

Abstract
Background
Sleep disturbances are common in smoking cessation attempts and are predictive of relapse. Despite this knowledge, there is no established animal model to study the effect of nicotine abstinence on sleep and EEG parameters.
Objectives
The present study was conducted to characterize sleep and wakefulness in male C57BL/6J mice during periods of oral nicotine administration and abstinence.
Methods
Male C57BL/6J mice were implanted with EEG/EMG recording devices. EEG/EMG data were recorded continuously for a period of 4 weeks. At the beginning of week 2, 200μg/ml of nicotine was added to the 0.2% saccharin vehicle drinking solution. Following a two-week period of oral nicotine administration, abstinence was initiated by excluding the nicotine from the 0.2% saccharin vehicle drinking solution. EEG/EMG were analyzed at pre-nicotine baseline, during nicotine administration, and on days 1, 2 and 5 of abstinence from nicotine.
Results
Oral nicotine administration decreased total sleep time during the active phase, consistent with the stimulant actions of nicotine. In contrast, NREM sleep quantity was increased during the active phase on nicotine abstinence day one and REM sleep was decreased during days two and five of abstinence. Further, sleep fragmentation was increased during the inactive phase on all days of abstinence. Oral nicotine administration and abstinence from nicotine also altered EEG relative power frequencies during the inactive and active phase.
Conclusions
Both oral nicotine administration and abstinence lead to sleep disturbances in mice. Similarities between this model and human reports on the effect of nicotine/nicotine withdrawal on sleep support its utility in examining the molecular mechanisms that modulate the relationship between sleep, nicotine, and nicotine abstinence/withdrawal.
Keywords: Nicotine, Nicotine withdrawal, Nicotine abstinence, Sleep, EEG
Cigarette smoking remains the leading cause of preventable death, accounting for more than 6 million deaths each year worldwide. Despite warnings on cigarette packages, and known health risks, people continue to smoke. Around 70% of smokers express a desire to quit (CDC, 2011), yet the overall quitting prevalence is only 6.2% (CDC, 2011). Among the factors that contribute to the poor quit rate is nicotine withdrawal. Negative physical and affective withdrawal symptoms are a reliable and persistent consequence of smoking cessation. To avoid the distress and impairment associated with withdrawal from chronic nicotine, many individuals reinitiate smoking (Aguirre Madrid, & Leventhal, 2015). Several symptoms of withdrawal are thought to contribute to relapse including the presence of adverse and long-lasting sleep disturbances (Prosise et al, 1994; Wetter et al, 1999; Colrain, Trinder, & Swan, 2004; Hamidovic & de Wit, 2009; Okun et al, 2001; Jaehne et al., 2015).
It is estimated that disturbed sleep occurs during 25-39% of cessation attempts (Jaehne et al.2009; Okun et al., 2011). Distinct from other withdrawal symptoms, sleep disturbances often do not diminish in severity within the first 21 days after cessation (Hughes, 2007). Several studies have observed an increased likelihood of relapse in those reporting cessation-related sleep disturbances (Hamidovic & de Wit, 2009; Zhou et al., 2009; Jaehne et al., 2014). Disrupted sleep may contribute to relapse through several mechanisms. Insufficient sleep might increase the relapse potential by intensifying the value of and cravings for cigarettes. Two days of sleep deprivation during forced withdrawal has been shown to increase smoking behavior, independent of attentional impairment and negative mood states. (Hamidovic & de Wit, 2009). Alternatively, insufficient and disrupted sleep during withdrawal may lead to drowsiness and fatigue. Nicotine, the main psychoactive ingredient in cigarettes, acts as a stimulant in the central nervous system producing arousal. Although smoking directly contributes to a condition of drowsiness and chronic fatigue (Hughes & Hatsukami, 1986; Corwin et al., 2002), smokers report near immediate alleviation of this with smoking (Parrott, 1993).
Another important consideration is the significant overlap between symptoms of disturbed or reduced sleep and nicotine withdrawal syndrome. Independent of withdrawal, insufficient sleep is linked to increased irritability, impaired attention and cognition, daytime sleepiness, anxiety, depression, increased appetite and weight gain (Institute of Medicine (US) Committee on Sleep Medicine and Research, 2006; Medic et al., 2017). Smokers in withdrawal also report these behavioral and cognitive impairments (Hughes, Higgins, & Bickel, 1994; Wesnes et al., 2013). Thus, disturbed sleep may not only be a symptom of withdrawal, it may also exacerbate other withdrawal symptoms.
Self-report studies reliably and consistently find that smoking cessation is related to perceived difficulty falling asleep, increased frequency and duration of awakenings, and daytime sleepiness (Cummings et al., 1985; Hatsukami et al., 1985; Hatsukami et al., 1998; Shiffman et al., 1995; Grove et al., 2006). Reports from polysomnography (PSG) studies are less consistent, but confirm the existence of sleep disturbances during withdrawal. Initial investigations aimed at objectively assessing sleep disturbances during quit attempts were small in sample size, failed to include follow up data, and produced contradictory results (Soldatos et al., 1980; Prosise et al., 1994; Zhang et al., 2006). PSG observed sleep disturbances include reduced sleep latency and total sleep time, as well as increased sleep stage changes, sleep fragmentation, and wake after sleep onset (WASO) (Soldatos et al., 1980; Prosise et al., 1994) Zhang et al., 2006). The most recent and comprehensive PSG study indicates a robust negative effect of nicotine withdrawal on sleep (Jaehne et al., 2014). In this study, sleep in smokers was measured and subjectively assessed before, during, and 3 months after withdrawal. Analyses of PSG data showed significant changes in REM latency, the percentage of time spent in stage 2 non-REM and slow-wave sleep (SWS), and WASO. No changes in sleep efficacy, non-REM latency, or total sleep time (TST) were observed. Interestingly, analysis of the self-report data reveal a noted change in sleep quality, efficiency, latency, and duration. These results suggest that perceived sleep disturbances differ greatly from measured sleep disturbances. The significance of this discrepancy has yet to be investigated.
Preliminary evidence strongly suggests an effect of nicotine withdrawal on sleep and a potential causative role of sleep disturbances in relapse. While most aspects of nicotine withdrawal have been well studied and characterized, the relationship between smoking cessation and sleep remains one of the least understood. Rodent models of nicotine abstinence have proven reliable predictors of human behavior during cessation attempts (Malin & Goyarzu, 2009) and are essential for determining the mechanisms through which nicotine abstinence impacts behavior and physiology. To our knowledge, sleep disturbances are the only common symptom of human nicotine withdrawal that have not been modeled in the rodent. Although sleep architecture varies between rodents and humans, the brain mechanisms controlling sleep timing, structure, depth and duration are comparable across species (Patterson, Nutt, & Wilson, 2011). Analogous brain circuitry and similar sleep EEG features make rodents a suitable model for investigating the behavioral and pharmacological effects of chronic nicotine exposure and abstinence on sleep and wakefulness. The goal of the current study was to develop a mouse model to investigate the effects of oral nicotine administration and abstinence from nicotine on sleep and electroencephalography (EEG) spectral patterns.
Methods
Animals and Experimental Design
All procedures were approved by the University of Colorado’s Institutional Animal Care and Use Committee and followed the National Institutes of Health guide for the care and use of laboratory animals. A total of 10 individually housed, 9-week old male C57BL/6J mice were used in the following experiment. One animal was excluded from statistical analyses for being more than two standard deviations away from the mean on all reported variables. During the experimental protocol, mice were maintained on a 12-hour light-dark schedule, with lights on at 0700, and had ad libitum access to food and either a vehicle solution containing 0.2% saccharin in water or 0.2% saccharin in water supplemented with 200 μg/ml nicotine as described below. Standard laboratory nesting squares were provided. Weights were collected prior to all experimental procedures and at the midway point. Nicotine was chronically administered via the oral nicotine administration paradigm as described previously (Sparks & Pauly, 1999). Briefly, mice received a 0.2% saccharin vehicle drinking solution for a one-week baseline period. Immediately following this baseline period, 200μg/ml of free base nicotine (Sigma Aldrich, St. Louise, MO) was added to the vehicle drinking solution and mice were provided with this solution for two weeks. Nicotine concentrations of 200μg/ml have previously been shown to induce upregulation of nAChR’s as well as physical withdrawal (Grabus et al., 2005; Sparks & Pauly, 1999). The nicotine solution was changed every 3-4 days, for a total of 4 times throughout the condition. The volume of remaining fluid was recorded at each solution change. Individual weights were used to calculate individual mg/kg/day of nicotine consumption. After fourteen days of nicotine exposure, nicotine abstinence was initiated by replacing the nicotine solution with 0.2% saccharin vehicle solution at the beginning of day 15 between ZT (Zeitgeber time) 2-3.
