

SUMMARY
The medial prefrontal cortex (mPFC) modulates a range of behaviors, including responses to noxious stimuli. While various pain modalities alter mPFC function, our understanding of changes to specific cell types underlying pain-induced mPFC dysfunction remains incomplete. Proper activity of cortical GABAergic interneurons is essential for normal circuit function. We find that nerve injury increases excitability of layer 5 parvalbumin-expressing neurons in the prelimbic (PL) region of the mPFC from male, but not female, mice. Conversely, nerve injury dampens excitability in somatostatin-expressing neurons in layer 2/3 of the PL region; however, effects are differential between males and females. Nerve injury slightly increases the frequency of spontaneous excitatory post-synaptic currents (sEPSCs) in layer 5 parvalbumin-expressing neurons in males but reduces frequency of sEPSCs in layer 2/3 somatostatin-expressing neurons in females. Our findings provide key insight into how nerve injury drives maladaptive and sex-specific alterations to GABAergic circuits in cortical regions implicated in chronic pain.
Graphical Abstract
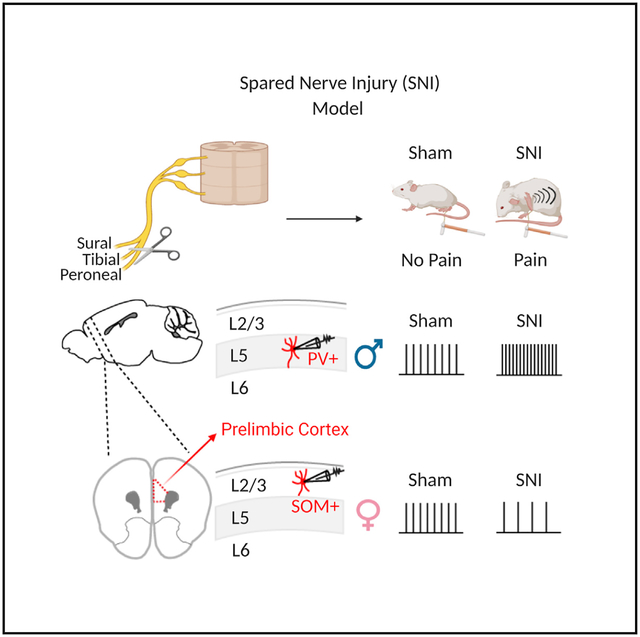
In Brief
Jones and Sheets show that a nerve injury model of chronic pain produces distinct changes to separate, but major, subclasses of GABAergic neurons in the prelimbic cortex. These changes are different between male and female mice and provide insight into how chronic pain alters brain circuits in a sex-specific manner.
INTRODUCTION
Inhibitory interneurons are essential for normal activity of cortical microcircuits. Given their sparse expression in the cortex (~20%–30%), minor alterations to their function are believed to drive significant changes to circuit activity. Numerous pain studies report structural and functional alterations to the medial prefrontal cortex (mPFC), a region implicated in affective and cognitive disturbances associated with chronic pain (Cardoso-Cruz et al., 2013; Cheriyan and Sheets, 2018; Huang et al., 2019; Ji et al., 2010; Kelly et al., 2016; Lee et al., 2015; Metz et al., 2009; Millecamps et al., 2007; Mitrić et al., 2019; Shiers et al., 2018; Zhang et al., 2015). Aspects of cellular and circuit mechanisms underlying these disturbances have emerged. For instance, cognitive deficits elicited by arthritic pain are associated with increased polysynaptic inhibition of layer 5 (L5) pyramidal neurons in the prelimbic (PL) region of the mPFC (Ji et al., 2010; Sun and Neugebauer, 2011). A subsequent study corroborated this finding by showing that nerve injury increases excitatory input to parvalbumin-positive inhibitory neurons (PVINs) in the PL cortex, consequentially decreasing the excitability of L5 pyramidal neurons (Zhang et al., 2015). This implies that chronic pain evokes an imbalance in inhibition/excitation in the PL cortex. Indeed, our lab has shown that the chronic constriction injury model of neuropathic pain enhances the inhibitory-excitatory balance of local inputs onto PL pyramidal neurons that project to the periaqueductal gray (PAG), a key structure in the descending analgesic system (Cheriyan and Sheets, 2018). A recent study demonstrated that glutamatergic basolateral amygdala (BLA) inputs onto PL PVINs are enhanced after chronic nerve injury (Huang et al., 2019). Collectively, these results provide compelling evidence for a pain circuit in which nerve injury increases the excitation of PL PVINs, which subsequently attenuate normal output of neighboring pyramidal neurons targeting subcortical structures involved in pain modulation (i.e., PAG). However, important questions remain regarding the impact of nerve injury between subclasses of inhibitory neurons across different lamina in distinct regions of the mPFC.
In the mPFC, PVINs are active throughout working memory (WM) trials on a delayed non-match-to-place task, except for the reward period when they are strongly suppressed (Kim et al., 2016). Disruption of GABAergic signaling in the mPFC drives deficits in WM and cognitive flexibility (Abbas et al., 2018; Bañuelos et al., 2014; Enomoto et al., 2011; Murray et al., 2015; Yu et al., 2019), which are deficits commonly comorbid with chronic pain (Dick and Rashiq, 2007; Povedano et al., 2007; Ryan et al., 1993; Schiltenwolf et al., 2014). Conversely, another class of cortical inhibitory neurons, somatostatin-expressing (SOM) interneurons, exhibit phasic activity during the delay period of the same task with no change from baseline during the reward period (Kim et al., 2016). Therefore, changes to the excitability of PVINs or SOM neurons in the mPFC are poised to be a critical factor in cognitive deficits associated with chronic pain. To date, electrophysiological analyses of mPFC PVINs or SOM neurons in a model of chronic pain have not been performed.
Traditionally, pain research has focused primarily on males, but it is clear that sex differences in pain exist (Mogil, 2012; Mogil and Bailey, 2010). It remains unknown whether pain-induced alterations to mPFC circuits are sexually dimorphic. Both women and female rodents demonstrate greater sensitivity to painful stimuli than males (Fillingim et al., 2009; Vacca et al., 2014). Additionally, chronic pain drives sexually dimorphic cognitive dysfunction, with male mice showing more significant deficits in set-shifting tasks post-injury than female mice (Shiers et al., 2018). However, functional alterations to defined mPFC GABAergic neurons in male and female mice in neuropathic pain models remain unclear. Here, we used transgenic breeding strategies to identify either PVINs or SOM neurons in both male and female mice. Our goal was to measure changes to the excitability of PVINs and SOM neurons in PL and infralimbic (IL) regions of the mPFC in the spared nerve injury (SNI) model of neuropathic pain.
RESULTS
SNI Sensitizes PL PVINs Only in L5 of Male Mice
We observed significant mechanical allodynia in both male and female PVIN-tdTomato transgenic mice at post-operative day (POD) 7 following SNI compared to that in sham mice (Figure 1A). Mechanical allodynia between male and female mice did not significantly differ (Figure 1A). Following behavioral assessments, we measured the firing properties of fluorescently labeled PVINs in the PL cortex of sham and SNI animals using whole-cell electrophysiology in acute brain slices (Figures 1B and 1C). Recordings revealed that sham female PVINs have significantly higher firing frequency compared to that of sham male PVINs, which manifested as greater action potential (AP) firing in response to depolarizing step currents (Figures 1D and 1E; Table S1). Because PVINs are expressed diffusely throughout the lamina of the mPFC (Figure 1B), we examined changes to PVINs as a function of the precise position of the soma along the radial axis of the PL region. Analysis of all sham PVINs recorded revealed that female PVINs had a significantly greater input resistance than male PVINs (Figure 1F; Table S1). However, this disparity was driven by the significant difference in input resistance between L5 PVINs (Figure 1F; Table S1). Our recording data revealed that SNI enhanced excitability of PVINs in the PL cortex of male mice (Figures 1G and 1H; Table S2). Laminar analysis showed that SNI significantly increased input resistance only in L5 PVINs (Figure 1I; Table S3). We did not detect any significant differences in the excitability of PVINs between sham and SNI female mice (Figures 1J–1L; Table S2). Together, these data indicate that 7–8 days following SNI, PVINs in a specific lamina of PL cortex are sensitized in male, but not female, mice.
Figure 1. SNI Increases PV Excitability in PL Cortex of Male, but Not Female, Mice.
