myelodysplastic syndromes and acute myeloid leukemia is similar to de novo diseases. J Hematol Oncol. 2015;8:45. doi: 10.1186/s13045-015-0139-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haferlach C, Dicker F, Herholz H, Schnittger S, Kern W, Haferlach T. Mutations of the TP53 gene in acute myeloid leukemia are strongly associated with a complex aberrant karyotype. Leukemia. 2008;22:1539–1541. doi: 10.1038/leu.2008.143. [DOI] [PubMed] [Google Scholar]
- 63.Rucker FG, Schlenk RF, Bullinger L, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119:2114–2121. doi: 10.1182/blood-2011-08-375758. [DOI] [PubMed] [Google Scholar]
- 64.Bowen D, Groves MJ, Burnett AK, et al. TP53 gene mutation is frequent in patients with acute myeloid leukemia and complex karyotype, and is associated with very poor prognosis. Leukemia. 2009;23:203–206. doi: 10.1038/leu.2008.173. [DOI] [PubMed] [Google Scholar]
- 65.Hou HA, Chou WC, Kuo YY, et al. TP53 mutations in de novo acute myeloid leukemia patients: longitudinal follow-ups show the mutation is stable during disease evolution. Blood Cancer J. 2015;5:e331. doi: 10.1038/bcj.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reuther GW, Der CJ. The Ras branch of small GTPases: Ras family members don’t fall far from the tree. Curr Opin Cell Biol. 2000;12:157–165. doi: 10.1016/s0955-0674(99)00071-x. [DOI] [PubMed] [Google Scholar]
- 67.Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: ‘it ain’t over ‘til it’s over’. Trends Cell Biol. 2000;10:147–154. doi: 10.1016/s0962-8924(00)01740-2. [DOI] [PubMed] [Google Scholar]
- 68.Wittinghofer A. Signal transduction via Ras. Biol Chem. 1998;379:933–937. [PubMed] [Google Scholar]
- 69.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 70.Bos JL, Verlaan-de Vries M, van der Eb AJ, et al. Mutations in N-ras predominate in acute myeloid leukemia. Blood. 1987;69:1237–1241. [PubMed] [Google Scholar]
- 71.Farr CJ, Saiki RK, Erlich HA, McCormick F, Marshall CJ. Analysis of RAS gene mutations in acute myeloid leukemia by polymerase chain reaction and oligonucleotide probes. Proc Natl Acad Sci U S A. 1988;85:1629–1633. doi: 10.1073/pnas.85.5.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Senn HP, Tran-Thang C, Wodnar-Filipowicz A, et al. Mutation analysis of the N-ras proto-oncogene in active and remission phase of human acute leukemias. Int J Cancer. 1988;41:59–64. doi: 10.1002/ijc.2910410112. [DOI] [PubMed] [Google Scholar]
- 73.Toksoz D, Farr CJ, Marshall CJ. Ras genes and acute myeloid leukaemia. Br J Haematol. 1989;71:1–6. doi: 10.1111/j.1365-2141.1989.tb06265.x. [DOI] [PubMed] [Google Scholar]
- 74.Neubauer A, Maharry K, Mrozek K, et al. Patients with acute myeloid leukemia and RAS mutations benefit most from postremission high-dose cytarabine: a cancer and Leukemia Group B study. J Clin Oncol. 2008;26:4603–4609. doi: 10.1200/JCO.2007.14.0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hou HA, Tien HF. Mutations in epigenetic modifiers in acute myeloid leukemia and their clinical utility. Expert Rev Hematol. 2016;9:447–469. doi: 10.1586/17474086.2016.1144469. [DOI] [PubMed] [Google Scholar]
- 76.Chou WC, Lei WC, Ko BS, et al. The prognostic impact and stability of Isocitrate dehydrogenase 2 mutation in adult patients with acute myeloid leukemia. Leukemia. 2011;25:246–253. doi: 10.1038/leu.2010.267. [DOI] [PubMed] [Google Scholar]
- 77.Hou HA, Kuo YY, Liu CY, et al. DNMT3A mutations in acute myeloid leukemia: stability during disease evolution and clinical implications. Blood. 2012;119:559–568. doi: 10.1182/blood-2011-07-369934. [DOI] [PubMed] [Google Scholar]
- 78.Chou WC, Hou HA, Chen CY, et al. Distinct clinical and biologic characteristics in adult acute myeloid leukemia bearing the isocitrate dehydrogenase 1 mutation. Blood. 2010;115:2749–2754. doi: 10.1182/blood-2009-11-253070. [DOI] [PubMed] [Google Scholar]
- 79.Chou WC, Chou SC, Liu CY, et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood. 2011;118:3803–3810. doi: 10.1182/blood-2011-02-339747. [DOI] [PubMed] [Google Scholar]
- 80.Chou WC, Huang HH, Hou HA, et al. Distinct clinical and biological features of de novo acute myeloid leukemia with additional sex comb-like 1 (ASXL1) mutations. Blood. 2010;116:4086–4094. doi: 10.1182/blood-2010-05-283291. [DOI] [PubMed] [Google Scholar]