To obtain electroencephalography (EEG) and electromyography (EMG) signals during pre-nicotine baseline, oral nicotine administration, and abstinence conditions, animals were implanted with cortical EEG and intramuscular EMG recording electrodes. Briefly, animals were anesthetized with isoflurane and placed in a stereotactic apparatus. A pre-fabricated headmount (8mm x 5mm) (Pinnacle Technology Inc., Lawrence, KS), was affixed to the skull with stainless steel screws acting as EEG electrodes. The anterior portion of the headmount was placed approximately 2mm anterior to bregma (Anterior screw placement: AP + 2.0mm, ML 2.5mm; Posterior screw placement AP - 6.0mm, ML 2.5mm). Electrodes have “posterior placement for increased hippocampal theta detection and bilateral placement to maximize recordings of delta activity during sleep” (Pinnacle Technology Inc., Lawrence, KS). Two flexible, stainless steel electrodes were inserted into the nuchal muscles to obtain the EMG signal. The headmount (Pinnacle Technology Inc., Lawrence, KS) was secured and insulated with dental acrylic. Upon completion of implantation, a (0.1mg/kg) intraperitoneal injection of Buprenorphine was given for pain management. Mice were subsequently individually housed in a specially designed recording chamber (Pinnacle Technology Inc., Lawrence, KS). Following a seven-day recovery period, mice were attached to recording cables (Pinnacle Technology Inc., Lawrence, KS) via an overhead swivel commutator system (Pinnacle Technology Inc., Lawrence, KS). Mice were habituated to the cables for a period of 4-5 days before initiation of EEG/EMG recording. The recording paradigm was as follows: one-week pre-nicotine baseline, two weeks 200μg/ml nicotine drinking solution and one week of abstinence from nicotine. A timeline for the experiment is shown in Fig 1.
Fig. 1.
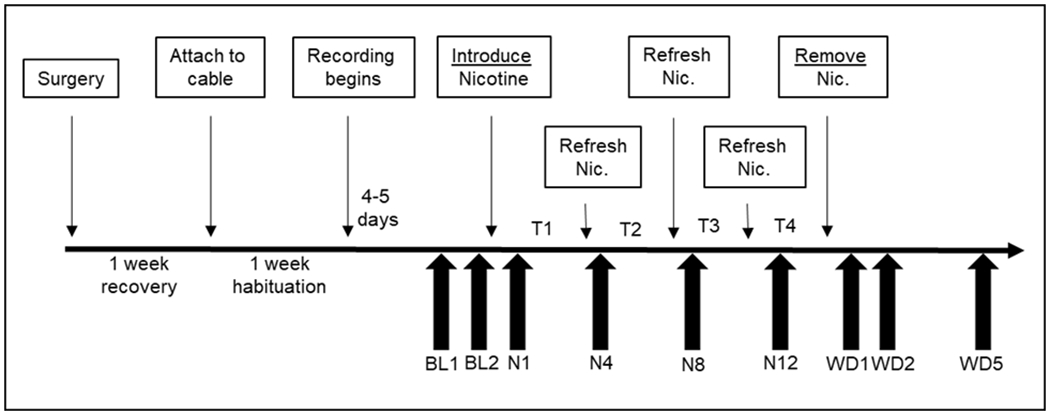
Experimental protocol schedule. Timeline of events (upper boxes) and scored/reported recording days (lower arrows).
Data Acquisition and Analysis
Two cortical EEG and one intramuscular EMG channels were continuously acquired over the entire four-week experiment using the Sirenia Acquisition system (Pinnacle Technology Inc., Lawrence, KS) at a sampling frequency of 500Hz. The EEG data were then band-pass filtered from 0.5-25 Hz and the EMG data were filtered from 0-100 Hz. The acquired EEG/EMG signals were condensed into nine 24-hour periods: the last two days of pre-nicotine baseline, days 1, 4, 8, and 12 of oral nicotine administration, and days 1, 2, and 5 of abstinence from nicotine.
The sleep scoring procedure consisted of an initial automated step using Sirenia Sleep Pro (Pinnacle Technology Inc., Lawrence, KS), followed by a manual review of all epochs to confirm accuracy. Epochs with high-amplitude, low frequency, delta-band activity (.05-4Hz) accompanied by low EMG activity were scored as NREM; those with low-amplitude, high frequency theta-band activity accompanied by very low EMG activity were scored as REM; and epochs with varied theta-band amplitude and high EMG activity were scored as wake. Vigilance states were classified in 4-second epochs as non-rapid eye movement sleep (NREM), rapid eye movement (REM), and wake. Days within the baseline and nicotine conditions were averaged.
To assess sleep quantity, percent time spent in each state and a combined NREM/REM (sleep) state, was obtained from each manually verified 24-hour period, as well as the 12-hour lights off (active phase) and 12-hour lights on (inactive phase) periods. Sleep architecture was evaluated using sleep bout frequency (number of sleep bouts) and sleep bout duration (average length of sleep bout, in seconds) for each 24-hour period, as well as each 12-hour lights off/on period. Another sleep parameter that was assessed was latency to persistent sleep during the active phase, the length of the first wake bout. Sleep bout frequency, sleep bout duration and latency to persistent sleep are all measures that are automatically calculated from the EEG data by the Sirenia Sleep Pro software.
The EEG spectral analysis was performed using Spike2 (Cambridge electronic Design Limited, Cambridge, England). Each 24-hour data file was converted into an EDF and transformed using the Fast Fourier Transform (FFT) algorithm (with a resolution of .7812 Hz). Spectral output was analyzed in 4-hour bins. To account for individual differences, the relative power for frequencies 0-25 Hz was derived by expressing each frequency as a proportion of the total (absolute) power. Individual frequencies were summed to represent Delta (0-4Hz), Theta (4-8.5Hz), Alpha (8.5-15Hz), and low Beta (15-25Hz) power bands. Similar to sleep/wake patterns, days within the baseline and nicotine conditions were averaged.
Statistical Analysis
Analyses were performed with SPSS version 24 or Graph Pad Prism 6. Sleep-wake profiles, bout frequency and duration, and latency to persistent sleep during the active phase, were compared using a repeated-measures analysis of variance (ANOVA) with Sidak corrections. For any analyses where sphericity was violated, the Greenhouse-Geisser correction was applied to reduce the possibility of type 1 error. Two-tailed Student’s t-tests were performed when a significant effect of treatment was observed. Spectral data were compared using a one-way or two-way (Time X Treatment) repeated-measures analysis of variance (ANOVA) with Sidak corrections. Sidak’s multiple comparisons test was performed when a significant effect of time, treatment, or an interaction was observed.
Results
Mouse weights were collected prior to all experimental procedures and at the midway point. Average pre-surgical weight was 24.16 (± 0.65) and average weight at the midway point was 24.01 (± 0.59). During the nicotine administration phase of the experiment, mice had access to a drinking solution consisting of 0.2% saccharin and 200 μg/ml nicotine/as the sole source of fluid for a period of two weeks. The solution was changed four times across the two week period and remaining volume measured to confirm intake. Mice initially consumed an average of 88.8 mg/kg/day (Fig. 2). Consumption decreased during the second measurement (54.0 mg/kg/day), and slightly increased during the last week of exposure (65.2 mg/kg/day and 64.8 mg/day/day, respectively). Overall, average daily consumption was 68.2 mg/kg/day. These nicotine intake values are consistent with previous reports where nicotine is the sole fluid source (Pietila & Ahtee, 2000). Average daily fluid consumption across the two-week period was 8 ± 0.87 ml/day (Fig. 2). This level of consumption is at/above the normal fluid intake for C57BL/6J mouse (4.82 mL daily) (Mouse Phenome Database, Jackson Laboratory, https://phenome.jax.org/measureset/40401).
Fig. 2.
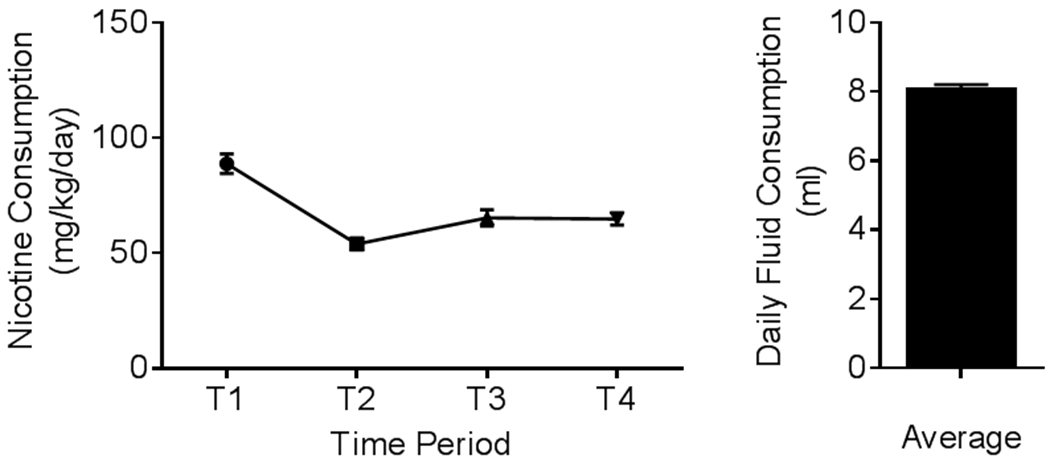
Average (n = 9; ± SEM) consumption of 200μg/ml of free-base nicotine (Sigma-Aldrich, St. Louis, MO) in a .02% saccharin drinking solution.
