

Abstract
Glycoside hydrolases (GHs) are found in all domains of life, and at least 87 distinct genes encoding proteins related to GHs are found in the human genome. GHs serve diverse functions from digestion of dietary polysaccharides to breakdown of intracellular oligosaccharides, glycoproteins, proteoglycans and glycolipids. Congenital disorders of GHs (CDGHs) represent more than 30 rare diseases caused by mutations in one of the GH genes. We previously used whole-exome sequencing of a homogenous Danish population of almost 2000 individuals to probe the incidence of deleterious mutations in the human glycosyltransferases (GTs) and developed a mutation map of human GT genes (GlyMAP-I). While deleterious disease-causing mutations in the GT genes were very rare, and in many cases lethal, we predicted deleterious mutations in GH genes to be less rare and less severe given the higher incidence of CDGHs reported worldwide. To probe the incidence of GH mutations, we constructed a mutation map of human GH-related genes (GlyMAP-II) using the Danish WES data, and correlating this with reported disease-causing mutations confirmed the higher prevalence of disease-causing mutations in several GH genes compared to GT genes. We identified 76 novel nonsynonymous single-nucleotide variations (nsSNVs) in 32 GH genes that have not been associated with a CDGH phenotype, and we experimentally validated two novel potentially damaging nsSNVs in the congenital sucrase-isomaltase deficiency gene, SI. Our study provides a global view of human GH genes and disease-causing mutations and serves as a discovery tool for novel damaging nsSNVs in CDGHs.
Keywords: congenital disorders of glycoside hydrolysis, nsSNV, WES
Introduction
Glycoside hydrolases (GHs) catalyze the breakdown of glycans and serve unique functions in the shaping and maintenance of the human glycome, the most complex nontemplate-derived macromolecules in living cells. GH-mediated glycan breakdown takes place both within and outside of cells. GHs are found in all living organisms and are classified in sequence-based families in the carbohydrate-active enzymes (CAZy) database (Henrissat 1991; Lombard et al. 2014). Over 160 GH families have been identified, and 87 members from 28 of these families are encoded in the human genome (Table I). The functional importance of efficient hydrolysis of glycans in cells is evident from the many diseases caused by deficiencies in GHs designated congenital disorders of GHs (CDGHs), which to date include mutations in 30 out of the 87 human GH genes. By far, most of these CDGHs are dysfunctions of lysosomal enzymes that cause the accumulation of unprocessed intermediate glycan structures in the lysosome, leading to clinical features (Brady and Schiffmann 2000; Bahr and Bendiske 2002). Seventeen lysosomal storage diseases (LSDs) are known for the GH genes (Futerman and van Meer 2004). LSDs have an estimated incidence around 1:5000 live births (Platt et al. 2012), with Gaucher disease being the most prevalent having an incidence around 1:57,000, followed by Fabry disease with 1:117,000 (Meikle et al. 1999). Disease severity for many of the LSDs is dependent on the nature of the gene impairment and the degree of residual enzyme activity present. More than 900 distinct mutations have been reported in the α-galactosidase gene GLA underlying Fabry disease and more than 460 mutations in GBA causing Gaucher disease (data from The Human Gene Mutation Database, HGMD, Stenson et al. 2017). Currently, the most effective treatments for LSDs are costly enzyme-replacement therapies (ERTs), and these provide in most cases only partial alleviation of the detrimental disease outcomes, for example, for Gaucher, Fabry and Pompe diseases and mucopolysaccharidosis (Wenger et al. 1997; Schiffmann and Moore 2006; Gritti 2011; Arvio and Mononen 2016; Kelley et al. 2016; Stirnemann et al. 2017), likely due to limited biodistribution of the infused enzymes.
Table I.
Human GH and GH-like genes
| CAZy GH family | Gene namea | Protein nameb | Substratec | Expression | SPd | TMd | Domain structuree | Compartmentf | Disorderg |
|---|---|---|---|---|---|---|---|---|---|
| GH1 | GBA3 | Cytosolic β-glucosidase | β-Glc | Reg | — | — | Soluble | Cytosol | — |
| GH1 | KL | Klotho | Inactive | Reg | X | X | Type-I TMD | Cell membrane | Tumoral calcinosis |
| GH1 | KLB | β-klotho | Inactive | Reg | — | X | Type-I TMD | Cell membrane | — |
| GH1 | LCT | Lactase-phlorizin hydrolase | β-Glc/β-Gal | Reg | X | X | Type-I TMD | Cell membrane | Lactase deficiency |
| GH1 | LCTL | Lactase-like protein | Inactivei | Reg | X | X | Type-I TMD | Cell membrane | — |
| GH2 | GUSB | β-glucuronidase | β-GlcA | Non-reg | X | — | Soluble | Lysosome | MPS VII |
| GH2 | MANBA | β-Mannosidase | β-Man | Non-reg | X | — | Soluble | Lysosome | Mannosidosis, beta |
| GH13, GH133 | AGL | Glycogen debranching enzyme | α-Glc/glycogen | Reg | — | — | Soluble | Cytosol | GSD IIIa/IIIb; Forbes disease |
| GH13, CBM48 | GBE1 | 1,4-α-glucan-branching enzyme | α-Glc/glycogen | Reg | — | — | Soluble | Cytosol | GSD IV |
| GH13 | AMY1A | α-Amylase | Starch/glycogen | Reg | X | — | Soluble | Secretory | — |
| GH13 | AMY1B | α-Amylase | Starch/glycogen | Reg | X | — | Soluble | Secretory | — |
| GH13 | AMY1C | α-Amylase | Starch/glycogen | Reg | X | — | Soluble | Secretory | — |
| GH13 | AMY2A | Pancreatic α-amylase | Starch/glycogen | Reg | X | — | Soluble | Secretory | — |
| GH13 | AMY2B | α-Amylase 2B | Starch/glycogen | Reg | X | — | Soluble | Secretory | — |
| GH13 | SLC3A1 | Neutral and basic amino acid transport protein | Inactivej | Reg | — | X | Type-II TMD | Cell membrane | Cystinuria |
| GH13 | SLC3A2 | 4F2 cell-surface antigen heavy chain | Inactivej | Non-reg | — | X | Type-II TMD | Cell membrane | — |
| GH18 | CHI3L1 | Chitinase-3-like protein 1 | Inactive (misses catalytic residues)i | Non-reg | X | — | Soluble | Secretory | — |
| GH18 | CHI3L2 | Chitinase-3-like protein 2 | Inactive (misses catalytic residues)i | Reg | X | — | Soluble | Secretory | — |
| GH18, CBM14 | CHIA | Acidic mammalian chitinase | Chitin/β-GlcNAc | Reg | X | — | Soluble | Secretory | — |
| GH18, CBM14 | CHIT1 | Chitotriosidase-1 | Chitotriose/β-GlcNAc | Reg | X | — | Soluble | Secretory | — |
| GH18 | CTBS | di-N-acetylchitobiase | Chitobiose/β-GlcNAc | Non-reg | X | — | Soluble | Lysosome | — |
| GH18 | OVGP1 | Oviduct-specific glycoprotein | Inactive (misses catalytic residues)i | Non-reg | X | — | Soluble | Secretory | — |
| GH20 | HEXA | β-Hexosaminidase subunit α | β-GalNAc/β-GlcNAc | Non-reg | X | — | Soluble | Lysosome | GM2 gangliosidosis, type I; Tay–Sachs disease |
| GH20 | HEXB | β-Hexosaminidase subunit β | β-GalNAc/β-GlcNAc | Non-reg | X | — | Soluble | Lysosome | GM2 gangliosidosis, type II; Sandhoff disease |
| GH20 | HEXDC | Hexosaminidase D | β-GalNAc/β-GlcNAc | Non-reg | — | — | Soluble | Cytosol | — |
| GH22 | LYZ | Lysozyme C | Peptidoglycan | Reg | X | — | Soluble | Secretory | Amyloidosis, renal |
| GH22 | LYZL1 | Lysozyme-like protein 1 | Peptidoglycan (catalytic residue present)i | Reg | X | — | Soluble | Secretory | — |
| GH22 | LYZL2 | Lysozyme-like protein 2 | Peptidoglycan (catalytic residue present)i | Reg | X | — | Soluble | Secretory | — |
| GH22 | LYZL4 | Lysozyme-like protein 4 | Peptidoglycan (inactive, catalytic residue missing)i | Reg | X | — | Soluble | Secretory | — |
| GH22 | LYZL6 | Lysozyme-like protein 6 | Peptidoglycan (catalytic residue present)i | Reg | X | — | Soluble | Secretory | — |