- 81.Chen TC, Hou HA, Chou WC, et al. Dynamics of ASXL1 mutation and other associated genetic alterations during disease progression in patients with primary myelodysplastic syndrome. Blood Cancer J. 2014;4:e177. doi: 10.1038/bcj.2013.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lin CC, Hou HA, Chou WC, et al. IDH mutations are closely associated with mutations of DNMT3A, ASXL1 and SRSF2 in patients with myelodysplastic syndromes and are stable during disease evolution. Am J Hematol. 2014;89:137–144. doi: 10.1002/ajh.23596. [DOI] [PubMed] [Google Scholar]
- 83.Lin ME, Hou HA, Tsai CH, et al. Dynamics of DNMT3A mutation and prognostic relevance in patients with primary myelodysplastic syndrome. Clin Epigenetics. 2018;10:42. doi: 10.1186/s13148-018-0476-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Whitman SP, Ruppert AS, Marcucci G, et al. Long-term disease-free survivors with cytogenetically normal acute myeloid leukemia and MLL partial tandem duplication: a cancer and Leukemia Group B study. Blood. 2007;109:5164–5167. doi: 10.1182/blood-2007-01-069831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chen Y, Kantarjian H, Pierce S, et al. Prognostic significance of 11q23 aberrations in adult acute myeloid leukemia and the role of allogeneic stem cell transplantation. Leukemia. 2013;27:836–842. doi: 10.1038/leu.2012.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pigneux A, Labopin M, Maertens J, et al. Outcome of allogeneic hematopoietic stem-cell transplantation for adult patients with AML and 11q23/MLL rearrangement (MLL-r AML) Leukemia. 2015;29(12):2375–2381. doi: 10.1038/leu.2015.143. [DOI] [PubMed] [Google Scholar]
- 87.Markova J, Michkova P, Burckova K, et al. Prognostic impact of DNMT3A mutations in patients with intermediate cytogenetic risk profile acute myeloid leukemia. Eur J Haematol. 2012;88:128–135. doi: 10.1111/j.1600-0609.2011.01716.x. [DOI] [PubMed] [Google Scholar]
- 88.Xu Y, Sun Y, Shen H, et al. Allogeneic hematopoietic stem cell transplantation could improve survival of cytogenetically normal adult acute myeloid leukemia patients with DNMT3A mutations. Am J Hematol. 2015;90(11):992–997. doi: 10.1002/ajh.24135. [DOI] [PubMed] [Google Scholar]
- 89.Paschka P, Schlenk RF, Gaidzik VI, et al. ASXL1 mutations in younger adult patients with acute myeloid leukemia: a study by the German-Austrian Acute Myeloid Leukemia Study Group. Haematologica. 2015;100:324–330. doi: 10.3324/haematol.2014.114157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yamaguchi S, Iwanaga E, Tokunaga K, et al. IDH1 and IDH2 mutations confer an adverse effect in patients with acute myeloid leukemia lacking the NPM1 mutation. Eur J Haematol. 2014;92:471–477. doi: 10.1111/ejh.12271. [DOI] [PubMed] [Google Scholar]
- 91.Xu X, Zhao J, Xu Z, et al. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J Biol Chem. 2004;279:33946–33957. doi: 10.1074/jbc.M404298200. [DOI] [PubMed] [Google Scholar]
- 92.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Figueroa ME, Abdel-Wahab O, Lu C, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shlush LI, Zandi S, Mitchell A, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–333. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 97.Lund K, Adams PD, Copland M. EZH2 in normal and malignant hematopoiesis. Leukemia. 2014;28:44–49. doi: 10.1038/leu.2013.288. [DOI] [PubMed] [Google Scholar]
- 98.Morin RD, Johnson NA, Severson TM, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nikoloski G, Langemeijer SM, Kuiper RP, et al. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010;42:665–667. doi: 10.1038/ng.620. [DOI] [PubMed] [Google Scholar]
- 100.Ernst T, Chase AJ, Score J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010;42:722–726. doi: 10.1038/ng.621. [DOI] [PubMed] [Google Scholar]
- 101.Tanaka S, Miyagi S, Sashida G, et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood. 2012;120:1107–1117. doi: 10.1182/blood-2011-11-394932. [DOI] [PubMed] [Google Scholar]
- 102.Basheer F, Giotopoulos G, Meduri E, et al. Contrasting requirements during disease evolution identify EZH2 as a therapeutic target in AML. J Exp Med. 2019;216:966–981. doi: 10.1084/jem.20181276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741–754. doi: 10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478:64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 105.Kihara R, Nagata Y, Kiyoi H, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia. 2014;28:1586–1595. doi: 10.1038/leu.2014.55. [DOI] [PubMed] [Google Scholar]