Effect of Oral Nicotine Administration and Abstinence on 24hr Sleep Quantity and Architecture
The effects of oral nicotine administration and abstinence from nicotine on sleep quantity and architecture were determined using EEG and EMG recordings. Preliminary analysis indicated no significant difference for any measure between the 4 days of nicotine administration evaluated. Therefore, data for nicotine treatment were averaged for days 1, 4, 8, and 12 to examine general effect of nicotine rather than an acute daily effect. Nicotine abstinence days are reported individually (Abstinence day one: AD1, Abstinence day 2: AD2, abstinence day five: AD5). Analyses revealed a main effect of treatment on total sleep time (TST) F4, 32 = 9.59, p = .000032) (Fig. 3a) (Table 1). During oral nicotine administration TST was reduced relative to pre-nicotine baseline (p = 0.001). During abstinence, there were no observed changes in 24hr TST. Among sleep states, a main effect of treatment was observed for NREM (F1.878, 15.021 = 9.926, p = .002) and REM (F4, 32 = 5.384, p = .002) (Fig. 3a) (Table 1). Follow-up pairwise comparisons revealed that NREM decreased significantly during nicotine administration (p = 0.001), whereas no change was detected for REM. Conversely, there were no observed changes in NREM quantity during abstinence from nicotine, but REM quantity was decreased on AD2 (p = 0.007) and AD5 (p = 0.010).
Fig. 3.
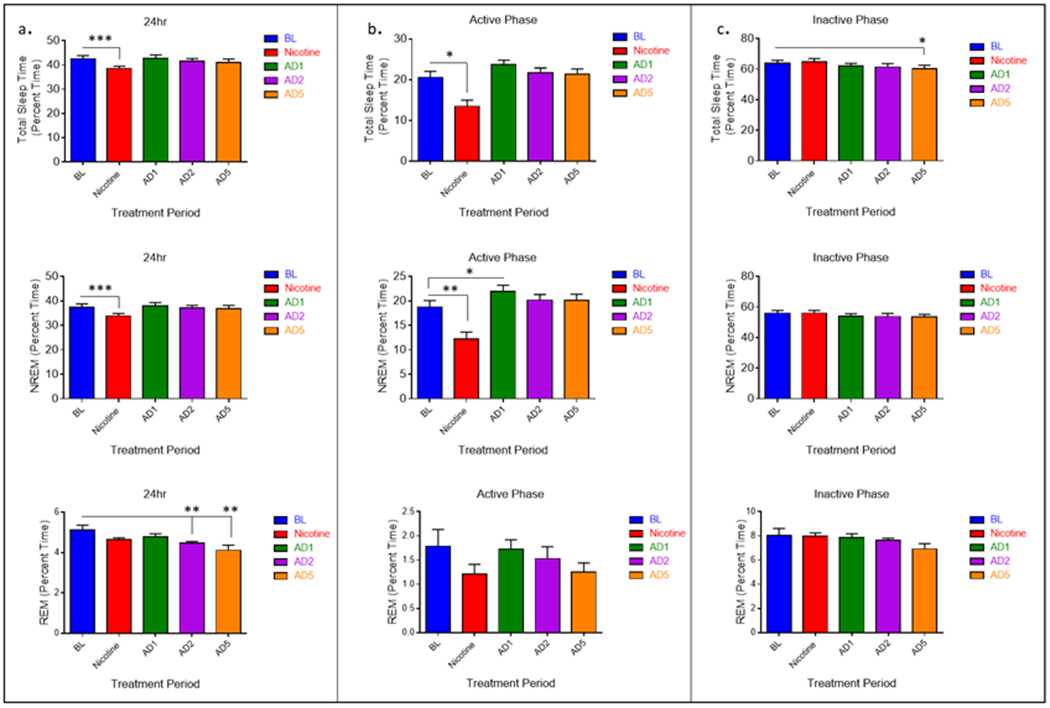
Average (n = 9; ± SEM) total sleep time (TST), NREM, and REM sleep quantity across treatment periods during 24 hours and separated by 12hr light-dark phase. * Indicates p < .05 ** Indicates p < .01
Table 1.
Average Daily Sleep Quantity in Minutes
| 24hr | Active Phase | Inactive Phase | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BL | NIC | AD1 | AD2 | AD5 | BL | NIC | AD1 | AD2 | AD5 | BL | NIC | AD1 | AD2 | AD5 | |
| Sleep | 612.60 | ***555.50 | 616.10 | 598.00 | 588.60 | 539.00 | ***488.20 | 547.30 | 598.00 | 529.44 | 73.62 | 67.01 | 68.76 | 64.23 | *59.18 |
| (± 19.12) | (± 13.69) | (± 18.98) | (± 16.02) | (± 22.20) | (± 19.11) | (± 13.98) | (± 18.52) | (± 16.02) | (± 19.99) | (± 3.60) | (± 1.13) | (± 2.28) | (± 1.38) | (± 3.72) | |
| NREM | 296.00 | ***194.00 | 340.20 | 312.70 | 307.30 | 270.30 | **176.60 | *315.40 | 290.70 | 289.70 | 25.69 | 17.34 | 24.84 | 22.02 | 18.04 |
| (± 21.39) | (± 21.81) | (± 16.66) | (± 16.56) | (± 18.63) | (± 18.85) | (± 19.52) | (± 18.73) | (± 16.32) | (± 18.58) | (± 5.00) | (± 2.99) | (± 2.7) | (± 3.51) | (± 2.62) | |
| REM | 922.40 | 933.40 | 892.00 | **884.3 | **869.60 | 807.00 | 806.70 | 779.20 | 774.70 | 769.70 | 115.50 | 114.7 | 112.8 | 109.6 | 99.62 |
| (± 25.47) | (± 29.73) | (± 25.92) | (± 31.83) | (± 30.93) | (± 26.18) | (± 25.42) | (± 23.00) | (± 29.84) | (± 25.84) | (± 8.41) | (± 3.94) | (± 4.66) | (± 2.803) | (± 6.37) | |
Indicates p < .05,
Indicates p < .01,
Indicates p < .001
Analyses of sleep architecture measures revealed a main effect of treatment for sleep bout frequency (SBF) (F4, 32 = 8.358, p = .000098) and sleep bout duration (SBD) (F4, 32 = 5.51, p = .001) (Fig. 4a). Post hoc analysis revealed a significant reduction in sleep bout frequency (p = 0.004) and no change in duration during the period of nicotine administration. Sleep architecture was greatly affected during abstinence from nicotine. Sleep bout duration was decreased on all days of abstinence, this effect was significant on days one (p = 0.023) and five (p = 0.028). Further, sleep bout frequency was increased significantly on all days of abstinence (p = 0.017 AD1, p = 0.045 AD2, p = 0.021 AD5). In summary, the reduction in TST during nicotine administration is the result of decreased NREM sleep quantity arising from a reduction in sleep bout frequency. The lack of observed changes in TST during the abstinence condition is due to contrasting changes in sleep bout duration and frequency.
Fig. 4.