| GH22 | SPACA3 | Sperm acrosome membrane-associated protein 3 | Inactive | Reg | — | X | Type-II TMD | Cell membrane | — |
| GH22 | SPACA5 | Sperm acrosome-associated protein 5 | β-GlcNAc (catalytic residue present)i | Reg | X | — | Soluble | Secretory | — |
| GH23 | LYG1 | Lysozyme g-like protein 1 | Peptidoglycan/inactive? (misses catalytic residue)i | Reg | X | — | Soluble | Secretory | — |
| GH23 | LYG2 | Lysozyme g-like protein 2 | Peptidoglycan (catalytic residue present)i | Reg | X | — | Soluble | Secretory | — |
| GH27 | GLA | α-Galactosidase A | α-Gal/glycolipid/glycoprotein | Non-reg | X | — | Soluble | Lysosome | Fabry disease |
| GH27 | NAGA | α-N-acetylgalactosaminidase | α-GalNAc/glycolipid/glycoprotein | Non-reg | X | — | Soluble | Lysosome | Schindler disease |
| GH29 | FUCA1 | Tissue α-L-fucosidase | α-Fuc/glycolipid/glycoprotein | Non-reg | X | — | Soluble | Lysosome | Fucosidosis |
| GH29 | FUCA2 | Plasma α-L-fucosidase | α-Fuc/glycolipid/glycoprotein | Non-reg | X | — | Soluble | Secretory | — |
| GH30 | GBA | Glucosylceramidase | β-Glc-Cer/glycolipid | Non-reg | - | — | Soluble | Lysosome | Gaucher disease |
| GH31 | GANAB | Neutral α-glucosidase AB | α-Glc | Non-reg | X | — | Soluble | ER/Golgi | Polycystic kidney disease 3 |
| GH31 | GANC | Neutral α-glucosidase C | α-Glc | Non-reg | — | — | Soluble | Cytosol | — |
| GH31 | GAA | Lysosomal α-glucosidase | α-Glc/glycolipid/glycoprotein | Non-reg | X | — | Soluble | Lysosome | GSD II, Pompe disease |
| GH31 | MYORG (KIAA1161) | Myogenesis-regulating glycosidase | Glycogen/possibly inactive (catalytic acid/base may be missing)i | Non-reg | — | X | Type-II TMD | ER | Basal ganglia calcification, idiopathic, 7k |
| GH31 | MGAM | Maltase-glucoamylase, intestinal | α-Glc | Reg | — | X | Type-II TMD | Cell membrane | — |
| GH31 | MGAM2 | Probable maltase-glucoamylase 2 | α-Glc | Reg | — | X | Type-II TMD | Cell membrane | — |
| GH31 | SI | Sucrase-isomaltase, intestinal | α-Glc | Reg | — | X | Type-II TMD | Cell membrane | Sucrase-isomaltase deficiency |
| GH33 | NEU1 | Sialidase-1 | α-Sia/glycolipid/glycoprotein | Non-reg | (−) | (X) | Soluble | Lysosomeh | Sialidosis, type I/II |
| GH33 | NEU2 | Sialidase-2 | α-Sia/glycolipid/glycoprotein | Reg | — | — | Soluble | Cytosolh | — |
| GH33 | NEU3 | Sialidase-3 | α-Sia/glycolipid/glycoprotein | Non-reg | — | — | Soluble | Cytosolh | — |
| GH33 | NEU4 | Sialidase-4 | α-Sia/glycolipid/glycoprotein | Reg | — | — | Soluble | Cytosolh | — |
| GH35 | GLB1 | β-Galactosidase | β-Gal/glycolipid/glycoprotein | Non-reg | X | — | Soluble | Lysosome | GM1 gangliosidosis, type I/II/III; MPS IVB (Morquio) |
| GH35 | GLB1L | β-Galactosidase-1-like protein | Active (catalytic residues present) | Reg | X | — | Soluble | Secretory | — |
| GH35 | GLB1L2 | β-Galactosidase-1-like protein 2 | Active (catalytic residues present) | Reg | — | X | Type-II TMD | Cell membrane | — |
| GH35 | GLB1L3 | β-Galactosidase-1-like protein 3 | Active (catalytic residues present) | Reg | — | — | Soluble | Cytosol | — |
| GH37 | TREH | Trehalase | α-Glc/trehalose (digestion) | Reg | X | — | GPI anchor | Cell membrane | — |
| GH38 | MAN2A1 | 1,6/1,3-α-Mannosidase 2A1 | α-Man/glycoprotein (processing) | Non-reg | — | X | Type-II TMD | Golgi | — |
| GH38 | MAN2A2 | 1,6/1,3-α-Mannosidase 2A2 | α-Man/glycoprotein (processing) | Non-reg | — | X | Type-II TMD | Golgi | — |
| GH38 | MAN2B1 | 1,2/1,6/1,3-α-Mannosidase 2B1 | α-Man/glycoprotein | Non-reg | X | — | Soluble | Lysosome | α-Mannosidosis, type I/II |
| GH38 | MAN2B2 | α-Mannosidase 2B2, Epididymis-specific | α-Man/glycoprotein | Non-reg | X | — | Soluble | Secretory | — |
| GH38 | MAN2C1 | α-Mannosidase 2C1 | α-Man/glycoprotein | Non-reg | — | — | Soluble | Cytosol | — |
| GH39 | IDUA | α-L-iduronidase | α-IduA/glycoprotein | Non-reg | X | — | Soluble | Lysosome | MPS I, Hurler/Scheie syndrome |
| GH47 | EDEM1 | ER degradation-enhancing α-mannosidase-like protein 1 | α-Man/glycoprotein | Non-reg | — | X | Type-II TMD | ER | — |
| GH47 | EDEM2 | ER degradation-enhancing α-mannosidase-like protein 2 | α-Man/glycoprotein | Non-reg | X | — | Soluble | ER | — |
| GH47 | EDEM3 | ER degradation-enhancing α-mannosidase-like protein 3 | α-Man/glycoprotein | Non-reg | — | — | Soluble | ER | — |
| GH47 | MAN1A1 | 1,2-α-Mannosidase IA | α-Man/N-glycan processing | Non-reg | — | X | Type-II TMD | Golgi | — |
| GH47 | MAN1A2 | 1,2-α-Mannosidase IB | α-Man/N- glycan processing | Non-reg | — | X | Type-II TMD | Golgi | — |
| GH47 | MAN1B1 | 1,2-α-Mannosidase | α-Man/N-glycan processing | Non-reg | — | X | Type-II TMD | ER | MRT15 |
| GH47 | MAN1C1 | 1,2-α-Mannosidase IC | α-Man/N-glycan processing | Non-reg | — | X | Type-II TMD | Golgi | — |
| GH56 | HYAL1 | Hyaluronidase-1 | β-GalNAc/hyaluronic acid | Reg | X | — | Soluble | Lysosome | MPS IX |
| GH56 | HYAL2 | Hyaluronidase-2 | β-GalNAc/hyaluronic acid | Reg | X | — | GPI anchor | Cell membrane | — |
| GH56 | HYAL3 | Hyaluronidase-3 | β-GalNAc/hyaluronic acid | Reg | X | — | Soluble | Secretory | — |
| GH56 | HYAL4 | Hyaluronidase-4 | β-GalNAc/hyaluronic acid | Reg | — | X | MPM | Cell membrane | — |
| GH56 | SPAM1 | Hyaluronidase PH-20 | β-GalNAc/hyaluronic acid | Reg | X | — | GPI anchor | Cell membrane | — |
| GH59 | GALC | Galactocerebrosidase | β-Gal-Cer/glycolipid | Non-reg | X | — | Soluble | Lysosome | Krabbe disease |
| GH63 | MOGS | Mannosyl-oligosaccharide glucosidase | α-Glc/processing glucosidase | Non-reg | — | X | Type-II TMD | ER | CDG IIb |
| GH65 | PGGHG | Acid trehalase-like protein 1 | α-Glc/protein-glucosylgalactosylhydroxylysine a-glucosidase (collagen) | Reg | — | — | Soluble | Cytosol | — |
| GH79 | HPSE | Heparanase | α-Glc | Reg | X | — | Soluble | Lysosome/secretory | — |
| GH79 | HPSE2 | Inactive heparanase-2 | Inactive (heparin binding) (missing nucleophile)i | Reg | X | — | Soluble | Secretory | Urofacial syndrome 1 |
| GH84 | MGEA5 | Bifunctional protein NCOAT (OGA) | β-GlcNAc/glycoprotein | Non-reg | — | — | Soluble | Cytosol | — |
| GH85 | ENGASE | Endo-β-N-acetylglucosaminidase | β-GlcNAc/glycoprotein | Non-reg | — | — | Soluble | Cytosol | — |
| GH89 | NAGLU | α-N-acetylglucosaminidase | α-GlcNAc/glycoprotein/glycolipid | Non-reg | X | — | Soluble | Lysosome | MPS IIIB; Sanfilippo B |
| GH99 | MANEA | Endo-α-1,2-mannosidase | α-Man/glycoprotein processing | Non-reg | — | X | Type-II TMD | Golgi | — |
| GH99 | MANEAL | Endo-α-1,2-mannosidase-like protein | α-Man/glycoprotein processing | Non-reg | — | X | Type-II TMD | Golgi | — |
| GH116 | GBA2 | Nonlysosomal glucosylceramidase | β-Glc-Cer/glycolipid | Non-reg | — | — | Soluble | Cytosol | Spastic paraplegia 46 |
| GHnc | CHID1 | Chitinase domain-containing protein 1 | Most likely catalytically inactivei | Non-reg | X | — | Soluble | Secretory | — |
| — | AGA | N(4)-(beta-N-acetylglucosaminyl)-L-asparaginase | β-GlcNAc | Non-reg | X | — | Soluble | Lysosome | Aspartyl-glucosaminuria |
| — | NGLY1 | Peptide-N(4)-(N-acetyl-beta-glucosaminyl)asparagine amidase | β-GlcNAc | Non-reg | — | — | Soluble | Cytosol | CDDG |
aGene name according to HGNC.
bThe protein name according UniProt entry.
cThe sugar substrate for the individual GHs is manually curated from UniProt, CAZy and literature.
dSPs are predicted by SignalP, transmembrane domain structures are predicted by TMHMM, in both cases based on UniProt protein sequence isoform 1; for details, see Supplementary Table SI.