- 106.Taskesen E, Havermans M, van Lom K, et al. Two splice-factor mutant leukemia subgroups uncovered at the boundaries of MDS and AML using combined gene expression and DNA-methylation profiling. Blood. 2014;123:3327–3335. doi: 10.1182/blood-2013-07-512855. [DOI] [PubMed] [Google Scholar]
- 107.Hou HA, Liu CY, Kuo YY, et al. Splicing factor mutations predict poor prognosis in patients with de novo acute myeloid leukemia. Oncotarget. 2016;7:9084–9101. doi: 10.18632/oncotarget.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lee SC, Abdel-Wahab O. Therapeutic targeting of splicing in cancer. Nat Med. 2016;22:976–986. doi: 10.1038/nm.4165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Agrawal AA, Yu L, Smith PG, Buonamici S. Targeting splicing abnormalities in cancer. Curr Opin Genet Dev. 2018;48:67–74. doi: 10.1016/j.gde.2017.10.010. [DOI] [PubMed] [Google Scholar]
- 110.Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111:2505–2515. doi: 10.1182/blood-2007-07-102798. [DOI] [PubMed] [Google Scholar]
- 111.Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 112.Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377:454–464. doi: 10.1056/NEJMoa1614359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schlenk RF, Weber D, Fiedler W, et al. Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood. 2019;133:840–851. doi: 10.1182/blood-2018-08-869453. [DOI] [PubMed] [Google Scholar]
- 114.Brunner AM, Li S, Fathi AT, et al. Haematopoietic cell transplantation with and without sorafenib maintenance for patients with FLT3-ITD acute myeloid leukaemia in first complete remission. Br J Haematol. 2016;175:496–504. doi: 10.1111/bjh.14260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Burchert A, Bug G, Finke J, Stelljes M, Rolling C, et al. Sorafenib as maintenance therapy post allogeneic stem cell transplantation for FLT3-ITD positive AML: results from the randomized, double-blind, placebo-controlled multicentre sormain trial. Blood. 2018;132(Supplement 1):661. [Google Scholar]
- 116.Ke YY, Singh VK, Coumar MS, et al. Homology modeling of DFG-in FMS-like tyrosine kinase 3 (FLT3) and structure-based virtual screening for inhibitor identification. Sci Rep. 2015;5:11702. doi: 10.1038/srep11702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3-mutated AML. N Engl J Med. 2019;381:1728–1740. doi: 10.1056/NEJMoa1902688. [DOI] [PubMed] [Google Scholar]
- 118.Wu M, Li C, Zhu X. FLT3 inhibitors in acute myeloid leukemia. J Hematol Oncol. 2018;11:133. doi: 10.1186/s13045-018-0675-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marcucci G, Geyer S, Laumann K, et al. Combination of dasatinib with chemotherapy in previously untreated core binding factor acute myeloid leukemia: CALGB 10801. Blood Adv. 2020;4:696–705. doi: 10.1182/bloodadvances.2019000492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bykov VJ, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med. 2002;8:282–288. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 121.Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-specific p53 mutant reactivation. Cancer Cell. 2012;21:614–625. doi: 10.1016/j.ccr.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18:89–102. doi: 10.1038/nrc.2017.109. [DOI] [PubMed] [Google Scholar]
- 123.Zhang Q, Bykov VJN, Wiman KG, Zawacka-Pankau J. APR-246 reactivates mutant p53 by targeting cysteines 124 and 277. Cell Death Dis. 2018;9:439. doi: 10.1038/s41419-018-0463-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sallman DA, DeZern AE, Garcia-Manero G, et al. Phase 2 results of APR-246 and Azacitidine (AZA) in Patients with TP53 mutant Myelodysplastic Syndromes (MDS) and Oligoblastic Acute Myeloid Leukemia (AML) Blood. 2019;134:676. [Google Scholar]
- 125.Konstantinopoulos PA, Karamouzis MV, Papavassiliou AG. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat Rev Drug Discov. 2007;6:541–555. doi: 10.1038/nrd2221. [DOI] [PubMed] [Google Scholar]
- 126.Karp JE, Lancet JE, Kaufmann SH, et al. Clinical and biologic activity of the farnesyltransferase inhibitor R115777 in adults with refractory and relapsed acute leukemias: a phase 1 clinical-laboratory correlative trial. Blood. 2001;97:3361–3369. doi: 10.1182/blood.v97.11.3361. [DOI] [PubMed] [Google Scholar]