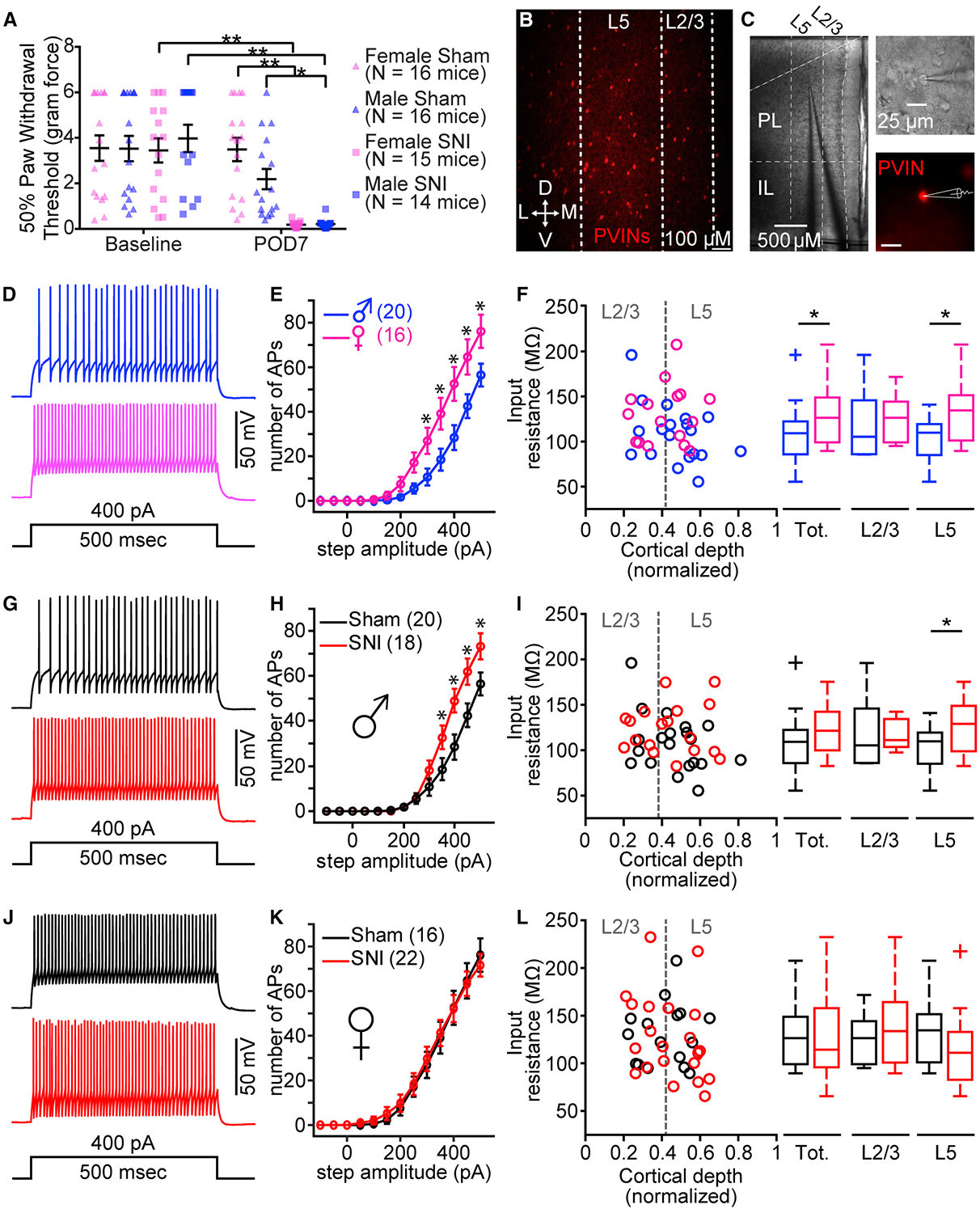
(A) Mechanical allodynia at baseline and post-operative day 7 (POD7) measured in male and female sham and SNI mice. Two-way repeated-measures ANOVA: interaction, F(1, 59) = 26.87, p < 0.0001; injury, F(1, 59) = 10.11, p = 0.0024; time, F(1, 59) = 60.31, p < 0.0001. Sidak post hoc tests revealed no significant differences between males and females at baseline and POD7. *p < 0.05; **p < 0.0001.
(B) Fluorescent confocal 10× image displaying distribution of PV-tdTomato neurons (PVINs) in the PL cortex (D, dorsal; V, ventral; M, medial; L, lateral).
(C) Representative images of a PVIN recording in PL cortex at 4× (left) and 60× (right).
(D and E) Example traces of action potential (AP) firing (D) and mean (±SEM) number of APs elicited in response to increasing step current from male and female sham mice (E) (two-way ANOVA: interaction, F(12, 408) = 5.00, p < 0.0001; sex, F(1, 34) = 1.34, p = 0.01; current, F(12, 408) = 108.9, p < 0.0001; *p < 0.05, Sidak post hoc test).
(F) Raw data and boxplot representation for input resistance versus normalized soma location for total (left: Mann-Whitney U = 90, n1 = 20, n2 = 16, p = 0.03), L2/3 (middle: t(12) = 0.27, p = 0.79), and L5 (right: t(20) = 2.35, p = 0.03) PVINs from sham males (total, n = 20 neurons; 9 mice) and females (total, n = 16 neurons; 6 mice). *p < 0.05.
(G and H) Example traces of AP firing (G) and mean (±SEM) number of APs elicited from male sham and SNI mice (H) (two-way ANOVA: interaction, F(12, 432) = 5.03, p < 0.0001; injury, F(1, 36) = 4.49, p = 0.04; current, F(12, 432) = 143.5, p < 0.0001; *p < 0.05, Sidak post hoc test).
(I) Input resistance versus normalized soma location for total (left: Mann-Whitney U = 123, n1 = 18, n2 = 20, p = 0.10), L2/3 (middle: t(11) = 0.15, p = 0.89), and L5 (right: t(23) = 2.10, p = 0.05) PVINs from male sham (total, n = 20 neurons; 9 mice) and SNI (total, n = 18 neurons; 6 mice). *p = 0.05.
(J and K) Example traces of AP firing (J) and mean (±SEM) number of APs elicited from female sham and SNI mice (K) (two-way ANOVA: interaction, F(12, 444) = 0.13, p = 0.99; injury, F(1, 37) = 0.04, p = 0.87; current, F(12, 444) = 128.1, p < 0.0001).
(L) Input resistance versus normalized soma location for total (t(37) = 0.31, p = 0.76), L2/3 (t(16) = 0.71, p = 0.49), and L5 (Mann-Whitney U = 39, n1 = 13, n2 = 8, p = 0.37) PVINs from female sham (total, n = 16 neurons; 6 mice) and SNI (total, n = 22 neurons; 7 mice). Plus symbol indicates outlier in MATLAB.
Data represent mean ± SEM.
SNI Increases Frequency of Synaptic Input to L5 PL PVINs Only in Male Mice
We next recorded spontaneous excitatory post-synaptic currents (sEPSCs) from L5 PVINs in the PL cortex from male and female mice. We found that frequency, but not amplitude, of sEPSCs was significantly greater in female PL PVINs (Figures 2A–2D; Table S4). To assess potential changes to synaptic activity driven by nerve injury, we next recorded sEPSCs from PVINs in the PL cortex from male sham and SNI mice (Figure 2E; Table S4). We found that SNI increased sEPSC frequency recorded in L5 PVINs of male mice but did not change sEPSC amplitude (Figure 2F; Table S4). While the increase in sEPSC frequency was not significant (t(22) = 1.58, p = 0.06, Student’s unpaired t test), it is consistent with findings of a previous study in male mice showing significantly increased sEPSC frequency in PL PVINs recorded 10 days after SNI (Zhang et al., 2015). Plots of mean cumulative distribution curves revealed an enhanced probability for shorter inter-EPSC intervals in L5 PVINs from male SNI mice (Figure 2G; Table S4). Mean cumulative distribution curves for sEPSC amplitude showed no noticeable differences (Figure 2H; Table S4). We observed no significant differences in the frequency or amplitude of sEPSCs recorded in L5 PVINs from female sham and SNI mice (Figures 2I–2L; Table S4).
Figure 2. SNI Reduces sEPSCs in L5 PL PVINs from Male Mice Only.