eDomain structures: type-II TMD and type-II TMDI represent single-pass type-I or type-II transmembrane proteins; MPM, multipass transmembrane proteins; GPI, GPI-anchored proteins; cytosolic soluble proteins are translated without a SP, organelle or secreted soluble proteins are translated with a cleavable SP.
fThe subcellular compartmentation is manually curated from UniProt, SignalP and TMHMM predictions and primary literature. Golgi, Golgi apparatus; ER, endoplasmic reticulum.
gCDGH, congenital disorder of GHs: MPS, mucopolysaccharidosis; GSD, glucagon storage diseases; CDG, congenital disorder of glycosylation; CDDG, congenital disorder of deglycosylation; for more details including gene, protein and OMIM IDs, see Supplementary Table SI.
hThe TMHMM browser predicts a type-II transmembrane domain, but NEU1 has experimentally been shown to be cleaved at position 48 and likely attached to the inner site of the lysosomal membrane by a C-terminal hydrophobic motif (Vinogradova et al. 1998; Bonardi et al. 2014). NEU2/3/4 have no predicted SP or TM; but NEU2 is described as cytosolic, NEU3 as membrane associated and NEU4 isoform 1 as membrane associated and isoform 2 as a lysosomal GH (Monti et al. 2000; Seyrantepe et al. 2004).
iInferred active or inactive GHs.
j SLC3A1 and SPACA5 are amino acid transporters evolved from ancient hydrolases.
k MYORG has been reported as a disease gene associated with Basal ganglia calcification, idiopathic, 7 (OMIM 618255) (Yao et al. 2018)
Although the majority of reported CDGHs affect the lysosome function, glycan hydrolysis also takes place in other intracellular compartments including the endoplasmic reticulum (ER), Golgi apparatus, cytosol and the extracellular space (Figure 1 and Table I). Congenital intestinal disorders caused by mutations in the extracellular GH genes are rare and generally cause mild clinical phenotypes, presumably because the endogenous GHs only provide a minor contribution to the hydrolytic capacity to degrade carbohydrates within the digestive tract (El Kaoutari et al. 2013). These CDGHs include congenital sucrase-isomaltase deficiency (CSID) caused by mutations in SI and congenital lactase deficiency caused by mutations in LCT.
Fig. 1.
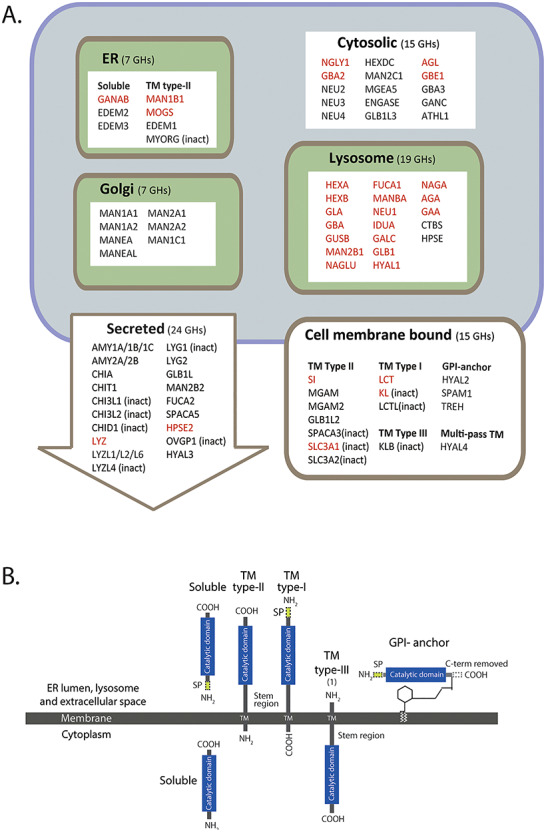
Overview of human GH genes classified in CAZy. (A) Classification of GHs according to predicted primary subcellular topology. GH genes known to be associated with a disorder are shown in red text and inactive (inact) GHs are denoted. (B) Schematic illustration of the different types of domain structures of GH proteins. The cytosolic GHs (15) are soluble and translated without a SP. The largest class of enzymes (45) are soluble proteins with a cleavable SP; other enzymes include type-I transmembrane proteins (3), type-II transmembrane proteins (18), one type-III transmembrane protein and one multipass membrane protein (not shown), in addition to GPI-anchored proteins (3). This figure is available in black and white in print and in color at Glycobiology online.
Studying a rather homogenous population allows for probing the nature and frequencies of known disease-causing mutations and obtains a global view of classes of related genes like the human GH genes. We previously probed the 208 human glycosyltransferase (GT) genes for functional deleterious mutant alleles in a homogenous population of 2000 Danes and reported rare known disease-causing alleles and importantly identified novel, albeit rare, null alleles in several GT genes without identified associated diseases (Hansen et al. 2015). Here, we analyzed the GH and GH-related genes for genetic variants using the same method and Danish whole-exome sequencing (WES) data and demonstrate that the allele frequencies (AFs) for known disease-causing variants in the GH genes were similar to those found in the ExAC and gnomAD European populations (Lek et al. 2016; Karczewski et al. 2019). We did find two mutations in the GH-related gene NGLY1 (p.Arg401* and p.Glu311Lys) with substantially higher AFs in the Danish cohort (2.8x and 33x, respectively) than in the European populations, which are classified as congenital disorders of deglycosylation (CDDGs) (OMIM 615273) (Enns et al. 2014). We identified and experimentally tested two novel predicted damaging nonsynonymous single-nucleotide variations (nsSNVs) in the sucrase-isomaltase (SI) gene, of which one had a 2.5× higher frequency in the Danish cohort. In general, the analyses of the Danish population revealed a higher percentage of reported pathogenic mutations in the GH genes compared to reported mutations in the GT genes (Hansen et al. 2015), and the majority of these pathogenic GH mutations were found in the LSD genes. Our study designated GlyMAP-II serves as a global view of the genetic variation in GH genes in a relatively small homogenous population of 2000 Danes.
Results
Human glycoside hydrolase genes analyzed
All human protein sequences related to GHs were collected from the CAZy database and represented 85 proteins belonging to 28 GH families. Although not encoding GHs, two additional genes, NGLY1 and AGA, were included so the total number of genes studied was 87 (Table I). NGLY1 and AGA both encode aspartylglucosaminidases that are involved in the degradation of N-linked glycans by cleaving of the β-aspartyl-glycosylamine linkage between the sugar moiety and the amide side chain of asparagine (Asn-β-GlcNAc). AGA is a lysosomal enzyme, and mutations in the gene results in the LSD aspartyl-glucosaminuria (Ikonen et al. 1991), and NGLY1 (N-glycanase 1) is a cytosolic enzyme that takes part in the endoplasmic reticulum-associated degradation (ERAD) quality control and deglycosylation of misfolded N-linked glycoproteins (Takahashi 1977), and mutations in NGLY1 cause CDDG (Lam et al. 2016). The majority (81/87) of the human GH genes encode for a protein with a single GH domain, one gene (AGL) encodes for a protein with two GH domains, and three genes (GBE1, CHIA and CHIA1) encode enzymes having both a GH domain and a carbohydrate-binding module (CBM). In the following, the 85 GH genes and AGA and NGLY1 will be referred to as the GH genes. The analysis of these 87 genes included only the annotated isofom-1 in UniProt and not alternative splice variants and protein isoforms. Disease-causing mutation are known for 30 of the 87 GH genes, and the term CDGH will be used for those known to underlie diseases, while GH genes without known cause of CDGHs represent genes without a monogenic disease association. The GH genes analyzed for signal peptide (SP), membrane-spanning domain(s) and glycophosphatidylinositol (GPI)-anchoring motifs employing public prediction servers (SignalP and TMHHM), gene databases and literature revealed that GH proteins are predicted to be targeted to six different subcellular compartments as shown in Figure 1A. The GHs include soluble proteins, single-pass type-I, type-II and type-III transmembrane proteins or GPI-anchored proteins (Figure 1B), and include 67 GHs with biochemically characterized GH activity, eight with inferred activities based the conservation of the catalytic residues, and 12 with sequence relatedness to GHs without known activities that may have developed other functions or lost their catalytic capability. The eight GHs with inferred hydrolytic activity and the 13 inactive or potentially inactive GHs are all cell membrane or secreted GHs and represent paralogs in the GH families GH1, GH13, GH18, GH22/GH23, GH31 and GH79 (Table I and Supplementary Table SI).
The group of 87 GHs comprises enzymes targeted to intracellular compartments (48), including the ER, Golgi, lysosome and the cytosol, and extracellular compartments (39) including the cell membrane and intercellular and extracellular space (Figure 1). The ER-located GHs are soluble or single-pass type-II proteins and include GANAB, MOGS and MAN1B1 involved in the initial trimming of the high-mannose N-glycan and the α-mannosidases like genes EDEM1-3 that recognize misfolded glycoproteins and target them for degradation and MYORG, a putative inactive glycosidase (Datta et al. 2009). The Golgi GHs are single-pass type-II α-mannosidases involved in trimming and processing of the immature high-mannose N-glycan; the lysosome GHs are soluble proteins with a cleavable SP responsible for degradation of all kinds of glycoconjugates including glycolipids and glycoproteins. The extracellular GHs comprise 15 cell membrane GHs that are predicted to represent single-pass membrane type-I, type-II and type-III, GPI-anchored and multipass membrane proteins. Nine are active or predicted active GHs involved in the degradation of dietary polysaccharides (SI, LCT, MGAM and TREH), and six are inactive GHs involved in amino acid transport (SLC3A1/A2) and calcium and phosphor homeostasis (KL) (Urakawa et al. 2006, Imura et al. 2007). The secreted GH genes encode for 17 biological active or inferred active GHs and 7 inactive GHs that all are soluble secreted proteins with a SP. The active GHs include polysaccharide degrading amylases (AMY1A/1B/1C/2A/2B), chitinases (CHIA/T1) and lysozyme and lysozyme-like GHs (LYZ/L1/L2/L6, LYG2), and the inactive GHs include members of the chitinase and lysozyme families (Table I and Supplementary Table SII).