- 127.Zimmerman TM, Harlin H, Odenike OM, et al. Dose-ranging pharmacodynamic study of tipifarnib (R115777) in patients with relapsed and refractory hematologic malignancies. J Clin Oncol. 2004;22:4816–4822. doi: 10.1200/JCO.2004.03.200. [DOI] [PubMed] [Google Scholar]
- 128.Lancet JE, Gojo I, Gotlib J, et al. A phase 2 study of the farnesyltransferase inhibitor tipifarnib in poor-risk and elderly patients with previously untreated acute myelogenous leukemia. Blood. 2007;109:1387–1394. doi: 10.1182/blood-2006-04-014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Maiti A, Naqvi K, Kadia TM, et al. Phase II trial of MEK inhibitor Binimetinib (MEK162) in RAS-mutant acute myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2019;19:142–148. doi: 10.1016/j.clml.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 130.Ragon BK, Odenike O, Baer MR, et al. Oral MEK 1/2 inhibitor Trametinib in combination with AKT inhibitor GSK2141795 in patients with acute myeloid leukemia with RAS mutations: a phase II study. Clin Lymphoma Myeloma Leuk. 2019;19:431–440. doi: 10.1016/j.clml.2019.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yen K, Travins J, Wang F, et al. AG-221, a first-in-class therapy targeting acute myeloid leukemia harboring oncogenic IDH2 mutations. Cancer Discov. 2017;7:478–493. doi: 10.1158/2159-8290.CD-16-1034. [DOI] [PubMed] [Google Scholar]
- 132.Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130:722–731. doi: 10.1182/blood-2017-04-779405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Stein EM, DiNardo CD, Fathi AT, et al. Molecular remission and response patterns in patients with mutant-IDH2 acute myeloid leukemia treated with enasidenib. Blood. 2019;133:676–687. doi: 10.1182/blood-2018-08-869008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Popovici-Muller J, Lemieux RM, Artin E, et al. Discovery of AG-120 (Ivosidenib): a first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med Chem Lett. 2018;9:300–305. doi: 10.1021/acsmedchemlett.7b00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with Ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378:2386–2398. doi: 10.1056/NEJMoa1716984. [DOI] [PubMed] [Google Scholar]
- 136.Roboz GJ, Dinardo CD, Stein EM, et al. Ivosidenib (IVO; AG-120) in IDH1-mutant newly-diagnosed acute myeloid leukemia (ND AML): updated results from a phase 1 study. J Clin Oncol. 2019;37:7028. [Google Scholar]
- 137.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J Clin Oncol. 2010;28:562–569. doi: 10.1200/JCO.2009.23.8329. [DOI] [PubMed] [Google Scholar]
- 138.Silverman LR, McKenzie DR, Peterson BL, et al. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J Clin Oncol. 2006;24:3895–3903. doi: 10.1200/JCO.2005.05.4346. [DOI] [PubMed] [Google Scholar]
- 139.Issa JJ, Roboz G, Rizzieri D, et al. Safety and tolerability of guadecitabine (SGI-110) in patients with myelodysplastic syndrome and acute myeloid leukaemia: a multicentre, randomised, dose-escalation phase 1 study. Lancet Oncol. 2015;16:1099–1110. doi: 10.1016/S1470-2045(15)00038-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Roboz GJ, Döhner H, Gobbi M, et al. Results from a global randomized phase 3 study of guadecitabine (G) Vs treatment choice (TC) in 815 patients with treatment naïve (TN) AML unfit for intensive chemotherapy (IC) ASTRAL-1 study: analysis by number of cycles. Blood. 2019;134:2591. [Google Scholar]
- 141.Wei AH, Döhner H, Pocock C, et al. The QUAZAR AML-001 maintenance trial: results of a phase III international, randomized, double-blind, placebo-controlled study of CC-486 (Oral Formulation of Azacitidine) in patients with acute myeloid leukemia (AML) in first remission. Blood. 2019;134:LBA-3-LBA. [Google Scholar]
- 142.Okada Y, Feng Q, Lin Y, et al. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 143.Bernt KM, Zhu N, Sinha AU, et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell. 2011;20:66–78. doi: 10.1016/j.ccr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Daigle SR, Olhava EJ, Therkelsen CA, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Stein EM, Garcia-Manero G, Rizzieri DA, et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood. 2018;131:2661–2669. doi: 10.1182/blood-2017-12-818948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13:337–356. doi: 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- 147.Zuber J, Shi J, Wang E, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wingelhofer B, Somervaille TCP. Emerging epigenetic therapeutic targets in acute myeloid leukemia. Front Oncol. 2019;9:850. doi: 10.3389/fonc.2019.00850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Berthon C, Raffoux E, Thomas X, et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol. 2016;3:e186–e195. doi: 10.1016/S2352-3026(15)00247-1. [DOI] [PubMed] [Google Scholar]