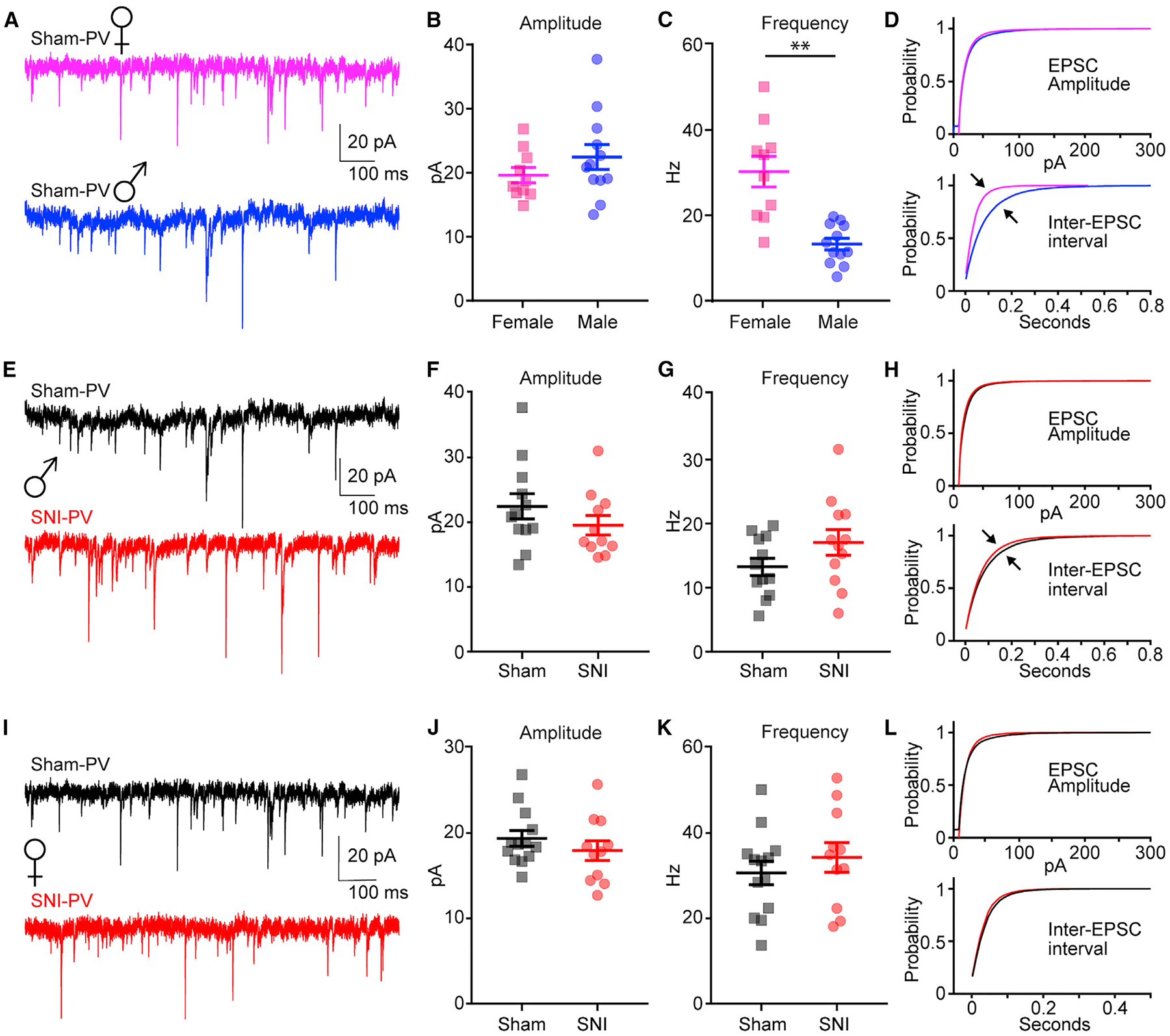
(A–D) Sample sEPSC traces (A); sEPSC amplitude (B), t(20) = 1.19, p = 0.23; sESPC frequency (C), t(20) = 4.73, **p = 0.0001; and mean cumulative distribution curves (D) (top, amplitude; bottom, inter-EPSC interval) for male (n = 12 neurons; 5 mice) and female (n = 13 neurons; 5 mice) sham mice.
(E–H) Sample sEPSC traces (E); sEPSC amplitude (F), t(21) = 1.17, p = 0.26; sESPC frequency (G), t(22) = 1.58, p = 0.06); and mean cumulative distribution curves
(H) for male sham (n = 12 neurons; 5 mice) and SNI (n = 12 neurons; 5 mice).
(I–L) Sample sEPSC traces (I); sEPSC amplitude (J), t(22) = 0.98, p = 0.34; sESPC frequency (K), t(22) = 0.83, p = 0.41; and mean cumulative distribution curves (L) for female sham (n = 13 neurons; 5 mice) and SNI (n = 11 neurons; 4 mice). Data represent mean ± SEM.
SNI Reduces Input Resistance of PL-SOM Neurons Only in Layer 2/3 of Female Mice
Both male and female SOM-tdTomato transgenic mice at POD7 following SNI displayed significant mechanical allodynia compared to sham mice (Figure 3A; Table S3). As in PVIN-tdTomato mice, mechanical allodynia measures between male and female mice did not significantly differ (Figure 3A; Table S3). In contrast to PVINs, recordings from fluorescently labeled SOM neurons in the PL cortex (Figures 3B and 3C; Table S3) revealed no significant differences in AP firing or input resistance between female and male mice (Figures 3D–3F; Table S3). Recordings showed no significant differences in the excitability of SOM neurons from SNI male mice (Figures 3G and 3H; Table S3). However, we found that input resistance was not statistically different between male sham and SNI SOM neurons (Figure 3I; Table S3). We found that AP firing in SOM neurons from female sham and SNI mice did not differ, but we observed a decreased input resistance in layer 2/3 (L2/3) SOM neurons from SNI mice (Figures 3J–3L; Table S3).
Figure 3. SNI Decreases Excitability of SOM Neurons in PL Cortex.
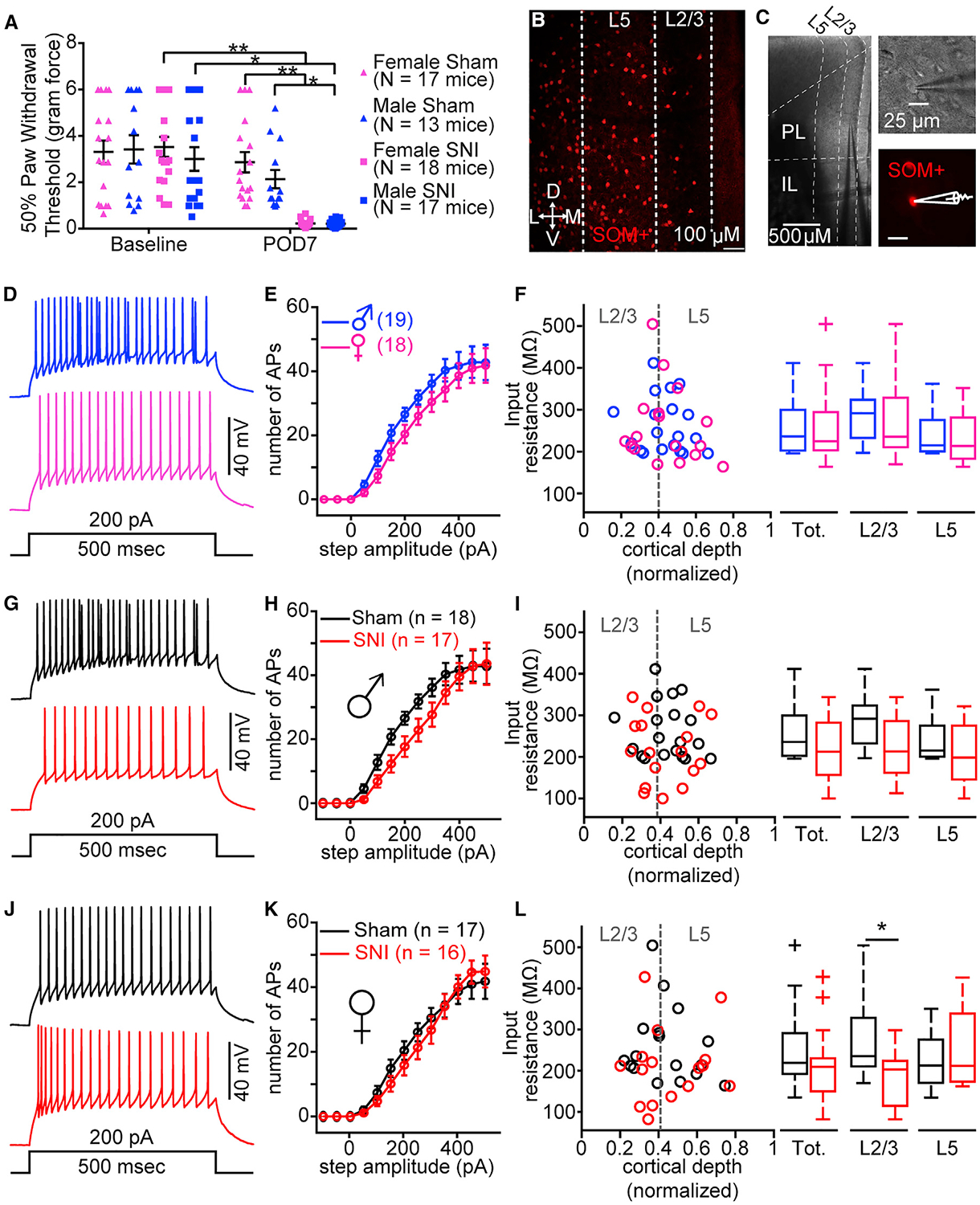
(A) Mechanical allodynia at baseline and post-operative day 7 (POD7) measured in male and female sham and SNI mice. Two-way repeated-measures ANOVA: interaction, F(1, 63) = 20.39, p < 0.0001; treatment, F(1, 63) = 14.25, p = 0.0004; time, F(1, 63) = 60.29, p < 0.0001. Sidak post hoc tests revealed no significant differences between males and females at baseline and POD7. *p < 0.05; **p < 0.0001.
(B) Example fluorescent confocal 10× image of a brain slice displaying distribution of SOM-tdTomato (SOM) neurons in the PL cortex.
(C) Representative images of a SOM neuron recording in PL cortex at 4× (left) and 60× (right).
(D and E) Example traces of action potential (AP) firing (D) and mean (±SEM) number of APs elicited in response to increasing step current from sham male and female mice (E) (two-way ANOVA: interaction, F(12, 420) = 0.52, p = 0.90; sex, F(1, 35) = 1.76, p = 0.19; current, F(12, 420) = 92.62, p < 0.0001).
(F) Raw data and boxplots representation for input resistance versus normalized soma location for total (Mann-Whitney U = 144, n1 = 19, n2 = 17, p = 0.59), L2/3 (t(15) = 0.11, p = 0.91), and L5 t(15) = 0.62, p = 0.54) SOM neurons from sham females (total, n = 17 neurons; 8 mice) and males (total, n = 18 neurons; 7 mice).