Assembly of nsSNVs for GH genes in the LuCamp exome dataset
The LuCamp cohort comprises 1965 individuals representing 983 healthy individuals and 982 individuals diagnosed with type 2 diabetes, overweight and hypertension, but otherwise healthy (Albrechtsen et al. 2013, Lohmueller et al. 2013). Analysis of the LuCamp cohort (Lohmueller et al. 2013) has shown that no significant gene association with the disease risk could be shown, and we considered the LuCamp population as 1965 healthy individuals in the subsequent single-nucleotide variation (SNV) analyses. The deep exome data contained only 83 of the 87 GH genes that SNVs for the genes AMY1A/1C/1C and MGAM2 were missing likely due to the design of the WES capture protocol (Li et al. 2010). We applied the same strategy as previously used with the GT genes (Hansen et al. 2015) for analyses of the GH genes with the aim to catalog predictable damaging nsSNVs. The strategy included assembly of the GH nsSNV data (Phase 1); collecting nsSNVs with three or more alleles (AF > 0.001); prediction of damaging nsSNVs employing the servers PolyPhen, Provean and SIFT (Phase 2); and finally experimental validation of selected predicted damaging nsSNVs (Phase 3) (Figure 2).
Fig. 2.

The selection process for finding putative damaging nsSNVs included three phases: Phase 1, assembly an SNV database for the 87 human GHs from the LuCamp and the ExAC/gnomAD public database; Phase 2, filtering the SNV database for potentially damaging nsSNVs using knowledge-based predictions and Phase 3, experimental validation of selected damaging nsSNVs in two GH genes.
Phase 1
A total of 4058 SNVs (Figures 2 and 3 and Table II) were identified in the 83 GH genes analyzed, of these were 57% intergenic, intronic and UTR SNVs, 15% were synonymous SNVs (sSNVs), and 28% (1144) were nsSNVs in the coding regions. The SNV distribution was similar to what we found for the GT genes (Figure 3A) (Hansen et al. 2015). The majority of the nsSNVs (66%) had AF < 0.001 as singletons or doubletons (Figure 3B). These rare nsSNVs were not included in the Phase 2 analysis as they were only found in one or few families. The remaining 34% of the nsSNVs with ≥3 alleles were analyzed for predictable damaging effect on protein function.
Fig. 3.

Global analysis of SNVs in GH genes from WES data from 1965 Danes. (A) Distribution of GH SNVs in intergenic, intronic and exonic regions of GH genes with comparative data from our previous analysis of SNVs in GT genes. (B) Distribution of AF values for SNVs, nsSNVs and predicted damaging nsSNVs in the GH genes. (C) Illustrates the Phase 2 filtration of data.
Table II.
SNVs in the GH and GT genes
| GH (83 genes)a | GT (204 genes)b | |
|---|---|---|
| Total number of SNVs, sSNVs and nsSNVs | 4058 | 6582 |
| nsSNVs | 1144 | 1875 |
| nsSNVs ≥3 alleles | 383 | 553 |
| Phase 2 nsSNVs ≥3 alleles, predicted damaging | ||
| nsSNVs in nondisease genes | 76 | 34 |
| nsSNVs in disease genes, not reported | 29 | 18 |
| In total | 105 | 52 |
| Reported mutations (HGMD) | 61 | 19 |
aNo LuCamp WES data for AMY1A/1B/1C, MGAM2; no LuCamp WES CDS data for AMY2A/2B, KIAA1161 and SPACA5, TREH.
bData from GlyMAP-I (Hansen et al. 2015).
Phase 2
Out of the 1144 nsSNVs in the LuCamp dataset 383 had at minimum of three alleles, and a total of 105 nsSNVs were predicted to be damaging applying the Phase II prediction servers PolyPhen-2, Provean and SIFT. These 105 nsSNVs included 76 nsSNVs in GH genes without known cause of CDGHs and 29 novel nsSNVs in known CDGH genes (Figure 3 and Supplementary Tables SII–SIV).
The 76 nsSNVs in the GH genes not implicated in CDGHs represented 23 with an AF > 0.01 and 53 with an AF < 0.01. Variant with AF values above 0.01 are generally considered as common a single nucleotide polymorphisms (SNPs), and the 23 nsSNVs with AF > 0.01 were considered as common SNPs and not as potential deleterious variants. Three of these common SNPs found in CHIA and CHIT1 have been reported associated with increased enzymatic activity, asthma and colorectal cancer (Supplementary Table SII). The remaining 53 nsSNVs with AF < 0.01 comprised 19 nsSNVs in the secreted GHs, 12 in the cell membrane GHs, 16 nsSNVs in the cytosolic GHs, three in the lysosome GHs and three nsSNVs in the Golgi GHs and included nine stop-gain nsSNVs, one stop-loss nsSNV, two splice site nsSNVs and 41 amino acid substitutions (Supplementary Table SII). Comparison of the AF values for all 76 Phase 2 nsSNVs in the GH genes without known cause of CDGHs with AF values in the ExAC and gnomAD European population revealed similar AFs, except for eight nsSNVs that had AF values that were 5× higher, one nsSNV that had an AF value 5× lower and seven nsSNVs that were absent in the European population. The nsSNVs with higher AF values or were absent in the European populations are putative candidates for being Danish or Scandinavian founder variants (Supplementary Table SII).
The 29 novel predicted damaging nsSNVs were found in 19 of the 30 CDGH genes. Nine of these nsSNVs had AF > 0.01, suggesting these are common SNPs, and they were therefore excluded as potential pathogenic mutations. The remaining 21 nsSNVs represented nine lysosomal CDGH genes (GUSB, HEXA/B, FUCA1, GAA, MAN2B1, IDUA and GALC), four cytosolic CDGH genes (GBE1, AGL, GBA2 and NGLY1), one ER/Golgi CDGH gene (GANAB), two cell membrane CDGH genes (SI and SLC3A1) and one secretory CDGH gene (HPSE2). All 29 novel Phase 2 nsSNVs in the CDGH genes were amino acid substitutions, and 25 had AF values similar to the ExAC and gnomAD European population, two had >5× higher AF values, three had >10× higher AF values, and one was absent (Supplementary Table SIII).
We found 61 nsSNVs in the LuCamp dataset that were annotated as mutations in HGMD. These represented 42 nsSNVs in 13 lysosomal LSD genes, six nsSNVs in three cytosolic CDGH genes, 12 nsSNVs in two cell membrane CDGH genes SI (three nsSNVs) and SLC3A1 (eight nsSNVs) and one nsSNV in a secreted CDGH gene (LYZ). Eight of the 61 nsSNVs had an AF > 0.01 in the LuCamp, the ExAC and gnomAD European populations suggesting these as common SNPs likely misclassified as mutations in the genes GBA, GLB1, HEXB, NAGA, AGL and LYZ (Supplementary Table SIV).
The annotated mutations in the LSD CDGH genes represented six in GBA associated with Gaucher disease and two associated with Parkinson disease, seven in HEXA associated with Tay–Sachs disease, five in GLB1 associated with GM1 gangliosidosis and four in HEXB associated with Sandhoff disease. The remaining annotated mutations found in the LSD CDGH genes were in AGA (2), FUCA1 (1), GLA (4), GUSB (1), GAA (2), MAN2B1 (2), MANBA (1), NAGA (3) and NAGLU (2) in the cytosolic CDGH genes AGL (three nsSNVs) and GBE1 (one nsSNV) associated with glycogen storage disease 3 or 4, in NGLY1 (two nsSNVs) associated with CDDG, in the cell membrane CDGH genes SI (three nsSNVs) and SLC3A1 (eight nsSNVs), and in the secreted CDGH gene LYZ (one nsSNV). Comparison of the AF values in the LuCamp population revealed that 42 of the annotated mutations had AF values similar to the ExAC and gnomAD European population, 12 had AF values 5× higher, one had an AF values 5× lower, and five were absent in the ExAC and gnomAD European population (Supplementary Table SIV).
Phase 3
The Phase 2 filtration process resulted in one nsSNV in NGLY1 and two in SI that were selected for experimental validation.
NGLY1
The LuCamp data included 13 nsSNVs in NGLY1, whereof 11 were predicted to be damaging. Two nsSNVs were reported pathogenic; p.Glu311Lys (He et al. 2015) with an AF value 34× higher than in ExAC and gnomAD and p.Arg401* (Enns et al. 2014) with an AF 2.8× higher than in the European population in ExAC and gnomAD. The Phase 2 filtration predicted p.Gly456Arg to be damaging having three alleles in the LuCamp population and an AF of 7.6e-4, which is 12× higher compared to the European population in ExAC and gnomAD (Figure 4A and Supplementary Tables SIII and SIV). The p.Gly456Arg nsSNV was experimentally validated employing a previously described method (He et al. 2015). Briefly, this method is based on intracellular ERAD sorting of misfolded glycoproteins and a Venus fluorescent protein assay for measuring N-glycan deglycosylation in the cytosol (Grotzke et al. 2013). The Venus protein assay showed that the p.Gly456Arg variant exhibits similar activity as the wild-type (WT) protein, and the mutation was therefore considered benign.