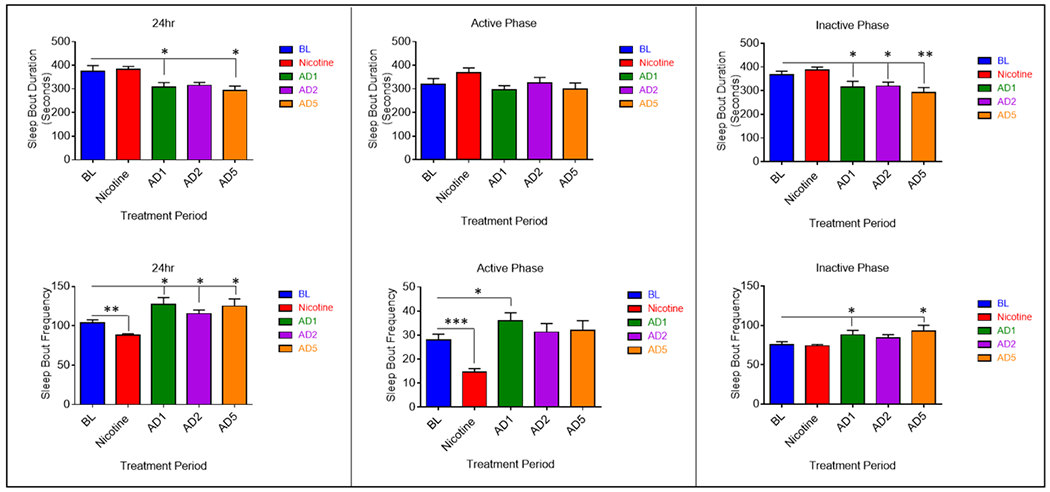
Average (n = 9; ± SEM) sleep bout duration (SBD) and sleep bout frequency (SBF) across treatment periods during 24 hours and separated by 12hr light-dark phase. * Indicates p < .05
Effect of Oral Nicotine Administration and Abstinence on Sleep Quantity and Architecture during the Active and Inactive Phases
The data were further analyzed to determine whether the effects of oral nicotine administration and abstinence from nicotine occurred during the active (lights off) or inactive (lights on) phase of the light cycle. A main effect of treatment on TST was observed for the 12-hour active phase (F4, 32 = 11.611, p = .000006) (Fig. 3b) (Table 1). Compared to pre-nicotine baseline, TST was reduced during the active phase of nicotine administration (p = 0.013). TST during the active phase of AD1 was increased, this effect was approaching significance (p = 0.051). No changes were observed for abstinence days two or five. Among sleep states, a main effect of treatment was seen for NREM (F4, 32 = 13.151, p = 0.000002), but not REM (Fig. 3b (Table 1)). Like the 24-hour period, nicotine administration reduced active phase NREM sleep (p = 0.008). An increase was observed on AD1 (p = .018).
Analyses of active phase sleep architecture revealed a main effect of treatment on sleep bout frequency (SBF) (F4, 32 = 11.617, p = .000006), but not sleep bout duration (SBD) (Fig. 4b). Sleep bout frequency was decreased during nicotine administration (p = .00044) and increased on AD1 (p = 0.032). Like the 24-hour period, reduced TST during the active phase is due to decreased NREM sleep quantity and reduced sleep bout frequency. The increase in active phase TST on AD1 is likely due to the significant increase in sleep bout frequency and NREM on that day. An additional variable assessed during the active phase was latency to persistent sleep, measured as the time (in seconds) it took to enter the first bout of sleep after lights off. A main effect of treatment on sleep latency was observed (F4, 32 = 10.767, p = 0.00012). An increase in latency to persistent sleep was seen during nicotine administration (p = 0.002) (Fig. 5). Conversely, latency to persistent sleep was decreased on AD1 (p = 0.018). This is suggestive of decreased sleepiness during nicotine administration and increased sleepiness on the first day of abstinence.
Fig. 5.
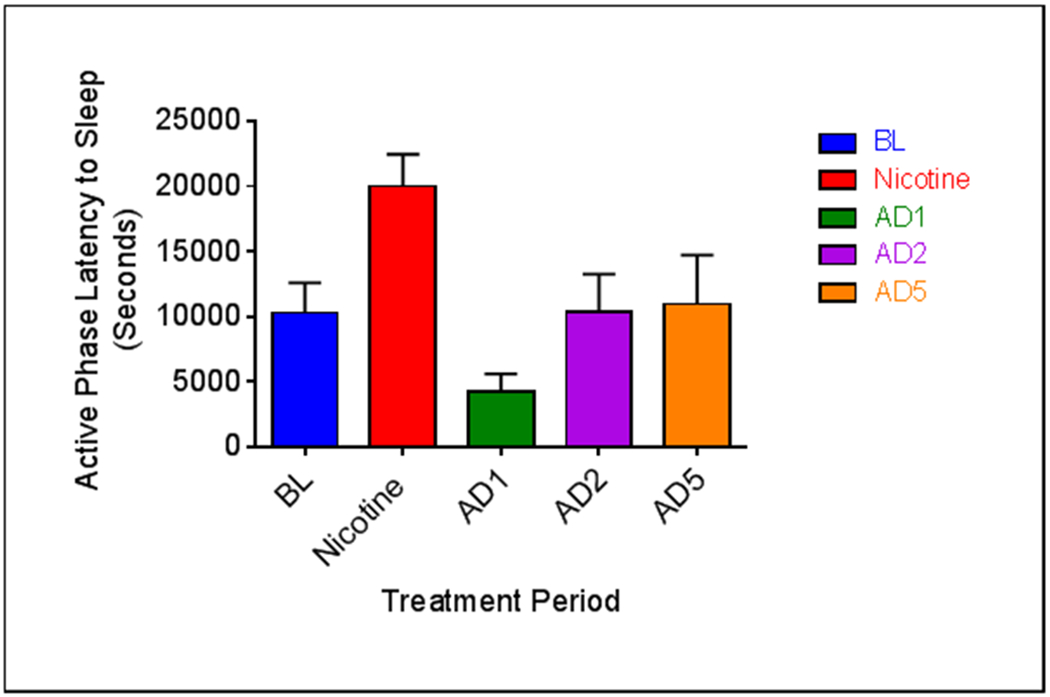
Average (n = 9; ± SEM) active phase sleep latency across treatment periods. * Indicates p < .05.
A main effect of treatment on TST also was observed for the 12-hour inactive phase (F4, 32 = 3.756, p = 0.013) (Fig 3c). Oral nicotine administration did not affect inactive phase TST. However, relative to pre-nicotine baseline, inactive phase TST was reduced on AD5 (p = .013). Among sleep states, no main effect was seen for REM or NREM (Fig 3c), although NREM was approaching significance (F4, 32 = 2.566, p = .057). No change in NREM was observed during the inactive phase of nicotine administration. Pairwise comparison revealed a significant decrease in inactive phase NREM on AD5 (p 0.038). This likely explains the significant decrease in inactive phase TST.
Analyses of inactive phase sleep architecture showed a main effect of treatment on both sleep bout frequency (SBF) (F4,32 = 5.084, p = .003) and sleep bout duration (SBD) (F4, 32 = 7.756, p = .00017) (Fig. 4c). No changes were seen for either variable during nicotine administration. Inactive phase sleep bout duration was significantly decreased on all days of abstinence (p = 0.044 AD1, p = 0.050 AD2, p = 0.002 AD5). Inactive phase sleep bout frequency was increased on all days of abstinence, this effect was significant on days one (p = 0.028) and five (0.004). Analyses of 12-hour data from the active (lights off) and inactive (lights on) phases indicate 24-hour effects of nicotine administration are due solely to changes during the active phase, whereas 24-hour effects of abstinence are due to changes in sleep quantity and architecture during both 12-hour phases. Changes in sleep bout frequency and duration during the inactive phase of abstinence are suggestive of increased sleep fragmentation during the primary sleep period.
Effect of Oral Nicotine Administration and Abstinence on EEG spectrum
The effects of oral nicotine administration and abstinence from nicotine on EEG power spectra were determined via fast Fourier transformation of cortically derived EEG frequencies. Analyses indicate an effect of treatment on relative delta power (0-4Hz) during both the active (F4, 32 = 9.303, p = < 0.0001) and inactive phases (F4, 32 = 5.620, p = 0.0015) (Fig. 6). Compared to pre-nicotine baseline, relative delta power was decreased during the active phase of nicotine administration (Fig. 6a). This effect was significant at ZT (Zeitgeber time)16-20 (p = 0.0003). Conversely, relative delta power was increased during the inactive phase. This effect was significant at ZT0-4 (p = 0.021). No changes were seen in relative delta power during the active phase of any abstinence day (Fig. 6b). However, an increase in relative delta power was observed during the inactive phase. This effect was significant at ZT0-4 on AD2 (p = 0.0083) and at ZT4-8 on all days of abstinence (p = 0.01 AD1, p = 0.007 AD2, p = 0.0025 AD5). Effects of nicotine administration on relative delta power primarily occurred during the middle of the active phase and the beginning of the inactive phase. Effects of abstinence from nicotine on relative delta power largely occurred during the beginning and middle of the inactive phase.
Fig. 6.