(G and H) Example traces of AP firing (G) and mean (±SEM) number of APs elicited from male sham and SNI mice (H) (two-way ANOVA: interaction, F(12, 408) = 1.03, p = 0.42; injury, F(1, 34) = 2.46, p = 0.13; current injection, F(12, 408) = 78.76, p < 0.0001).
(I) Input resistance versus normalized soma location for total (t(34) = 1.83, p = 0.08), L2/3 (t(15) = 1.64, p = 0.12), and L5 (Mann-Whitney U = 31, n1 = 8, n2 = 9, p = 0.67) SOM neurons from male sham (total, n = 20 neurons; 7 mice) and SNI (total, n = 18 neurons; 8 mice).
(J and K) Example traces of AP firing (J) and mean (±SEM) number of APs elicited from female sham and SNI mice (K) (two-way ANOVA: interaction, F(12, 384) = 0.67, p = 0.78; injury, F(1, 32) = 0.16, p = 0.69; current, F(12, 384) = 95.05, p < 0.0001).
(L) Input resistance versus normalized soma location for total (Mann-Whitney U = 90, n1 = 16, n2 = 17, p = 0.10), L2/3 (t(16) = 2.39, p = 0.03), and L5 (Mann-Whitney U = 27, n1 = 7, n2 = 8, p = 0.96) SOM neurons from female sham (total, n = 17 neurons; 8 mice) and SNI (total, n = 16 neurons; 7 mice). Plus symbol indicates outlier in MATLAB. *p < 0.05.
Data represent mean ± SEM.
SNI Significantly Reduces Frequency of sEPSCs in L2/3 PL-SOM Neurons of Female Mice
To assess SNI-induced changes in synaptic activity, we recorded sEPSCs from layer 2/3 of the PL cortex in SOM male and female mice. We found that neither the frequency nor the amplitude of sEPSCs from SOM neurons differed between male and female mice (Figures 4A–4D; Table S4). We also found that SNI did not alter amplitude or frequency of sEPSCs in male mice (Figures 4E–4G; Table S4). Mean cumulative distribution curves for amplitude and inter-EPSC intervals were also similar (Figure 4H; Table S4). Contrary to those in males, L2/3 SOM neurons from female SNI mice demonstrated significantly reduced sEPSC frequency and noticeable differences in the mean cumulative distribution curves for inter-EPSC intervals compared to sham mice (Figures 4I–4L; Table S4). However, no differences in amplitude of sEPSCs from L2/3 SOM neurons were detected between female SNI and sham mice (Figures 4I–4L; Table S4).
Figure 4. SNI Significantly Reduces Frequency of sEPSCs in L2/3 SOM Neurons of Female Mice.
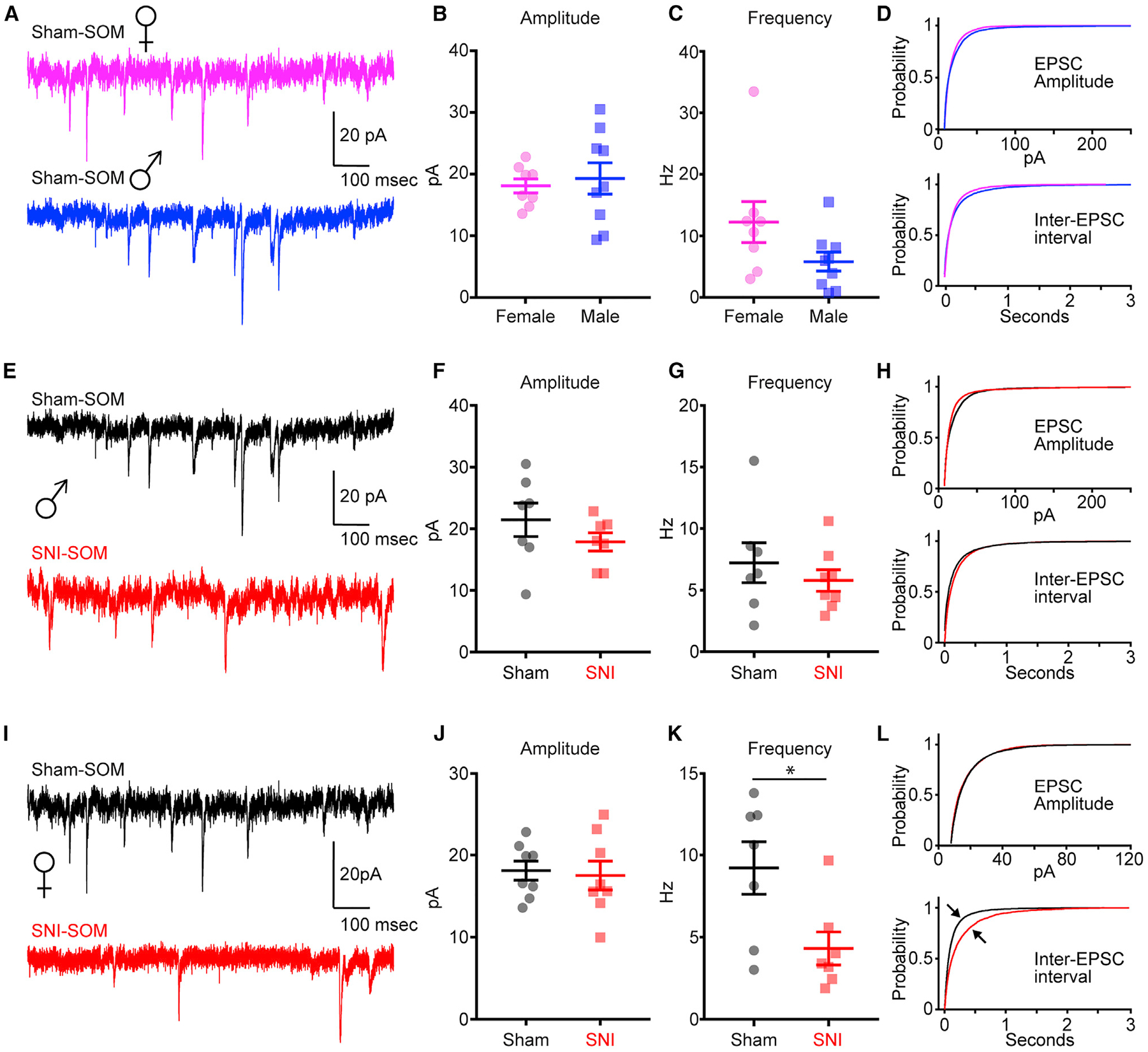
(A–D) Sample sEPSC traces (A), sEPSC amplitude (B) (t(13) = 1.20, p = 0.25), (C) sESPC frequency (t(12) = 0.87, p = 0.40), and mean cumulative distribution curves
(D) (top, amplitude; bottom, inter-EPSC interval) for L2/3 SOM neurons from sham males (n = 7 neurons; 3 mice) and females (n = 7 neurons; 4 mice).
(E–H) Sample sEPSC traces (E), sEPSC amplitude (F) (t(12) = 1.16, p = 0.27), sEPSC frequency (G) (t(13) = 0.81, p = 0.43), and mean cumulative distribution curves
(H) for L2/3 SOM neurons from male sham (n = 7 neurons; 3 mice) and SNI (n = 8 neurons; 3 mice).
(I–L) Sample sEPSC traces (I), sEPSC amplitude (J) (t(14) = 0.28, p = 0.78), sEPSC frequency (K) (t(12) = 2.59, p = 0.02), and mean cumulative distribution curves (L) for L2/3 SOM neurons from female sham (n = 7 neurons; 4 mice) and SNI (7 neurons; 5 mice). *p < 0.05.
Data represent mean ± SEM.
SNI Does Not Alter Excitability of Either PVINs or SOM Neurons in IL Cortex
Our previous work shows that sciatic nerve injury decreases the excitability of L5 pyramidal neurons in the PL, but not the IL, region of the mPFC (Cheriyan and Sheets, 2018). Therefore, we tested whether SNI altered the firing frequencies of PVINs and SOM neurons in the IL cortex again using whole-cell electrophysiology in acute brain slices. We found that L2/3 IL-SOM neurons fired fewer APs in female sham mice compared to sham males (U = 13.5, n1 = 7, n2 = 10, p = 0.03) and that SNI reduced the AP height of L2/3 IL-SOM neurons in female SNI mice compared to sham (t(13) = 2.51, p = 0.03; data not shown). Therefore, SNI does not significantly alter the intrinsic profile of either PVINs or SOM+ neurons in the IL cortex at POD7–POD8.
DISCUSSION
Our data indicate that SNI leading to neuropathic pain behavior evokes changes to mPFC inhibitory neurons not only in a sub-type-specific manner (PVINs versus SOM neurons) but also in a regional, laminar-specific, and sex-specific manner. Disparities in SNI-induced changes between PVINs and SOM neurons have implications for distinct changes to mPFC inhibition based on the local targeting of these two major subtypes of inhibitory neurons. In other cortical regions, PVINs target soma and basal dendrites of intralaminar cortical pyramidal neurons, thereby altering AP output (Markram et al., 2004; Marlin and Carter, 2014). Therefore, our data indicate that SNI-evoked hyperexcitability of L5 PVINs in male mice dampens L5 pyramidal neuron output, consistent with previous findings (Zhang et al., 2015). Indeed, we have found that chronic constriction of the sciatic nerve enhances excitation of L5, but not L2/3, PVINs stimulated with uncaged glutamate (Cheriyan and Sheets, 2018). Explanations for why the sensitization of PVINs is specific to L5 following nerve injury have yet to emerge. Previous findings indicate that nerve injury elicits hyperactivity of ascending BLA inputs, which drives increased feed-forward inhibition of L5 pyramidal neurons in the PL cortex (Ji et al., 2010; Zhang et al., 2015). Additionally, one study showed that targeting of BLA is strongest to PVINs compared to SOM and pyramidal neurons in the mPFC (McGarry and Carter, 2016). This suggests that SNI sensitization of BLA-mPFC inputs has the greatest impact on PVIN activity, but preferential targeting of BLA to L5 versus L2/3 PVINs remains unknown.