Fig. 4.
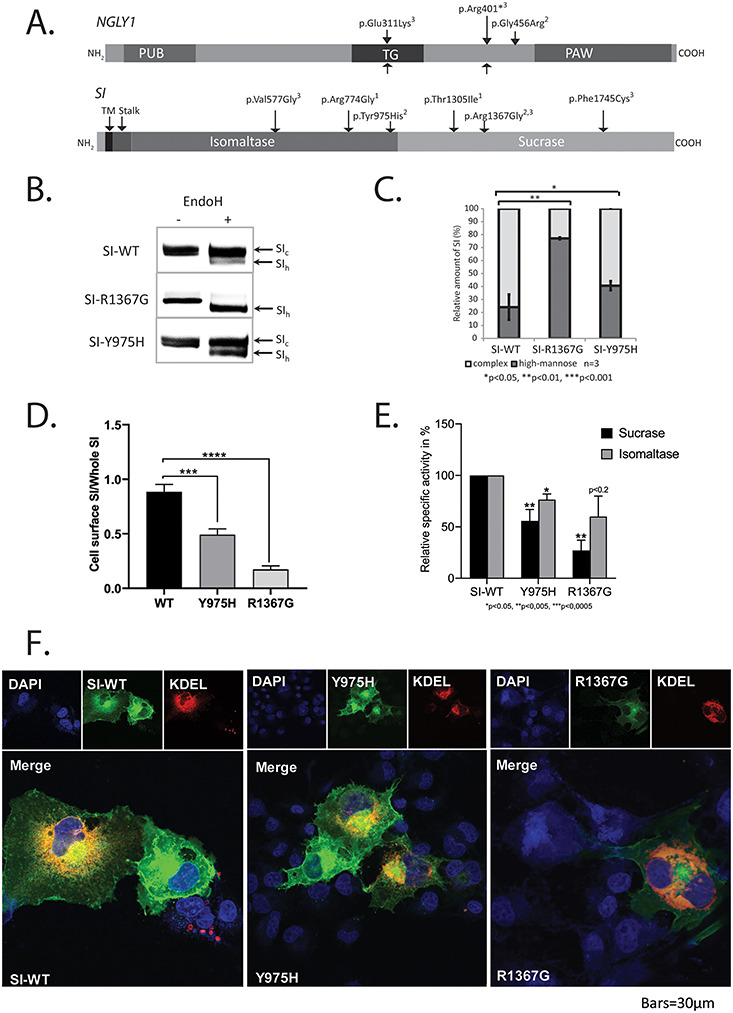
Experimental validation of predicted damaging nsSNVs in the NGLY1 and SI genes. (A) Graphic presentation of the NGLY1 and SI genes showing the Phase 2 predicted damaging nsSNVs1, the experimental validated nsSNVs2 and the reported mutations3. (B) The intracellular sorting of the SI mutant and wild-type (WT) proteins was analyzed by the glycosylation status in COS-1 cells. After transient expression of mutant constructs SI-R1367G and SI-Y975H and WT controls, SI was immunoprecipitated from cell extracts 48 h posttransfection and treated with endo H and analyzed by 6% SDS-PAGE and western blotting showing the complex glycosylated (SIc) and the endo H-digested form of mannose-rich (SIh). (C) Graphic presentation of the western blot band intensities of SIh/SIc of mutant and WT SI as means of ± SD of three independent experiments. (D) WT SI and mutants transiently expressed in COS-1 cells were posttransfection biotinylated and after cells lysis immunoprecipitated with anti-SI antibodies and analyzed by SDS-PAGE and WB. The protein bands in the first sample were visualized by streptavidin-HRP and corresponded to the cell-surface-localized WT SI or SI mutants; the proteins in the second sample were visualized by anti-SI antibodies (primary) and anti-IgG (secondary) and corresponded to total cellular SI. Cell-surface WT and mutant SI was quantified vs. total cellular SI (WT SI was set as 100% (n ≥ 3), data for SI-Y975H is adapted from Husein and Naim 2019). (E) Graphic presentation of the specific sucrase and isomaltase activities of the SI mutant and WT proteins. Immunoprecipitated SI from transfected COS-1 cells was assayed for enzymatic activity by colorimetric measurement of released glucose after incubation with either sucrose or isomaltose as a substrate, and the activities were normalized relative to SI amounts detected by immunoblotting (Supplementary Table SVI). Results are means of ± SD of three independent experiments. (F) Intracellular localization of the SI mutants and the WT proteins. WT and mutant SI were transiently expressed in COS-1 cells together with a protein carrying the ER retention signal Lys Asp Glu Leu (KDEL) coupled to ds Red. The cells were grown on coverslips, fixed, prepared for indirect immunofluorescence using anti-SI antibodies and analyzed by confocal microscopy (blue (DAPI), nuclei; red, KDEL-ds Red as an ER marker; green, SI). Representative fluorescence micrographs of the intracellular localization of SI WT and mutant proteins and KDEL-ds Red are shown, as well as merged images with staining of nuclei. PM, plasma membrane; G, Golgi apparatus; ER, endoplasmic reticulum; N, nuclei. This figure is available in black and white in print and in color at Glycobiology online.
SI
A total of 41 nsSNVs were found in the LuCamp dataset; six of these fulfilled the Phase 2 criteria and included three reported and three novel nsSNVs (Figure 4A and Supplementary Tables SIII and SIV). One reported nsSNV, p.Arg1367Gly, had not been functionally validated and therefore selected for experimental validation together with p.Tyr975His. Both nsSNVs were nonconservative substitutions of phylogenetically conserved amino acids and were predicted to affect catalytic domains of the isomaltase (p.Tyr975His) and the sucrase (p.Arg1367Gly) (Figure 4A). The nsSNVs were tested by transient expression of full coding length mutant and WT SI proteins in COS-1 cells followed by immunoprecipitation, endo H treatment and western blot analysis (Figure 4B and C). The p.Tyr975His SI mutant protein has been recently analyzed and revealed biosynthetic forms similar to their WT SI counterparts (Husein and Naim 2019). Both the p.Tyr975His SI mutant and the WT SI are similarly N-glycosylated and contained both endo H-sensitive mannose-rich glycosylated species and endo H-resistant mature complex N-glycosylated forms compatible with normal intracellular processing and trafficking. By contrast, the majority of the p.Arg1367Gly SI mutant protein revealed the endo H-sensitive mannose-rich glycosylated form (Figure 4B and C), suggesting that this mutant is retained in the ER. Utilizing cell-surface biotinylation showed that almost 50% of p.Tyr975His SI (Husein and Naim 2019) and almost 80% of p.Arg1367Gly SI (new data) (Figure 4D) localized to the cell surface as compared to the WT counterparts, indicating that despite the normal intracellular processing of these mutants, substantial proportions are retained intracellularly. The biochemical data were supported by analyses of the intracellular distribution of the mutants by confocal microscopy (Figure 4F), which revealed WT SI and p.Tyr975His SI at the cell surface in fluorescein isothiocyanate (FITC)-labeled structures or in the ER co-localizing with a ds Red conjugated protein carrying the ER retention signal, Lys Asp Glu Leu (KDEL) (Figure 4F). On the other hand, the p.Arg1367Gly SI mutant was found mainly in the ER co-localizing with KDEL-ds Red (Figure 4F). Analysis of the enzymatic function of the SI mutant proteins demonstrated that the mutations p.Arg1367Gly and p.Tyr975His elicited an overall reduction in sucrase and isomaltase activities to variable extent, with the p.Arg1367Gly mutation most severely affected the sucrase activity (Figure 4E and Supplementary Table SVI), overall showing that these SI mutations are hypomorphic.
Known disease-causing mutations
Manual examination of HGMD revealed 4447 annotated mutations in the 30 CDGH genes, and by comparison 2272 annotated mutations in the 57 congenital disorders of glycosylation (CDG) GT genes (excluding the blood group genes) (Figure 5). These mutations included missense, stop-gain, stop-loss, splice site mutations, indels, large rearrangements, inversions, duplications and deletions. Almost half of the CDGH genes (12/30) had 100 or more mutations annotated, 11 CDGH genes had 20–100 mutations annotated, six had 5–20 mutations, and one gene had less than five mutations annotated (Figure 5A and Supplementary Table SV). A majority of the CDGH genes with 100 or more mutations represented LSD genes, and two CDGH genes in the cytosol (AGL and GBE1, both glycogen storage disease) and one inactive cell membrane GH (SLC3A1 associated with cystinuria) had more than 50 mutations annotated (Supplementary Table SV). Comparing these findings to our previous study of GTs with updated numbers of annotated mutations (Feb 2019) (Hansen et al. 2015), only five CDG GT genes had more than 100 annotated mutations, 15 had 20–100, 26 had between 5 and 20, and 13 had less than 5 annotated mutations (Figure 5A). The five GTs with 100 or more annotated mutations were found in FKRP in the O-Man POMT glycosylation pathway, in EXT1 and EXT2 in the O-Xyl pathway for glycosaminoglycan (GAG) biosynthesis and in two GT genes with other functions (PYGM and UGT1A1). The present analysis revealed substantially more annotated pathogenic mutations in CDGH genes than in CDG GT genes, which suggests that the phenotypic consequences of defects in the CDGH genes are less severe and mutations are maintained in the population.