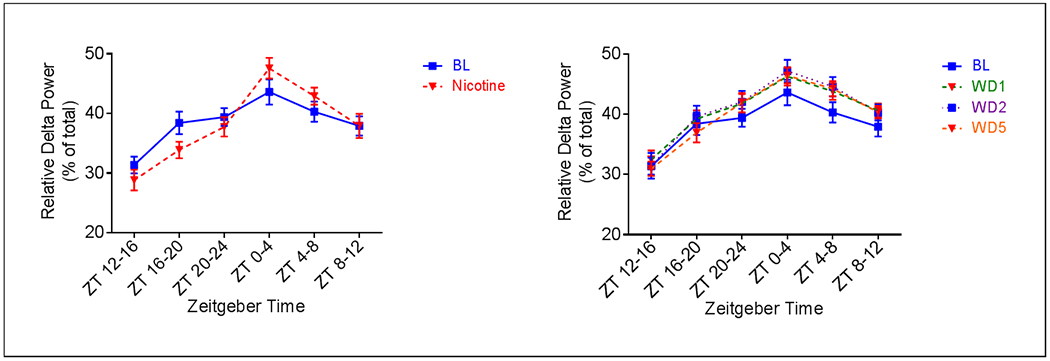
Average (n = 9; ± SEM) relative delta power across treatment periods. ** Indicates p < 0.01, *** Indicates p < 0.001, * Indicates p < .05 relative to AD1, ## indicates p < 0.01 relative to AD2, ### indicates p < 0.001 relative to AD2, $$ indicates p < 0.01 relative to AD5
A main effect of treatment was also observed for relative power in the theta (4.0-8.5Hz) (Fig. 7) and alpha (8.5-15Hz) (Fig. 8) ranges during both the active (F4, 32 = 4.855, p = 0.0036 for Theta; F4, 32 = 5.242, p = 0.0023 for Alpha) and inactive phases (F4, 32 = 5.820, p = 0.0012 for Theta; F4, 32 = 2.845, p = 0.0400). Relative theta power was increased during the active phase of nicotine administration, compared to pre-nicotine baseline (Fig. 7a). This effect was significant at ZT 16-20 (p = .04). Similarly, relative alpha was also increased during the active phase of nicotine administration (Fig. 8a). This effect was significant at ZT 12-16 (p < 0.001) and ZT 16-20 (p < 0.001). Relative theta power was decreased during the active phase of abstinence from nicotine (Fig. 7b). This effect was significant at ZT 20-24 for all days of abstinence (p = 0.0018 AD1, p = 0.0039 AD2, p = 0.006 AD5). Relative alpha power during the active phase of abstinence was comparable to that of pre-nicotine baseline, with the exception that an increase was seen at ZT 16-20 on AD5 (p = .0057) (Fig. 8b). Effects of nicotine administration on active phase theta power primarily occurred during the middle the 12hr period, whereas effects on alpha occurred during both the beginning and middle of the 12hr period. Abstinence from nicotine affected active phase theta only during the end of the 12hr period. Alpha power, however, was altered during the middle of the 12hr active phase.
Fig. 7.
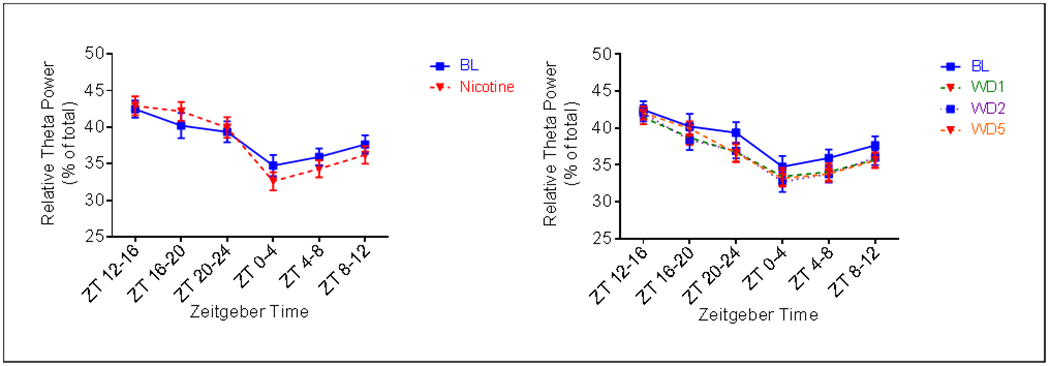
Average (n = 9; ± SEM) relative theta power per four hours across treatment periods. * Indicates p < 0.05, ** Indicates p < 0.01 relative to AD1, ## indicates p < 0.01 relative to AD2, $$$ indicates p < 0.001 relative to AD5
Fig. 8.
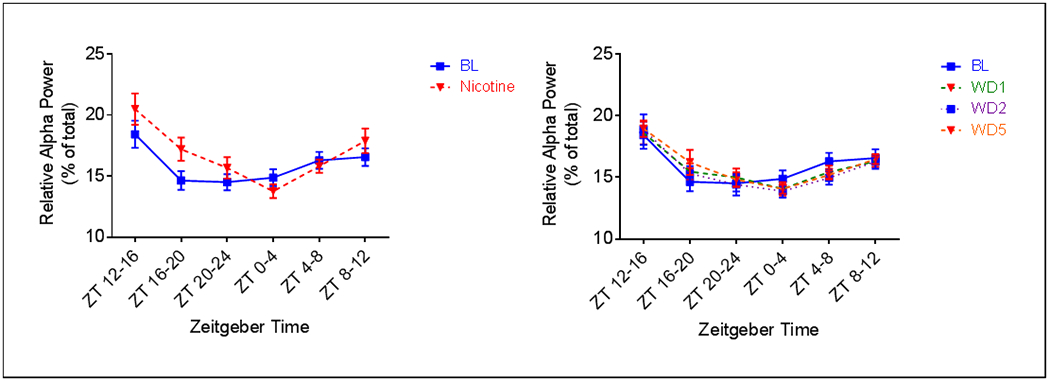
Average (n = 9; ± SEM) relative alpha power per four hours across treatment periods. * Indicates p < 0.05, # indicates p < 0.05 relative to AD2, $$ indicates p < 0.01 relative to AD5
Compared to pre-nicotine baseline, a decrease in relative theta power was observed across the entire inactive period during nicotine administration (Fig. 7a). This effect was significant at ZT 0-4 (p = 0.0176). At the start of the inactive period, alpha power was decreased relative to pre-nicotine baseline during nicotine administration (Figure 8a). However, during the last four hours (ZT8-12), alpha was increased (p = .0332). Relative theta power was greatly affected during abstinence (Fig. 7b). A decrease was seen on all days, this effect was significant at ZT 0-4 on AD2 (p = .0278), at ZT 4-8 on AD2 (p = 0.0160) and AD5 (p = 0.0176), and at ZT 8-12 on AD1 (p = .0477) and AD5 (p = 0.0313). Finally, although relative alpha was decreased on all days of abstinence, compared to pre-nicotine baseline, the effect was only significant at ZT 4-8 on AD2 (p = 0.0306) (Fig. 8b). Effects of nicotine administration on relative theta power during the inactive phase occurred primary during the beginning of the 12hr period, whereas effects on alpha power only occurred during the end of the 12hr period. A large effect of abstinence from nicotine on theta was observed during the inactive phase, with time of effect dependent upon day. A lesser effect was observed for inactive phase alpha.
A main effect of treatment on relative beta power was not observed during either the phase (Fig. 9). However, an effect of time and a time by treatment interaction was detected. Pairwise comparisons reveal that nicotine administration reduced relative beta power at ZT 0-4 (p = 0.0041), the beginning of the inactive phase (Fig. 9a). A reduction was also observed at ZT 0-4 on AD1 (p = 0.0487) and at ZT 4-8 on all days of abstinence (p = 0.0021 AD1, p = 0.0008 AD2, p = 0.0055 AD5) (Fig. 9b).
Fig. 9.
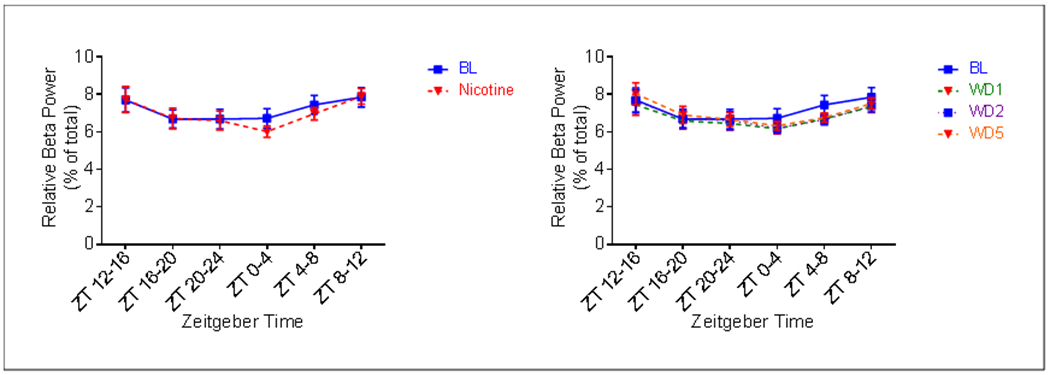
Average (n = 9; ± SEM) relative beta power per four hours across treatment periods.
Finally, to further examine differences in spectral power during oral nicotine administration and abstinence from nicotine, the percent change between each 4hr period was calculated for each relative spectral power band. Significant differences were observed only for the transition between the active phase to the inactive phase (Fig. 10). Compared to pre-nicotine baseline, relative delta power during nicotine administration showed a greater increase (F8, 32 = 9.083, p < 0.001), whereas relative theta showed a greater decrease (F8, 32 = 5.079, p = 0.004). Relative alpha and beta each showed greater decreases, although not significant. These data suggest that oral nicotine administration greatly affects the power spectrum at the time of sleep onset. No differences between pre-nicotine baseline and abstinence days were observed.