Local inputs from SOM neurons, also known as Martinotti cells (Kawaguchi and Kubota, 1996, 1997; Wang et al., 2004), target both basal and apical dendrites of cortical pyramidal neurons (Fino and Yuste, 2011; Gupta et al., 2000; Kawaguchi and Kubota, 1997; Marlin and Carter, 2014). Inhibition targeted to dendrites regulates the probability and wider timing window for dendritic events (Kim et al., 1995), including Ca2+ influx, Ca2+ APs, and NMDA spikes (Doron et al., 2017; Hayama et al., 2013; Larkum et al., 2001; Marlin and Carter, 2014). Inhibition of L2/3 SOM neurons increases both dendritic excitability and burst firing of excitatory pyramidal neurons in somatosensory cortex (Gentet et al., 2012). Here, we show that SNI reduced synaptic excitability and input resistance of L2/3 SOM neurons in female mice. This suggests that neuropathic pain may enhance the excitability of PL pyramidal neurons in female mice via reduced L2/3 SOM activity. Why attenuated sEPSC frequency and input resistance are observed only in the L2/3 neurons of female, but not male, SNI mice remains unresolved. Why SNI drives the hypoexcitability of SOM neurons and hyperexcitability of PVINs in the PL cortex is also unclear. PVINs target SOM neurons in the somatosensory cortex (Walker et al., 2016), which suggests that the hypoexcitability of the PL-SOM neurons from SNI mice may be driven, at least in part, by enhanced local inhibitory inputs from hyperexcitable PVINs.
Previous work shows that the local input strength of PVINs and SOM neurons onto cortical pyramidal neurons differs based on projection target (Lee et al., 2014; McGarry and Carter, 2016; Rock and Apicella, 2015). In the mPFC, PVINs preferentially target subcortically projecting pyramidal neurons defined by thick-tufted apical dendrites and robust expression of h-current (Lee et al., 2014). We have shown that PL neurons with subcortical projections to the PAG are hypoexcitable following chronic constriction injury (CCI) of the sciatic nerve in male mice (Cheriyan and Sheets, 2018). Based on our present data, this suggests that SNI sensitizes PVINs in male mice, which are preferentially targeting PAG-projecting PL neurons and dampening their excitability. In fact, a recent study showed that nerve injury augments feed-forward inhibition of mPFC output to the PAG (Huang et al., 2019). Because SNI had no effect on PL PVIN excitability in female mice, it is likely that SNI also does not dampen PL output to the PAG in female mice. Recent work supports this notion by showing that nerve injury does not alter AP firing in L5 pyramidal neurons in the mPFC of female mice (Gadotti et al., 2019).
The effects of SNI on PVINs in mPFC may underlie specific aspects of cognitive dysfunction associated with chronic pain. The activity of PVINs drives cortical gamma frequency in the cortex (Lewis et al., 2012; Sohal et al., 2009), which is associated with WM in humans (Howard et al., 2003). Reduced function of PVINs is implicated in impaired cognition driven by attenuated gamma oscillations in cortical activity (Lewis et al., 2012). Although our data indicate an enhanced function of PVINs after SNI, increased excitability of PVINs indicates a shift in excitation-inhibition balance, which likely alters the prevalence of cortical gamma frequency associated with cognitive control. Indeed, gamma oscillations in the PFC are positively associated with both chronicity and severity of pain in back pain patients (May et al., 2019). Cognitive control, or executive control, is defined as flexible adaptation of mental resources in order to achieve a particular goal and includes behaviors such as attention, decision making, and memory (Mackie et al., 2013). These cognitive behaviors are impaired in rodent models of chronic pain (Leite-Almeida et al., 2009; Low et al., 2012; Shiers et al., 2018). A recent report showed impaired set-shifting, a test of cognitive flexibility, in male, but not female, injured mice (Shiers et al., 2018). While cellular mechanisms underlying sexually dimorphic cognitive deficits associated with chronic pain remain unresolved, our present data showing increased AP firing in PL PVINs from male, but not female, SNI mice provide new insight into sex-specific changes within cortical circuits implicated in executive control.
Our results showing that SNI does not alter the excitability of PVINs or SOM neurons in the IL region of the mPFC are consistent with studies showing that IL pyramidal neurons are unchanged in chronic pain models (Cheriyan and Sheets, 2018; Mitrić et al., 2019). However, this result is also intriguing given previous findings in other pain models. In a model of arthritic pain, inhibition of IL pyramidal neurons is enhanced due to a reduction in retrograde endocannabinoid depression of presynaptic inhibitory inputs (Kiritoshi et al., 2016). However, there is evidence that cannabinoid receptors 1 (CB1s) are not expressed in PVINs (Speed et al., 2015; Wedzony and Chocyk, 2009) but are detected in SOM neurons within the somatosensory cortex (Hill et al., 2007). Expression of CB1s in SOM neurons within mPFC has yet to be delineated. Therefore, our reported lack of SNI-induced changes to IL PVINs and IL-SOM neurons may reflect an absence of CB1 expression in these neurons. Studies report CB1 expression on cholecystokinin (CCK)-expressing interneurons in hippocampus and somatosensory cortex (Bodor et al., 2005; Tsou et al., 1999); however, functional studies of mPFC-CCK+ neurons have not been explored in any pain model. Reduced PV expression and shorter axon initial segments are observed in L5/6 IL neurons of male mice 3 weeks following SNI (Shiers et al., 2018), which suggests that our recordings 7–8 days after SNI may be too early to detect significant changes to IL PVINs and IL-SOM neurons. Nonetheless, consistent with our present findings in the PL region, this same study shows that SNI-induced alterations to IL neurons were specific to male mice (Shiers et al., 2018).
Overall, our data show complex and differential changes to mPFC inhibitory neurons of male and female mice in the SNI model of neuropathic pain. We hope that this research will update our knowledge of brain permutations associated with chronic pain so as to lead to more targeted pharmacotherapeutic strategies for cognitive deficits between male and female pain patients. Next steps will involve optogenetic or chemogenetic stimulation of SOM neurons and PVINs in the PL region of male and female SNI mice during measurements of cognitive flexibility and anxiety behaviors. These experiments will provide key insight into the role of specific inhibitory neuron subclasses in various disorders comorbid with chronic pain.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the Lead Contact, Patrick L. Sheets (plsheets@iu.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The datasets supporting the current study have not been deposited in a public repository because all data collected are included in the study. Data are available from the Lead Contact upon reasonable request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All experiments were approved by the Indiana University School of Medicine Animal Care and Use Committee (protocol number 11234) and adhered to the animal welfare guidelines of the National Institutes of Health. Transgenic mice expressing the red fluorescent protein TdTomato in cortical neurons expressing somatostatin or parvalbumin were obtained by mating female SST-IRES-Cre (SST < tm2.1(cre)Zjh > /J; stock #013044; Jackson Labs; (Taniguchi et al., 2011) or PV-IRES-Cre (B6;129P2-Pvalbtm1(cre)Arbr/J;stock #008069; Jackson Labs; (Hippenmeyer et al., 2005) mice with male Ai14 (B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J; stock #007914;Jackson Labs; (Madisen et al., 2010) mice. Using immunohistochemistry, an overlap rate of PVIN with SOM+, and vice versa, of 2%–6% was identified; however, electrophysiological signatures for each cell type have been identified (Hu et al., 2013; Nassar et al., 2015). Further inclusion/exclusion criteria is detailed in the electrophysiology subsection of the methods. Female and male SOM (n = 65; 33 males and 32 females) and PVIN mice (n = 61; 30 males and 31 females) aged 36–60 days and weighing 15–22 g were kept on a 24-hour light/dark cycle (lights on 7AM and off 7PM) and fed ad libitum for the duration of the experiments. All behavioral experiments and surgeries were conducted during the light phase (typically 7 am- 10am). Mice were group housed until after the SNI surgery, after which time they were single-housed to prevent possible development of empathic responses to pain in sham animals and to reduce the incidence of wound reopening.