Fig. 5.

Graphic presentation of number of annotated mutations in HGMD (Feb 2019) and annotated mutations in the LuCamp population for the CDGH and the CDG genes. (A) Number of genes with [1:5], [5–20], [20–100] and [100,] annotated mutations in HGMD and number of mutations annotated in HGMD and number of annotated mutations found in the LuCamp population (B) for the CDGH genes and (C) for the CDG GT genes.
We did not identify and validate any obvious deleterious nsSNVs in GHs genes not already assigned to CDGHs, similar to what we previously did with two galactosaminyltransferases (GALNTs) in the GlyMAP-I study (Hansen et al. 2015), but the rare nsSNVs identified may serve as a discovery base for future studies.
Discussion
The GlyMAP-II study presents a catalog of human GH genes and an analysis of genetic variations in a homogenous population of almost 2000 Danes. We surveyed the known human GHs and summarized predicted subcellular localizations, substrate specificities and biological functions, which illustrate that this is a more diversified class of enzymes compared to GTs with greater variation in domain structures, synthesis and intracellular trafficking, membrane retention mode and cytolocalization (Figure 1A). The GlyMAP-II study applied the same three-phase approach to analysis of genetic variations using the same WES data as our previous GlyMAP-I study for human GT genes (Figure 2) (Hansen et al. 2015), and as predicted from the higher prevalence of congenital disorders associated with GHs compared to GT genes, we found higher prevalence of known deleterious GH gene allele variants in particular for the lysosomal GHs in the study population. The study indicates that mutations in GH genes as an enzyme class may be better tolerated than mutations in GT genes. Many of the known disease-causing GT genes (currently 59) display severe phenotypes due to global effects on glycosylation pathways (Freeze et al. 2017; Ng and Freeze 2018; Jaeken and Péanne 2017; Hansen et al. 2015); however, more recently GT genes that underlie more subtle phenotypes are emerging, and these often represent GT genes that encode members of larger isoenzyme families with predicted functional redundancies (Joshi et al. 2018). Similarly, experiences with GT gene knockout studies in mice suggest that many GT genes are essential for development and often result in embryonic lethality (Stanley 2016), while knockout of some GT genes and in particular members of isoenzyme families produce more subtle or no apparent phenotype (Lowe and Mart 2003; Mizumoto et al. 2014; Furukawa et al. 2017). Moreover, in agreement with our previous analysis of GT genes, the study shows that the widely used prediction algorithms (PolyPhen-2, Provean and SIFT) performed poorly with GHs.
The prevalence of LSDs is estimated to range from 7.5 to 23.5 per 100,000 live births depending on the population (Kingma et al. 2015), whereas mutations in the GT genes are so rare that the incidence for most of the CDGs are unknown, but suggested to approach 0.1–0.5 per 100,000 among Europeans (Jaeken et al. 2001, Péanne et al. 2018). Our analysis of the LuCamp WES data revealed a similar distribution of genetic variants of GH and GT genes with 28/29% of the variants being nsSNVs in the coding regions (Figure 3), and the two gene sets exhibited similar distributions of AFs with 67%/71% of the nsSNVs being rare variants (singletons or doubletons) with AF values < 0.001. The latter corresponds to what is found in larger populations including the ExAC and the gnomAD databases (Li et al. 2010, Fu et al. 2013, Lappalainen et al. 2019, Karczewski et al. 2019). The Phase 2 analyses of the nsSNVs in GH genes resulted in twice as many predicted damaging nsSNVs in the 83 GH genes compared to what we found previously for the 204 GT genes (Hansen et al. 2015), and the number of predicted damaging nsSNVs (≥3 alleles) was also substantially higher for all GHs including those without known disease-causing mutations (Table II). DNA variants with AF values > 0.01 are in general considered as common polymorphisms (Lappalainen et al. 2019), and about 1/3 of the predicted damaging nsSNVs in GHs were above this threshold and therefore likely not deleterious mutations. This points to weaknesses of bioinformatic prediction tools, as also discussed in our previous study of GTs where we selected 14 predicted damaging nsSNVs in GALNT genes for experimental validation and found that only two were actually inactive (Hansen et al. 2015).
The WES data contained 61 previously reported mutations in 19 of the 30 GH genes underlying CDGHs (Figure 5B), while we only found 19 reported mutations in 9 of the 59 GT genes causing CDGs (Figure 5C) (Hansen et al. 2015). Interestingly, eight of these CDGH mutations and four of the CDG mutations had AF > 0.01 (Supplementary Tables SIV and SVII) and thus represent relatively common variants. Potential misclassification of reported disease-causing mutations was recently raised as a concern (Shah et al. 2018), because analyses of WES data identified mutations with AFs that were implausible compared to the incidence of the disorder, and upon further review the authors found insufficient documentation for the pathogenicity of some of the reported mutations. We therefore reexamined the eight identified GH mutations with AF > 0.01 and found that six of these were shown to represent tolerant polymorphisms and the pathogenicity of the residual two was uncertain (Supplementary Table SIV). These eight mutations included variants in the lysosomal genes GBA, GLB1, HEXB and NAGA. The four reported mutations in the GT genes with AF > 0.01 were found in the ALG6, POMGNT1 and FKTN genes and reported to be associated with a milder phenotype or for one of the ALG6 mutations as a modifying variant (Supplementary Table SVII).
Our Phase 3 experimental validation of selected predicted damaging nsSNVs uncovered two hypomorphic mutations and one nsSNVs without functional consequence. The hypomorphic nsSNVs found in the SI gene included one nsSNV that affected the activity of the isomaltase domain and one nsSNV in the sucrase domain without functional consequence for the sucrase activity, but both nsSNVs were shown to cause defect protein sorting. Experimental validation of the NGLY1 nsSNV (p.Gly456Arg) showed it was functionally active and hence likely represented a polymorphism. While we only identified and validated one novel damaging nsSNVs here with the limited efforts devoted to Phase 3, the GlyMAP approach offers insight into pathogenic heritable mutations harbored in a defined population. Although there are limitations in bioinformatic tools for predicting damaging nsSNVs (Hansen et al. 2015) and the number of nsSNVs that practically can be experimental validated, the GlyMAP provides an overview of both known pathogenic mutations and predicted damaging putative pathogenic mutations that can be found in the Danish population. We also compared the AF values for the Phase 2 GH nsSNVs to the ExAC and gnomAD dataset for the European populations. Approximately 75% of all the Phase 2 nsSNVs had AF values similar to those found in the ExAC and gnomAD European population within a 5× difference margin. The remaining 25% of the LuCamp nsSNVs had AF values that were substantially higher or completely absent in the ExAC and gnomAD data. This is in agreement with the common ancestry of the LuCamp population and the ExAC and gnomAD European populations and further suggests the existence of nsSNVs with specific Nordic founder origin. This includes the validated NGLY1 nsSNV p.Gly456Arg with an AF value 10× higher than in the ExAC and gnomAD European population and the reported NGLY1 mutation p.Glu311Lys with an AF value 34× higher than in the ExAC and gnomAD European population, suggesting these as Scandinavian founder mutation.
The 61 previously reported pathogenic mutations in GH genes found in the LuCamp population comprised 42 mutations in 13 of the 17 LSD CDGH genes. The substantial number of mutations in the lysosomal CDGH genes is in line with lysosomal GHs representing the majority of CDGHs with higher incidences as found for Gaucher (GBA) and Fabry (GLA) diseases. The majority of the annotated mutations in the 30 CDGH genes (3690 out of 4437) were found in the LSD GH genes with GLA and GBA as the two top scores.
The GH group of genes includes 12 genes encoding proteins that appear to have lost their catalytic active residues. These inactive GH-like proteins are all targeted to the extracellular space and include the cell membrane genes KL, KLB, LCTL (CAZy family GH1), SLC3A1/A2 (CAZy family GH13), the secreted chitinases CHI3L1/L2 (CAZy family GH18) and the lysozyme LYZL4 gene (CAZy family GH22). Two of these genes have developed new functions; KL as a part of the calcium and phosphate homeostasis where it interacts with FGF23 and a mutation reported in a case with tumoral calcinosis HFTC3 (Ichikawa et al. 2010, OMIM 604824). SLC3A1 has developed into an amino acid transporter with more than 200 mutation reported in combination with cystinuria (Calonge et al. 1994, OMIM 104614) (Table I).
Interestingly, surveying the expression patterns of the 85 GHs using RNAseq data from 32 different tissues (Human Protein Atlas, Uhlén et al. 2015) revealed that these 12 extracellular GHs exhibit varying degree of tissue-specific expression patterns (Supplementary Figure S2). Applying a defined τ-value for analysis of the RNAseq data (Joshi et al. 2018), we classified the GH genes as regulated for τ > 0.82 and not-regulated for τ < 0.82 (Supplementary Figure S1) and found that 37 GH genes, primarily cell membrane and secreted GHs, were predicted to be under high transcriptional regulation, while the residual 50 GH genes, primarily intracellular GH genes, were not substantially influenced by transcriptional regulation (Table I and Supplementary Figure S2). This suggests that intracellular GHs are generally ubiquitously expressed, whereas the cell membrane and the secreted GHs are under regulation and expressed in a tissue-specific manner, for example, as the GHs in the gastrointestinal tract participating in the degradation of dietary polysaccharides.