Fig. 10.
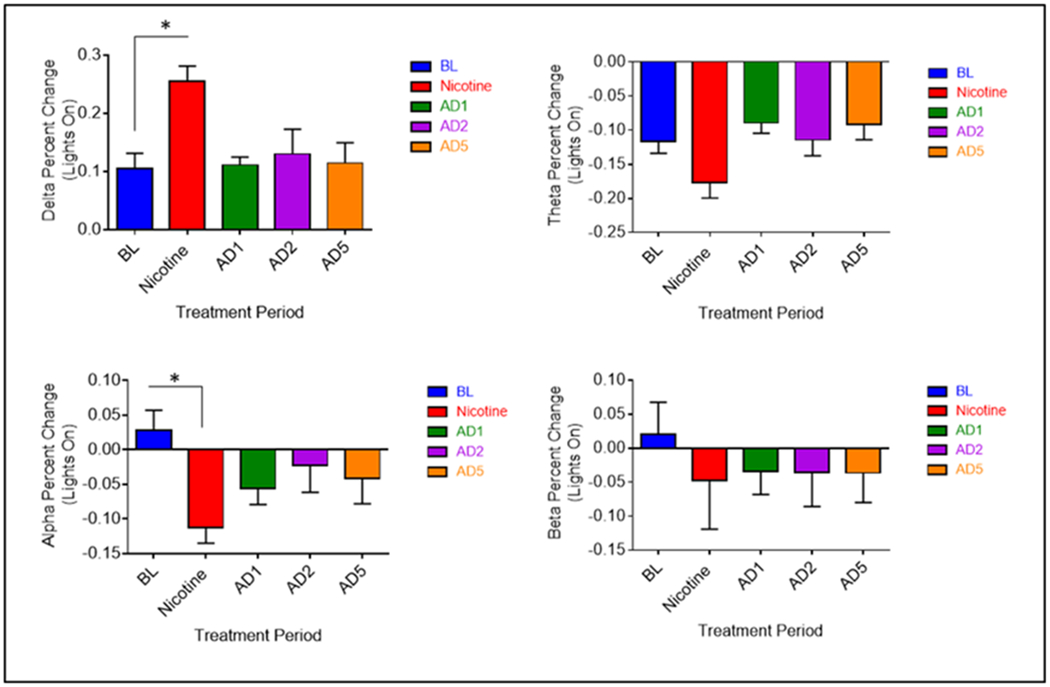
Average (n = 9; ± SEM) relative spectral power percent change at lights on. * Indicates p < 0.05
Discussion
Together, the data in this study are consistent with literature showing a stimulating effect of nicotine during periods of consumption and a negative effect of abstinence on sleep quantity and architecture. Numerous reports in both rodents and humans have detailed measurable increases in arousal and attention with nicotine administration and tobacco smoking. Human studies consistently demonstrate the increased presence of sleep disruptions during periods of withdrawal from chronic nicotine. To our knowledge, this is the first study to examine the chronic general effects of nicotine and abstinence from nicotine on sleep and wakefulness in a rodent model of oral nicotine administration and abstinence. The reports on EEG frequencies during the active and inactive phases are novel and provide a foundational description of the physiological correlates to nicotine and abstinence induced behavioral changes in sleep and wakefulness.
The present study shows that oral nicotine administration decreases total sleep time (TST) by reducing NREM sleep and sleep bout frequency (SBF). Analyses of the 12hr active (lights off) and inactive (lights on) phases show the 24hr effect is due to changes occurring only during the active phase, the period in which a rodent drinks the majority of their daily fluids (Johnson et al, 2003). Overall, these findings are consistent with studies of acute administration of nicotine in rodents and comparable to studies of nicotine exposure on human arousal. Similar decreases in NREM have been reported in the first hour following one intraperitoneal (i.p) injection of nicotine in C57BL/6J mice (Lena et al., 2004) and in the four hours following a single subcutaneous (s.c.) injection of nicotine in rats (Salin-Pascual et al., 1999). Human self-report measures indicate that smoking induces increases in alertness and concentration (Knott et al., 1998), suggesting a key motive for smoking is the psychoactive, stimulant effect that results from nicotine exposure (Crocq, 2003). Behavioral studies confirm that exposure to nicotine results in measurable increases in stimulation. Nicotine administration improves selective attention and arousal (Foulds et al., 1996; Ernst et al., 2001; Kumari et al., 2003), while tobacco smoking improves performance on some cognitive tasks (Swan and Lessov-Schlaggar, 2007). Finally, although reports in humans also detail the increased presence of sleep disruptions in smokers versus non-smokers, no effect of nicotine administration was observed during the rodent inactive period. This may be due to differences in route of administration and/or nicotine metabolism rates or the generally more consolidated sleep period in humans versus mice. Nonetheless, these data suggest that daily oral nicotine intake causes increased arousal during the rodent active phase with little effect on the normal rodent sleep period.
Reduced active phase NREM quantity and sleep bout frequency during nicotine administration were accompanied by increased relative power in the alpha frequency bands and reduced relative power in the slow delta frequency bands, an EEG profile consistent with arousal and attention. In humans, the effect of nicotine exposure is similar. Two subcutaneous injections of 0.6 mg nicotine, 40 mins apart, is sufficient to produce an increase in mean dominant alpha frequency (Foulds et al., 1994), whereas cigarette smoking has been shown to increase relative and dominant alpha frequencies (Knott et al., 1998; Domino et al., 2009) while simultaneously decreasing relative power in the delta frequencies (Knott et al., 1998). Increases in relative theta during the active period are also consistent with increases in time spent awake. Theta rhythms are prominent during periods of wake associated with movement in rodents (Brown et al., 2012). Theta amplitude increases have also been observed within periods of quite awake during sleep deprivation paradigms (Vyazovskiy & Tobler, 2005). These increases may represent a marker of sleep propensity during extended periods of wake, such as that observed during the active phase of nicotine administration. Finally, delta power is homestatically regulated and represents an index of sleep pressure during periods of wakefulness. Decreased delta during the active phase suggests reduced drive for sleep. This is consistent with the reduced number of sleep bouts and amount of NREM.
Although no changes to sleep quantity or architecture were observed during the inactive phase of nicotine administration, alterations in the EEG power spectrum were seen. The primary effect of nicotine administration on the inactive phase power spectrum was to increase relative power in the delta range and decrease relative power in the theta range. Delta oscillations during periods of sleep are characteristic of the deeper stages of NREM sleep (Brown et al., 2012; Bjornvatn et al., 1998). Although relative depth of NREM was not characterized, increased inactive phase delta may suggest changes to NREM sleep depth. Theta activity is prevalent in both active awake and REM sleep. Since no reductions in REM sleep were observed during the inactive phase of nicotine administration, reduced theta might suggest that animals were less active while awake during the inactive period. Finally, in this study, changes to inactive phase relative alpha power were time dependent. Relative alpha was decreased at the beginning of the inactive phase, unchanged in the middle, and significantly increased during the last four hours. Alpha rhythms are prevalent during quiet awake, but also occur at the beginning of the sleep period, during light NREM, and may provide a means of inducing sleep (Kim et al., 2013). Increases in alpha power have been reported in the sleep of smokers versus nonsmokers (Zhang et al., 2008). In this study, the progression from reduced to elevated alpha across the inactive period along with elevated delta may suggest deeper sleep during the early part of the inactive period that transitions to lighter sleep during the later parts of the inactive period.
Abstinence from nicotine had a profound effect on sleep quantity and architecture. Regarding sleep quantity, an increase in NREM sleep was observed during the active phase on abstinence day one (AD1). Although the increase in NREM sleep might suggest an abstinence induced increase in sleep drive, taken together with the literature, it might also be the consequence of accumulated homeostatic sleep pressure, defined by a compensatory increase in sleep depth and duration resulting for preceding sleep loss. Independent of abstinence, sleep loss in rodents consistently produces subsequent rebounds of NREM sleep (Rechtschaffen et al., 1999). In mice, chronic sleep restriction of 6 hours for a period of 3 days produced increases in NREM sleep during the immediately proceeding a recovery period (Clasadonte et al., 2014). In the current study, mice experienced a 9.4% reduction in 24hr TST during the two-week nicotine administration period. This reduction is equivalent to an average loss of 58mins of sleep per day. Relative to nicotine administration, active phase NREM sleep was increased by 78.7% (139mins) on AD1. NREM sleep on AD1 was also 16.1% (45mins) greater than NREM sleep during pre-nicotine baseline, suggesting some degree of NREM sleep rebound.