METHOD DETAILS
Spared Nerve Injury (SNI) Model
Mice were typically tested in groups of 2. For each group, the experimenter was blinded to the baseline von Frey results and randomly selected one mouse to be SNI and one to be sham. Mice were weighed and briefly anesthetized in an anesthesia box with 1.5%–2.5% isoflurane in 100% O2 at a flow rate of 0.8–1.0 L/min (SurgiVet Isotech 4, Smith). The snout of the mouse was then placed into a flexible nose cone (Vetamac, Rossville, IN) connected to the isoflurane vaporizer allowing for continued anesthesia. Body temperature was maintained at 37°C using a feedback-controlled heating pad (FHC; Bowdoin, ME).The lateral surface of the left hind leg was shaved and disinfected using betadine and isopropyl alcohol. An approximately 4 mm incision through the skin was made and the underlying muscle layers were separated by blunt dissection using saline moistened sterile wooden dowels. The trifurcation of the left sciatic nerve was visualized. For SNI mice, an approximately 2 mm section of the tibial and common peroneal nerves distal to the trifurcation was removed, leaving the sural nerve intact. For sham mice, the trifurcation was exposed and visualized but not manipulated. The muscle layers of both SNI and sham mice were replaced and the outer skin layers were glued together using Vetbond (3M, MN). Approximately half of the mice were given a subcutaneous injection of meloxicam (1mg/kg) immediately after closing the incision. We found that meloxicam did not affect the development of allodynia. All mice recovered in a clean home cage with ad libitum water and wet feed on a heat pad for at least 30 min prior to being returned to the vivarium. Mice were monitored 4 days post-operation for signs of excessive pain such as reduced eating, drinking, activity or grooming.
Pain Assessment
Following surgery, the experimenter was blinded to SNI and sham groups. These animals were randomly assigned a number one or two and the animal assigned number one was always tested first by the blinded experimenter. The behavior testing apparatus was an elevated wire-mesh platform that held 4-inch wide, 4.5 inch long and 4.5-inch tall glass cubicles. On testing days, mice acclimated to the apparatus for 30–60 min prior to testing. Assessment of mechanical allodynia via paw-withdrawal threshold (PWT) was performed using von Frey filaments (Touch Test Sensory Evaluator, North Coast Medical Inc., Morgan Hills, CA) in combination with the up-down method to quantify the responses at 50% gram force (Bonin et al., 2014; Chaplan et al., 1994). PWTs were measured at baseline (prior to SNI or sham surgery), and at postoperative day 7 (POD7). The experimenter was blinded to the surgical group of the mice throughout the duration of the experiment. Mice that were outliers as determined by the ROUT method with Q = 1 were excluded from further analysis. This included 2 SNI and 1 sham from the PV-tdTomato mice and 2 SNI and 2 sham from the SOM-tdTomato mice.
Slice Preparation
Mice were euthanized on either POD7 or POD8 for subsequent recordings. For both sham and SNI groups, mice were briefly (~15–20s) anesthetized with isoflurane and decapitated Brains were rapidly dissected and placed in ice-cold carbogenated choline (in mM-25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 0.5 CaCl2, 7 MgCl2, 25 D-glucose, 110 C5H14ClNO, 11.60 C6H7NaO6, 3.09 C3H3NaO3) for sectioning. Three hundred micrometer thick coronal slices containing mPFC were prepared using a VT1200s Leica Vibratome and transferred to artificial cerebrospinal fluid (ACSF) solution (in mM- 127 NaCl, 25 NaHCO3, 1,25 NaH2PO4, 2.5 KCl, 25 d-glucose, 2 CaCl, 1 MgCl) for 30 min at 37°C then at room temperature until recording.
Electrophysiology
Recordings in the mPFC contralateral to injury took place on POD7 or POD8.Borosilicate glass pipettes were fabricated using a horizontal puller (P-97 Sutter) to have series resistances between 2 and 5 MΩ and pipette capacitance was compensated. A cesium-based internal was used for sEPSC in SOM mice (in mM- cesium methanesulfonate, 10 HEPES, 10 sodium phosphocreatine, 4 magnesium chloride, 4 sodium ATP, 0.4 sodium GTP, 3 ascorbic acid, 1 EGTA, 0.5 QX-314) while a potassium-based internal solution was used for all other recordings including sEPSC in PVINs (in mM- 128 K-gluconate, 10 HEPES, 10 sodium phosphocreatine, 4 magnesium chloride, 4 sodium ATP, 0.4 sodium GTP, 3 ascorbic acid, 1 EGTA, and ~4mg/ml biocytin). We found that K-internal solution allowed for more stable recordings in PVINs. Recordings targeted labeled neurons 50–80 μm deep within in the slice. Series resistance for each neuron was recorded in voltage clamp mode. It was not compensated but was required to be < 35 MU for inclusion in the analysis. Neuronal properties were recorded in current-clamp mode at resting membrane potential except for voltage sag. Voltage sag was measured from a membrane potential of −70 ± 3 mV by presenting multiple one second hyperpolarizing current steps (−200 pA, −150 pA, −100 pA, −50 pA). Percentage voltage sag was calculated using the peak voltage (Vpeak) and steady-state voltage (Vss) using the Equation 100 × (Vpeak–Vss)/Vpeak. Input resistance was measured from the steady-state responses to a series of hyperpolarizing and subthreshold depolarizing current steps (duration 1.0 s, amplitude −200 to 100 pA, 50 pA steps), as the slope of a linear least-squares fit to the resulting voltage–current relationship. Current threshold for action potentials (APs) was defined as the magnitude of current step that produced at least one AP. Voltage threshold (in mV) for APs was defined as the point when dV/dt exceeded 10% of its maximum value, relative to a dV/dt baseline measured 2 ms before the AP peak, which was measured as the maximum membrane potential reached after threshold. All sEPSC recordings were filtered at 2 kHZ and digitized at 10 kHZ while all other recordings were filtered at 4 kHZ and digitized at 10 kHZ. For sEPSC recordings, Gabazine (Tocris, Bristol, UK; 10 or 25 μM) was added to the ACSF but blockers were not added to the ACSF for other recordings. Cells were allowed to equilibrate for 3–5 min prior to the onset of recordings for sEPSC. Resting membrane potential, series resistance, and input resistance were collected prior to the onset of sEPSC recordings. Means for amplitude and frequency were collected and analyzed for sEPSC recordings using custom MATLAB (MathWorks, Natick, MA) analysis routines. Spontaneously firing SOM+ neurons have been previously identified in the presubicular cortex and amygdala (Nassar et al., 2015; Wilson et al., 2019). We detected spontaneously firing SOM+ neurons (total = 14), but excluded them from analysis due to variable waveforms, significant rundown at higher current injections and instability of leak currents. Thus, the fidelity of the recordings could not be guaranteed. To be confident that the cells we recorded were indeed PVIN and SOM+ neurons, we conducted principal component analysis (PCA). Similar to Hu et al. (2013), we used 4 electrophysiological parameters (action potential half-width, slow afterhyperpolarization, fast afterhyperpolarization, and spike frequency adaptation). This generated 3 main clusters of cells (cluster 1 was predominately SOM+ neurons, cluster 2 was predominately PVINs and cluster 3 was a mix of both cell types), which is similar to Nassar et al. (2015). For each cluster identified we further grouped the individual cell types into sub-clusters and conducted t tests on various electrophysiological variables. These comparisons resulted in significant differences between PVIN and SOM+ neurons, the most consistent of which was a difference in input resistance. Therefore, we are confident that the cells we have identified as PVIN and SOM+ neurons are correct.