In summary, the GlyMAP-II study presents a global view of the large class of GH genes classified in the CAZy database with diverse functions in hydrolysis of carbohydrates and other substrates and defines the genetic variations found in a small homogenous population of almost 2000 Danes. Analysis of the LuCamp WES data revealed that ~5% of the GH nsSNVs and ~1% of the GT nsSNVs represented known disease-associated mutations. Our analysis revealed a higher incidence of known deleterious mutations in the GH genes compared to the GT genes, which was related to concentration in the lysosomal GH genes. The results serve as a discovery platform for evaluation and discovery of novel damaging nsSNVs in CDGHs.
Methods and materials
The human GH proteome
The human GH protein sequences were extracted and manually curated from the CAZy database (CAZy.org), and the UniProt and GenBank annotations were retrieved for the resulting proteins. Two additional genes, AGA and NGLY1, involved in degradation of N-linked glycans by removing the GlcNAc residue bound to the peptide Asn residue were added to the GHs. Table I with Supplementary Table SII list HGNC gene and protein names, GenBank and UniProt IDs, length of longest protein isoform, primary sugar substrate specificities, data for subcellular localization and when described, disease disorders.
GH WES data and prediction of damaging variants
The WES data (Lohmueller et al. 2013) were obtained from The Lundbeck Foundation Centre for Applied Medical Genomics in Personalized Disease Prediction, Prevention and Care (LuCamp,www.lucamp.org) and mined for genetic variants and AF values for the 87 GH genes. AF values for the European population were downloaded from the ExAC browser/Exome Aggregation Consortium (Lek et al. 2016) and the gnomAD browser (Karczewski et al. 2019). The LuCamp WES data included single-nucleotide variants, whereas indels were not included in the dataset. Standard nomenclature for nucleotide and protein variants follows the recommendation for sequence variant nomenclature (http://varnomen.hgvs.org/).
Phase 2 prediction of damaging nsSNVs was done employing the WEB-based algorithms PolyPhen-2 (pph2) (Adzhubei et al. 2010), SIFT (Kumar et al. 2009) and Provean (Choi et al. 2012), as previously described for the GT GlyMAP (Hansen et al. 2015). A predicted damaging score of 0–4 was calculated for each nsSNV by adding scores from the three prediction servers. For PolyPhen-2 “benign” = 0, “possibly damaging” = 1, “probably damaging” = 2; for SIFT and Provean “tolerated/neutral” = 0 and “damaging/deleterious” = 1. A total score of 3 or 4 was considered damaging. Stop-gain or stop-loss nsSNV and splice site SNV were considered as damaging and given a score of 4.
Experimental validation of SI and NGLY1 mutants
SI
Experimental validation of SI nsSNVs was performed for the two selected mutants and the WT gene introduced into the cDNA of SI in an SI-pSG8 vector by site-directed mutagenesis PCR using the oligonucleotides p.Arg1367Gly forward: 5′-GCTTTCCCAGATTTCTTCGGGACTTCCACGGCCGAGTGGTGGGCCAG-3′ and reverse: 5′-CTGGC CCACCACTCGGCCGTGGAAGTCCCGAAGAAAATCTGGGAAAGC-3′ and p.Tyr975His forward: 5′-CTCTATCCAAGGCGCCTGAGTGTCACTTTCCCAGACAAGATAACTC-3′ and reverse: 5′-GAGTTATCTTGTCTGGGAAAGTGACACTCAGGCGCCTTGGATAGAG-3′. Analysis of the glycosylation status was performed by transient expression of the SI mutants and WT in COS-1 cells followed by immunoprecipitation using mAb anti-SI from detergent extracts of the transfected cells 48 h posttransfection (Alfalah et al. 2009). The western blot band intensities were subsequently quantified by Quantity One 1-D Analysis Software (Bio-Rad Laboratories, Feldkirchen, Bavaria, Germany), and activities of mutant and WT proteins were performed in triplicates as previously described (Alfalah et al. 2009). Fluorescence images were generated in a Leica SP5 confocal microscope using anti-SI antibodies (primary) and goat anti-mouse IgG (secondary) carrying DyLight 488. KDEL-ds Red was the ER marker. Experimental validation of the NGLY1 p.Gly456Arg allele was carried out using the Venus assay according to He et al. (2015).
Supplementary Material
{kind=link}
Acknowledgements
The Novo Nordisk Foundation Center for Basic Metabolic Research is an independent Research Center at the University of Copenhagen partially funded by an unrestricted donation from the Novo Nordisk Foundation (www.metabol.ku.dk).
Conflict of interest statement
None declared.
Funding
This work was supported by Læge Sofus Carl Emil Friis og hustru Olga Doris Friis’ Legat (H.C.); The Danish National Research Foundation (grant number DNRF107); The Lundbeck Foundation (grant numbers R223-2016-563 and R317-2019-225); the National Institutes of Health (grant number R01 DK099551 to H.H.F.); the Bertrand Might Research Foundation (to H.H.F); QOL Medical LLC, Vero Beach, Florida, USA (H.Y.N) and The Lundbeck Foundation that funded The Lundbeck Foundation Centre for Applied Medical Genomics in Personalized Disease Prediction, Prevention and Care (LuCamp, www.lucamp.org).
Web sources
CAZy, Carbohydrate-Active enZYmes Database (www.cazy.org).
ExAC Browser (Beta) (http://exac.broadinstitute.org/about).
gnomAD (http://gnomad-beta.broadinstitute.org/).
HGMD, The Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/).
Human Protein Atlas (https://www.proteinatlas.org/).
RNAseq data downloaded from EMBL-EBI Expression Atlas (https://www.ebi.ac.uk/gxa/home).
LuCamp (http://www.LuCamp.org).
PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/).
Provean (http://provean.jcvi.org/index.php).
SIFT (http://sift.jcvi.org/).
SignalP (http://www.cbs.dtu.dk/services/SignalP/).
TMHMM (http://www.cbs.dtu.dk/services/TMHMM/).
UniProt (http://www.uniprot.org/).
Abbreviations
AF, allele frequency; CAZy, carbohydrate-active enzymes; CBM, carbohydrate-binding module; CDDGs, congenital disorders of deglycosylation; CDG, congenital disorders of glycosylation; CDGH, congenital disorders of glycoside hydrolase; CSID, congenital sucrase-isomaltase deficiency; ER, endoplasmic reticulum; ERT, enzyme-replacement therapy; FITC, fluorescein isothiocyanate; GAG, glycosaminoglycan; GALNT, galactosaminyltransferase; GH, glycoside hydrolase; GPI, glycophosphatidylinositol; GT, glycosyltransferase; HGMD, Human Gene Mutation Database; LSD, lysosomal storage disease; nsSNV, nonsynonymous single-nucleotide variation; SI, sucrase-isomaltase; SNP, single-nucleotide polymorphism; SNV, single-nucleotide variation; SP, signal peptide; sSNV, synonymous single-nucleotide variation; WT, wild type.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Meth. 7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfalah M, Keiser M, Leeb T, Zimmer KP, Naim NY. 2009. Compound heterozygous mutations affect protein folding and function in patients with congenital sucrase-isomaltase deficiency. Gastroenterology. 136:883–892. [DOI] [PubMed] [Google Scholar]
- Albrechtsen A, Grarup N, Li Y, Sparsø T, Tian G, Cao H, Jiang T, Kim SY, Korneliussen T, Li Q et al. 2013. Exome sequencing-driven discovery of coding polymorphisms associated with common metabolic phenotypes. Diabetologia. 56:298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvio M, Mononen I. 2016. Aspartylglycosaminuria: A review. Orphanet J Rare Dis. 11:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahr BA, Bendiske J. 2002. The neuropathogenic contributions of lysosomal dysfunction. J Neurochem. 83:481–489. [DOI] [PubMed] [Google Scholar]
- Bonardi D, Ravasio V, Borsani G, d'Azzo A, Bresciani R, Monti E, Giacopuzzi E. 2014. In silico identification of new putative pathogenic variants in the NEU1 sialidase gene affecting enzyme function and subcellular localization. PLoS One. 9:e104229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady RO, Schiffmann R. 2000. Clinical features of and recent advances in therapy for Fabry disease. JAMA. 284:2771–2775. [DOI] [PubMed] [Google Scholar]
- Calonge MJ, Gasparini P, Chillaron J, Chillon M, Gallucci M, Rousaud F, Zelante L, Testar X, Dallapiccola B, Di Silverio F et al. 1994. Cystinuria caused by mutations in rBAT, a gene involved in the transport of cystine. Nat Genet. 6:420–425. [DOI] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. 2012. Predicting the functional effect of amino acid substitutions and Indels. PLoS One. 7:e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta K, Guan T, Gerace L. 2009. NET37, a nuclear envelope transmembrane protein with glycosidase homology, is involved in myoblast differentiation. J Biol Chem. 284:29666–29676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. 2013. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol. 11:497–504. [DOI] [PubMed] [Google Scholar]
- Enns GM, Shashi V, Bainbridge M, Gambello MJ, Zahir FR, Bast T, Crimian R, Schoch K, Platt J, Cox R et al. 2014. Mutations in NGLY1 cause an inherited disorder of the endoplasmic reticulum-associated degradation pathway. Genet Med. 16:751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeze HH, Kinoshita T, Schnaar RL. 2017. Genetic disorders of glycan degradation In: Varki A, editor. Executive Editor.Essentials of Glycobiology. Chapter 44 3rd ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory Press. [Google Scholar]