Consistent with other studies of the effect of sleep loss on sleep latencies (McKenna et al., 2008), a decreased sleep latency was observed during the active phase on AD1. This result lends further support for a homestatically regulated increase in sleep drive. However, the increases in NREM quantity and sleep latency were not accompanied by increased relative delta power, an EEG correlate of homestatically driven sleep pressure. This suggests the increased sleep drive may not be driven by homeostatic sleep pressure and may therefore be an independent effect of abstinence from nicotine. Notably, increased sleep drive, suggestive of increased sleepiness, is consistent with human reports of increased daytime sleepiness during quitting attempts (Prosise et al., 1994).
Changes in sleep quantity observed on later days of abstinence differ from those observed on AD1. 24hr REM sleep was decreased on abstinence days two (AD2) and five (AD5). Inactive phase TST was reduced on AD5. These findings are consistent with reports of prolonged sleep disturbance during withdrawal (Jorenby et al., 1996; Shiffman et al., 2006). Reduced relative theta across the entire 24hr period of AD2 and AD5 is consistent with reduced REM sleep quantity. Further, the modest reduction in TST during the inactive phase may be the result of reduced REM.
Although the effects of abstinence from nicotine on sleep quantity were dependent upon the day, a consistent disruption of sleep architecture was observed across all days of abstinence. On all days of abstinence, relative to pre-nicotine baseline, sleep bout duration (SBD) was decreased (this effect was significant on AD1 and AD5), whereas sleep bout frequency (SBF) was increased. These changes are characteristic of increased sleep fragmentation, or an increase in arousals between periods of sleep. These finding are highly consistent with the human literature. During periods of abstinence, smokers commonly report an increase in the number and duration of awakenings (Hatsukami et al., 1984; Hatsukami et al., 1988; Shiffman et al., 1995; Grove et al., 2006; Jaehne et al., 2015). PSG studies confirm increased sleep fragmentation following nicotine cessation (Prosise et al., Jaehne et al., 2015). In the current study, during the time in which sleep is fragmented, an increase in relative delta power is observed. Increased slow wave activity has previously been associated with sustained sleep fragmentation (Baud et al., 2015) and is likely a function of increased sleep pressure and a more rapid entrance into the deeper stages of NREM sleep.
Overall these data suggest a negative impact of nicotine administration and abstinence from nicotine on measures of sleep quantity and architecture. The primary effect of nicotine administration was to reduce sleep quantity during the active phase. Additionally, numerous changes to the power spectrum were observed during nicotine administration. An increase in NREM sleep was observed during the active phase on AD1, but the primary effect of abstinence was to fragment sleep during the inactive phase. Fragmented sleep during abstinence was accompanied by a number of changes to the power spectrum.
Importantly, this study describes the first animal model of alterations to behavioral and physiological correlates of sleep and wakefulness during chronic nicotine administration and abstinence from nicotine. It should be noted that this study specifically utilizes male mice and, therefore, does not address known sex differences in sleep disturbances between men and women in nicotine withdrawal (Wetter et al., 1999; Weinberger et al., 2016). Nonetheless, this model closely resembles overall human reports and thus provides a novel method to help address the confounds and conflicts between self-report and polysomnography (PSG) measurers of sleep disturbances. Additionally, similarities between this mouse model and human reports along with comparable brain mechanisms controlling sleep timing, structure, depth and duration (Patterson, Nutt, & Wilson, 2011) support the utility of this model to examine the molecular and genetic mechanisms that modulate the relationship between sleep, chronic nicotine exposure and nicotine abstinence, knowledge that is lacking in the current literature. This model also provides a framework to examine the efficacy of current and novel sleep aids on abstinence and withdrawal induced sleep disturbances. Independent of withdrawal, reduced and disturbed sleep leads to many of the same symptoms that define the nicotine withdrawal syndrome. This raises the question: do sleep disturbances during withdrawal cause or exacerbate other symptoms of withdrawal, and if we improve sleep during withdrawal will we simultaneously improve the other symptoms? Finally, an important implication of the current study is that efforts aimed at understanding or improving the nicotine withdrawal syndrome should evaluate and consider the presence of disturbed sleep.
Acknowledgments
Funding Sources:
Grant #: T32DA017637-12, National Institute on Drug Abuse
Title: Research Training – Genetics of Substance Abuse.
PI: John K. Hewitt.
Footnotes
Conflict of Interest Statement
On behalf of all authors, the corresponding author states there is no conflict of interest.
Contributor Information
Hunter L. Mathews, The University of Colorado Boulder, Department of Psychology and Neuroscience, Institute for Behavioral Genetics, 1480 30th Street, Boulder, CO 80309.
Jerry A. Stitzel, The University of Colorado Boulder, Department of Integrative Physiology, Institute for Behavioral Genetics, 1480 30th Street, Boulder, CO 80309.
References
- Aguirre CG., Madrid J, & Leventhal AM (2015) Tobacco withdrawal symptoms mediate motivation to reinstate smoking during abstinence. J Abnorm Psychol 124(3):623–634. http://doi:10.1037/abn0000060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud MO, Magistretti PJ, Petit JM (2015). Sustained sleep fragmentation induces sleep homeostasis in mice. Sleep, 38(4) 567–579. doi: 10.5665/sleep.4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW (2012). Control of sleep and wakefulness. Physiol Rev, 92(3) 1087–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorvatn B, Fagerland S, Ursin R (1998). EEG power densities (0.5–20 Hz) in different sleep-wake stages in rats. Physiol. Behav 63, 413–417 10.1016/S0031-9384(97)00460-5 [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (2011). Quitting Smoking Among Adults – United States – 2001-2010. Morbidity and Mortality Weekly Report, 60(44), 1513–1519. [PubMed] [Google Scholar]
- Clasadonte J, McIver SR., Schmitt LI, Halassa MM, Haydon PG (2014). Chronic sleep restriction disrupts sleep homeostasis and behavioral sensitivity to alcohol by reducing the extracellular accumulation of adenosine. J Neurosci, 34(5) 1879–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colrain IM, Trinder J, & Swan GE (2004). The impact of smoking cessation on objective and subjective markers of sleep: review, synthesis, and recommendations. Nicotine Tob Res, 9(6), 913–925. [DOI] [PubMed] [Google Scholar]
- Corwin EJ, Klein LC, Rickelman K (2002) Predictors of fatique in healthy young adults: moderating effects of cigarette smoking and gender. Biological Research for Nursing, 3(4) 163–164. [DOI] [PubMed] [Google Scholar]
- Cummings KM, Giovino G, Jaén CR, & Emrich LJ (1985). Reports of smoking withdrawal symptoms over a 21 day period of abstinence. Addictive Behaviors, 10(4), 373–81. [DOI] [PubMed] [Google Scholar]
- Crocq MA (2003). Alcohol, nicotine, caffeine, and mental disorders. Dialogues Clin Neurosci, 5(2) 175–185). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domino et al. (2009). Tobacco smoking produces widespread dominant brain wave alpha frequency increases. Int J Psychophysiol, 74(3) 192–198. doi: 10.1016/j.ijpsycho.2009.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst M, Heishman SJ, Spurgeon L, London ED (2001). Smoking history and nicotine effects on cognitive performance. Neuropsychopharmacology, 25(3) 313–319. [DOI] [PubMed] [Google Scholar]
- Foulds J, McSorley K, Sneddon J, Feyerabend C, Jarvis MJ, Russell MA (1994) Effect of subcutaneous nicotine injections on EEG alpha frequency in non-smokers: a placebo-controlled pilot study. Psychopharmacology (Berl), 115(1-2), 163–166. [DOI] [PubMed] [Google Scholar]
- Foulds J, Stapleton J, Swettenham J, Bell N, McSorley K, Russell MAH (1996). Cognitive performance effects of subcutaneous nicotine in smokers and never-smoker. Pyschopharmacology, 127, 31–38. [DOI] [PubMed] [Google Scholar]