QUANTIFICATION AND STATISTICAL ANALYSES
Power was set at 0.80, alpha of 0.05. For EPSCs, a priori power analysis was based off of the work of Zhang et al. (Zhang et al., 2015). For all other data, power analysis was completed post hoc. Sex was not included as a variable for power analysis as we did not initially compare males and females. We conducted separate power analysis for behavioral and electrophysiological data but did not conduct power analysis for every neuronal property that we report. All data were first analyzed for normality and outliers using the D’Agostino-Pearson normality test and the ROUT method with Q set to 1% in Prism (GraphPad Software, San Diego, CA). Two-way repeated-measures ANOVA was used for behavioral and FI curve analysis. Unpaired t tests or Mann-Whitney U tests were used for excitability and sEPSC data. Sidak’s multiple comparisons test was used for all multiple comparisons. Cells that exhibited series resistance >35 mOhm were excluded from analysis. For sEPSC, cells that had greater than 15% change in series resistance from baseline to the end of recording were removed from further analysis. Power and sample size were calculated using GPower (Heinrich-Heine-Universität, Düsseldorf, Germany).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Streptavidin Alexa Fluor 488 conjugate | Invitrogen | Ref# S32354; RRID: AB_2315383 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SR95531 hydrobromide: Gabazine | Tocris | Cat # 1262; PubChem CID: 107896 |
| Biocytin | Tocris | Cat # 3349; PubChem CID: 83814 |
| QX314 chloride | Tocris | Cat # 2313; PubChem CID: 21462 |
| HEPES | Sigma-Aldrich | Cat# H4034; PubChem CID: 23831 |
| Potassium Gluconate | Sigma-Aldrich | Cat# P1847; PubChem CID: 16760467 |
| Phosphocreatine disodium salt hydrate | Sigma-Aldrich | Cat# P7936; PubChem CID: 43835030 |
| Magnesium Chloride Solution | Sigma-Aldrich | Cat# M1028; PubChem CID: 24584 |
| Adenosine 5’-triphosphate disodium salt hydrate | Sigma-Aldrich | Cat# A6419; PubChem CID: 16218877 |
| Guanosine 5’-triphosphate sodium salt hydrate | Sigma-Aldrich | Cat# G8877; PubChem CID: 13544577 |
| Ascorbic Acid | Sigma-Aldrich | Cat# 5960; PubChem CID: 54670067 |
| Cesium Methanesulfonate | Sigma-Aldrich | Cat# C1426; PubChem CID: 5148066 |
| Ethylene glycol-bis(2-amino-ethylether)-N,N,N’,N’- tetraacetic acid (EGTA) | Sigma-Alrdich | Cat# E3889; PubChem CID: 6207 |
| Sodium Chloride | Sigma-Aldrich | Cat# S7653; PubChem CID: 5234 |
| Sodium Bicarbonate | Sigma-Aldrich | Cat# S6014; PubChem CID: 24192197 |
| Sodium Phosphate Monohydrate Monobasic | Sigma-Aldrich | Cat# S9638; PubChem CID: 516949 |
| Potassium Chloride Solution | Sigma-Aldrich | Cat# 60137; PubChem SID: 24881800 |
| D-glucose | Sigma-Aldrich | Cat# G8270; PubChem CID: 79025 |
| Calcium Chloride | Sigma-Aldrich | Cat# 793639; PubChem CID: 24854 |
| Choline Chloride | Sigma-Aldrich | Cat# C1879; PubChem CID: 19844489 |
| Sodium Pyruvate | Sigma-Aldrich | Cat# P8574; PubChem CID: 10154024 |
| Isoflurane USP | Patterson Veterinary | Cat # 07-893-2374; NDC: 14043-704-05 |
| Normal Saline | McKesson | MFR# 37-6240 |
| Experimental Models: Organisms/Strains | ||
| Mouse: SOM+: SST < tm2.1(cre)Zjh > /J | Jackson Laboratories | Stock # 013044; RRID: IMSR_JAX:013044 |
| Mouse: PV+: B6;129P2-Pvalbtm1(cre)Arbr/J | Jackson Laboratories | Stock # 008069; RRID: IMSR_JAX:008069 |
| Mouse: Ai14: B6.Cg-Gt(ROSA) 26Sortm14(CAG-tdTomato)Hze/J | Jackson Laboratories | Stock # 007914; RRID: IMSR_JAX:007914 |
| Software and Algorithms | ||
| Prism version 7.04 | GraphPad Software | RRID: SCR_002798 |
| MATLAB version R2018b | MathWorks | RRID: SCR_001622 |
| GPower | Heinrich-Heine- Universität | RRID: SCR_013726 |
| Adobe Illustrator CC 2018 | Adobe Inc. | RRID: SCR_010279 |
| BioRender | BioRender | https://biorender.com |
| Other | ||
| Touch Test Sensory Evaluator: von Frey Filaments | North Coast Medical Inc. | Item #s: NC12775- 01, 02, 04, 05, 06, 08, 10,12 |
| Nikon Confocal | Nikon | A1R |
| Vibratome | Leica | VT1200S |
| Axon CNS Multiclamp Amplifier | Molecular Devices | 700B |
| Cool LED | Scientifica | pE-4000 |
| Upright Microscope | Olympus | U-CMDPTS |
| Microscope Objectives | Olympus | 4X and 60X |
| Inline Heater and Monitor | Warner Instruments | Cat# 64–0102 and TC-324B |
| Anesthesia System | Vetamac | VAD Compact II |
| Feedback Rodent Warmer | Stoelting | X2 |
| Micropipette Puller | Sutter Instruments | Flaming/Brown P-97 |
| Borosilicate Glass Capillaries with Filaments | Warner | G150-F |
Highlights.
Nerve injury enhances excitability of prelimbic PV+ neurons in male mice
Nerve injury reduces EPSC frequency in prelimbic SOM+ neurons in female mice
These changes to PV+ and SOM+ neurons are laminar specific
Nerve injury does not affect PV+ or SOM+ neurons in infralimbic cortex
ACKNOWLEDGMENTS
The work was supported by National Institutes of Health grant R01-NS094389 (to P.L.S.). The graphical abstract was created using BioRender (Toronto, ON, Canada). The authors thank Christopher C. Lapish (IUPUI) for technical help in data analysis, Kevin D. Chen for assistance with processing slices and confocal imaging, and Briana Mork for assistance with graphical design.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107729.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abbas AI, Sundiang MJM, Henoch B, Morton MP, Bolkan SS, Park AJ, Harris AZ, Kellendonk C, and Gordon JA (2018). Somatostatin Interneurons Facilitate Hippocampal-Prefrontal Synchrony and Prefrontal Spatial Encoding. Neuron 100, 926–939.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bañuelos C, Beas BS, McQuail JA, Gilbert RJ, Frazier CJ, Setlow B, and Bizon JL (2014). Prefrontal cortical GABAergic dysfunction contributes to age-related working memory impairment. J. Neurosci 34, 3457–3466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor AL, Katona I, Nyíri G, Mackie K, Ledent C, Hájos N, and Freund TF (2005). Endocannabinoid signaling in rat somatosensory cortex: laminar differences and involvement of specific interneuron types. J. Neurosci 25, 6845–6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonin RP, Bories C, and De Koninck Y (2014). A Simplified Up-Down Method (SUDO) for Measuring Mechanical Nociception in Rodents Using Von Frey Filaments. Mol. Pain [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso-Cruz H, Lima D, and Galhardo V (2013). Impaired spatial memory performance in a rat model of neuropathic pain is associated with reduced hippocampus-prefrontal cortex connectivity. J. Neurosci 33, 2465–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, and Yaksh TL (1994). Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63. [DOI] [PubMed] [Google Scholar]
- Cheriyan J, and Sheets PL (2018). Altered Excitability and Local Connectivity of mPFC-PAG Neurons in a Mouse Model of Neuropathic Pain. J. Neurosci 38, 4829–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick BD, and Rashiq S (2007). Disruption of attention and working memory traces in individuals with chronic pain. Anesth. Analg 104, 1223–1229. [DOI] [PubMed] [Google Scholar]
- Doron M, Chindemi G, Muller E, Markram H, and Segev I (2017). Timed Synaptic Inhibition Shapes NMDA Spikes, Influencing Local Dendritic Processing and Global I/O Properties of Cortical Neurons. Cell Rep. 21, 1550–1561. [DOI] [PubMed] [Google Scholar]
- Enomoto T, Tse MT, and Floresco SB (2011). Reducing prefrontal gamma-aminobutyric acid activity induces cognitive, behavioral, and dopaminergic abnormalities that resemble schizophrenia. Biol. Psychiatry 69, 432–441. [DOI] [PubMed] [Google Scholar]
- Fillingim RB, King CD, Ribeiro-Dasilva MC, Rahim-Williams B, and Riley JL 3rd. (2009). Sex, gender, and pain: a review of recent clinical and experimental findings. J. Pain 10, 447–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, and Yuste R (2011). Dense inhibitory connectivity in neocortex. Neuron 69, 1188–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadotti VM, Zhang Z, Huang J, and Zamponi GW (2019). Analgesic effects of optogenetic inhibition of basolateral amygdala inputs into the prefrontal cortex in nerve injured female mice. Mol. Brain 12, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentet LJ, Kremer Y, Taniguchi H, Huang ZJ, Staiger JF, and Petersen CC (2012). Unique functional properties of somatostatin-expressing GABAergic neurons in mouse barrel cortex. Nat. Neurosci 15, 607–612. [DOI] [PubMed] [Google Scholar]
- Gupta A, Wang Y, and Markram H (2000). Organizing principles for a diversity of GABAergic interneurons and synapses in the neocortex. Science 287, 273–278. [DOI] [PubMed] [Google Scholar]
- Hayama T, Noguchi J, Watanabe S, Takahashi N, Hayashi-Takagi A, Ellis-Davies GC, Matsuzaki M, and Kasai H (2013). GABA promotes the competitive selection of dendritic spines by controlling local Ca2+ signaling. Nat. Neurosci 16, 1409–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill EL, Gallopin T, Férézou I, Cauli B, Rossier J, Schweitzer P, and Lambolez B (2007). Functional CB1 receptors are broadly expressed in neocortical GABAergic and glutamatergic neurons. J. Neurophysiol 97, 2580–2589. [DOI] [PubMed] [Google Scholar]
- Howard MW, Rizzuto DS, Caplan JB, Madsen JR, Lisman J, Aschen-brenner-Scheibe R, Schulze-Bonhage A, and Kahana MJ (2003). Gamma oscillations correlate with working memory load in humans. Cereb. Cortex 13, 1369–1374. [DOI] [PubMed] [Google Scholar]
- Hu H, Cavendish JZ, and Agmon A (2013). Not all that glitters is gold: off-target recombination in the somatostatin-IRES-Cre mouse line labels a subset of fast-spiking interneurons. Front. Neural Circuits 7, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Gadotti VM, Chen L, Souza IA, Huang S, Wang D, Ramakrishnan C, Deisseroth K, Zhang Z, and Zamponi GW (2019). A neuronal circuit for activating descending modulation of neuropathic pain. Nat. Neurosci 22, 1659–1668. [DOI] [PubMed] [Google Scholar]