- 150.Harris WJ, Huang X, Lynch JT, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21:473–487. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 151.Feng Z, Yao Y, Zhou C, et al. Pharmacological inhibition of LSD1 for the treatment of MLL-rearranged leukemia. J Hematol Oncol. 2016;9:24. doi: 10.1186/s13045-016-0252-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Maes T, Mascaro C, Tirapu I, et al. ORY-1001, a potent and selective covalent KDM1A inhibitor, for the treatment of acute leukemia. Cancer Cell. 2018;33:495–511. doi: 10.1016/j.ccell.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 153.Bots M, Verbrugge I, Martin BP, et al. Differentiation therapy for the treatment of t(8;21) acute myeloid leukemia using histone deacetylase inhibitors. Blood. 2014;123:1341–1352. doi: 10.1182/blood-2013-03-488114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mims AS, Mishra A, Orwick S, et al. A novel regimen for relapsed/refractory adult acute myeloid leukemia using a KMT2A partial tandem duplication targeted therapy: results of phase 1 study NCI 8485. Haematologica. 2018;103:982–987. doi: 10.3324/haematol.2017.186890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 156.Certo M, Del Gaizo Moore V, Nishino M, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 157.Chao DT, Korsmeyer SJ. BCL-2 family: regulators of cell death. Annu Rev Immunol. 1998;16:395–419. doi: 10.1146/annurev.immunol.16.1.395. [DOI] [PubMed] [Google Scholar]
- 158.Campos L, Rouault JP, Sabido O, et al. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood. 1993;81:3091–3096. [PubMed] [Google Scholar]
- 159.Lagadinou ED, Sach A, Callahan K, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell. 2013;12:329–341. doi: 10.1016/j.stem.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Konopleva M, Zhao S, Hu W, et al. The anti-apoptotic genes Bcl-X(L) and Bcl-2 are over-expressed and contribute to chemoresistance of non-proliferating leukaemic CD34+ cells. Br J Haematol. 2002;118:521–534. doi: 10.1046/j.1365-2141.2002.03637.x. [DOI] [PubMed] [Google Scholar]
- 161.Konopleva M, Pollyea DA, Potluri J, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6:1106–1117. doi: 10.1158/2159-8290.CD-16-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Wang YW, Tsai CH, Lin CC, et al. Cytogenetics and mutations could predict outcome in relapsed and refractory acute myeloid leukemia patients receiving BCL-2 inhibitor venetoclax. Ann Hematol. 2020;99:501–511. doi: 10.1007/s00277-020-03911-z. [DOI] [PubMed] [Google Scholar]
- 163.Wei AH, Strickland SA, Jr, Hou JZ, et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II study. J Clin Oncol. 2019;37:1277–1284. doi: 10.1200/JCO.18.01600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.DiNardo CD, Pratz K, Pullarkat V, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood. 2019;133:7–17. doi: 10.1182/blood-2018-08-868752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lancet JE, Cortes JE, Hogge DE, et al. Phase 2 trial of CPX-351, a fixed 5:1 molar ratio of cytarabine/daunorubicin, vs cytarabine/daunorubicin in older adults with untreated AML. Blood. 2014;123:3239–3246. doi: 10.1182/blood-2013-12-540971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Castaigne S, Pautas C, Terre C, et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): a randomised, open-label, phase 3 study. Lancet. 2012;379:1508–1516. doi: 10.1016/S0140-6736(12)60485-1. [DOI] [PubMed] [Google Scholar]
- 167.Cortes JE, Heidel FH, Hellmann A, et al. Randomized comparison of low dose cytarabine with or without glasdegib in patients with newly diagnosed acute myeloid leukemia or high-risk myelodysplastic syndrome. Leukemia. 2019;33:379–389. doi: 10.1038/s41375-018-0312-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Non applicable.