- Fu W, O'Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, Gabriel S, Rieder MJ, Altshuler D, Shendure J et al. 2013. NHLBI exome sequencing project, Akey JM. 2012. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 493:216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K, Ohmi Y, Ji S, Zhang P, Bhuiyan RH, Ohkawa Y, Tajima O, Hashimoto N, Furukawa K. 2017. Glycolipids: Essential regulator of neuro-inflammation, metabolism and gliomagenesis. Biochim Biophys Acta Gen Subj. 1861:2479–2484. [DOI] [PubMed] [Google Scholar]
- Futerman AH, Meer G. 2004. The cell biology of lysosomal storage disorders. Nat Rev Mol Cell Biol. 5:554–565. [DOI] [PubMed] [Google Scholar]
- Grotzke JE, Lu Q, Cresswell P. 2013. Deglycosylation-dependent fluorescent proteins provide unique tools for the study of ER-associated degradation. Proc Natl Acad Sci U S A. 110:3393–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritti A. 2011. Gene therapy for lysosomal storage disorders. Expert Opin Biol Ther. 11:1153–1167. [DOI] [PubMed] [Google Scholar]
- Hansen L, Lind-Thomsen A, Joshi HJ, Pedersen NB, Have CT, Kong Y, Wang S, Sparso T, Grarup N, Vester-Christensen MB et al. 2015. A glycogene mutation map for discovery of diseases of glycosylation. Glycobiology. 25:211–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P, Grotzke JE, Ng BG, Gunel M, Jafar-Nejad H, Cresswell P, Enns GM, Freeze HH. 2015. A congenital disorder of deglycosylation: Biochemical characterization of N-glycanase 1 deficiency in patient fibroblasts. Glycobiology. 25:836–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrissat B. 1991. A classification of glycosyl hydrolases based on amino-acid sequence similarities. Biochem J. 280:309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husein DM, Naim HY. 2019. Impaired cell surface expression and digestive function of sucrase-isomaltase gene variants are associated with reduced efficacy of low FODMAPs diet in patients with IBS-D. Gut pii: gutjnl-2019-319411. doi: 10.1136/gutjnl-2019-319411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa S, Baujat G, Seyahi A, Garoufali AG, Imel EA, Padgett LR, Austin AM, Sorenson AH, Pejin Z, Topouchian V et al. 2010. Clinical variability of familial tumoral calcinosis caused by novel GALNT3 mutations. Am J Med Genet. 152A:896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen E, Baumann M, Grön K, Syvänen AC, Enomaa N, Halila R, Aula P, Peltonen L. 1991. Aspartylglucosaminuria: cDNA encoding human aspartylglucosaminidase and the missense mutation causing the disease. EMBO J. 10:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imura A, Tsuji Y, Murata M, Maeda R, Kubota K, Iwano A, Obuse C, Togashi K, Tominaga M, Kita N et al. 2007. Alpha-Klotho as a regulator of calcium homeostasis. Science. 316:1615–1618. [DOI] [PubMed] [Google Scholar]
- Jaeken J, Matthijs G. 2001. Congenital disorders of glycosylation. Annu Rev Genomics Hum Genet. 2:129–151. [DOI] [PubMed] [Google Scholar]
- Jaeken J, Péanne R. 2017. What is new in CDG? J Inherit Metab Dis. 40:569–586. [DOI] [PubMed] [Google Scholar]
- Joshi HJ, Hansen L, Narimatsu Y, Freeze HH, Henrissat B, Bennett EP, Wandall HH, Clausen H, Schjoldager KT. 2018. Glycosyltransferase genes that cause monogenic congenital disorders of glycosylation are distinct from glycosyltransferase genes associated with complex diseases. Glycobiology. 28:284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP et al. 2019. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of function intolerance across human protein-coding genes. bioRxiv Jan 28. doi: 10.1101/531210. [DOI] [Google Scholar]
- Kelley ML, Strezoska Ž, He K, Vermeulen A, Av S. 2016. Versatility of chemically synthesized guide RNAs for CRISPR-Cas9 genome editing. J Biotechnol. 233:74–83. [DOI] [PubMed] [Google Scholar]
- Kingma SD, Bodamer OA, Wijburg FA. 2015. Epidemiology and diagnosis of lysosomal storage disorders; challenges of screening. Best Pract Res Clin Endocrinol Metab. 29:145–157. [DOI] [PubMed] [Google Scholar]
- Kumar P, Henikoff S, Ng PC. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 4:1073–1081. [DOI] [PubMed] [Google Scholar]
- Lam C, Ferreira C, Krasnewich D, Toro C, Latham L, Zein WM, Lehky T, Brewer C, Baker EH, Thurm A et al. 2016. Prospective phenotyping of NGLY1-CDDG, the first congenital disorder of deglycosylation. Genet Med. 19:160–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Scott AJ, Brandt M, Hall IM. 2019. Genomic analysis in the age of human genome sequencing. Cell. 177:70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O'Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB et al. 2016. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Vinckenbosch N, Tian G, Huerta-Sanchez E, Jiang T, Jiang H, Albrechtsen A, Andersen G, Cao H, Korneliussen T et al. 2010. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat Genet. 42:969–972. [DOI] [PubMed] [Google Scholar]
- Lohmueller KE, Sparsø T, Li Q, Andersson E, Korneliussen T, Albrechtsen A, Banasik K, Grarup N, Hallgrimsdottir I, Kiil K et al. 2013. Whole-exome sequencing of 2,000 Danish individuals and the role of rare coding variants in type 2 diabetes. Am J Hum Genet. 93:1072–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, Henrissat B. 2014. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42:D490–D495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe JB, Mart JD. 2003. A genetic approach to mammalian glycan function. Annu Rev Biochem. 72:643–691. [DOI] [PubMed] [Google Scholar]
- Meikle PJ, Hopwood JJ, Clague AE, Carey WF. 1999. Prevalence of lysosomal storage disorders. JAMA. 281:249–254. [DOI] [PubMed] [Google Scholar]
- Mizumoto S, Yamada S, Sugahara K. 2014. Human genetic disorders and knockout mice deficient in glycosaminoglycan. Biomed Res Int. 2014:495764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti E, Bassi MT, Papini N, Riboni M, Manzoni M, Venerando B, Croci G, Preti A, Ballabio A, Tettamanti G et al. 2000. Identification and expression of NEU3, a novel human sialidase associated to the plasma membrane. Biochem J. 349:343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng BG, Freeze HH. 2018. Perspectives on glycosylation and its congenital disorders. Trends Genet. 34:466–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Péanne R, Lonlay P, Foulquier F, Kornak U, Lefeber DJ, Morava E, Pérez B, Seta N, Thiel C, Van Schaftingen E et al. 2018. Congenital disorders of glycosylation (CDG): Quo vadis? Eur J Med Genet. 61:643–663. [DOI] [PubMed] [Google Scholar]
- Platt FM, Boland B, Spoel AC. 2012. The cell biology of disease: Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J Cell Biol. 199:723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann R, Moore DF. 2006. Neurological effects of enzyme replacement therapy in Fabry disease In: Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry disease: Perspectives from 5 years of FOS. Chapter 39 Oxford: Oxford PharmaGenesis. [PubMed] [Google Scholar]
- Shah N, Hou YC, Yu HC, Sainger R, Caskey CT, Venter JC, Telenti A. 2018. Identification of misclassified ClinVar variants via disease population prevalence. Am J Hum Genet. 102:609–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyrantepe V, Landry K, Trudel S, Hassan JA, Morales CR, Pshezhetsky AV. 2004. Neu4, a novel human lysosomal lumen sialidase, confers normal phenotype to sialidosis and galactosialidosis cells. J Biol Chem. 279:37021–37029. [DOI] [PubMed] [Google Scholar]
- Stanley P. 2016. What have we learned from glycosyltransferase knockouts in mice? J Mol Biol. 428:3166–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, Hussain M, Phillips AD, Cooper DN. 2017. The human gene mutation database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet. 136:665–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirnemann J, Belmatoug N, Camou F, Serratrice C, Froissart R, Caillaud C, Levade T, Astudillo L, Serratrice J, Brassier A et al. 2017. A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci. 18:pii:E441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N. 1977. Demonstration of a new amidase acting on glycopeptides. Biochem Biophys Res Commun. 76:1194–1201. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A et al. 2015. Proteomics. Tissue-based map of the human proteome. Science. 347:1260419. [DOI] [PubMed] [Google Scholar]
- Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T. 2006. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature. 444:770–774.17086194 [Google Scholar]
- Vinogradova MV, Michaud L, Mezentsev AV, Lukong KE, El-Alfy M, Morales CR, Potier M, Pshezhetsky AV. 1998. Molecular mechanism of lysosomal sialidase deficiency in galactosialidosis involves its rapid degradation. Biochem J. 330:641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger DA, Rafi MA, Luzi P. 1997. Molecular genetics of Krabbe disease (globoid cell leukodystrophy): Diagnostic and clinical implications. Hum Mutat. 10:268–279. [DOI] [PubMed] [Google Scholar]
- Yao X-P, Cheng X, Wang C, Zhao M, Guo X-X, Su H-Z, Lai L-L, Zou X-H, Chen X-J, Zhao Y et al. 2018. Biallelic mutations in MYORG cause autosomal recessive primary familial brain calcification. Neuron 98: 1116–1123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.