- Ji G, Sun H, Fu Y, Li Z, Pais-Vieira M, Galhardo V, and Neugebauer V (2010). Cognitive impairment in pain through amygdala-driven prefrontal cortical deactivation. J. Neurosci 30, 5451–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, and Kubota Y (1996). Physiological and morphological identification of somatostatin- or vasoactive intestinal polypeptide-containing cells among GABAergic cell subtypes in rat frontal cortex. J. Neurosci 16, 2701–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, and Kubota Y (1997). GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb. Cortex 7, 476–486. [DOI] [PubMed] [Google Scholar]
- Kelly CJ, Huang M, Meltzer H, and Martina M (2016). Reduced Glutamatergic Currents and Dendritic Branching of Layer 5 Pyramidal Cells Contribute to Medial Prefrontal Cortex Deactivation in a Rat Model of Neuropathic Pain. Front. Cell. Neurosci 10, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HG, Beierlein M, and Connors BW (1995). Inhibitory control of excitable dendrites in neocortex. J. Neurophysiol 74, 1810–1814. [DOI] [PubMed] [Google Scholar]
- Kim D, Jeong H, Lee J, Ghim JW, Her ES, Lee SH, and Jung MW (2016). Distinct Roles of Parvalbumin- and Somatostatin-Expressing Interneurons in Working Memory. Neuron 92, 902–915. [DOI] [PubMed] [Google Scholar]
- Kiritoshi T, Ji G, and Neugebauer V (2016). Rescue of Impaired mGluR5-Driven Endocannabinoid Signaling Restores Prefrontal Cortical Output to Inhibit Pain in Arthritic Rats. J. Neurosci 36, 837–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, and Sakmann B (2001). Dendritic mechanisms underlying the coupling of the dendritic with the axonal action potential initiation zone of adult rat layer 5 pyramidal neurons. J. Physiol 533, 447–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AT, Gee SM, Vogt D, Patel T, Rubenstein JL, and Sohal VS (2014). Pyramidal neurons in prefrontal cortex receive subtype-specific forms of excitation and inhibition. Neuron 81, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Manders TR, Eberle SE, Su C, D’amour J, Yang R, Lin HY, Deisseroth K, Froemke RC, and Wang J (2015). Activation of corticostriatal circuitry relieves chronic neuropathic pain. J. Neurosci 35, 5247–5259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite-Almeida H, Almeida-Torres L, Mesquita AR, Pertovaara A, Sousa N, Cerqueira JJ, and Almeida A (2009). The impact of age on emotional and cognitive behaviours triggered by experimental neuropathy in rats. Pain 144, 57–65. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Curley AA, Glausier JR, and Volk DW (2012). Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 35, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low LA, Millecamps M, Seminowicz DA, Naso L, Thompson SJ, Stone LS, and Bushnell MC (2012). Nerve injury causes long-term attentional deficits in rats. Neurosci. Lett 529, 103–107. [DOI] [PubMed] [Google Scholar]
- Mackie MA, Van Dam NT, and Fan J (2013). Cognitive control and attentional functions. Brain Cogn. 82, 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, and Wu C (2004). Interneurons of the neocortical inhibitory system. Nat. Rev. Neurosci 5, 793–807. [DOI] [PubMed] [Google Scholar]
- Marlin JJ, and Carter AG (2014). GABA-A receptor inhibition of local calcium signaling in spines and dendrites. J. Neurosci 34, 15898–15911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May ES, Nickel MM, Ta Dinh S, Tiemann L, Heitmann H, Voth I, Tölle TR, Gross J, and Ploner M (2019). Prefrontal gamma oscillations reflect ongoing pain intensity in chronic back pain patients. Hum. Brain Mapp 40, 293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGarry LM, and Carter AG (2016). Inhibitory Gating of Basolateral Amygdala Inputs to the Prefrontal Cortex. J. Neurosci 36, 9391–9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz AE, Yau HJ, Centeno MV, Apkarian AV, and Martina M (2009). Morphological and functional reorganization of rat medial prefrontal cortex in neuropathic pain. Proc. Natl. Acad. Sci. USA 106, 2423–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millecamps M, Centeno MV, Berra HH, Rudick CN, Lavarello S, Tkatch T, and Apkarian AV (2007). D-cycloserine reduces neuropathic pain behavior through limbic NMDA-mediated circuitry. Pain 132, 108–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrić M, Seewald A, Moschetti G, Sacerdote P, Ferraguti F, Kummer KK, and Kress M (2019). Layer- and subregion-specific electrophysiological and morphological changes of the medial prefrontal cortex in a mouse model of neuropathic pain. Sci. Rep 9, 9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS (2012). Sex differences in pain and pain inhibition: multiple explanations of a controversial phenomenon. Nat. Rev. Neurosci 13, 859–866. [DOI] [PubMed] [Google Scholar]
- Mogil JS, and Bailey AL (2010). Sex and gender differences in pain and analgesia. Prog. Brain Res 186, 141–157. [DOI] [PubMed] [Google Scholar]
- Murray AJ, Woloszynowska-Fraser MU, Ansel-Bollepalli L, Cole KL, Foggetti A, Crouch B, Riedel G, and Wulff P (2015). Parvalbumin-positive interneurons of the prefrontal cortex support working memory and cognitive flexibility. Sci. Rep 5, 16778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar M, Simonnet J, Lofredi R, Cohen I, Savary E, Yanagawa Y, Miles R, and Fricker D (2015). Diversity and overlap of parvalbumin and somatostatin expressing interneurons in mouse presubiculum. Front. Neural Circuits 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povedano M, Gascón J, Gálvez R, Ruiz M, and Rejas J (2007). Cognitive function impairment in patients with neuropathic pain under standard conditions of care. J. Pain Symptom Manage 33, 78–89. [DOI] [PubMed] [Google Scholar]
- Rock C, and Apicella AJ (2015). Callosal projections drive neuronal-specific responses in the mouse auditory cortex. J. Neurosci 35, 6703–6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan CM, Williams TM, Finegold DN, and Orchard TJ (1993). Cognitive dysfunction in adults with type 1 (insulin-dependent) diabetes mellitus of long duration: effects of recurrent hypoglycaemia and other chronic complications. Diabetologia 36, 329–334. [DOI] [PubMed] [Google Scholar]
- Schiltenwolf M, Akbar M, Hug A, Pfüller U, Gantz S, Neubauer E, Flor H, and Wang H (2014). Evidence of specific cognitive deficits in patients with chronic low back pain under long-term substitution treatment of opioids. Pain Physician 17, 9–20. [PubMed] [Google Scholar]
- Shiers S, Pradhan G, Mwirigi J, Mejia G, Ahmad A, Kroener S, and Price T (2018). Neuropathic Pain Creates an Enduring Prefrontal Cortex Dysfunction Corrected by the Type II Diabetic Drug Metformin But Not by Gabapentin. J. Neurosci 38, 7337–7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS, Zhang F, Yizhar O, and Deisseroth K (2009). Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 459, 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speed HE, Masiulis I, Gibson JR, and Powell CM (2015). Increased Cortical Inhibition in Autism-Linked Neuroligin-3R451C Mice Is Due in Part to Loss of Endocannabinoid Signaling. PLoS ONE 10, e0140638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, and Neugebauer V (2011). mGluR1, but not mGluR5, activates feed-forward inhibition in the medial prefrontal cortex to impair decision making. J. Neurophysiol 106, 960–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi H, He M, Wu P, Kim S, Park R, Sugino K, Kvitsiani D, Fu Y, Lu J, Lin Y, et al. (2011). A Resource of Cre Driver Lines for Genetic Targeting of GABAergic Neurons in Cerebral Cortex. Neuron 71, 995–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou K, Mackie K, Sañudo-Peña MC, and Walker JM (1999). Cannabinoid CB1 receptors are localized primarily on cholecystokinin-containing GABAergic interneurons in the rat hippocampal formation. Neuroscience 93, 969–975. [DOI] [PubMed] [Google Scholar]
- Vacca V, Marinelli S, Pieroni L, Urbani A, Luvisetto S, and Pavone F (2014). Higher pain perception and lack of recovery from neuropathic pain in females: a behavioural, immunohistochemical, and proteomic investigation on sex-related differences in mice. Pain 155, 388–402. [DOI] [PubMed] [Google Scholar]
- Walker F, Möck M, Feyerabend M, Guy J, Wagener RJ, Schubert D, Staiger JF, and Witte M (2016). Parvalbumin- and vasoactive intestinal polypeptide-expressing neocortical interneurons impose differential inhibition on Martinotti cells. Nat. Commun 7, 13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Toledo-Rodriguez M, Gupta A, Wu C, Silberberg G, Luo J, and Markram H (2004). Anatomical, physiological and molecular properties of Martinotti cells in the somatosensory cortex of the juvenile rat. J. Physiol 561, 65–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedzony K, and Chocyk A (2009). Cannabinoid CB1 receptors in rat medial prefrontal cortex are colocalized with calbindin- but not parvalbumin- and calretinin-positive GABA-ergic neurons. Pharmacol. Rep 61, 1000–1007. [DOI] [PubMed] [Google Scholar]
- Wilson TD, Valdivia S, Khan A, Ahn HS, Adke AP, Martinez Gonzalez S, Sugimura YK, and Carrasquillo Y (2019). Dual and Opposing Functions of the Central Amygdala in the Modulation of Pain. Cell Rep. 29, 332–346.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W, Yen YC, Lee YH, Tan S, Xiao Y, Lokman H, Ting AKT, Ganegala H, Kwon T, Ho WK, and Je HS (2019). Prenatal selective serotonin reuptake inhibitor (SSRI) exposure induces working memory and social recognition deficits by disrupting inhibitory synaptic networks in male mice. Mol. Brain 12, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Gadotti VM, Chen L, Souza IA, Stemkowski PL, and Zamponi GW (2015). Role of Prelimbic GABAergic Circuits in Sensory and Emotional Aspects of Neuropathic Pain. Cell Rep. 12, 752–759. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the current study have not been deposited in a public repository because all data collected are included in the study. Data are available from the Lead Contact upon reasonable request.