- Grabus SD, Martin BR, Batman AM, Tyndale RF, Sellers E, Damaj MI (2005). Nicotine physical dependence and tolerance in the mouse following chronic oral administration. Psychpharmacology (Berl), 178(102) 183–192. [DOI] [PubMed] [Google Scholar]
- Grove JR, Wilkinson A, Dawson B, Eastwood P, & Heard P (2006). Effects of exercise on aspects of sleep during tobacco withdrawal. Australian Psychologist, 41(1), 69–76.doi: 10.1080/00050060500395127 [DOI] [Google Scholar]
- Hamidovic A, de Wit H (2009). Sleep deprivation increases cigarette smoking. Pharmacol Biochem Behav, 93(3):263–9. http://doi:10.1016/j.pbb.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatsukami D, Hughes JR, Pickens R (1985). Characterization of tobacco withdrawal: physiological and subjective effects. NIDA Res Monogr, 53, 56–67. [PubMed] [Google Scholar]
- Hatsukami DK., Dahlgren L, Zimmerman R, Hughes JR (1988). Symptoms of tobacco withdrawal from total cigarette cessation versus partial cigarette reduction. Psychopharmacology, 94(2), 242–247. [DOI] [PubMed] [Google Scholar]
- Hughes JR, Hatsukami DK, Mitchell JE, & Dahlgren LA (1986). Prevalence of smoking among psychiatric outpatients. American Journal of Psychiatry, 143, 993–997. [DOI] [PubMed] [Google Scholar]
- Hughes JR (2007). Effects of abstinence from tobacco: Valid symptoms and time course. Nicotine and Tobacco Research 9(3), 315–327. http://doi:10.1080/14622200701188919 [DOI] [PubMed] [Google Scholar]
- Institute of Medicine (US) Committee on Sleep Medicine and Research (2006). Sleep disorders and sleep deprivation: an unmet public health problem. National Academic Press, Washington (DC). [PubMed] [Google Scholar]
- Jaehne A, Loessl B, Bárkai Z, Riemann D, & Hornyak M (2009). Effects of nicotine on sleep during consumption, withdrawal and replacement therapy. Sleep Medicine Reviews, 13(5), –377. 10.1016/j.smrv.2008.12.003 [DOI] [PubMed] [Google Scholar]
- Jaehne A, Unbehaun T, Feige B, Cohrs S, Rodenbeck A, & Riemann D (2015). Sleep changes in smokers before, during and 3 months after nicotine withdrawal. Addiction Biology, 20(4), 747–755. 10.1111/adb.12151 [DOI] [PubMed] [Google Scholar]
- Jorenby DE, Hatsukami DK, Smith SS, Fiore MC, Allen., S, Jensen, J, Baker, TB (1996). Psychopharmacology (Berl). 128(2) 130–138. [DOI] [PubMed] [Google Scholar]
- Johnson RF, Beltz TG, Thunhorst RL, Johnson AK (2003). Investigations on the physiological controls of water and saline intake in C57BL/6 mice. Am J Phyiol Regul Integr Comp Physiol, 285(2), 394–403. [DOI] [PubMed] [Google Scholar]
- Kim SC., Lee MH, Jang C, Kwon JW, Park JW (2013). The effect of alpha rhythm sleep on EEG activity and individuals’ attention. J Phys Ther Sci, 25(12) 1515–1518. doi: 10.1589/jpts.25.1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott VJ, Harr A, Ilivitsky V, Mahoney C (1998). The cholinergic basis of the smoking induced EEG activation profile. Neuropsyschobiology, 38(2), 97–107. [DOI] [PubMed] [Google Scholar]
- Kumari V et al. (2003). Cognitive effects of nicotine in humans: an fMRI study. Neuroimage, 19(3), 1003–1013. 10.1016/S1053-8119(03)00110-1 [DOI] [PubMed] [Google Scholar]
- Lena C, Popa D, Grailhe R, Escourrou P, Changeux JP, Adrien J (2004). Beta2 containing nicotinic receptors contribute to the organization of sleep and regulate putative micro-arousals in mice. J Neurosci 24: 5711–5718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin DH, & Goyarzu P (2010). Rodent models of nicotine withdrawal In Henningfield JE et al. (Eds.). Nicotine psychopharmacology, handbook of experimental pharmacology (pp. 401–434). Berlin: Springer-Verlag. [DOI] [PubMed] [Google Scholar]
- McKenna et al. , (2008). Assessing sleepiness in the rat: a multiple sleep latencies test compared to polysomnographic measurers of sleepiness. J Sleep Res, 17(4) 365–375. doi: 10.1111/j.13652869.2008.00686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medic G Wille M, Hemels MEH (2017). Short- and long-term health consequences of sleep disruption. Nat Sci Sleep, 9, 151–161. http://doi:10.2147/NSS.S134864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun ML, Levine MD, Patricia H, Perkins KA, Marcus MD (2011). Subjective sleep disturbance during a smoking cessation program: associations with relapse. Addict Behav 36(8), 861–864. 10.1016/j.addbeh.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrott AC (1993) Cigarette smoking: effects upon self-rated stress and arousal over the day. Addict Behav, 18(4), 389–395. [DOI] [PubMed] [Google Scholar]
- Patterson LM, Nutt DJ, & Wilson SJ (2011). Sleep and its disorders in translational medicine. J Psychopharmacology, 25(9) 1226–1234. doi: 10.1177/0269881111400643. [DOI] [PubMed] [Google Scholar]
- Pietila K, Ahtee L (2000). Chronic nicotine administration in the drinking water affects the striatal dopamine in mice. Pharmacol Biochem Beha, 66(1), 95–103. [DOI] [PubMed] [Google Scholar]
- Prosise GL, Bonnet MH, Berry RB, & Dickel MJ (1994) Effects of abstinence from smoking on sleep and daytime sleepiness. Chest, 105, 1136–1141. [DOI] [PubMed] [Google Scholar]
- Rechtschaffen A, Bergmann BM, Gilliand MA, Bouer K (1999). Effects of method, duration, and sleep stage on rebound from sleep deprivation in the rat. Sleep, 22(1) 11–31. [DOI] [PubMed] [Google Scholar]
- Salin-Pascual RJ, Moro-Lopez ML, Gonzalez-Sanchez H, & Blanco-Centurion C (1999). Changes in sleep after acute and repeated administration of nicotine in the rat. Psychopharmacology, 145(2), 133–138. 10.1007/s002130051041 [DOI] [PubMed] [Google Scholar]
- Shiffman S, Paty JA, Gnys M, Kassel JD, & Elash C (1995). Nicotine withdrawal in chippers and regular smokers: Subjective and cognitive effects. Health Psychology, 14(4), 301–309. [DOI] [PubMed] [Google Scholar]
- Shiffman S et al. (2006). Natural history of nicotine withdrawal. Addiction (Abingdon, England), 101(12), 1822–32. https://doi:10.1111/j.1360-0443.2006.01635.x [DOI] [PubMed] [Google Scholar]
- Soldatos CR , Kales JD, Scharf MB, & Bixler EO, Kales A (1980). Cigarette smoking associated with sleep difficulty. Science, 207(4430), 551–553. [DOI] [PubMed] [Google Scholar]
- Sparks JA, & Pauly JR (1999) Effects of continuous oral nicotine administration on brain nicotinic receptors and responsiveness to nicotine in C57Bl/6 mice. Psychopharmacology (Berl), 141(2) 145–153. [DOI] [PubMed] [Google Scholar]
- Swan GE & Lessov-Schlaggar CN (2007). The effects of tobacco smoke on nicotine cognition and the brain. Neuropsychol Rev, 17(3) 259–273. [DOI] [PubMed] [Google Scholar]
- Vyazovskiy VV, Tobler I (2005). Theta activity in the waking EEG is a marker of sleep propensity in the rat. Brain Res, 1050(1–2), 64–71. [DOI] [PubMed] [Google Scholar]
- Weinberger AH, Platt J, Shuter J, Goodwin RD (Gender Differences in self-reported withdrawal symptoms and reducing or quitting smoking three years later: A prospective, longitudinal examination of U.S. adults. Drug Alcohol Depend, 165, 253–259. doi: 10.1016/j.drugalcdep.2016.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesnes KA, Edgar CJ, Kezic I, Salih HM, de Boer P (2013) Effect of nicotine withdrawal on cognition in a clinical trial setting. Psychopharmacology (Berl), 229(1) 133–140. doi: 10.1007/s00213-013-3089-6 [DOI] [PubMed] [Google Scholar]
- Wetter DW, & Young TB (1994). The relation between cigarette smoking and sleep disturbance. Preventive Medicine 23, 328–334. 10.1006/pmed.1994.1046 [DOI] [PubMed] [Google Scholar]
- Wetter DW, Fiore MC, Young TB, McClure JB, de Moor CA (1999). Gender differences in response to nicotine replacement therapy: Objective and subjective indexes of tobacco withdrawal. Experimental and Clinical Psychopharamacology. 7(2) 135–144. doi: 10.1037/1064-1297.7.2.135 [DOI] [PubMed] [Google Scholar]
- Zhang L, Samet J, Caffo B, Punjabi NM (2006). Cigarette smoking and nocturnal sleep architecture. American Journal of Epidemiology,164(6):529–37 [DOI] [PubMed] [Google Scholar]
- Zhang L, Samet J, Caffo B, Bankman I, Punjabi NM (2008). Power spectral analysis of EEG activity during sleep in cigarette smokers. Chest, 133(2) 427–32. [DOI] [PubMed] [Google Scholar]
- Zhou X, Nonnemaker J, Sherrill B, Gilsenan AW, Coste F, & West R (2009). Attempts to quit smoking and relapse: Factors associated with success or failure from the ATTEMPT study. Addictive Behaviors, 34(4), 365–373. 10.1016/j.addbeh.2008.11.013 [DOI] [PubMed] [Google Scholar]